
Ultra-robust, high-adhesive, self-healing, and photothermal zwitterionic hydrogels for multi-sensory applications and solar-driven evaporation†
Youyou
Chen
abc,
Chen
Zhang
 *bc,
Rui
Yin
bc,
Minghan
Yu
bc,
Yijie
Liu
d,
Yaming
Liu
abc,
Haoran
Wang
bc,
Feihua
Liu
bc,
Feng
Cao
d,
Guoqing
Chen
*a and
Weiwei
Zhao
*abc
*bc,
Rui
Yin
bc,
Minghan
Yu
bc,
Yijie
Liu
d,
Yaming
Liu
abc,
Haoran
Wang
bc,
Feihua
Liu
bc,
Feng
Cao
d,
Guoqing
Chen
*a and
Weiwei
Zhao
*abc
aState Key Laboratory of Advanced Welding & Joining, Harbin Institute of Technology, Harbin 150001, People's Republic of China. E-mail: chenguoqing@hit.edu.cn; wzhao@hit.edu.cn
bSauvage Laboratory for Smart Materials, The School of Materials Science and Engineering, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, People's Republic of China. E-mail: zhangchen2020@hit.edu.cn
cShenzhen Key Laboratory of Flexible Printed Electronics Technology, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, People's Republic of China
dSchool of Science, Harbin Institute of Technology (Shenzhen), Shenzhen 518055, People's Republic of China
First published on 7th July 2023
Abstract
Zwitterionic hydrogels have received considerable attention owing to their characteristic structures and integrating multifunctionality. However, the superhydrophilicity-induced poor mechanical properties severely hinder their potential applications. Besides, from the perspective of wide applications, zwitterionic hydrogels with integrated high mechanical properties, conductivity and multifunctionalities including self-adhesive, self-healing, and photothermal properties are highly desirable yet challenging. Herein, a new class of high-performance and multifunctional zwitterionic hydrogels are designed based on the incorporation of polydopamine-coated liquid metal nanoparticles (LM@PDA). Due to the efficient energy dissipation endowed by the isotropically extensible deformation of LM@PDA and the multiple interactions within the hydrogel matrix, the resultant hydrogels exhibited ultrahigh robustness with tensile strength of up to 1.3 MPa, strain of up to 1555% and toughness of up to 7.3 MJ m−3, superior or comparable to those of most zwitterionic hydrogels. The introduced LM@PDA also endows the hydrogels with high conductivity, versatile adhesion, autonomous self-healing, excellent injectability, three-dimensional printability, degradability, and photothermal conversion performance. These preferable properties enable the hydrogels promising as wearable sensors with multiple sensory capabilities for a wide range of strain values (1–500%), pressures (0.5–200 kPa) and temperatures (20–80 °C) with an impressive temperature coefficient of resistance (up to 0.15 °C−1). Moreover, these hydrogels can be also applied as solar evaporators with a high water evaporation rate (up to 2.42 kg m−2 h−1) and solar-thermal conversion efficiency (up to 90.3%) for solar desalination and wastewater purification. The present work can pave the way for the future development of zwitterionic hydrogels and beyond.
New conceptsZwitterionic hydrogels have received considerable attention in the next-generation electronics, biomedical engineering, and energy and environmental areas due to their characteristic structures and multifunctionality. However, the superhydrophilicity-induced poor mechanical properties severely hinder their potential applications. This study presents a new class of high-performance and multifunctional zwitterionic hydrogels by incorporating polydopamine-coated liquid metal nanoparticles (LM@PDA) into poly(sulfobetaine methacrylate) (PSBMA) hydrogels. Different from traditional rigid fillers, the extensible LM@PDA filler can deform isotropically and form multiple interactions with the hydrogel matrix to efficiently dissipate energy, yielding ultra-robust hydrogels. Also, LM@PDA can act as multifunctional fillers to simultaneously enhance the conductivity and photothermal conversion performance of hydrogels via the quantum tunneling effect and combined surface plasmons and lattice vibrations, respectively. Moreover, the synergetic effect between the filler and the matrix endows our hydrogel with versatile adhesion, autonomous self-healing, remarkable photothermal conversion, multi-sensory capacities, and excellent solar-driven evaporation performance. This work not only inspires the design of novel elastic and interactive fillers to enhance the hydrogel's mechanical strength but also paves the way for the fabrication of high-performance all-round hydrogels for applications in next-generation electronic, energy and environmental fields. |
Introduction
Hydrogels are soft materials with three-dimensional (3D) networks formed by hydrophilic polymer chains.1–3 Their combined advantages of chemical and structural versatility, biocompatibility, functionality, and mechanical properties have led to many novel applications, including wearable sensors,4,5 energy storage/conversion,6,7 and environmental protection.8,9 Recently, zwitterionic hydrogels, which bear equal positively and negatively charged groups in one unit, have received considerable attention owing to their characteristic structures and integrated multifunctionality, such as water retention capacity, conductivity, adhesiveness, antifouling and biocompatibility.10 However, zwitterionic hydrogels generally suffer from poor mechanical properties due to their superhydrophilicity, which severely hinders their potential applications.11To date, significant efforts have been made to develop high-strength zwitterionic hydrogels by designing double/triple networks and incorporating nanomaterials.12–14 For example, Dong et al.15 and Li et al.11 developed double-network and triple-network poly(sulfobetaine)-based zwitterionic hydrogels, respectively, which show superior mechanical strength compared with their single-network counterparts. Pei et al.16 and Sun et al.17 incorporated dopamine-modified nanoclay and gelatin-dispersed carbon nanotubes into sulfobetaine methacrylate-based zwitterion hydrogels, respectively, thereby enhancing the mechanical properties via abundant physical cross-linking. However, all the above-mentioned zwitterionic hydrogels still show unsatisfactory tensile properties, i.e., an obvious seesaw effect between strength and strain (strain < 200% at a strength of 1 MPa, or strength <100 kPa at a strain of 900%), which may be caused by the limited network malleability or restricted filler orientation. Moreover, from the perspective of wide applications, zwitterionic hydrogels with integrated high mechanical strength, stretchability, conductivity and multifunctionalities including self-adhesive, self-healing, and photothermal properties are highly desirable yet challenging.
Recently, liquid metals (LMs) have emerged as ideal flexible fillers for hydrogels, considering their fluidic nature, metallic conductivity, chemical stability, biocompatibility, and negligible toxicity.18–23 As compared to traditional rigid nanofillers, soft LMs can isotropically deform along with hydrogels to release the internal stress concentrations caused by the mechanical mismatch at the polymer–filler interface. Small-sized LM nanoparticles (LMNPs) are especially desirable for developing LM-embedded hydrogels not only because of their easier incorporation into hydrogels but also for their attractive properties to endow the hydrogels with additional functionalities, including excellent photothermal conversion performance, stimuli-responsive coalescence, catalytic potential, and alloying capacity.24 However, it remains challenging to fabricate an LM-filled hydrogel with satisfactory mechanical performance due to the interfacial incompatibility between LMNPs and the hydrogel matrix, which leads to severe macrophase separation and thus poor mechanical properties.25
Surface modification has recently been exploited to address the above issues by decorating graphene,26 silica,27 and a synthetic polymer28 onto the surface of LMNPs to prevent coalescence and improve stability. However, these encapsulated LMNPs suffer from complicated fabrication processes and chemistry, broad size distribution, potential environmental threats from the utilization of non-degradable materials, and a lack of sufficient functional groups to interact with the hydrogel matrix.19,29,30 Polydopamine (PDA), inspired by natural melanin and mussel adhesive proteins, has intrinsic versatile surface adhesion properties and abundant functional groups.31 On one hand, its catechol groups can coordinate with Ga3+ in LMNPs to form a stable shell, preventing LMNPs from fracture and exudation to the hydrogel network.32 On the other hand, its plentiful functional groups can actively interact with the hydrophilic polymer network via multiple noncovalent interactions to enhance the mechanical performance.4 Given these merits, PDA is supposed to effectively encapsulate LMNPs to improve their stability and form strong interaction with the hydrogel matrix to avoid phase separation. In addition, PDA's intrinsic broadband light absorption across the entire solar spectrum33 together with LMNPs’ broad surface plasmon resonance34 can endow the LM@PDA with remarkable photothermal conversion performance.
In this work, we incorporated core–shell structured LM@PDA as multifunctional fillers (reinforcing filler, conductive filler and photothermal conversion filler) into poly(sulfobetaine methacrylate) (PSBMA) zwitterionic hydrogels to simultaneously enhance the mechanical properties, conductivity, and photothermal conversion performance of the zwitterionic hydrogels (Fig. 1a). Due to the isotropic and extensible deformability of LM@PDA as well as the multiple noncovalent interactions formed within the hydrogel matrix, the resultant PSBMA-LM@PDA zwitterionic hydrogels exhibited ultrahigh robustness with a tensile strength of up to 1.3 MPa, strain of up to 1555% and toughness of up to 7.3 MJ m−3, superior or comparable to those of most zwitterionic hydrogels. Besides, LM@PDA addition also endowed hydrogels with high conductivity (up to 0.45 S m−1), strong adhesion (adhesion strength of up to 1.3 MPa on aluminum), autonomous self-healing (recovery efficiency up to 90%), and remarkable photothermal conversion performance. These preferable properties enabled PSBMA-LM@PDA hydrogels as wearable sensors with multi-sensory capabilities (Fig. 1b), including strain (1–500%), pressure (0.5–200 kPa) and temperature (20–80 °C), with an impressive temperature coefficient of resistance (up to 0.15 °C−1). Benefiting from the porous hydrophilic networks that enabled stable water transport, high light absorption (ca. 95% over the entire solar spectrum) and efficient photothermal conversion, PSBMA-LM@PDA hydrogels could also be applied as solar evaporators with high water evaporation rate (up to 2.42 kg m−2 h−1) and solar-thermal conversion efficiency (up to 90.3%). This work can not only inspire the design of high-performance and multifunctional zwitterionic hydrogels but also expand their application scenarios from flexible electronics to energy and environmental areas.
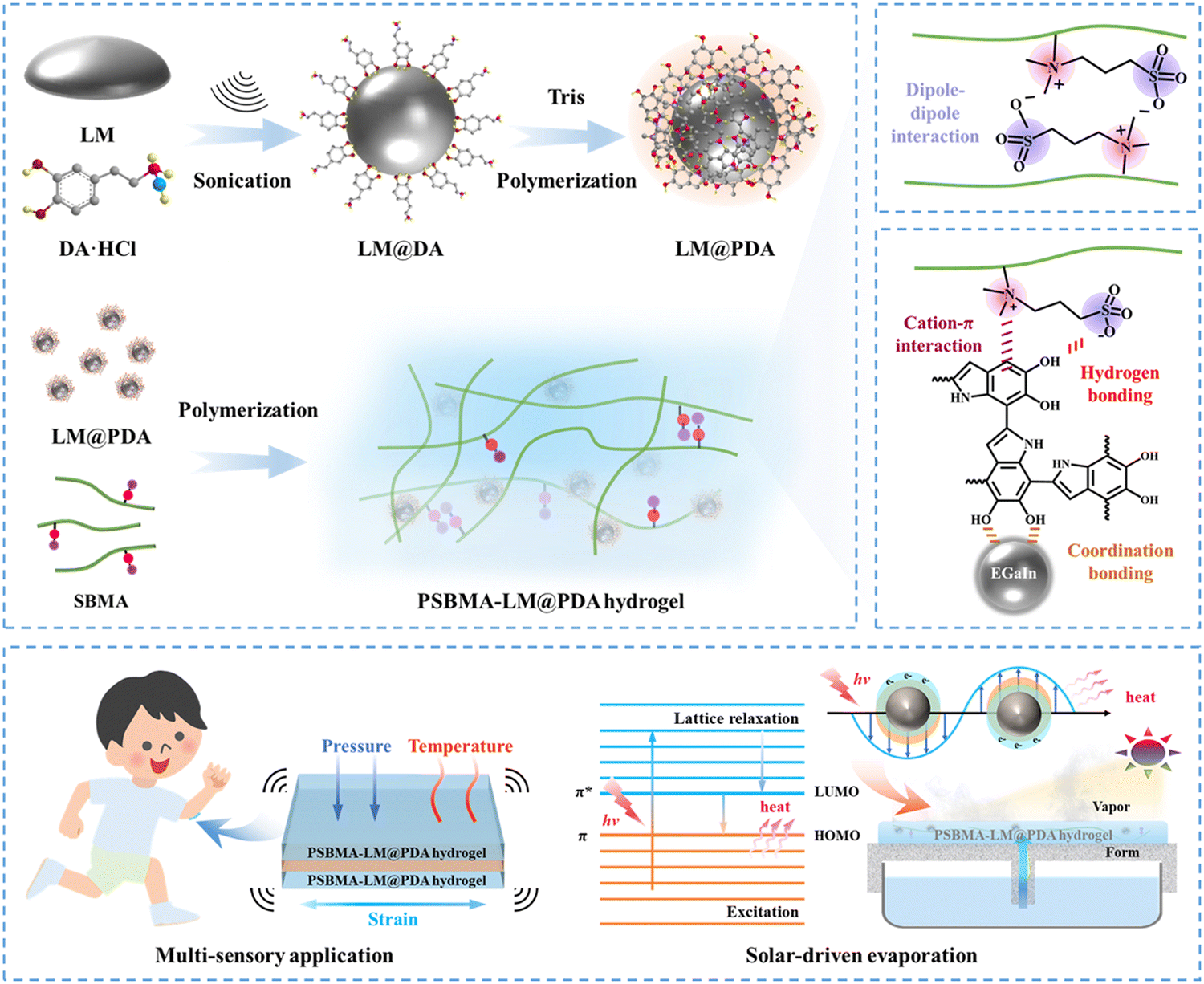 | ||
| Fig. 1 Schematic illustration of (a) synthesis and (b) applications of PSBMA-LM@PDA hydrogels. | ||
Results and discussion
The preparation process of LM@PDA involved the following two steps (Fig. 1a): Bulk EGaIn was first sonicated in an aqueous dopamine hydrochloride solution, resulting in dopamine-assisted dispersion of LM (LM-DA). During the sonication, ultrathin oxide layers were formed spontaneously on the surface of LMNPs, and the dopamine molecules were anchored to the oxide layer via coordination of their oxygen-containing groups with Ga3+. The Tris reagent was then added to the LM-DA suspension to trigger the self-polymerization of dopamine on the surfaces of LMNPs, resulting in LM@PDA. A transmission electron microscopy (TEM) micrograph (Fig. 2a) was presented to distinctly demonstrate the core–shell structure of the LM@PDA. The thickness of the PDA shell was about 12 nm (Fig. S1, ESI†). The energy-dispersive X-ray spectroscopy (EDS) mapping further validated the successful and uniform coating of PDA on the surface of LM@PDA (Fig. 2b). Fourier transform infrared (FTIR) spectra confirmed the interactions between PDA and LMNPs. As shown in Fig. S2, ESI,† typical stretching vibrations of benzene ring skeletons and C–OH red-shifted from 1529 and 1284 cm−1 in PDA to 1492 and 1269 cm−1 in LM@PDA, respectively, illustrating the energy transfer and bond formation between them, i.e., the decoration of PDA on LMNPs.35,36 The obtained LM@PDA suspension showed satisfactory stability after more than 24 h standing (Fig. S3, ESI†). In sharp contrast, the LMNPs sonicated in DI water exhibited severe phase separation within 6 h. The size distribution and Z-average size of LM@PDA (225.1 ± 1.7 nm, Fig. S4, ESI†) were also narrower and smaller than those of the LMNPs sonicated in DI water (351.6 ± 2.4 nm), ascribing to the stabilizing effect of the PDA coating.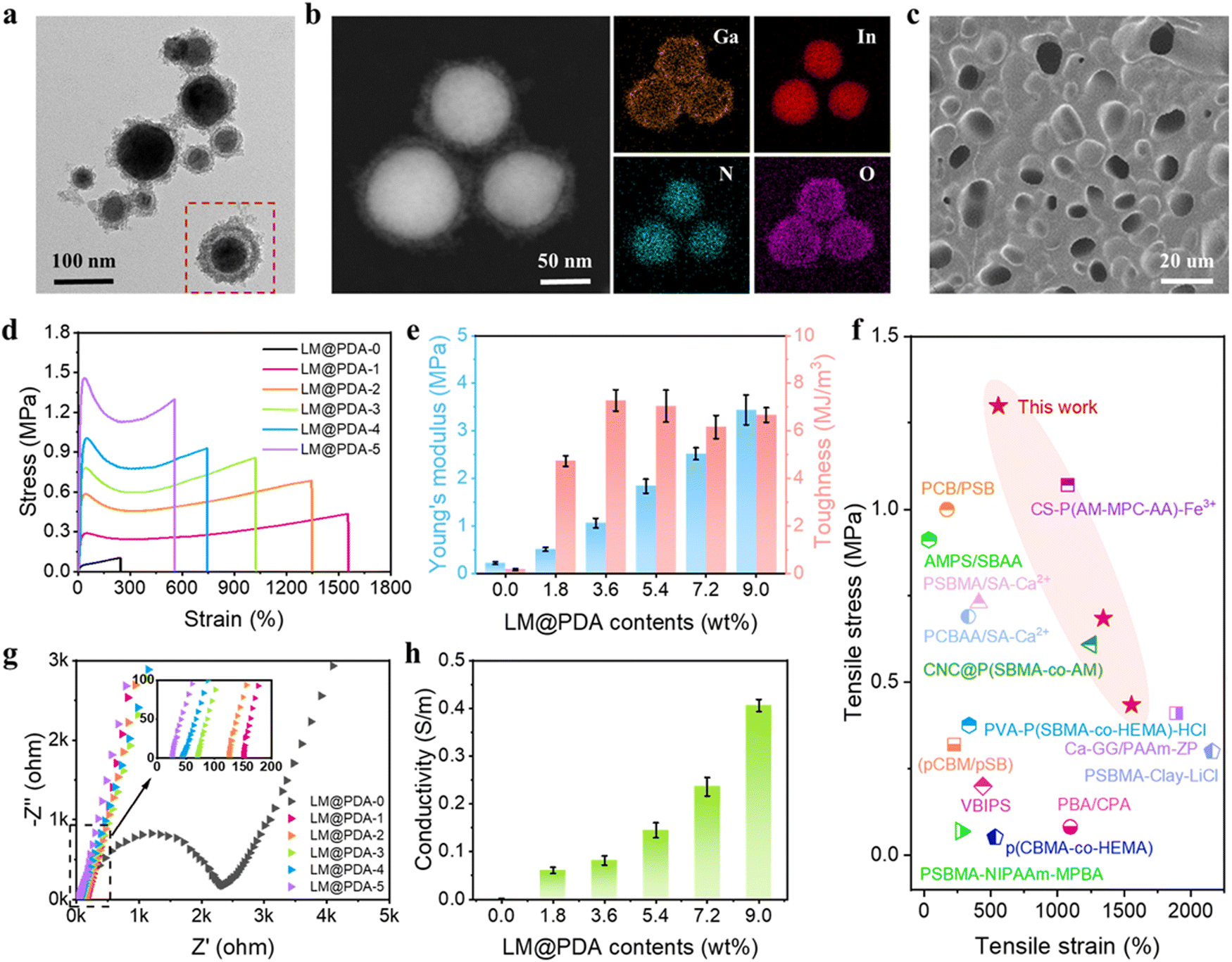 | ||
| Fig. 2 TEM image of LM@PDA with (a) core–shell structure and (b) corresponding elemental mapping by EDS. (c) Scanning electron microscope (SEM) image of the surface section of the freeze-dried PSBMA-LM@PDA hydrogel. (d) Tensile stress–strain curves and (e) Young's modulus and toughness of PSBMA-LM@PDA hydrogels with different LM@PDA contents. (f) Comparison of tensile stress and strain of PSBMA-LM@PDA hydrogels with previously reported zwitterionic hydrogels. (g) EIS plots and (h) conductivity of PSBMA-LM@PDA hydrogels with different LM@PDA contents. | ||
The PSBMA-LM@PDA hydrogels were fabricated by the in situ polymerization of SBMA in the presence of LM@PDA, with no additional crosslinkers. The interactions among LMNPs, PDA, and PSBMA chains are illustrated in Fig. 1a, which included dipole–dipole interactions between the SBMA moieties, hydrogen bonding between the sulfonate groups on SBMA and phenolic hydroxyl groups on PDA, cation–π interactions between the quaternary ammoniums on SBMA and aromatic benzene rings of PDA, and coordination bonding between the Ga3+ and the catechol groups of PDA. All of these dynamic bonds can act as sacrificial bonds, contributing to effective energy dissipation. Microstructural characterization (Fig. 2c, SEM) showed that the freeze-dried PSBMA-LM@PDA hydrogel network appeared as a 3D porous structure. EDS mapping (Fig. S5, ESI†) indicated the uniform distribution of LM@PDA in the hydrogel matrix, which could be attributed to the multiple hydrogen bonding and cation–π interactions between LM@PDA fillers and the PSBMA hydrogel matrix.
Mechanical properties are the primary characteristics for many practical applications of hydrogels. The homogeneous distribution of isotropically extensible LM@PDA as well as the multiple dynamic interactions within the hydrogel matrix endowed our PSBMA-LM@PDA hydrogels with extraordinary mechanical properties. The reinforcing mechanism of our PSBMA-LM@PDA hydrogels is illustrated in Fig. S6, ESI.† The LM@PDA served as a physical crosslinker in the PSBMA hydrogel matrix, forming multiple dynamic interactions. During stretching, the firm interfacial interactions between LM@PDA and PSBMA could facilitate the deformation and orientation of the LMNPs, enhancing stress transfer and energy dissipation. Tensile tests of pure PSBMA, PSBMA-PDA and PSBMA-LM@PDA hydrogels with the same PDA content were carried out to verify the reinforcement effect of LM@PDA. As shown in Fig. S7, ESI,† the PSBMA-LM@PDA hydrogel exhibited optimum mechanical properties. Specifically, the pure PSBMA hydrogel exhibited a tensile strength of 0.10 MPa, a strain of 243%, a Young's modulus of 0.22 MPa, and a toughness of 0.17 MJ m−3. After the introduction of PDA, the tensile strength, strain, Young's modulus and toughness of the PSBMA-PDA hydrogel were dramatically improved due to the formation of hydrogen bonding and cation–π interactions within the hydrogel matrix. Furthermore, due to the synergistic effect of the deformability of LMNPs and the multiple dynamic interactions, the PSBMA-LM@PDA hydrogel exhibited the highest tensile strength of 0.86 MPa, a strain of 1022%, a Young's modulus of 1.84 MPa, and a toughness of 7.04 MJ m−3, which were about 8.6, 4.2, 8.4, and 41.4 times those of the pure PSBMA hydrogel. Compared with the PSBMA-PDA hydrogel, PSBMA-LM@PDA hydrogel exhibited higher strength and ductility, demonstrating the important role of LM@PDA in enhancing mechanical properties.
Different contents of LM@PDA were introduced to explore the effect of LM@PDA content on the mechanical properties of the hydrogels. As shown in Fig. 2d and e, with increasing LM@PDA contents from 1.8 to 9.0 wt%, both the tensile stress and Young's modulus increased monotonically from 0.4 MPa to 1.3 MPa and from 0.5 MPa to 3.4 MPa, respectively. The monotonic increase of mechanical strength and Young's modulus of the PSBMA-LM@PDA hydrogels can be explained as follows: LM@PDA can serve as a physical crosslinker within the PSBMA hydrogel matrix to enhance stress transfer and energy dissipation. Higher LM@PDA can increase the crosslinking density through the hydrogen bonding, cation–π interactions and coordination bonding between the Ga3+ PDA and PSBMA. The tensile strain exhibited a declining trend, decreasing from 1555% to 556% with LM@PDA increasing from 1.8 to 9.0 wt%. Due to the balanced stress and strain values, the toughness of PSBMA-LM@PDA hydrogels was well maintained and a maximum toughness of 7.3 MJ m−3 (PSBMA-LM@PDA-2) can be achieved. Overall, our PSBMA-LM@PDA hydrogels exhibited extraordinary mechanical properties with a tensile strength up to 1.3 MPa, a strain up to 1555% and a toughness up to 7.3 MJ m−3, superior or comparable to those exhibited by most zwitterionic hydrogels15,37–49 (Fig. 2f and Table S1, ESI†).
Rheology tests were also carried out to investigate the influence of LM@PDA content on the mechanical performance of the PSBMA-LM@PDA hydrogels, and the results are shown in Fig. S8, ESI.†. As expected, all the PSBMA-LM@PDA hydrogels displayed a dominant elastic solid behavior, with the storage modulus (G′) obviously higher than the loss modulus (G′′) over the entire frequency range. The storage modulus was directly related to the crosslinking density of the hydrogel.48 With the increase of the LM@PDA content, the storage modulus of the PSBMA-LM@PDA increased from ca. 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa to 400000 Pa, which was due to the positive relationship between LM@PDA content and the crosslinking density. The loss tangent (tanδ = G′′/G′) was further calculated to evaluate the elastic properties of the hydrogels, where the gradually lower tanδ indicated a tougher performance25 (Fig. S9, ESI†).
000 Pa to 400000 Pa, which was due to the positive relationship between LM@PDA content and the crosslinking density. The loss tangent (tanδ = G′′/G′) was further calculated to evaluate the elastic properties of the hydrogels, where the gradually lower tanδ indicated a tougher performance25 (Fig. S9, ESI†).
Electrochemical impedance spectroscopy (EIS) was carried out to evaluate the conductivity of PSBMA-LM@PDA hydrogels with different LM@PDA contents. As shown in Fig. 2g, the Nyquist plot of the pure PSBMA hydrogel exhibited a semicircular arc at higher frequencies and a spike at lower frequencies. The semicircle in the high-frequency region represented the charge-transfer impedance, which indicated the difficulty of charge transfer at the electrode/electrolyte interface.50 The calculated conductivity of the pure PSBMA hydrogel was 0.002 S m−1. With the incorporation of conductive LMNPs, the conductivity of PSBMA-LM@PDA hydrogels dramatically increased. As shown in the inset of Fig. 2g, all the Nyquist plots of PSBMA-LM@PDA hydrogels with LM@PDA addition showed a linear trend without the semicircle, indicating fast electron transfer occurring at the electrode–hydrogel interface. The intercepts of EIS plots on the Z′-axis represented the impedance of the hydrogels. As the LM@PDA content increased, the impedance of the PSBMA-LM@PDA hydrogels shifted to a lower value, indicating the enhancement in conductivity. The calculated conductivity of PSBMA-LM@PDA hydrogels with varied LM@PDA contents is shown in Fig. 2h, exhibiting an increasing trend from 0.002 to 0.45 S m−1, which can be attributed to the denser conductive network and the stronger quantum tunneling effect.19 Considering the excellent mechanical properties and high conductivity of the PSBMA-LM@PDA-5 hydrogel, it was chosen for the following experiments, unless otherwise stated.
Self-adhesiveness is another exceptional property of our PSBMA-LM@PDA hydrogels. The PSBMA-LM@PDA-5 hydrogel exhibited a universal and robust adhesion to a wide range of substrates including glass, filter paper, aluminum (Al), copper (Cu), nitrile rubber, and silicone rubber, as demonstrated in Fig. S10, ESI.† The adhesion strength of the hydrogel to diverse substrates was evaluated via lap shear tests (Fig. 3a). The results are displayed in Fig. 3b and c, with the strength values of 1318 kPa for Al, 717 kPa for glass, 554 kPa for Cu, 369 kPa for filter paper, 249 kPa for nitrile rubber, and 40 kPa for silicone rubber. As comparison, the adhesion strength of the pure PSBMA hydrogel to Al was only 72 kPa, counting only 5.5% of that for the PSBMA-LM@PDA-5 hydrogel, as shown in Fig. S11, ESI.†
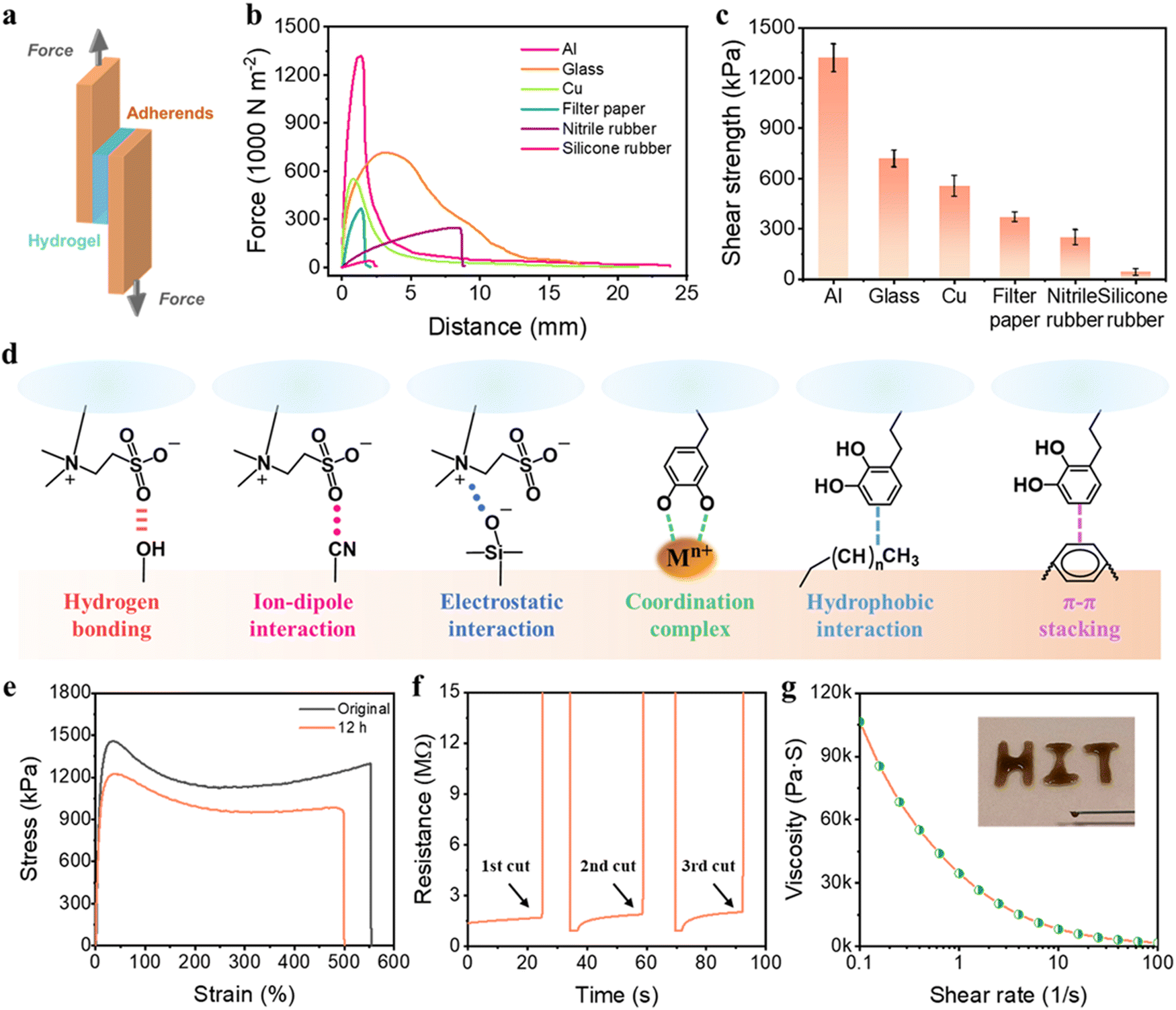 | ||
| Fig. 3 (a) Schematic illustration of the lap shear test. (b) Lap shear curves of the PSBMA-LM@PDA-5 hydrogel adhered on Al, glass, Cu, filter paper, nitrile rubber, and silicone rubber. (c) Shear strength of the PSBMA-LM@PDA-5 hydrogel adhered to the above substrates. (d) Schematic adhesion mechanisms of the PSBMA-LM@PDA-5 hydrogel. (e) Stress–strain curves of the original hydrogel and the healed hydrogel after healing for 12 h. (f) Electrical self-healing properties of the PSBMA-LM@PDA-5 hydrogel. (g) Viscosity of PSBMA-LM@PDA-5 measured as a function of the shear rate; inset: printing patterns of the injectable PSBMA-LM@PDA-5 hydrogel through a syringe. | ||
Such outstanding adhesive behaviors of the PSBMA-LM@PDA-5 hydrogel can be attributed to the synergistic interactions of SBMA and PDA with the substrates (Fig. 3d). The polar zwitterion groups can provide plentiful active sites to form noncovalent bonding, including hydrogen bonding, ion–dipole interaction, and electrostatic attraction to various substrates.41,51 For example, the sulfonic acid groups on PSBMA chains can form hydrogen bonding with filter paper, glass, silicone rubber, and human skin and ion–dipole interaction with nitrile rubber; the quaternary ammonium groups on SBMA can form electrostatic attraction with glass. Besides, the functional groups of PDA can interact with diverse substrates through multiple covalent/noncovalent bond interactions such as hydrogen bonding, coordination complex, hydrophobic interaction, and π–π stacking.52,53 For instance, the catechol groups on PDA can interact with glass via hydrogen bonding and form coordination bonds with various metal ions, and the aromatic rings on PDA can interact with polystyrene via hydrophobic interaction and π–π stacking.
Specifically, upon contact with human skin, the zwitterionic groups on the PSBMA chains can form dipole–dipole interaction with the –COOH and –NH2 on the tissue surface, while PDA can interact with the tissue surface via hydrogen bonding, Schiff base reaction, or Michael addition.16 The synergistic effect of SBMA and PDA enabled a robust adhesion to human skin (Fig. S10, ESI†), avoiding the interfacial debonding between the PSBMA-LM@PDA-5 hydrogel and human skin even under intensive body movements, which was favorable in collecting stable and precise signals of the human movements for flexible sensors.
PSBMA-LM@PDA hydrogels also displayed outstanding self-healing properties, thanks to the presence of multiple noncovalent interactions such as hydrogen bonding, dipole–dipole interactions, cation–π interactions, and coordination bonding. The macroscopic self-healing properties of the PSBMA-LM@PDA-5 hydrogel are demonstrated in Fig. S12, ESI.† When two fractured PSBMA-LM@PDA-5 hydrogel segments were in contact, they could autonomously merge and heal into an integral bulk within seconds. The healed hydrogel could maintain integrity even under stretching. The self-healing efficiency of the PSBMA-LM@PDA-5 hydrogel was measured via tensile tests. As shown in Fig. 3e, after self-healing at room temperature for 12 h, the healed hydrogel could be stretched to 500% with a high self-healing efficiency of 90%. Compared to the original sample, the healed hydrogel showed a lower elongation at break, which can be attributed to the breakage of the non-reversible chemical association in the polymer matrix. Microscopic self-healing properties of the PSBMA-LM@PDA-5 hydrogel were further observed, as shown in Fig. S13, ESI.† The crack almost disappeared after 12 h. In contrast, the pure PSBMA hydrogel failed to heal even under a long-term incubation (24 h), as illustrated in Fig. S14, ESI.†
Fig. 3f shows the real-time resistance changes during the successive cutting and contacting of the PSBMA-LM@PDA-5 hydrogel. When the PSBMA-LM@PDA-5 hydrogel was cut into two parts, the resistance increased instantaneously and recovered to the initial value along with an autonomous healing process. The electrically self-healing properties could be ascribed to the reconstruction of the conductive pathways among neighbouring LM@PDA in the PSBMA-LM@PDA-5 hydrogel network.
In addition, due to the physical crosslinking of the hydrogel network, the PSBMA-LM@PDA-5 hydrogel also possessed shear thinning properties, which was favorable for the injectability and 3D printability of the hydrogel. As shown in Fig. 3g, the viscosity of the PSBMA-LM@PDA-5 hydrogel dramatically declined from 106540 to 1699 Pa s as the shear rate increased from 0.1 to 100 s−1, indicating the shear-thinning behavior. Such behavior allowed the direct formation of our hydrogels in specific shapes and sizes. Therefore, the PSBMA-LM@PDA-5 hydrogel could be easily injected by using a syringe to write the letters “HIT”. This was beneficial for the green, rapid, and economical development of large-scale material fabrication.
Our PSBMA-LM@PDA-5 hydrogel also exhibited excellent degradability, as shown in Fig. S15, ESI;† the PSBMA-LM@PDA-5 hydrogel exhibited excessive swelling and was largely degraded within 60 min. This can be explained as follows: the abundant hydrophilic groups of PDA and PSBMA, including hydroxyl groups, catechol groups, sulfonic acid groups and quaternary ammonium groups, endowed the PSBMA-LM@PDA-5 hydrogel with excessive water absorption capacity. Meanwhile, the porous structure of the hydrogels could provide more locations for free water to penetrate. When the PSBMA-LM@PDA-5 hydrogel was placed in water, the water molecule could quickly combine with PDA and PSBMA, resulting in the swelling of the hydrogels. With the swelling, the dynamic bonds in the fully physically crosslinked PSBMA-LM@PDA-5 hydrogel could be easily broken, leading to the collapse of the hydrogel structure and making the hydrogel easily degradable.
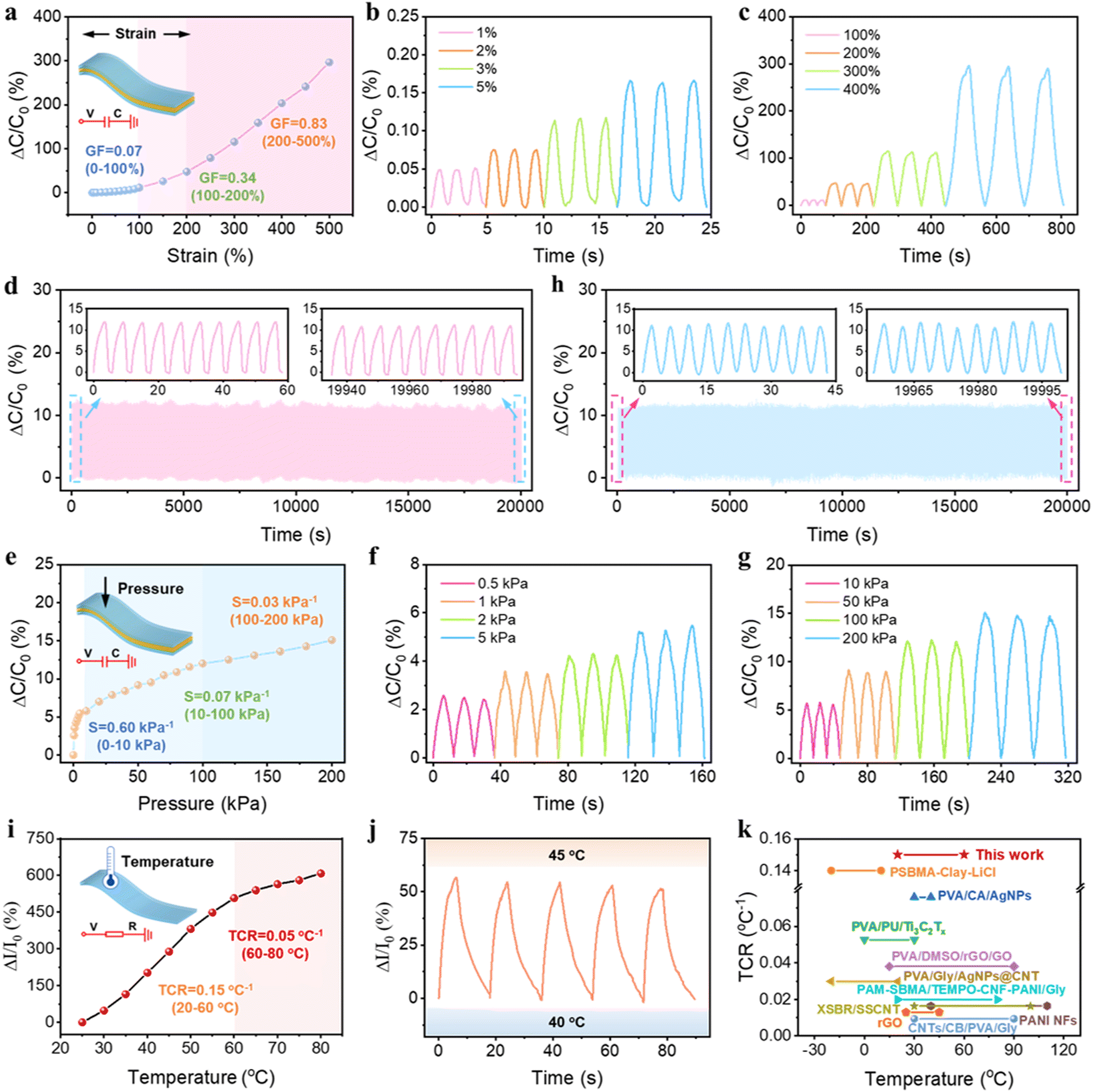
Benefiting from the excellent mechanical properties and high conductivity, our PSBMA-LM@PDA-5 hydrogel-based sensors exhibited sensitive responses to multiple physical stimuli, including tensile strain, pressure, and temperature. Since the capacitive devices have high sensitivity and no inherent temperature sensitivity,54 a capacitive strain/pressure sensor composed of a dielectric layer sandwiched by two PSBMA-LM@PDA-5 hydrogel layers was assembled, as illustrated in the insets of Fig. 4a and d. The relationship between the deformation and capacitance in a parallel-plate capacitor is shown as follows: 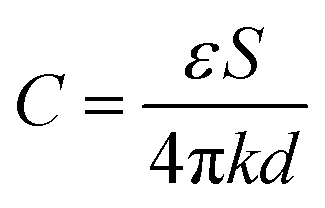 , where C is the capacitance, ε is the dielectric constant of the dielectric layer, S is the effective area of the conductive layer, k is the electrostatic constant, and d is the thickness of the dielectric layer. When strain/pressure external stimuli are applied to the capacitor, the expanded working area and decreased thickness in the capacitor would lead to an increase in capacitance, thus allowing for diverse detection of strain/pressure-induced deformation. Fig. 4a shows that the relative capacitance change (ΔC/C0) of our PSBMA-LM@PDA-5 hydrogel-based capacitor increased monotonously with an increase in strain. The calculated gauge factor (defined as δ(ΔC/C0)/δγstrain) was 0.07 (R2 > 0.95) in the low-strain regime (0–100%), 0.34 (R2 > 0.99) in the middle-strain regime (100–200%), and 0.83 (R2 > 0.99) in the high-strain regime (200–500%). Obviously, the as-prepared PSBMA-LM@PDA-5 hydrogel-based strain sensor possessed a wide strain sensing range with high sensitivity. In addition to the merit of sensitivity, the reversibility and long-term stability of the hydrogel-based strain sensor are also of great importance for practical applications. Fig. 4b and c display the discriminate strain sensing of the PSBMA-LM@PDA-5 hydrogel-based strain sensor in the small strain range of 1–5% and the large strain range of 100–400%, respectively. It can be seen that the relative capacitance change under stepped cyclic stretched strain almost completely recovered to the initial value, indicating excellent strain-sensing reversibility. Fig. 4d shows that the PSBMA-LM@PDA-5 hydrogel-based strain sensor could work stably for 20000 s (3500 cycles) at a fixed strain of 100%, demonstrating excellent long-term stability.
, where C is the capacitance, ε is the dielectric constant of the dielectric layer, S is the effective area of the conductive layer, k is the electrostatic constant, and d is the thickness of the dielectric layer. When strain/pressure external stimuli are applied to the capacitor, the expanded working area and decreased thickness in the capacitor would lead to an increase in capacitance, thus allowing for diverse detection of strain/pressure-induced deformation. Fig. 4a shows that the relative capacitance change (ΔC/C0) of our PSBMA-LM@PDA-5 hydrogel-based capacitor increased monotonously with an increase in strain. The calculated gauge factor (defined as δ(ΔC/C0)/δγstrain) was 0.07 (R2 > 0.95) in the low-strain regime (0–100%), 0.34 (R2 > 0.99) in the middle-strain regime (100–200%), and 0.83 (R2 > 0.99) in the high-strain regime (200–500%). Obviously, the as-prepared PSBMA-LM@PDA-5 hydrogel-based strain sensor possessed a wide strain sensing range with high sensitivity. In addition to the merit of sensitivity, the reversibility and long-term stability of the hydrogel-based strain sensor are also of great importance for practical applications. Fig. 4b and c display the discriminate strain sensing of the PSBMA-LM@PDA-5 hydrogel-based strain sensor in the small strain range of 1–5% and the large strain range of 100–400%, respectively. It can be seen that the relative capacitance change under stepped cyclic stretched strain almost completely recovered to the initial value, indicating excellent strain-sensing reversibility. Fig. 4d shows that the PSBMA-LM@PDA-5 hydrogel-based strain sensor could work stably for 20000 s (3500 cycles) at a fixed strain of 100%, demonstrating excellent long-term stability.
 | ||
| Fig. 4 Relative capacitance changes of the PSBMA-LM@PDA-5 hydrogel-based strain sensor (a) as a function of strain, (b) under small strains (1%, 2%, 3% and 5%), and (c) under large strains (100%, 200%, 300% and 400%). (d) Long-term stability of the PSBMA-LM@PDA-5 hydrogel-based strain sensor under 3500 cycles of 100% strain; insets: magnified curves of the initial and final 10 cycles. Relative capacitance changes of the PSBMA-LM@PDA-5 hydrogel-based pressure sensor (e) as a function of pressure, (f) under small pressures (0.5 kPa, 1 kPa, 2 kPa and 5 kPa), and (g) under large pressures (10 kPa, 50 kPa, 100 kPa and 200 kPa). (h) Long-term stability of the PSBMA-LM@PDA-5 hydrogel-based pressure sensor under 4500 cycles of 100 kPa pressure; insets: magnified curves of the initial and final 10 cycles. Relative current changes of the PSBMA-LM@PDA-5 hydrogel-based temperature sensor (i) as a function of temperature from 20 to 80 °C and (j) under temperature cycling between 40 and 45 °C. (k) Comparison of performance parameters with various temperature sensors reported in the literature. | ||
In addition to the responses to tensile strain, the PSBMA-LM@PDA-5 hydrogel-based capacitor also displayed a high pressure sensitivity. As displayed in Fig. 4e, the PSBMA-LM@PDA-5 hydrogel-based pressure sensor presented a wide sensing range from a low limit of detection of 0.5 kPa to a high pressure of 200 kPa, and its pressure sensitivity, defined as δ(ΔC/C0)/δp, was calculated to be 0.6 kPa−1 (from 0 to 10 kPa), 0.07 kPa−1 (from 10 to 100 kPa) and 0.03 kPa−1 (from 100 to 200 kPa). The pressure sensor could also fully recover to the initial state under both low (0.5 kPa, Fig. 4f) and high (200 kPa, Fig. 4g) pressing releasing. Notably, this pressure sensor also exhibited outstanding stability and reliability over 4500 consecutive pressing-releasing cycles upon a cycle pressure of 100 kPa, as shown in Fig. 4h.
Furthermore, we found that the PSBMA-LM@PDA-5 hydrogel possessed stimuli-responsive resistance that was highly temperature-sensitive. Temperature can influence the concentration of charge carriers as well as the migrating rate of both electrons and ions within the hydrogel matrix, thus altering the conductivity. At elevated temperatures, the Ga3+ ions may dissociate due to the weakened interaction between Ga3+ ions and the catechol groups on the PDA layer, resulting in an increased concentration of charge carriers.55 Also, higher temperatures boost the ion migration facilitation from the zwitterionic groups on PSBMA chains.56 Therefore, rising temperatures enhanced the migrating rate of both electrons and Ga3+ ions, i.e., improved conductivity in our PSBMA-LM@PDA-5 hydrogel. Then we designed a resistance-based temperature sensor based on the temperature responsivity of the PSBMA-LM@PDA-5 hydrogel, as illustrated in the inset of Fig. 4i. The thermal response of the temperature sensor was monitored during the heating process. As expected, the relative current changes increased with the increasing temperature and exhibited a high-sensitivity TCR (defined as δ(ΔI/I0)/δT) of 0.15 °C−1 under temperatures of 20–60 °C−1 and 0.05 °C−1 under temperatures of 60–80 °C−1. Significantly, the TCR value of the PSBMA-LM@PDA-5 hydrogel-based temperature sensor exceeded that of many temperature sensors, which adopted other reported thermosensitive materials,45,47,57–64 as shown in Fig. 4k and Table S2, ESI.†Fig. 4j shows the relative current changes of the temperature sensor when the temperature was cyclically switched between 40 and 45 °C for five cycles. A nearly constant response was obtained, indicating good repeatability and stability of our temperature sensor.
Due to the exceptional mechanical properties, satisfactory conductivity, outstanding sensing sensitivity, and signal stability of the PSBMA-LM@PDA-5 hydrogel, the hydrogel-based capacitor was harnessed as a wearable sensor to detect diverse physiological activities of the human body. The sensor was attached onto the finger joint to monitor the finger movements. As shown in Fig. 5a, when the bending angle of the finger changed from 0° to 30°, 60°, and 90°, the hydrogel-based sensor could rapidly and accurately respond to the bending angles via relative capacitance change in real time. When the finger was straightened, the relative capacitance of the sensor was able to fully recover to its pristine state. The cyclic bending of the finger, wrist, and elbow could also be accurately monitored as shown in Fig. 5b–d. Besides the large mechanical deformations, the capacitance waveforms can distinguish subtle movements such as swallowing and speaking, thanks to the low detection limit and high sensitivity of our hydrogel-based sensor. As shown in Fig. 5e–h, the hydrogel sensor fixed on the throat exhibited distinct and repeatable relative capacitance signals during the swallowing and speaking of words such as “Liquid”, “Metal”, and “Hydrogel”, respectively. All the above results suggested that our PSBMA-LM@PDA-5 hydrogel possessed great prospects and potential as a flexible wearable sensor for human motion monitoring and speech recognition.
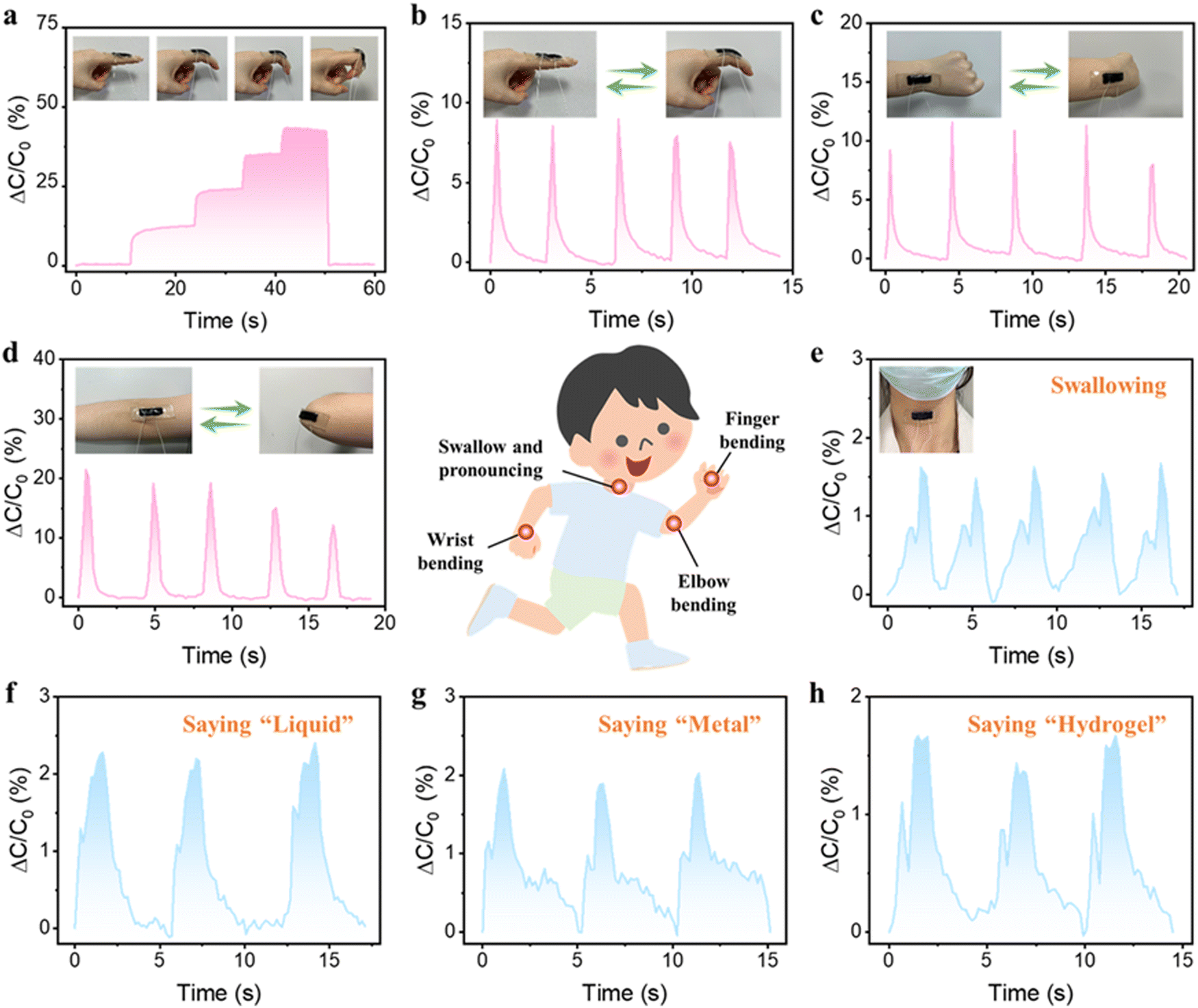 | ||
| Fig. 5 Real-time monitoring of human motions and subtle movements by the PSBMA-LM@PDA-5 hydrogel-based capacitor: (a) finger bending at different angles, (b) cyclic finger bending, (c) cyclic wrist bending (d) cyclic elbow bending, and (e) swallowing and speaking; (f) ‘Liquid’, (g) ‘Metal’ and (h) ‘Hydrogel’. | ||
Another appealing feature of our PSBMA-LM@PDA hydrogels was the excellent photothermal conversion performance, which can be ascribed to the combination of the surface plasmons and the lattice vibration of LM@PDA. As shown in Fig. 6a, when the wavelength of the incident light is longer than the dimension of the interacting LMNPs, localized surface plasmon resonance (LSPR) will occur as a result of the interaction between the surface electrons and the incident light. At the resonant frequency, the incident light can be absorbed and converted into heat.34,65 Additionally, PDA can also contribute to the photothermal conversion through lattice vibration as a result of abundant conjugated π bonds. Under incident light illumination, the loosely bonded electrons in PDA can be excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). Then, with the electron relaxation from the LUMO to the HOMO, energy can be transferred from the excited electrons to vibrational modes within the atomic lattices, resulting in heat release.66
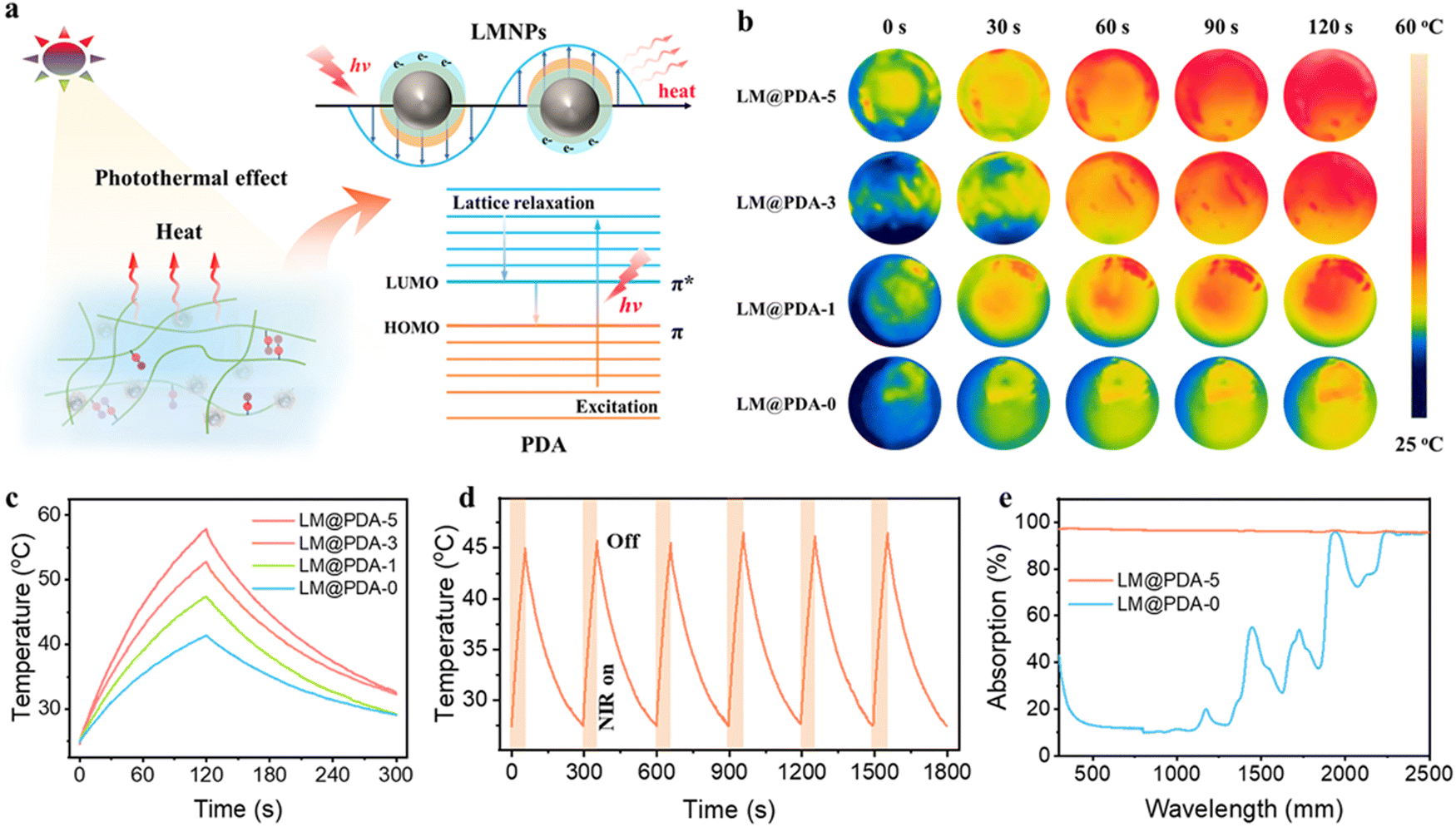 | ||
| Fig. 6 (a) Photothermal effect and photothermal conversion mechanism of PSBMA-LM@PDA hydrogels. (b) Corresponding thermal imaging of PSBMA-LM@PDA-0, PSBMA-LM@PDA-1, PSBMA-LM@PDA-3, and PSBMA-LM@PDA-5 hydrogels recorded using an IR camera. (c) The temperature change curves versus time of PSBMA-LM@PDA-0, PSBMA-LM@PDA-1, PSBMA-LM@PDA-3, and PSBMA-LM@PDA-5 hydrogels during on–off cycling under NIR irradiation. (d) Cycling stable performance of the PSBMA-LM@PDA-5 hydrogel during six cycles of heating and cooling under NIR irradiation. (e) UV-vis-NIR spectra of PSBMA-LM@PDA-0 and PSBMA-LM@PDA-5 hydrogels. | ||
To deeply understand the photothermal conversion performance of LM@PDA, the IR images of PSBMA-LM@PDA hydrogels with different LM@PDA contents were recorded using an infrared camera under near-infrared NIR irradiation. As illustrated in Fig. 6b and c, the surface temperature of the PSBMA-LM@PDA hydrogels rose faster and higher with the increment in LM@PDA content. Notably, the temperature increase of the PSBMA-LM@PDA-5 hydrogel was 33.1 °C within 2 min of irradiation, which was significantly higher than that of the pure PSBMA hydrogel (16.7 °C), demonstrating the excellent photothermal conversion properties of LM@PDA. Importantly, the PSBMA-LM@PDA-5 hydrogel also demonstrated outstanding photothermal stability under six on/off cycles of NIR irradiation, as shown in Fig. 6d. All these results demonstrated that LM@PDA could effectively and repeatedly convert the absorbed light into thermal energy. Then UV-vis-NIR spectra were recorded to investigate the light absorption ability of LM@PDA. As shown in Fig. 6e, the PSBMA-LM@PDA-5 hydrogel exhibited a high light absorption (ca. 95%) over the entire 300–2500 nm wavelength range, remarkably higher than that of the pure PSBMA hydrogel, which can be evidently attributed to the presence of LM@PDA with desirable light-absorbing ability.
With the combined advantages of robust broadband light absorption, excellent photothermal conversion performance and strong water transportation abilities, the PSBMA-LM@PDA hydrogels possess tremendous potential in solar-driven evaporation. As illustrated in Fig. 7a, the PSBMA-LM@PDA hydrogel-based interfacial evaporator was composed of three structural components: the photothermal hydrogel to convert the absorbed light into thermal energy for vapor generation, the air-laid paper to transfer water to the hydrogel, and the polystyrene (PS) foam with a water supply hole in the center to localize the hydrogel surface at the air/water interface as well as inhibit the heat loss to bulk water.
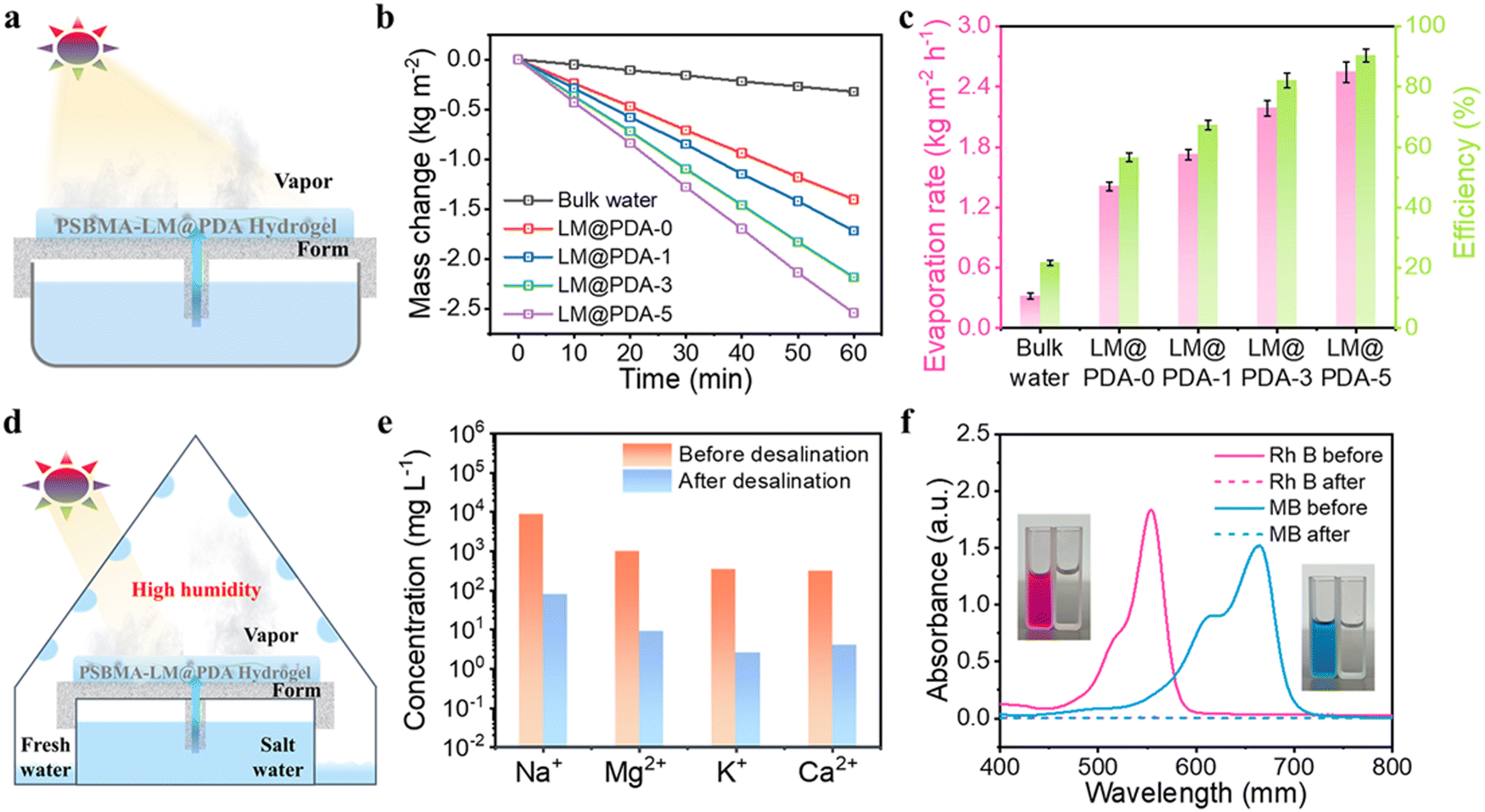 | ||
| Fig. 7 (a) Schematic illustration of the PSBMA-LM@PDA hydrogel-based interfacial evaporator. (b) Mass change of bulk water and PSBMA-LM@PDA-0, PSBMA-LM@PDA-1, PSBMA-LM@PDA-3, and PSBMA-LM@PDA-5 hydrogels under 1 sun. (c) Water evaporation rate and energy efficiency of bulk water and PSBMA-LM@PDA-0, PSBMA-LM@PDA-1, PSBMA-LM@PDA-3, and PSBMA-LM@PDA-5 hydrogels under 1 sun. (d) Schematic illustration of solar-driven clear water collection. (e) Ion concentration of simulated seawater before and after evaporation. (f) UV-vis spectra and photographs of RhB solution and MB solution before and after evaporation. | ||
To verify the solar-driven evaporation performance, the water mass change due to evaporation under 1 sun irradiation (1.0 kW m−2) using the PSBMA-LM@PDA hydrogel-based evaporators was tracked. Control experiments with pure water were carried out for comparison purposes. The evaporation rates were calculated from the slope of water mass change curves. As shown in Fig. 7b, the water evaporation rates of PSBMA-LM@PDA hydrogels were much faster than that of pure water. After 60 min illumination, the water evaporation rate of the PSBMA-LM@PDA-5 hydrogel (2.54 kg m−2 h−1) was much higher than those of the PSBMA-LM@PDA-3 hydrogel (2.18 kg m−2 h−1), the PSBMA-LM@PDA-1 hydrogel (1.72 kg m−2 h−1), the pure PSBMA hydrogel (1.40 kg m−2 h−1), and pure water (0.32 kg m−2 h−1) on account of the excellent light absorption ability of LM@PDA.
The solar energy conversion efficiency (η) was used to evaluate the overall solar-to-vapor efficiency, which can be calculated by the following equation: 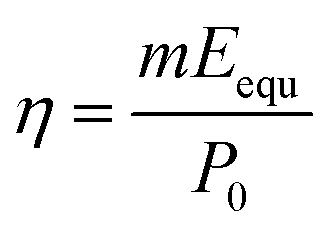 , where m is the net evaporation rate that subtracts the evaporation rate under dark evaporation conditions and P0 is the solar irradiation power (1 kW m−2). Eequ refers to the equivalent evaporation enthalpy of water in the hydrogel, which can be calculated by mass change under dark evaporation conditions assuming the unchanged power input (Uin): Uin = Eequmh = E0m0, where mh is the mass change of the hydrogel under dark evaporation conditions and E0 and m0 correspond to the evaporation enthalpy and mass change of pure water under the same conditions.67,68 As shown in Fig. S16, ESI,† the enthalpy of water evaporation in PSBMA-LM@PDA hydrogels (1278 J g−1 for the PSBMA-LM@PDA-5 hydrogel) was much smaller than that of bulk water (2450 J g−1), suggesting the effective reduction in water vaporization energy. The energy efficiency of the PSBMA-LM@PDA-5 hydrogel was calculated to be 90.3% under 1 sun illumination, which was 4.2 times that of pure water (21.7%) (Fig. 7c). Our PSBMA-LM@PDA-5 hydrogel-based evaporator achieved a considerable evaporation rate with a highly efficient energy utilization compared with the recently reported materials, including hydrogels,69,70 coatings,71,72 photothermal aerogels,73–75 and biomass carbonizations76,77 (Table S3, ESI†).
, where m is the net evaporation rate that subtracts the evaporation rate under dark evaporation conditions and P0 is the solar irradiation power (1 kW m−2). Eequ refers to the equivalent evaporation enthalpy of water in the hydrogel, which can be calculated by mass change under dark evaporation conditions assuming the unchanged power input (Uin): Uin = Eequmh = E0m0, where mh is the mass change of the hydrogel under dark evaporation conditions and E0 and m0 correspond to the evaporation enthalpy and mass change of pure water under the same conditions.67,68 As shown in Fig. S16, ESI,† the enthalpy of water evaporation in PSBMA-LM@PDA hydrogels (1278 J g−1 for the PSBMA-LM@PDA-5 hydrogel) was much smaller than that of bulk water (2450 J g−1), suggesting the effective reduction in water vaporization energy. The energy efficiency of the PSBMA-LM@PDA-5 hydrogel was calculated to be 90.3% under 1 sun illumination, which was 4.2 times that of pure water (21.7%) (Fig. 7c). Our PSBMA-LM@PDA-5 hydrogel-based evaporator achieved a considerable evaporation rate with a highly efficient energy utilization compared with the recently reported materials, including hydrogels,69,70 coatings,71,72 photothermal aerogels,73–75 and biomass carbonizations76,77 (Table S3, ESI†).
The practical applications of the PSBMA-LM@PDA-5 hydrogel for solar desalination and water treatment were further explored. As a proof-of-concept demonstration, a custom-designed solar water evaporation collection device was used to collect the condensed water from seawater and contaminated water. As illustrated in Fig. 7d, the PSBMA-LM@PDA-5 hydrogel-based evaporator was placed in a glass container with an inclined surface to absorb light and generate water vapor. Then the generated water vapor condensed into droplets on the inner surface, which further flowed along the wall and was collected at the condenser bottom.
To demonstrate the solar desalination capability of the PSBMA-LM@PDA-5 hydrogel, natural seawater (Yellow Sea, China) was used as the water source. As shown in Fig. 7e, after desalination by our solar water evaporation system, the Na+, Mg2+, K+, and Ca2+ concentrations characterized by inductively coupled plasma mass spectroscopy (ICP-MS) were significantly reduced by two to three orders of magnitude, which met the drinking water standard of the World Health Organization (WHO). In addition, rhodamine B (RhB) and methylene blue (MB) aqueous solutions were selected to verify the wastewater treatment behavior of our solar water evaporation system. As presented in Fig. 7f, the collected condensed water turned colorless and transparent after purification. According to UV-vis absorption characterization, the characteristic absorption peaks of both RhB and MB solutions disappeared, and the purification efficiency in the purified collected water reached 100%, which could be ascribed to the combined adsorption mechanism of PDA on dye molecules via chelation and hydrogen bonding and the distillation mechanism. The PSBMA-LM@PDA-5 hydrogel-based evaporator demonstrated potential application in wastewater treatment.
Finally, to demonstrate the advantages of our PSBMA-LM@PDA hydrogels in next-generation hydrogel devices, we compared our PSBMA-LM@PDA-5 hydrogel with previously reported representative hydrogels in terms of tensile strength, self-adhesion, self-healing, strain sensing, pressure sensing, temperature sensing, photothermal, and solar evaporation performance, substantiating that our PSBMA-LM@PDA-5 hydrogel exhibited overwhelmingly comprehensive features over other counterparts4,19,23,25,78–87 (Table S4, ESI†).
Conclusions
In summary, a class of high-performance and multifunctional zwitterionic hydrogels were successfully prepared, where LM@PDA with a stable core–shell structure was proposed as multifunctional fillers to impart the hydrogels with ultrahigh robustness, high conductivity, versatile adhesion, autonomous self-healing, remarkable photothermal conversion, multi-sensory capacities and solar-driven evaporation performance. These preferable properties enabled PSBMA-LM@PDA hydrogels as wearable sensors for human motion monitoring and speech recognition and also as evaporators for solar desalination and wastewater purification. We believe this work would provide novel insights into developing all-round zwitterionic hydrogels and broaden their applications from flexible electronics to solar energy utilization and water purification.Experimental section
Materials
Liquid metal EGaIn (gallium 75 wt% and indium 25 wt%) was purchased from Shenyang Jiabei Trading Co. Ltd (Shenyang, China). [2-(Methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl) ammonium hydroxide (SBMA), dopamine hydrochloride, tris(hydroxymethyl)aminomethane (Tris), ammonium persulfate (APS), rhodamine B, and methylene blue were purchased from Aladdin (Shanghai, China). VHB 4910 tape (1 mm thickness) was obtained from 3M Corporation.Preparation of LM@PDA
Bulk EGaIn (0.75 g) was added into 15 mL dopamine hydrochloride solution (10 mg mL−1). The mixture was sonicated using a probe ultrasonicator (TL-250Y, Jiangsu Tianling) for 40 min in an ice-water bath. After sonication, 0.18 g of Tris was added into the solution to trigger the self-polymerization of dopamine. The spontaneous oxidation and self-polymerization process was performed at room temperature for 4 h with constant stirring. The resultant suspension was further diluted to different LM@PDA nanoparticle concentrations of 0.5, 1.0, 1.5, 2.0 and 2.5 wt% for the subsequent preparation of hydrogels.Preparation of PSBMA-LM@PDA hydrogels
2.8 g of SBMA and 0.02 g APS were added into LM@PDA suspensions (10 g, with different LM@PDA concentrations of 0.5, 1.0, 1.5, 2.0 and 2.5 wt%) with stirring for 2 h. The resulting hydrogel precursors were transferred into silicone molds and subsequently incubated at 70 °C for 2 h of polymerization. Based on this method, hydrogels with 0, 1.8, 3.6, 5.4, 7.2, and 9.0 wt% of LM@PDA relative to SBMA content were prepared and denoted as PSBMA-LM@PDA-0, PSBMA-LM@PDA-1, PSBMA-LM@PDA-2, PSBMA-LM@PDA-3, PSBMA-LM@PDA-4, and PSBMA-LM@PDA-5, respectively, and also referred to simply as LM@PDA-0, LM@PDA-1, LM@PDA-2, LM@PDA-3, LM@PDA-4, LM@PDA-5.Mechanical tests
Mechanical properties of the hydrogel samples (20 mm in length, 10 mm in width, and 2 mm in thickness) were measured using a tensile testing machine (Mark-10, ESM301 system, USA) at a speed of 50 mm min−1. The Young's modulus was calculated from the slope of the initial linear region of the stress–strain curves (5–20% strain). Lap shear tests were performed to evaluate the adhesive strength of the hydrogels through the universal testing machine at a speed of 50 mm min−1. The hydrogels were applied between two substrates with a sandwiched bonding area of 20 mm × 10 mm. The adhesion strength was calculated by dividing the maximum force by the adhesion area. Shear thinning test was performed using a rotary rheometer (Anton Paar MCR301, Austria) to characterize the injectability of hydrogels; the viscosity of hydrogels was monitored at different shear rates from 0.1 to 100 s−1 at a fixed strain of 1%. A Mark-10 machine was also used in strain and pressure performance evaluations.Electrical tests
The electrochemical impedance spectroscopy of the hydrogels was performed using an electrochemical workstation (CHI600E, China) in the frequency range of 0.01 to 105 Hz with an open circuit voltage of 5 mV. The real-time capacitance of the strain and pressure sensors was recorded using a capacitance meter (LCR, Agilent E4980A, USA). The real-time resistance of the temperature sensor was measured using a digital sourcemeter (Keithley 2450), equipped with a hot plate with a controllable temperature to perform temperature measurements. The consent of all participants involved in the sensor experiments has been obtained.Solar evaporation performance
The solar evaporation performance of the hydrogels was measured using a solar simulator (Newport Oriel Sol3A, USA) outputting the simulated solar flux at 1000 W m−2 (1 sun).Characterization
The morphologies of the LM@PDA were obtained by TEM (JEM-3200FS, JEOL, Japan) equipped with EDS mapping. The morphologies of the freeze-dried PSBMA-LM@PDA hydrogels were obtained by SEM (Zeiss Supra 55, Germany) equipped with EDS. The microscopic self-healing process was observed on an optical microscope (Leica DM2500, Germany). The size distribution of the LM@PDA was characterized by dynamic light scattering (Zetasizer Nano ZS, Malvern instruments). The chemical structure was analyzed by FTIR (Nicolet is5, USA). The UV-vis-NIR spectra of the hydrogel samples were recorded using a spectrophotometer (Lambda 950, PerkinElmer) in the range of 300–2500 nm, and the absorbance (A) was calculated according to the formula A = 1 − T − R, where T and R are the transmittance and reflectance of the as-prepared hydrogels, respectively. An infrared camera (FLIR/T630sc, USA) was used to capture the digital photos. The concentrations of salt ions were tracked by ICP (MDTC-EQ-M29-01, USA).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Natural Science Foundation of China (No. 52073075), the Shenzhen Science and Technology Program (Grant No. KQTD201708110344233), and the Initial Scientific Research Foundation of Overseas High-level Talents of Harbin Institute of Technology (Shenzhen) (No. DB11409008).Notes and references
- Y. S. Zhang and A. Khademhosseini, Science, 2017, 356, 139–148 CrossRef PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- C. Zhang, Y. Zhou, H. Han, H. Zheng, W. Xu and Z. Wang, ACS Nano, 2021, 31, 1785–1794 CrossRef PubMed.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS PubMed.
- Q. Fu, S. Hao, L. Meng, F. Xu and J. Yang, ACS Nano, 2021, 15, 18469–18482 CrossRef CAS PubMed.
- J. Yang, Z. Xu, J. Wang, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Funct. Mater., 2021, 31, 2009438 CrossRef CAS.
- F. Yu, P. Yang, Z. Yang, X. Zhang and J. Ma, Chem. Eng. J., 2021, 426, 131900 CrossRef CAS.
- Y. Guo, H. Lu, F. Zhao, X. Zhou, W. Shi and G. Yu, Adv. Mater., 2020, 32, 1907061 CrossRef CAS PubMed.
- Y. Zhao, N. Yang, X. Chu, F. Sun, M. U. Ali, Y. Zhang, B. Yang, Y. Cai, M. Liu, N. Gasparini, J. Zheng, C. Zhang, C. F. Guo and H. Meng, Adv. Mater., 2023, 2211617 CrossRef CAS.
- X. Li, C. Tang, D. Liu, Z. Yuan, H. C. Hung, S. Luozhong, W. Gu, K. Wu and S. Jiang, Adv. Mater., 2021, 33, 2102479 CrossRef CAS PubMed.
- Y. Yu, F. Xie, X. Gao and L. Zheng, Soft Matter, 2021, 17, 4352–4362 RSC.
- L. Wang, G. Gao, Y. Zhou, T. Xu, J. Chen, R. Wang, R. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 3506–3515 CrossRef CAS PubMed.
- G. Lee, H. Seo, D. Kim, S. Shin and K. Kwon, RSC Adv., 2023, 13, 1672–1683 RSC.
- D. Dong, C. Tsao, H. C. Hung, F. Yao, C. Tang, L. Niu, J. Ma, J. MacArthur, A. Sinclair, K. Wu, P. Jain, M. R. Hansen, D. Ly, S. G. H. Tang, T. M. Luu, P. Jain and S. Jiang, Sci. Adv., 2021, 7, eabc5442 CrossRef CAS PubMed.
- X. Pei, H. Zhang, Y. Zhou, L. Zhou and J. Fu, Mater. Horiz., 2020, 7, 1872–1882 RSC.
- X. Sun, S. He, Z. Qin, J. Li and F. Yao, Compos. Commun., 2021, 26, 100784 CrossRef.
- Y. Xu, R. Rothe, D. Voigt, S. Hauser, M. Cui, T. Miyagawa, M. Patino Gaillez, T. Kurth, M. Bornhäuser, J. Pietzsch and Y. Zhang, Nat. Commun., 2021, 12, 2407 CrossRef CAS PubMed.
- Y. Ye and F. Jiang, Nano Energy, 2022, 99, 107374 CrossRef CAS.
- Y. Su, J. Zhao, W. Zhan, H. Yuan, L. Wu and G. Sui, Chem. Eng. J., 2022, 435, 135018 CrossRef CAS.
- J. Wu, J. Hu, Y. Feng, H. Fan, K. Wang and Z. Deng, Chem. Eng. J., 2023, 458, 141400 CrossRef CAS.
- Y. Chen, Z. Chen, C. Chen, H. Ur Rehman, H. Liu, H. Li and M. S. Hedenqvist, Chem. Eng. J., 2021, 421, 127762 CrossRef CAS.
- M. Sun, P. Li, H. Qin, N. Liu, H. Ma, Z. Zhang, J. Li, B. Lu, X. Pan and L. Wu, Chem. Eng. J., 2023, 454, 140459 CrossRef CAS.
- Y. Liu, W. Zhang and H. Wang, Mater. Horiz., 2021, 8, 56–77 RSC.
- Y. Hu, H. Zhuo, Y. Zhang, H. Lai, J. Yi, Z. Chen, X. Peng, X. Wang, C. Liu, R. Sun and L. Zhong, Adv. Funct. Mater., 2021, 31, 2106761 CrossRef CAS.
- M. A. Creighton, M. C. Yuen, N. J. Morris and C. E. Tabor, Nanoscale, 2020, 12, 23995–24005 RSC.
- P. Zhu, S. Gao, H. Lin, X. Lu, B. Yang, L. Zhang, Y. Chen and J. Shi, Nano Lett., 2019, 19, 2128–2137 CrossRef CAS PubMed.
- T. Liu, X. Wu, S. Zhu, F. Lorandi, L. Ni, S. Li, M. Sun, B. P. Bloom, D. H. Waldeck, V. Viswanathan, J. F. Whitacre and K. Matyjaszewski, ACS Appl. Energy Mater., 2022, 5, 3615–3625 CrossRef CAS.
- Q. Wei, M. Sun, Z. Wang, J. Yan, R. Yuan, T. Liu, C. Majidi and K. Matyjaszewski, ACS Nano, 2020, 14, 9884–9893 CrossRef CAS PubMed.
- B. He, P. Wang, B. Wang, Y. Du, L. Ji, S. Liu, Q. Ye and F. Zhou, Prog. Org. Coat., 2022, 169, 106922 CrossRef CAS.
- W. Cheng, X. Zeng, H. Chen, Z. Li, W. Zeng, L. Mei and Y. Zhao, ACS Nano, 2019, 13, 8537–8565 CrossRef CAS.
- T. Gan, W. Shang, S. Handschuh-Wang and X. Zhou, Small, 2019, 15, 1804838 CrossRef PubMed.
- W. Chen, H. Miao, G. Meng, K. Huang, L. Kong, Z. Lin, X. Wang, X. Li, J. Li, X. Y. Liu and N. Lin, Small, 2022, 18, 2107196 CrossRef CAS PubMed.
- E. Petryayeva and U. J. Krull, Anal. Chim. Acta, 2011, 706, 8–24 CrossRef CAS PubMed.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- X. Liu, J. Cao, H. Li, J. Li, Q. Jin, K. Ren and J. Ji, ACS Nano, 2013, 7, 9384–9395 CrossRef CAS PubMed.
- D. Chen, X. Zhao, H. Gao, G. Ren, J. Luo, H. Wang, C. Zha, K. Yang and P. Jia, ACS Biomater. Sci. Eng., 2022, 8, 2624–2635 CrossRef CAS PubMed.
- K. T. Huang, W. H. Hung, Y. C. Su, F. C. Tang, L. D. Linh, C. J. Huang and L. H. Yeh, Adv. Funct. Mater., 2023, 2211316 CrossRef CAS.
- J. Zhang, S. Qian, L. Chen, L. Chen, L. Zhao and J. Feng, J. Mater. Sci. Technol., 2021, 85, 235–244 CrossRef CAS.
- J. Zhang, L. Chen, L. Chen, S. Qian, X. Mou and J. Feng, Carbohydr. Polym., 2021, 257, 117627 CrossRef CAS PubMed.
- B. Yang and W. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 40620 CrossRef CAS PubMed.
- Q. Jiao, L. Cao, Z. Zhao, H. Zhang, J. Li and Y. Wei, Biomacromolecules, 2021, 22, 1220–1230 CrossRef CAS PubMed.
- J. Ren, Y. Liu, Z. Wang, S. Chen, Y. Ma, H. Wei and L. Shaoyu, Adv. Funct. Mater., 2022, 32, 2107404 CrossRef CAS.
- M. Yao, Z. Wei, J. Li, Z. Guo, Z. Yan, X. Sun, Q. Yu, X. Wu, C. Yu, F. Yao, S. Feng, H. Zhang and J. Li, Nat. Commun., 2022, 13, 5339 CrossRef CAS PubMed.
- Y. Chen, C. Zhang, R. Yin, A. Yin, Q. Feng, F. Liu, J. Shao, T. Su, H. Wang, G. Chen and W. Zhao, Chem. Eng. J., 2022, 449, 137907 CrossRef CAS.
- S. Y. Zheng, S. Mao, J. Yuan, S. Wang, X. He, X. Zhang, C. Du, D. Zhang, Z. L. Wu and J. Yang, Chem. Mater., 2021, 33, 8418–8429 CrossRef CAS.
- S. Hao, L. Meng, Q. Fu, F. Xu and J. Yang, Chem. Eng. J., 2022, 431, 133782 CrossRef CAS.
- H. Guo, M. Bai, Y. Zhu, X. Liu, S. Tian, Y. Long, Y. Ma, C. Wen, Q. Li, J. Yang and L. Zhang, Adv. Funct. Mater., 2021, 31, 2106406 CrossRef CAS.
- X. Hu, P. Zhang, J. Liu, H. Guan, R. Xie, L. Cai, J. Guo, L. Wang, Y. Tian and X. Qiu, Chem. Eng. J., 2022, 446, 136988 CrossRef CAS.
- M. Chen, J. Chen, W. Zhou, X. Han, Y. Yao and C. P. Wong, Adv. Mater., 2021, 33, 2007559 CrossRef CAS PubMed.
- G. Gao, F. Yang, F. Zhou, J. He, W. Lu, P. Xiao, H. Yan, C. Pan, T. Chen and Z. L. Wang, Adv. Mater., 2020, 32, 2004290 CrossRef CAS.
- C. Zhang, M. Q. Ma, T. T. Chen, H. Zhang, D. F. Hu, B. H. Wu, J. Ji and Z. K. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 34356–34366 CrossRef CAS.
- Z. Wang, H. C. Yang, F. He, S. Peng, Y. Li, L. Shao and S. B. Darling, Matter, 2019, 1, 115–155 CrossRef.
- X. Jing, H. Li, H. Y. Mi, Y. J. Liu, P. Y. Feng, Y. M. Tan and L. S. Turng, Sens. Actuators, B, 2019, 295, 159–167 CrossRef CAS.
- L. Zhou, Y. Li, J. Xiao, S. W. Chen, Q. Tu, M. Sen Yuan and J. Wang, Anal. Chem., 2023, 95, 3811 CrossRef CAS PubMed.
- F. Mo, Z. Chen, G. Liang, D. Wang, Y. Zhao, H. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2020, 10, 2000035 CrossRef CAS.
- L. Chen, X. Chang, H. Wang, J. Chen and Y. Zhu, Nano Energy, 2022, 96, 107077 CrossRef CAS.
- H. Liu, C. Du, L. Liao, H. Zhang, H. Zhou, W. Zhou, T. Ren, Z. Sun, Y. Lu, Z. Nie, F. Xu, J. Zhu and W. Huang, Nat. Commun., 2022, 13, 3420 CrossRef CAS PubMed.
- H. Chen, J. Huang, J. Liu, J. Gu, J. Zhu, B. Huang, J. Bai, J. Guo, X. Yang and L. Guan, J. Mater. Chem. A, 2021, 9, 23243–23255 RSC.
- X. Yu, W. Qin, X. Li, Y. Wang, C. Gu, J. Chen and S. Yin, J. Mater. Chem. A, 2022, 10, 15000–15011 RSC.
- G. Ge, Y. Lu, X. Qu, W. Zhao, Y. Ren, W. Wang, Q. Wang, W. Huang and X. Dong, ACS Nano, 2020, 14, 218–228 CrossRef CAS PubMed.
- M. Lin, Z. Zheng, L. Yang, M. Luo, L. Fu, B. Lin and C. Xu, Adv. Mater., 2022, 34, 2107309 CrossRef CAS.
- Q. Liu, H. Tai, Z. Yuan, Y. Zhou, Y. Su and Y. Jiang, Adv. Mater. Technol., 2019, 4, 1800594 CrossRef.
- J. Gu, J. Huang, G. Chen, L. Hou, J. Zhang, X. Zhang, X. Yang, L. Guan, X. Jiang and H. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 40815–40827 CrossRef CAS PubMed.
- F. Ding, L. Zhang, X. Chen, W. Yin, L. Ni and M. Wang, Front. Bioeng. Biotechnol., 2022, 10, 1–19 Search PubMed.
- C. Lei, Y. Guo, W. Guan and G. Yu, J. Polym. Sci., 2021, 59, 3084–3099 CrossRef CAS.
- C. Lei, W. Guan, Y. Guo, W. Shi, Y. Wang, K. P. Johnston and G. Yu, Angew. Chem., Int. Ed., 2022, 61, e202208487 CAS.
- L. Zhao, Z. Yang, J. Wang, Y. Zhou, P. Cao and J. Zhang, Chem. Eng. J., 2023, 451, 138676 CrossRef CAS.
- L. Hao, N. Liu, H. Bai, P. He, R. Niu and J. Gong, J. Colloid Interface Sci., 2022, 608, 840–852 CrossRef CAS PubMed.
- H. Lu, M. Li, X. Wang, Z. Wang, M. Pi, W. Cui and R. Ran, Chem. Eng. J., 2022, 450, 138257 CrossRef CAS.
- C. Wen, H. Guo, J. Yang, Q. Li, X. Zhang, X. Sui, M. Cao and L. Zhang, Chem. Eng. J., 2021, 421, 130344 CrossRef CAS.
- X. Su, D. Hao, M. Sun, T. Wei, D. Xu, X. Ai and X. Guo, Adv. Funct. Mater., 2022, 32, 2108135 CrossRef CAS.
- L. Song, L. Geng, Y. P. Tian, P. Mu and J. Li, J. Mater. Chem. A, 2021, 9, 23117–23126 RSC.
- Z. Wei, C. Cai, Y. Huang, Y. Wang and Y. Fu, Nano Energy, 2021, 86, 106138 CrossRef CAS.
- X. Hu, W. Xu, L. Zhou, Y. Tan, Y. Wang, S. Zhu and J. Zhu, Adv. Mater., 2017, 29, 1604031 CrossRef PubMed.
- N. Xu, X. Hu, W. Xu, X. Li, L. Zhou, S. Zhu and J. Zhu, Adv. Mater., 2017, 29, 1606762 CrossRef PubMed.
- M. Zhu, Y. Li, F. Chen, X. Zhu, J. Dai, Y. Li, Z. Yang, X. Yan, J. Song, Y. Wang, E. Hitz, W. Luo, M. Lu, B. Yang and L. Hu, Adv. Energy Mater., 2018, 8, 1701028 CrossRef.
- Z. Wei, Y. Wang, C. Cai, Y. Zhang, S. Guo, Y. Fu and S. C. Tan, Adv. Funct. Mater., 2022, 32, 2206287 CrossRef CAS.
- C. Liu, F. Li, G. Li, P. Li, A. Hu, Z. Cui, Z. Cong and J. Niu, ACS Appl. Mater. Interfaces, 2022, 14, 9608–9617 CrossRef CAS PubMed.
- Z. Bai, X. Wang, M. Zheng, O. Yue, M. Huang, X. Zou, B. Cui, L. Xie, S. Dong, J. Shang, G. Gong, A. M. Blocki, J. Guo and X. Liu, Adv. Funct. Mater., 2023, 33, 2212856 CrossRef CAS.
- B. Zhang, L. Rong, Z. Zhou and W. Yuan, Chem. Eng. J., 2023, 462, 142305 CrossRef CAS.
- C. Ma, Q. Liu, Q. Peng, G. Yang, M. Jiang, L. Zong and J. Zhang, ACS Nano, 2021, 15, 19877–19887 CrossRef CAS PubMed.
- Y. Peng, S. Tang, X. Wang and R. Ran, Macromol. Mater. Eng., 2021, 306, 2100309 CrossRef CAS.
- C. Y. Lo, Y. Zhao, C. Kim, Y. Alsaid, R. Khodambashi, M. Peet, R. Fisher, H. Marvi, S. Berman, D. Aukes and X. He, Mater. Today, 2021, 50, 35–43 CrossRef CAS.
- X. Qu, J. Liu, S. Wang, J. Shao, Q. Wang, W. Wang, L. Gan, L. Zhong, X. Dong and Y. Zhao, Chem. Eng. J., 2023, 453, 139785 CrossRef CAS.
- F. Li, N. Li, S. Wang, L. Qiao, L. Yu, P. Murto and X. Xu, Adv. Funct. Mater., 2021, 31, 2104464 CrossRef CAS.
- L. Xu, Y. Chen, M. Yu, M. Hou, G. Gong, H. Tan, N. Li and J. Xu, Nano Energy, 2023, 107, 108119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00629h |
| This journal is © The Royal Society of Chemistry 2023 |