
A universal synthesis of ultrathin Pd-based nanorings for efficient ethanol electrooxidation†
Yu
Wang‡
a,
Mengfan
Li‡
a,
Zhilong
Yang‡
a,
Wenchuan
Lai
a,
Jingjie
Ge
bc,
Minhua
Shao
 bc,
Yu
Xiang
*d,
Xuli
Chen
*a and
Hongwen
Huang
*a
bc,
Yu
Xiang
*d,
Xuli
Chen
*a and
Hongwen
Huang
*a
aCollege of Materials Science and Engineering, Hunan University, Changsha, Hunan 410082, People's Republic of China. E-mail: chenxuli@hnu.edu.cn; huanghw@hnu.edu.cn
bDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
cEnergy Institute, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
dResearch Institute of Chemical Defense, Beijing, 100191, China. E-mail: drxiangyu2016@126.com
First published on 4th February 2023
Abstract
Metallic nanorings (NRs) with open hollow structures are of particular interest in energy-related catalysis due to their unique features, which include the high utilization of active sites and facile accessibility for reactants. However, there is still a lack of general methods for synthesizing Pd-based multimetallic NRs with a high catalytic performance. Herein, we develop a template-directed strategy for the synthesis of ultrathin PdM (M = Bi, Sb, Pb, BiPb) NRs with a tunable size. Specifically, ultrathin Pd nanosheets (NSs) are used as a template to steer the deposition of M atoms and the interatomic diffusion between Pd and M, subsequently resulting in the hollow structured NRs. Taking the ethanol oxidation reaction (EOR) as a proof-of-concept application, the PdBi NRs deliver a substantially improved activity relative to the Pd NSs and commercial Pd/C catalysts, simultaneously showing outstanding stability and CO tolerance. Mechanistically, density functional theory (DFT) calculations disclose that the incorporation of Bi reduces the energy barrier of the rate-determining step in the EOR C2-path, which, together with the high ratio of exposed active sites, endows the PdBi NRs with an excellent EOR activity. We believe that our work can illuminate the general synthesis of multimetallic NRs and the rational design of advanced electrocatalysts.
New conceptsUltrathin NSs have been regarded as efficient catalysts due to their high atomic utilization associated with their unique structure. Owing to the effective exposure of atoms at both exterior and interior surfaces, NRs with a hollow structure can provide a promising approach to transcend the atomic utilization ceiling of ultrathin NSs, which can further enhance the electrocatalytic performance. However, a general strategy for synthesizing Pd-based multimetallic NRs with a high catalytic performance remains lacking because of the intrinsically isotropic growth behavior of metals. In this work, we report a universal route for the synthesis of ultrathin PdM (M = Bi, Sb, Pb, BiPb) NRs with tunable sizes that uses ultrathin Pd NSs as the template. To the best of our knowledge, the prepared PdBi, PdSb, and PdBiPb multimetallic NRs in our work are new ultrathin Pd-based NRs, which can expand the library of this unique Pd-based nanostructure. We anticipate that our universal strategy can be extended to synthesize other multimetallic NRs, which can advance the design of remarkable electrocatalysts. |
Introduction
Designing catalysts with both high activity and durability is of crucial importance to advance the development of electrochemical energy conversion devices.1–4 So far, noble-metal-based materials (e.g., Pt, Pd, Rh) are still regarded as the most effective candidates for various electrocatalytic reactions, such as the ethanol oxidation reaction (EOR),5–7 the oxygen reduction reaction (ORR),8–10 and the methanol oxidation reaction (MOR).11,12 However, the high cost that arises from the unaffordable use of noble metals seriously retards the commercial applications of devices.13,14 Accordingly, it is of great importance to explore electrocatalysts with an improved catalytic activity and stability to enhance the utilization efficiency of noble metals.One promising solution is to alloy the noble metal with a second metal, which can tune the electronic structure and thus improve the intrinsic activity.15–18 In addition, fabricating favorable nanostructures, such as 0D (e.g., polyhedral),19,20 1D (e.g., nanowires),21,22 2D (e.g., nanosheets),23,24 and 3D structures (e.g., nanoflowers),25,26 has also been found to be an effective way of increasing the number of active sites, which can promote the catalytic performance. Among those nanostructures, ultrathin NSs have received widespread academic interest due to their high utilization efficiency and electron mobility.27,28 To further enhance the catalytic performance of noble metals, many studies have been focused on ultrathin NSs to maximize the number of active sites. Decreasing the thickness of ultrathin NSs is an effective strategy to achieve this target but leads to insufficient stability during the reaction.28–32 An alternative strategy is to construct hollow structures, such as NRs. NRs, which have a hollow structure, can utilize more catalytic sites at both exterior and interior surfaces compared with those with a solid structure, and is expected to boost the catalytic activity.32–36 For instance, the PtCu NRs have been successfully produced through the galvanic replacement approach, and delivered a superior MOR performance owing to the hollow NR structure and the ligand effect.34 In another notable case, Guo and co-workers recently reported a sequential reduction strategy for generating ultrathin PdPtCu NRs, and the boosted the ORR performance, which can be ascribed to the combination of the unique structure and the alloyed Cu atoms.32 Despite some achievements in the synthesis of metallic NRs, a general method for generating the ultrathin Pd-based NRs is still lacking.
Herein, we report a template-directed method for the general synthesis of ultrathin Pd-based (Pd-M, M = Bi, Sb, Pb, BiPb) NRs with a tunable size. Selecting PdBi NRs as a typical example, a time-dependent study was conducted to reveal the formation mechanism of the NR structure. As a proof-of-concept reaction, the PdBi NRs were applied as EOR electrocatalysts, and exhibited significantly enhanced activity, stability and CO-tolerance towards the EOR. Moreover, based on both experimental results and density functional theory (DFT) calculations, the optimized electronic structure arising from the introduced Bi atoms and the greater number of active sites offered by the NR structure account for the enhanced EOR performance.
Results and discussion
Synthesis and structural characterization of PdBi NRs
The ultrathin PdBi NRs were prepared via a two-step template-directed approach using Pd NSs as the template. In the first step, ultrathin Pd NSs were produced according to the previously reported method35 (see the Experimental section in the ESI† for details). Transmission electron microscopy (TEM) images revealed the successful production of ultrathin NSs with an average lateral dimension of 31.5 nm and a thickness of 1.0 nm (Fig. S1 and S2, ESI†), where the thickness was determined through measuring the vertical standing NSs directly. Subsequently, the PdBi NRs were obtained by heating a mixture solution containing the pre-synthesized Pd NSs, bismuth neodecanoate (Bi(C10H19O2)3), polyvinylpyrrolidone (PVP), ascorbic acid (AA), ethylene glycol (EG) and N,N-dimethylacetamide (DMAC) at 140 °C for 3 h (see ESI† for details).
Fig. 1a and b show the morphology of the PdBi NRs based on TEM and high-angle annular dark-filed scanning transmission electron microscopy (HAADF-STEM), respectively, demonstrating the good uniformity of the as-synthesized NRs. According to direct counting, the average lateral dimension and thickness of the resulting products was estimated to be 23.3 and 2.1 nm, respectively (Fig. S3, ESI†). To verify the formation of PdBi NRs, the chemical composition of the NRs was analyzed. The atomic ratio of Pd/Bi was estimated to be 88.3/11.7 using inductively coupled plasma-atomic emission spectroscopy (ICP-AES), in line with the value of 90.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.85 obtained via STEM energy-dispersive X-ray spectroscopy (STEM-EDX), manifesting the successful introduction of Bi. The X-ray diffraction (XRD) pattern in Fig. 1c shows that the diffraction features are similar to those of typical Pd (JCPDS No. 46-1043), indicating the face centered cubic (fcc) crystal structure. Relative to the ultrathin Pd NSs, the diffraction peaks of the PdBi NRs present an overall shift towards a lower angle, which can be ascribed to the lattice expansion upon incorporation of the larger Bi atoms. It should be noted that, owing to the ultrathin diameter and strain effect of the ultrathin 2D nanostructures, the diffraction peaks of pure ultrathin Pd NSs are relatively broad and exhibit a slightly negative shift compared with those of typical Pd.37,38 Besides, no characteristic diffraction peaks of Bi or Bi oxides were observed, confirming the formation of the alloy structure instead of a heterostructure in the PdBi NRs.39,40
:9.85 obtained via STEM energy-dispersive X-ray spectroscopy (STEM-EDX), manifesting the successful introduction of Bi. The X-ray diffraction (XRD) pattern in Fig. 1c shows that the diffraction features are similar to those of typical Pd (JCPDS No. 46-1043), indicating the face centered cubic (fcc) crystal structure. Relative to the ultrathin Pd NSs, the diffraction peaks of the PdBi NRs present an overall shift towards a lower angle, which can be ascribed to the lattice expansion upon incorporation of the larger Bi atoms. It should be noted that, owing to the ultrathin diameter and strain effect of the ultrathin 2D nanostructures, the diffraction peaks of pure ultrathin Pd NSs are relatively broad and exhibit a slightly negative shift compared with those of typical Pd.37,38 Besides, no characteristic diffraction peaks of Bi or Bi oxides were observed, confirming the formation of the alloy structure instead of a heterostructure in the PdBi NRs.39,40
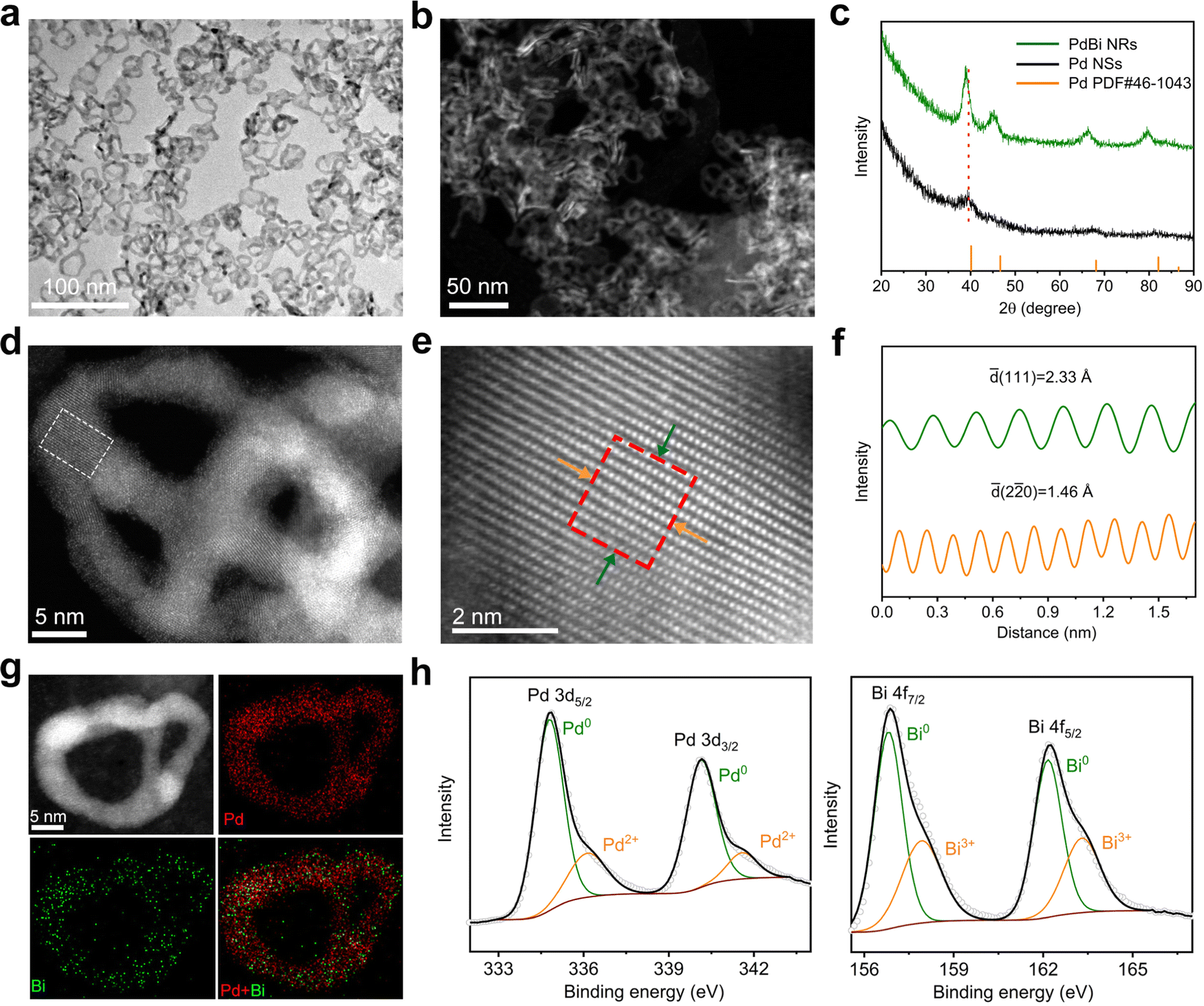 | ||
| Fig. 1 Structural and compositional characterization of the ultrathin PdBi NRs. (a) Low-magnification TEM image. (b) HAADF-STEM image. (c) XRD patterns. (d) Atomic-resolution aberration-corrected HAADF-STEM image. (e) Enlarged atomic-resolution HAADF-STEM image taken from the area marked by the white rectangular box in (d). (f) Intensity profiles taken along the atomic layers marked by the corresponding green and orange arrows in (e). (g) STEM-EDX elemental mapping images of a representative PdBi NR. High-resolution XPS spectra for (h) Pd 3d and (i) Bi 4f. | ||
Atomic-resolution aberration-corrected HAADF-STEM was performed to reveal the NR atomic-scale structural information (Fig. 1d and e). The atomic-resolution HAADF-STEM images display the well-defined crystalline nature of the PdBi NRs. As depicted in Fig. 1f, the corresponding intensity profiles of the NRs show lattice spacings of 2.33 Å and 1.46 Å, which can be indexed to the (111) and (220) planes, respectively, of fcc Pd. Note that the larger lattice spacings of the PdBi NRs in contrast to standard Pd (2.25 and 1.38 Å) prove the incorporation of Bi atoms into the Pd lattice, consistent with the observations from XRD.41Fig. 1g depicts the STEM-EDX elemental mapping profiles gathered from individual PdBi NRs, demonstrating the uniform distribution of Pd and Bi atoms throughout the NRs. To further investigate the chemical states in the NRs, high-resolution X-ray photoelectron spectroscopy (XPS) was carried out. The high-resolution XPS spectra of Pd 3d and Bi 4f clearly display that most of the Pd and Bi is in their metallic states, while a small amount of the oxidized state co-exists because of the surface oxidation of ultrathin PdBi NRs after exposure to air (Fig. 1h).42 Note that the Pd 3d peaks exhibit a negative shift compared with the Pd NSs (Fig. S4, ESI†), suggesting electron transfer from Bi to Pd in the PdBi NRs.43 Such an electron transfer process may regulate the electronic structures of Pd, which can optimize the adsorption behavior of the reaction intermediates and thus be expected to improve the catalytic activity.43,44 In addition, we also used Pd NSs with diverse lateral dimensions as templates to synthesize the PdBi NRs. As illustrated in Fig. S5 and S6 (ESI†), PdBi NRs with lateral sizes of 11.9 and 32.7 nm can be derived from 18.2 nm- and 43.0 nm-sized Pd NSs, respectively, demonstrating the suitability of the template-directed method for flexibly controlling the structure of the NRs.
Formation mechanism of the PdBi NRs
To understand the formation mechanism of the PdBi NRs, we investigated the time-dependent structure evolution from the Pd NSs to the PdBi NRs during the template-directed synthesis. During the first stage of the reaction (Fig. 2a, 1 min), the as-prepared ultrathin Pd NSs broke up into smaller NSs. When the reaction time was prolonged to 5 min, the fragile NSs became thicker, and some tiny pores were formed on the NSs (Fig. 2b). As the reaction time was extended to 15 min, the pores were gradually enlarged and merged to create the hollow feature and some NRs structures (Fig. 2c). After reaction for 180 min, the porous NSs were totally transformed into the NRs with high uniformity (Fig. 2d). Based on TEM images of the intermediates, we find that the resultant NRs inherited well the two-dimensional structure of the Pd NS seeds, demonstrating that the Pd NSs acted as templates to guide the evolution of the morphology. The compositions of the NRs obtained at different stages were also analyzed based on ICP-AES (Fig. 2e), where the atomic percentage of Bi started from zero, and gradually increased to the final composition of the PdBi NRs after 180 min. Taking the homogeneous distributions of Bi and Pd in the PdBi NRs into consideration, the results demonstrate the interatomic diffusion behavior between Bi and Pd during the reaction process. Moreover, time-dependent XRD patterns (Fig. 2f) also showed the continuous negative shift of the diffraction peaks with increasing reaction time, caused by the gradual incorporation of Bi to expand the Pd lattice.43,44 Taking these results together, we can conclude that the template-directed growth and the subsequent interatomic diffusion contribute to the formation of ultrathin NRs.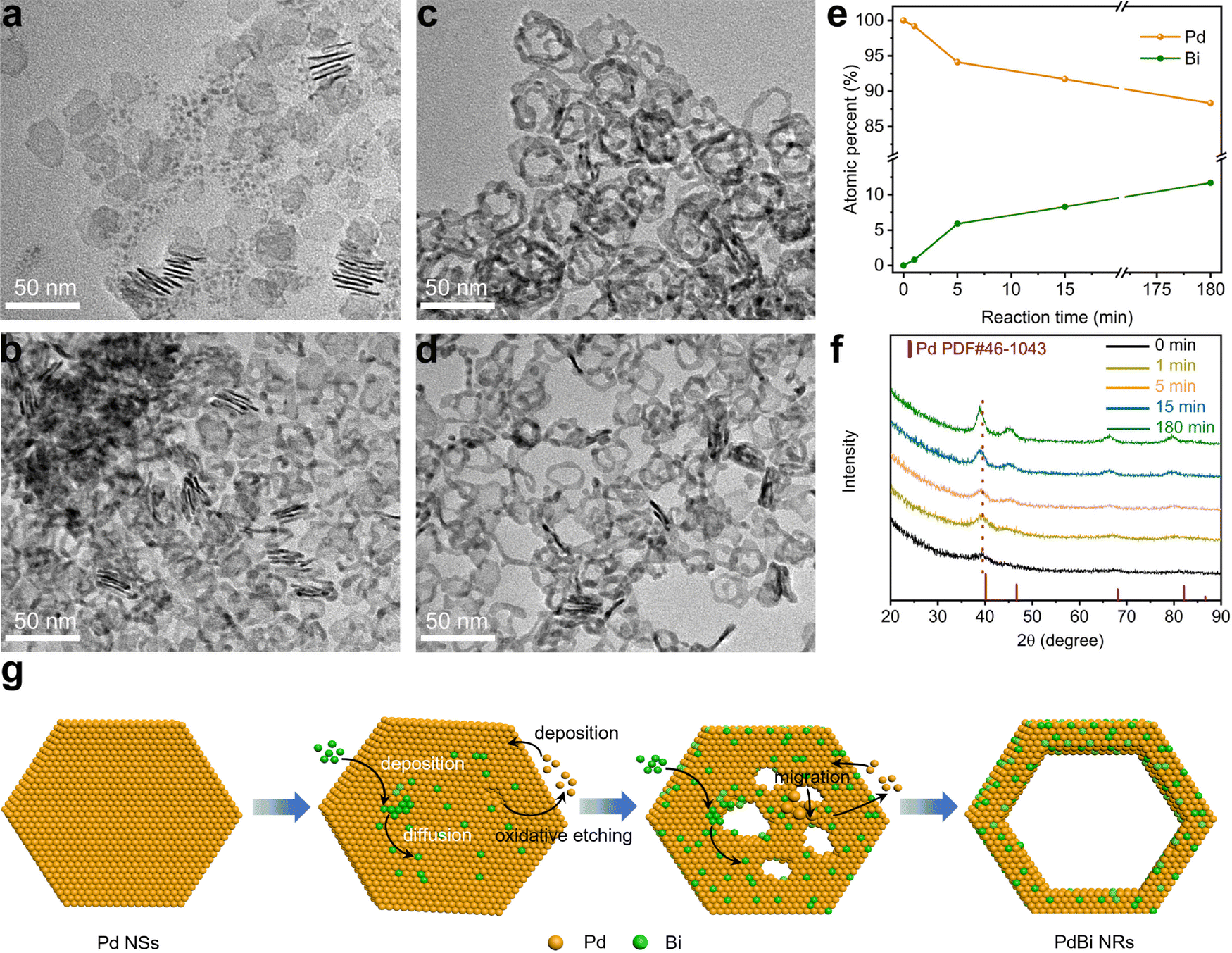 | ||
| Fig. 2 Formation mechanism for the PdBi NRs. Representative TEM images of the PdBi NRs obtained at various reaction times of (a) 1 min, (b) 5 min, (c) 15 min, and (d) 180 min at 140 °C. (e) Plot showing the time-dependent compositional evolution for formation of the PdBi NRs, determined via ICP-AES. (f) XRD patterns of the PdBi NRs collected at various reaction times. (g) Schematic illustration showing the synthesis mechanism of the PdBi NRs using the template-directed strategy. | ||
However, one question for understanding the formation mechanism still remains: how is the hollow structure constructed? We thus conducted a set of control experiments that used the standard procedure except for the absence of the Bi precursor, to investigate the effect of the incorporated Bi atoms on the morphological evolution. Interestingly, as shown in Fig. S7 (ESI†), only fragile NSs with few pores were obtained. Because the Br− ions selectively absorbed on the side facets, and protected them from oxidation, the oxidative etching initially began at the surface region.36 Therefore, the fragmentation of the NSs and the formation of surface pores can be ascribed to oxidative etching by the residual O2 during synthesis.32 By comparing the morphology in the presence and absence of Bi, we noticed that the Bi atoms played an important role in the formation of the NR structure. It was speculated that the deposited Bi atoms on the Pd NSs may lower the diffusion barrier of Pd and thus accelerate the migration of Pd atoms.45 To test this hypothesis, we conducted a control experiment using the standard procedure by increasing the amount of Bi precursor from 2 mg to 3 mg. Interestingly, the obtained PdBi possessed a nanowire-like structure (Fig. S8, ESI†). This may be attributed to the excessive migration of Pd atoms, thus resulting in severe fragmentation of the Pd NSs. On the basis of these results, we can reasonably give a picture of the PdBi NR formation mechanism as illustrated in Fig. 2g. To be specific, at the initial stage, fragmentation and small pores appeared on the ultrathin Pd NSs. Meanwhile, due to the selective absorption of Br− ions on the side facets, the Bi atoms mainly deposited and diffused on the surface region of the Pd NSs. The deposited Bi atoms accordingly lowered the diffusion barrier, and thus promoted interatomic diffusion including the migration of Pd atoms. With the migration of Pd atoms, the pores were continuously enlarged and finally the PdBi NRs were formed.
Generality of template-directed approach for synthesizing the PdM NRs
The synthetic approach was generalized to obtain other ultrathin PdM bimetallic NRs, including PdSb NRs and PdPb NRs, where we replaced the corresponding metal precursor in the standard synthesis. The morphology, composition and structure of the PdSb NRs and PdPb NRs were characterized via TEM, ICP-AES, XRD and STEM-EDX, as shown in Fig. 3 and Table S1 (ESI†). The TEM images clearly demonstrate that the synthesized PdM (M = Sb and Pb) NRs all show typical a NR structure (Fig. 3a and d). According to the ICP-AES results in Table S1 (ESI†), the atomic ratio of Pd/Sb and Pd/Pb is 93.7/6.3 and 75.9/24.1, respectively, indicating the successful incorporation of Sb and Pb atoms. As illustrated in Fig. 3b and e, the XRD diffraction peaks of the PdSb NRs and the PdPb NRs match well with the fcc structure of Pd, and show a shift compared with pure Pd NSs due to the incorporation of Sb, and Pb atoms, demonstrating the formation of the alloy structure in each case.46–48 Furthermore, the uniform distribution of Pd and Sb/Pb throughout the NRs was confirmed in the STEM-EDX elemental mapping profiles (Fig. 3c and f). Beyond bimetallic NRs, the synthetic approach can be further generalizable for trimetallic systems. The successful synthesis of PdBiPb trimetallic NRs was verified through structural and compositional characterization (Fig. 3g–i and Table S1, ESI†). Taken together, we confirm the generality of our template-guided synthetic approach to obtain Pd-based NRs. It is believed that this universal method can be extended to other Pd-based multimetallic NRs beyond our work.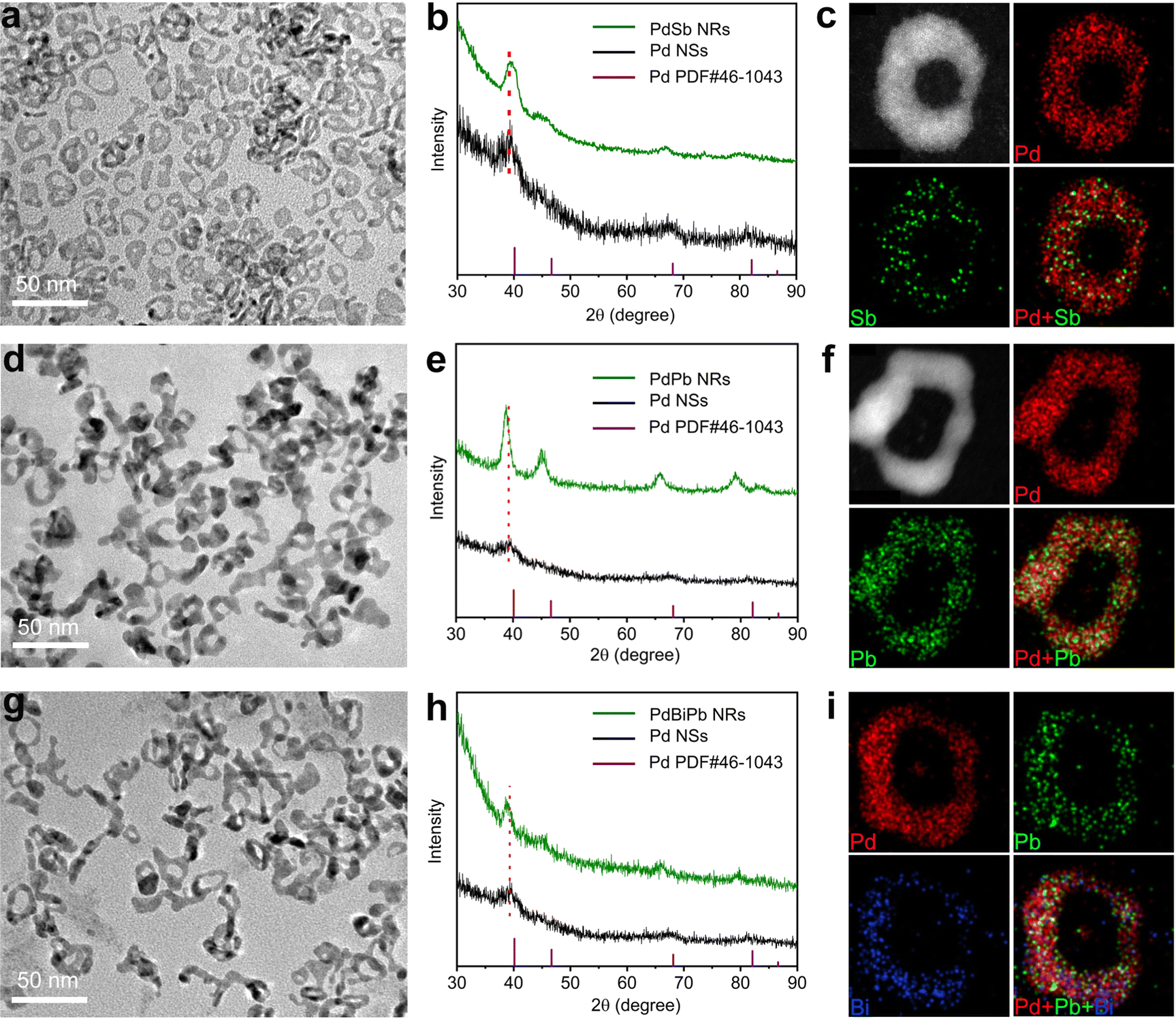 | ||
| Fig. 3 Structural and compositional characterization of the obtained PdSb NRs, PdPb NRs and PdBiPb NRs. (a–c) TEM image, XRD pattern and STEM-EDX elemental mapping images of the PdSb NRs. (d–f) TEM image, XRD pattern and STEM-EDX elemental mapping images of the PdPb NRs. (g–i) TEM image, XRD pattern and STEM-EDX elemental mapping images of the PdBiPb NRs. | ||
EOR performance
Taking the EOR as a model reaction, the electrocatalytic activity of the PdBi NRs was then inspected. The Pd NSs and commercial Pd/C were also investigated as reference catalysts. Before electrochemical testing, the as-obtained PdBi NRs and Pd NSs were loaded on Vulcan XC-72R carbon to prepare carbon-supported catalysts (20 wt%). Fig. 4a presents the cyclic voltammograms (CVs) recorded in N2-saturated 1 M KOH electrolyte at a scan rate of 50 mV s−1. The electrochemically active surfaces areas (ECSAs) of the three catalysts were determined based on integration of the PdO reduction peak. Owing to the NRs having a high ratio of exposed atoms, the PdBi NR/C possesses the largest ECSA (74.14 m2 g−1Pd), compared with Pd NS/C (35.34 m2 g−1Pd) and commercial Pd/C (40.37 m2 g−1Pd). The ECSA of the Pd NS/C is lower than expected, probably on account of the aggregation of two-dimensional ultrathin NSs. Moreover, to evaluate the EOR catalytic activity, the CVs were obtained at room temperature in N2-saturated 1 M KOH containing 1 M CH3CH2OH at a scan rate of 50 mV s−1. The Pd mass-normalized CV curves show that the PdBi NR/C catalyst exhibits the lowest onset potential compared with Pd NS/C and Pd/C, suggesting that the EOR over PdBi NR/C has a decreased activation barrier (Fig. 4b).49Fig. 4c and Fig. S9 (ESI†) display that the PdBi NR/C catalyst shows the highest mass activity (MA) of 3.21 A mg−1Pd, which is 3.41 and 4.52 times higher than that of Pd NS/C (0.94 A mg−1Pd) and the commercial Pd/C catalyst (0.71 A mg−1Pd). Moreover, the EOR mass activity of PdBi NR/C exceeds most of the reported catalysts (Table S2, ESI†). For the specific activity (SA; normalized to the corresponding ECSA), it is demonstrated by Fig. S10 (ESI†) that the PdBi NR/C catalyst also delivers an SA of 4.33 mA cm−2, 2.47 times higher than that of commercial Pd/C (1.75 mA cm−2). These results together confirm the good catalytic activity of PdBi NRs toward the EOR in an alkaline medium, which can be traced back to the NR features and Bi incorporation.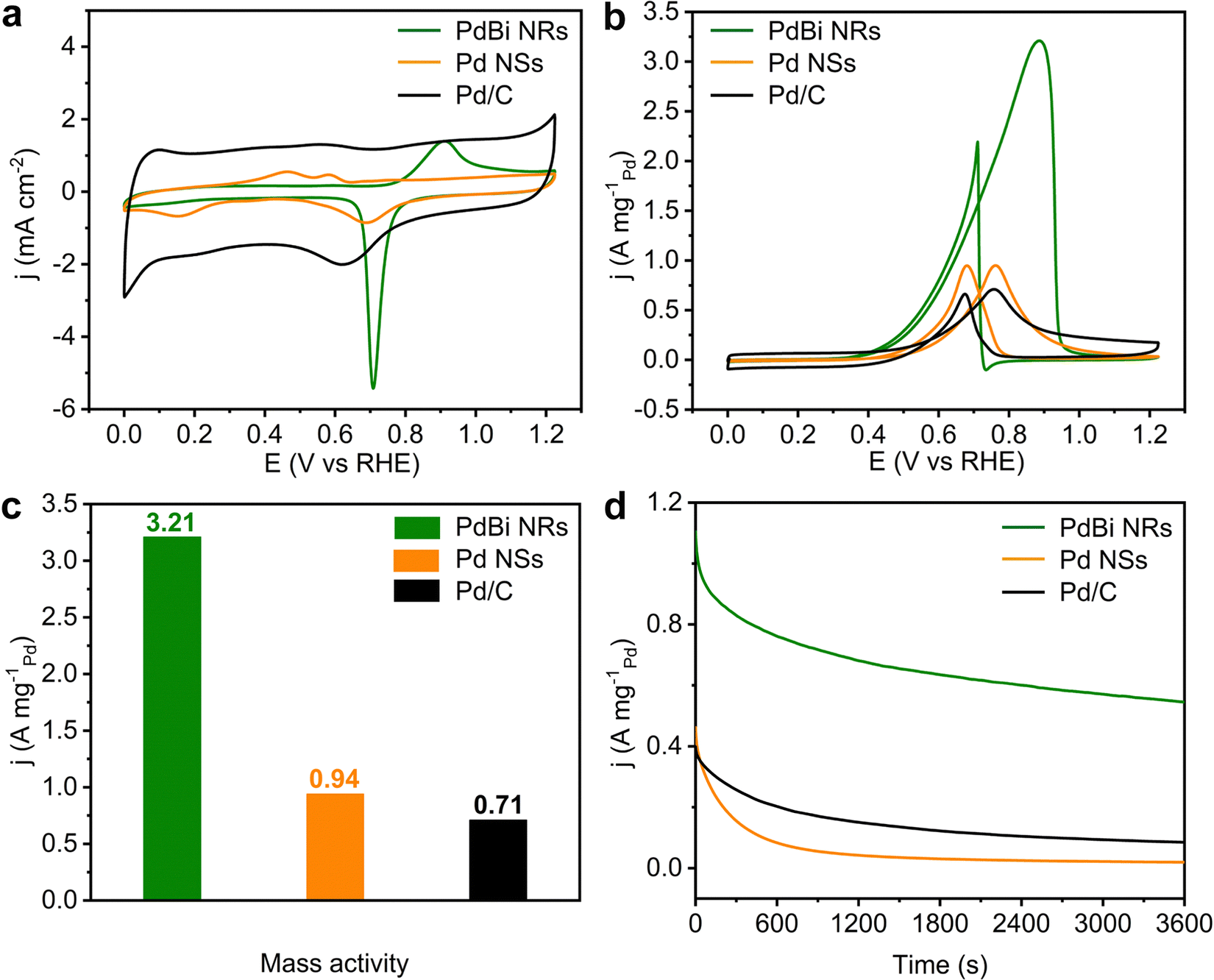 | ||
| Fig. 4 Electrocatalytic EOR performance of the PdBi NRs, Pd NSs and commercial Pd/C. (a) CV curves recorded at room temperature in N2-saturated 1 M KOH solution at a scan rate of 50 mV s−1. (b) CV curves recorded at room temperature in N2-saturated 1 M KOH solution with 1 M ethanol at a scan rate of 50 mV s−1. (c) Mass activities recorded at the corresponding peak potentials in the CV curves in (b). (d) Chronoamperometry stability testing in 1 M KOH with 1 M ethanol at a potential of 0.725 VRHE. | ||
Considering the significance of the stability term for practical applications, we then conducted chronoamperometry tests at 0.725 VRHE to assess the EOR stability of the developed catalysts. As shown in Fig. 4d, the PdBi NR/C maintains the highest activity among all the catalysts. Specifically, the current density of PdBi NR/C retains 49.4% of its initial value, while commercial Pd/C and Pd NS/C retain only 21.2% and 4.2%, respectively, after 3600 s of stability testing. Besides, the morphology and composition of PdBi NR/C show a negligible change after the aforementioned stability measurement (Fig. S11 and Table S3, ESI†), in line with the retained activity. Interestingly, the PdBi NR/C catalyst can easily be reactivated by sweeping for several cycles using a fresh KOH electrolyte. Fig. S12 (ESI†) depicts five consecutive reactivation cycles for PdBi NR/C, where this catalyst shows only a slight drop in EOR activity after five cycles. The aforementioned results reveal the enhanced EOR stability of the PdBi NRs/C catalyst.
CO-tolerance is another important parameter for an ideal catalyst towards the EOR because CO will absorb strongly on the active sites and thus will have a negative impact on the EOR performance of the catalyst.49,50 As is well known, the ratio between the forward current density (If) and the backward current density (Ib) reflects the CO anti-poisoning ability of a catalyst.6,51 From the CV curves in Fig. 4b, the If/Ib value of PdBi NR/C was calculated to be 1.46, which is higher than that for Pd NSs (1.00) and the commercial Pd/C (1.05), indicating that the PdBi NRs may have an enhanced tolerance to CO poisoning. We also conducted CO-stripping experiments to further study the CO-tolerance performance of PdBi NR/C, as shown in Fig. S13 (ESI†). In the case of commercial Pd/C, the peak ranging from 0.76 to 1.0 VRHE in the first scan can be ascribed the CO oxidative removal.52 For the PdBi NR/C, the peak ranging from 0.70 to 0.82 VRHE in the first scan can be attributed to the CO oxidative removal, and the peak around 0.9 VRHE can be recognized as a combination of CO oxidative removal and oxidation of the PdBi surface.44 It can be seen that the PdBi NR/C shows a lower onset potential compared with commercial Pd/C, indicating that CO can be easily removed from the surface of such a catalyst. The doping of the Bi atoms may weak the adsorption of CO, and thus enhance the CO-tolerance performance.40 Taken together, the PdBi NR/C possesses excellent activity, stability and CO-tolerance towards the EOR owing to the incorporation of Bi atoms and the efficient utilization of Pd atoms in the NR structure.
Mechanistic understanding
To further provide in-depth insight into the enhanced EOR activity of the PdBi NR/C catalyst, density functional theory (DFT) calculations were performed on Pd (111) and PdBi (111) slabs, which represent the corresponding catalysts.42,43 Fig. S14 (ESI†) and Fig. 5a exhibit configurations of the reaction intermediates involved in both the C1- and C2-paths of the EOR. The free energies of the intermediates on the Pd (111) and PdBi (111) slabs were calculated to gain further insight. As shown in Fig. 5b and c, the Gibbs free energy difference in the *CH3CO → *CH2CO branch (C1-path, Pd: 0.97 eV, PdBi: 1.27 eV) is much higher than that in the *CH3CO → *CH3COOH branch (C2-path, Pd: 0.17 eV, PdBi: 0.08 eV), indicating that the C2 pathway should be the dominant reaction pathway for all catalysts.51,53 Therefore, we specifically analyzed the C2 pathway reaction in the following. Based on the calculated free energy profiles, it is observed that the second dehydrogenation of ethanol (*CH3CHOH → *CH3COH) was the rate-determining step (RDS) of the C2-path of the EOR.54–56 Notably, PdBi exhibits a lower free energy demand (0.51 eV) in the RDS relative to that of pure Pd (0.88 eV), verifying that the incorporation of Bi atoms can thermodynamically promote the RDS and thus the C2 pathway in the EOR. This can be attributed to the redistribution of electron charge and the optimized electronic structure of Pd arising from the alloyed Bi.42,43 Taking all of these analyses together, the ligand effect from the incorporated Bi atoms consequently endows PdBi with an excellent intrinsic activity, which, together with the unique structure of the NRs and the high percentage of exposed atoms, ultimately results in the enhanced electrocatalytic EOR performance of the PdBi NRs.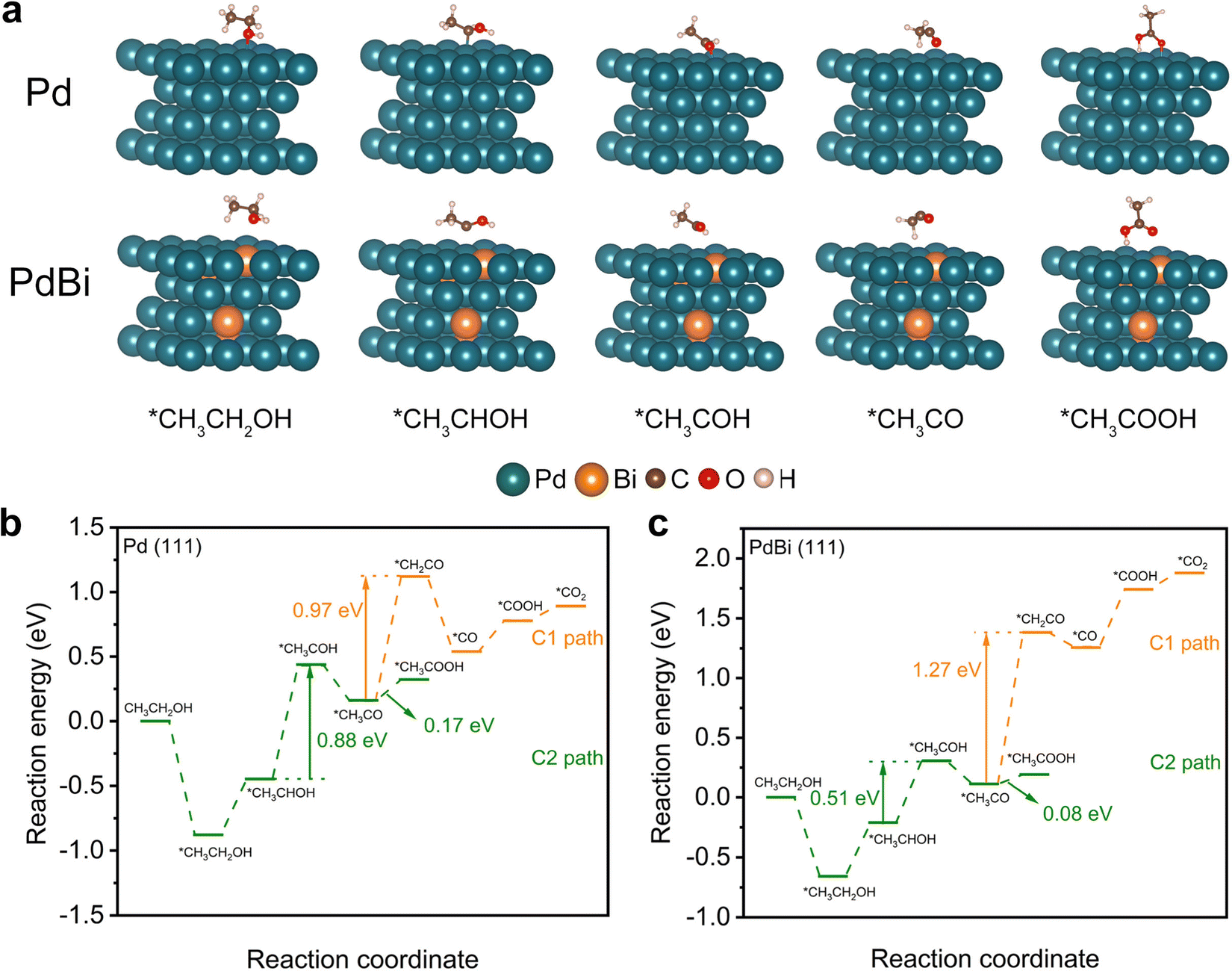 | ||
| Fig. 5 Mechanistic understanding based on DFT calculations. (a) Configurations of the adsorbed intermediates for the C2-path of the EOR on Pd (111) and PdBi (111) slabs. (b) Calculated C1- and C2-path EOR free energy profiles on Pd (111). (c) Calculated EOR free energy profiles of C1- and C2-paths on PdBi (111). | ||
Conclusions
In summary, we have developed a universal method for the synthesis of ultrathin PdM (M = Bi, Sb, Pb and BiPb) NRs with tunable sizes through a template-directed strategy. The formation of the NR structure relies on the deposition of foreign atoms on the Pd NS templates and subsequent interatomic diffusion to create hollow structures and the final NRs. As a proof-of-concept application, the PdBi NR/C catalyst exhibits a substantially enhanced activity compared with Pd NS/C and the commercial Pd/C catalyst for the electrocatalytic EOR in an alkaline medium. Meanwhile, the PdBi NR/C also shows good stability and CO-tolerance, indicating its good suitability for practical applications. Mechanistically, it is disclosed that the incorporation of Bi can reduce the energy demand of the RDS in the EOR C2-path, which, together with the abundant active sites exposed by unique NR structure, endows the PdBi NRs with an excellent catalytic EOR performance. We believe this work provides a general and effective strategy for the synthesis of Pd-based multimetallic NRs, and can further illuminate the design of advanced electrocatalysts for energy-related applications.Author contributions
H. H. guided and supervised the project. Y. W. and M. L. prepared the samples, carried out experiments and analyzed the experimental data. Z. Y. undertook the DFT simulations and theoretical analyses. Y. X., W. L., J. G. and M. S. assisted the experimental analysis. Y. W. and X. C. wrote the paper. All authors contributed to the discussion and analysis of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2021YFA1502000), NSFC (No. U2032149 and 22102052), the Science and Technology Innovation Program of Hunan Province (No. 2021RC3065 and 2021RC2053), and Shenzhen Science and Technology Program (No. JCYJ20210324120800002).References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- X. Zhao, H. Zhao, J. Sun, G. Li and R. Liu, Chin. Chem. Lett., 2020, 31, 1782–1786 CrossRef CAS.
- H. Fan and V. Maheshwari, Adv. Funct. Mater., 2021, 31, 2106149 CrossRef CAS.
- J. Zhang, Y. Yuan, L. Gao, G. Zeng, M. Li and H. Huang, Adv. Mater., 2021, 33, 2006494 CrossRef CAS PubMed.
- S. H. Han, H. M. Liu, P. Chen, J. X. Jiang and Y. Chen, Adv. Energy Mater., 2018, 8, 1801326 CrossRef.
- L. Pu, H. Fan and V. Maheshwari, Catal. Sci. Technol., 2020, 10, 2020–2028 RSC.
- B.-W. Zhang, W.-H. Lai, T. Sheng, X.-M. Qu, Y.-X. Wang, L. Ren, L. Zhang, Y. Du, Y.-X. Jiang and S.-G. Sun, J. Mater. Chem. A, 2019, 7, 5214–5220 RSC.
- G. Niu, M. Zhou, X. Yang, J. Park, N. Lu, J. Wang, M. J. Kim, L. Wang and Y. Xia, Nano Lett., 2016, 16, 3850–3857 CrossRef CAS PubMed.
- Y. Zhang, B. Huang, G. Luo, T. Sun, Y. Feng, Y. Wang, Y. Ma, Q. Shao, Y. Li and Z. Zhou, Sci. Adv., 2020, 6, eaba9731 CrossRef CAS PubMed.
- L. Gao, T. Sun, X. Tan, M. Liu, F. Xue, B. Wang, J. Zhang, Y.-F. Lu, C. Ma and H. Tian, Appl. Catal., B, 2022, 303, 120918 CrossRef CAS.
- Y.-C. Jiang, H.-Y. Sun, Y.-N. Li, J.-W. He, Q. Xue, X. Tian, F.-M. Li, S.-B. Yin, D.-S. Li and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 35767–35776 CrossRef CAS PubMed.
- A. Ali and P. K. Shen, J. Mater. Chem. A, 2019, 7, 22189–22217 RSC.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, 1802234 CrossRef PubMed.
- H. Huang, K. Li, Z. Chen, L. Luo, Y. Gu, D. Zhang, C. Ma, R. Si, J. Yang and Z. Peng, J. Am. Chem. Soc., 2017, 139, 8152–8159 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- Y. Wu, Y. Zhao, J. Liu and F. Wang, J. Mater. Chem. A, 2018, 6, 10700–10709 RSC.
- Y. Sun, H. Xiang, H. Li, G. Yu, H. Chen and S. Liu, Chin. Chem. Lett., 2020, 31, 2491–2494 CrossRef CAS.
- X. Li, X. Li, C. Liu, H. Huang, P. Gao, F. Ahmad, L. Luo, Y. Ye, Z. Geng and G. Wang, Nano Lett., 2020, 20, 1403–1409 CrossRef CAS PubMed.
- X. Wang, M. Vara, M. Luo, H. Huang, A. Ruditskiy, J. Park, S. Bao, J. Liu, J. Howe and M. Chi, J. Am. Chem. Soc., 2015, 137, 15036–15042 CrossRef CAS PubMed.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- L. Gao, X. Li, Z. Yao, H. Bai, Y. Lu, C. Ma, S. Lu, Z. Peng, J. Yang and A. Pan, J. Am. Chem. Soc., 2019, 141, 18083–18090 CrossRef CAS PubMed.
- J. Fan, S. Yu, K. Qi, C. Liu, L. Zhang, H. Zhang, X. Cui and W. Zheng, J. Mater. Chem. A, 2018, 6, 8531–8536 RSC.
- J. Liang, S. Li, Y. Chen, X. Liu, T. Wang, J. Han, S. Jiao, R. Cao and Q. Li, J. Mater. Chem. A, 2020, 8, 15665–15669 RSC.
- L. Y. Zhang, F. Wang, S. Wang, H. Huang, X. Meng, Y. Ouyang, W. Yuan, C. X. Guo and C. M. Li, Adv. Funct. Mater., 2020, 30, 2003933 CrossRef CAS.
- F. Gao, Y. Zhang, F. Ren, Y. Shiraishi and Y. Du, Adv. Funct. Mater., 2020, 30, 2000255 CrossRef CAS.
- H. Xu, H. Shang, C. Wang and Y. Du, Small, 2021, 17, 2005092 CrossRef CAS PubMed.
- F. Lin, F. Lv, Q. Zhang, H. Luo, K. Wang, J. Zhou, W. Zhang, W. Zhang, D. Wang and L. Gu, Adv. Mater., 2022, 34, 2202084 CrossRef CAS PubMed.
- F. Saleem, Z. Zhang, B. Xu, X. Xu, P. He and X. Wang, J. Am. Chem. Soc., 2013, 135, 18304–18307 CrossRef CAS PubMed.
- F. Saleem, B. Xu, B. Ni, H. Liu, F. Nosheen, H. Li and X. Wang, Adv. Mater., 2015, 27, 2013–2018 CrossRef CAS PubMed.
- M. Li, F. Tian, T. Lin, L. Tao, X. Guo, Y. Chao, Z. Guo, Q. Zhang, L. Gu and W. Yang, Small Methods, 2021, 5, 2100154 CrossRef CAS PubMed.
- Y. Li, W. Wang, K. Xia, W. Zhang, Y. Jiang, Y. Zeng, H. Zhang, C. Jin, Z. Zhang and D. Yang, Small, 2015, 11, 4745–4752 CrossRef CAS PubMed.
- K. Cao, H. Yang, S. Bai, Y. Xu, C. Yang, Y. Wu, M. Xie, T. Cheng, Q. Shao and X. Huang, ACS Catal., 2021, 11, 1106–1118 CrossRef CAS.
- Y. Shi, Y. Fang, G. Zhang, X. Wang, P. Cui, Q. Wang and Y. Wang, J. Mater. Chem. A, 2020, 8, 3795–3802 RSC.
- W. Wang, X. Zhang, Y. Zhang, X. Chen, J. Ye, J. Chen, Z. Lyu, X. Chen, Q. Kuang and S. Xie, Nano Lett., 2020, 20, 5458–5464 CrossRef CAS PubMed.
- Y. Sun, X. Zhang, M. Luo, X. Chen, L. Wang, Y. Li, M. Li, Y. Qin, C. Li and N. Xu, Adv. Mater., 2018, 30, 1802136 CrossRef PubMed.
- L. Tian, Z. Li, M. Song and J. Li, Nanoscale, 2021, 13, 12088 RSC.
- C. He, J. Tao and P. K. Shen, ACS Catal., 2018, 8, 910–919 CrossRef CAS.
- C. Hu, Z. Chen, F. Han, Z. Lin and X. Yang, Chem. Commun., 2019, 55, 13566–13569 RSC.
- X. Li, H. You, C. Wang, D. Liu, R. Yu, S. Guo, Y. Wang and Y. Du, J. Colloid Interface Sci., 2021, 591, 203–210 CrossRef CAS PubMed.
- J. Bai, Q. Xue, Y. Zhao, J.-X. Jiang, J.-H. Zeng, S.-B. Yin and Y. Chen, ACS Sustainable Chem. Eng., 2018, 7, 2830–2836 CrossRef.
- X. Lao, M. Yang, X. Sheng, J. Sun, Y. Wang, D. Zheng, M. Pang, A. Fu, H. Li and P. Guo, ACS Appl. Energy Mater., 2022, 5, 1282–1290 CrossRef CAS.
- H. Wang, L. Jiao, L. Zheng, Q. Fang, Y. Qin, X. Luo, X. Wei, L. Hu, W. Gu and J. Wen, Adv. Funct. Mater., 2021, 31, 2103465 CrossRef CAS.
- X. Yuan, Y. Zhang, M. Cao, T. Zhou, X. Jiang, J. Chen, F. Lyu, Y. Xu, J. Luo and Q. Zhang, Nano Lett., 2019, 19, 4752–4759 CrossRef CAS PubMed.
- J. Guo, L. Gao, X. Tan, Y. Yuan, J. Kim, Y. Wang, H. Wang, Y. J. Zeng, S. I. Choi and S. C. Smith, Angew. Chem., Int. Ed., 2021, 60, 10942–10949 CrossRef CAS PubMed.
- Q. Wang, J. Liu, W. Zhang, T. Li, Y. Wang, H. Li and A. Cabot, Inorg. Chem., 2022, 61, 6337–6346 CrossRef CAS PubMed.
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed.
- W. Niu, Y. Gao, W. Zhang, N. Yan and X. Lu, Angew. Chem., Int. Ed., 2015, 54, 8271–8274 CrossRef CAS PubMed.
- C. Liu, Y. Shen, J. Zhang, G. Li, X. Zheng, X. Han, L. Xu, S. Zhu, Y. Chen and Y. Deng, Adv. Energy Mater., 2022, 12, 2103505 CrossRef CAS.
- R. M. Altarawneh, Energy Fuels, 2021, 35, 11594–11612 CrossRef CAS.
- S. H. Han, H. M. Liu, P. Chen, J. X. Jiang and Y. Chen, Adv. Energy Mater., 2018, 8, 1801326 CrossRef.
- M. Chu, J. Huang, J. Gong, Y. Qu, G. Chen, H. Yang, X. Wang, Q. Zhong, C. Deng and M. Cao, Nano Res., 2022, 15, 3920–3926 CrossRef CAS.
- I. Kim, O. H. Han, S. A. Chae, Y. Paik, S. H. Kwon, K. S. Lee, Y. E. Sung and H. Kim, Angew. Chem., Int. Ed., 2011, 50, 2270–2274 CrossRef CAS PubMed.
- M. Liu, M. Xie, Y. Jiang, Z. Liu, Y. Lu, S. Zhang, Z. Zhang, X. Wang, K. Liu and Q. Zhang, J. Mater. Chem. A, 2021, 9, 15373–15380 RSC.
- M. Zhou, J. Liu, C. Ling, Y. Ge, B. Chen, C. Tan, Z. Fan, J. Huang, J. Chen and Z. Liu, Adv. Mater., 2022, 34, 2106115 CrossRef CAS PubMed.
- Y. Zhang, X. Liu, T. Liu, X. Ma, Y. Feng, B. Xu, W. Cai, Y. Li, D. Su and Q. Shao, Adv. Mater., 2022, 2202333, DOI:10.1002/adma.202202333.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh01363k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |