
Self-reported and self-facilitated theranostic oxygen nano-economizer for precise and hypoxia alleviation-potentiated photodynamic therapy†
Shumeng
Li
a,
Fujun
Yang
b,
Yongdan
Wang
a,
Linshan
Jia
a and
Xiaohong
Hou
 *c
*c
aDepartment of Pharmaceutical Analysis, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, P. R. China
bDepartment of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, P. R. China
cSchool of Pharmaceutical Engineering, Shenyang Pharmaceutical University, Shenyang, Liaoning Province 110016, P. R. China. E-mail: syphu_houxiaohong@163.com; Fax: +86-024-43520212
First published on 28th September 2023
Abstract
Photodynamic therapy (PDT) has been extensively investigated for cancer treatment by virtue of singlet oxygen-induced oxidative damage to tumors. Nevertheless, the therapeutic efficiency of PDT is still limited by the low singlet oxygen yield attributed to the improper irradiation duration and the tumor hypoxic microenvironment. To tackle these challenges, we elaborately design a theranostic oxygen nano-economizer to self-report the optimal irradiation duration and alleviate tumor hypoxia simultaneously, which is engineered by fluorescent 9,10-anthracenyl bis (benzoic acid) (DPA)-MOF, tetrakis (4-carboxyphenyl) porphyrin (TCPP), triphenyl phosphine (TPP) and redox-responsive lipid-PEG (DSPE-SS-PEG2k). Upon laser irradiation, the fluorescence of DPA-MOF could be quenched, thereby self-reporting the optimal irradiation duration for sufficient PDT. The decoration of DSPE-SS-PEG2k and TPP endows the theranostic oxygen nano-economizer with a tumor-specific response and mitochondrial targeting capability, respectively. Notably, singlet oxygen generated from TCPP reduces oxygen consumption by disrupting the entire oxidative phosphorylation (OXPHOS) pathway in the mitochondria of tumor cells, further improving the level of singlet oxygen in a self-facilitated manner for hypoxia alleviation-potentiated PDT. As expected, such a self-reported and self-facilitated theranostic oxygen nano-economizer exhibits potent antitumor activity in the 4T1 tumor-bearing mouse model. This study offers a theranostic paradigm for precise and hypoxia alleviation-potentiated cancer therapy.
New conceptsPhotodynamic therapy (PDT) has been recognized as a promising therapeutic option for cancer with extraordinary therapeutic selectivity and security. However, determining when to irradiate and how long to expose to the laser of photosensitizers still confuse clinicians. In addition, the hypoxic niches in most solid tumors nearly become the “lands beyond treatment”, demonstrating strong resistance to oxygen-dependent PDT. Herein, we propose a self-reported and self-facilitated theranostic oxygen nano-economizer based on co-delivery of singlet oxygen probes and therapeutic agents for precise and hypoxia alleviation-potentiated photodynamic tumor eradication. It is engineered by fluorescent 9,10-anthracenyl bis (benzoic acid) (DPA)-MOF, tetrakis (4-carboxyphenyl) porphyrin (TCPP), triphenyl phosphine (TPP) and redox-responsive lipid-PEG (DSPE-SS-PEG2k). Upon laser irradiation, the fluorescence of DPA-MOF could be quenched, thereby self-reporting the optimal irradiation duration for sufficient PDT. Irradiated with the self-reported time duration, DSPE-SS-PEG2k@TPP@TCPP@DPA-MOF (PTTD NPs) effectively suppressed cellular oxygen consumption by destroying the entire mitochondrial oxidative phosphorylation (OXPHOS) machinery in tumor cells for self-facilitating PDT. Such a uniquely engineered nano-economizer drives a conceptual step forward in theranostic nanoplatform design and provides a new paradigm for photo-therapeutic modality toward clinical treatment of cancer. |
1. Introduction
Photodynamic therapy (PDT), as a promising therapeutic option for cancer, has been approved in the clinic for decades due to its minimal invasiveness and high therapeutic selectivity.1–6 Under near-infrared (NIR) laser irradiation, photosensitizers (PSs) at the ground singlet state can be excited to the triplet state, thereby generating abundant cytotoxic singlet oxygen for inducing tumor cell apoptosis or necrosis.7–10 However, determining when to irradiate and how long to expose to a laser still confuse clinicians.11 Given that irradiating at an improper time duration during PDT would result in insufficient therapy or overtreatment, various fluorescent chemoprobes have been reported to monitor singlet oxygen for precision PDT.12–14 Among them, 9,10-anthracenyl bis (benzoic acid) (DPA) with blue fluorescence as an anthracene derivative can specifically react with singlet oxygen, thereby forming a nonfluorescent endoperoxide. Despite the excellent stability and controllability, the application of DPA in singlet oxygen monitoring is still restricted by structural amorphism and poor solubility.15,16 Obviously, more efficient DPA-based singlet oxygen monitoring systems for precise PDT are certainly warranted.Hypoxia has been recognized as a major microenvironmental factor in solid tumors due to malignant proliferation features, abnormal tumor vasculatures and inadequate oxygen diffusion.17–19 Furthermore, the mitochondrion-associated oxidative phosphorylation (OXPHOS) metabolic pathway of tumor cells also contributes significantly to a hypoxic microenvironment by constantly consuming oxygen to produce energy for cell survival.20–23 In recent decades, many strategies have been exploited to overcome the hypoxic limitation in PDT, mainly including in situ oxygen generation and oxygen exogenous supply.24–26 Nonetheless, most of these strategies are limited by intrinsically low intracellular hydrogen peroxide concentrations or premature oxygen leakage, which greatly compromised their capability of improving tumor oxygenation. As a comparison, suppressing cellular oxygen consumption may be a better approach to effectively alleviate hypoxia for decreasing the resistance to oxygen-dependent PDT. On this basis, several mitochondrial complex inhibitors such as metformin, tamoxifen and atovaquone have been widely used to reduce the oxygen consumption rate of solid tumors.27–29 Despite some success, it remains a challenge to completely impair OXPHOS machinery through inhibiting the activity of a single mitochondrial complex. Given this, we believe that only strategies capable of destroying the entire OXPHOS metabolic pathway can best deal with tumor hypoxia to improve the efficacy of PDT.
With the development of biomedical nanotechnology, great efforts have been devoted to the delivery of multiple drugs.30,31 Reasonable nano-drug delivery systems (nano-DDSs) have shown many advantages, such as improving the physicochemical properties, enhancing accumulation in tumors and promoting the security of therapeutic agents in biological systems.31–33 Nevertheless, most nano-DDSs are constructed by encapsulating therapeutic agents into inorganic or organic nanomaterials, which have the risk of inefficient loading and premature leakage of therapeutic agents due to poor affinity between nanomaterials and therapeutic agents.34–36 In recent decades, metallic organic framework materials (MOFs) constructed with metal ions/clusters and organic linkers (or bridging ligands) have been widely used for biosensing, catalysis and cancer therapy.37–40 Due to the high porosity, structural diversity of crystals and stable coordination of metal clusters and organic linkers, therapeutic agents could be adsorbed on the surface of MOFs or turned into multiple ligands in MOFs, which could show unique advantages for the synchronous co-delivery of therapeutic agents.41–43 Therefore, MOFs constructed for the synchronous co-delivery of therapeutic agents have emerged as promising nanoplatforms for precise PDT.
In the present study, we propose a self-reported and self-facilitated theranostic oxygen nano-economizer for precise and hypoxia alleviation-potentiated photodynamic tumor eradication (Scheme 1). Specifically, photosensitive TCPP@DPA-MOF (TD NPs) with the co-delivery of tetrakis (4-carboxyphenyl) porphyrin (TCPP) and DPA were constructed by incorporating the TCPP into the DPA-MOF, which was expected to trap and monitor singlet oxygen to self-report the optimal irradiation duration for precise PDT. With high porosity and gas adsorption ability, such a nanosystem displayed a more sensitive and accurate response to singlet oxygen when compared to the equivalent DPA solution. Moreover, inspired by the excellent post-synthetic modification capability of MOFs, TTD NPs with mitochondrial targeting features were further constructed by decorating the TD NPs with a typical mitochondrion-targeting molecule triphenylphosphine (TPP). Subsequently, a small amount of disulfide bond-containing lipid-PEG (DSPE-SS-PEG2k) was further leveraged for fabricating the theranostic oxygen nano-economizer (PTTD NPs) with long blood circulation and tumor-specific drug release. Notably, modification of TPP and DSPE-SS-PEG2k had negligible effects on the singlet oxygen-monitoring efficiency of TD NPs. TTD NPs could be readily released from PTTD NPs following the GSH-triggered breakage of disulfide bonds in DSPE-SS-PEG2k. Irradiated for the self-reported time duration, PTTD NPs effectively suppressed cellular oxygen consumption by destroying the entire mitochondrial OXPHOS machinery in tumor cells. More importantly, the saved oxygen was further utilized by TCPP to generate more singlet oxygen for self-facilitating PDT. As expected, such PTTD NPs significantly relieved tumor hypoxia and suppressed tumor growth. To our knowledge, this is the first attempt to develop a theranostic oxygen nano-economizer based on the combination of the singlet oxygen-monitoring feature of DPA-MOF and the mitochondrion-targeting feature of TPP. Such a self-reported and self-facilitated theranostic oxygen nano-economizer would drive a conceptual step forward in precise and hypoxia relief-potentiated PDT.
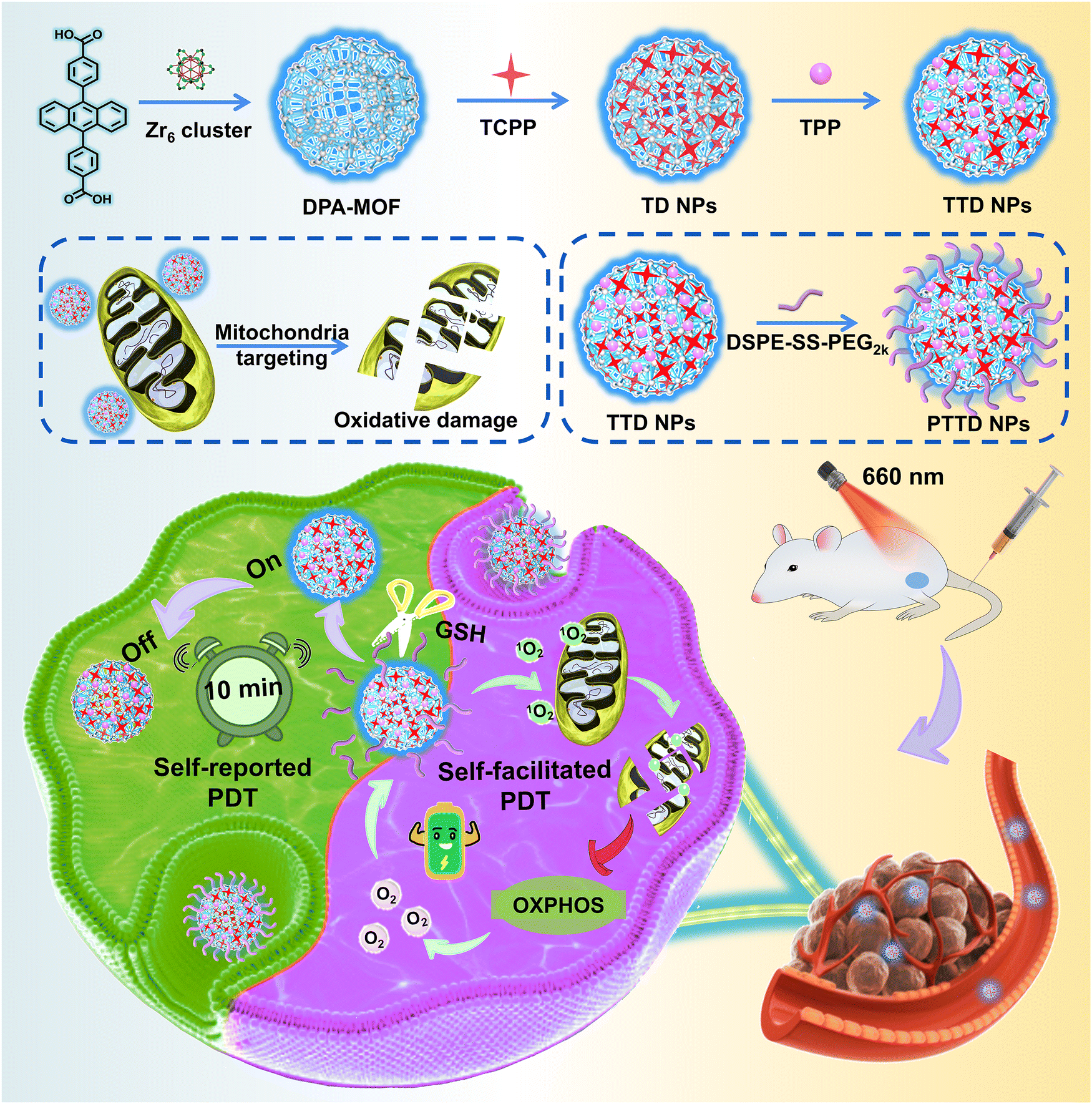 | ||
| Scheme 1 Schematic representation of the self-reported and self-facilitated theranostic oxygen nano-economizer for precise and hypoxia alleviation-potentiated photodynamic tumor eradication. PTTD NPs were precisely engineered by DPA-MOF, TCPP, mitochondrial targeting agent TPP and DSPE-SS-PEG2k. After the nanoassembly was precisely delivered into tumors, GSH triggered breakage of disulfide bonds in DSPE-SS-PEG2k, which exposed TPP for targeting mitochondria of tumor cells. DPA-MOF were expected to self-report the optimal irradiation duration for precise PDT via fluorescence quenching. Irradiated for the self-reported time duration, PTTD NPs effectively suppressed cellular oxygen consumption by destroying the entire mitochondrial OXPHOS machinery to facilitate PDT. | ||
2. Results and discussion
2.1. Singlet oxygen self-reported MOF optimization
DPA-MOF with high adsorption of gas was designed (Fig. 1A) and synthesized (Fig. S1, ESI†) for improving the low sensitivity of DPA. As expected, in the presence of singlet oxygen, DPA-MOF not only retained the fluorescence quenching properties, but also showed a larger linear range of singlet oxygen detection than DPA from 3 mM to 4 mM (Fig. 1B and C). At the same time, the singlet oxygen detection limitation was promoted by an order of magnitude in DPA-MOF (Table S1, ESI†), which proved the higher trapping capacity of singlet oxygen of MOFs due to the good lattice structure and gas adsorption manner.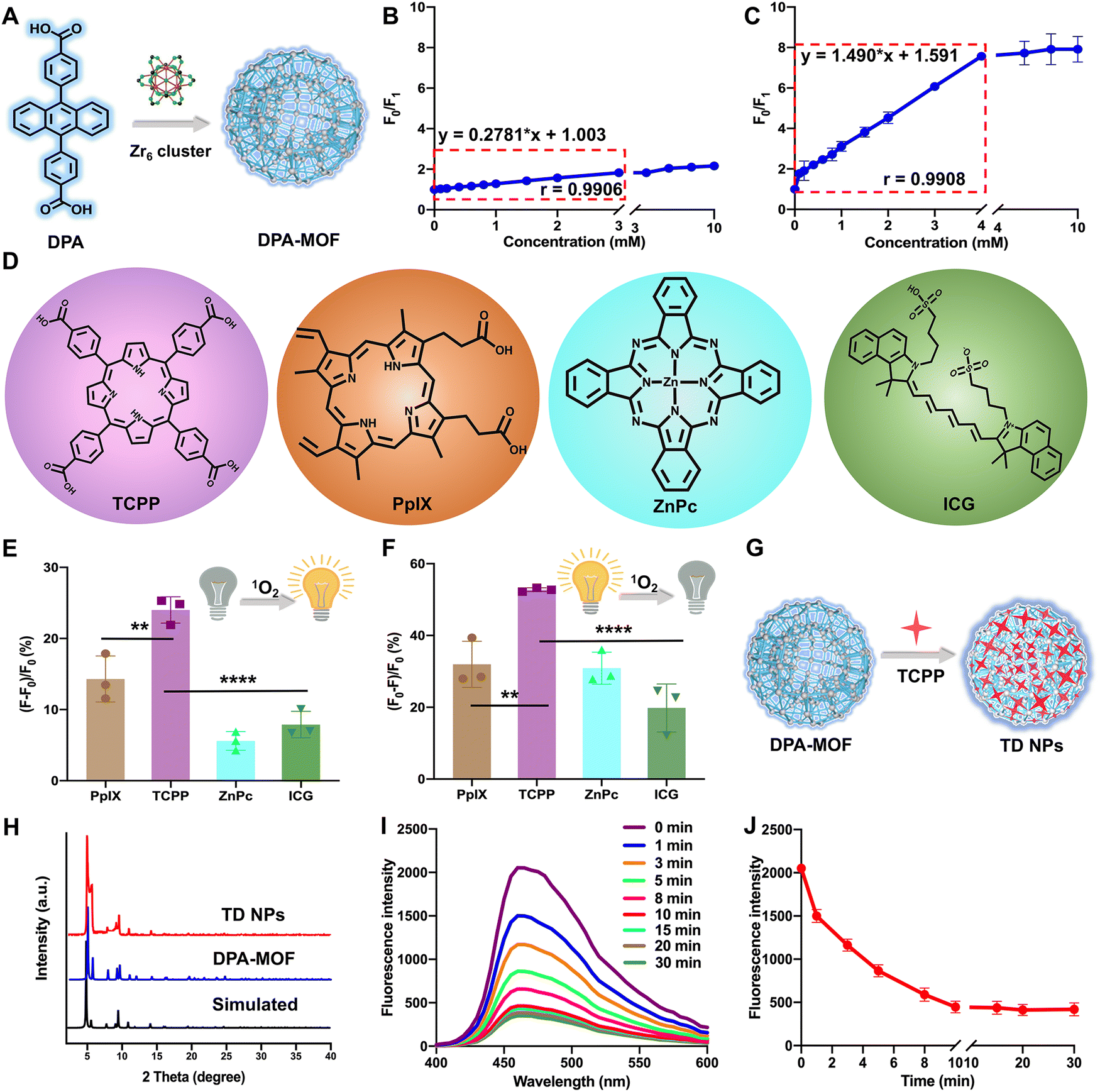 | ||
| Fig. 1 (A) Schematic diagram of the DPA-MOF. The linear range of (B) DPA and the (C) DPA-MOF for singlet oxygen detection. (D) Chemical structures of various PSs. Singlet oxygen was reported via the (E) SOSG probe and (F) DPA-MOF under laser irradiation (660 nm, 50 mW cm−2). (G) Schematic diagram of TD NPs. (H) XRD of the simulated DPA-MOF, DPA-MOF and TD NPs. (I) and (J) Singlet oxygen was reported by TD NPs via fluorescence intensity changes from 0 min to 30 min under laser irradiation (660 nm, 50 mW cm−2). Statistics: **P < 0.01,****P < 0.0001. | ||
Then, MOFs constructed with photodynamic PSs and DPA were designed to achieve precise PDT. In order to figure out the optimal PSs for PDT, six commonly used photosensitizers were utilized for singlet oxygen production (Fig. 1D and Fig. S2, ESI†), including three porphyrin derivatives (TCPP, chlorin e6 (Ce6) and protoporphyrin (PpIX)), zinc phthalocyanine (ZnPc), indocyanine green (ICG) and methylene blue (MB). First, singlet oxygen generation from PSs was detected using the SOSG probe, among which TCPP had the maximum changes of fluorescence intensity than other PSs (Fig. 1E and Fig. S2, ESI†). And, the TCPP group also showed the maximum fluorescence decay while detecting by DPA-MOF in Fig. 1F and Fig. S2 (ESI†), which demonstrated the highest singlet oxygen generation of TCPP under laser irradiation. In addition, various degrees of fluorescence decay caused by multifarious PSs indicated that DPA-MOF had the potential to be universal nanoplatforms for singlet oxygen generation. The photostability of PSs was also evaluated due to the problem of photobleaching. As shown in Fig. S3 (ESI†), the UV spectra of PSs were scanned, and the maximum absorption peak was selected to observe the photostability of PSs. As shown in Fig. S3 (ESI†), the absorbance of the TCPP solution at 420 nm remained above 95% after exposure to strong irradiation (660 nm, 50 mW cm−2), suggesting a much better photobleaching resistance than other PSs. The superior singlet oxygen generation efficacy and good photostability together suggested that TCPP is an ideal photosensitizer candidate for PDT applications. Taken together, TCPP was selected as the photosensitizer to carry out theranostic precise PDT through incorporating into DPA-MOF by coordination with Zr6 cluster and adsorption on the surface of the MOF (Fig. 1G and H). As expected, TCPP@DPA-MOF (TD NPs) showed excellent capacity of singlet oxygen generation. As shown in Fig. 1I and J, the fluorescence intensity of TD NPs decreased to the lowest level after 10 min of illumination, and no more declined when prolonging the illumination time. Thus, the fluorescence intensity of TD NPs could monitor the singlet oxygen generation and remind the optimal time for irradiation.
2.2. Preparation and characterization of NPs
The preparation process of PTD NPs, TTD NPs and PTTD NPs is displayed in Fig. 2A. The inner core TD NPs was modified with TPP and DSPE-SS-PEG2k. And the formulation by mixing TD NPs with different concentrations of TPP solution were optimized though stirring at room temperature for 12 h. After centrifugation, the TTD NPs of each group were washed with fresh DMF for removing free TPP. As shown in Fig. S4 and Table S2 (ESI†), the highest loading efficiency (34.61%) and encapsulated efficiency (10.61%) were showed when 20 mg of TPP was used to form TTD NPs. And then DSPE-SS-PEG2k was also added for PTTD NP formation. As shown in scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 2B and C and Fig. S1, ESI†), well-distributed DPA-MOF, PTD NPs, TTD NPs and PTTD NPs with circle-like shapes were prepared successfully. The HADDF-STEM image (Fig. 2D) indicated the composition of Zr, N, P and S elements in the obtained NPs, which further confirmed the successful preparation of PTTD NPs. As shown in Fig. 2E and Fig. S5, Table S3 (ESI†), the hydrodynamic sizes of TTD NPs and PTTD NPs were 155.8 nm (PDI = 0.12) and 178.9 nm (PDI = 0.16), which indicated that the hydration radius of the PEG shell slightly increased the particle size of NPs. And, the drug loadings of DPA, TCPP and TPP in TTD NPs were 40.77%, 18.38% and 34.64% in TTD NPs, and 32.62%, 14.70% and 27.71% in PTTD NPs, respectively (Table S3, ESI†). Moreover, PTD NPs and PTTD NPs incubated in PBS containing 10% FBS displayed good colloidal stability for 72 h at 37 °C, while the particle size of TTD NPs significantly increased after 8 h under the same conditions (Fig. 2F). And the long-term stability of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs was explored, and as shown in Fig. S6 (ESI†), nanoparticles displayed good long-term storage stability at 4 °C for 7 days. These results demonstrated that PEGylation modification helped NPs in resisting the interference of proteins, which would be beneficial to deliver NPs to tumors.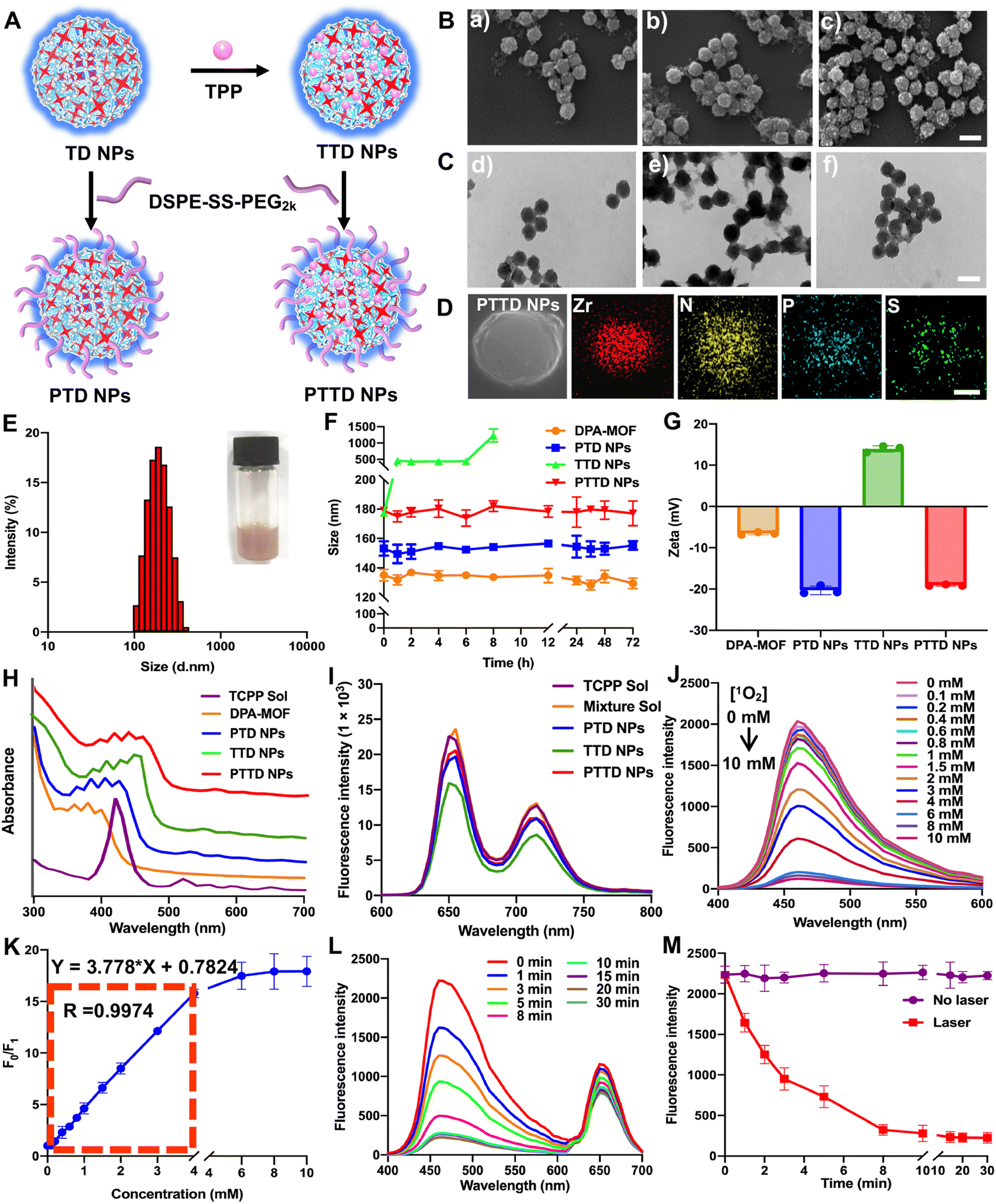 | ||
| Fig. 2 The characterization of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs. (A) Schematic diagram of PTD NPs, TTD NPs and PTTD NPs. (B) SEM images of (a) PTD NPs, (b) TTD NPs and (c) PTTD NPs. (C) TEM images of (d) PTD NPs, (e) TTD NPs and (f) PTTD NPs, scale bar = 200 nm. (D) Energy-dispersive X-ray mapping images of PTTD NPs, scale bar = 50 nm. (E) Appearance photographs and particle size distribution profiles of PTTD NPs. (F) Colloidal stability of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs incubated in PBS containing 10% FBS for 72 h at 37 °C. (G) Zeta potentials of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs. (H) UV absorption spectra of TCPP solution, DPA-MOF, PTD NPs, TTD NPs and PTTD NPs from 300 nm to 700 nm. (I) The fluorescence emission spectra of the TCPP solution, mixture solution of DPA-MOF and TCPP, PTD NPs, TTD NPs and PTTD NPs from 600 nm to 800 nm. (J) Fluorescence spectral changes of PTTD NPs in the presence of singlet oxygen with concentrations from 0 to 10 mM. (K) Singlet oxygen detection limitation of PTTD NPs. (L) Fluorescence intensity changes of PTTD NPs under different laser duration conditions. (M) Singlet oxygen detection of PTTD NPs with or without a laser (660 nm, 50 mW cm−2). | ||
As shown in Fig. 2G and Table S3 (ESI†), a positive zeta potential of 14.0 mV in TTD NPs showed that TPP with the positive charge was modified on the surface of NPs successfully. To further verify the formation mechanisms of TTD NPs and PTTD NPs, the intermolecular interactions and forces of TCPP and TPP were figured out. As depicted in Fig. S7 (ESI†), three intermolecular forces were identified in NPs, including hydrogen bonds, hydrophobic forces and π–π stacking interactions. TTD NPs were incubated with sodium dodecyl sulfate (SDS) and urea to figure out hydrophobic forces and hydrogen bonds. As shown in Fig. S8 (ESI†), the zeta potential of TTD NPs changed from the positive zeta potential to the negative zeta potential in the presence of SDS and urea, indicating that the hydrophobic forces and hydrogen bonds between TCPP and TPP were destroyed. And, as shown in Fig. 2H, the ultraviolet spectra of TTD NPs and PTTD NPs were visibly red shifted when compared to those of PTD NPs, confirming the existence of the π–π stacking interactions in the NPs. These results revealed that multiple interactions and forces drove the nanoassembly of TD NPs and TPP. Moreover, the redox-specific response of PTTD NPs was also monitored by zeta potentials. As shown in Fig. S9 (ESI†), the potential of PTTD NPs was changed from −19.1 mV to 14.1 mV in 4 h with 1 mM DTT incubation, showing that DSPE-SS-PEG2k would be broken with DTT due to the high reduction of disulfide groups and exposing TPP of PTTD NPs. These results suggested that PEG-ylated nanoparticles with excellent colloidal stability would be beneficial to deliver to tumors, and the positive potential from TPP after PEG disintegration was capable of mitochondria targeting, which provided favorable conditions for the construction of oxygen nano-economizers.
Then the spectral properties of NPs were investigated. As shown in Fig. 2I and Fig. S10 (ESI†), PTD NPs, TTD NPs and PTTD NPs showed a similar fluorescence intensity of TCPP (∼660 nm) to that of the TCPP solution, suggesting the relatively weak aggregation-induced quenching (ACQ) effect of TCPP in NPs. The possible reason may be attributed to the ordered crystal composition of MOFs helping in increasing the distance between the TCPP molecules, thereby significantly relieving the ACQ effect. More excitingly, as shown in Fig. 2J and K, the fluorescence changes of PTTD NPs were consistent with DPA-MOF in the presence of singlet oxygen (0–10 mM). And, there was a good linear relationship between the fluorescence change and the concentration of singlet oxygen in 0–4 mM with a quantification limit of 20.6 μM, which showed non-affection of singlet oxygen detection after PTTD NP synthesis. In addition, under the same illumination conditions, PTTD NPs showed similar fluorescence changes to TD NPs. As shown in Fig. 2L and M, the fluorescence stopped declining after 10 min of illumination, indicating that the synthesis of PTTD NPs had no effect on the singlet oxygen generation for PDT.
2.3. In vitro fluorescence colocalization and cellular uptake
High-efficiency co-delivery of drugs is extremely important for precise PDT. As shown in Fig. S11 (ESI†), we verified the co-delivery of TCPP and DPA by cell fluorescence colocalization experiments. Completely fused TCPP (red fluorescence) and DPA (blue fluorescence) proved the successful synchronous co-delivery of TCPP and DPA, which provided favorable conditions for capturing and self-reporting the singlet oxygen more sensitively. In addition, both fluorescence without an interference were conducive to the observation of singlet oxygen capture and uptake behavior. The blue fluorescence intensity of DPA would decrease with the singlet oxygen, which was favorable to observe real-time changes of singlet oxygen intracellularly. And the fluorescence intensity of TCPP was not affected by singlet oxygen, so the stronger red fluorescence changes could be used to track the location and observe the uptake behavior of NPs.The cellular uptake of NPs was explored by evaluating the intracellular fluorescence signals of TCPP in 4T1 cells. As shown in Fig. 3A and Fig. S12 (ESI†), the cellular uptake of PTD NPs, TTD NPs and PTTD NPs increased in a time-dependent manner at 4, 8 and 12 h compared to 0 h, and PTD NPs and PTTD NPs showed much higher cellular uptake efficiency than that of TTD NPs at 4, 8 and 12 h. The unsatisfactory cellular uptake of TTD NPs should be attributed to poor colloidal stability (Fig. 2F), especially in the presence of buffer salts and FBS in cell culture media. However, there was no significant difference between PTD NPs and PTTD NPs according to the fluorescence quantitative results (Fig. 3A(d)). The reason was that PTD NPs and PTTD NPs had similar colloidal stability and weak ACQ, so both actual cellular uptake efficiencies were comparable. These results revealed that PEG modification significantly improved the cellular uptake efficiency of NPs (Fig. 3A).
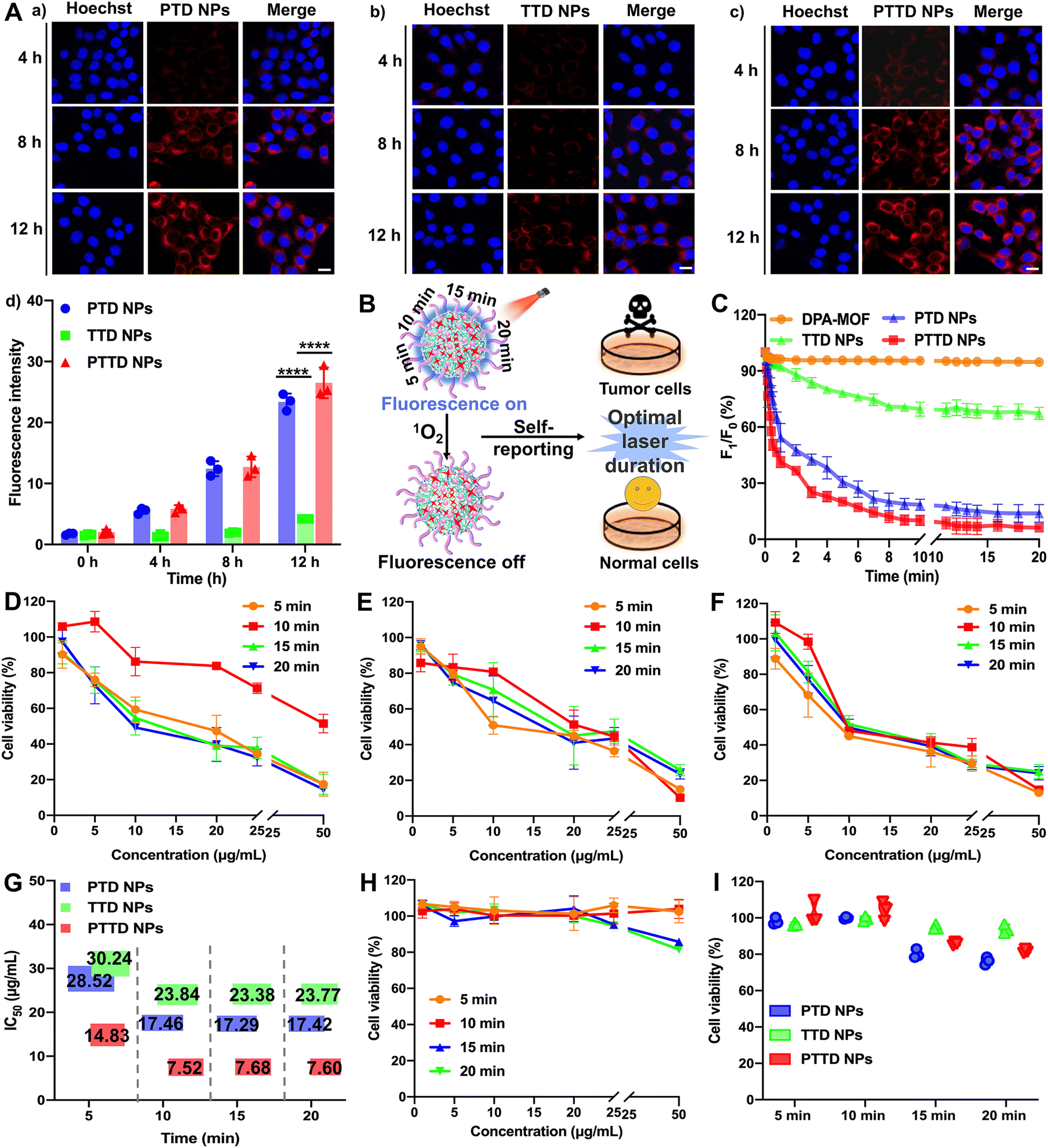 | ||
| Fig. 3 Screening of the optimal treatment scheme in vitro. (A) Cellular uptake: (a)–(c) confocal imaging of 4T1 cells treated with PTD NPs, TTD NPs and PTTD NPs for 4 h, 8 h and 12 h, respectively, scale bar = 10 μm; (d) quantitative analysis of fluorescence intensity. (B) Schematic diagram of optimal laser duration screening. (C) Dynamic changes of fluorescence intensity within 0–20 min of illumination in 4T1 cells treated with DPA-MOF, PTD NPs, TTD NPs and PTTD NPs. (D)–(F) Cytotoxicity against 4T1 cells under different laser conditions. Cytotoxicity against 4T1 cells treated with (D) PTD NPs, (E) TTD NPs and (F) PTTD NPs after 5, 10, 15 and 20 min irradiation (660 nm, 50 mW cm−2). (G) IC50 values of PTD NPs, TTD NPs and PTTD NPs at different laser durations in 4T1 cells. (H) Cytotoxicity against 3T3 cells treated with PTTD NPs after 5, 10, 15 and 20 min irradiation (660 nm, 50 mW cm−2). (I) Ultimate cell survival of 3T3 cells treated with PTD NPs, TTD NPs and PTTD NPs under 5, 10, 15 and 20 min irradiation (660 nm, 50 mW cm−2). Statistics: ****P < 0.0001. | ||
2.4. Self-reported PDT in vitro
PTD NPs, TTD NPs and PTTD NPs could capture singlet oxygen instantaneously due to the superior gas adsorption capacity of MOFs, which visually reported the singlet oxygen yield through fluorescence intensity attenuation. Therefore, we further explored the singlet oxygen self-reporting of NPs to screen the optimal laser duration for precise PDT (Fig. 3B). The dynamic fluorescence intensity changes of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs were observed at different exposure laser durations (Fig. 3C and Fig. S13, ESI†). The fluorescence of PTD NPs and PTTD NPs was rapidly quenched to 50% of the initial fluorescence value within 5 min when exposed to a laser (660 nm, 50 mW cm−2), and the fluorescence of both NPs decreased to less than 10% of the initial fluorescence value with 10 min laser irradiation. However, there were no significant fluorescence changes of all NPs when prolonging the laser durations to 15 min or 20 min, indicating that the PSs in four NPs were fully excited after 10 min of illumination, in which singlet oxygen production reached the maximum to quench the fluorescence of DPA basically. However, the fluorescence of TTD NPs had a tiny decrease by only about 20% after 10 min of illumination, which due to TTD NPs with low uptake efficiency generated less singlet oxygen. These results had proved the singlet oxygen self-reported potential of PTTD NPs, which encouraged us to further explore whether the singlet oxygen generation at an exposure laser duration of 10 min guaranteed the curative effect of PDT.Therefore, the cytotoxicity against 4T1 and 3T3 cells was further used to screen the optimal laser duration for precise PDT. As shown in Fig. 3D–F, the cytotoxicity of PTD NPs, TTD NPs and PTTD NPs reached the maximum after 10 min of irradiation, which was 2 times greater than that in 5 min. The reason was that the singlet oxygen generated by incomplete excitation of PSs at insufficient laser durations was insufficient to kill tumor cells. However, the cytotoxicity had little changes with the illumination of 15 min and 20 min compared to 10 min (Fig. 3G and Table S4, ESI†), which proved that the maximum concentration of singlet oxygen was produced in 10 min. In addition, as shown in Fig. 3H and I and Fig. S14 (ESI†), the cytotoxicity against 3T3 cells was used to screen the most security laser duration to normal cells. We were surprised to find that the survival rate of 3T3 cells was above 95% after an illumination of 10 min, while the viability of 3T3 cells dropped to 85% and 80% after prolonging illumination to 15 min and 20 min. It might be because the non-disintegration of DSPE-SS-PEG2k in normal cells with a low GSH content limited the diffusion of singlet oxygen at appropriate laser durations. However, the prolonged laser duration triggered the disintegration of NPs, which resulted in the diffusion of singlet oxygen from the NPs to 3T3 cells for producing stronger cytotoxicity. These results indicated that the fluorescence of PTTD NPs was quenched after exposing to a laser duration of 10 min, in which time singlet oxygen had the most potent killing effect to tumor cells and sufficient security to normal cells. Therefore, 10 min irradiation were selected as the optimal laser duration to facilitate PDT for cancer therapy.
2.5. Mitochondria targeting ability
As mentioned above, encouraged by the results of singlet oxygen self-reporting for optimal laser duration screening, the tumor hypoxia-alleviation function of the nano-economizer was further explored. As shown in Fig. 4A, the key to kill tumor cells was mitochondrial damage caused by singlet oxygen, which destroyed entire OXPHOS due to complex enzyme inhibition via blocking of electron and proton transfer. Therefore, the mitochondria targeting ability of PTD NPs, TTD NPs and PTTD NPs should be investigated by CLSM first. TPP in both TTD NPs and PTTD NPs exhibited perfect mitochondria-targeting performance (Fig. 4B), which was reflected in the perfect match between NPs (red fluorescence) and mitochondrial markers (green fluorescence). However, PTTD NPs showed superior mitochondrial targeting function with strong red fluorescence in mitochondria while the red fluorescence of TTD NPs was weak, since only a few TTD NPs could reach mitochondria for low cellular uptake efficiency (Fig. 3A). In contrast, the fluorescence of PTD NPs without TPP did not match with the mitochondrial markers on account of no mitochondria targeting function. Therefore, the nanoparticles with TPP had superior mitochondrial targeting ability to provide the possibility for enhancing PDT.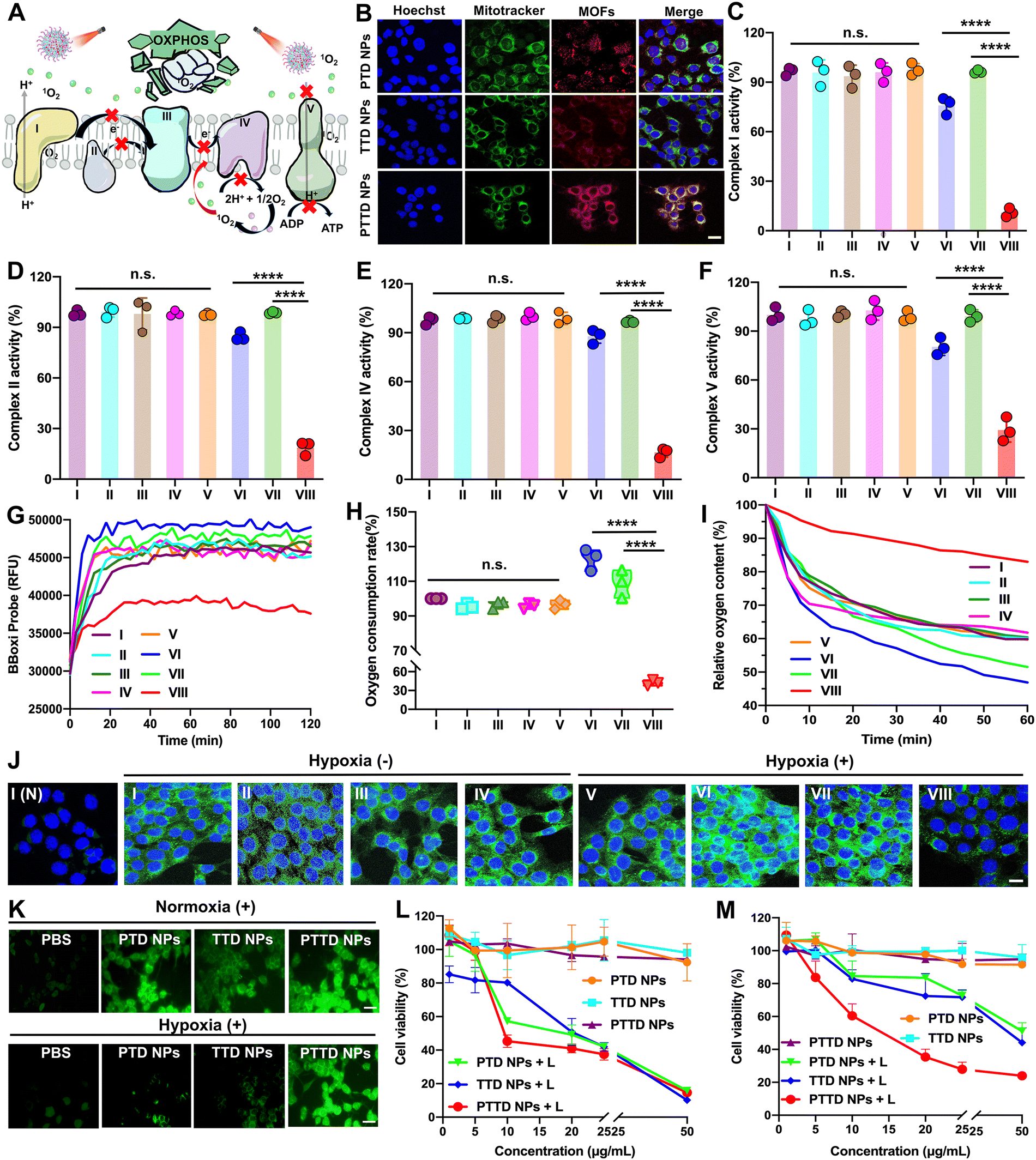 | ||
| Fig. 4 In vitro hypoxia alleviation and antitumor effect of NPs. (A) Schematic diagram of the antitumor mechanism via OXPHOS damage. (B) CLSM images about mitochondria-targeting of PTD NPs, TTD NPs and PTTD NPs, scale bar = 10 μm. (C)–(F) The mitochondrial respiration-related enzyme relative activity of (C) complex I, (D) complex II, (E) complex IV, and (F) complex V. (G) OCR change in 4T1 cells of PBS, PTD NPs, TTD NPs and PTTD NPs with or without laser irradiation (50 mW cm−2, 10 min) at 0–120 min. (H) Quantitative analysis of OCR at 120 min. (I) Relative oxygen content in the 4T1 cells of PBS, PTD NPs, TTD NPs and PTTD NPs with or without laser irradiation (50 mW cm−2, 10 min) at 0–60 min. (J) Immunofluorescence images of HIF-1α with or without laser (50 mW cm−2, 10 min), scale bar = 10 μm. (K) Cellular ROS generation in the 4T1 cells of PBS, PTD NPs, TTD NPs and PTTD NPs with laser irradiation (50 mW cm−2, 10 min) under normoxic or hypoxic conditions, scale bar = 20 μm. (L and M) Cytotoxicity against 4T1 cells with or without laser irradiation (50 mW cm−2, 10 min) under (L) normoxic and (M) hypoxic conditions. (I (N), PBS (Normoxia); I, PBS; II, PTD NPs; III, TTD NPs; IV, PTTD NPs; V, PBS + L; VI, PTD NPs + L; VII, TTD NPs + L; VIII, PTTD NPs + L). Statistics: ****P < 0.0001. | ||
2.6. Multimodal mitochondrial damage assay
Singlet oxygen damaged mitochondria by inactivating complex enzymes, which finally lead to OXPHOS destroyed for oxygen consumption inhibition. After confirming the mitochondria targeting ability of NPs, we further explored PTD NP, TTD NP and PTTD NP-initiated singlet oxygen burst affecting the activity of complex enzymes. As shown in Fig. 4C, compared with the control group (∼100), PTTD NPs displayed a significant complex I inactivation with the lowest activity values of 6.9-fold lower than that of the PTD NP group and 8.8-fold lower than that of the TTD NP group under laser irradiation. Similarly, as shown in Fig. 4D, PTTD NPs reduced the activity of complex II, which was 4.5-fold lower than that of the PTD NP group and 5.3-fold lower than that of the TTD NP group, displaying a similar tendency to complex I. Complex I and complex II are two different respiratory initiation sites on the ETC chain, and the blocking of the two complex enzymes by PTTD NPs demonstrated that the OXPHOS was blocked at the starting point. Whereafter, electron sources were transferred from complexes I and II to complex III, and cytochrome c in complex III was reduced and in turn shuttled to complex IV to donate its electron for the final reduction of oxygen. Therefore, inhibiting complex I and complex II finally damaged complex IV, which would block electrons transferring to restrain cellular respiration for inhibiting oxygen consumption. As shown in Fig. 4E, complex IV was inactivated by singlet oxygen, showing the inhibitory effect of PTTD NPs on complex IV up to 82.3% compared with the control group.Complex V availed the proton gradient produced by complexes I, II and IV to phosphorylate ADP for generating ATP.44–46 The inhibitory effect on complexes I, II and IV would further inhibit electron and proton transfer, which further damaged complex V. As shown in Fig. 4F, PTTD NPs displayed significant passivation to complex V with the lowest activity values of 2.7-fold lower than that of the PTD NP group and 3.4-fold lower than that of the TTD NP group under laser irradiation. The inhibition of complex V significantly caused mitochondrial damage to blocked proton transmission, which resulted in the disappearance of the proton gradient to inhibit ATP synthesis. As shown in Fig. S15 (ESI†), PTTD NPs showed strong resistance to ATP synthesis. Compared with the control group (∼100), the ATP content in the PTTD NP group significantly decreased to 4.56%, 16.25-fold lower than that in the PTD NP group and 20-fold lower than that in the TTD NP group. These results proved that PTTD NPs significantly inhibited the activity of complexes due to excellent mitochondria targeting ability and high uptake capacity, and a large of singlet oxygen generated from PTTD NPs under optimal laser duration conditions would destroy entire OXPHOS to reduce oxygen consumption for alleviating tumor hypoxia.
2.7. Hypoxia alleviation in vitro
The inhibition of mitochondrial complexes destroyed the entire OXPHOS, which could effectively reduce the respiratory rate of tumor cells. As shown in Fig. 4G, the mitochondrial oxygen consumption rate (OCR) of 4T1 cells was evaluated using a multimode microplate reader. After treatment with PTTD NPs under laser irradiation (660 nm, 50 mW cm−2) for 10 min, the OCR of 4T1 cells was greatly decreased, which proved an effective mitochondrial respiratory inhibition. And the quantitative statistics provided more visually powerful results (Fig. 4H). Notably, the PTTD NPs + L group always kept the lowest OCR, which reduced 17.8% than the PBS group due to great respiration inhibition caused by mitochondrial damage. However, the PTD NPs under laser irradiation showed higher OCR than any other groups, which due to PTD NPs could not target to mitochondria but consume massive oxygen during PDT, exacerbating tumor hypoxia to accelerate cellular respiration. Moreover, to further monitor the oxygen consumption intracellularly, the oxygen content of the cell medium was measured using dissolved oxygen meters. As shown in Fig. 4I, compared with other groups, the relative oxygen content of the PTTD NPs + L group could keep 85% of the initial value, which was 30% more than the other groups. Therefore, PTTD NPs under laser irradiation significantly decreased oxygen consumption caused by mitochondrial respiratory, suggesting great potential to provide oxygen for self-facilitating PDT in hypoxic tumors.Moreover, the results of hypoxia-inducible factor-1α (HIF-1α) could also prove the increase of the oxygen content in tumor cells obviously. The expression of HIF-1α in 4T1 cells was detected by immunofluorescence staining, and the green fluorescence intensity of cells was directly proportional to the expression of HIF-1α. As shown in Fig. 4J and Fig. S16 (ESI†), there is a low HIF-1α expression in 4T1 cells of the PBS group under normoxic conditions, while the obvious green immunofluorescence of HIF-1α treated with PBS was observed under hypoxic conditions. Moreover, the overexpression of HIF-1α in the cells treated with PTD NPs and TTD NPs under laser irradiation (660 nm, 50 mW cm−2, 10 min) showed obvious green fluorescence, indicating the aggravated hypoxia by PDT. Among them, the PTTD NPs + L group showed obvious advantages to reverse the overexpression of HIF-1α under hypoxia conditions (Fig. S16, ESI†), which could significantly alleviate cell hypoxia and improve the oxygen content almost up to normoxia conditions. Therefore, these results revealed that PDT could significantly up-regulate cellular HIF-1α, while the inhibition of tumor cell respiration by destroying OXPHOS to improve the oxygen content could effectively fight against the process.
2.8. Cellular singlet oxygen generation
The oxygen consumption inhibition caused by entire OXPHOS damage proved an important role of the singlet oxygen burst in destroying mitochondria. Therefore, singlet oxygen production in tumor cells needed to be further explored. Herein, DCFH-DA (a cell-permeable probe) is used to detect intracellular singlet oxygen generation via changes of fluorescence. As shown in Fig. 4K and Fig. S17, S18 (ESI†), under normoxic conditions, the cellular fluorescence signals treated with PTD NPs, TTD NPs and PTTD NPs under laser irradiation sharply enhanced compared with the cells without laser treatment. And the fluorescence intensity of PTD NPs and PTTD NPs was stronger than that of TTD NPs under laser irradiation, which was probably caused by excellent colloidal stability (Fig. 2F) and high cellular uptake efficiency (Fig. 3A) of PEGylated nanoparticles. Notably, such an oxygen nano-economizer with oxygen consumption inhibition also showed great bursts of singlet oxygen even at a hypoxic environment. As shown in Fig. 4K and Fig. S18 (ESI†), the fluorescence intensity of PTTD NPs with a laser under hypoxia conditions was similar to that in the normoxic environment, while the fluorescence intensity of PTD NPs and TTD NPs was very weak. These results proved that PTTD NPs at appropriate laser durations would self-facilitate PDT by providing more available oxygen to generate singlet oxygen due to OXPHOS inhibition even under hypoxic conditions.2.9. In vitro cytotoxicity
Then, we further evaluated cytotoxicity in vitro in two tumor cell lines (4T1 and CT26 cells) under normoxic and hypoxic conditions using the MTT assay. As shown in Fig. 4L and M and Fig. S19, Table S5 (ESI†), PTD NPs, TTD NPs and PTTD NPs demonstrated significantly enhanced cytotoxicity under laser irradiation while NPs without laser irradiation showed tiny cytotoxicity against both 4T1 and CT26 cells, suggesting the anticancer effects of singlet oxygen generation by excited TCPP. Moreover, NPs with laser irradiation under normoxic conditions showed stronger cytotoxicity than that under hypoxic conditions, indicating an adversarial effect of hypoxia on PDT (Fig. 4M and Fig. S19, ESI†). However, PTTD NPs revealed similar cytotoxicity under hypoxic conditions compared to that under normoxic conditions. This was attributed to the unique characteristics of PTTD NPs in mitochondria targeting and oxygen consumption inhibition, which provided more oxygen to generate singlet oxygen for tumor cell killing. These results showed that PTTD NPs with excellent photodynamic cytotoxicity could alleviate the adversarial effect of hypoxia on PDT. Taken together, PTTD NPs generated singlet oxygen to destroy entire OXPHOS, which decreased oxygen consumption and provided more oxygen for enhancing PDT. Such an elaborately engineered oxygen nano-economizer showed promising integration of diagnosis and treatment advantages by screening the optimal exposure laser duration and alleviating hypoxia for self-reporting and self-facilitating PDT.2.10. Biodistribution
We then studied the biodistribution of PTD NPs, TTD NPs and PTTD NPs in 4T1 breast tumor-bearing BALB/c mice. At 48 h post-administration, all the mice were sacrificed and the main organs and tumors were taken out to further quantify the ex vivo fluorescence intensity. As shown in Fig. 5A and B, all three NPs showed the maximum accumulation of TCPP in tumors at 12 h, and the fluorescence intensity gradually decreased from 12 h to 48 h at tumor sites. PTD NPs and PTTD NPs showed strong fluorescence signals at the peak fluorescence signals of 12 h. By contrast, lower fluorescence signals were found in tumors of TTD NP-treated mice, which indicated that the PEG-ylated nanoparticles could promote the specific accumulation of tumors due to excellent stability in blood and the enhanced permeability and retention (EPR) effect. Moreover, after 48 h injection, the fluorescence of ex vivo was obtained, which was similar to the results in vivo, and PTTD NPs and PTD NPs had stronger fluorescence at tumor sites compared to TTD NPs ex vivo (Fig. 5C−E). Obviously, improving the stability of NPs by PEG modification in the blood would no doubt significantly facilitate the accumulation of nanoparticles in tumors. Moreover, these biodistribution results figured out a beginning laser irradiation time after injection for the in vivo therapeutic scheme.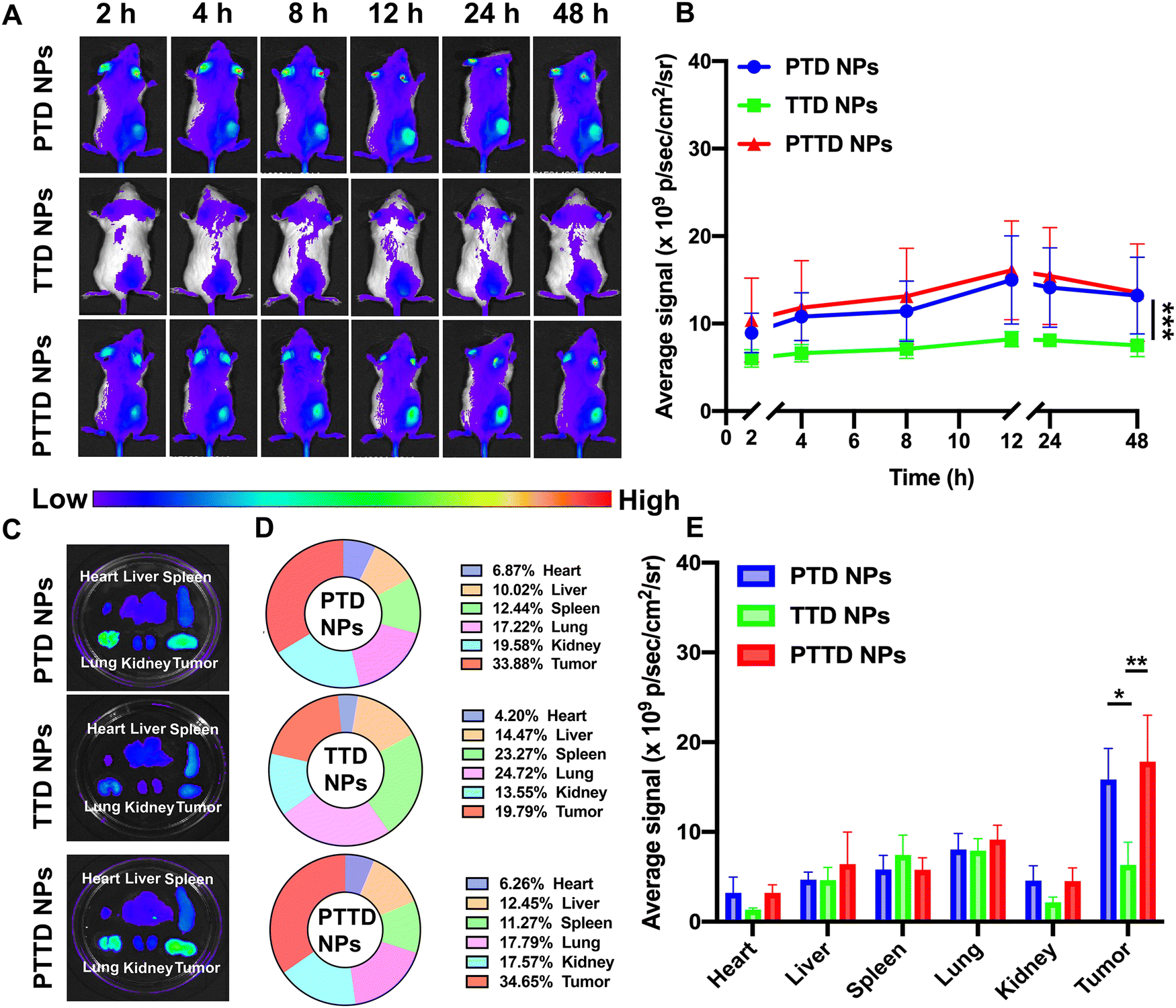 | ||
| Fig. 5 In vivo and ex vivo biodistributions of PTD NPs, TTD NPs and PTTD NPs (n = 4). (A) Fluorescence images in vivo at different time intervals. (B) Quantitative results of the TCPP distribution at the tumor sites in different times in vivo. (C) Fluorescence images of the TCPP distribution in the major organs and tumors ex vivo at 48 h. (D) Fluorescence percentages of each organ and tumor ex vivo at 48 h. (E) Quantitative results of the TCPP distribution in the major organs and tumor ex vivo at 48 h. Data were analyzed using an IVIS spectrum small-animal in vivo imaging system, *P < 0.05, **P < 0.01 and ***P < 0.001. | ||
2.11. Self-reported optimal laser duration in vivo
Subsequently, PTTD NPs were utilized to screen the optimal laser duration in vivo due to the best tumor accumulation. As shown in Fig. 6A, after 12 h intravenous injection of PTTD NPs (10 mg kg−1) in 4T1 breast tumor-bearing mice, different illumination times (5, 10, 15, 20 min) were applied at the tumor sites with a 660 nm laser (50 mW cm−2). The optimal laser duration was screened by the blue fluorescence changes of PTTD NPs (Fig. 6B and Fig. S20, ESI†). Compared to the PTTD NPs with no laser (0 min), the blue fluorescence quenched to 50.55% of the initial value in the PTTD NPs + L (5 min) group. And the fluorescence of the PTTD NPs + L (10 min) group was almost completely quenched to the 1.91% of initial value. However, when prolonging the laser duration to 15 min or 20 min, the fluorescence of PTTD NPs did not change any more, which indicated that the TCPP in PTTD NPs was completely excited, and the maximum singlet oxygen was generated to induce fluorescence self-quenching under 10 min laser radiation. Then, to further determine the optimal time of PTTD NPs, tumor growth at different laser durations was observed. Consistent with the laser exposed time that caused the fluorescence self-quenching of PTTD NPs, after 10 min laser duration, PTTD NPs showed high tumor growth inhibition. However, there had no obviously increased effect on the tumor therapy when increasing the exposure laser time to 15 min and 20 min. As shown in Fig. 6C–F and Fig. S21 (ESI†), the tumor weight and tumor burden were also no longer changed in the groups of PTTD NPs + L (10 min), PTTD NPs + L (15 min) and PTTD NPs + L (20 min). Furthermore, the HE and TUNEL assay results also proved the PTTD NPs + L (10 min) group with the strongest lesions of tumors, and no significant lesions increased when prolonged the exposure time to 15 min and 20 min (Fig. 6G and Fig. S22, ESI†). These results proved that 10 min of illumination could cause the fluorescence of PTTD NPs to be basically quenched, achieving the maximum tumor inhibition and damage effect at the same time. However, when the illumination time was extended to 15 min or 20 min, the fluorescence intensity of PTTD NPs was unchanged and the tumor inhibition effect was no longer increased. More importantly, the prolonging laser duration resulted in side effects such as the weight loss (Fig. 6H) and abnormal liver function (Fig. 6I), which are attributed to the long-term thermal radiation caused by high-energy photons resulted in harmful impacts on normal tissues and organs. Therefore, consistent with the results in vitro, the PTTD NPs self-reported the optimal laser duration to be 10 min successfully via blue fluorescence quenching, which could provide the optimal therapeutic scheme for self-facilitated PDT in vivo.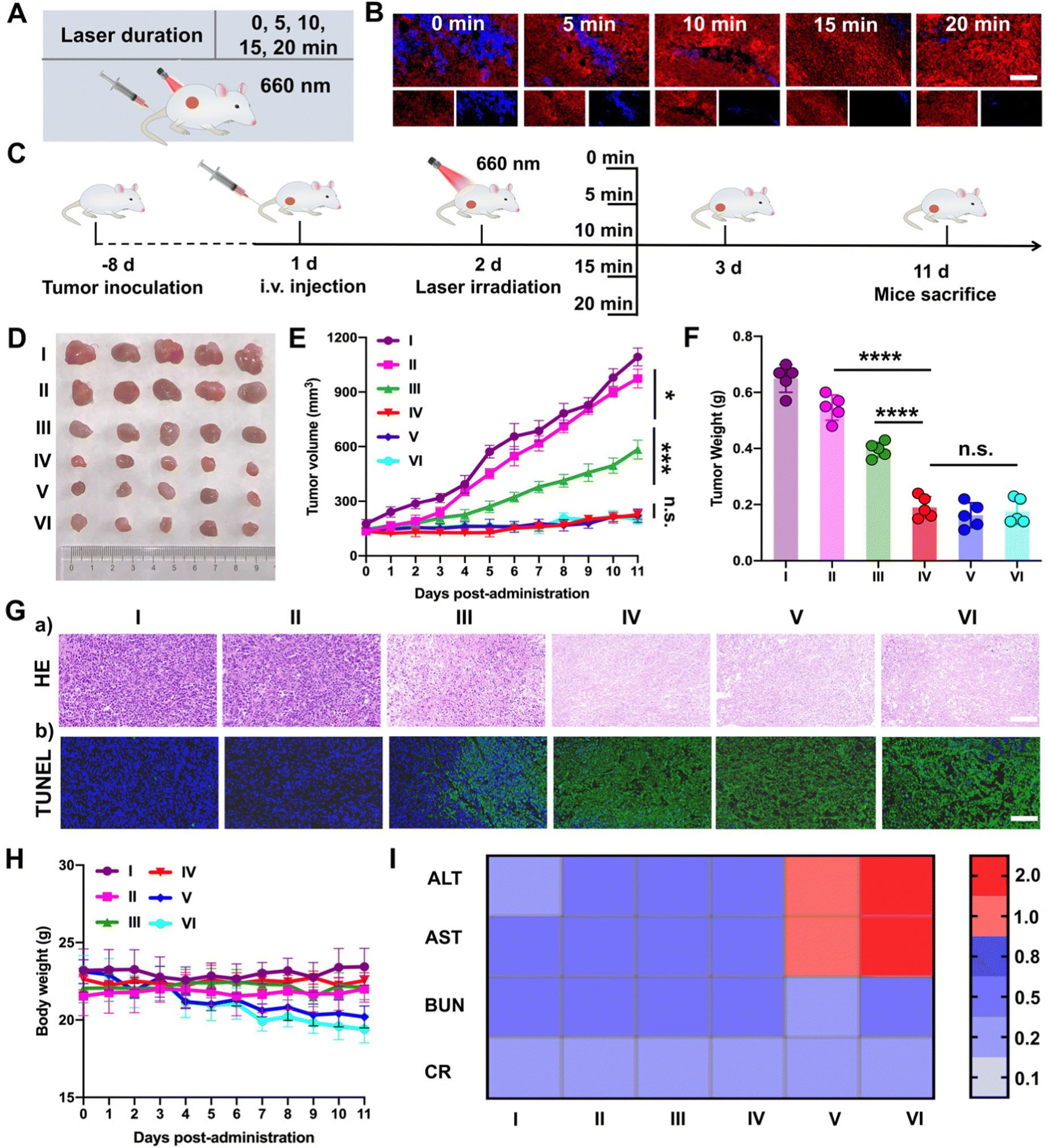 | ||
| Fig. 6 Self-reported laser duration in 4T1 breast xenograft tumors in vivo (n = 5). (A) Scheme of optimal laser duration screening. (B) Fluorescence changes of PTTD NPs at different laser durations (blue: PTTD NPs, red: DAPI, scale bars = 50 μm). (C) Screening of the optimal laser duration via tumor treatment in vivo. (D) Tumors images. (E) Tumor growth profiles and (F) tumor weight at 11 days post-administration. (G) (a) H&E assay and (b) immunofluorescence staining of the TUNEL assay, scale bars: 50 μm. (H) Changes in the body weight. (I) Liver and kidney functions in the blood of Balb/c mice (the maximum healthy value is 1.0). Groups: I, saline; II, PTTD NPs + L (0 min); III, PTTD NPs + L (5 min); IV, PTTD NPs + L (10 min); V, PTTD NPs + L (15 min); VI, PTTD NPs + L (20 min). *P < 0.05, ***P < 0.001, ****P < 0.0001. | ||
2.12. In vivo multimodal synergetic cancer therapy
Furthermore, the results above encouraged us to further investigate the hypoxia-alleviation in tumors and the synergetic antitumor activity in vivo under optimal laser duration conditions. Saline, PTD NPs, TTD NPs and PTTD NPs were intravenously administrated to the 4T1 breast tumor-bearing mice every other day for a total of five injections at doses of 10 mg kg−1. According to the results of the distribution and optimal laser duration (Fig. 5 and 6), laser-treated groups received irradiation (50 mW cm−2) for 10 min at 12 h post-injection (Fig. 7A). After five treatments, a hypoxyprobe (pimonidazole, hypoxyprobe-1 plus kit) was used to observe hypoxia-alleviation. The mice were sacrificed after 1.5 h intraperitoneal injection of pimonidazole on the 11th day, and the main organs and tumor tissues were collected. As shown in Fig. 7B, the immunofluorescence of hypoxyprobe-1 proved that PTTD NPs significantly alleviated tumor hypoxia under laser irradiation through the red fluorescence expression was weaker over other formulations. In addition, the hypoxia-alleviation in tumors was also explored by the expression of HIF-1α. In the groups without laser, the expression of HIF-1α was similar to that in the saline group. However, an increased HIF-1α expression was observed in PTD NPs and TTD NPs with laser (groups VI and VII), indicating the aggravated hypoxia by PDT. In contrast, an obviously decreased HIF-1α expression was detected in the PTTD NPs with laser (group VIII), and the downregulation of HIF-1α suggesting that PTTD NPs showed efficient hypoxia alleviation in tumors (Fig. 7B).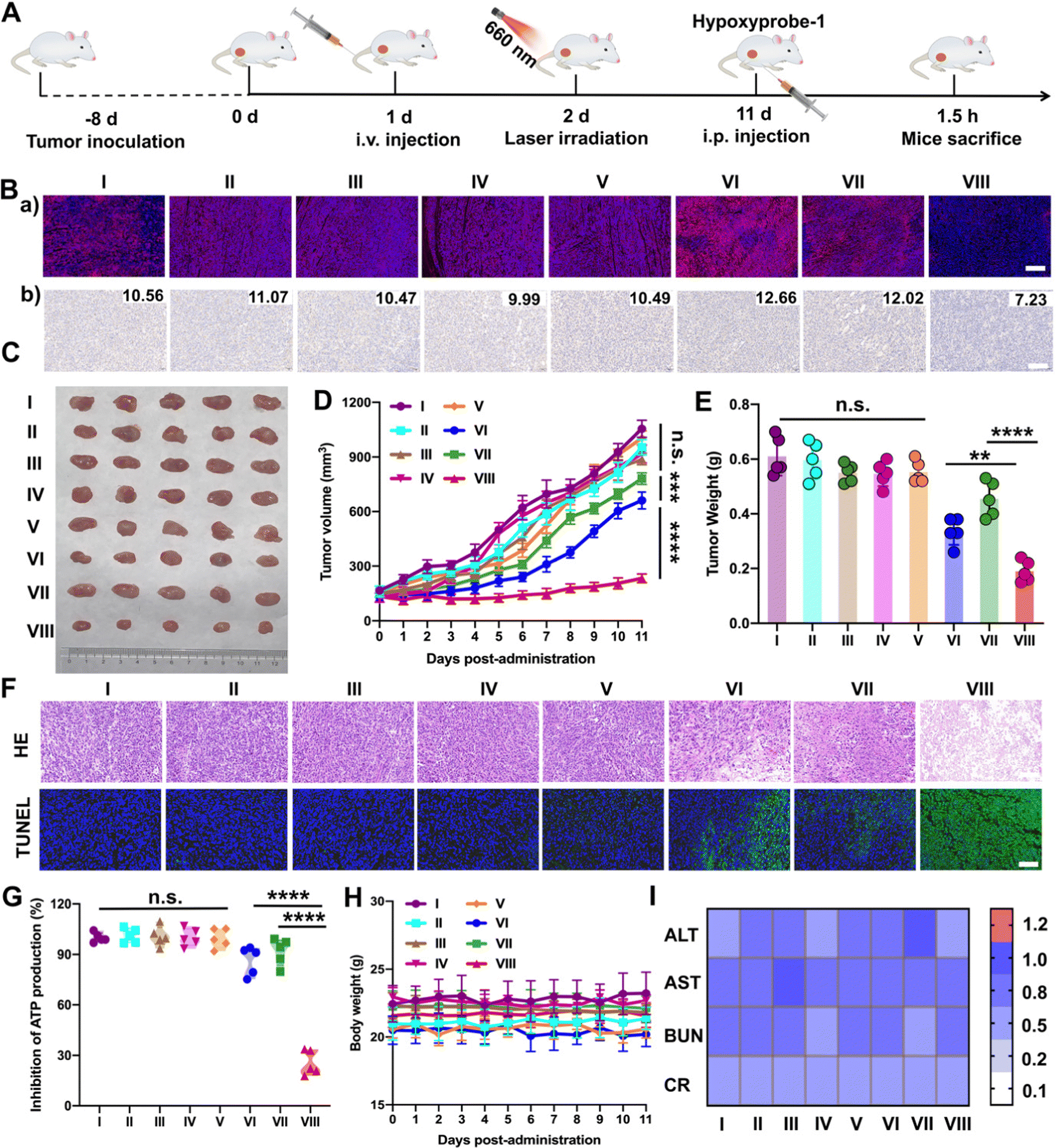 | ||
| Fig. 7 In vivo antitumor activity against the 4T1 breast xenograft tumor (n = 5). (A) Treatment scheme for photodynamic therapy. (B) (a) Immunofluorescence staining of hypoxyprobe-1 and (b) immunohistochemical staining and H-SCORE analysis of HIF-1α after different treatments. Scale bars, 50 μm. (C) Tumors images. (D) Tumor growth profiles and (E) tumor weight at 11 days post-administration. (F) H&E assay and immunofluorescence staining of the TUNEL assay. Scale bars, 50 μm. (G) Inhibition of ATP in tumors. (H) Changes in the body weight. (I) Liver and kidney functions in the blood of Balb/c mice (the maximum healthy value is 1.0). Groups: I, saline; II, PTD NPs; III, TTD NPs; IV, PTTD NPs; V, saline + L; VI, PTD NPs + L; VII, TTD NPs + L; VIII, PTTD NPs + L. **P < 0.01, ***P < 0.001, ****P < 0.0001. | ||
Hypoxia-alleviation in tumors could obviously improve the effect of PDT by increasing the oxygen content for photoactive oxygen conversion. As shown in Fig. 7C–E, similar to saline, NPs without laser showed little tumor suppressive effects due to tiny singlet oxygen generation. PTD NPs and TTD NPs also showed inferior antitumor activity even under laser irradiation, while the PTTD NPs with laser demonstrated distinct therapeutic advantages over other formulations. The results of the HE and TUNEL assays also confirmed the potent tumor suppression capacity of PTTD NPs (Fig. 7F), which should be ascribed to PTTD NPs alleviate tumor hypoxia through the damaging OXPHOS pathway. As shown in Fig. 7G, the decrease of ATP in the PTTD NP group proved tumor damage via complex inhibition in mitochondria, which caused by singlet oxygen generation by PTTD NPs under laser irradiation. Taken together, the mitochondrial damage blocked entire OXPHOS pathway, which further increased oxygen consumption inhibition to relieve tumor hypoxia and provided more singlet oxygen to promote PDT. The potent antitumor activity of PTTD NPs should be ascribed to multiple advantages throughout the delivery process: (i) synchronous co-delivery of DPA and TCPP (Fig. 2); (ii) favorable colloidal stability (Fig. 2); (iii) efficient cellular uptake and the self-reporting of optimal laser duration (Fig. 3); (iv) excellent mitochondrial targeting capacity after TPP modification (Fig. 4); (v) definite mechanisms of tumor hypoxia-alleviation caused by OXPHOS inhibition (Fig. 4); (vi) abundant accumulation in tumors after PEGylation modification (Fig. 5); and (vii) the optimal laser duration caused by precise self-reported PDT in vivo (Fig. 6). Moreover, the H&E staining results (Fig. S23, ESI†) revealed no distinct damage in the major organs (heart, liver, spleen, lung and kidney). And the sustained stability of the hemolysis test, body weight and normal liver and kidney function index also indicated that nanoparticles had high biosafety (Fig. 7H and I and Fig. S24, ESI†). These results indicated that PTTD NPs with significant antitumor synergistic effects demonstrated no obvious systemic toxicity to the animals under at the optimal exposure laser duration.
3. Conclusions
In summary, an oxygen nano-economizer consisting of DPA-MOF, TCPP, TPP and DSPE-SS-PEG2k was rationally engineered by a comprehensive consideration of several important issues for improving PDT, mainly including the (i) high spatiotemporal consistency of singlet oxygen self-generation and self-reporting via the efficient and synchronous co-delivery of DPA and TCPP; (ii) self-reported optimal laser duration of PDT according to fluorescence self-quenching of DPA; (iii) self-facilitated PDT via hypoxia alleviation in tumors caused by the entire OXPHOS pathway damage; (iv) efficient antitumor treatment in vitro and in vivo via tumor-specific accumulation and definite synergistic therapeutic mechanism with high security. As a result, the self-reported and self-facilitated oxygen nano-economizer demonstrated multiple therapeutic advantages in cancer therapy. This uniquely engineered nano-economizer drives a conceptual step forward in theranostic nanoplatform design and provides a new paradigm for photo-therapeutic modality toward clinical treatment of cancer.4. Experimental
4.1. Materials
DPA was purchased from Shanghai Damas Chemical Technology Co. Ltd, China. TCPP and TPP were purchased from Shanghai Leyan Biomedical Co. Ltd, China. DSPE-SS-PEG2k was purchased from Ruixi Bio (Xian, China). The RPMI 1640 and DMEM cell culture medium, penicillin–streptomycin and fetal bovine serum were obtained from GIBCO, Invitrogen Corp., USA. Cell culture dishes and plates were bought from NEST Biotechnology Co., Ltd (Wuxi, China). Mitochondrial Complex I, Complex II, Complex IV, micromitochondrial respiratory chain complex V activity assay kit and ATP content assay kit were purchased from Beijing Solarbio Science & Technology Co., Ltd. HIF-1α, Triton X-100, bovine serum albumin and cellular singlet oxygen detection assay kit (DCFH-DA) were purchased from Dalian Meilun Biotech Co. Ltd, China. 3-(4,5-Dimthyl-2-thiazolyl)-2,5-dipphenyl-2H-terazolium bromide (MTT) and trypsin–EDTA were obtained from Sigma-Aldrich, USA. Hoechst 33342 was purchased from BD Biosciences, USA. Other chemicals and solvents were of analytical or HPLC grade.4.2. Preparation of DPA-MOF
DPA (20.9 mg, 0.05 mmol), ZrOCl2·8H2O (10 mg, 0.03 mmol) and benzoic acid (100 mg, 0.82 mmol) were ultrasonically dissolved in 50 mL of dimethyl formamide (DMF), and the mixture was stirred (500 rpm) at 100 °C for 5 h. The reaction solution was cooled down to room temperature after completion, and the formed NPs were collected by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min) and washed with DMF to remove excess chemicals. The formed products were then washed with ultrapure water to remove residual DMF.
000 rpm, 10 min) and washed with DMF to remove excess chemicals. The formed products were then washed with ultrapure water to remove residual DMF.
4.3. Singlet oxygen detection sensitivity of DPA and DPA-MOF
H2O2 and sodium hypochlorite (NaClO) were employed to produce singlet oxygen. Concentrations of H2O2 from 0 to 10 mM were added into the DPA solution and DPA-MOF and then 10 mM NaClO was added into the mixture solution. After 10 min incubation, the fluorescence of DPA was recorded to detect singlet oxygen (F0/F1, where F0 is the initial fluorescence intensity of DPA and F1 is the fluorescence intensity of DPA measured at various time points with laser).4.4. Photodynamic efficiency of PSs
The singlet oxygen production of different PSs under laser irradiation (660 nm, 50 mW cm−2) was determined using a singlet oxygen sensor green reagent (SOSG) kit and DPA-MOF simultaneously. After mixing TCPP, PpIX, Ce6, MB, ZnPc and ICG (2 μg mL−1) with 2 μmol mL−1 SOSG or 30 μg mL−1 DPA-MOF, the mixed solution was irradiated for 10 min. The fluorescence changes of SOSG (ex: 504 nm and em:525 nm) and DPA-MOF (ex: 365 nm and em: 460 nm) were evaluated using a Varioskan LUX multimode microplate reader.4.5. Preparation and singlet oxygen self-reporting of TD NPs
The preparation of TD NPs was performed in a similar manner with DPA-MOF. DPA, ZrOCl2·8H2O and benzoic acid were stirred (500 rpm) at 100 °C for 3 h, and TCPP (2 mg, 0.0026 mmol) was added into the mixture and stirred for another 3 h. After successful synthesis, TD NPs (30 μg mL−1) were irradiated with laser (660 nm, 50 mW cm−2), and the corresponding fluorescence emission spectra were recorded in turn at 0, 1, 3, 5, 8, 10, 15, 20 and 30 min using a Varioskan LUX multimode microplate reader.4.6. Preparation and characterization of PTD NPs, TTD NPs and PTTD NPs
TPP (20 mg) and TD NPs (4 mg) were dissolved in 20 mL of DMF. After ultrasonication, two solutions were mixed and stirred at room temperature (1000 rpm) for 12 h. The formed TTD NPs (TPP@TCPP@DPA-MOF) were collected by centrifugation (10000 rpm, 10 min) and washed with DMF and ultrapure water, respectively. The PEGylated nanoparticles (PTTD NPs) were prepared by a very similar fabrication process to TTD NPs, except for using DSPE-SS-PEG2k (20 wt%) dissolved in DMF as the PEGylation modifier. And PTD NPs (DSPE-SS-PEG2k@TCPP@DPA-MOF) were prepared from TD NPs by using DSPE-SS-PEG2k (20 wt%) as the PEGylation modifier directly.
The morphologies of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs were observed by SEM and TEM (TESCAN MIRA LMS, Czech Republic and JEOL 100CX II, Japan) after staining with 2% phosphotungstic acid. The particle size and zeta potential of NPs were measured using a Zetasizer (NanoZS, Malvern Co., UK). The size changes of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs were recorded while incubated in PBS (pH 7.4) consisting of 10% FBS in a shaking box (37 °C) at 0, 1, 2, 4, 6, 8, 12, 24, 36, 48 and 72 h. Moreover, the long-term stability of four particles was also determined once a day during storage in a refrigerator (4 °C) for 7 days. Furthermore, the zeta changes of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs were determined during incubation in PBS (pH 7.4) consisting of 1 mM DTT in a shaking box (37 °C) at 0, 0.5, 1, 2 and 4 h.
4.7. Co-assembly mechanisms
The three-dimensional molecular structures of TCPP in TD NPs and TPP were obtained through the Autodock Vina software. The results were analyzed using a discovery Studio 2017 Visualizer software. The intermolecular forces were investigated by incubating the TTD NPs with SDS (200 mM) and urea (200 mM), and the zeta potentials were determined using a Zetasizer (NanoZS, Malvern Co., UK). Moreover, the ultraviolet spectra of TCPP solution, DPA-MOF, TD NPs, PTD NPs and PTTD NPs (30 g mL−1) were scanned from 300 nm to 700 nm and analyzed using a multimode microplate reader (Thermo Scientific, USA) to examine the existence of π–π stacking interactions.4.8. Fluorescence spectra
The fluorescence spectra of TCPP solution, mixture solution of DPA-MOF and TCPP, PTD NPs, TTD NPs and PTTD NPs (equal to 10 μg mL−1 of TCPP) were scanned at an excitation wavelength of 415 nm and an emission wavelength of 675 nm using a Varioskan LUX multimode microplate reader (Thermo Scientific, USA).4.9. Singlet oxygen reporting of PTTD NPs
H2O2 at concentrations from 0 to 10 mM was added into PTTD NPs (30 μg mL−1) and then, 10 mM NaClO was added into the mixture. After 10 min incubation, the fluorescence of PTTD NPs was recorded to reflect on the quantitation of the singlet oxygen detection limit (F0/F1, where F0 is the initial fluorescence intensity of DPA and F1 is the fluorescence intensity of DPA measured at various time points using a laser). Then, PTTD NPs were irradiated to observe fluorescence quenching under laser irradiation (660 nm, 50 mW cm−2), and the corresponding fluorescence emission spectra of NPs were recorded in turn at 0, 1, 3, 5, 8, 10, 15, 20 and 30 min using a Varioskan LUX multimode microplate reader.4.10. Cell culture
Cells were gained from American Type Culture Collection (ATCC) and cultured in the RPMI 1640 medium or the DMEM medium containing 10% FBS, penicillin (100 units per mL) and streptomycin (100 μg mL−1). The cells were incubated in a humid atmosphere (5% CO2) at 37 °C.4.11. Co-localization of DPA and TCPP
4T1 cells (5 × 104 cells per well) were seeded into 24-well plates and cultured under dark conditions. After 12 h incubation, DPA-MOF, PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1) were added into the wells. After uptake for another 12 h, the cells were washed with ice-cold PBS, and the intracellular fluorescence intensity of DPA and TCPP was recorded using an Eclipse Ti-U inverted microscope (Nikon Corp., Tokyo, Japan).4.12. Cellular uptake
4T1 cells (5 × 104 cells per well) were seeded into 24-well plates and incubated for 12 h to study the uptake manner of NPs. After incubation, PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1) were added in each well, and the cells were cultured for another 0, 4, 8 and 12 h, respectively. Then, the drug-containing media were removed, and the cells were washed three times with ice-cold PBS and fixed with 4% paraformaldehyde. At last, the cell nuclei would be stained for 10 min by using Hoechst 33342. The intracellular fluorescence signals were observed by confocal laser scanning microscopy (CLSM, C2, Nikon, Japan).4.13. Self-reporting of the optimal laser duration in vitro
4T1 cells (5 × 104 cells per well) were seeded into 24-well plates and cultured for 12 h. PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1) were added into wells and cells were incubated with NPs for another 12 h. After incubation, the media containing nanoparticles were removed, and the cells were exposed to laser irradiation (660 nm, 50 mW cm−2) for different durations (5, 10, 15, 20 min), separately. The intracellular fluorescence intensity of DPA in DPA-MOF, PTD NPs, TTD NPs and PTTD NPs was recorded over time by using an Eclipse Ti-U inverted microscope during illumination (Nikon Corp., Tokyo, Japan).The cytotoxicity of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs with different laser durations was evaluated by the MTT assay in both 4T1 cells and 3T3 cells. Cells (2 × 103 cells per well) were seeded into 96-well plates before drug treatment. After incubation for 12 h, blank media were replaced by concentrations of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs, respectively. After the uptake of nanoparticles for 12 h, the groups with laser were exposed to a laser source (660 nm, 50 mW cm−2) for 5, 10, 15 and 20 min. Afterward, the cells were further cultured for 24 h under the dark conditions. After this, the cells were incubated for another 4 h at 37 °C after adding 25 μL of MTT (5 mg mL−1). Finally, the entire media were removed and replaced by 200 μL of DMSO, and the absorbency was measured using a Varioskan LUX multimode microplate reader (Thermo Scientific, USA).
4.14. Photodynamic action on mitochondria
The 4T1 cellular mitochondria targeting was assessed using a CLSM. 4T1 cells (5 × 104 cells per well) were seeded into 24-well plates and cultured for 12 h. Fresh culture media containing PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1) were added in each well. Then, the drug-containing media were removed, and the cells were stained with Mito-tracker Green for 30 min to label mitochondria. Then, Hoechst 33342 was used to stain the cell nuclei for 15 min. Finally, the intracellular fluorescence signals were observed by confocal laser scanning microscopy (CLSM, C2, Nikon, Japan). The cells without any treatment were used as the control.Mitochondria complexes I, II, IV and ATPsynthase (mitochondrial Complex V) activity intracellular were detected using the mitochondria complexes I, II, IV and V testing kits (Solarbio, Beijing, China). 4T1 cells were seeded in 10 cm culture dishes (106 cells per dish) for 24 h and then treated with PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1). After 12 h incubation, the cells were irradiated under laser irradiation (660 nm, 50 mW cm−2) for 10 min. Subsequently, cells were collected to extract mitochondria, and then the complex I, II, IV and V activity were evaluated using the mitochondrial testing kits according to the protocol. Similarly, adenosine triphosphate (ATP) content intracellular was evaluated using an ATP Detection kit by the same methods.
4.15. Hypoxia-alleviation assay in vitro
The oxygen consumption rate (OCR) could be tested using an oxygen consumption probe. In brief, 4T1 cells were seeded in 96-well plates (2 × 103 cells per well) with 12 h incubation. Then, PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1) were incubated with cells for another 12 h. The cells were divided into eight groups: blank control (PBS), PTD NPs, TTD NPs, PTTD NPs, PBS + L, PTD NPs + L, TTD NPs + L, PTTD NPs+ L. The laser-irradiated groups were treated with laser (660 nm, 50 mW cm−2) for 10 min. Then, 10 μL of the oxygen consumption probe and 100 μL of the oxygen sealing solution were added into the cells, and the fluorescence of the plates was measured using a Varioskan LUX multimode microplate reader every 3 min (Thermo Scientific, USA).The intracellular oxygen concentration could be tested using an oxygen electrode. 4T1 cells were seeded in 6-well plates (1 × 105 cells per well) for the intracellular oxygen concentration test with 12 h incubation. Then, PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1) were incubated with cells for another 12 h. The laser-irradiated groups were treated with laser (660 nm, 50 mW cm−2) for 10 min post-incubation. The oxygen electrode was inserted into the cell culture and sealed with a liquid paraffin, followed by recording oxygen concentration every 10 min.
The decreased expression of HIF-1α under hypoxia conditions was investigated on 4T1 cells (5 × 104 cells per well) seeded into 24-well plates. After the uptake of PTD NPs, TTD NPs and PTTD NPs (50 μg mL−1) for 12 h, the laser-irradiated groups were exposed to a laser source (660 nm, 50 mW cm−2) for 10 min post-incubation. Then, the drug-containing media were removed, and the cells were washed three times with ice-cold PBS. After this, the cells were fixed with 4% paraformaldehyde and PBS with 0.1% Triton X-100 and 1% bovine serum albumin (1 mL per well) were utilized to preincubate the fixed cells for 15 min. The primary antibody (HIF-1α) was diluted in the PBS solution (1:500), and the cells were incubated with HIF-1α for 12 h at 4 °C under dark conditions. After incubation, the cells were incubated with a secondary antibody (diluting by PBS solution (1:100)) for 2 h at room temperature. Whereafter, Hoechst 33342 was used to stain the cell nuclei for 10 min. Finally, all the fluorescence images were obtained by confocal laser scanning microscopy (CLSM, C2, Nikon, Japan).
4.16. Cellular singlet oxygen detection
4T1 cells (5 × 104 cells per well) were seeded into 24-well plates and cultured for 12 h under normoxic or hypoxic conditions. After removing the old culture media, cells were incubated with PTD NPs, TTD NPs and PTTD NPs (30 μg mL−1). After uptake for 12 h, the drug-containing media were replaced by DCFH-DA (20 μM) and incubated for 30 min at 37 °C. Then, the laser-treated groups were irradiated using a laser (660 nm, 50 mW cm−2) for 10 min. Finally, the cells were washed with ice-cold PBS for three times, and the intracellular fluorescence intensity of DCF was recorded using an Eclipse Ti-U inverted microscope (Nikon Corp., Tokyo, Japan).4.17. Cytotoxicity
The cytotoxicity of PTD NPs, TTD NPs and PTTD NPs with or without laser treatment was evaluated using the MTT assay in 4T1 cells and CT26 cells under normoxic and hypoxic conditions. After cells (2 × 103 cells per well) were incubated in 96-well plates for 12 h and further cultured with serial concentrations of PTD NPs, TTD NPs or PTTD NPs for another 12 h, the laser-treated groups were exposed to a laser source (660 nm, 50 mW cm−2) for 10 min. Afterward, the cells were further cultured for 24 h in the dark. Then, 25 μL of MTT (5 mg mL−1) was added to the wells and the cells were incubated for another 4 h at 37 °C. After this, 200 μL of DMSO was added into wells, and the absorbency was measured using a Varioskan LUX multimode microplate reader (Thermo Scientific, USA).4.18 Animal studies
Balb/c mice were utilized and conformed to the Animal Laboratory Ethics Committee of Shenyang Pharmaceutical University. The living environment of animals was remained at a temperature of ∼25 °C, and the standard food and water were supplied abundantly. All the animal experiments were conducted according to the Guidelines for the Care and Use of Laboratory Animals approved by the Institutional Animal Ethical Care Committee (IAEC) of Shenyang Pharmaceutical University.4.19. Biodistribution
The distribution of NPs was studied in 4T1 breast tumors on female Balb/c mice. Simply, 100 μL of PBS containing 4T1 cells (5 × 106 cells) was inoculated subcutaneously into the right back of mice. When the tumor volume approached about 400 mm3, PTD NPs, TTD NPs and PTTD NPs (10 mg kg−1) were injected into mice through the tail vein, respectively. The fluorescence of tumors at 2, 4, 8, 12, 24 and 48 h was reported using the IVIS spectrum small-animal in vivo imaging system. The mice were sacrificed at 48 h for further quantitative analysis of tumors and major organs (heart, liver, spleen, lung and kidney). Fluorescence imaging and intensity of major organs and tumors were also analyzed using the IVIS spectrum small-animal in vivo imaging system (n = 4).4.20. Self-reported optimal laser duration in vivo
The self-reported optimal laser duration in vivo was studied in 4T1 breast tumors on the female Balb/c mice. After the tumor volume was about 150 mm3, the mice were randomly divided into 6 groups (n = 5): control (saline) and PTTD NPs + L (L = 0, 5, 10, 15 and 20 min). To observe the singlet oxygen self-reporting of PTTD NPs at different laser durations, PTTD NPs (10 mg kg−1) were intravenously administrated into the tumor-bearing mice via the tail vein, and the mice with laser treated were exposed to laser (660 nm, 50 mW cm−2) for 0, 5, 10, 15 and 20 min at 12 h post-injection. After irradiation, the mice were killed immediately, and the tumors were collected and frozen in liquid nitrogen. Finally, the immunofluorescence imaging of tumor tissues was observed by CLSM (C2, Nikon, Japan).Then, the tumor inhibition effects at different laser durations were further verified. After the tumor volume was about 150 mm3, the 6 groups of mice (control (saline), PTTD NPs + L (L = 0, 5, 10, 15 and 20 min), n = 5) were intravenously administrated via the tail vein at an interval of one day with saline and PTTD NPs (10 mg kg−1) for five injections total, respectively. The mice in the laser-treated groups were irradiated for 5, 10, 15 and 20 min at 12 h post-injection (660 nm, 50 mW cm−2). The tumor volume and the body weight were measured every day. The mice were sacrificed at 11 days to collect the blood samples, major organs (heart, liver, spleen, lung, kidney) and tumors. The serum samples were obtained from the blood via centrifugation (1.3 × 104 rpm, 5 min) for hepatic and renal function analyses. The organs and tumors were stained with H&E to evaluate the pathological variations. Tumor sections were processed for the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and the fluorescence change detection of PTTD NPs at the tumor site.
4.21. Hypoxia-alleviation and synergetic antitumor activity in vivo
The hypoxia alleviation in tumors and antitumor effects of nanoparticles in vivo was also investigated on the 4T1 breast tumor-bearing female Balb/c mice. The mice were randomly divided into 8 groups (n = 5): control (saline), PTD NPs, TTD NPs, PTTD NPs, saline + L, PTD NPs + L, TTD NPs + L and PTTD NPs + L. These formulations were intravenously administrated into the tumor-bearing mice via the tail vein at an interval of one day with doses of 10 mg kg−1 for five injections total, respectively. The mice in the laser-treated groups were exposed to a laser source (660 nm, 50 mW cm−2) for 10 min at 12 h post-injection. After five injections of therapeutic agents, a hypoxia-specific probe (pimonidazole; hypoxyprobe-1 plus kit, Hypoxyprobe Inc.) was intraperitoneally injected into mice with a dose of 60 mg kg−1 for 11 days. After 1.5 h, the mice were sacrificed and the blood samples, major organs (heart, liver, spleen, lung and kidney) and tumors were collected. The serum samples were collected for hepatic and renal function analyses. And the organs and tumors were stained with H&E to evaluate the pathological variations. Tumor sections were weighed and processed for immunofluorescence of the hypoxyprobe and the tissue chemical protein of HIF-1α to verify hypoxia-alleviation in tumors. The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was conducted and the tumor damage and the ATP content in tumors were evaluated using an ATP Detection kit.4.22. Hemolysis assay
Red blood cells (RBCs) were centrifuged (3000 rpm for 5 min), washed with PBS 3 times, and then diluted to 10-fold with PBS. 100 μL of the RBC solution was added into 900 μL of DPA-MOF, PTD NPs, TTD NPs and PTTD NPs with different concentrations. 100 μL of the RBC solution incubated in water was utilized as the positive control. After incubation at 37 °C for 30 min, the mixture was centrifuged at 3000 rpm for 10 min. The absorption of the supernatant was detected at 570 nm using a Varioskan LUX multimode microplate reader (Thermo Scientific, USA).4.23. Statistical analysis
All the data were calculated and presented as mean ± SD. The significant differences between groups were identified by the t-test or one-way analysis of variance (ANOVA), and P < 0.05 was considered as statistically significant.Author contributions
All authors gave approval to the final version of the manuscript. X. H., S. L., and F. Y. conceived this work. S. L., F. Y., and Y. W. designed, prepared, and characterized the materials. S. L., F. Y., and L. J. carried out the in vitro, ex vivo, and in vivo experiments. X. H., S. L., F. Y., and Y. W. analyzed the data and discussed the results. X. H., S. L., F. Y., and L. J. wrote and edited the manuscript. X. H. supervised the project.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 82273898).Notes and references
- S. Li, F. Yang, X. Sun, Y. Wang, X. Zhang, S. Zhang, H. Zhang, Q. Kan, J. Sun and Z. He, Chem. Eng. J., 2021, 426, 130838 CrossRef CAS.
- D. Wang, H. Wu, S. Z. F. Phua, G. Yang, W. Qi Lim, L. Gu, C. Qian, H. Wang, Z. Guo and H. Chen, Nat. Commun., 2020, 11, 357 CrossRef CAS PubMed.
- X. Zhang, J. Xiong, K. Wang, H. Yu, B. Sun, H. Ye, Z. Zhao, N. Wang, Y. Wang and S. Zhang, Bioact. Mater., 2021, 6, 2291–2302 CrossRef CAS PubMed.
- Z. Shenwu, J. Wang, Z. Kong, X. Sun, Z. He, B. Sun, C. Luo and J. Sun, Biomaterials, 2022, 121433 Search PubMed.
- S. Li, X. Shan, Y. Wang, Q. Chen, J. Sun, Z. He, B. Sun and C. Luo, J. Controlled Release, 2020, 326, 510–522 CrossRef CAS PubMed.
- F. Yang, Q. Ji, R. Liao, S. Li, Y. Wang, X. Zhang, S. Zhang, H. Zhang, Q. Kan and J. Sun, Chin. Chem. Lett., 2022, 33, 1927–1932 CrossRef CAS.
- S. Li, Y. Wang, T. Du and X. Hou, Chem. Eng. J., 2022, 138621, DOI:10.1016/j.cej.2022.138621.
- D. Wei, Y. Huang, B. Wang, L. Ma, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2022, e202201486 CAS.
- Y. Wang, S. Xu, L. Shi, C. Teh, G. Qi and B. Liu, Angew. Chem., Int. Ed., 2021, 60, 14945–14953 CrossRef CAS PubMed.
- J. Zhu, W. Wang, X. Wang, L. Zhong, X. Song, W. Wang, Y. Zhao and X. Dong, Adv. Healthcare Mater., 2021, 10, 2002038 CrossRef CAS PubMed.
- Z. Zhang, R. Wang, R. Luo, J. Zhu, X. Huang, W. Liu, F. Liu, F. Feng and W. Qu, ACS Nano, 2021, 15, 5366–5383 CrossRef CAS PubMed.
- Y. Yuan, C. J. Zhang, R. T. Kwok, S. Xu, R. Zhang, J. Wu, B. Z. Tang and B. Liu, Adv. Funct. Mater., 2015, 25, 6586–6595 CrossRef CAS.
- W. Fudickar and T. Linker, Chem. – Eur. J., 2006, 12, 9276–9283 CrossRef CAS PubMed.
- J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed.
- N. Umezawa, K. Tanaka, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 2899–2901 CrossRef CAS PubMed.
- X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783–4804 RSC.
- F. Yang, Z. Zhao, B. Sun, Q. Chen, J. Sun, Z. He and C. Luo, Trends Cancer, 2020, 6, 645–659 CrossRef CAS PubMed.
- E. B. Rankin and A. J. Giaccia, Science, 2016, 352, 175–180 CrossRef CAS PubMed.
- E. B. Rankin, J.-M. Nam and A. J. Giaccia, Trends Cancer, 2016, 2, 295–304 CrossRef PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- W. Lv, Z. Zhang, K. Y. Zhang, H. Yang, S. Liu, A. Xu, S. Guo, Q. Zhao and W. Huang, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 CrossRef CAS PubMed.
- E. M. Klosowski, B. T. L. de Souza, M. S. Mito, R. P. Constantin, G. C. Mantovanelli, J. M. Mewes, P. F. V. Bizerra, P. V. M. da Costa Menezes, E. H. Gilglioni and K. S. Utsunomiya, Free Radical Biol. Med., 2020, 153, 34–53 CrossRef CAS PubMed.
- X. Luo, X. Gong, L. Su, H. Lin, Z. Yang, X. Yan and J. Gao, Angew. Chem., Int. Ed., 2021, 60, 1403–1410 CrossRef CAS PubMed.
- Z. Liu, Y. Xue, M. Wu, G. Yang, M. Lan and W. Zhang, Biomacromolecules, 2019, 20, 4563–4573 CrossRef CAS PubMed.
- D. A. Belcher, A. Lucas, P. Cabrales and A. F. Palmer, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- L. Feng, R. Zhao, B. Liu, F. He, S. Gai, Y. Chen and P. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 41047–41061 CrossRef CAS PubMed.
- H. Zuo, J. Tao, H. Shi, J. He, Z. Zhou and C. Zhang, Acta Biomater., 2018, 80, 296–307 CrossRef CAS PubMed.
- C. Hu, X. He, Y. Chen, X. Yang, L. Qin, T. Lei, Y. Zhou, T. Gong, Y. Huang and H. Gao, Adv. Funct. Mater., 2021, 31, 2007149 CrossRef CAS.
- Z. Yang, Q. Chen, J. Chen, Z. Dong, R. Zhang, J. Liu and Z. Liu, Small, 2018, 14, 1803262 CrossRef PubMed.
- C. Luo, J. Sun, B. Sun and Z. He, Trends Pharmacol. Sci., 2014, 35, 556–566 CrossRef CAS PubMed.
- S. Zhang, J. Guan, M. Sun, D. Zhang, H. Zhang, B. Sun, W. Guo, B. Lin, Y. Wang and Z. He, Drug Delivery, 2017, 24, 1460–1469 CrossRef CAS PubMed.
- X. Shan, S. Li, B. Sun, Q. Chen, J. Sun, Z. He and C. Luo, J. Controlled Release, 2020, 319, 322–332 CrossRef CAS PubMed.
- F. Yang, Z. Kong, Q. Ji, S. Li, J. Sun, Z. He, S. Zhang and C. Luo, ACS Mater. Lett., 2023, 5, 429–449 CrossRef CAS.
- B. Sun, C. Luo, W. Cui, J. Sun and Z. He, J. Controlled Release, 2017, 264, 145–159 CrossRef CAS PubMed.
- G. Shafirstein, N. R. Rigual, H. Arshad, M. T. Cooper, D. A. Bellnier, G. Wilding, W. Tan, M. Merzianu and B. W. Henderson, Head Neck, 2016, 38, E377–E383 CrossRef PubMed.
- C. Luo, J. Sun, B. Sun, D. Liu, L. Miao, T. J. Goodwin, L. Huang and Z. He, Small, 2016, 12, 6353–6362 CrossRef CAS PubMed.
- J. Park, Q. Jiang, D. Feng, L. Mao and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 3518–3525 CrossRef CAS PubMed.
- X. Fu, Z. Yang, T. Deng, J. Chen, Y. Wen, X. Fu, L. Zhou, Z. Zhu and C. Yu, J. Mater. Chem. B, 2020, 8, 1481–1488 RSC.
- W. Zhang, J. Lu, X. Gao, P. Li, W. Zhang, Y. Ma, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2018, 57, 4891–4896 CrossRef CAS PubMed.
- S. Haddad, I. Abánades Lázaro, M. Fantham, A. Mishra, J. Silvestre-Albero, J. W. Osterrieth, G. S. Kaminski Schierle, C. F. Kaminski, R. S. Forgan and D. Fairen-Jimenez, J. Am. Chem. Soc., 2020, 142, 6661–6674 CrossRef CAS PubMed.
- W. Ma, H. Zhang, S. Li, Z. Wang, X. Wu, R. Yan, F. Geng, W. Mu and Y. Jin, ACS Biomater. Sci. Eng., 2022, 8, 1354–1366 CrossRef CAS PubMed.
- H.-J. Cai, T.-T. Shen, J. Zhang, C.-F. Shan, J.-G. Jia, X. Li, W.-S. Liu and Y. Tang, J. Mater. Chem. B, 2017, 5, 2390–2394 RSC.
- Y. Ma, X. Li, A. Li, P. Yang, C. Zhang and B. Tang, Angew. Chem., Int. Ed., 2017, 56, 13752–13756 CrossRef CAS PubMed.
- I. Vercellino and L. A. Sazanov, Nat. Rev. Mol. Cell Biol., 2022, 23, 141–161 CrossRef CAS PubMed.
- P. R. Rich and A. Maréchal, Essays Biochem., 2010, 47, 1–23 CrossRef CAS PubMed.
- J. S. Sousa, E. D’Imprima and J. Vonck, Membr. Proteins, 2018, 167–227 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01244a |
| This journal is © The Royal Society of Chemistry 2023 |