
Profiling protein targets of cellular toxicant exposure
Joseph C.
Genereux

Department of Chemistry, University of California, Riverside, CA 92521, USA. E-mail: josephg@ucr.edu
First published on 13th January 2023
Abstract
Environmental agents of exposure can damage proteins, affecting protein function and cellular protein homeostasis. Specific residues are inherently chemically susceptible to damage from individual types of exposure. Amino acid content is not completely predictive of protein susceptibility, as secondary, tertiary, and quaternary structures of proteins strongly influence the reactivity of the proteome to individual exposures. Because we cannot readily predict which proteins will be affected by which chemical exposures, mass spectrometry-based proteomic strategies are necessary to determine the protein targets of environmental toxins and toxicants. This review describes the mechanisms by which environmental exposure to toxins and toxicants can damage proteins and affect their function, and emerging omic methodologies that can be used to identify the protein targets of a given agent. These methods include target identification strategies that have recently revolutionized the drug discovery field, such as activity-based protein profiling, protein footprinting, and protein stability profiling technologies. In particular, we highlight the necessity of multiple, complementary approaches to fully interrogate how protein integrity is challenged by individual exposures.
 Joseph C. Genereux | Dr Joseph C. Genereux is an Assistant Professor of Chemistry at the University of California, Riverside. His research group develops mass spectrometry-based bioanalytical assays to characterize misfolding and mistrafficking of cellular proteins in response to stress. He received his PhD in Chemistry from California Institute of Technology, followed by NIH and AHA postdoctoral fellowships at The Scripps Research Institute investigating protein homeostasis pathways. |
I. Environmental exposure and protein integrity
The world is a dangerous place. A living cell is constantly exposed to molecules that challenge its ability to maintain its function. Exogenous toxins and toxicants that bind to or chemically modify lipids, nucleic acids, and proteins can deprive the cell of the endogenous functions of these biomolecules, while simultaneously introducing new deleterious functions. We know the general mechanisms by which exposure can affect protein function, or introduce new functions. For many of the prominent exposure agents, including herbicides, pesticides, tobacco byproducts, and industrial chemicals, we have good understanding of their chemical reactivity. However, the chemical diversity of proteins that enables their functional diversity also makes it challenging to determine which proteins will be affected by which chemicals. This complication is exacerbated by the potential for indirect effects mediated by secondary chemical, metabolites, and engagement of cellular signaling pathways. In this sense, the sensitivity of proteins to environmental exposure is more idiosyncratic than the better understood effects on the genome. This problem is in many ways similar to that faced by the drug screening community, wherein they want to discover the protein target of a bioactive molecule, or to identify potential off-target proteins. The omic technologies that have been developed in the pharmaceutical community thus offer an opportunity for the toxicological community. In this review, I discuss how environmental agents can affect protein integrity, and the emerging methods to identify the susceptible proteome.II. Types of environmental toxicants
Environmental toxins and toxicants can affect the proteome through direct and indirect mechanisms (Fig. 1). In the direct mechanism, physical binding or conjugation to a protein modulates its activity (Fig. 1A). For example, competitive inhibitors prevent binding of chemical substrates and ligands, agonists and antagonists directly modulate protein function, and allosteric binders modulate activity and substrate binding affinities. Chemical modifications can also affect protein stability. Misfolded protein is associated with loss of function due to deprivation of the folded protein, and gain of function through the production of toxic states, such as oligomers and aggregates. Environmental toxins can also perturb the cell through indirect mechanisms. Both microsomal and non-microsomal processes modify toxins to generate metabolites, some of which have enhanced chemical reactivity and greater potential to damage proteins.1 Heavy metals and redox cyclers generate reactive oxygen species (ROS), such as superoxide, that can then directly react with biomolecules to change their chemical structure (Fig. 1B). Binding of a small molecule to one member of a complex can deprive the rest of the complex of that member (Fig. 1C). The structures, stabilities, and functions of the other protein members of that complex will then be changed, despite none of those proteins directly binding to the exposure agent. If an exposure severely destabilizes a population of proteins, the misfolded proteins can titrate chaperones away from the native clients (Fig. 1D). These client proteins will then lose stability, inhibiting both their native functions and potentially contributing to disrupted cellular proteostasis. If proteostasis is unable to recover, persistent stress response pathway activation can lead to apoptosis.2–4 Another indirect mechanism is the engagement of endogenous signaling pathways (Fig. 1E). As an example, halogenated aromatics and other endocrine disruptors affect signaling pathways far downstream of their directing binding targets.5 | ||
| Fig. 1 Mechanisms of protein dysregulation subsequent to environmental exposure. (A) A protein's activity can be modulated by direct binding to an exposure agent. (B–E) Indirect mechanisms by which environmental exposure can modulate protein function and levels in the cell. | ||
For each of these mechanisms, toxicity is often mediated by the persistent engagement of cellular stress responses. Similarly to how high levels of DNA adducts lead to persistent activation of the DDR,6 the continued presence of misfolded protein in the endoplasmic reticulum (ER) induces chronic UPR activation.7 Cellular remodeling during these responses can protect the cell by upregulating biomolecular repair and degradation machinery. In the absence of resolving the underlying stress, stress responses eventually activate apoptosis leading to cell death.8,9
III. Protein damage
A. Ligand binding
Proteins have evolved to bind. Enzymes bind their substrates, trafficking proteins bind their clients, and scaffolds regulate protein localization through protein–protein interactions. Environmental chemicals that mimic substrates or protein binding partners can exploit these binding pockets and surfaces. This is the mechanism by which organochlorine and pyrethroid insecticides primarily act, binding channel proteins and inhibiting their activities.10 Hormone receptors are prone to activation or inhibition by a variety of exogenous molecules, to the extent that screening for deleterious hormone receptor interactions has become part of standard toxicity panels. Commercial binding assay panels typically offer several dozen receptors with known side effects associated with small molecule binding. Even in the absence of activation or inhibition, binding interactions can impact protein stability.11B. Protein adducts
Proteins harbor multiple nucleophilic sites, including amines at the N-termini and lysine residues, thiols at cysteine residues, carboxylates at C-termini and aspartate and glutamate residues, alcohols at serine, threonine, and tyrosine, and endocyclic nitrogens in histidine and tryptophan.1,12 Endogenous modification of these sites is highly regulated and plays a central role in cellular signaling pathways and establishing the functional state of a cell.13 However, exogenous agents can also react with and modify these sites, generating protein adducts.14 A few examples are in Fig. 2A. For example, tobacco smoke contains a wide range of potent electrophiles, including conjugated aldehydes, polycyclic aromatics, nitrosamines, and epoxides.15 Enzymes rely upon pK-perturbation of active site residues,16 increasing the population of nucleophilic ionization states for cysteine, lysine, and serine. Such sites and their enzymatic function are particularly sensitive towards exposure to electrophiles17 (Fig. 2B). As a consequence, enzymes with strongly nucleophilic active site cysteines feature prominently in known protein targets of environmental chemicals. | ||
| Fig. 2 (A) Examples of common electrophilic exposure agents. BDPE is a metabolic product of benzo[a]pyrene. (B) Mechanism of chloroacetamide modification at cysteines. Note that the ionized thiolate is the active nucleophile. (C) Mechanism for oxidation of thiols by hydrogen peroxide to the sulfenate. | ||
Pesticides, insecticides, and herbicides are used on a wide scale in support of global agriculture, and thus need to be highly potent at inhibiting their biological targets, while simultaneously cheap to produce. Not surprisingly, some of the most popular herbicides and pesticides tend to be covalent inhibitors featuring potent electrophilic activity. The most widely used insecticides include organophosphates and carbamates, while popular herbicides include glyphosate (metabolizes to the aldehyde glyoxylate), atrazine, and chloroacetanilides (e.g. alachlor). All of these molecules are potent electrophiles that can modify proteins.18–21
C. Protein oxidation
Proteins harbor multiple sites that are subject to oxidation. The most sensitive positions are the cysteine thiols and methionine thioethers. Oxidation of cysteine thiols to sulfenic (RSOH; transient and reversible), sulfinic (RSO2H) or sulfonic (RSO3H) acids alters their chemistry through an increase in size, pH-dependent loss of sulfur nucleophilicity, and increased local electron density.22 Oxidation of two cysteine residues to a disulfide forms a crosslink, sharply reducing protein entropy and shifting the conformational space available to the protein.23 Methionine does not form oxidative crosslinks, but is readily oxidized to the sulfoxide (RS(O)R′). These oxidations can be performed directly by the exposure agent, by ROS generated by the exposure agent (e.g. Fenton chemistry), or by ROS produced from metabolic enzymes perturbed by the exposure agent (e.g. paraquat) (Fig. 2C). As is the case with other protein post-translational modifications, protein oxidation is exploited by well-regulated signaling pathways.24,25 For example, hydrogen peroxide activates ERK1/2 by inhibiting its dephosphorylation, which then engages the MAPK pathway to promote cellular growth.26–29 In addition to cysteines and methionine, protein co-factors such as iron–sulfur clusters and zinc finger motifs are subject to chemical modification by oxidation.30,31 For example, oxidation of high-potential iron–sulfur clusters can lead to decomposition of the cluster and subsequent complete inactivation of the protein.32To protect cellular redox balance, dedicated pathways maintain redox buffering molecules such as glutathione and oxidoreductases.33,34 Endogenous and exogenous oxidants can perturb this balance. Endogenous oxidants include byproducts of metabolic redox pathways, including respiration.35 Detoxification enzymes such as monoamine oxidases and cytochrome C oxidases generate peroxides.36,37 Activated neutrophils generate peroxide and hypochlorous acid as part of the immune response.38 Exogenous oxidants include heavy metals and redox cyclers. Through direct protein binding,39,40 arsenic and cadmium dysregulate cellular mechanisms that prevent the accumulation of ROS.41,42 Iron and copper catalyze Fenton chemistry, directly producing superoxide from peroxide.43 The popular herbicide paraquat is a redox cycler that both inhibits metabolic enzymes and generates superoxide.44
D. Protein homeostasis
Protein function does not solely depend on chemical structure. Rather, the three-dimensional arrangement of atoms has a profound effect on protein activity. Nascent proteins typically fold to a thermodynamically stable structure with well-defined conformational distributions and dynamics.45,46 It is this specific combination of structure and dynamics that enables the protein to bind its ligands or partners and perform its chemistry. Chemical modifications that do not affect the active site can still disrupt protein function if they change the thermodynamics between the folded and misfolded states, or change the conformational dynamics of the protein.In addition to loss of function, protein misfolding can induce proteotoxic gain of function. Misfolded proteins present non-native binding surfaces that can affect the function of other proteins, or disrupt the integrity of cellular membranes. If these surfaces are hydrophobic, misfolded proteins can aggregate. Aggregates can titrate chaperones, leading to disfunction in pathways that rely on those chaperones, and ultimately proteostasis collapse.47,48 As a defense, cells are adept at segregating aggregates and degrading misfolded proteins through quality control pathways.49 Although these processes defend against toxic gain of function, they can exacerbate loss of function. For example, destabilizing mutations in CFTR that only moderately decrease protein activity are sufficient for quality control pathways to degrade nearly all of the protein, leading to profound loss of function and consequent disease.50 The relationship between protein thermodynamic stability and degradation is so strong that degradation rate and stability are frequently used interchangeably in the literature.51
IV. Methods for characterizing protein damage
The simplest way to assess the potential of an agent of exposure to directly modify biomolecules is to incubate isolated lipids, oligonucleotides, and peptides with the agent and look for reaction products. For greater physiological relevance, proteins can be extracted from tissue following either organismal or tissue exposure, digested to peptides, and the modification products quantified by LC-MS.52 The results of such studies are generally consistent with the known reactivity of functional groups. Three properties contribute to the relative reactivity of protein residues: nucleophilicity, hardness, and reversibility. Nucleophilicity depends on pKa. Cysteine has a lower pKa than lysine, such that the cysteine thiolate/thiol ratio at neutral pH is much greater than the lysine amine/ammonium ratio. Glutamate and aspartate have lower pKas, and are almost solely present as oxyanions at neutral pH, but are poor nucleophiles due to resonance stabilization.53 Hardness/softness theory postulates that hard (less polarizable) electrophiles react most readily with hard nucleophiles, while soft (more polarizable) electrophiles react most readily with soft nucleophiles.54,55 Cysteine is soft, as are maleimides, leading to selective conjugation. Diols are hard, as are amines, leading to selective conjugation to form pyrroles. Finally, partially reversible reactions will have a lower steady state yield of protein adduction.For DNA, chemical reactivity of short sequence motifs in small oligonucleotides strongly predicts reactivity inside long double stranded regions. However, chemical reactivity of a functional group inside proteins or in RNA has a large dynamic range, driven largely by local differences in solvent accessibility and pKa. In a seminal study, lysine was systematically placed at 25 different positions of a staphylococcal nuclease,56 and its pKa measured. The apparent pKa varied by 4 units, but importantly the technique could not detect extreme values, implying that the true variation in just a single protein is far greater. In another illustration, the Nomura group measured the proteome-wide reactivity of several NHS-esters.57 They found that labeling was highly dependent on ancillary regions of the molecule, demonstrating that even molecules with highly reactive functional groups have proteome-wide selectivity. Similar selectivity has been found for epoxides,17 fluorosulfonates,58,59 and a variety of aminophiles.60 In the face of the incredible chemical diversity of biomolecules in living cells, the only way to evaluate protein sensitivity to an exposure agent is through direct application of omic methods.
A. Transcriptional methods
To avoid the risk of proteotoxicity, all subcellular environments have mechanisms to sense the accumulation of misfolded proteins.8 In most cases, a protein sensor is maintained in an inactive state by direct binding to one or more chaperones.7,61,62 If misfolded protein accumulates, then the chaperones are titrated from the sensor, initiating a signaling cascade that ultimately results in a transcriptional program. Monitoring these transcriptional targets of misfolded protein response pathways can thus serve as a proxy for measuring the presence of misfolded proteins. This approach is the most established method for determining whether and under what conditions protein misfolding occurs in the cell.63There are four general approaches to measure stress responses at the transcriptional level. Insertion of a luminescent or fluorescent reporter downstream of a stress responses activator reports on increased transcriptional activity at that activator.64,65 RT-PCR or Northern Blotting of a set of transcriptional reporter genes specific to the stress response can be performed.66 These two approaches can incorrectly assign stress responses due to overlap between individual stress responses and target genes.67 Full transcriptomics through RNAseq can be better matched with the individual transcriptional programs of individual stress responses, at a far higher cost. Recently, a targeted RNAseq approach has been described that effectively deconvolutes multiple transcriptional programs to identify which stress responses are activated following cellular exposure.68 In this approach, 150 genes that together reflect orthogonal fingerprints between major cellular stress responses are amplified from cDNA, prior to next generation sequencing. Although this approach has so far been primarily used to precisely identify the stress pathways activated by pharmacological compounds,69 it has great promise to serve the same purpose for environmental toxicants.
Transcriptional approaches have been used to characterize stress response activation in response to cellular exposure to environmental agents.70–74 For example, cadmium (10 μM, 4 h on LLC-PK1 tubular epithelial cells) induces transcriptional programs downstream of the UPR-associated transcription factors PERK, ATF6, and XBP1s;73 similar results are seen in other experiments.75,76 Transcription of heat shock genes, as measured by RNAseq, was used to determine the stress pathways activated by honey bee exposure to paraquat.77 A more targeted approach using RT-PCR assessed the effect of midge exposure to chlorpyrifos and endosulfan on heat shock response activation.78,79 It is surprising that there is a relative lack of systematic studies on which common environmental toxins induce stress responses in human cells. It is possible that emergence of the target-identification approaches described below will increase the interest in identifying which exposures, and under which conditions, activate misfolded protein stress responses.
B. Activity-based protein profiling
For exposure agents that directly modify a protein target (Fig. 1A), activity-based protein profiling (ABPP) is an effective way to identify both the modified proteins and the site of the adduct.80 Two elements are necessary for an ABPP probe: a reactive group that irreversibly conjugates to the protein target, and an affinity handle for purification. To minimize perturbation of protein binding to the native molecule, the most common affinity handle is a terminal alkyne. Cellular or organismal lysate, live cells, isolated tissue, or live animals are exposed to the probe, biotinylated azide clicked to the probe through copper-assisted cyclization, and the affinity purified proteome identified and quantified through mass spectrometry. Isotope-coded biotin-azides and desthiobiotin-azides are widely available to improve quantification.81,82 Digestion prior to avidin purification enables concentration of peptides harboring the modification, and hence greater identification of modified sites on proteins, as opposed to modified proteins. An alternative setup uses a generic probe that promiscuously targets a specific residue or structural motif (isoTOP-ABPP). In this setup, large number of molecules can be screened for their ability to compete with the probe for binding, and the molecules screened neither need chemical modifications nor do they need to bind covalently.Both setups have been used by Nomura and colleagues to identify protein targets of diverse herbicides, pesticides, and other toxicants.83,84 Because ABPP does not require genetic encoding, it is readily performed in vivo, ex vivo, in cellulo, and in vitro. In one study, they identified a series of fatty acid metabolic enzymes as hepatic targets of the herbicide acetochlor that was intraperitoneally administered to mice85 (Fig. 3A). Conjugation of acetochlor to active-site cysteine residues leads to enzymatic inhibition and consequent perturbation of the metabolome, which was validated in living mice (Fig. 3B). Some of these same targets are also inhibited by intraperitoneal injection of the herbicide glyphosate, again due to conjugation at active site cysteines with consequent metabolic disruption.86 In this case, however, the reactive species is a metabolite of glyphosate, glyoxylate. It is notable that the use of a promiscuous cysteine probe in competition with glyphosate, rather than the conversion of glyphosate into a probe, meant that natural metabolism of glyphosate was unperturbed. In this context, use of a probe-modified glyphosate might have not found these targets. On the other hand, modification of other residues besides those targeted by the promiscuous probe cannot be profiled using the indirect competition approach. Once targets have been determined, ABPP can be used to characterize target activity following treatment.87 If an antibody is available for adducts, then modified proteins can be directly identified by immunostaining following 2-dimensional gel electrophoresis.18,19 That approach was used to determine proteins modified by murine exposure to the atrazine metabolite diaminochlorotriazine, followed by mass spectrometric identification of protein spots. Modified peptides were not directly observed in this case.
 | ||
| Fig. 3 (A) The isoTOP-ABPP approach as applied by Counihan et al. to determine the hepatic targets of acetochlor in living mice. (B) Their identification of fatty acid oxidation enzymes Acaa1, Acaa2, and Scp2 allowed them to construct specific hypotheses as to how metabolite levels should be perturbed by acetochlor exposure. These hypotheses were validated by isotope tracing studies and metabolomic studies of livers from treated mice. Reprinted (adapted) with permission from Counihan et al., ACS Chem. Biol., 12, 635. Copyright 2017 American Chemical Society. | ||
Competitive electrophilic ABPP is not limited to promiscuous cysteine probes. Using probes that are specific for an individual class of proteins simplifies the recovered proteome, allowing greater proteomic coverage of the target class. A probe that is specific to glutathione S-transferase (GST) was thus used to identify which GSTs are inhibited by exposure to the polycyclic aromatic hydrocarbon benzo[a]pyrene.88 Similarly, probes against CYP enzymes have been effective for identifying which CYPs are sensitive to individual PAHs and to tobacco smoke exposure.89–91 A notable study found that the murine gut microbiome strongly impacted the sensitivity of microsomal CYP expression and activity to PAH exposure.91 This library approach was also successfully used to identify CYPs responsible for metabolism of pyrethroid insecticides.92
The activity based-protein profiling approach is not limited to electrophiles. Several groups have developed probes to discover protein targets of metal complexes harboring gold,93–95 arsenic,96–98 and platinum.99 An interesting discovery found using this approach in A549 cells is that strongly arsenic-binding proteins are dominated by anti-oxidant proteins such as thioredoxin and peroxiredoxin.96 It should be noted that metal binding to protein targets can be non-covalent or weakly covalent. While the use of non-clickable probe controls can help lower false positives, ABPP metal probes have not yet demonstrated the scope of paired electrophoresis-ICP-MS or microarray approaches.100,101
The identification of cysteine oxidation products is another ABPP approach that is relevant to understanding the effects of environmental exposure on proteins. These technologies build off of earlier enrichment-free approaches for profiling cysteine oxidation states.102,103 The Carroll group and others have developed a series of chemical probes that are specific between thiols, sulfinic acids, sulfonic acids, sulfenic acids, disulfides and other sulfur oxidation products.22,104–107 In a recent application, three click probes specific to thiols, sulfenic, and sulfinic acids were used to isolate peptides with the specific cysteine states from C. elegans lysates (Fig. 4A).105 The authors found that 1537 proteins showed a meaningful increase in oxidized thiol sites when the animals were exposed to peroxide prior to lysis (Fig. 4B). GO ontological analysis confirms that metabolic pathways are among the most enriched (Fig. 4C), consistent with the typical targets of cysteine-targeting ABPP experiments.
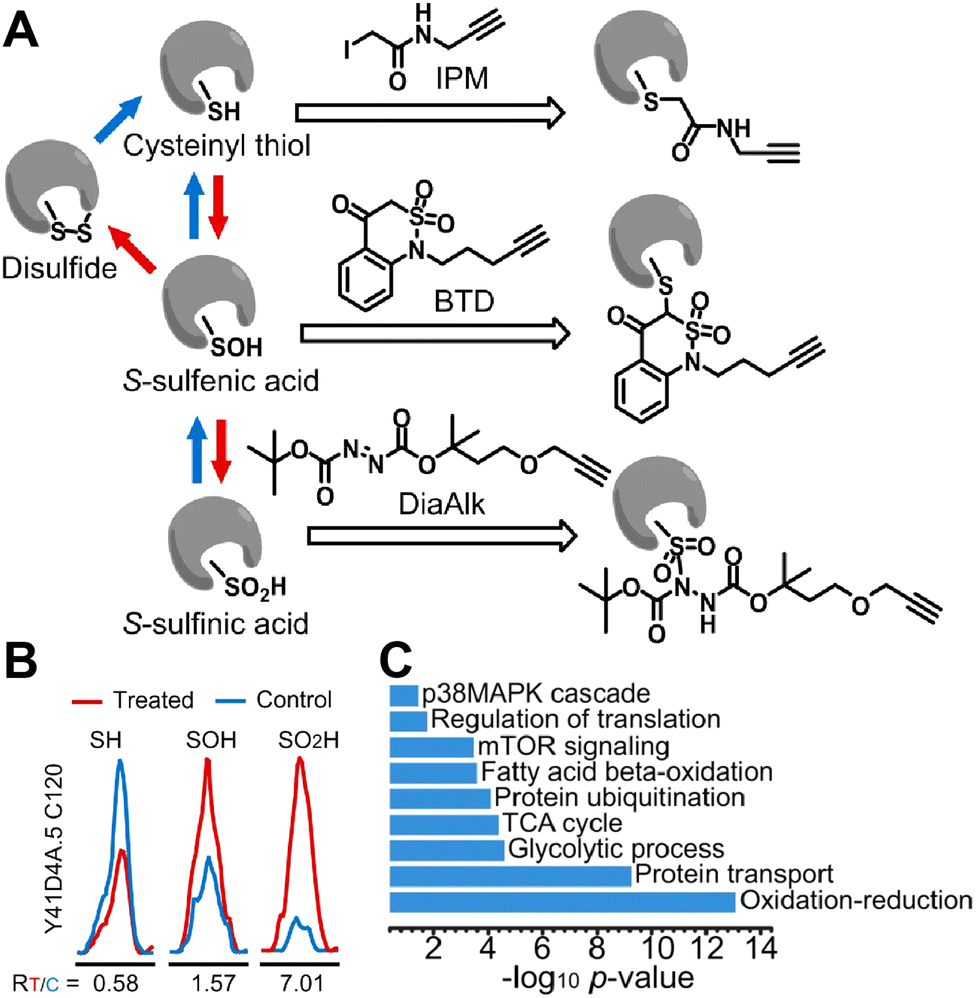 | ||
| Fig. 4 Redox profiling of cysteines. (A) Chemical probes specific to cysteine oxidation species. (B) Change in levels of SH, SOH, and SO2H states of Y41D4A.5C120 in C. elegans lysate from animals treated with H2O2 (5 mM, 5 min) vs. control. (C) GO annotations for pathways enriched in proteins that show redox sensitivity. Adapted from Meng et al., Nat. Commun., 2021, 12, 1415. The Creative Commons license may be viewed at https://creativecommons.org/licenses/by/4.0/. | ||
C. Footprinting methods
If the structure of a protein is changed, the solvent accessibility changes as well. Profiling solvent accessibility through exposure to reactive species provides a map of protein structure108,109 (Fig. 5A). The details of this map depend on the method employed and the depth of coverage. Proteomics is well-suited to these structural experiments due to the low sample mass requirements. The most widely used method for characterizing solvent accessibility is hydrogen deuterium exchange (HDX).110 HDX is mostly limited to relatively purified samples due to the complexity of the data, though increasingly progress has been made in applying this technique to more complicated systems.111–113 | ||
| Fig. 5 (A) Protein footprinting techniques chemically mark solvent accessible regions of the protein. Any changes in protein structure or stability can have a local event on solvent accessibility. (B) Protein fractionation techniques separate proteins based on their destabilization. Solubility profiling techniques determine protein aggregation susceptibility in response to denaturing conditions, while Hsp40 affinity profiling identifies destabilized proteins through co-IP with a chaperone. | ||
Rather than targeting the backbone, reactive species can target the amino acid residues instead. The broadest coverage is achieved with reactive species that can modify all available amino acid side-chains, such as hydroxyl radicals or carbenes.108 The most developed version of this approach is Fast Photochemical Oxidation of Proteins (FPOP).114,115 Hydroxyl radicals are generated using laser photolysis of hydrogen peroxide, and rapidly quenched in a flow set-up combined with actinometry to ensure exquisite control of radical yield and kinetics.116,117 The irreversibility of the modification products, and the ability to identify modification sites using tandem mass spectrometry fragmentation spectra, enable amino-acid localization of modifications more readily than can be done with HDX spectra. Because the spectra are highly complex, identifying peptides from biologically complex media is challenging. Recently, Jones and colleagues developed the technological and computational advances necessary to adapt FPOP to complex biological samples. Experimentally, a robotic controlled flow system ensures precise, reproducible and robust delivery of hydroxyl radicals to living cells (or organisms!).118–120 Computationally, a hierarchical approach allows high-confidence assignment of diverse side chain modifications, overcoming the challenge presented by a spectral library with potential modifications on every amino acid side chain.121 Although this approach has the potential to eventually be the complete solution to most questions about cellular protein structure, currently depth of coverage is still limited and most proteins will only have a few peptides quantified in a single experiment. Given the rapid pace of advance, it is likely that in the next decade, intracellular FPOP could be as standard for characterizing in cellulo and in vivo protein structure as HDX already is for characterizing in vitro structure.
The technical challenges associated with complex proteome analysis are largely alleviated by using methionine oxidation by hydrogen peroxide to measure solvent accessibility, as used in SPROX.109,122–124 Methionine oxidation is irreversible. Because methionine is a relatively rare residue, there are only a few sites per protein, and unmodified methionines can be enriched using bromoacetyl resin,125 allowing much greater depth of proteomic coverage. SPROX is the most mature of the technologies used for characterizing protein structure proteome-wide, having been applied to numerous systems over the course of many years, and benchmarked against alternative technologies.126–130 SPROX is well-validated for providing reliable unfolding free energies for at least proteins that follow two-state unfolding models. Even for proteins that defy two-state transitions, oxidation kinetics are a valid proxy for destabilization.131
Electrophilic species have also been used for protein footprinting in biologically complex samples, including thioimidates,126,132–134 aldehydes,135,136 maleimides,137 and activated esters.138 These probes are limited to nucleophilic residues such as cysteine, lysine, or histidine. With activating agents, the carboxylates aspartate and glutamate can also be profiled,108,139 while carbocations expand labeling to tryptophan, methionine, and serine.140 Dimethyl labeling of lysine with formaldehyde is particularly effective, due to the small size of the reagent and the rapid reaction with lysine.135,136 The product Schiff base can be immortalized through external reductant, and allows straightforward introduction of 13C and 2D isotope labels for multiplexed quantitative proteomics.141
Limited proteolysis is another powerful footprinting method.142,143 Rather than using a small molecule as the reactive species, a promiscuous protease is used. Peptide bonds that are better protected from the protease are cleaved more slowly, or in lower yield. Either fully tryptic peptides or semi-tryptic peptides can be quantified. Conditions that decrease yield of fully tryptic peptides or increase yield of semi-tryptic peptides associated with a protein can then be inferred to destabilize that protein in that specific region. It is important to note that not all peptides are proteotypic; if a protein changes its conformation or dynamics in one region, peptides in unaffected regions will not reflect those changes. The quantification method is critical to the feasibility of this approach as well. Pulse proteolysis was first developed thirty years ago, with protein quantification by gel being used to determine proteolytic susceptibility over a range of denaturants.144 Attempts to translate this approach to mass spectrometry were successful for interrogating the stability of abundant or specific proteins,145–148 but the high chemical complexity challenged the ability to achieve adequate chemical depth using untargeted mass spectrometry. It wasn’t until the development of DIA that pulse proteolysis was extended to a deep-coverage omics method by Picotti and colleagues.143,149 Recently, Fried and colleagues successfully used LiP and DDA to quantify proteolytic susceptibility of over 1000 proteins in E. coli lysate with ≥2 peptides per protein.150,151 To increase depth of coverage while still using DDA, a fractionation method was developed by Fitzgerald and colleagues whereby semi-tryptic peptides are selective labeled by TMT, allowing TMT-based enrichment and isobaric quantification, termed Semi-Tryptic Enrichment for Proteolysis Procedures with Pulse Proteolysis (STEPP-PP).152
This STEPP-PP approach was used to identify changes in protein stability subsequent to environmental exposure to copper (Fig. 6).153 To increase experimental throughput, the one-pot method was used. Bacterial (E. coli) cells were treated with copper (10 μM for 15 min) in the presence of the ionophore pyrithione, followed by proteomics on whole lysate (determining altered protein expression) and STEPP (to determine changes in proteolytic susceptibility and hence protein stability). Far more proteins showed significantly changed stability than expression levels. Two metabolic targets with increased stability, GAPDH and IDH, showed decreased function following copper exposure both in cellular lysates and as recombinant proteins. Furthermore, the ionophore pyrithione was found to destabilize ribosomal proteins.
 | ||
| Fig. 6 (A) Outline of the STEPP-PP approach. TMT labeling of N-termini exposed by the pulse proteolysis protects those semi-tryptic peptides, allowing fully tryptic peptides to be depleted by the NHS-bead incubation. This simplifies the overall sample by removing peptides that are insensitive to pulse proteolysis. (B) Although the brief exposure to copper and ionophore has little effect on protein expression, dozens of proteins show enhanced or decreased tryptic susceptibility, showing profound effects of copper exposure on the stability of the E. coli proteome. Reprinted (adapted) with permission from Weibelhaus et al., ACS Chem. Biol., 16, 214. Copyright 2021 American Chemical Society. | ||
D. Protein stability through fractionation
Fractionation methods serve as an alternative to footprinting methods (Fig. 5B). The most widely applied approaches to fractionate proteins on the basis of their stability exploit the propensity of misfolded proteins to aggregate.154,155 Biological samples can be homogenized or lysed and ultracentrifuged to separate soluble and insoluble proteins. Changes in the homogenization/lysis method or the ultracentrifugation methods strongly influence this fractionation. Because most proteins are soluble, the dynamic range of aggregation in naïve lysates is small. This range can be increased by heating the sample. If a protein's stability is different between two different conditions (e.g. with or without exposure to a ligand), then the extent to which it misfolds and aggregates at different temperatures will also be different. This approach has been widely used to profile individual proteins for several decades, with several different names including Cellular Thermal Shift Assay (CETSA). Building off of recent improvements in mass spectrometric quantitative precision, CETSA has been applied to determine melting temperatures proteome-wide, and termed CETSA by Mass Spectrometry (CETSA-MS) or Thermal Proteome Profiling (TPP).156,157 In the similar method Target Identification by Ligand Stabilization (TILS), the precipitate is profiled as well.158 In most applications of CETSA-MS and TPP, lysate or intact cells are treated, aliquoted, incubated at a range of temperatures, soluble protein recovered and prepped for bottom-up MS, and each sample labeled with a different isobaric tag. Samples corresponding to each temperature are pooled and analyzed in the same instrument run, allowing melting curves to be reconstructed with high precision. Depending on the TMT plex used (currently available up to 18plex) and the number of points desired in the melting curve, up to two conditions can usually be profiled in one run. The major application has been to identify protein targets of drugs, metabolites, and other ligands, but CETSA-MS has been effective at determining protein–protein interactions as well.159 A recent report found that data independent analysis (DIA) also provides good quantitative precision.160 Although the melting temperature is a proxy for protein stability, it does not represent an equilibrium measurement of unfolding. Hence, unlike the critical chaotrope concentration in SPROX or other footprinting techniques, melting temperature does not allow direct determination of ΔGunfolding. In a single example, TILS was successfully applied to identify E. coli protein targets of the polybrominated diphenyl ether metabolite 6-OH-BDE-47.158 The authors validated that one of the targets, FabI, mediates the antibacterial activity of the molecule. A similar thermal shift assay was also reported to validate the protein target of the mycotoxin insecticide Destruxin A in moth ovary cells.161 A challenge with any profiling method is the prioritization of target hits. Fang and colleagues have integrated CETSA analysis of monoethylhexyl phthalate (MEHP) with molecular docking to identify and prioritize hepatic target proteins.162,163 They found that ANAPC5 is a direct target of MEHP, and possibly explains G1 cell cycle arrest induced by this toxin.The large number of temperatures that must be sampled to construct a thermal melting curve, as well as the failure of the melting curves for many proteins to satisfy two-state assumptions, represents a significant challenge to TPP. Gaetani et al. demonstrated that the number of temperatures sampled and experimental throughput can both be dramatically increased through temperature-based Proteome Integral Solubilty Alteration (PISA).164,165 In this approach, intact cells or lysates are treated, aliquoted and challenged against many temperatures. The samples are pooled prior to centrifugation and sample processing for MS. Rather than individually TMT labeling each temperature condition, the entire pool is labeled for a single TMT channel, allowing each TMT to represent a distinct biological condition or replicate. The increase in sample throughput enabled more conditions and biological replicates to be sampled, while decreasing sample requirements. Furthermore, this method avoids the need to curve fit against a predetermined model, as the area beneath the entire melting curve is integrated. This innovation is critical to profiling the complete proteome, as failure to fit to a two-state model excludes a substantial fraction of the proteome from analysis in TPP.166 Because each temperature condition is pooled, two dozen temperatures can be readily sampled, increasing precision without sacrificing throughput.
An alternative approach for determining proteins aggregation propensity is a recently described kosmotropic shift assay.167 Rather than heating the proteome, Beusch et al. varied the divalent zinc concentration. Proteins are salted out with addition of ions such as zinc, and the susceptibility of individual proteins to aggregation serves as a proxy for stability. To maximize throughput and precision, PISA was used for this application. This ion-based approach is notably not redundant with the temperature-based approach. Rather, for the ligand binding cases studied, some common targets are found, while some are found only by one method. A similar approach was taken using solvent-induced denaturation. Van Vranken et al. measured protein aggregation susceptibility in the presence of an acetone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol:acetic acid mixture, termed solvent proteome profiling (SPP).168 SPP denaturation curves and SPP-PISA were both highly effective at determining protein targets of drugs. Hundreds of proteins were identified that are responsive to TPP or SPP but not both, further demonstrating the inherent complementarity of different aggregation susceptibility protein profiling schemes. A recent report by Zhang et al. found that varying pH is a similarly effective method for profiling protein target engagement.169
:ethanol:acetic acid mixture, termed solvent proteome profiling (SPP).168 SPP denaturation curves and SPP-PISA were both highly effective at determining protein targets of drugs. Hundreds of proteins were identified that are responsive to TPP or SPP but not both, further demonstrating the inherent complementarity of different aggregation susceptibility protein profiling schemes. A recent report by Zhang et al. found that varying pH is a similarly effective method for profiling protein target engagement.169
Our laboratory has pursued an alternative fractionation method that relies upon the ability of chaperones to recognize misfolded proteins irrespective of the structural motifs associated with the folded proteins. Over a third of the proteome relies upon the Hsp70 chaperoning system even under basal conditions, with a greater proportion being protected by Hsp70 under stress. Hsp70, however, is involved in many different pathways, and strongly interacts with a large network of co-chaperones. J-Domain (Hsp40) co-chaperones identify misfolded substrates and deliver them to Hsp70. This handoff can be easily prevented with a single site-mutation to the J-domain. After surveying various J-domain proteins, we found that the human J-domain protein mutant, DNAJB8H31Q, strongly binds destabilized proteins allowing their affinity purification.170,171 This binding is resistant to strong detergent buffer, allowing stringent purification conditions prior to quantitative proteomics. The DNAJB8H31Q associated proteome is destabilized compared to the bulk proteome.170 Even brief, mild heat shock induces monotonic increased association, consistent with DNAJB8H31Q affinity reflecting protein destabilization.171
We have applied our assay to several major agents of exposure.146,172 Just 15 min of cellular exposure to arsenic (sodium arsenite; 100 μM) is adequate to induce destabilization of several proteins, primarily RNA binding proteins such as TDP43 and HNRNPA0 and including the pyruvate dehydrogenase component PDHA1146 (Fig. 7). These mostly represent proteins known to decrease function or aggregate in response to arsenic exposure, and were validated by limited proteolysis. Cadmium is like arsenic in that both ligate thiols and induce reactive oxygen species. However, the cadmium profile is markedly different, with the RNA-binding proteins apparently unaffected. We also used this approach to profile several herbicides from the electrophilic chloroacetanilide class: propachlor, alachlor, and acetochlor.172 These each share a highly reactive chloroacetamide motif, differing only slightly in the distribution of methyl/ethyl groups. Despite the profound methodological difference between this assay (Hsp40 affinity in HEK239T cells) and the previous ABPP study (conjugation to liver proteins in living mice),85 the targets of acetochlor show a high degree of overlap. By contrast, Hsp40 affinity following cellular propachlor exposure is not well-correlated with protein aggregation. Although a few proteins are highly sensitive to each herbicide, the general profile is distinct, with propachlor specifically targeting the GAPDH and PARK7/DJ-1. These studies demonstrate that Hsp40 affinity profiling is a powerful omic method for easily identifying proteins that are destabilized by cellular exposure.
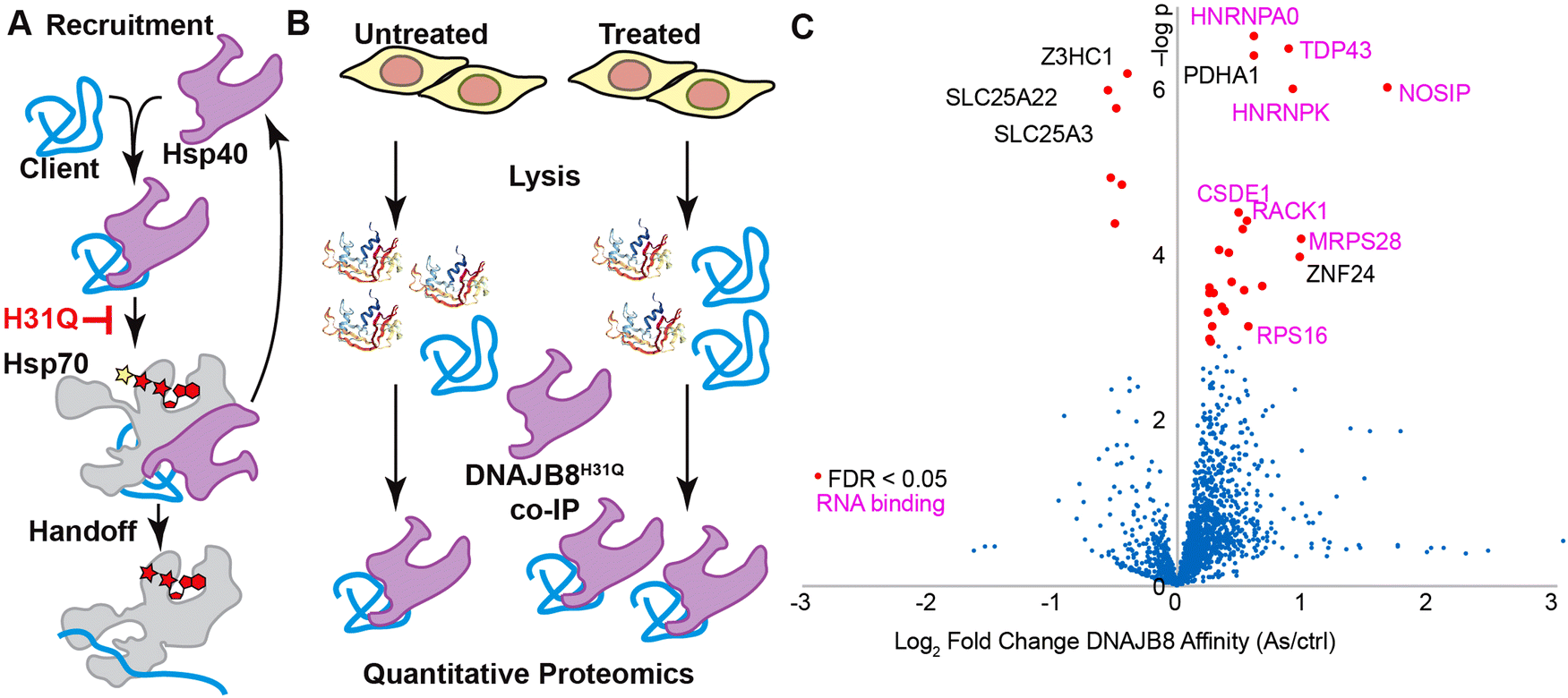 | ||
| Fig. 7 (A) The H31Q mutation blocks client handoff from Hsp40s, making them a thermodynamic sink for destabilized client proteins. (B) If a treatment destabilizes a client protein, then the affinity for DNAJB8 will increase, leading to increased recovery by affinity purification. (C) 15 min exposure of HEK293T cells to 100 μM sodium arsenite increases Hsp40 affinity of several proteins, particularly RNA binding proteins which as a class are known to be arsenite-sensitive. Reprinted (adapted) with permission from Quanrud et al., Anal. Chem., 93, 16940. Copyright 2021 American Chemical Society. | ||
E. Perspective
The need for complementary methods is underscored by the wildly differing results found using different assays. Cox et al. recently compared 20 censuses of protein stabilities, based on a limited proteolysis dataset, five footprinting studies, and fourteen thermal profiling studies.166 The correlation between thermal protein profiling studies is outstanding, as is the correlation between footprinting studies that use the same footprinting approach. The correlations between experiments that use different techniques, however, is poor. The authors ascribe this disconnect to, in part, the unavoidable over-reliance of data analysis on a two-state unfolding model that does not accurately reflect misfolding dynamics for most of the proteome. Even beyond challenges associated with model fitting, the extent of aggregation correlates poorly with protein misfolding.173 Limited proteolysis and SPROX rely on the successful identification and quantification of proteotypic peptides. Hsp40 affinity profiling relies upon recognition by the Hsp40 and is not as competent for identifying stabilization as destabilization. FPOP, despite having the highest potential of all these methods, still lags in terms of the depth of coverage. In contrast, ABPP is limited to the detection of direct adducts or strong binders but remains the most mature technology.Combining multiple strategies allows more rapid prioritization and validation of targets.174 This can be particularly useful when the validation assay uses target proteomics to ensure that the primary hits from the initial assay are successfully profiled. For example, an ABPP study of perfluorooctanoic acid identified the fatty acid lipases Acaca and Acacb as likely targets, and then used targeted PRM with the thermal shift assay to validate that PFOA directly binds both enzymes.175 Similarly, we used PRM with limited proteolysis to validate metal and chloroacetanilide targets first identified by the Hsp40 affinity assay.146,172 The ready adaptation of mass spectrometry-based protein profiling technologies to targeted experiments has another potential but unrealized benefit. Laboratory toxicity studies frequently employ toxin concentrations much higher than natural exposures. Higher concentrations can enable acute effects to be observed on an experimentally convenient time-scale,176 but can also lead to observations that are not physiologically relevant to environmental exposures. While higher concentrations of toxins during screening experiments enables protein target discovery, targeted validation experiments using physiologically relevant concentrations can be used to confirm with greater sensitivity that the same effects on protein biochemistry are observed. In this way, greater confidence in protein targets can be generated prior to resource-intensive evaluation of functional consequences.
Despite these challenges, it is clear that there are now a variety of viable technologies available to characterize how environmental toxins impact protein integrity and stability following cellular exposure. The continued application of these technologies will provide greater insight into how cells are affected by chemical exposures, and narrow our gap in understanding how the exposome threatens the proteome.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Society for Analytical Chemists of Pittsburgh Starter Grant and the University of California, Riverside.References
- R. M. LoPachin and A. P. DeCaprio, Toxicol. Sci., 2005, 86, 214–225 CrossRef CAS PubMed.
- A. Fribley, K. Zhang and R. J. Kaufman, in Apoptosis, ed. P. Erhardt and A. Toth, Humana Press, Totowa, NJ, 2009, vol. 559, pp. 191–204 Search PubMed.
- N. S. Anderson and C. M. Haynes, Trends Cell Biol., 2020, 30, 428–439 CrossRef CAS PubMed.
- K. Pakos-Zebrucka, I. Koryga, K. Mnich, M. Ljujic, A. Samali and A. M. Gorman, EMBO Rep., 2016, 17, 1374–1395 CrossRef CAS PubMed.
- S. H. Safe, Crit. Rev. Toxicol., 1994, 24, 87–149 CrossRef CAS PubMed.
- W. P. Roos and B. Kaina, Cancer Lett., 2013, 332, 237–248 CrossRef CAS PubMed.
- P. Walter and D. Ron, Science, 2011, 334, 1081–1086 CrossRef CAS PubMed.
- S. Fulda, A. M. Gorman, O. Hori and A. Samali, Int. J. Cell. Biol., 2010, 2010, 214074 Search PubMed.
- B.-J. Guan, V. van Hoef, R. Jobava, O. Elroy-Stein, L. S. Valasek, M. Cargnello, X.-H. Gao, D. Krokowski, W. C. Merrick, S. R. Kimball, A. A. Komar, A. E. Koromilas, A. Wynshaw-Boris, I. Topisirovic, O. Larsson and M. Hatzoglou, Mol. Cell, 2017, 68, 885–900.e6 CrossRef CAS PubMed.
- D. B. Sattelle, Advances in Insect Physiology, Elsevier, 1990, vol. 22, pp. 1–113 Search PubMed.
- J. Ha, H. Park, J. Park and S. B. Park, Cell Chem. Biol., 2021, 28, 394–423 CrossRef CAS.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto, Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS PubMed.
- Y. Kumagai and Y. Abiko, Chem. Res. Toxicol., 2017, 30, 203–219 Search PubMed.
- D. H. Phillips and S. Venitt, Int. J. Cancer, 2012, 131, 2733–2753 CrossRef CAS PubMed.
- B. Honig and A. Nicholls, Science, 1995, 268, 1144–1149 CrossRef CAS PubMed.
- D. Greenbaum, K. F. Medzihradszky, A. Burlingame and M. Bogyo, Chem. Biol., 2000, 7, 569–581 CrossRef CAS PubMed.
- G. P. Dooley, K. F. Reardon, J. E. Prenni, R. B. Tjalkens, M. E. Legare, C. D. Foradori, J. E. Tessari and W. H. Hanneman, Chem. Res. Toxicol., 2008, 21, 844–851 Search PubMed.
- G. P. Dooley, A. K. Ashley, M. E. Legare, R. J. Handa and W. H. Hanneman, Toxicol. Lett., 2010, 199, 17–21 CrossRef CAS PubMed.
- X. Yang and M. G. Bartlett, Rapid Commun. Mass Spectrom., 2016, 30, 652–664 CrossRef CAS PubMed.
- I. Jablonkai and K. K. Hatzios, J. Agric. Food Chem., 1993, 41, 1736–1742 CrossRef CAS.
- Y. Shi and K. S. Carroll, Acc. Chem. Res., 2020, 53, 20–31 CrossRef CAS PubMed.
- D. Fass, Annu. Rev. Biophys., 2012, 41, 63–79 CrossRef CAS PubMed.
- J. R. Stone and S. Yang, Antioxid. Redox Signaling, 2006, 8, 243–270 CrossRef CAS PubMed.
- C. Lennicke and H. M. Cochemé, Mol. Cell, 2021, 81, 3691–3707 CrossRef CAS PubMed.
- W. Chadwick, A. Keselman, S.-S. Park, Y. Zhou, L. Wang, R. Brenneman, B. Martin and S. Maudsley, J. Signal Transduction, 2011, 2011, 1–15 CrossRef PubMed.
- R. Sugiura, R. Satoh and T. Takasaki, Cells, 2021, 10, 2509 CrossRef CAS PubMed.
- C. Lennicke, J. Rahn, R. Lichtenfels, L. A. Wessjohann and B. Seliger, Cell Commun Signal, 2015, 13, 39 CrossRef PubMed.
- J. D. Keyes, D. Parsonage, R. D. Yammani, L. C. Rogers, C. Kesty, C. M. Furdui, K. J. Nelson and L. B. Poole, Free Radical Biol. Med., 2017, 112, 534–543 CrossRef CAS PubMed.
- K. A. Webster, H. Prentice and N. H. Bishopric, Antioxid. Redox Signaling, 2001, 3, 535–548 CrossRef CAS PubMed.
- H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- J. C. Crack, J. Green, A. J. Thomson and N. E. L. Brun, Acc. Chem. Res., 2014, 47, 3196–3205 CrossRef CAS PubMed.
- L. Ellgaard, C. S. Sevier and N. J. Bulleid, Trends Biochem. Sci., 2018, 43, 32–43 CrossRef CAS PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS PubMed.
- B. Chance, H. Sies and A. Boveris, Physiol. Rev., 1979, 59, 527–605 CrossRef CAS PubMed.
- A. Maurel, C. Hernandez, O. Kunduzova, G. Bompart, C. Cambon, A. Parini and B. Francés, Am. J. Physiol.: Heart Circ. Physiol., 2003, 284, H1460–H1467 CrossRef CAS PubMed.
- M. E. Albertolle and F. Peter Guengerich, J. Inorg. Biochem., 2018, 186, 228–234 CrossRef CAS PubMed.
- M. B. Hampton, A. J. Kettle and C. C. Winterbourn, Blood, 1998, 92, 3007–3017 CrossRef CAS PubMed.
- S. Shen, X.-F. Li, W. R. Cullen, M. Weinfeld and X. C. Le, Chem. Rev., 2013, 113, 7769–7792 CrossRef CAS PubMed.
- C. D. Klaassen, J. Liu and S. Choudhuri, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 267–294 CrossRef CAS PubMed.
- J. J. V. Branca, C. Fiorillo, D. Carrino, F. Paternostro, N. Taddei, M. Gulisano, A. Pacini and M. Becatti, Antioxidants, 2020, 9, 492 CrossRef CAS PubMed.
- Y. Hu, J. Li, B. Lou, R. Wu, G. Wang, C. Lu, H. Wang, J. Pi and Y. Xu, Biomolecules, 2020, 10, E240 CrossRef PubMed.
- C. C. Winterbourn, Toxicol. Lett., 1995, 82–83, 969–974 CrossRef CAS PubMed.
- J. S. Bus, S. D. Aust and J. E. Gibson, Biochem. Biophys. Res. Commun., 1974, 58, 749–755 CrossRef CAS PubMed.
- H. Yang, G. Luo, P. Karnchanaphanurach, T.-M. Louie, I. Rech, S. Cova, L. Xun and X. S. Xie, Science, 2003, 302, 262–266 CrossRef CAS PubMed.
- W. Chen, W. Lu, P. G. Wolynes and E. A. Komives, Nucleic Acids Res., 2021, 49, 11211–11223 CrossRef CAS PubMed.
- A. Yu, Y. Shibata, B. Shah, B. Calamini, D. C. Lo and R. I. Morimoto, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1481–1490 CAS.
- M. S. Hipp, S.-H. Park and F. U. Hartl, Trends Cell Biol., 2014, 24, 506–514 CrossRef CAS PubMed.
- B. Chen, M. Retzlaff, T. Roos and J. Frydman, Cold Spring Harbor. Perspect. Biol., 2011, 3, a004374 Search PubMed.
- N. Vij, S. Fang and P. L. Zeitlin, J. Biol. Chem., 2006, 281, 17369–17378 CrossRef CAS PubMed.
- E. McShane, C. Sin, H. Zauber, J. N. Wells, N. Donnelly, X. Wang, J. Hou, W. Chen, Z. Storchova, J. A. Marsh, A. Valleriani and M. Selbach, Cell, 2016, 167, 803–815.e21 CrossRef CAS PubMed.
- Y. Zhuo, Y. Zhang, M. Li, H. Wu, S. Gong, X. Hu, Y. Fu, X. Shen, B. Sun, J.-L. Wu and N. Li, Food Chem. Toxicol., 2021, 153, 112257 CrossRef CAS PubMed.
- T. B. Phan, M. Breugst and H. Mayr, Angew. Chem., Int. Ed., 2006, 45, 3869–3874 CrossRef CAS PubMed.
- R. M. LoPachin, B. C. Geohagen and L. U. Nordstroem, Toxicology, 2019, 418, 62–69 CrossRef CAS PubMed.
- J. Zhang, C. Wang, L. Ji and W. Liu, Chem. Res. Toxicol., 2016, 29, 841–850 Search PubMed.
- D. G. Isom, C. A. Castaneda, B. R. Cannon and B. Garcia-Moreno E., Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- C. C. Ward, J. I. Kleinman and D. K. Nomura, ACS Chem. Biol., 2017, 12, 1478–1483 CrossRef CAS PubMed.
- W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 CrossRef CAS PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T.-A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. Am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- J. Anckar and L. Sistonen, Annu. Rev. Biochem., 2011, 80, 1089–1115 CrossRef CAS PubMed.
- T. Shpilka and C. M. Haynes, Nat. Rev. Mol. Cell Biol., 2018, 19, 109–120 CrossRef CAS PubMed.
- P. M. Conn, The unfolded protein response and cellular stress, Elsevier/Academic Press, San Diego, CA, 1st edn, 2011 Search PubMed.
- D. Wirth, E. Christians, C. Munaut, C. Dessy, J.-M. Foidart and P. Gustin, Cell Stress Chaperones, 2002, 7, 387–395 CrossRef CAS PubMed.
- V. Ortner, A. Ludwig, E. Riegel, S. Dunzinger and T. Czerny, Cell Stress Chaperones, 2015, 20, 277–288 CrossRef CAS PubMed.
- M. E. Feder and G. E. Hofmann, Annu. Rev. Physiol., 1999, 61, 243–282 CrossRef CAS PubMed.
- K. Mahmood, S. Jadoon, Q. Mahmood, M. Irshad and J. Hussain, BioMed Res. Int., 2014, 2014, 1–17 Search PubMed.
- J. M. D. Grandjean, L. Plate, R. I. Morimoto, M. J. Bollong, E. T. Powers and R. L. Wiseman, ACS Chem. Biol., 2019, 14, 784–795 CrossRef CAS PubMed.
- J. D. Rosarda, K. R. Baron, K. Nutsch, G. M. Kline, C. Stanton, J. W. Kelly, M. J. Bollong and R. L. Wiseman, ACS Chem. Biol., 2021, 16, 2852–2863 CrossRef CAS PubMed.
- C. Steurer, N. Eder, S. Kerschbaum, C. Wegrostek, S. Gabriel, N. Pardo, V. Ortner, T. Czerny and E. Riegel, PLoS One, 2018, 13, e0209077 CrossRef CAS PubMed.
- Y. Zhang, Y.-H. Ahn, I. J. Benjamin, T. Honda, R. J. Hicks, V. Calabrese, P. A. Cole and A. T. Dinkova-Kostova, Chem. Biol., 2011, 18, 1355–1361 CrossRef CAS PubMed.
- J. D. West and L. J. Marnett, Chem. Res. Toxicol., 2005, 18, 1642–1653 Search PubMed.
- M. Yokouchi, N. Hiramatsu, K. Hayakawa, A. Kasai, Y. Takano, J. Yao and M. Kitamura, Cell Death Differ., 2007, 14, 1467–1474 CrossRef CAS PubMed.
- P. M. Quirós, M. A. Prado, N. Zamboni, D. D’Amico, R. W. Williams, D. Finley, S. P. Gygi and J. Auwerx, J. Cell Biol., 2017, 216, 2027–2045 CrossRef PubMed.
- B. Luo, Y. Lin, S. Jiang, L. Huang, H. Yao, Q. Zhuang, R. Zhao, H. Liu, C. He and Z. Lin, Cell Death Dis., 2016, 7, e2251–e2251 CrossRef CAS PubMed.
- A. Chargui, S. Zekri, G. Jacquillet, I. Rubera, M. Ilie, A. Belaid, C. Duranton, M. Tauc, P. Hofman, P. Poujeol, M. V. El May and B. Mograbi, Toxicol. Sci., 2011, 121, 31–42 CrossRef CAS PubMed.
- S. R. Shih, D. M. Bach, N. C. Rondeau, J. Sam, N. L. Lovinger, A. J. Lopatkin and J. W. Snow, Sci. Rep., 2021, 11, 22087 CrossRef PubMed.
- A.-B. Muñiz-González, F. Paoli, J.-L. Martínez-Guitarte and V. Lencioni, Environ. Pollut., 2021, 290, 118061 CrossRef PubMed.
- A.-B. Muñiz-González, M. Novo and J.-L. Martínez-Guitarte, Environ. Sci. Pollut. Res., 2021, 28, 31431–31446 CrossRef PubMed.
- D. R. Hall and H. Peng, Comp. Biochem. Physiol., Part D: Genomics Proteomics, 2020, 34, 100655 CAS.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- P. R. A. Zanon, L. Lewald and S. M. Hacker, Angew. Chem., Int. Ed., 2020, 59, 2829–2836 CrossRef CAS PubMed.
- D. K. Nomura and J. E. Casida, J. Agric. Food Chem., 2011, 59, 2808–2815 CrossRef CAS PubMed.
- D. Medina-Cleghorn, L. A. Bateman, B. Ford, A. Heslin, K. J. Fisher, E. D. Dalvie and D. K. Nomura, Chem. Biol., 2015, 22, 1394–1405 CrossRef CAS PubMed.
- J. L. Counihan, M. Duckering, E. Dalvie, W. Ku, L. A. Bateman, K. J. Fisher and D. K. Nomura, ACS Chem. Biol., 2017, 12, 635–642 CrossRef CAS PubMed.
- B. Ford, L. A. Bateman, L. Gutierrez-Palominos, R. Park and D. K. Nomura, Cell Chem. Biol., 2017, 24, 133–140 CrossRef CAS PubMed.
- A. Zulet, M. Gil-Monreal, J. G. Villamor, A. Zabalza, R. A. L. van der Hoorn and M. Royuela, PLoS One, 2013, 8, e73847 CrossRef CAS PubMed.
- E. G. Stoddard, B. J. Killinger, S. A. Nag, J. Martin, R. Corley, J. N. Smith and A. T. Wright, Chem. Res. Toxicol., 2019, 32, 1259–1267 Search PubMed.
- E. G. Stoddard, S. Nag, J. Martin, K. J. Tyrrell, T. Gibbins, K. A. Anderson, A. K. Shukla, R. Corley, A. T. Wright and J. N. Smith, Chem. Res. Toxicol., 2021, 34, 2145–2156 Search PubMed.
- N. C. Sadler, B.-J. M. Webb-Robertson, T. R. Clauss, J. G. Pounds, R. Corley and A. T. Wright, Chem. Res. Toxicol., 2018, 31, 308–318 Search PubMed.
- W. L. Garcia, C. J. Miller, G. X. Lomas, K. A. Gaither, K. J. Tyrrell, J. N. Smith, K. R. Brandvold and A. T. Wright, Chem. Res. Toxicol., 2022, 35, 585–596 Search PubMed.
- H. M. Ismail, P. M. O’Neill, D. W. Hong, R. D. Finn, C. J. Henderson, A. T. Wright, B. F. Cravatt, J. Hemingway and M. J. I. Paine, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19766–19771 CrossRef CAS PubMed.
- X.-F. Cheng, N. Wang, Z. Jiang, Z. Chen, Y. Niu, L. Tong, T. Yu and B. Tang, Bioconjugate Chem., 2022, 33, 1131–1137 CrossRef CAS PubMed.
- S. K. Fung, T. Zou, B. Cao, P.-Y. Lee, Y. M. E. Fung, D. Hu, C.-N. Lok and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS PubMed.
- D. Hu, Y. Liu, Y.-T. Lai, K.-C. Tong, Y.-M. Fung, C.-N. Lok and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 1387–1391 CrossRef CAS PubMed.
- X. Yan, J. Li, Q. Liu, H. Peng, A. Popowich, Z. Wang, X.-F. Li and X. C. Le, Angew. Chem., Int. Ed., 2016, 55, 14051–14056 CrossRef CAS PubMed.
- K. Nan, M. He, B. Chen and B. Hu, Anal. Chim. Acta, 2021, 1183, 339007 CrossRef CAS PubMed.
- X. Zhang, F. Yang, J.-Y. Shim, K. L. Kirk, D. E. Anderson and X. Chen, Cancer Lett., 2007, 255, 95–106 CrossRef CAS PubMed.
- R. M. Cunningham and V. J. DeRose, ACS Chem. Biol., 2017, 12, 2737–2745 CrossRef CAS PubMed.
- X. Xu, H. Wang, H. Li and H. Sun, Chem. Lett., 2020, 49, 697–704 CrossRef CAS.
- H. Zhang, L. Yang, J. Ling, D. M. Czajkowsky, J.-F. Wang, X.-W. Zhang, Y.-M. Zhou, F. Ge, M. Yang, Q. Xiong, S.-J. Guo, H.-Y. Le, S.-F. Wu, W. Yan, B. Liu, H. Zhu, Z. Chen and S. Tao, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15084–15089 CrossRef CAS PubMed.
- L. I. Leichert, F. Gehrke, H. V. Gudiseva, T. Blackwell, M. Ilbert, A. K. Walker, J. R. Strahler, P. C. Andrews and U. Jakob, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8197–8202 CrossRef CAS PubMed.
- M. Sethuraman, M. E. McComb, H. Huang, S. Huang, T. Heibeck, C. E. Costello and R. A. Cohen, J. Proteome Res., 2004, 3, 1228–1233 CrossRef CAS PubMed.
- S. Akter, L. Fu, Y. Jung, M. L. Conte, J. R. Lawson, W. T. Lowther, R. Sun, K. Liu, J. Yang and K. S. Carroll, Nat. Chem. Biol., 2018, 14, 995–1004 CrossRef CAS PubMed.
- J. Meng, L. Fu, K. Liu, C. Tian, Z. Wu, Y. Jung, R. B. Ferreira, K. S. Carroll, T. K. Blackwell and J. Yang, Nat. Commun., 2021, 12, 1415 CrossRef CAS PubMed.
- J. D. Majmudar, A. M. Konopko, K. J. Labby, C. T. M. B. Tom, J. E. Crellin, A. Prakash and B. R. Martin, J. Am. Chem. Soc., 2016, 138, 1852–1859 CrossRef CAS PubMed.
- L. J. Alcock, B. L. Oliveira, M. J. Deery, T. L. Pukala, M. V. Perkins, G. J. L. Bernardes and J. M. Chalker, ACS Chem. Biol., 2019, 14, 594–598 CrossRef CAS PubMed.
- X. R. Liu, M. M. Zhang and M. L. Gross, Chem. Rev., 2020, 120, 4355–4454 CrossRef CAS PubMed.
- U. Kaur, H. Meng, F. Lui, R. Ma, R. N. Ogburn, J. H. R. Johnson, M. C. Fitzgerald and L. M. Jones, J. Proteome Res., 2018, 17, 3614–3627 CrossRef CAS PubMed.
- E. I. James, T. A. Murphree, C. Vorauer, J. R. Engen and M. Guttman, Chem. Rev., 2022, 122, 7562–7623 CrossRef CAS PubMed.
- A. M. Zmyslowski, M. C. Baxa, I. A. Gagnon and T. R. Sosnick, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119436119 CrossRef CAS PubMed.
- M. Kaldmäe, A. Leppert, G. Chen, M. Sarr, C. Sahin, K. Nordling, N. Kronqvist, M. Gonzalvo-Ulla, N. Fritz, A. Abelein, S. Lain, H. Biverstål, H. Jörnvall, D. P. Lane, A. Rising, J. Johansson and M. Landreh, FEBS J., 2020, 287, 2823–2833 CrossRef PubMed.
- M. Fang, Z. Wang, K. A. Cupp-Sutton, T. Welborn, K. Smith and S. Wu, Anal. Chim. Acta, 2021, 1143, 65–72 CrossRef CAS PubMed.
- B. C. Gau, J. S. Sharp, D. L. Rempel and M. L. Gross, Anal. Chem., 2009, 81, 6563–6571 CrossRef CAS PubMed.
- D. T. Johnson, L. H. Di Stefano and L. M. Jones, J. Biol. Chem., 2019, 294, 11969–11979 CrossRef CAS PubMed.
- J. S. Sharp, S. K. Misra, J. J. Persoff, R. W. Egan and S. R. Weinberger, Anal. Chem., 2018, 90, 12625–12630 CrossRef CAS PubMed.
- K. S. Li, L. Shi and M. L. Gross, Acc. Chem. Res., 2018, 51, 736–744 CrossRef CAS PubMed.
- A. Rinas, V. S. Mali, J. A. Espino and L. M. Jones, Anal. Chem., 2016, 88, 10052–10058 CrossRef CAS PubMed.
- U. Kaur, D. T. Johnson and L. M. Jones, J. Am. Soc. Mass Spectrom., 2020, 31, 1372–1379 CrossRef CAS PubMed.
- J. A. Espino and L. M. Jones, Anal. Chem., 2019, 91, 6577–6584, DOI:10.1021/acs.analchem.9b00244.
- A. Rinas, J. A. Espino and L. M. Jones, Anal. Bioanal. Chem., 2016, 408, 3021–3031 CrossRef CAS PubMed.
- J. Adhikari, G. M. West and M. C. Fitzgerald, J. Proteome Res., 2015, 14, 2287–2297 CrossRef CAS PubMed.
- E. J. Walker, J. Q. Bettinger, K. A. Welle, J. R. Hryhorenko and S. Ghaemmaghami, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6081–6090 CrossRef CAS PubMed.
- G. M. West, L. Tang and M. C. Fitzgerald, Anal. Chem., 2008, 80, 4175–4185 CrossRef CAS PubMed.
- P. D. Dearmond, Y. Xu, E. C. Strickland, K. G. Daniels and M. C. Fitzgerald, J. Proteome Res., 2011, 10, 4948–4958 CrossRef CAS PubMed.
- Y. Xu, I. N. Falk, M. A. Hallen and M. C. Fitzgerald, Anal. Chem., 2011, 83, 3555–3562 CrossRef CAS PubMed.
- N. Wiebelhaus, N. Singh, P. Zhang, S. L. Craig, D. N. Beratan and M. C. Fitzgerald, J. Am. Chem. Soc., 2022, 144, 3925–3938 CrossRef CAS PubMed.
- Y. Xu, G. M. West, M. Abdelmessih, M. D. Troutman and R. A. Everley, ACS Chem. Biol., 2021, 16, 1445–1455 CrossRef CAS PubMed.
- H. Meng and M. C. Fitzgerald, J. Proteome Res., 2018, 17, 1129–1137 CrossRef CAS PubMed.
- J. Adhikari and M. C. Fitzgerald, J. Am. Soc. Mass Spectrom., 2014, 25, 2073–2083 CrossRef CAS PubMed.
- E. J. Walker, J. Q. Bettinger, K. A. Welle, J. R. Hryhorenko, A. M. Molina Vargas, M. R. O’Connell and S. Ghaemmaghami, J. Biol. Chem., 2022, 298, 101872 CrossRef CAS PubMed.
- K. K. Nguyen, S. Thurmond, R. Hai and J. C. Genereux, Anal. Bioanal. Chem., 2019, 411, 4987–4998 CrossRef CAS PubMed.
- E. G. Jaffee, M. A. Lauber, W. E. Running and J. P. Reilly, Anal. Chem., 2012, 84, 9355–9361 CrossRef CAS PubMed.
- G. M. West, C. L. Tucker, T. Xu, S. K. Park, X. Han, J. R. Yates and M. C. Fitzgerald, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9078–9082 CrossRef CAS.
- C. Bamberger, S. Pankow, S. Martínez-Bartolomé, M. Ma, J. Diedrich, R. A. Rissman and J. R. Yates, J. Proteome Res., 2021, 20, 2762–2771 CrossRef CAS PubMed.
- C. Bamberger, J. Diedrich, S. Martìnez-Bartholomé and J. R. Yates, J. Proteome Res., 2022, 21, 1017–1028 CrossRef CAS PubMed.
- G. Shen, W. Cui, H. Zhang, F. Zhou, W. Huang, Q. Liu, Y. Yang, S. Li, G. R. Bowman, J. E. Sadler, M. L. Gross and W. Li, Nat. Struct. Mol. Biol., 2017, 24, 69–76 CrossRef CAS PubMed.
- K. Yu, M. Niu, H. Wang, Y. Li, Z. Wu, B. Zhang, V. Haroutunian and J. Peng, J. Am. Soc. Mass Spectrom., 2021, 32, 936–945 CrossRef CAS PubMed.
- J. Wen, H. Zhang, M. L. Gross and R. E. Blankenship, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6134–6139 CrossRef CAS PubMed.
- J. Sun, S. Li, W. Li and M. L. Gross, Anal. Chem., 2021, 93, 13101–13105 CrossRef CAS PubMed.
- C. Bamberger, S. Pankow, S. K. R. Park and J. R. Yates, J. Proteome Res., 2014, 13, 1494–1501 CrossRef CAS PubMed.
- Y. Feng, G. De Franceschi, A. Kahraman, M. Soste, A. Melnik, P. J. Boersema, P. P. de Laureto, Y. Nikolaev, A. P. Oliveira and P. Picotti, Nat. Biotechnol., 2014, 32, 1036–1044 CrossRef CAS PubMed.
- S. Schopper, A. Kahraman, P. Leuenberger, Y. Feng, I. Piazza, O. Müller, P. J. Boersema and P. Picotti, Nat. Protoc., 2017, 12, 2391–2410 CrossRef CAS PubMed.
- C. Park and S. Marqusee, Nat. Methods, 2005, 2, 207–212 CrossRef CAS PubMed.
- B. Lomenick, R. Hao, N. Jonai, R. M. Chin, M. Aghajan, S. Warburton, J. Wang, R. P. Wu, F. Gomez, J. A. Loo, J. A. Wohlschlegel, T. M. Vondriska, J. Pelletier, H. R. Herschman, J. Clardy, C. F. Clarke and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21984–21989 CrossRef CAS PubMed.
- G. M. Quanrud, M. R. Montoya, L. Mei, M. R. Awad and J. C. Genereux, Anal. Chem., 2021, 93, 16940–16946 CrossRef CAS PubMed.
- Y. Chang, J. P. Schlebach, R. A. VerHeul and C. Park, Protein Sci., 2012, 21, 1280–1287 CrossRef CAS PubMed.
- P.-F. Liu, D. Kihara and C. Park, J. Mol. Biol., 2011, 408, 147–162 CrossRef CAS PubMed.
- P. Leuenberger, S. Ganscha, A. Kahraman, V. Cappelletti, P. J. Boersema, C. von Mering, M. Claassen and P. Picotti, Science, 2017, 355, eaai7825 CrossRef PubMed.
- P. To, B. Whitehead, H. E. Tarbox and S. D. Fried, J. Am. Chem. Soc., 2021, 143, 11435–11448 CrossRef CAS PubMed.
- P. To, Y. Xia, S. O. Lee, T. Devlin, K. G. Fleming and S. D. Fried, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2210536119 CrossRef CAS PubMed.
- R. Ma, H. Meng, N. Wiebelhaus and M. C. Fitzgerald, Anal. Chem., 2018, 90, 14039–14047 CrossRef CAS PubMed.
- N. Wiebelhaus, J. M. Zaengle-Barone, K. K. Hwang, K. J. Franz and M. C. Fitzgerald, ACS Chem. Biol., 2021, 16, 214–224 CrossRef CAS PubMed.
- W. Colon and J. W. Kelly, Biochemistry, 1992, 31, 8654–8660 CrossRef CAS PubMed.
- D. R. Booth, M. Sunde, V. Bellotti, C. V. Robinson, W. L. Hutchinson, P. E. Fraser, P. N. Hawkins, C. M. Dobson, S. E. Radford, C. C. F. Blake and M. B. Pepys, Nature, 1997, 385, 787–793 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef CAS PubMed.
- M. M. Savitski, F. B. M. Reinhard, H. Franken, T. Werner, M. F. Savitski, D. Eberhard, D. M. Molina, R. Jafari, R. B. Dovega, S. Klaeger, B. Kuster, P. Nordlund, M. Bantscheff and G. Drewes, Science, 2014, 346, 1255784 CrossRef PubMed.
- H. Peng, H. Guo, O. Pogoutse, C. Wan, L. Z. Hu, Z. Ni and A. Emili, Environ. Sci. Technol., 2016, 50, 11329–11336 CrossRef CAS PubMed.
- L. Dai, T. Zhao, X. Bisteau, W. Sun, N. Prabhu, Y. T. Lim, R. M. Sobota, P. Kaldis and P. Nordlund, Cell, 2018, 173, 1481–1494.e13 CrossRef CAS PubMed.
- C. Ruan, Y. Wang, X. Zhang, J. Lyu, N. Zhang, Y. Ma, C. Shi, G. Qu and M. Ye, Anal. Chem., 2022, 94, 6482–6490 CrossRef CAS PubMed.
- J. Wang, W. Hu and Q. Hu, Toxins, 2019, 11, 67 CrossRef CAS PubMed.
- T. Xu, Y. T. Lim, L. Chen, H. Zhao, J. H. Low, Y. Xia, R. M. Sobota and M. Fang, Environ. Sci. Technol., 2020, 54, 15925–15934 CrossRef CAS PubMed.
- T. Xu, L. Chen, Y. T. Lim, H. Zhao, H. Chen, M. W. Chen, T. Huan, Y. Huang, R. M. Sobota and M. Fang, Environ. Sci. Technol., 2021, 55, 1842–1851 CrossRef CAS PubMed.
- M. Gaetani, P. Sabatier, A. A. Saei, C. M. Beusch, Z. Yang, S. L. Lundström and R. A. Zubarev, J. Proteome Res., 2019, 18, 4027–4037 CrossRef CAS PubMed.
- M. Gaetani and R. A. Zubarev, in Cell-Wide Identification of Metabolite-Protein Interactions, ed. A. Skirycz, M. Luzarowski and J. C. Ewald, Springer US, New York, NY, 2023, vol. 2554, pp. 91–106 Search PubMed.
- D. Cox, C.-S. Ang, N. B. Nillegoda, G. E. Reid and D. M. Hatters, Nat. Commun., 2022, 13, 1992 CrossRef CAS PubMed.
- C. M. Beusch, P. Sabatier and R. A. Zubarev, Anal. Chem., 2022, 94, 7066–7074 CrossRef CAS PubMed.
- J. G. Van Vranken, J. Li, D. C. Mitchell, J. Navarrete-Perea and S. P. Gygi, eLife, 2021, 10, e70784 CrossRef CAS PubMed.
- X. Zhang, K. Wang, S. Wu, C. Ruan, K. Li, Y. Wang, H. Zhu, X. Liu, Z. Liu, G. Li, L. Hu and M. Ye, Chem. Sci., 2022, 13, 12403–12418 RSC.
- L. Mei, M. R. Montoya, G. M. Quanrud, M. Tran, A. Villa-Sharma, M. Huang and J. C. Genereux, J. Proteome Res., 2020, 19, 1565–1573 CrossRef CAS PubMed.
- M. R. Montoya, G. M. Quanrud, L. Mei, J. L. Montaño and J. C. Genereux, Evaluating Client Protein Recovery by the Hsp40s DNAJB8 and DNAJB1 with AP-MS, bioRxiv, 2022 DOI:10.1101/2022.03.31.485989.
- G. Quanrud, S. Balamurugan, C. Canizal and J. Genereux, Cellular Exposure to Chloroacetanilide Herbicides Induces Distinct Protein Destabilization Profiles, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-bmqkk.
- X. Sui, D. E. V. Pires, A. R. Ormsby, D. Cox, S. Nie, G. Vecchi, M. Vendruscolo, D. B. Ascher, G. E. Reid and D. M. Hatters, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2422–2431 CrossRef CAS PubMed.
- A. Cabrera, N. Wiebelhaus, B. Quan, R. Ma, H. Meng and M. C. Fitzgerald, J. Am. Soc. Mass Spectrom., 2020, 31, 217–226 CrossRef CAS PubMed.
- X. Shao, F. Ji, Y. Wang, L. Zhu, Z. Zhang, X. Du, A. C. K. Chung, Y. Hong, Q. Zhao and Z. Cai, Anal. Chem., 2018, 90, 11092–11098 CrossRef CAS PubMed.
- D. W. Gaylor, Toxicology, 2000, 149, 17–19 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |