
Superstructures of water-dispersive hydrophobic nanocrystals: specific properties
M. P.
Pileni

Sorbonne Université département de chimie, 4 Place Jussieu, 75005 Paris, France. E-mail: mppileni@orange.fr; Tel: +33 (0) 683 028 206
First published on 6th September 2023
Abstract
Here, we describe water-soluble superstructures of hydrophobic nanocrystals that have been developed in recent years. We will also report on some of their properties which are still in their infancy. One of these structures, called “cluster structures”, consists of hydrophobic 3D superlattices of Co or Au nanocrystals, covered with organic molecules acting like parachutes. The magnetic properties of Co “cluster structures” a retained when the superstructures is dispersed in aqueous solution. With Au “cluster structures”, the longer wavelength optical scattered spectra are very broad and red-shifted, while at shorter wavelengths the localized surface plasmonic resonance of the scattered nanocrystals is retained. Moreover, the maximum of the long-wavelength signal spectra is linearly dependent on the increase in assembly size. The second superstructure was based on liquid–liquid instabilities favoring the formation of Fe3O4 nanocrystal shells (colloidosomes) filled or unfilled with Au 3D superlattices and also spherical solid crystal structures are called supraballs. Colloidosomes and supraballs in contact with cancer cells increase the density of nanocrystals in lysosomes and near the lysosomal membrane. Importantly, the structure of their organization is maintained in lysosomes for up to 8 days after internalization, while the initially dispersed hydrophilic nanocrystals are randomly aggregated. These two structures act as nanoheaters. Indeed, due to the dilution of the metallic phase, the penetration depth of visible light is much greater than that of homogeneous metallic nanoparticles of similar size. This allows for a high average heat load overall. Thus, the organic matrix acts as an internal reservoir for efficient energy accumulation within a few hundred picoseconds. A similar behavior was observed with colloidosomes, supraballs and “egg” structures, making these superstructures universal nanoheaters, and the same behavior is not observed when they are not dispersed in water (dried and deposited on a substrate). Note that colloidosomes and supraballs trigger local photothermal damage inaccessible to isolated nanocrystals and not predicted by global temperature measurements.
 M. P. Pileni | Materials Horizons is one of the best journals likely to report on developments in materials research at different scales. Publications related to advanced manufacturing and unique material measurements have placed the author at the forefront of materials science. Professor Marie-Paule Pileni is Emeritus Professor at the Sorbonne University. She chaired the Institut Universitaire de France, an institute promoting cutting-edge research in France. She was a pioneer in the field of nanotechnology where she brought important advances. Her major advances concern 3D superlattices of nanocrystals. These structures have potential applications ranging from biomedicine to energy conversion. She has received numerous international awards. |
Wider impactHere, we show new 3D superstructures which are assemblies of water-dispersed hydrophobic nanocrystals. These superstructures function as universal nanoheaters: individual nanocrystals absorb light, followed by rapid heating of the surrounding organic matrix. Therefore, the last step of the light–heat conversion process ultimately results in the heating of the organic matrix of the ligand. This new photothermal property promotes certain technological advances for the development and exploitation of energy release processes, a vital element for our modern society. Moreover, they possess the properties of both nanocrystals and their (collective) assemblies without the disadvantages of dispersed nanoparticles (toxicity, stability, and uncontrolled aggregation). As is known, the control of interactions between nanomaterials and cells remains a biomedical challenge. Here, we demonstrate that these superstructures self-assemble in tumor cells with a marked increase in nanocrystal density near the lysosome membrane. These internalized superstructures form long chains when subjected to a magnetic field. They target various compartments of the tumor microenvironment and trigger local photothermal damage inaccessible to dispersed nanocrystals and not predicted by global temperature measurements. In conclusion, these superstructures have potential applications ranging from biomedicine (hyperthermia, imaging, and drug targeting) to energy conversion (water splitting, H2 dissociation, and production of new molecules and new materials). |
Introduction
More than two decades ago, controlling the size of colloidal nanocrystals initiates their assemblies into long-range ordered arrays.1,2 This field of research has widened considerably because of their potential applications in various research areas. To maintain their integrity and isolate themselves from each other, these metals, semiconductors and other functional nanomaterials are coated with molecules of organic surfactants. Spontaneously, the nanocrystals self-assemble into 3D superlattices. Due to the interdigitation of alkyl chains, the average interparticle distance is close to the length of the surfactant molecule. The size of the nanocrystals and the solvent used to disperse them determine the final morphology of the 3D superlattices with either a solution nucleation process or layer-by-layer growth. Nucleation of 3D superlattices is mainly driven by solvent-mediated interactions.3 A conceptual analogy has been observed between atomic crystals and 3D superlattices of nanocrystals: atoms and atomic bonds are replaced by incompressible nanocrystals and coating agents acting as mechanical springs. The structures of the 3D superlattices are similar to those obtained with atomic crystals (fcc, hcp or bcc structures, negative crystals, quasi-crystalline structures, and binary crystals).4–10 These 3D superlattices exhibit collective properties that are mainly due to dipole interactions or intrinsic properties related to the order of the nanocrystals.9–24 Thus, for example, -the STM-induced photon emission depends on the degree of integration of the nanocrystals in the lattice site25 whereas, according to the nanocrystal ordering, the electronic behavior evolves from Coulomb blockade to ohmic resistance.16,26–29 The intrinsic vibrational properties of nanocrystals in the 3D superlattices have been demonstrated.30,31 Nanocrystallinity, atomic packing in nanocrystals with the formation of single domains or polycrystalline nanocrystals, plays a major role in the chemical and physical properties of these 3D superlattices.32 Most of these studies were obtained with dried superlattices deposited on a substrate.To increase the potential applications, these 3D superlattices with such magnificent properties must be dispersed in aqueous solution. This would open applications towards the release of energy which is a vital element for our modern society. These new hybrids could be good candidates not only for solar power generation and waste heat recovery, but also for water heating, cooling or air conditioning, as well as for space research and catalysis. Moreover, if we consider that cancer is still one of the main causes of death in the world today, the exploitation of heat release for medical treatments and more precisely for cancer therapies represents another major human challenge in our societies. Here, we give an overview of hydrophilic superstructures of self-assembled hydrophobic nanocrystals and describe some of their specific properties.
Preparation and specific properties of various hybrid superstructures dispersed in aqueous solutions
To create “cluster” structures, two steps must be carried out:(i) Generate Au-shaped 3D superlattices. This is achieved by controlling the affinity between the solvent and the coating agent. Au nanocrystals coated with octadecane-thiol (C18H37SH) are dispersed in toluene at 60 °C. The colloidal solution is stored in a beaker under an atmosphere saturated with isopropanol. The trace of isopropanol acts as a non-solvent3,10,35,36 inducing a marked increase in attractive interactions between nanocrystals. Consequently, this favors the precipitation of shaped aggregates with a flat surface, well-defined facets in hexagonal symmetry and a fairly wide particle size distribution (Fig. 1b and c).
 | ||
| Fig. 1 TEM image of 5.5 nm Au nanocrystals coated with hexadecanthiol and deposited on a substrate (a). SEM images of 3D-shaped superlattices (b) and (c) and “cluster” structures (d) and (e). TEM images of “cluster” structures (f) and (g). Note the attachment of the vesicles forming cap-like structures (f) and the highly ordered Au nanocrystals (g) observed at the edge of superlattices and multi-layers of lipids attached parallel to the superlattice surface. Source: N. Yang et al.,34ACS. | ||
(ii) Interdigitation processes bridging vesicles to prevent shaped assemblies for precipitation. These processes promote the dispersion of hydrophobic 3D superlattices in aqueous solutions. The best interdigitation process is obtained with long-tailed surfactant molecules. Therefore, 3D-shaped superlattices (C18) are mixed with a methanol solution of dipalmitoylphosphatidylcholine ([DPPC] = 10 mg mL−1)- and PEG2000-DOPE ([DPPC]/[PEG2000-DOPE] = 95.5) both characterized by long alkyl chains (C16 and C18, respectively). PEG2000-DOPE is characterized not only by a C18 alkyl chain but also by a very long hydrophilic chain (C91) which bridges from one vesicle to another and therefore prevents precipitation and acts as “parachutes” to prevent gravity processes (Fig. 1d). The methanol is removed using a rotary evaporator machine and a uniform film is obtained covering the wall of the flask. Then it is kept under vacuum overnight to remove the residual solvent. By adding water, the 3D superlattice was dispersed in solution. The TEM image (Fig. 1f) reveals the structure of individual composites, with the presence of large vesicles attached to the surface of the superlattice and drape-like structures attached to them. Such a large-scale TEM image shows that nanocrystals are highly ordered at the edge of superlattices and parallel multilayers of lipids attached to the Au superlattice surface (Fig. 1g).
The small-angle X-ray diffraction pattern (SAXRD) of the shaped aggregates (Fig. 2a) indicates that these aggregates are fcc-3D Au superlattices while that of the “cluster” Au structure (Fig. 2b) shows a new diffraction ring compared to the 3D-shaped superlattices (Fig. 2a). The SAXRD pattern of the vesicle bilayers shows a well-defined signal (Fig. 2c). Fig. 2d shows the 1D profiles of these different models illustrating that the diffraction peaks of the “cluster” structure (orange curve) are the combination of the peaks of the 3D-shaped superlattices (green curve) and the vesicles (blue curve). The edge-to-edge interparticle distance is 2.8 nm and remains unchanged after vesicular treatment. The calculated distance of the lipid bilayer (5.7 nm) is in good agreement with the thickness of the reported liposome shell.37 From these data, we claimed that organic molecules (DPPC/PEG2000-DOPE) are strongly attached to the surfaces of 3D-shaped superlattices.
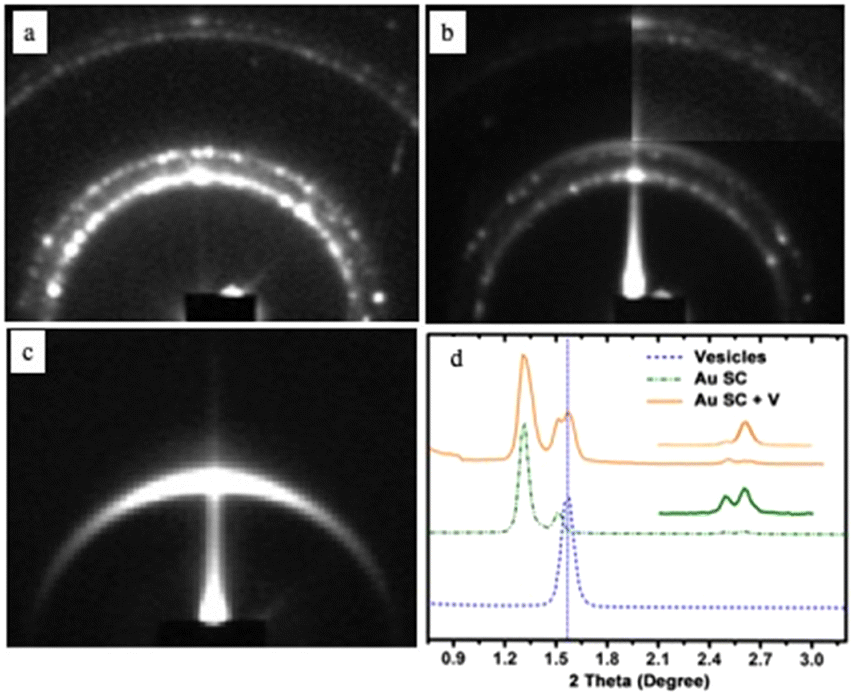 | ||
| Fig. 2 SAXRD patterns of (a) well-defined aggregates, (b) “cluster structures”, and (c) vesicles. (d) Profiles of the various patterns. Source: N. Yang et al.,33 John Wiley & Sons. Inc. | ||
Similar data are obtained with Co-shaped 3D superlattices. Due to CoO nanocrystal undefined layers formed at the 3D superlattice edges, their integrity are retained for several weeks under air.
By controlling the nucleation and growth time processes, the size of the Au “cluster structure” is controlled. Their absorption spectra are red-shifted increasing their average size with a shoulder around 530 nm. Using Mie theory, the calculated absorption spectra of Au “cluster” structures are in good agreement with the sum of two spectra: at a short wavelength, one is at a fixed wavelength (530 nm) and the second is red-shifted with an increase in the aggregate size (Fig. 3a). At a shorter wavelength, the peak fulfills the resonance condition of spherical Au nanocrystals in a ligand-based medium. The scattered spectra at a longer wavelength are very broad and red-shifted linearly with wavelength as the assembly size increases (Fig. 3b). Such a long wavelength optical response is attributable to a photonic (i.e. collective) mode of the assembly, since its spectral position scales linearly with the size of the superlattices when the fingerprint of the dispersed nanocrystals used as building blocks is retained.
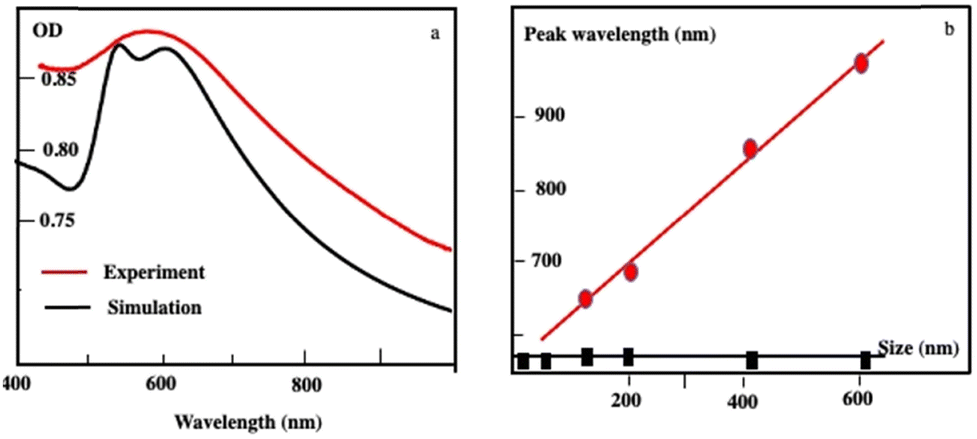 | ||
| Fig. 3 (a) Extinction spectra of Au “cluster” structures characterized an average size of 198 nm ± 84 nm recorded experimentally (red curve) and the simulated spectrum (black curve). (b) Variation of the peak wavelength as a function of the assembly size: The black curve illustrates the absorption peak of dispersed Au nanocrystals used as building blocks. The scattering peak represented in the red curve red shifts as the assembly size increases. Source: N. Yang et al.,34ACS. | ||
To investigate the energy flow upon light irradiation, pump–probe experiments with ultrashort laser pulses were performed. A 100 fs pump pulse at 400 nm wavelength is used to excite the Au “cluster”.38 Differential transmission (ΔT/T) spectra are recorded as a function of time, t, and the probe wavelength, λ (Fig. 4)
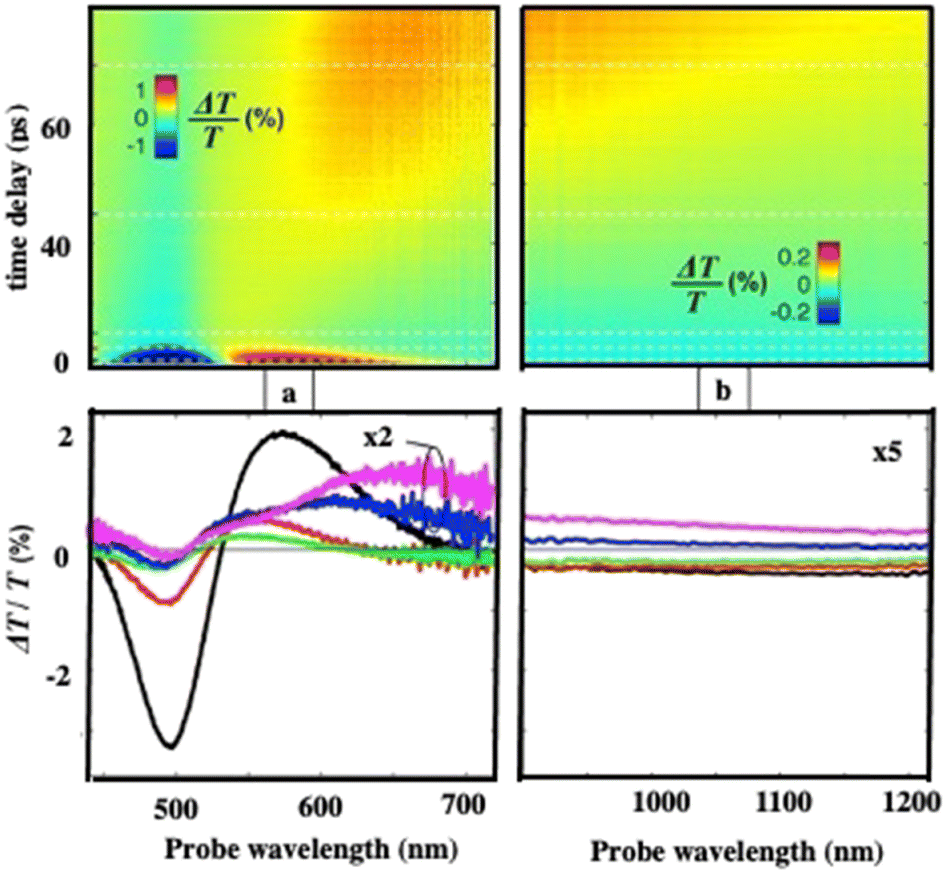 | ||
| Fig. 4 Measured (a) and (b) differential transmission maps ΔT/T (top) and map cross-sections at selected time delays (bottom) for the Au “cluster” structure. | ||
At an initial timescale of ten picoseconds, the transient signal resembles the typical ΔT/T map recorded for plasmonic nanoparticles. In particular, cross-sections of the map at early delays show the characteristic transient spectra of isolated nanocrystals that are dominated by shifting and broadening of plasmon resonances as previously observed.39 Moreover, these spectra exhibit a decay constant of a few picoseconds which is the timescale of electron–phonon scattering in noble metals.40Fig. 5a shows the dynamics at selected probe wavelengths for delay times down to 10 ps. This demonstrates that in the ultra-fast optical response, Au assemblies retain the imprint of their building blocks. Both dynamic and static data confirm that Au “cluster” structures at room temperature retain the fingerprint of isolated nanocrystals used as building blocks. This was already observed by scanning tunneling microscopy/scanning tunneling spectroscopy (STM/STS) experiments at low temperature, with dried and thick Au 3D superlattices.27
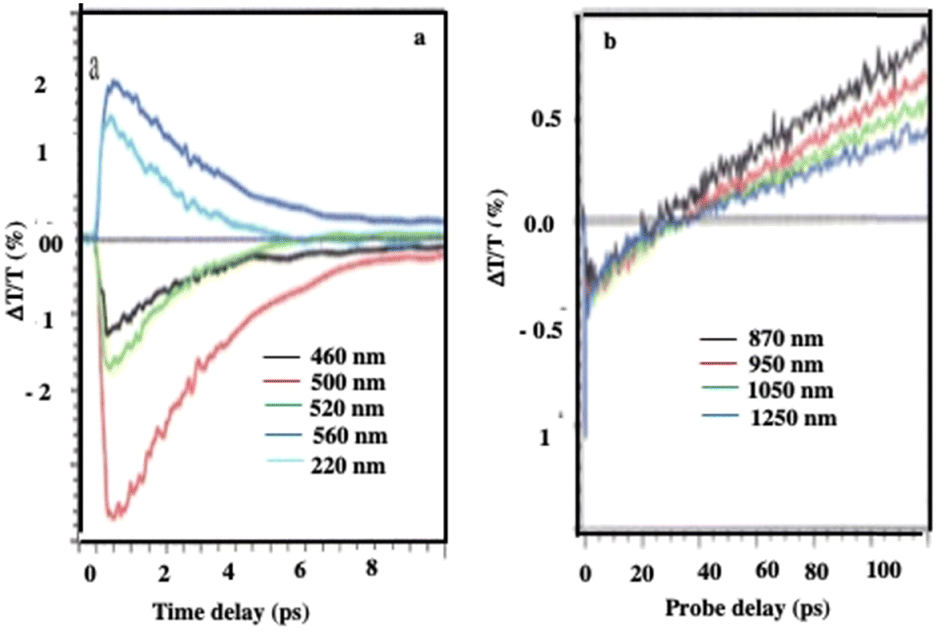 | ||
| Fig. 5 Dynamics of ΔT/T at selected probe wavelengths, at the two different time scales of energy flow in the 198 nm Au “cluster” structures. Source: A. Mazzanti et al.,38PNAS. | ||
At longer time scales, the ΔT/T map (Fig. 4b and 5b) exhibits very distinct features which, to the best of our knowledge, have not been reported neither for the nanostructures nor for their building blocks. Indeed, a monotonic decay over time has always been observed reaching zero ΔT/T at and above 10 ps. Here, instead of a monotonic decrease over time, we observe the build-up of a positive signal that is red-shifted by about 100 nm from the early spectra. The broad transient signal is observed between about 100 and 150 ps and then remains constant at the nanosecond scale. This novel phenomenon cannot be ascribed to mechanical oscillations that, in Au nanocrystals, are well known to have a strong impact on the ultrafast optical response at this time scale.41 To explain the origin of such an unexpected transient optical response at the 100 ps time scale which remains constant at the nanosecond time scale, a model based on the largest penetration depth light in the 3D superlattice (about 60 nm for Au nanocrystals) was proposed.38 Thus, despite the thickness of the 3D superlattices, the power absorbed per unit volume is more homogeneous compared to that of a nanosphere of the same diameter. The pump pulse is absorbed by the metallic phase of the 3D superlattice and initiates a chain of energy transfer processes. According to this model, the delayed accumulation of the ΔT/T signal on the time scale of a few hundred picoseconds is the signature of the final step of the light-heat conversion process which ultimately results in the heating of the ligand organic matrix. The very good agreement between the experiments and the model confirms that: (i) due to the dilution of the metal phase, the penetration depth of visible light is much greater than in homogeneous metal nanocrystals of a similar size, thus enabling a high average thermal load in the whole assembly, (ii) the organic matrix, even though not directly absorbing, acts as an internal reservoir for the efficient accumulation of energy within few hundred picoseconds; (iii) a collective regime of photo-temperature generation allowed by the 3D superlattice. From this, we claim that the Au “cluster structures” behave like nanoheaters. We must point out that such a specific property is not observed when the same 3D superlattices are deposited on a surface, i.e. under dried conditions.42
At very long-time scales (several hundred ps), an oscillation is observed with a period of 300 ps (Fig. 6). Taking into account the average diameter of Au “cluster structures” (D = 198 nm ± 84 nm) and the speed of sound inside the superlattices, estimated to νσ = 1235 ± 12 m s−1 at ambient temperature for the fcc crystalline structure of 7.1 nm Co nanocrystals used as building blocks,31 the oscillation period (T = 2D/νσ) is found to be 236 ± 77 ps. Good agreement is obtained between the calculated (236 ps) period and that measured (300 ps). This oscillation is attributed to collective breathing modes of the Au nanocrystals used as building blocks in the crystalline structure. Indeed, these data have been related to those obtained fifteen years ago from which we discovered that Ag nanocrystals breathing coherently in the 3D superlattice, when subjected to light.30
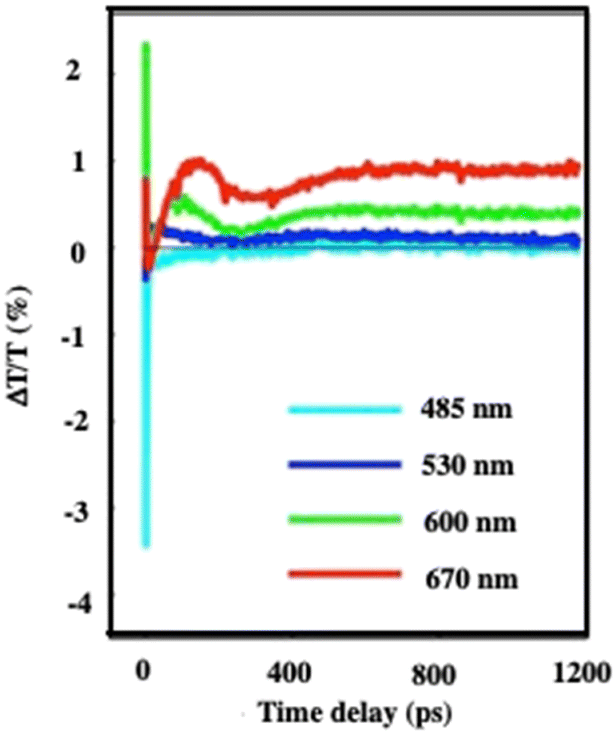 | ||
| Fig. 6 Dynamics of ΔT/T at selected probe wavelengths, on nanometer time scales of energy flow in the 198 nm Au “cluster structures”. Source: A. Mazzanti et al.,38PNAS. | ||
From the data presented above, we identify that the collective respiration process of self-assembled nanocrystals into 3D superlattices is observed in both dry and aqueous environments. This greatly supports such behavior, which has been contested in the past. However, conversely, we found that the unexpected property related to energy release is observed when the assemblies are dispersed in aqueous solutions and not in dried systems. To ensure that this last property can be considered as unique and due to self-assemblies of nanocrystals, we need to develop new hybrid structures dispersed in aqueous solutions.38
Based on previous research,43–49 we know that microscopic particles are found at the interface between two immiscible fluids. The force, pushing the micrometric particles to adsorb at the interfaces between immiscible liquids, decreases markedly with the size of the particles to reach at the nanometric scale a thermal fluctuation energy compared to the liquid–liquid interfacial energy. At the nanometric scale, the nanocrystals diffuse freely at the interface. To block them at the interface, a cross-linking agent could be used. Instead, we proposed to produce “Janus” nanocrystals (Fig. 7) at the chloroform–water interface:50 oleic acid molecules covalently bound to the surface nanocrystals interact selectively with octadecene (ODE, C18H36) and water-soluble dodecyltrimethylammonium bromide (DTAB) in the chloroform and aqueous phases, respectively. The specific interactions between aliphatic octadecene, ODE, (C18) and oleic acid (C18) induce the formation of a monolayer–nanocrystal membrane promoting the adsorption of nanocrystals at the chloroform/water interface and consequently their blocking at the liquid/liquid interface,51,52 water on the hydrophilic side. This approach avoids complicated procedures, such as those found in nanocrystalline polymers.53,54
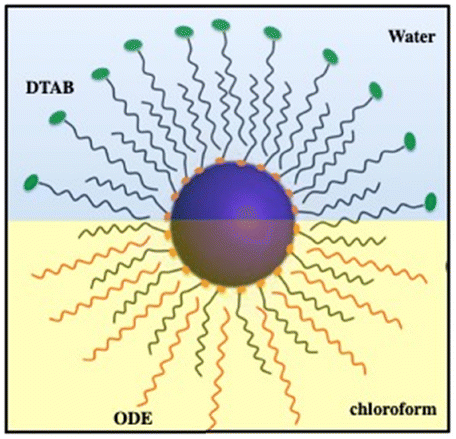 | ||
| Fig. 7 Schematic illustration of the formation of Janus nanocrystals. Source: Z. Yang et al.50ACS. | ||
Thus, 3 mg of Fe3O4 nanocrystals are dispersed in a mixture of 200 μL of chloroform and 8 μL of ODE. An aqueous solution (1 mL) containing 18 mg mL−1 dodecyltrimethylammonium bromide (DTAB) was added. The resulting emulsion is produced by strong vortex stirring for 30 s. To stabilize the emulsion, 5 mL of an ethylene glycol solution containing 0.4 g of polyvinylpyrrolidone (PVP K30, Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was added rapidly and vortexed for 30 s. The emulsion was then heated to 70 °C under an N2 atmosphere and maintained at this temperature for 15 min to evaporate the internal phase of chloroform. The suspension was left under air to gradually reach room temperature.
000) was added rapidly and vortexed for 30 s. The emulsion was then heated to 70 °C under an N2 atmosphere and maintained at this temperature for 15 min to evaporate the internal phase of chloroform. The suspension was left under air to gradually reach room temperature.
The resulting nanocrystal assemblies were washed twice with ethanol and dispersed in deionized water to form stable colloids. Spherical colloidosomes, envelope(s) of Fe3O4 nanocrystals, are produced: Fig. 8a shows the shell(s) of hexagonal packed Fe3O4 nanocrystals (inset Fig. 8a).
 | ||
| Fig. 8 TEM images of Fe3O4 colloidosomes (a), Au “egg” structures (b) and (c), Au/Fe3O4 “egg” structure (d) and supraballs (e) and Fe3O4 nanocrystals (2 mg mL−1). With Au “egg” structures, the amount of 5 nm nanocrystals differs by their amounts such as 0.6 mg mL−1 (b) and 6 mg mL−1 (c). With Au/Fe3O4 “egg” structures, the amount of 3.5 nm Au nanocrystals is 0.6 mg mL−1. Source: Z. Yang,50ACS. | ||
By doing the same experiments using a mixture of 9.6 m Fe3O4 and 5 nm Au nanocrystals coated with oleic acid and dodecanthiol respectively, Au “egg” structures are produced (Fig. 8b and c):55 The Fe3O4 nanocrystals retain the structure of the colloidosome as already observed in the absence of Au nanocrystals (Fig. 8a) and, in the center, a more contrasting structure is attributed to the Au nanocrystal assemblies. By increasing the relative amount of Au nanocrystals, the internal structure allows to obtain well-defined fcc superlattices (Fig. 8c). By decreasing the size of the nanocrystals from 9.6 nm to 6.5 nm and from 5 nm to 3.5 nm of the Fe3O4 and Au nanocrystals, the internal structures of the “eggs” change markedly. Instead of having in the center of the superstructure a 3D fcc Au superlattice as shown in Fig. 8c, self-assembled Au and Fe3O4 nanocrystals have a NaZn13-like structure, i.e. (Fe3O4)Au13 (Fig. 8d). As described above to produce the colloidosomes, the presence of ODE allows specific interactions with oleic acid favoring the locking of Fe3O4 nanocrystals at the liquid–liquid interface. By keeping the same experimental conditions to produce colloidosomes in the absence of ODE, the high mobility of the Fe3O4 nanocrystals at the interface induces the formation of large spherical solid dense-packed assemblies in the fcc structure of Fe3O4 nanocrystals called supraballs (Fig. 8e).
The absorption spectrum of Fe3O4 nanocrystals self-assembled in colloidosomes and supraballs drops markedly compared to the same nanocrystals dispersed in solution (Fig. 9a). This change of the absorption spectra of Fe3O4 nanocrystals is clearly due to the assemblies. Indeed, the addition of ethanol to the superstructures dispersed in aqueous solutions induces the destruction of these assemblies and the absorption spectrum of the dispersed nanocrystals is recovered. With “egg” structures, the absorption spectra of Au assemblies remain in the visible range (Fig. 9b) and as expected is red shifted with respect to the localized surface plasmon resonance of dispersed Au nanocrystals in solution.
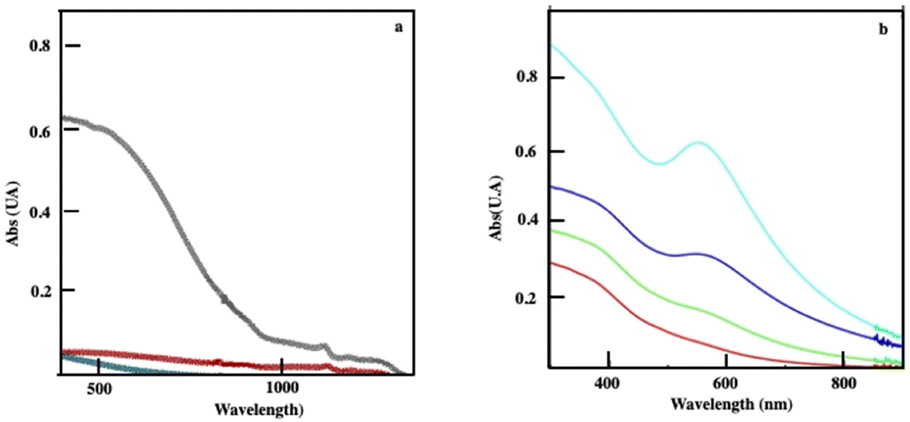 | ||
| Fig. 9 (a) Absorption spectra of Fe3O4 nanocrystals (01 mg mL−1) dispersed in aqueous solutions (black) of the corresponding colloidosomes (red) and supraballs (green). (b) Absorption spectra of “egg” structures of Fe3O4 (0.1 mg mL−1) with various amounts of Au aggregates with a rather Au/Fe3O4 of 0.1 (red), 0.3 (green), 1 (blue) and 2 (turquoise). Source: A. Mazzanti,38PNAS. | ||
The same pump–probe experiments are performed with the different superstructures as shown in Fig. 5. The differential transmission (ΔT/T) spectra recorded as a function of time and probe wavelength, Δ at short time(1–10 ps), confirm behavior similar to that observed with the Au “cluster” structure corresponding to electron–phonon scattering (Fig. 10). This confirms the fingerprint of isolated nanocrystals used as building blocks.
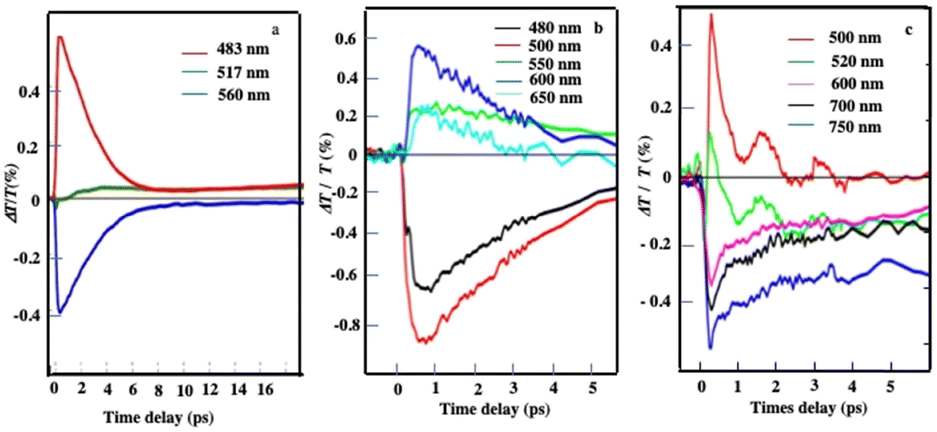 | ||
| Fig. 10 Dynamics of ΔT/T at selected probe wavelengths, at short time scales of energy flow in the supraballs, (a) Au “egg” structure (b) and Au/Fe3O4 “egg” structures (c). Source: A. Mazanti et al.,38PNAS and Schirato et al.,56ACS. | ||
Similarly, on a longer time scale, a positive signal appears with colloidosomes, supraballs, and Au “egg” and Au/Fe3O4 “egg” structures as shown in Fig. 11. This clearly confirms that superstructures dispersed in aqueous solutions behave as nanoheaters. Note that such “nanoheaters” cannot be detected with dried superstructures.
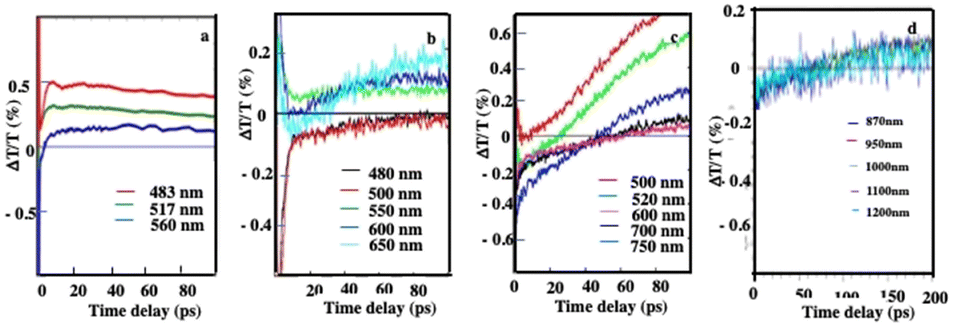 | ||
| Fig. 11 Dynamics of ΔT/T at selected probe wavelengths, at long-time scales of energy flow in the Au “egg” structure (a), Au/Fe3O4 “egg” structures (b), supraballs (c) and colloidosomes (d). (a)–(c) The pump laser excitation is 800 nm and (d) 400 nm. Source: A. Mazanti et al.,38PNAS and Schirato et al.,56ACS. | ||
The above characteristics are due to the collective regime of photo-temperature generation allowed by the assembly, since even though the nanoheater is valid for the Au “cluster” structure and also with a rather wide variety of superstructures made of self-assembled hydrophobic nanocrystals dispersed in aqueous solution. These similar behaviors have been experimentally observed with Fe3O4 nanocrystals self-assembled in fcc spherical superlattices (called supraballs) and binary [(Fe3O4)Au13] or Au 3D colloidal crystals trapped in colloidosomes and in colloidosomes themselves less extended. These results clearly indicate that these superstructures act as universal nanoheaters.56 Such a specific property requires the presence of an aqueous medium. Another specific property of such hybrid structures is related to the evolution of the absorption spectra of the Au superlattices with respect to their building blocks. Thus, with 3D Au superlattices, their photonic collective modes are size-dependent and red-shifted relative to the localized surface plasmon resonance. However, the major change is the disappearance of the visible absorption of colloidosomes and supraballs.
Potential biological applications57,58
At this stage, one can ask the following question: Could these hybrid structures be used in biomedical research? To answer this vast question, let us first compare the intimate interactions of, with A431 squamous cell carcinoma cells, Fe3O4 nanocrystals dispersed in the aqueous solution (coated with dopamine) and self-assembled into colloidosomes or supraballs.The cells are exposed to the nanomaterials in the serum-complemented culture medium for one day. In order to elucidate the intracellular distribution and fate of nanocrystals in tumor cells, the nanocrystal's uptake is monitored by TEM at different points of time for up to one week.
Dispersed Fe3O4 nanocrystals are rapidly internalized in a dispersed state in closed-membrane endosomes. They are concentrated in lysosomes and appear as aggregates surrounded by an electron-dense lysosomal matrix (Fig. 12a).
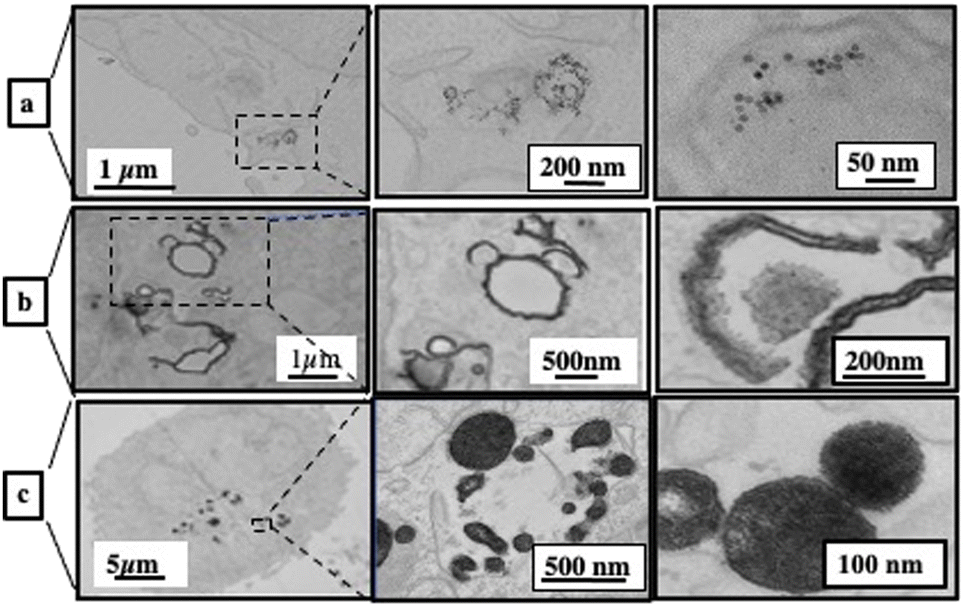 | ||
| Fig. 12 2D TEM slice images at various magnifications of A431 cells incubated for one day with Fe3O4 nanocrystals coated with dopamine (a), self-assembled in colloidosomes (b) and self-assembled in supraballs (c) ([Fe] = 28 μg L−1). Source: A. Nicolas-Boluda et al.,57 John Wiley & Sons. Inc. | ||
Unlike dispersed nanocrystals, colloidosomes show a very distinct intracellular distribution. The most striking difference is that the nanocrystalline shells of colloidosomes are close to cell membranes, fitting the shape of intracellular vesicles (Fig. 12b).
The size of the nanocrystal self-assemblies increases markedly in cells, suggesting cell-induced deformation, rearrangement and/or fusion of assemblies. This significant reorganization of the nanocrystal assemblies in contact with the cell membrane suggested competing interactions between the oleic acid surfactant molecules used to coat the nanocrystals and the hydrophobic parts of the cell membrane.
The supraballs collectively interact with the plasma membrane and retain mostly their spherical structure after entering intracellular compartments (Fig. 12c). Similar to colloidosomes, but to a lesser extent, the apparent size of supraballs is found to be larger in cells compared to that in the aqueous suspension, suggesting, again, fusion and reorganization.
The structural organization are studied by small-angle X-ray scattering (SAXS) experiments. Fig. 13 shows the structure factor, S(q). It is characterized by a power law decay with exponents of −1.7, −2.3 and −2.9 for dispersed nanocrystals, colloidosomes and supraballs, respectively. These changes in the exponent indicate that the dispersed nanocrystals are internalized as loose fractal aggregates while, with colloidosomes and supraballs, the nanocrystals are densely packed. These data confirm those shown by TEM (see above).
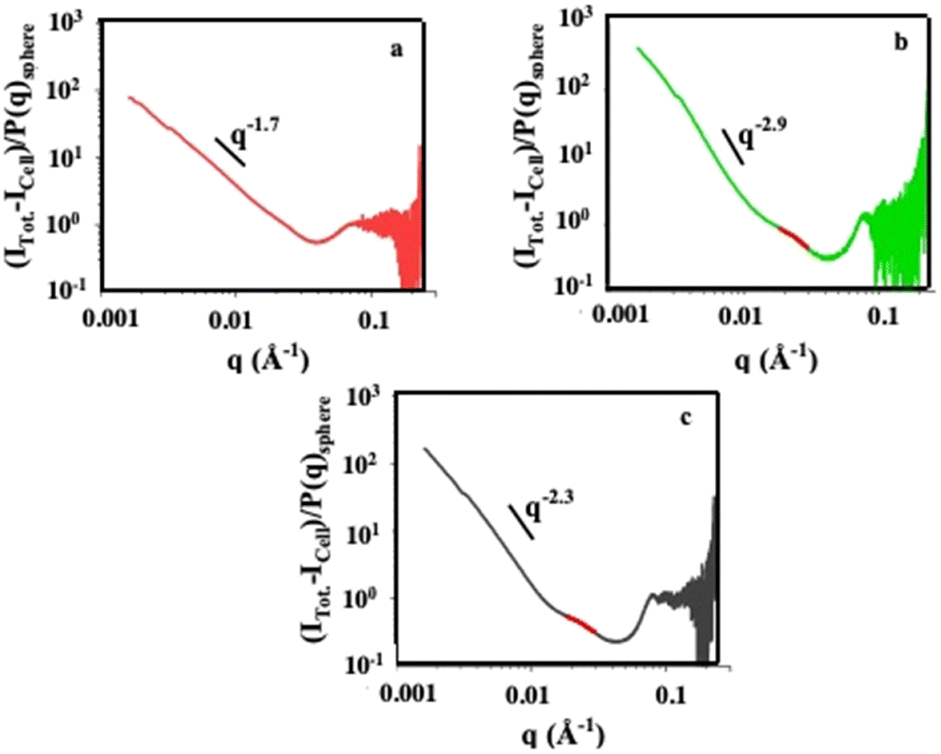 | ||
| Fig. 13 Structure factor, S(q), of the nanocrystals in cells incubated at different time points: to isolate the contribution of the nanocrystals to the overall scattering intensities, the unlabeled cells (Icell) is subtracted from the total scattering intensity of the incubated cells (Itotal). At a high value of q, the form factor P(q) is consistent with a diameter of nanocrystals around 8 nm slightly lower than that deduced by TEM of individual nanocrystals. Therefore, the structure factor, S(q), of the nanocrystals in the incubated cells is: S(q) = [Itotal − Icell]/P(q)sphere. The incubation periods are 30 min (green), 4 hours (yellow), 1 day (orange) and 2 days (red). Source: A. Nicolas-Boluda et al.,57 John Wiley & Sons. Inc. | ||
The surface density of nanocrystals in lysosomes, i.e. the ratio of the surface occupied by the nanocrystals to the surface of the lysosome, is evaluated according to the electron density on the TEM images. On day 1 of incubation, the local density of the nanocrystals represents for the colloidosomes and supraballs respectively 34% and 52% of the lysosome whereas that of the dispersed nanocrystals is very low (4%). In addition, the surface density of the nanocrystals in the vicinity of the membrane (the percentage of the surface occupied by the nanocrystals within 100 nm of the lysosome membrane) reaches average values of 35% and 41%, respectively, and is more than 10 times higher to that of dispersed nanocrystals (3.3%).
As stated above, supraballs act as a nanoheater as well as in low efficiency colloidosomes. Here, we assess thermal damage on cells. Whatever the experimental conditions, the cells having internalized the different nanostructures without laser irradiation did not show any significant improvement in the level of apoptosis or necrosis compared to the control cells, irradiated or not. This shows that the nanostructures or the exposure laser did not affect cell viability. Furthermore, it confirms no significant toxicity in A431 cells down to the concentration.
As shown in Fig. 9a, the absorption spectra of colloidosomes and supraballs drop markedly compared to those of dispersed nanocrystals. Consequently, the number of photons absorbed by the superstructures, when they are subjected to a laser power of 808 nm (1 W cm−2), drops markedly. This effect could be partially compensated by the fact that these hybrid structures behave like nanoheaters.
Let us compare the overall heating efficiency induced by illumination of dispersed Fe3O4 nanocrystals and superstructures either dispersed in solution or internalized tumor cells incubated for one day. In solution, the temperature increases by 10.9 °C, 2 °C and 8.8 °C while, in the cells, it is 38.4 °C, 28 °C and 33.5 °C of dispersed nanocrystals, colloidosomes and supraballs, respectively (Fig. 14). The larger value obtained with dispersed nanocrystals compared to superstructures is attributed to the difference in the number of photons absorbed. Moreover, in solution, the difference in temperature increment between the supraballs and the colloidosomes is attributed to a lower energy transfer efficiency from the nanocrystals to the alkyl chain (mechanism of the nanoheaters). In cells, due to the large assemblies of nanocrystals, the efficiency of colloidosomes markedly increases. Fig. 14 clearly shows that the global heating of the tissue by superstructures is not efficient compared to that by dispersed nanocrystals.
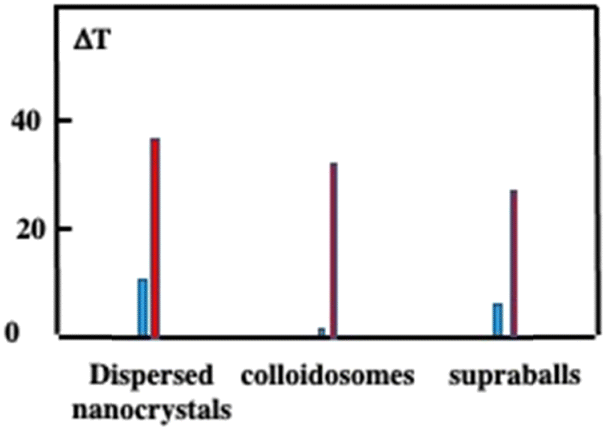 | ||
| Fig. 14 Temperature in solution induced by subjecting the superstructures and dispersed Fe3O4 nanocrystals (blue) and in a pellet of 1 million cells having internalized Fe3O4 nanocrystals, colloidosomes and supraballs (red). Dispersion in 100 μL of phosphate-buffered saline upon irradiation with an 808 nm laser (1 W cm−2) for 5 min. | ||
For therapy, cellular damage can come from global heating of the tissue, of the cell pellet or from local thermal damage induced in the vicinity of nanosources of heat. Indeed, it has been shown that the temperature increase near to the heat nanosources is much higher than the macroscopic temperature in the surrounding medium.59,60 Here, we assess thermal damage to cells in the nanocrystal-loaded cell pellet that combines local intracellular heating with the overall temperature increase in the pellet as measured by thermal imaging. Cell apoptosis and necrosis are assessed through the evaluation of the membrane permeability to fluorescent dyes. Fig. 15a shows that cells having internalized dispersed Fe3O4 nanocrystals show slightly enhanced level of apoptosis and necrosis after irradiation but the difference with non-irradiated cells is not statistically significant. In contrast, colloidosome- and supraball-loaded cells feature a significant enhancement of the apoptosis (more than 15% of the total cell number) and necrosis level (∼15%) after irradiation. This suggests that supraballs and to a larger extent colloidosomes could induce local thermal damages independently of the global temperature increment in the cell pellet. This is related to the nanoheater efficiency. As observed above, in vitro, due higher nanocrystal packing, the light absorption induces a larger chain energy transfer with supraballs than colloidosomes. However, in cells, the colloidosome shape is not retained and the nanocrystals self-assembled in large domains. Consequently, in the first approximation, we could assume, but not quantify, that the energy penetration is almost an even larger for colloidosomes than supraballs.
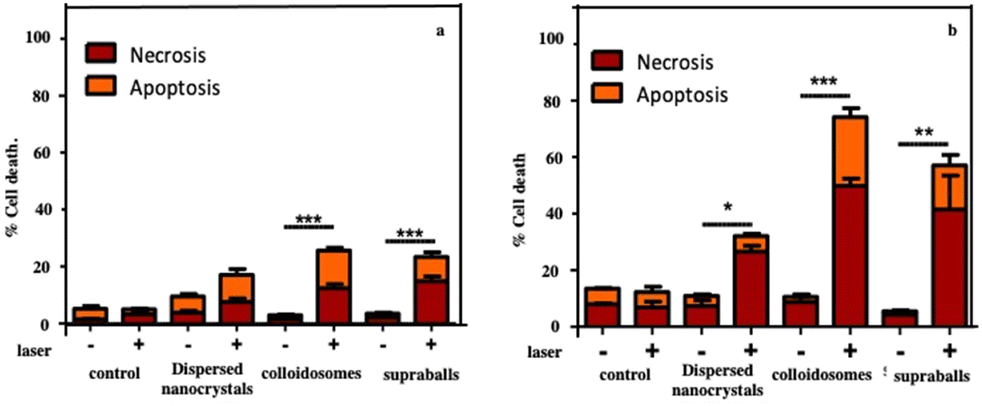 | ||
| Fig. 15 Evaluation of the local cellular damages induced by dispersed Fe3O4 nanocrystals, colloidosomes and supraball-mediated photothermal therapy in cell monolayers 4 h post-irradiation. The percentage of cell necrosis and apoptosis caused by dispersed Fe3O4 nanocrystals, colloidosomes and supraballs. Damages induced in the vicinity of the heat nanosources (a) and mediated photothermal therapy in cell monolayers (b). Cell apoptosis and necrosis are assessed through the evaluation of the membrane permeability to fluorescent dyes. Hoechst 33342 stains the condensed chromatin of live cells, and this allows the quantification of all cells. The YO-PRO™ dye can enter cells under apoptosis whilst, propidium iodide cannot, it only stains dead necrotic cells. The staining pattern resulting from the simultaneous use of these three dyes allows to distinguish alive, apoptotic and necrotic cell populations by fluorescence microscopy. Source: A. Nicolas-Boluda et al.,58 John Wiley & Sons. Inc. | ||
To isolate thermal damage resulting from local intracellular heating, monolayers of thermalized cells are exposed to laser. Colloidosome-mediated intracellular local absorption induces the highest levels of cell damage compared to dispersed Fe3O4 nanocrystals and supraballs (Fig. 15b). Overall, the two self-assemblies of Fe3O4 nanocrystals clearly induce massive cell death: 74% and 57% of cell damage (both necrosis and apoptosis) in comparison to less than 35% for dispersed Fe3O4 nanocrystals. These data clearly demonstrate that self-assembled Fe3O4 nanocrystals, once internalized by cells can cause massive local damage. This causes cell apoptosis and necrosis without the global warming of the environment. Thus, the local intracellular heating is combined with the global temperature.
The apparent contradiction between the overall heating efficiency and the biological outcome on cells could be due to the fact that self-assemblies of nanocrystals in tumor cells act as an energy reservoir involving heat transfer with preferential propagation within the assembly. Moreover, the large percentage of nanocrystals close to lysosomal membranes induces local damage. Hence, a direct biological consequence of the energy accumulation in the lysosome membrane due to the close proximity of the lysosome membrane to the absorbing Fe3O4 nanocrystals in colloidosomes and supraballs can induce lysosome swelling and membrane permeabilization much more efficiently than for isolated Fe3O4 nanocrystals, without significant global heating. Thus, it is concluded that the optimization of Fe3O4 nanocrystal density in lysosomes and the proximity to the lysosome membrane, together with the high energy accumulation within the hydrophobic shell/core of colloidosomes/supraballs, enhance the local hot spot inducing cell death. These data agree with a previous study showing that cancer cells can be particularly sensitive to agents which induce lysosomal cell death through lysosome membrane permeabilization or disruption.61,62 Hence, lysosome membrane permeabilization can be triggered by a wide variety of agents, one of them being local heating.63,64
Another thermal damage in the biological system is demonstrated by the deposition of colloidosomes or supraballs on the fresh and highly organized type I collagen sheep tendon. Fig. 16a shows flattened structures of colloidosomes whereas, as expected, more compact and rigid supraballs are observed in Fig. 16b. By subjecting these samples to the NRI laser (10 min at 2 W cm−2), “holes” appear on the dense collagen networks (Fig. 16c and d).
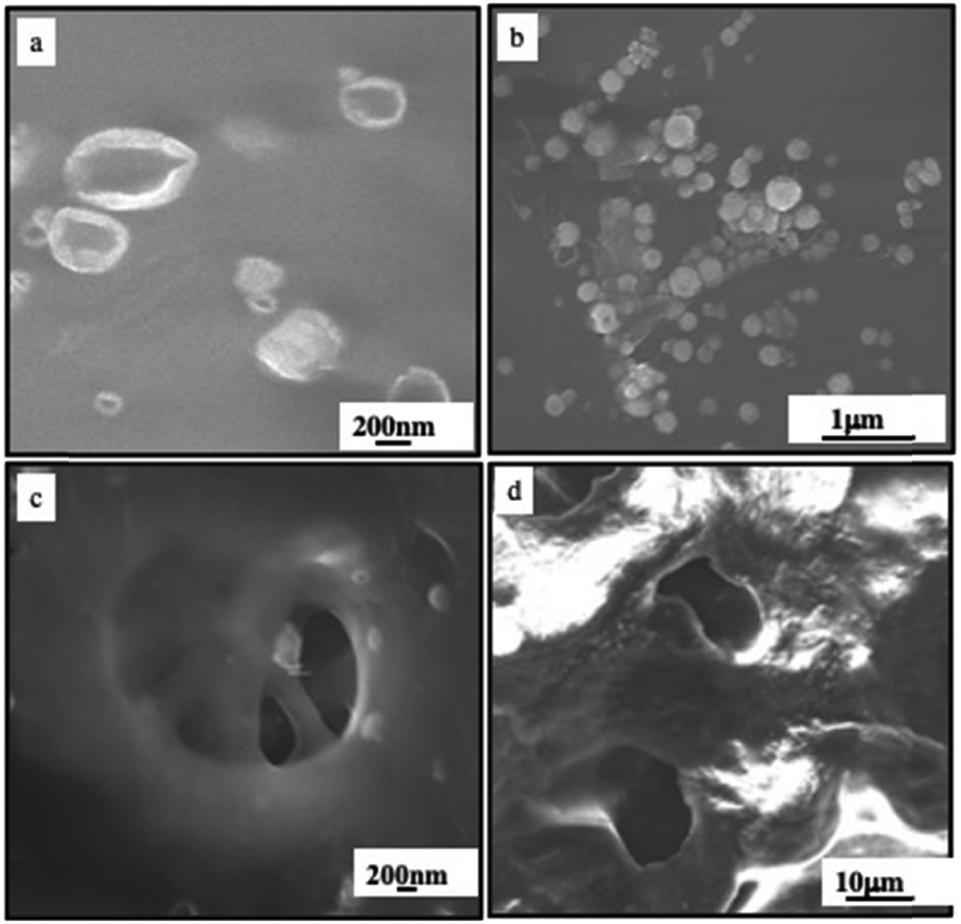 | ||
| Fig. 16 SEM image of the fresh sheep tendon incubated with colloidosomes (a) and (c) and supraballs (b) and (d) before (a) and (b) and after (c) and (d) irradiated with a NIR laser (10 min at 2 W cm−2). Source: A. Nicolas-Boluda et al.,58 John Wiley & Sons. Inc. | ||
Similar holes are observed in samples that underwent supraball-mediated PTT. This means that colloidosome-/supraball-mediated PTT induces collagen denaturation, but to a lesser extent with supraballs compared to colloidosomes. These experiments clearly reveal that colloidosomes are the most effective light-activated nano-heaters to induce local denaturation of collagen fibers under our experimental conditions. Similar data are obtained on the collagen matrix on the tumor.
Conclusions
Here, we give an overview of a fairly large series of novel water-soluble hybrid organic/inorganic supercrystalline structures.The “cluster structures” are 3D-shaped superlattices of hydrophobic nanocrystals (Co or Au) coated with phospholipids and a hydrophilic chain (C91) bridging from one vesicle to another and thus playing the role of “parachutes”. This was possible by controlling the growth process of the 3D superlattices and the packing parameters involving both the tail and the surface of the head group of the surfactant molecules.
Dense hydrophobic nanocrystals of Fe3O4 form either shells or fcc solid spherical particles called colloidosomes or supraballs, respectively. “Egg” structures are based on a shell of colloidosomes filled with 3D superlattices differing in size.
These superstructures and their physical properties are at an early stage. With “cluster structures”, the appearance of a photonic mode is observed. It is attributed to the formation of metamaterial retaining the imprint of the isolated nanocrystals. In addition, these water-dispersive hybrid structures subjected to light function as highly efficient universal nanoheaters.
Using colloidosomes and supraballs as nanocarriers in a cell model of human squamous cell carcinoma (A431), the cellular uptake is markedly increased compared to dispersed water-soluble nanocrystals. Referring to nanocrystals dispersed in the aqueous phase, these hybrids are mainly localized near the lysosome membrane with an increase of up to a factor of 10.
Over the past two decades, a number of specific intrinsic and collective properties of dried 3D superlattices deposited on a substrate have been discovered. The data presented here are essential for the search for new properties and their use for potential applications.
Nanocrystal self-assemblies retain the fingerprint of their building blocks. From there, all the specific properties observed with the dispersed nanocrystals could be used with self-assemblies. This makes it possible to combine the properties of both the dispersed nanocrystals and their assemblies. Additionally, toxicity issues can be reduced due to the micrometric scale of the self-assemblies.
To our knowledge, none of the researchers working in this field have discovered that such self-assembled nanocrystals in aqueous solution behave like a universal nanoheater. This result is important in the scientific fields related to the release of energy. Thus, these superstructures could be useful not only in biomedicine but also in the field of solar energy, photo-acoustics, catalysis, chemical reactions, etc.. The size and great stability of these superstructures are again an advantage for the environmental protection.
Moreover, we demonstrated that these superstructures are internalized in the cancer cells. Their size is either retained or increased in cancer cells. This maintains or increases the efficiency of the nanoheater. Thus, the properties discovered in the dispersed superstructures could be used and even improved in the cancer cells. The average location close to the lysosomal membrane makes them more fragile if they are subjected either to a magnetic field or to light and therefore weakens them.
These superstructures have several advantages related to the thermal effects induced either by a magnetic field or by light. Thus, they could help in the treatment of inoperable tumors, soft tissue cancer therapies without satisfactory therapies or with reduced success (breasts/small cell lungs/brain sarcomas/pancreas, and recurrences), strong tissue hyperthermia by ultrasound (medical treatment in oncology) and finally in rheumatism.
Magnetic resonance imaging (MRI) cannot be excluded for applications other than imaging practices for/or during surgical procedures.
Conflicts of interest
No conflicts of interest.Acknowledgements
My special thanks to Profs. Z. Yang from Shandong University (China), G. Cerullo, G. de La Vallée from Politecnico di Milano (Italy), and Dr F. Gazeau from the University of Paris.References
- L. Motte and M. P. Pileni, J. Phys. Chem., 1995, 99, 16425–16429 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Sciences, 1995, 270, 5240 Search PubMed.
- N. Goubet, J. Richardi, P. A. Albouy and M. P. Pileni, J. Phys. Chem. Lett., 2011, 2, 417–422 CrossRef CAS.
- L. Motte, F. Billoudet, J. Douin, E. Lacaze and M. P. Pileni, J. Phys. Chem., 1997, 101, 138 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, S. O’Brien and C. B. Murray, J. Am. Chem. Soc., 2005, 127, 8741–8747 CrossRef CAS PubMed.
- N. Goubet and M. P. Pileni, J. Phys. Chem. Lett., 2011, 2, 1024–1031 CrossRef CAS.
- L. Zhu, W. Huang, F. Yang, L. Yin, S. Liang, W. Zhao, L. Mao, X. Yu, R. Qiao and Y. Zhao, Adv. Biosyst, 2019, 3, 1800246 CrossRef PubMed.
- N. Goubet and M.-P. Pilenie, Nano Res., 2014, 7, 171–179 CrossRef CAS.
- M. A. Boles and D. V. Talapin, J. Am. Chem. Soc., 2015, 137, 4494–4502 CrossRef CAS PubMed.
- M. P. Pileni, J. Phys. Chem., 2001, 105, 3358–3372 CrossRef CAS.
- M. P. Pileni, Acc. of Chem. Res., 2007, 40, 685–693 CrossRef CAS PubMed.
- A. Fortini, E. Sanz and M. Dijkstra, Phys. Rev. E Stat. Nonlin. Soft Matter Phys., 2008, 78, 041402 CrossRef PubMed.
- M. P. Pileni, Acc. Chem. Res., 2008, 41, 1799–1809 CrossRef CAS PubMed.
- J. Chen, A. Dong, J. Cai, X. Ye, Y. Kang, J. M. Kikkawa and C. B. Murray, Nano Lett., 2010, 10, 5103–5108 CrossRef CAS PubMed.
- J.-S. Lee, M. V. Kovalenko, J. Huang, D. S. Chung and D. V. Talapin, Nat. Nanotech., 2011, 6, 348–352 CrossRef CAS PubMed.
- M. P. Pileni, Account Chem Res., 2012, 45, 1965–1972 CrossRef CAS PubMed.
- B. Li, C. Zhang, B. Jiang, W. Han and Z. Lin, Angew. Chem., 2015, 127, 4324–4328 CrossRef.
- H. Xu, Y. Xu, X. Pang, Y. He, J. Jung, H. Xia and Z. Lin, Sci. Adv., 2015, 1, 1500025 CrossRef PubMed.
- S. Neretina, R. A. Hughes, K. D. Gilroy and M. Hajfathalian, Acc. Chem. Res., 2016, 49, 2243–2250 CrossRef CAS PubMed.
- C. Lu and Z. Tang, Adv. Mater., 2016, 28, 1096–1108 CrossRef CAS PubMed; S. Zhang, C. I. Pelligra, X. Feng and C. O. Osuji, Adv. Mater., 2018, 30, 1705794 CrossRef PubMed.
- B. Luo, J. W. Smith, Z. Ou and Q. Chen, Acc. Chem. Res., 2017, 50, 1125–1133 CrossRef CAS PubMed.
- S. Zhang, C. I. Pelligra, X. Feng and C. O. Osuji, Adv. Mater., 2018, 30, 1705794 CrossRef PubMed.
- N. S. Mueller, Y. Okamura, B. G. M. Vieira, S. Juergensen, H. Lange, E. B. Barros, F. Schulz and S. Reich, Nature, 2000, 583, 780–784 CrossRef PubMed.
- A. O. Gusev, A. Taleb, F. Silly, F. Charraet and M. P. Pileni, Adv. Mater., 2000, 12, 1583 CrossRef CAS.
- A. Taleb, F. Silly, A. O. Gusev, F. Charra and M. P. Pileni, Adv. Mater., 2000, 12, 633–637 CrossRef CAS.
- P. Yang, I. Arfaoui, T. Cren, N. Goubet and M. P. Pileni, Nano Lett., 2012, 12, 2051–2055 CrossRef CAS PubMed.
- G. H. Carey, et al. , MRS Commun., 2013, 3, 83–90 CrossRef CAS.
- M. P. Boneschanscher, et al. , Science, 2014, 344, 1377–1380 CrossRef CAS PubMed.
- A. Courty, A. Mermet, P. A. Albouy, E. Duval and M. P. Pileni, Nat. Mater., 2005, 4, 395–398 CrossRef CAS PubMed.
- I. Lisiecki, D. Polli, C. Yan, G. Soavi, E. Duval, G. Cerullo and M. P. Pileni, Nano Lett., 2013, 13, 4914–4919 CrossRef CAS PubMed.
- M. P. Pileni, Acc. Chem. Res., 2017, 50, 1946–1955 CrossRef CAS PubMed.
- N. Yang, Z. Yang, M. Held, P. Bonville, P. A. Albouy, R. Lévy and M. P. Pileni, ACS Nano, 2016, 10, 2277–2286 CrossRef CAS PubMed.
- N. Yang, C. Deeb, J. L. Pelouard, N. Felidj and M. P. Pileni, ACS Nano, 2017, 11, 7797–7806 CrossRef CAS PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- M. Kranenburg, M. Veturoli and B. Smit, J. Phys Chem. B, 2003, 107, 11491–11501 CrossRef CAS.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, Rev. Biomembr., 2000, 1469, 159–195 CrossRef CAS PubMed.
- A. Mazzanti, Z. Yang, M. G. Silva, N. Yang, G. Rizza, P. E. Coulon, C. Manzoni, A. M. de Paula, G. Cerullo, G. Della Valle and M. P. Pileni, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8161–8166 CrossRef CAS PubMed.
- M. G. Silva, D. C. Teles-Ferreira, L. Siman, C. R. Chaves, L. O. Ladeira, S. Longhi, G. Cerullo, C. Manzoni, A. M. de Paula and G. Della Valle, Phys. Rev. B, 2018, 98, 115407 CrossRef CAS.
- C. Sun, F. Vallée, L. H. Acioli, E. P. Ippen and J. G. Fujimoto, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 15337–15348 CrossRef CAS PubMed.
- G. V. Hartland, Chem. Rev., 2011, 111, 3858–3887 CrossRef CAS PubMed.
- N. Goubet, I. Tempra, J. Yang, G. Soavi, D. Polli, G. Cerullo and M. P. Pileni, Nanoscale, 2015, 7, 3237–3246 RSC.
- S. U. Pickering, J. Chem. Soc. Trans, 1907, 91, 2001 RSC.
- O. D. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374 CrossRef CAS.
- O. D. Velev and K. Nagayama, Langmuir, 1997, 13, 1856 CrossRef CAS.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006 CrossRef CAS PubMed.
- F. Montanarella, J. J. Geuchies, T. Dasgupta, P. T. Prins, C. van Overbeek, R. Dattani, P. Baesjou, M. Dijkstra, A. V. Petukhov, A. van Blaaderen and D. Vanmaekelbergh, Nano Lett., 2018, 18, 3675 CrossRef CAS PubMed.
- R. G. Harrison, A. L. Washburn, A. Pickett and D. M. Call, J. Mater. Chem., 2008, 18, 3718 RSC.
- H. Duan, D. Wang, N. S. Sobal, M. Giersig, D. G. Kurth and H. Möhwald, Nano Lett., 2005, 5, 949 CrossRef CAS PubMed.
- Z. Yang, T. Altantzis, D. Zanaga, S. Bals, G. Van Tendeloo and M. P. Pileni, J. Amer. Chem. Soc., 2016, 138, 3493–3500 CrossRef CAS PubMed.
- J. Lacava, P. Born and T. Kraus, Nano Lett., 2012, 12, 3279–3328 CrossRef CAS PubMed.
- N. Glaser, D. J. Adams, A. Boeker and G. Krausch, Langmuir, 2006, 22, 5227–5229 CrossRef CAS PubMed.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS PubMed.
- R. J. Hickey, J. Koski, X. Meng, R. A. Riggleman, P. Zhang and S. J. Park, ACS Nano, 2014, 8, 495–502 CrossRef CAS PubMed.
- D. Wang, Z. Yang, G. Rizza, S. Balls and M. P. Pileni, personal communication.
- A. Schirato, L. Moretti, Z. Yang, A. Mazzanti, G. Cerullo, M. P. Pileni, M. Maiuri and G. Della Valle, J. Phys. Chem. C, 2022, 126(14), 6308–6317 CrossRef CAS PubMed.
- A. Nicolas-Boluda, Z. Yang, I. Dobryden, F. Carn, N. Winckelmans, C. Pechoux, P. Bonville, S. Bals, P. M. Claesson, F. Gazeau and M. P. Pileni, Adv. Funct. Mater., 2020, 2004274, 1–15 Search PubMed.
- A. Nicolas-Boluda, Z. Yang, T. Guilbern, L. Fouassier, F. Carn, F. Gazeau and M. P. Pileni, Adv. Funct. Mater., 2021, 2006824, 1–17 Search PubMed.
- J. T. Dias, M. Mros, P. del Pino, S. Rivera, V. Grazu and J. M. de la Fuente, Angew. Chem., 2013, 52, 11526–11529 CrossRef CAS PubMed.
- A. Riedinger, P. Guardia, A. Curcio, M. A. Garcia, R. Cingolani, L. Manna and T. Pellegrino, Nano Lett., 2013, 13, 2399–2406 CrossRef CAS PubMed.
- L. Groth-Pedersen and M. Jäättelä, Cancer Lett., 2013, 332, 265–274 CrossRef CAS PubMed.
- P. Boya and G. Kroemer, Oncogene, 2008, 27, 6434–6451 CrossRef CAS PubMed.
- P. Clerc, P. Jeanjean, N. Hallali, M. Gougeon, B. Pipy, J. Carrey, D. Fourny and V. Gigoux, J. Controlled Release, 2018, 270, 120–134 CrossRef CAS PubMed.
- D. Fourmy, J. Carrey and V. Gigoux, Nanomedicine, 2015, 10, 893–896 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |