
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Competition of polar and antipolar states hidden behind a variety of polarization switching modes in hydrogen-bonded molecular chains†
Sachio
Horiuchi‡
 *a,
Hiromi
Minemawari
a and
Shoji
Ishibashi‡
*b
*a,
Hiromi
Minemawari
a and
Shoji
Ishibashi‡
*b
aResearch Institute for Advanced Electronics and Photonics (RIAEP), National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, 305-8565, Japan. E-mail: s-horiuchi@aist.go.jp
bResearch Center for Computational Design of Advanced Functional Materials (CD-FMat), National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, 305-8568, Japan. E-mail: shoji.ishibashi@aist.go.jp
First published on 23rd March 2023
Abstract
Switchable π-electron systems are very powerful fragments to emphasize ferroelectric or antiferroelectric polarizations up to record-high levels among organic molecular crystals. According to the Cambridge Structural Database, many azole crystals such as imidazoles and tetrazoles contain polar and bistable hydrogen-bonded molecular sequences suitable for ferroelectricity or antiferroelectricity. Indeed, polarization hysteresis experiments on the 5-phenyl-1H-tetrazole (PHTZ) family combined with single crystal structural analysis have revealed one ferroelectric, two antiferroelectrics, and two hybrid-like dielectrics. Here, the rich variations for the interrelation between the hydrogen-bonding states and the polarization switching modes are interpreted by density functional theory (DFT) calculations with an excellent consistency. Large switchable polarizations are theoretically confirmed, and, as expected, the largest contribution is the switchable π-electron systems. By mapping the energy levels of polar/antipolar states, the disordered hydrogen bonds always appear when the ground state is accompanied by a nearly degenerate state. The straightforward case is the hybrid-like dielectric caused by the competition between the polar and antipolar states. However, contrastive behaviors are observed when the switchable dipoles are involved in competition between the different antipolar arrangement. For example, the PHTZ crystal exhibits typical antiferroelectric switching regardless of the hydrogen disorder, whereas polarization switching is silent in the imidazole derivatives. The latter is explained by the switching field increase with depth of the ground state relative to the energy level of the polar state.
New conceptsVarious electronic, mechanical, thermal, and electrooptical devices employ ferroelectrics/antiferroelectrics. A promising strategy to emphasize the performances in organic systems is to construct polar/antipolar order from bistable hydrogen-bonded sequences of switchable π-electron molecules. Although many hydrogen-bonded dielectrics contain hydrogen disorder, a comprehensive understanding of its origins and effects on polarization switching and structure–property relationships is lacking. The single crystal structural analysis, polarization hysteresis experiments, and theoretical evaluations herein reveal that disordered hydrogen bonds always occur when the polar or antipolar ground state is accompanied by a nearly degenerate state. This discovery also indicates two different origins of hydrogen disorder. The first case is accompanied by ferroelectric/antiferroelectric hybrid-like dielectric behaviors when the switchable dipoles reveal competition between the polar and antipolar ordering. In the second situations, which are often overlooked in disordered hydrogen-bonded crystals, the competition concerns the different antipolar arrangements. The polarization switching is simply either the normal antiferroelectric type or inactive, depending on the depth of these antipolar ground states relative to the energy level of the polar state. Consequently, density functional theory (DFT) calculations using available crystal structural data for many hydrogen-bonded crystals hold promise to predict the polarization switching modes, the performance, and the order/disorder of protons. |
Introduction
Various electronic, mechanical, thermal, and electrooptical devices exploit reversibly switchable electric polarizations in ferroelectrics/antiferroelectrics. Examples include capacitors, memories, sensors, photonic devices, actuators, and energy harvesting devices.1 Ferroelectrics show net (spontaneous) polarization at a zero-bias, whereas antiferroelectrics do not. Whether a net polarization is present depends on the arrangement of the switchable local dipoles. Lead-based perovskite oxides are often used in practical applications, despite their toxicity. However, organic materials should not only realize a lead-free alternative but also support innovative applications such as mechanically soft, wearable, portable, and/or biomedical devices.2,3 Extensive studies have identified about 100 organic solids suitable for ferroelectrics.4Theoretical calculations indicate that the loosely bound π-electrons strongly emphasize polarizations for record-high performances among organic molecular materials.5 Indeed, many prototropic ferroelectrics and acid–base supramolecular ferroelectrics have comparable or larger polarizations than ferroelectric polymers with much lower switching fields.6 Note that prototropy refers to the tautomerism of molecules by relocating hydrogen atoms. The large polarizability of π-electron systems can strengthen the nonlinear optical effects, which should maximize many useful functionalities such as the wavelength conversion, terahertz light emission, optical modulation, etc.7,8 Prototropy also delivers novel antiferroelectrics with very large polarizations, as demonstrated by squaric acid (SQA), which exhibits an excellent energy-storage performance among bulk antiferroelectric materials.9
Here, we modify the π-electron systems of imidazole to realize a new series of prototropic dielectrics. Imidazole is suitable building blocks for single-component electronic ferroelectrics and antiferroelectrics10 because they can build-up polar chains by extending the NH⋯N hydrogen-bonded molecular sequence. Each chain can spontaneously become bistable with respect the polarity, which is switchable by the prototropy (specifically called “annular tautomerism”), which induces the simultaneous relocation of the N–H hydrogen atom and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond position of the heterocyclic system (Fig. 1). There are several examples of benzimidazoles and their bridged dimers.10–12 Despite their analogous chemical structures to imidazoles, tetrazoles are not well researched as potential electronic ferroelectric/antiferroelectric candidates.
N double bond position of the heterocyclic system (Fig. 1). There are several examples of benzimidazoles and their bridged dimers.10–12 Despite their analogous chemical structures to imidazoles, tetrazoles are not well researched as potential electronic ferroelectric/antiferroelectric candidates.
 | ||
| Fig. 1 Chemical structures of azoles. (a) PHTZ (left), imidazoles (center), and ferroelectric/antiferroelectric benzimidazoles (right). (b) Polarization-reversal mechanism and a small polar domain on the hydrogen-bonded molecular chains. | ||
For conventional crystalline solids, the relationship between ferroelectricity/antiferroelectricity and the crystal symmetry is straightforward regarding the long-range-order. This relationship can be applied to materials informatics utilizing a crystal structural database to accelerate the development of functional materials. Indeed, structural assessments using the Cambridge Structural Database (CSD) is the methodology behind many of the latest successful discoveries of the hydrogen-bonded organic molecular ferroelectrics and the significantly improved performance.6
This research begins by revisiting several reported crystal structures of phenyltetrazoles (PHTZs), and examines the structural analysis and polarization hysteresis experiments on the single crystals. Several ferroelectric and antiferroelectric crystals have been developed with large polarization. Additionally, the polarization can be switched in diverse manners, even in PHTZs crystals that do not possess neither ferroelectric nor antiferroelectric long-range-order. Such disordered protons should induce relaxor ferroelectricity13 by embedding small polar domains on hydrogen-bonded molecular chains, as exemplified by the bottom scheme in Fig. 1(b). Although disordered hydrogen bonds have been discovered in numerous crystals such as those in the imidazole family,14–21 few reports have examined relaxor-like polarization switching (e.g., dabco salts).14 Here, we conduct density functional theory (DFT) calculations on the relative stabilities and the polarization amplitudes of possibly competing polar and antipolar crystal forms and demonstrate consistent explanations for various polarization switching behaviors of PHTZs crystals. Additionally, comparative studies using imidazoles provide a comprehensive understanding of the polarization switching behaviors, including inactive polarization switching despite disordered hydrogen bonds.
Results and discussion
Materials exploration from a database
The CSD entries were used to search for structural candidates of ferroelectric tetrazole crystals. There are two crystallographical requirements to realize ferroelectricity: a polar crystal symmetry and a pseudosymmetry requisite for bistability regarding the polarities. These requirements are modified in the case of antiferroelectrics as the antipolar symmetry is constructed by locally polar subunits, which are arranged with some pseudo-translational symmetry. Using the Cambridge Crystallographic Data Centre (CCDC) ConQuest program, the substructure search and basic queries were combined to find ferroelectric crystal structures composed of single-component compounds without metallic elements. Initially, we focused on polar crystal structural entries comprised of polar chains constructed by intermolecular NH⋯N hydrogen bonds. Using the program PLATON as part of the International Union of Crystallography (IUCr) checkCIF facility,22 two entries had polar but pseudo-centrosymmetric crystal structures: 5-methyl-1H-tetrazole (CSD refcode FIZZOD: Cc space group)23 and 2,2,3-trimethyl-3-(1H-tetrazol-5-yl)butanenitrile (CSD refcode EJEHEG: Pca21 space group).24 However, high-voltage experiments were avoided on these two compounds because tetrazoles are explosive or combustive. They are often used as gas generators such as blowing agents and automobile airbags.Phenyl-substitution at the 5-position can chemically stabilize tetrazole. 5-Phenyl-1H-tetrazoles (PHTZs) (Fig. 1(a)) can be handled in the typical manner near and above room temperature. It should be noted that both ferroelectric and paraelectric structural models have been demonstrated at room temperature for the unsubstituted PHTZ crystal. The first deposited dataset (CSD refcode: TOSJOA)25 shows a ferroelectric character. The pseudo-orthorhombic lattice with the monoclinic Cc space group reveals a global pseudo-mirror symmetry parallel to the (001) plane, where all the switchable NH hydrogen atoms are orientationally ordered toward the c-direction. In contrast, the second deposited dataset (TOSJOA01)26 concerns reinterpretation with the supergroup symmetry (orthorhombic Ama2), which treats this mirror as the proper symmetry by posing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 positional disorder of the NH hydrogen (i.e., paraelectric model). The polarization hysteresis property should provide insight for choosing the proper structural model.
:1 positional disorder of the NH hydrogen (i.e., paraelectric model). The polarization hysteresis property should provide insight for choosing the proper structural model.
Materials and thermal properties
A survey of the reported crystal structures for substituted PHTZ derivatives revealed similar structures with hydrogen-bonded chains (Fig. 1(b)). A characteristic observation is that the 1:1 positional disorder of the NH hydrogen atoms is observed at least seven entries: 5-(4-aminophenyl)-1H-tetrazole (abbreviation: APHTZ; CSD refcode: CITVIL),27 5-(4-chlorophenyl)-1H-tetrazole (CPHTZ; KUSLUG),28 5-(4-methylphenyl)-1H-tetrazole (MPHTZ; QUCJII),29 5-(4-methoxyphenyl)-1H-tetrazole (MOPHTZ; YOGCAZ),30 5-[3,5-bis(trifluoromethyl)phenyl]-1H-tetrazole (BTFPHTZ; FUQPUE),31 5-(3,5-dimethoxyphenyl)-1H-tetrazole (DMOPHTZ; FIRVOU), and 5-(4-ethynylphenyl)-1H-tetrazole (ENPHTZ; SIMMAF). Herein we prepared single crystals of PHTZ, APHTZ, CPHTZ, and MPHTZ, evaluated their polarization hysteresis, and reexamined their crystal structures. We also included new crystal structures and the electric properties for 5-(4-fluorophenyl)-1H-tetrazole (FPHTZ), and 5-(4-trifluoromethylphenyl)-1H-tetrazole (TFPHTZ).
Differential scanning calorimetry (DSC) can assess the high-temperature stability of the ferroelectric or antiferroelectric phase. Fig. S1 (ESI†) shows the DSC curves for the APHTZ, MPHTZ, PHTZ, CPHTZ, and FPHTZ crystals. Phase transitions are not observed over a wide temperature range up to 410 K.
Ferroelectric properties
The electric polarization versus electric field (P–E) hysteresis measurements were examined by applying a bipolar electric field of a triangular waveform on a single crystal. To maximize the measured polarizations, the direction of the applied field E was parallel or close to that of the hydrogen-bonded chain. Except for the TFPHTZ crystal, which exhibits a linear P–E curve, the observed loops indicate polarization switching in the measured field range. The polarization can be classified into three forms: ferroelectric (FE), antiferroelectric (AFE), and a hybrid.The polarization hysteresis of the APHTZ crystal reveals a single quasi-rectangular loop, which is a characteristic of the FE form (Fig. 2(a)). When an electric field E is applied parallel to the [100] direction, a remanent polarization of 7.0 μC cm−2 and a coercive field Ec of 29 kV cm−1 are observed at f = 1 Hz at room temperature. The polarization is comparable to those of ferroelectric polymers (such as polyvinylidene fluoride (PVDF))2 and benzimidazoles,10 whereas the switching field is less than 1/10 those of ferroelectric polymers.2 As mentioned above, the calorimetric experiments on the APHTZ suggested that the ferroelectric phase is stable over a wide temperature range of 170–410 K. This thermal stability is at least as excellent as that of ferroelectric polymers and benzimidazoles.2,10 Actually, the crystal heated in the oil bath retains the ferroelectric hysteresis loops but the coercive field decreases slightly at least up to 365 K (Fig. S2, ESI†).
 | ||
| Fig. 2 Polarization hystereses of the ferroelectric and antiferroelectric PHTZs at room temperature. (upper) P–E loops at various frequencies and (lower) the corresponding J–E curve at selected frequencies. (a) APHTZ crystal with the E‖[100] configuration. (b) PHTZ crystal with the E‖[100] configuration. (c) MPHTZ crystal with the E‖[100] configuration. (d) Energy-storage performance of PHTZ crystals compared with the reported organic molecular AFE crystals. For the abbreviations of AFE compounds, see Fig. S3 (ESI†). | ||
Antiferroelectric properties
The PHTZ and MPHTZ crystals exhibit double hysteresis loops and behave as typical antiferroelectrics. The steep polarization jumps in the P–E curve are identified at the sharp peaks in the corresponding current density (J)–E curves (Fig. 2(b) and (c)). At room temperature, the polarization jump ΔP is 6.9 μC cm−2 for PHTZ with an E‖[100] configuration and slightly larger (8.2 μC cm−2) for MPHTZ with an E‖[100] configuration. Note that these polarizations are close to the spontaneous polarization of APHTZ and are comparable to ΔP of the antiferroelectric benzimidazoles (7–8 μC cm−2).10A critical functionality of antiferroelectrics is their energy storage property because antiferroelectrics more effectively store energy at a higher density than linear dielectrics and ferroelectrics.32,33 A high performance has been achieved with inorganic antiferroelectrics, which have been commercially applied to dc link condensers. Eqn (1)–(3) show the P–E curves for the evaluations. The stored energy density Ws during the forward (antiferroelectric-to-ferroelectric phase) switching is given as
 | (1) |
 | (2) |
| η = Wr/Ws | (3) |
 of various organic antiferroelectric crystals. The MPHTZ crystal shows the second best
of various organic antiferroelectric crystals. The MPHTZ crystal shows the second best  and a moderately high η.
and a moderately high η.
Deformed hysteresis loops
Unlike the above cases, the CPHTZ and FPHTZ crystals display diffusive P–E hysteresis curves. Their maximum polarizations of about 5–6 μC cm−2 are slightly smaller than the spontaneous polarization of ferroelectric APHTZ. Although finite remanent polarization is a characteristic of ferroelectrics, its significant reduction from the field-induced maximum manifests as the poor retention of polarization. For the FPHTZ crystal with an E‖[100] configuration, the two-step switching processes, which are reminiscent of antiferroelectricity, are evident from the emergence of doublet peaks in each displacement current (i.e., the J–E curve) during the forward and backward bias sweeps (Fig. 3(a)). Because the doublet peaks partly overlap, the backward phase switching to the antiferroelectric state is incomplete in a zero field in these P–E and J–E curves. For the CPHTZ crystal, the doublet structure of the displacement current is further smeared into a very broad peak lying across the zero field (Fig. 3(b)). These results suggest that both crystals are likely a phase mixture of ferroelectric and antiferroelectric forms under a zero external bias. For convenience, this picture is called the “FE/AFE (F/A) hybrid” state hereafter. The F/A state appears to be very sensitive to the crystal specimens and the history of electric treatments accompanied by the significant changes of the hysteresis curves. | ||
| Fig. 3 Polarization hystereses with F/A hybrid behaviors in PHTZs at room temperature. (upper) P–E loops and (lower) the corresponding J–E curve. (a) FPHTZ crystal with the E‖[100] configuration. (b) CPHTZ crystal with the E⊥(100) configuration. | ||
Crystal structures
The crystal structures have been determined for six compounds. Table S1 (ESI†) summarizes their crystal data and refinement details. This study carefully redetermined the crystal structures for PHTZ, APHTZ, CPHTZ, and MPHTZ because the disordered NH hydrogen atoms in the reported datasets are incompatible with the FE or AFE long-range ordering. The bistable NH⋯N hydrogen bonds in all these crystals exhibit a moderately long N⋯N distance (2.8–2.9 Å), which is sufficient to prefer static disorder instead of dynamic disorder, as demonstrated in the 2-phenyl-1H-imidazole (PHIM) crystal.17The FE form of the APHTZ crystal adopts a monoclinic crystal lattice (Fig. 4(a)). The automatic process for the structural solution suggests that the crystal is in the nonpolar P21/n space group, and this analysis can reproduce the reported crystal structure (CSD refcode: CITVIL). In contrast, the polar Pn subgroup was manually set as the more proper symmetry upon considering the appearance of (0k0) reflections (k = odd) and ferroelectricity. This assignment is validated by the better structural refinement results (with the lower R-factors) in this study. The two crystallographically independent molecules are related by the pseudo-inversion center, except their NH hydrogen atoms. Since the aligned NH hydrogen atoms cause the dipole moment of the hydrogen-bonded coplanar molecular chains along the crystal [110] or [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction (Fig. 5(a), thick arrows), their net polarization gives rise to the uniaxial polarity parallel to the [100] direction (Fig. 5(a), open arrows). This observation is consistent with the ferroelectricity observed with an E‖[100] configuration.
0] direction (Fig. 5(a), thick arrows), their net polarization gives rise to the uniaxial polarity parallel to the [100] direction (Fig. 5(a), open arrows). This observation is consistent with the ferroelectricity observed with an E‖[100] configuration.
 | ||
| Fig. 4 Crystal structures and arrangements of switchable dipole moments (open arrows) of (a) APHTZ, (b) FPHTZ, (c) CPHTZ, (d) MPHTZ, and (e) PHTZ crystals. Orange balls represent the disordered NH hydrogen atoms. | ||
 | ||
| Fig. 5 Schematic arrangements of the switchable dipole moments (filled arrows) in H-ordered hypothetical (a) FE-I, (b) FE-II, and (c) AFE forms of the APHTZ or FPHTZ crystal. Open arrows represent the directions of the total polarization P. | ||
The crystal structures of MPHTZ and PHTZ belong to orthorhombic systems. The bistable NH⋯N hydrogen bonds construct mutually twisted molecular chains parallel to the [100] direction. Despite very similar antiferroelectric switching behaviors, these compounds exhibit critical structural differences. Our redetermination of the crystal structure of MPHTZ reveals additional weak Bragg spots, which imply a doubled periodicity of (a, b, c) = (c′, 2a′, b′) from the reported crystal structure (CSD refcode: QUCJII). The revised crystal structure adopts the Pbca space group (Fig. 4(d) and Fig. S4a, ESI†). Unit-cell doubling is caused by the antipolar ordering of asymmetrically located hydrogen atoms. The observed antiferroelectricity can be explained by the two sublattices with switchable polarizations P1 and P2 (= −P1) parallel to the a-axis, as discussed in the next section.
Ferroelectric and paraelectric structural models have been reported for the unsubstituted PHTZ crystal. Our reexamined structural analysis of the PHTZ crystal reproduces the paraelectric model (orthorhombic Ama2 space group) with a sufficient reliability rather than the ferroelectric one (monoclinic Cc space group) (Fig. 4(e) and Fig. S4b, ESI†). It should be noted that the c-direction polarity is fixed due to the permanent dipole along the longitudinal molecular axis whereas the switchable dipole components along the hydrogen-bonded chains is parallel to the a-axis. Therefore, the disorder or polar or antipolar order of dipole moments in such the special case will be argued hereafter only regarding the latter switchable direction component. The 1:1 positional disorder of the NH hydrogen atoms seems to conflict with the expectation that the three-dimensional antipolar arrangements of switchable dipoles are responsible for antiferroelectricity.
Both the CPHTZ and FPHTZ crystals in the F/A state can accommodate the disordered NH⋯N bonds. As detailed below, the X-ray diffraction did not indicate the loss of the corresponding inversion or translational symmetries, indicating no long-range ferroelectric or antiferroelectric ordering. The reexamined structural analysis on CPHTZ reproduced the reported crystal structure with the monoclinic P2/c space group. The bistable NH⋯N hydrogen bonds construct coplanar molecular chains parallel to the [101] direction (Fig. 4(c)). The asymmetric unit corresponds to two half units of the CPHTZ molecule, where each one is located on a twofold rotation axis. The overall crystal structure of FPHTZ (Fig. 4(b)) is nearly isomorphous to that of APHTZ (Fig. 4(a)). The critical difference is the presence or absence of the inversion symmetry on the NH⋯N bonds. These observations imply that the F/A hybrid behaviors of electric polarization may be related to the spatial mixture of short-range FE and AFE-ordered microregions.
The TFPHTZ crystal belongs to the monoclinic P21/c space group. Polar molecular chains are constructed by the ordered NH⋯N bonds, whereas the CF3 group is orientationally disordered (Fig. S5, ESI†). Despite the antipolar arrangement of the molecular chains, pseudo-translational symmetry is not apparent, which is consistent with the inactive polarization switching.
Theoretical polarizations
Since the Berry phase formalism of the electric polarization was theoretically established for a periodic lattice system,34,35 density functional theory (DFT) calculations can evaluate the spontaneous polarizations of many ferroelectrics and the sublattice polarizations of antiferroelectrics. Recently, organic systems have been evaluated theoretically due to the excellent accuracy of reproducing the experimentally optimized polarizations.5,36 Herein spontaneous polarizations are simulated for the ferroelectric form of APHTZ and the field-induced ferroelectric forms of the other PHTZs.The electric polarizations of APHTZ were evaluated by tracing the change in the Berry phase of the electronic structures during the transformation from the reference hypothetical paraelectric structure (λ = 0) to the target ferroelectric one (λ = 1) (Fig. S6a, ESI†). The λ = 1 structure was constructed from the X-ray diffraction data, and the core locations of all atoms were computationally relaxed to minimize the total energy. The optimized N–H bond lengths are reasonable (1.05 Å). Then the λ = 0 structure was constructed by averaging the atomic coordinates between λ = 1 and its inverted (λ = –1) structure. The theoretical polarization at λ = 1 is (Pa, Pb, Pc*) = (7.53, 0, −0.01) μC cm−2. It should be noted that the positional optimization of only the hydrogen atoms gave almost the same results: (7.46, 0, −0.19) μC cm−2. These values agree well with the observed one (7.0 μC cm−2). Both the experimental and theoretical polarizations are more than twice that (3.2 μC cm−2) obtained from the point-charge model, which is expressed as

The field-induced polarizations were estimated by the sublattice polarizations for antiferroelectric MPHTZ crystals. The antipolar structure was built by two kinds of sublattices, which interpenetrate each other. One was extracted as a periodic polar crystal lattice, and its sublattice polarization P1 was computed. The unit cell contains four sublattices: two with polarization P1 and two with antiparallel polarization P2 = −P1. Flipping either P1 or P2 gives rise to the theoretical polarization jump as ΔPcal = 2(P1 − P2) = 4P1. The theoretically obtained ΔPcal (7.64 μC cm−2 along the [100] direction) agrees with the experimental one ΔP (8.2 μC cm−2) within the experimental error (∼10%) for MPHTZ crystals. These values are also close to the spontaneous polarization (7.61 μC cm−2, Fig. S6b, ESI†) of the ferroelectric form, which was modeled by rearranging the NH hydrogen atoms from antipolar to polar order prior to the positional optimization of all atoms.
Phase stability
Static disorder is suggested on the NH⋯N bonds in three compounds with a long N⋯N separation (2.81–2.91 Å): the FPHTZ and CPHTZ crystals exhibiting a F/A hybrid behavior and the antiferroelectric PHTZ crystal. In the former two crystals, the competition between the emergence of polar and antipolar states may prevent their long-range-order. The AgNbO3 is an example in which the corresponding phase coexistence is demonstrated by both the observed hysteresis curvatures and the small energy difference between the ferroelectric and antiferroelectric phases (0.1–0.5 meV f.u.−1).37 For each PHTZ crystal, the polar (hypothetical ferroelectric) and antipolar (hypothetic antiferroelectric) structures are modeled by locating the ordered NH hydrogen, and then their energy difference was examined after the positional optimization of all atoms under the lattice parameters fixed at room-temperature values.The crystal symmetry with the Pn space group describes the hypothetical ferroelectric state of FPHTZ, as observed in the ferroelectric analogue, APHTZ crystal. This structure (called FE-I hereafter, Fig. 5(a)) has uniaxial polarity parallel to the a-axis with spontaneous polarization of 7.88 μC cm−2 (Fig. S6c, ESI†). The spontaneous polarization is slightly larger than the observed field-induced maximum (5.2 μC cm−2) (Fig. 3(a)). On the other hand, the a-direction components of the dipoles can be arranged in an antipolar manner by flipping the polarity of either dipolar hydrogen-bonded molecular chains along the crystal [110] direction or those along the [10] direction in FPHTZ and APHTZ crystals. This hypothetical structure corresponds to the P21 space group, and it produces a smaller uniaxial polarization (5.88 μC cm−2) along the b-direction (and is distinguished as an FE-II form, see Fig. 5(b)). This observation is reminiscent of the two different ferroelectric phases electrically interconverted in a 2-phenylmalondialdehyde (PhMDA) crystal possessing canted dipoles.9Fig. 5(c) shows the purely antipolar arrangement by allowing the fourfold lattice volume, which is denoted hereafter as the AFE form. Table S2 (ESI†) summarizes the phase stability relative to the FE-I form. FE-I is the stable form for APHTZ (Fig. 6, filled circles), which is consistent with the observations. In contrast, the F/A hybrid behavior observed along the crystal a-direction in the FPHTZ crystal can be reasonably explained by the observations that the FE-I, FE-II, and AFE forms (Fig. 6, open circles) have nearly degenerate energies. The separation between the first and second lowest energy levels in FPHTZ (0.12 meV per molecule) is much smaller than that in APHTZ (0.53 meV per molecule). Outwardly, these energy difference between the antipolar and polar phase appears to be insignificant per molecule at the thermal energy (∼26 meV) at room temperature (∼300 K). However, actual phase transformation requires additional cost for the nucleation and expansion of domains accompanied by a pair of domain walls with opposite bound charge on chains. In the Section S5 of the ESI,† this additional cost is estimated based on the ferroelectric switching of APHTZ crystal and then we evaluate the requirement for the F/A hybrid behavior regarding the energy ΔE of antipolar state relative to the ferroelectric form. The small ΔE of FPHTZ satisfies the required range (−0.16 to +0.04 meV per molecule), whereas ΔE of APHTZ is far above this ΔE range even considering the computational resolution (<0.3 meV per molecule). These simulated results are consistent with the experimentally observed switching modes.
 | ||
| Fig. 6 Ferroelectric (FE), antiferroelectric (AFE), and phase-coexisting (F/A hybrid and A/A hybrid) states in the PHTZs crystals. (a) Schematic illustrations of various states in a zero field. (b) Energy histogram of the candidate H-ordered states relative to each hypothetical FE (or FE-I) form. Filled and open circles point to the non-degenerate and nearly degenerate ground states, respectively. | ||
For the CPHTZ crystal, the hypothetical ferroelectric (or antiferroelectric) structure with spontaneous (or sublattice) polarization along the [101] direction can be constructed by reducing the crystal symmetry from the P2/c space group to the Pc (or P) subgroup symmetry (Fig. S7, ESI†). The observed field-induced maximum (∼6 μC cm−2) (Fig. 3(b)) is slightly smaller than the a*-direction component (8.21 μC cm−2) of the calculated spontaneous polarization of the FE form (Fig. S6d, ESI†). Its F/A hybrid behavior is again consistent with the very small energy separation between the polar (FE) and antipolar (AFE-I, II, and III) crystal forms (within 0.24 meV per molecule).
Extending the above argument can provide a rational explanation for the antiferroelectric switching behaviors observed in the hydrogen-ordered MPHTZ and disordered PHTZ crystals. The candidate ferroelectric and antiferroelectric structural forms examined herein (see Fig. 7) belong to the maximum subgroup of the prototypical space group (orthorhombic Pbcm for MPHTZ and Ama2 for PHTZ) that describes the paraelectric (PE)-like structure with disordered hydrogen bonds. For MPHTZ, the observed crystal form AFE-I is the most stable among the one hypothetical ferroelectric and three hypothetical antiferroelectric states (Fig. 6). The AFE-I and AFE-II forms display dipole-alternation along the c-axis, which are nearly degenerate (energy difference of only 0.05 meV per molecule) but more stable compared with the AFE-III and FE ones. Therefore, the coexistence of these forms on the mesoscopic scale (hereafter called the “A/A hybrid state”) should suppress the long-range-order of NH hydrogen atoms, whereas the observed steep polarization jumps ΔP (6.9 μC cm−2 along the [100] direction) can be attributed to the phase changes from the A/A hybrid state with zero-polarization to the FE state (calculated ΔP = 9.19 μC cm−2 (Fig. S6e, ESI†)) and vice versa. For the MPHTZ and PHTZ crystals, the AFE-I state is stabilized from the FE one by 1.78 and 0.96 meV per molecule, corresponding to energy densities of 1.45 and 0.90 J cm−3, respectively, considering their respective molecular volumes (196 and 170 Å3). These simulated results roughly agree with their respective energy densities Ws's (1.044 and 0.655 J cm−3) obtained from the polarization hysteresis experiments, although the simulations do not consider the effects of thermal fluctuation.
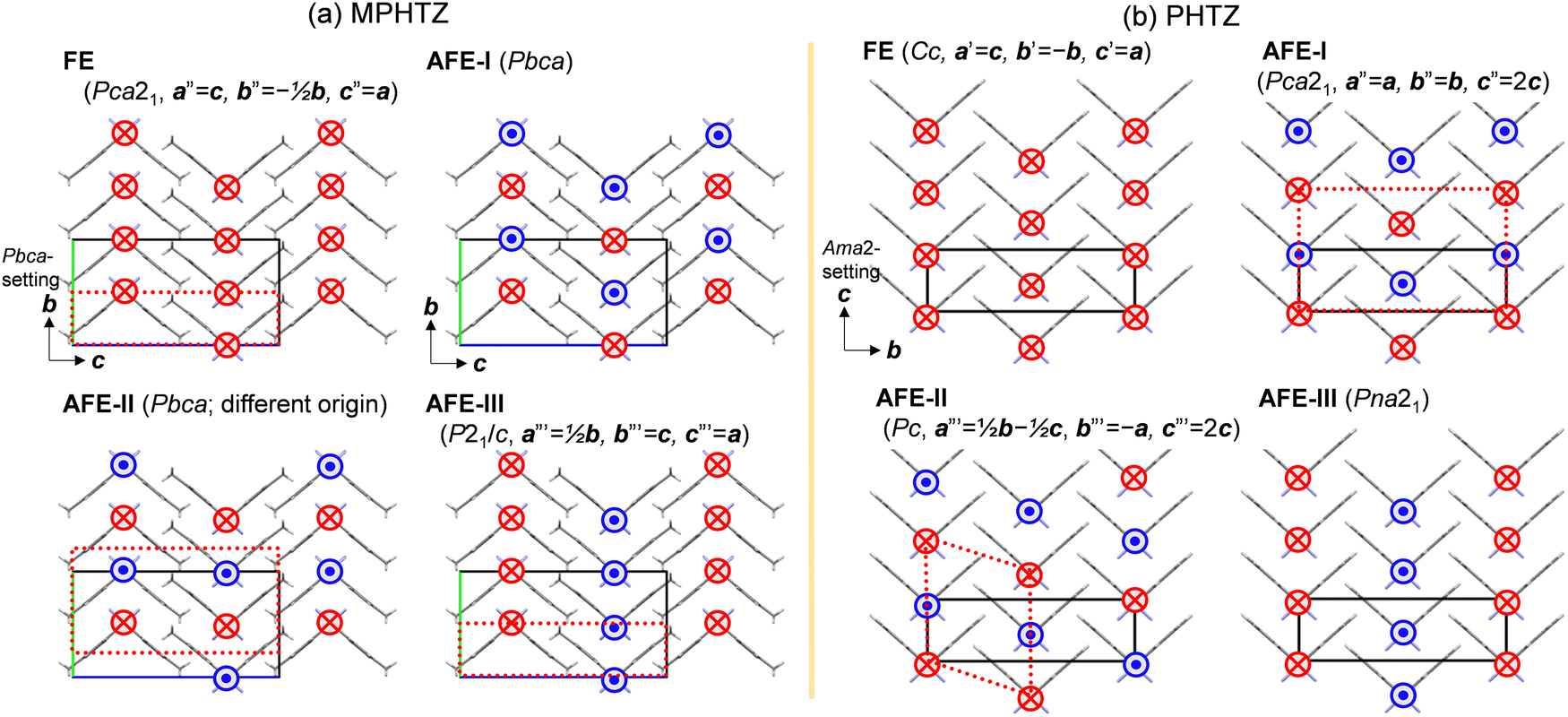 | ||
| Fig. 7 Schematic arrangements of switchable dipole moments (arrows) in candidate ferroelectric and antiferroelectric structural forms of (a) MPHTZ and (b) PHTZ crystals. Open rectangles define the a-axis projection of (a, b, c) axes of the fundamental orthorhombic (Pbca or Ama2) structure. New space group and required transformation (if necessary) to the new unit cell (red dotted squares) are given at the top of each panel. | ||
Imidazole family without polarization switching
From the structural resemblance, imidazoles should be compared with the tetrazoles because the hydrogen bonds are frequently reported to be disordered. For instance, 2-phenyl-1H-imidazole (PHIM) and PHTZ have isomorphous molecular and crystal structures (Ama2 space group) (see Fig. 4(e) and Fig. S8a, ESI†), but there are differences in the molecular tilting angles and lattice parameters. Ferro-/antiferro-electricity was not reported for PHIM.17 A series of 2,4,5-tri-substituted-1H-imidazoles such as 2,4,5-tribromo-1H-imidazole (TBIM; Fig. S8b, ESI†) shows analogous molecular packing with the same orthorhombic Ama2 symmetry,19 whereas the 2-bromo-4,5-dimethyl-1H-imidazole (DM-BIM) crystal adopts the same orthorhombic Pbcm symmetry (Fig. S8c, ESI†)18 as the prototypical paraelectric form of the MPHTZ crystal. In this study, neither ferroelectric nor antiferroelectric switching is observed even when a strong electric field up to 150–200 kV cm−1 is applied along the hydrogen-bonded chains for the PHIM, TBIM, and DM-BIM crystals.Next, we theoretically evaluated the relative stabilities of these imidazoles. The reported structural datasets at room temperature were used for PHIM and TBIM (CSD refcode: OBUQUZ and BOWROW, respectively),16,19 while the atomic coordinates of DM-BIM were experimentally obtained in this study at room temperature instead of employing the reported ones at 150 K (CSD refcode: KIPMON).18 Fig. S9 and S10 (ESI†) depict the hypothetical ferroelectric or antiferroelectric crystal forms. In each imidazole crystal, the two antipolar AFE-I and AFE-II forms compete (with small energy difference of 0–0.1 meV per molecule) as the most stable states (Fig. 6). This finding is consistent with the observed disordered hydrogen bonds. Although the spontaneous polarizations in the ferroelectric form, which ranges from 9.4–11.3 μC cm−2 (Fig. S11, ESI†) are slightly larger than those of PHTZs, the separation between these AFE states and FE state exceeds 3.4 meV per molecule and is much larger than those of PHTZ and MPHTZ (0.96 and 1.78 meV per molecule, respectively) as well as SQA (1.25 meV per molecule).35 The lack of polarization switching despite proton disorder is explained by the deep stabilization of the nearly degenerate antipolar states.
Conclusion
The tetrazole unit tends to construct hydrogen-bonded molecular sequences suitable for a variety of polarization switching modes. Five 5-phenyl-1H-tetrazoles exhibit ferroelectricity, antiferroelectricity, or a hybrid-like behavior. The ordered NH hydrogen atoms construct a polar (FE) structure for APHTZ and an antipolar (AFE) structure for MPHTZ. However, the NH hydrogen atoms remain disordered with a paraelectric-type crystal symmetry for FPHTZ, CPHTZ, and PHTZ crystals. Large switchable polarizations are experimentally and theoretically confirmed with a good consistency. The switching is mainly attributed to the switchable π-electron systems, which is consistent with the prototropic mechanism.DFT calculations for the relative stabilities of the polar and antipolar states explain both the observed variations in the polarization switching behaviors and the presence or absence of long-range-ordering. The hydrogen-bonded molecular dielectrics herein are classified into six classes according to the key features as summarized in Table 1. FPHTZ and CPHTZ exhibit nearly degenerate FE and AFE states. This competition causes the F/A hybrid behaviors and the disordered NH hydrogen locations as observed in the revised crystal structures. Another important scenario of proton disorder, which is often neglected in previous studies, causes the A/A hybrid state, when switchable dipoles in the ground states are involved in competition between the different antipolar arrangement. This mechanism well explains the difference between MPHTZ and PHTZ. The A/A hybrid state with hydrogen disorder itself has a negligible impact on the appearance of antiferroelectric polarization switching, but the depth of the antipolar ground states relative to the energy level of polar state well describe the corresponding stored energy density. Comparative studies on the imidazole families clarified that the disappearance of polarization switching in proton-disordered crystals arises from the deepened energy levels of the A/A hybrid states. In summary, the methodologies evaluated herein well predict the polarization switching modes, performance, and order/disorder of protons precisely from available crystal structural data for many hydrogen-bonded crystals. It should be noted that the dipole–dipole interactions may be critical to determine the observed phase stabilities. (See Fig. S12 for supplementary explanations analyzed based on the distribution of the interchain distance of parallel and antiparallel polarities, ESI.†)
| Type | Ground states and switchable dipoles | ΔEAFa | Protons | Switching mode | Examples | |
|---|---|---|---|---|---|---|
| Number of states & ordering mode | ||||||
| a Energy of the most stable antipolar (hypothetical antiferroelectric) state measured from that of the most stable polar (hypothetical ferroelectric) state. | ||||||
| I | 1 non-competing | Polar | >0 | Order | FE | APHTZ |
| II | 1 non-competing | Antipolar | <0 | Order | AFE | MPHTZ |
| III | 1 non-competing | Antipolar | ≪0 | Order | NA | — |
| IV | ≥2 competing | Polar & antipolar | ≈0 | Disorder | F/A hybrid | FPHTZ, CPHTZ |
| V | ≥2 competing | All antipolar | <0 | Disorder | AFE-like (A/A-hybrid) | PHTZ |
| VI | ≥2 competing | All antipolar | ≪0 | Disorder | NA | PHIM, TBIM, DM-BIM |
Methods
Preparation and electric measurements
Commercially available PHTZs and imidazoles were purified by recrystallization and vacuum sublimation. Single crystals of PHIM were grown by recrystallization from chloroform. All others were grown by slow evaporation of an alcohol solution in a stream of argon gas. Electric measurements were conducted using single crystals with painted silver electrodes. The P–E hysteresis were measured by applying a high-voltage triangular wave field and various alternating frequencies to single crystals immersed in silicone oil to prevent atmospheric discharge. The system used to evaluate ferroelectrics (Toyo, FCE-1) comprised a current/charge–voltage converter (model 6252), an arbitrary waveform generator (Biomation 2414B), an analog-to-digital converter (WaveBook 516), and a voltage amplifier (NF, HVA4321).Crystallographic studies
Table S1 (ESI†) summarizes the crystallographic data and experimental details. X-ray diffraction data were collected from single crystals at room temperature using graphite-monochromated Mo Kα radiation (0.7107 Å) and a four-circle diffractometer equipped with a two-dimensional detector [hybrid pixel detector (Rigaku AFC10 with PILATUS200K)]. Data were collected and processed using CrysAlisPro (Rigaku Oxford Diffraction, Tokyo). Crystal structure crystallographic software packages [Molecular Structure (MSC; Woodlands, TX) and Rigaku (Tokyo)] were used for the direct method and structure refinement. The final refinements of the nonhydrogen atoms were performed with anisotropic thermal factors. The hydrogen-bonded hydrogen atoms were found by differential Fourier synthesis and refined isotropically. The remaining hydrogen atoms were calculated in their ideal geometrical positions.Theoretical calculations
Polarization as a function of λ was calculated using the Berry phase approach.34,35 All the calculations were performed using the QMAS code38 based on the projector augmented-wave method39 and the plane-wave basis set. The Perdew–Burke–Ernzerhof (PBE) version of the generalized gradient approximation (GGA)40 was used to describe the electronic exchange–correlation energy. The atomic positions were optimized using the fast inertial relaxation engine (FIRE) algorithm.41 The lattice parameters were fixed at those determined by the diffraction studies at room temperature. These procedures are justified by the recent work, which reproduced the hydrogen bonding geometry as precise as the van der Waals DFT calculations.42 The convergence criterion was set to 5 × 10−5 Ha bohr−1 for the maximum force.Author contributions
S. H. and S. I. designed the study. S. H. prepared the samples and conducted the measurements. H. M. contributed to the crystal structural assessments. S. I. performed the DFT calculations. All the authors wrote and revised the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partially supported by JSPS KAKENHI (Grant Number 21H04679) and JST CREST (Grant Number JPMJCR18J2), Japan. Some of the computations in this work was carried out using the facilities at the Supercomputer Center, the Institute for Solid State Physics, The University of Tokyo and the supercomputer “Flow” at the Information Technology Center, Nagoya University.References
- M. E. Lines and A. M. Glass, Principles and Applications of Ferroelectrics and Related Materials, Oxford University Press, New York, 1977 Search PubMed.
- Ferroelectric Polymers, ed. H. S. Nalwa, CRC Press, 1995 Search PubMed.
- Woodhead Publishing Series in Electronic and Optical Materials, Organic Ferroelectric Materials and Applications, ed. K. Asadi, Woodhead Publishing, 2022 Search PubMed.
- C. Park, K. Lee, M. Koo and C. Park, Adv. Mater., 2021, 33, 2004999 CrossRef CAS PubMed.
- S. Horiuchi, K. Kobayashi, R. Kumai and S. Ishibashi, Nat. Commun., 2017, 8, 14426 CrossRef CAS PubMed.
- S. Horiuchi and S. Ishibashi, J. Phys. Soc. Jpn., 2020, 89, 051009 CrossRef.
- R. Sawada, H. Uemura, M. Sotome, H. Yada, N. Kida, K. Iwano, Y. Shimoi, S. Horiuchi and H. Okamoto, Appl. Phys. Lett., 2013, 102, 162901 CrossRef.
- M. Sotome, N. Kida, S. Horiuchi and H. Okamoto, Appl. Phys. Lett., 2014, 105, 041101 CrossRef.
- S. Horiuchi and S. Ishibashi, Chem. Sci., 2021, 12, 14198–14206 RSC.
- S. Horiuchi, F. Kagawa, K. Hatahara, K. Kobayashi, R. Kumai, Y. Murakami and Y. Tokura, Nat. Commun., 2012, 3, 1308 CrossRef PubMed.
- K. Kobayashi, S. Horiuchi, S. Ishibashi, R. Murakami and R. Kumai, J. Am. Chem. Soc., 2018, 140, 3842–3845 CrossRef CAS PubMed.
- S. Horiuchi, S. Ishibashi, R. Haruki, R. Kumai, S. Inada and S. Aoyagi, Chem. Sci., 2020, 11, 6183–6192 RSC.
- L. E. Cross, Ferroelectrics, 1987, 76, 241–267 CrossRef CAS.
- M. Szafrański and A. Katrusiak, J. Phys. Chem. B, 2008, 112, 6779–6785 CrossRef PubMed.
- M. Morimoto and M. Irie, Chem. Commun., 2011, 47, 4186–4188 RSC.
- M. M. Barforoush, S. Naderi, A. R. Ghanbarpour, A. A. Tehrani and H. R. Khavasi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o3248 CrossRef PubMed.
- M. Sikora, P. Bernatowicz, M. Szafrański and A. Katrusiak, J. Phys. Chem. C, 2014, 118, 7049–7056 CrossRef CAS.
- C. J. Serpell and P. D. Beer, Cryst. Growth Des., 2013, 13, 2866–2871 CrossRef CAS.
- M. Andrzejewski, J. Marciniak, K. W. Rajewski and A. Katrusiak, Cryst. Growth Des., 2015, 15, 1658–1665 CrossRef CAS.
- M. Owczarek, K. A. Hujsak, D. P. Ferris, A. Prokofjevs, I. Majerz, P. Szklarz, H. Zhang, A. A. Sarjeant, C. L. Stern, R. Jakubas, S. Hong, V. P. Dravid and J. F. Stoddart, Nat. Commun., 2016, 7, 13108 CrossRef CAS PubMed.
- N. Kuhn, M. Steimann and M. Walker, Z. Kristallogr. – New Cryst. Struct., 2001, 216, 328 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- Y. Ohno, Y. Akutsu, M. Arai, M. Tamura and T. Matsunaga, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1014–1016 CrossRef.
- A. S. Lyakhov, S. V. Voitekhovich, P. N. Gaponik and L. S. Ivashkevich, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o388–o389 CrossRef PubMed.
- T. M. Krygowski and M. Cyranski, Tetrahedron, 1996, 52, 10255–10264 CrossRef CAS.
- R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 252–253 CrossRef PubMed.
- Y. Zou, S. Hong, M. Park, H. Chun and M. S. Lah, Chem. Commun., 2007, 5182–5184 RSC.
- L. Xu, L.-Y. Gu, D.-Y. Zhao, B. Wang and T.-G. Kang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o742 CrossRef CAS PubMed.
- D.-Y. Hu, X.-W. Chu and Z.-R. Qu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o2463 CrossRef CAS PubMed.
- T. Jin, F. Kitahara, S. Kamijo and Y. Yamamoto, Chem. – Asian J., 2008, 3, 1575–1580 CrossRef CAS PubMed.
- H. Gerhards, A. Krest, P. J. Eulgem, D. Naumann, D. Rokitta, M. Valldor and A. Klein, Polyhedron, 2015, 100, 271–281 CrossRef CAS.
- G. Wang, Z. Lu, Y. Li, L. Li, H. Ji, A. Feteira, D. Zhou, D. Wang, S. Zhang and I. M. Reaney, Chem. Rev., 2021, 121, 6124–6172 CrossRef CAS PubMed.
- Z. Liu, T. Lu, J. Ye, G. Wang, X. Dong, R. Withers and Y. Liu, Adv. Mater. Technol., 2018, 3, 1800111 CrossRef.
- R. Resta, Rev. Mod. Phys., 1994, 66, 899–915 CrossRef CAS.
- R. D. King-Smith and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 1651–1654 CrossRef CAS PubMed.
- S. Ishibashi, S. Horiuchi and R. Kumai, Phys. Rev. B, 2018, 97, 184102 CrossRef CAS.
- H. Moriwake, A. Konishi, T. Ogawa, C. A. J. Fisher, A. Kuwabara and D. Fu, J. Appl. Phys., 2016, 119, 064102 CrossRef.
- S. Ishibashi, T. Tamura, S. Tanaka, M. Kohyama and K. Terakura, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 153310 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 170201 CrossRef PubMed.
- S. Ishibashi, S. Horiuchi and R. Kumai, Phys. Rev. Mater., 2021, 5, 094409 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details about experimental data and theoretical calculations. CCDC 2226486–2226492. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2mh01530g |
| ‡ S. H. and S. I. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |