
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence anisotropy using highly polarized emitting dyes confined inside BNNTs†
A.
Badon‡
 a,
J.-B.
Marceau‡
a,
C.
Allard
b,
F.
Fossard
c,
A.
Loiseau
c,
L.
Cognet
a,
E.
Flahaut
d,
G.
Recher
a,
N.
Izard
e,
R.
Martel
f and
E.
Gaufrès
*a
a,
J.-B.
Marceau‡
a,
C.
Allard
b,
F.
Fossard
c,
A.
Loiseau
c,
L.
Cognet
a,
E.
Flahaut
d,
G.
Recher
a,
N.
Izard
e,
R.
Martel
f and
E.
Gaufrès
*a
aLaboratoire Photonique Numérique et Nanosciences, Institut d’Optique, CNRS UMR5298, Université de Bordeaux, F-33400 Talence, France. E-mail: etienne.gaufres@cnrs.fr
bDépartement de Génie Physique, Polytechnique Montréal, Montréal, Québec H3C 3A7, Canada
cLaboratoire d'Étude des Microstructures, ONERA-CNRS, UMR104, Université Paris-Saclay, BP 72, 92322 Châtillon Cedex, France
dCIRIMAT, Université de Toulouse, CNRS, INPT, UPS, UMR CNRS-UPS-INP No. 5085, Université Toulouse 3 Paul Sabatier, Bât. CIRIMAT, 118, route de Narbonne, 31062 Toulouse cedex 9, France
eLaboratoire Charles Coulomb, UMR5221 CNRS-Université de Montpellier, 34095 Montpellier, France
fDépartement de Chimie, Université de Montréal, Montréal, Québec H3C 3J7, Canada
First published on 20th December 2022
Abstract
Polarized fluorescence emission of nanoscale emitters has been extensively studied for applications such as bioimaging, displays, and optical communication. Extending the polarization properties in large assemblies of compact emitters is, however, challenging because of self-aggregation processes, which can induce depolarization effects, quenching, and cancellations of molecular dipoles. Here we use α-sexithiophene (6T) molecules confined inside boron nitride nanotubes (6T@BNNTs) to induce fluorescence anisotropy in a transparent host. The experiments first indicate that individual 6T@BNNTs exhibit a high polarization extinction ratio, up to 700, at room temperature. Using aberration-corrected HRTEM, we show that the fluorescence anisotropy is consistent with a general alignment of encapsulated 6T molecules along the nanotube axis. The molecular alignment is weakly influenced by the nanotube diameter, a phenomenon ascribed to stronger molecule-to-sidewall interactions compared to intermolecular interactions. By stretching a flexible thin film made of transparent polymers mixed with 6T@BNNTs, we induce a macroscopic fluorescence anisotropy within the film. This work demonstrates that the dyes@BNNT system can be used as an easy-to-handle platform to induce fluorescence anisotropy in photonic materials.
New conceptsPolarized light emission from a single organic dye has been extensively studied in photonics and it is increasingly exploited in various fields such as bioimaging and quantum photonics. Extending polarization effects in fluorescent molecular assemblies through dipolar alignment is, however, extremely challenging due to depolarization and quenching effects induced by random and unstable molecular orientation, molecular self-aggregation or random positioning. Here we demonstrate that Boron Nitride Nanotubes can be used as a 1D dielectric template to align chains of aligned emitting molecules (Dyes@BNNT), almost independently of the BNNT diameter. At the same time, we use the BNNT as a nano container, which can be easily oriented within a flexible and transparent matrix by stretching. Shaping polarized emission from nano to macroscale with large assemblies of aligned Dyes@BNNTs enables the realisation of highly anisotropic fluorescent thin films. This concept paves the way toward new ordered and cooperative assemblies of organic emitters for advanced photonics. |
Polarized light-matter interaction in molecules offers advanced functionalities in various applications such as bio-imaging,1 solar energy harvesting,2,3 and quantum photonics,4 to name just a few. For instance, organic dyes such as rod-like polythiophenes show strong photon absorption/emission transitions between well-oriented dipoles and brightly polarized fluorescence at the single molecule level.5–9 Yet, the spontaneous self-organization of the dyes into aggregates generally prevents the macroscopic alignment of molecular dipoles. The pure polarization properties of single molecules are, as a result, difficult to translate in larger ensembles of bright emitters. This randomization of the molecular dipoles implies that additional control is required for the formation of a highly-polarized dye aggregate. Characterized by a head-to-tail ordered stacking of dyes, the J-aggregate is an emblematic molecular architecture that can be favoured by controlled assembly to brighten up the lower emission state. This aggregation state gives rise to higher fluorescence quantum yields, narrower red-shifted emission bands, and strong polarization dependencies.10–12 Supramolecular assemblies have been used to reinforce selective bonding between dyes in favour of the J-aggregate state, but only partial control has been achieved, thanks to specific adsorption mechanisms at surfaces or in solutions.13–15 Other approaches based on host/guest concepts or using nanoporous crystals, including zeolites and metal–organic frameworks (MOF), have also attracted significant interest for templating and aligning fluorescent molecular dipoles.16,17 While these approaches have greatly expanded our understanding of the polarization effects in molecular systems, the synthesis and manipulation of a large ensemble of highly polarized molecular emitters remain nowadays challenging for applications. Beyond the difficulty of making a large population of molecular aggregates, the challenge for application lies also in finding ways to organize them and upscale the optical anisotropy of photonic materials.
Our team has recently shown that fluorescent dye molecules encapsulated inside a BNNT exhibit surprisingly strong and stable fluorescence in normal conditions and a few examples of dyes have shown clear signatures of emitting J-like aggregation states.18 Here we explore the polarization properties of α-sexithiophene (6T) encapsulated inside a BNNT (6T@BNNT) on individual and on large assemblies of these nanohybrids. The results on individual 6T@BNNTs with 1–8 nm inner diameters show clear single dipole polarization fingerprints. Surprisingly, the 1D ordering is almost independent of the nanotube diameter, length and molecular filling level. Aberration-corrected High Resolution Transmission Electronic Microscopy (ac-HRTEM) and high-resolution fluorescence imaging of many individual 6T@BNNTs indicate instead that the alignment along the nanotube axis is strict for inner diameters below 3 nm, and it remains mostly aligned for larger diameters. This preferential molecular alignment is ascribed to stronger molecule-sidewall interactions. By capitalizing on the natural anisotropy of this 6T@BNNT system, we prepared a macroscopic thin film made of 6T@BNNTs dispersed in a poly(methyl methacrylate) (PMMA) matrix for stretching experiments and observed a strong fluorescence anisotropy in the stretching direction. This proof-of-concept highlights BNNTs as an easy-to-handle container of aligned aggregates of molecules for building fluorescence anisotropy in optical materials.
Results and discussion
Their preparation begins by opening raw BNNTs (BNNT LLC supplier and plasma torch BNNTs from the National Research Council Canada) using mechanical grinding and ultrasound treatments, followed by a thermal purification at 800 °C in air, which removes residual elemental boron and other h-BN sub-products. Purified BNNTs are then dispersed in dimethylformamide (DMF) by ultrasound and a small fraction of the solution is either spin-coated on a Si/SiO2 substrate (previously patterned with localisation marks) or drop cast onto a Molybdenum TEM grid covered with a holey SiO2 membrane. These BNNTs are then liquid-filled with 6T molecules by placing the entire sample in a 5 × 10−6 M solution of 6T in toluene at 115 °C. The last step of this liquid phase encapsulation consists of removing the non-encapsulated 6T molecules using toluene rinsing cycles at room temperature. Free 6T molecules are finally completely removed using an oxidative piranha solution (or in some experiments a mild oxygen plasma). More details about the sample preparation can be found in the supplementary information file.Fig. 1 presents a schematic representation of 6T molecules aligned inside a BNNT along with the diameter distribution of the nanotubes for this study. BNNTs have a similar structure to the SWCNT templates, but they are produced commercially with a slightly larger inner diameter distribution in the range of 1–5 nm. While these values of inner diameters in BNNTs open new perspectives for templating larger molecules, the confinement obtained is rather weak for common dyes whose size is typically 1–2 nm. The inner diameter, dinner, was statistically obtained using ac-HRTEM and the results indicate a relatively broad distribution, ranging from 0.7 nm to 7 nm, with an occurrence maximum centred at about 2 nm (see Fig. 1(b)). Considering that an elongated 6T molecule is ∼2.5 nm in length, the large distribution of dinner can therefore accommodate various stacking configurations, including the face to face and head-to-tail stackings.
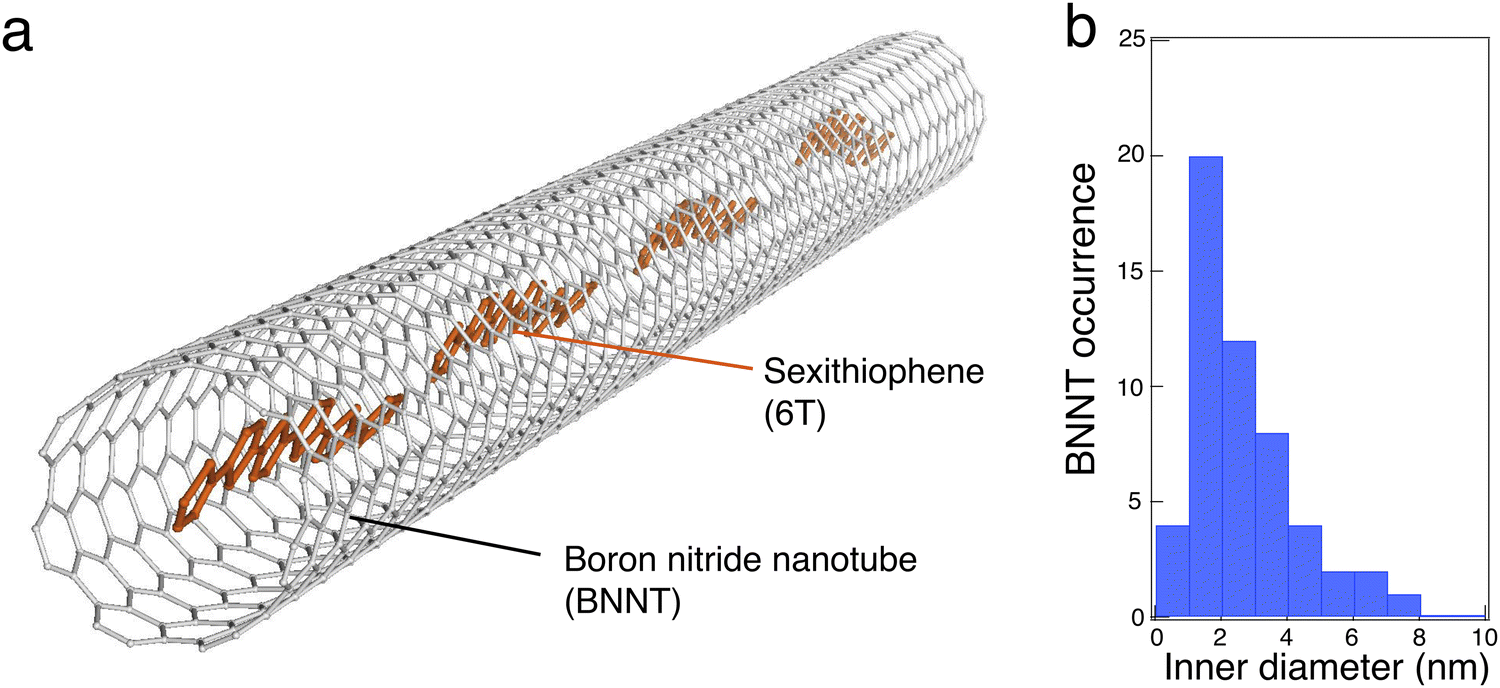 | ||
| Fig. 1 (a) Schematic illustration of α-sexithiophene (6T) molecules aligned inside a boron nitride nanotube (BNNT). (b) Inner diameter distribution of BNNTs probed on the grid using TEM. Note that the BNNTs have between 2 to 4 walls and rarely above 10 nm inner diameters (about 5 walls or more). | ||
To investigate the possible distribution of patterns, we performed polarized fluorescence imaging of individual 6T@BNNTs lying on a Si/SiO2 substrate (Fig. S1, ESI†). Briefly, a polarized 532 nm laser (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 polarization ratio) was collimated onto the sample using a 60× objective to homogeneously photo-excite individual 6T@BNNTs within a field-of-view of roughly 45 × 45 μm2. A combination of half- and quarter-wave plates allowed the excitation of the samples with a well-defined polarization state (linear or circular with control over the orientation). The emitted fluorescence was collected and analysed through a polarizing beam splitter prism cube and a 660 ± 5 nm band pass filter. Further details on the optical set up are available in the supplementary information. Fig. 2(a) shows a representative series of polarized fluorescence images obtained from individual 6T@BNNTs recorded using different excitation polarization conditions (blue arrows). The top series displays the fluorescence of randomly oriented 6T@BNNTs of lengths comparable to that of the optical resolution of our apparatus (∼1 μm), and excited with a circularly polarized light at a laser wavelength of 532 nm. Note that a circular polarization ensures that all orientations are simultaneously excited with our setup. The middle and bottom series present the responses of two in-plane orthogonal 6T@BNNTs having relatively longer lengths of ∼4 μm. The first presents a homogeneous fluorescence intensity along the BNNT axis, while the second displays spotted emission, probably due to a partial filling of 6T dyes in the nanotube. The two 6T@BNNTs are excited with a linearly polarized light that can be oriented approximately along the 6T@BNNT axis.
:1000 polarization ratio) was collimated onto the sample using a 60× objective to homogeneously photo-excite individual 6T@BNNTs within a field-of-view of roughly 45 × 45 μm2. A combination of half- and quarter-wave plates allowed the excitation of the samples with a well-defined polarization state (linear or circular with control over the orientation). The emitted fluorescence was collected and analysed through a polarizing beam splitter prism cube and a 660 ± 5 nm band pass filter. Further details on the optical set up are available in the supplementary information. Fig. 2(a) shows a representative series of polarized fluorescence images obtained from individual 6T@BNNTs recorded using different excitation polarization conditions (blue arrows). The top series displays the fluorescence of randomly oriented 6T@BNNTs of lengths comparable to that of the optical resolution of our apparatus (∼1 μm), and excited with a circularly polarized light at a laser wavelength of 532 nm. Note that a circular polarization ensures that all orientations are simultaneously excited with our setup. The middle and bottom series present the responses of two in-plane orthogonal 6T@BNNTs having relatively longer lengths of ∼4 μm. The first presents a homogeneous fluorescence intensity along the BNNT axis, while the second displays spotted emission, probably due to a partial filling of 6T dyes in the nanotube. The two 6T@BNNTs are excited with a linearly polarized light that can be oriented approximately along the 6T@BNNT axis.
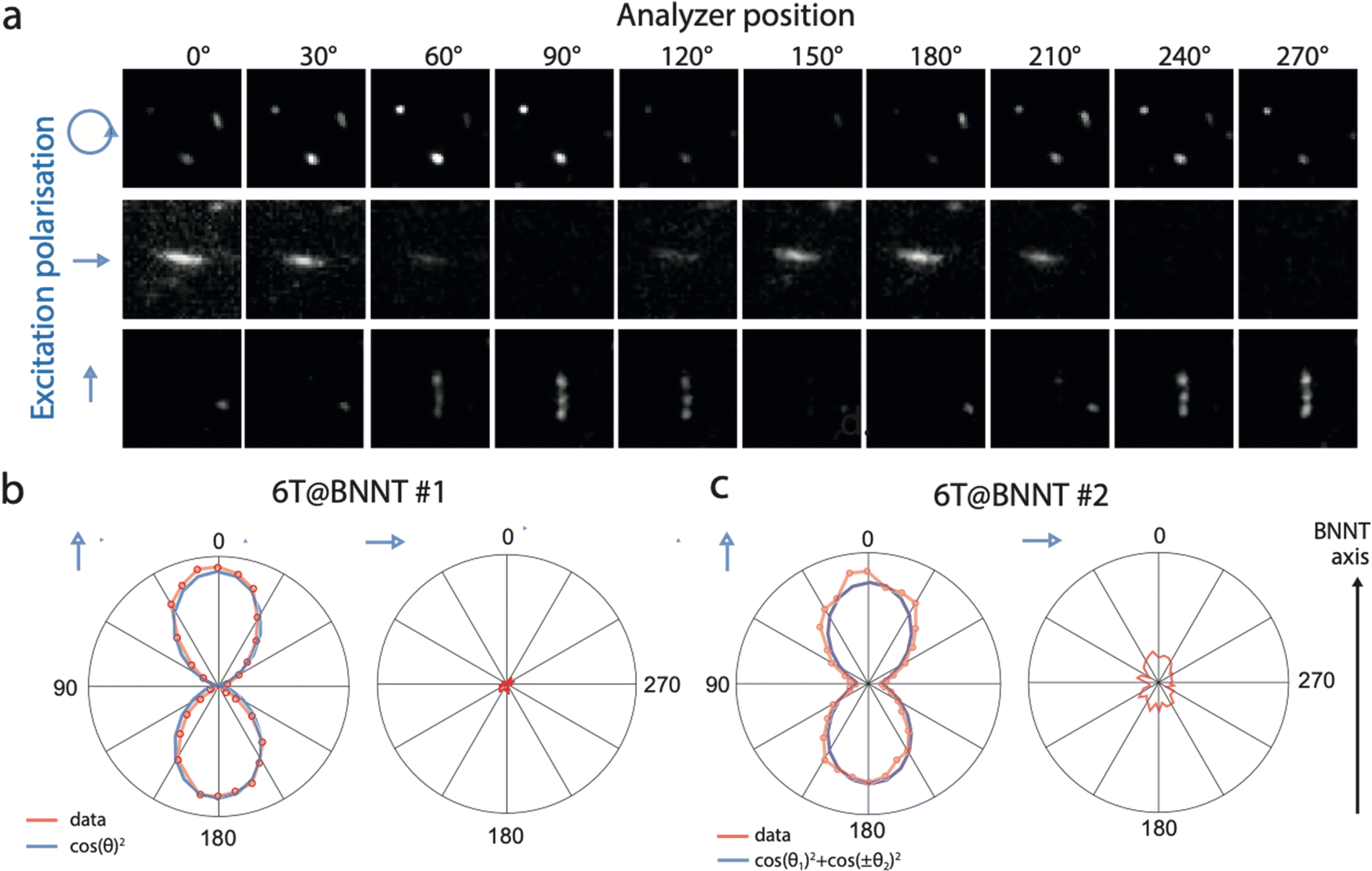 | ||
| Fig. 2 (a) Polarized fluorescence images of short (<1 μm) and long (∼4 μm) individual 6T@BNNTs deposited on a Si/SiO2 substrate. The polarization angle of the detected signal ranges from 0° to 270° with an incremental step of 30° between each image. The blue arrows indicate the state and direction of the excitation polarization for the series: circular, horizontal and vertical polarization from top, to middle, to bottom, respectively. (b) and (c) Polar plots of the fluorescence intensity for two 6T@BNNTs as a function of the analyser position and for two orthogonal excitation polarizations, i.e. along and orthogonal to the nanotube axis. The excitation wavelength is 532 nm. The fitting curves are plotted as a blue line. | ||
For all these cases, a periodic and severe extinction of the signal is unambiguously seen in the polar plots of the 6T@BNNTs, which is characteristic of a highly polarized fluorescence. These examples are in fact representative of what is typically observed in our samples (see other examples in the supplementary information file), namely a strong polarization effect is systematically observed, and there is no evidence of length, diameter, or filling level dependency. Considering that the molecular density is roughly 400 pairs of 6T molecules per micron of BNNT length, this result qualitatively demonstrates the good capacity of BNNTs to orientate large ensembles of elongated fluorescent organic dyes.
Fluorescence emission from molecular assemblies with BNNTs has already been reported by our group18 and by others.19 A highly polarized emission was also measured by Jakubek et al. from P3HT polymers wrapped along the BNNTs and used to image BNNTs under a microscope for quality assessments.20,21 Fluorescence emission from molecular assemblies of polymers and molecular dyes with single-walled carbon nanotubes (SWCNTs) has also been reported in the past.22–27 Thanks to their hollow 1D crystalline architecture, they offer narrow and uniform cavities for dye encapsulation along the whole nanotube. However, the following experiments have clearly shown that fast energy transfer processes between photo-excited dyes and the SWCNT host (Eg < 1 eV) readily quench the fluorescence emission of the dyes,28–32 which rather indicates that the fluorescence emission from dyes@SWCNTs is due to residual free dyes left in solution after rinsing and cleaning.
To better analyse the observed polarization dependencies, we plotted in Fig. 2(b) and (c) the integrated intensity of two typical 6T@BNNTs as a function of the analyser polarization angle, θ. The signal to noise (s/n) ratio is maximized when exciting a given candidate with a linearly polarized light roughly parallel to the nanotube axis, a condition that was selected for these plots. The first 6T@BNNT (Fig. 2(b), left) displays a highly polarized emission with an ultimate extinction ratio of 700 (Ipara/Iortho), which is only limited by the s/n ratio for Iortho. In cross-polarized excitation conditions (Fig. 2(b) right), the signal is near zero. This large extinction of signal combined with a good fit to the first term of the Legendre polynomial function (〈P2n(cosθ)〉) (blue line) is consistent with a strong and mono-dispersed alignment of emitters with uniaxial symmetry.33 This result highlights the ability of BNNTs to impose homogeneous molecular alignments along the nanotube axis for thousands of molecules.
While a strong alignment is noted for most 6T@BNNTs, we found, however, a few cases as in Fig. 2(c) of nanohybrids having slightly more complex polarization dependencies, yet still indicating aligned molecules. That is, the polar plots feature incomplete extinctions of the fluorescence at 90° and additional side lobes in the polar plots, which are symmetrically oriented at around ±30° with respect to the BNNT axis. Because of these lobes, the fit function to a polar plot (blue line) clearly shows that a more elaborate function than simple cos2θ functions is necessary to capture these lobes. The extra contributions to polarization are clearly beyond the simple case of aligned dipoles and would need at least the other 〈P4〉 coefficient of the Legendre polynomial for uniaxial symmetry, as noted in polarization experiments with polymer fibres.34,35 These cases of more complex polarization highlight the presence of a sub-population of nanohybrids for which a slightly more complex structure is observed.
To better understand these polarization patterns, we imaged many 6T@BNNTs supported on a TEM grid using ac-HRTEM imaging at 80 kV, which minimizes beam damage. Fig. 3(a) presents some representative TEM images of 6T@BNNT nanohybrids having few walls and diameters ranging from 0.8 nm to 3.5 nm. As already reported before,18 an overall ordering of the 6T molecules is clearly observed for BNNTs with the smallest inner diameter. While a complete modelling of these images is not yet available, one can readily see that a subset of the molecules adopts relatively large angles relative to the nanotube axis. This is more frequent when the inner tube diameter increases (see examples highlighted by white arrows). To quantify this trend, we measured the deviation angle for 757 molecules confined inside a statistical assembly of 50 different BNNTs of dinner ranging from 0.8 to 4.5 nm. The results of occurrences of deviation angles are reported in Fig. 3(b) for dinner < 1.2 nm; dinner < 2 nm; 2 nm < dinner < 3 nm; and dinner > 3 nm, respectively. Two main distributions appear in the statistics: the first has a sharp distribution close to 0°, which is representative of the smallest inner diameters. The second varies broadly in the range from 10° to 60° for dinner > 2 nm with a mean deviation angle of roughly 20° to 40°.
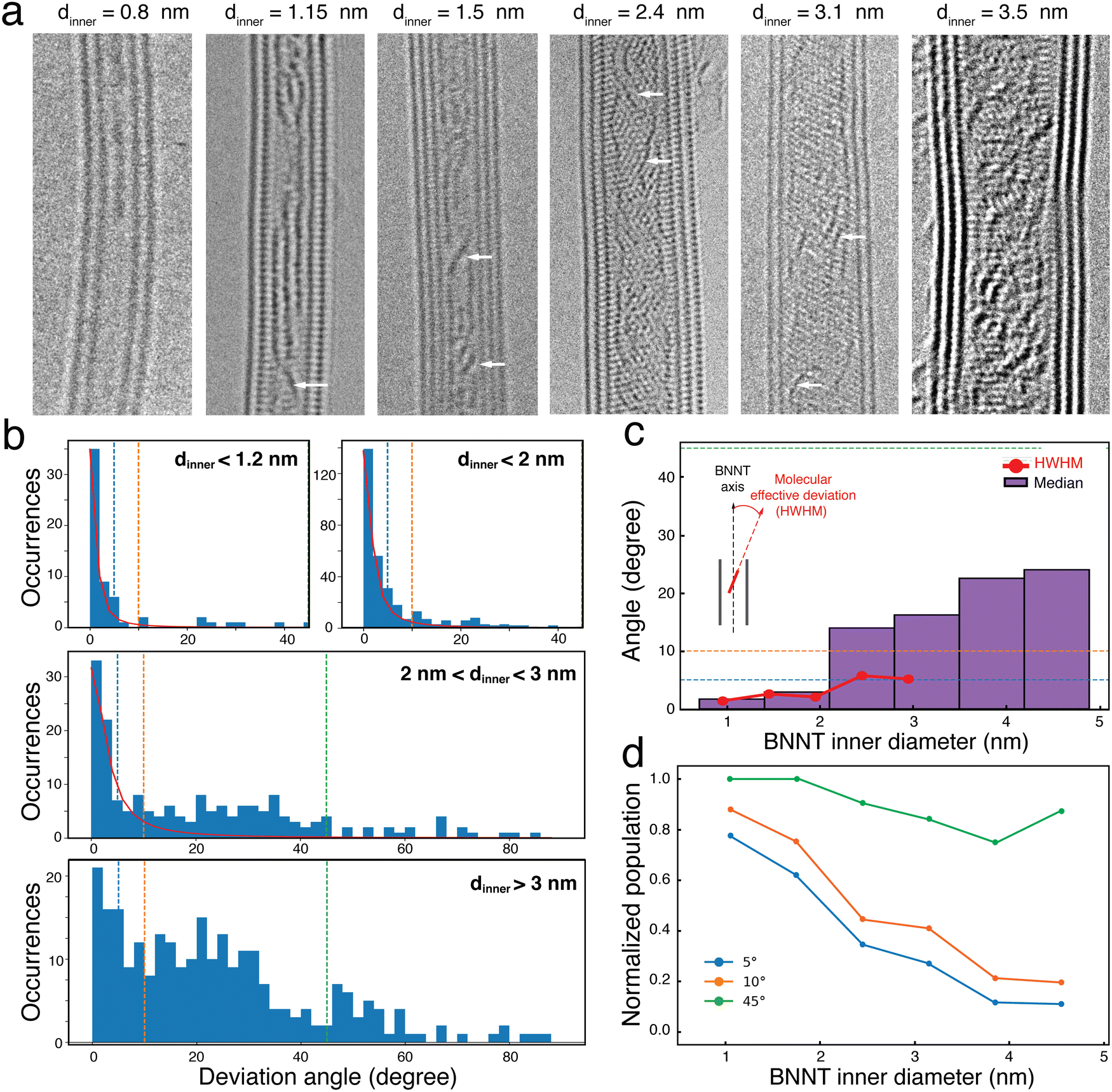 | ||
| Fig. 3 (a) HRTEM images recorded at 80 kV of 6T@BNNTs with inner diameters, dinner, between 0.8 and 4.5 nm. The white arrows highlight some molecules with a deviation angle in the range between 20° and 40°. (b) Distributions of the deviation angles (absolute values) between the molecules and the BNNT axis for different dinner subgroups. (c) Red line: evolution of the deviation angle distributions as a function of dinner extracted from a Lorentzian fit of the distribution of the angles in b. Histograms of the Half Width at Half Maximum (HWHM) in red and of the median value in purple. (d) Normalized evolution of the 6T populations having a deviation angle of less than 5°, 10° and 45°, as a function of dinner. | ||
While the observation of a preferential angle distribution in BNNTs of small dinner (compared to the molecule length) is not surprising, the results of a cross-over between perfectly aligned assemblies to more complex, yet still aligned, distributions of molecule sub-populations with 20–40° orientations relative to the nanotube axis is noteworthy. This evolution is clearly highlighted in the Half Width at Half Maximum (HWHM) plot in Fig. 3(c), which was calculated using Lorentzian fits applied to the deviation angle distributions for each 0.5 nm interval of dinner (see supporting information file for details). An interesting feature is the values of HWHM remaining below 7° up to dinner approaching 3 nm, this length being slightly larger than that of a 6T molecule. For dinner > 3 nm, a simple Lorentzian function can no longer capture the second distribution of 20–40° angles (see Fig. 3(b), middle) due to the limited statistics in this diameter range. To overcome this limitation, we also plotted in Fig. 3(c) the median value of the distribution, which shows that the value never exceeds 25° for 6T@BNNT of dinner < 5 nm.
Fig. 3(d) shows the normalized populations of the 6T molecule of orientations characterized by a maximum angle deviation of 5°, 10° and 45°, respectively. Two important trends in the distribution are identified in this plot. For BNNTs with dinner < 1.5 nm, we see that more than 80% of the molecules have deviation angles of less than 10°. For larger diameters, the distribution shows that 80% of the 6T molecules display deviation angles below 45°.
The orientation distributions become more complex at dinner > 2 nm, but the results are consistent with the two main signatures of polarization emission patterns observed in Fig. 2. Most of the population is first strictly polarized along the nanotube axis and the second population has a more complex polarization dependency with, nevertheless, an overall preferential distribution aligned in the direction of the nanotube axis. The BNNT appears to preferentially adsorb molecules on the sidewall, which forces a cross-over in the orientation distribution at dinner = 3 nm and above when the molecule–molecule spacing is large. This is particularly noticeable in the two HRTEM images in Fig. 3(a) of the 6T@BNNTs with dinner = 2.4 nm and 3.1 nm.
To better understand the nature of molecular interaction participating to this alignment within the cavity of BNNTs, we measured the lateral distances between the wall of the BNNTs and the adsorbed molecules (d6T-BNNT) and the distance between adjacent confined 6T molecules (d6T–6T). Fig. 4(a) reports the statistical map of these distances acquired on a large ac-HRTEM dataset of images as a function of the dinner. A colour code is used to separate different stacking configurations, as illustrated in the pictograms, i.e., single (blue), double (green), triple (red) and more than three rows (yellow) of molecules. A first observation is that the d6T-BNNT distribution is centred around a value of 0.35 nm, independently of the inner diameter and of the stacking configuration (see Fig. 4(a) and (c)). This value corresponds to the van der Waals distances (dvdW), which is similar to that of an interlayer in hBN and graphene. Differently, we observed that the d6T–6T distribution spreads over values ranging from 0.35 nm to 1 nm (see Fig. 4(c)) and is characterized by a clear dependence on the inner diameter and stacking configuration. More precisely, for a given inner diameter, d6T–6T generally explores the space available inside the BNNT. Considering the adsorption competition between 6T–6T and 6T-BNNT, these observations point toward a preferential adsorption of the 6T molecules onto the BNNT wall. Because of the tubular structure, this favours a molecular alignment of the 6T along the BNNT axis that is not correlated to the BNNT diameter, at least for the sample studied here (dinner between 0.7 and 4.5 nm).
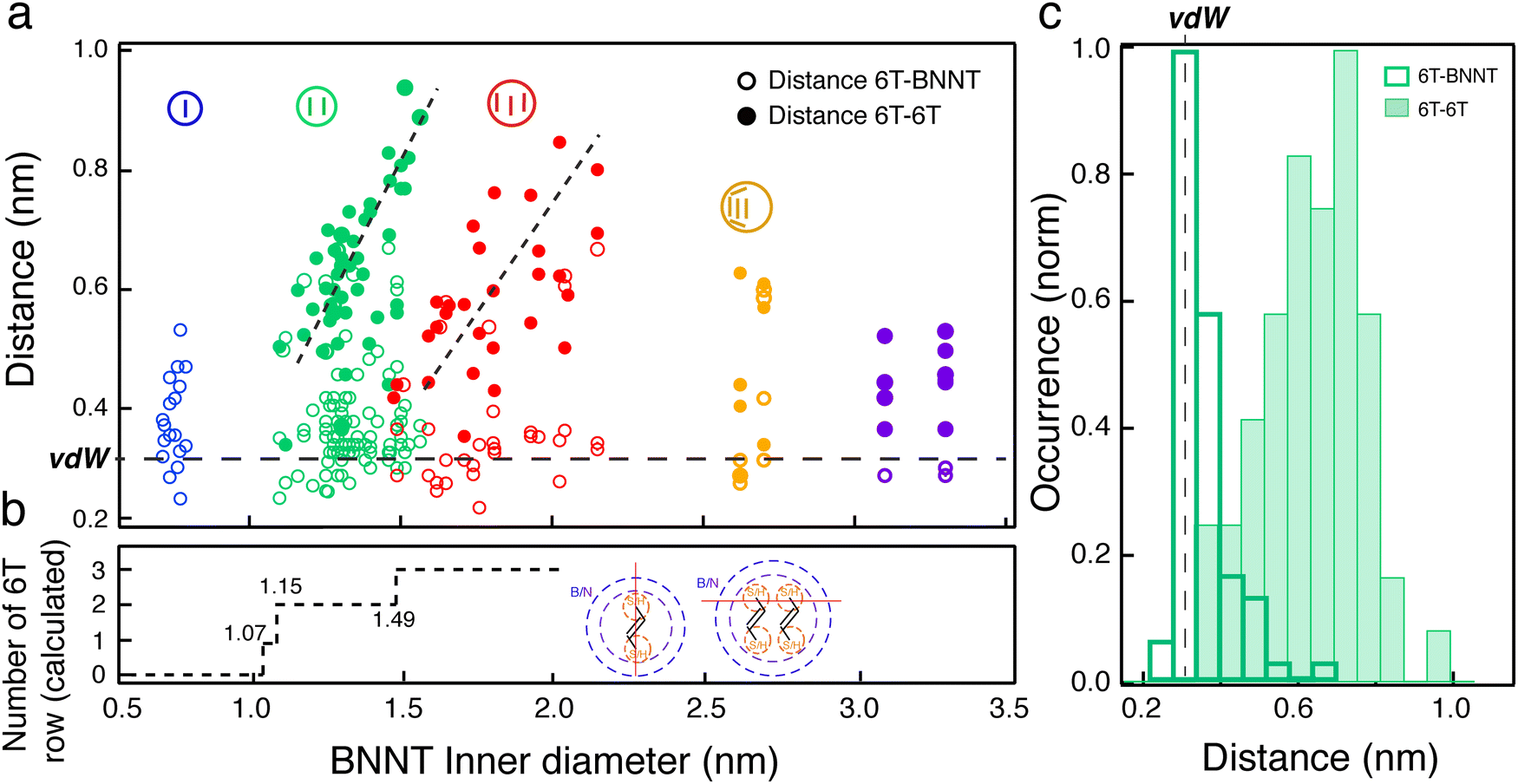 | ||
| Fig. 4 Statistics of measured lateral distances of 6T molecules confined inside BNNTs of inner diameters ranging from 0.7 to 3.3 nm. (a) Inter-distances between the BNNT wall and the adsorbed 6T molecules (6T-BNNT, open circle) and between adjacent 6T molecules (6T–6T, filled circle) inside BNNTs. The colour code indicates the stacking configuration of the confined 6T molecules, i.e. single (blue), pair array (green), triple array (red) and undefined (yellow and violet) aggregates. (b) The calculated number of 6T arrays allowed to stack parallel inside the BNNT as a function of the diameter of the BNNT, including the van der Waals (vdW) distance. Inset: Schematic representation of vdW radii (dotted purple lines) for the BN wall and for the S atoms (yellow dotted lines) of the 6T molecules placed inside the BNNT. The red line sets the spacing direction used for the estimation of a single array and of arrays with >2 molecules. (c) d6T–6T (filled green) and d6T-BNNT (open green) inter-distance distributions in the specific diameter range between 1.2 nm and 1.5 nm, favouring pair stacking of 6T molecules. | ||
The competitive interaction leading to molecular alignment is most likely the main cause of the highly polarized fluorescence patterns in individual 6T@BNNTs (Fig. 2). That is, the stronger 6T-BNNT interaction identified in these statistics is probably driving the polarization anisotropy along the nanotube axis. Another finding is the presence of clear thresholds in the d6T–6T distributions at dinner = 1.15 and 1.49 nm, which corresponds to the transitions from single to double array and from double to triple arrays of molecules, respectively. The transitions are clearly visible using ac-HRTEM as in Fig. 3, which are at values of minimum space required within the BNNT to accept an additional row of 6T molecules. To better understand these thresholds, we estimated the available volume for an additional row of 6Ts by considering the van der Waals radii of the BNNT wall and of the encapsulated 6T molecules (see ESI† for details). As highlighted in Fig. 4(b), the calculated thresholds are consistent with the measured single-to-double and double-to-triple thresholds. However, a discrepancy is noted for the minimum inner diameter required for the encapsulation of a single 6T array. According to this model, a minimum diameter of 1.07 nm would be required to accept a 6T molecule, whereas HRTEM observations unambiguously show 6T molecules encapsulated in inner diameters of 0.7 nm. Similar behaviours have been reported for quaterthiophene molecules (4T) inside single walled carbon nanotubes.36,37
The polarized fluorescence and statistical TEM analysis point toward a stacking mechanism resulting from a balance of competitive interactions to accommodate the 1D confinement of the inner cavity of the BNNTs. The molecules naturally tend to form by themselves a tilted stacking pattern called a herringbone, which is the preferred packing for example in polymorphic 6T crystals.38–40 Indeed, the weak interactions between 6T and repulsive forces along specific directions are balanced by various tilt angles ranging from 45° in the low temperature phase to 65° in the high temperature phase. Another driving force is the effect of the chiral angle of the BNNT itself, which can sway the 6T stacking in the tilted orientation relative to the nanotube axis. Preferential chirality angles of 15–30° have been reported in large multi-walled BNNTs,41 but a flat distribution from 0° to 30° is rather common in single- and double-walled BNNTs.42 Considering the length of 6T molecules and the space allowed by BNNTs with inner diameters considered here, most of these effects appear simply limited by the available space. It is clear, however, that the dominant interaction between the BNNT wall and the 6T molecules explains the high degree of alignment of 6Ts inside the BNNTs. Shin et al. reported that molecular dynamics calculations applied to the adsorption of poly(4-vinylpyridine) on the outer wall of boron nitride nanotubes and on the surface of hexagonal boron nitride show that decreasing curvature, i.e., increasing diameter, favors molecular adsorption on the BN wall.43 This trend may also explain the key role of the BNNT wall in shaping molecules beyond the purely geometric argument of 1D confinement in the large diameter BNNTs studied here.
The high anisotropy and polarization dependency of individual 6T@BNNT represents therefore a clear advantage to confer polarized properties in macroscopic fluorescent materials. Inspired by previous studies on the alignment of carbon nanotubes and other nanomaterials in a polymeric matrix,44,45 we have prepared a polymer stretchable matrix made of PMMA loaded with dispersed 6T@BNNTs in toluene. After 1 hour of stirring, the resulting solution was then dried on a hot plate to form a thin film of about 180 μm thickness. (Fig. 5(a) left). Next, the films were mechanically stretched at a temperature of 130 °C. After a deformation of the films ranging from 100% to 230% (Fig. 5(a)), the samples were sealed between a microscope slide and a coverslip with defined refractive index oil matching the immersion fluid of the objective (see supplementary file).
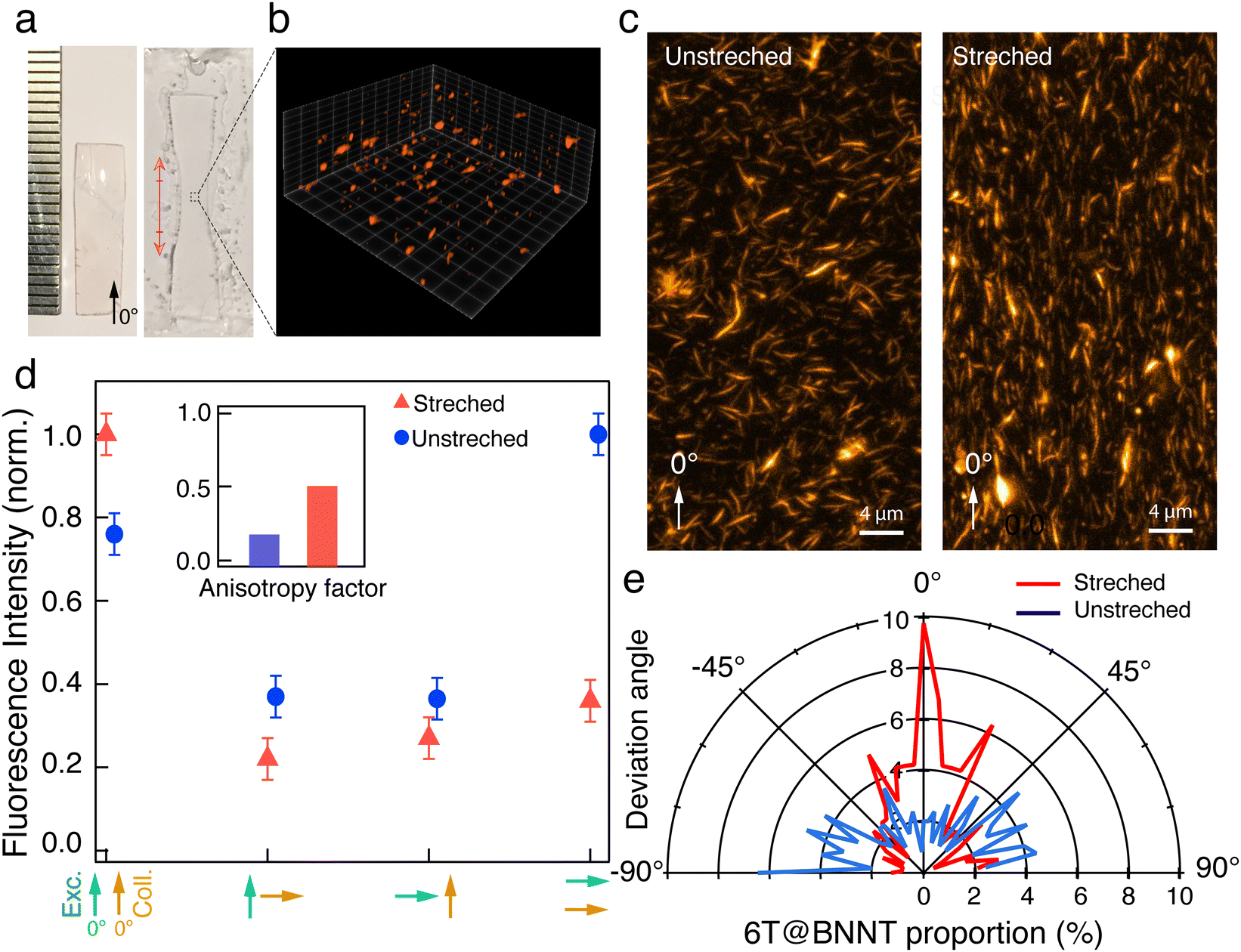 | ||
| Fig. 5 Polarized fluorescence from transparent thin films based on 6T@BNNT and PMMA. (a) Images of a centimetre wide PMMA film before and after stretching (left and right). (b) Partial rendering of a ‘z-stack’ of a PMMA-6T@BNNT film acquired with confocal fluorescence microscopy in a 40 μm × 40 μm × 30 μm volume. (c) z-Projection of the stretched and unstretched film datacubes. (d) Normalized fluorescence intensity recorded from the film, when unstretched and stretched, for different parallel, orthogonal and crossed polarization conditions of the excitation and collection, as highlighted by green and orange arrows, respectively. Inset: Anisotropy factor for the unstretched (left) and stretched sample (right) estimated from the data presented in the same panel. (e) Polar representation of the 6T@BNNT percentage as a function of the deviation angle from the direction of stretch (0°). | ||
We first characterized the thin film using 3D confocal fluorescence imaging (Leica SP8 WLL2 microscope) with a 530 nm excitation wavelength and a collection spectral window ranging from 600 nm to 750 nm and a z-step of 600 nm (Fig. 5(b)). A z-projection of the fluorescence stack is presented in Fig. 5(c) to map all the 6T@BNNTs in the whole volume. After denoising, the orientation statistic of the detected 6T@BNNTs within the datacube are analysed using MorphoLibJ and 3DSuite (Fig. 5(e) and supplementary file).46,47 As expected, the as-prepared film is composed of randomly oriented 6T@BNNTs (Fig. 5(c)), albeit the quantitative analysis of the angle repartition in Fig. 5(e) reveals a slight alignment of the 6T@BNNT along the 90° direction, which is most likely due to the presence of sheer force during the polymer liquid flow and drying steps on the hot plate. While the 6T@BNNT is slightly oriented before stretching, we clearly observed a strong realignment along the stretching direction (0°). More quantitatively, 40% of the detected 6T@BNNTs display a deviation angle from the stretching direction below 20° (Fig. 5(e)).
Finally, we performed statistical polarization measurements to study the impact of the polymer stretching upon the light polarization emitted by the thin films. Fig. 5(d) presents the normalized intensity of the fluorescence, parallelly (I0–0, I0–90) and orthogonally polarized (I90–0, I90–90) with respect to the incident polarization. For the as-prepared sample, the fluorescence polarization properties are consistent with what is expected for randomly dispersed immobilized uniaxial fluorophores. Nevertheless, we noticed that values of I0–0 and I90–90, which are supposed to be very similar, slightly differ due to the unintentional alignment mentioned above along the 90° direction. In clear contrast, the 6T@BNNT alignment within the stretched PMMA matrix, shown in Fig. 5(c) and (e), is revealed by the subsequent alignment of the fluorescence patterns, mainly along the stretching direction. To better highlight the polarizing effect induced by both the confinement of the 6T molecules inside BNNTs and the alignment of the BNNT within the PMMA matrix, we estimated the anisotropy factor, defined as 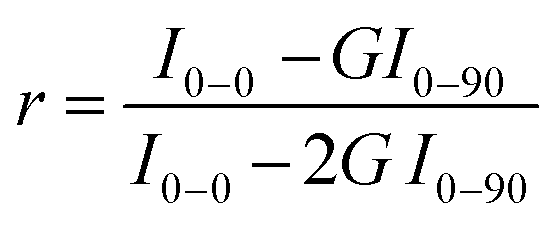 , with G, the optical factor correction of the set-up (G = 1.12). We found an enhanced anisotropy factor r = 0.500 ± 0.014 for the stretched film, compared to r = 0.185 ± 0.005 for the unstretched film. These results on films clearly demonstrate that the BNNTs act, because of their anisotropy and surface for molecular adsorption, as a powerful 1D template for aligning organic molecules from the nano to the macroscopic scales. They are easy-to-handle 1D transparent containers for the assembly of ordered and polarized molecules in advanced materials.
, with G, the optical factor correction of the set-up (G = 1.12). We found an enhanced anisotropy factor r = 0.500 ± 0.014 for the stretched film, compared to r = 0.185 ± 0.005 for the unstretched film. These results on films clearly demonstrate that the BNNTs act, because of their anisotropy and surface for molecular adsorption, as a powerful 1D template for aligning organic molecules from the nano to the macroscopic scales. They are easy-to-handle 1D transparent containers for the assembly of ordered and polarized molecules in advanced materials.
In summary, we have determined the polarization distribution of individual boron–nitride nanotube nanohybrids made of α-sexithiophene encapsulated inside BNNTs of inner diameter between 0.7 nm and 4.5 nm. The results underline a clear correlation between the preferential alignment of the molecules observed with transmission electron microscopy and the strong polarization dependency of the luminescence emission relative to the nanotube axis. The remarkable ability of BNNTs to create aligned 1D aggregates is highlighted by a complete extinction of the emission intensity of 6T at 90° in a cross-polarization configuration. The overall alignment of the molecules along the BNNT axis enables highly polarized patterns of fluorescence emission from all observed 6T@BNNTs. The samples reveal, however, a cross over between a perfectly aligned distribution to a mixed distribution having slightly larger angles for inner diameters in the 2–3 nm range. A detailed analysis of the polarization properties of these nanohybrids shows clear thresholds of inner diameter of BNNTs for which a slight shift in the arrangement of the molecules leads to higher polynomial terms in the distribution. Finally, we have used 6T@BNNTs as polarized light emitters in transparent and flexible thin films. The high fluorescence anisotropy conferred to these nanohybrids opens up new perspectives in the macroscopic preparation of polarized materials composed of 1D organic aggregates for photonics and nano-optics.
Author contributions
EG, RM and GR conceived the experiments, J-BM, EG and CA made the samples. AB, J-BM, CA, FF, NI, EF, GR, AL, LC, RM and EG performed the experiments, and analyzed and discussed the results. All authors contributed to writing the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors warmly acknowledge B. Simard and his team at the National Research Council Canada for donation of BNNT materials and G. Wang for support on the Cs-corrected TEM of MPQ – Paris Diderot University and METSA support for access to the Cs-corrected TEM of MPQ. E. G. acknowledges funding from CNRS starting package funding and the CNRS Tremplin program, GDRi Graphene and Co, the GDRi multifunctional nano for travel support. R. M. received financial support for this work from the Natural Sciences and Engineering Research Council of Canada (NSERC) under grants RGPIN-2019-06545 and RGPAS-2019-00050 and Canada Research Chairs (CRC) programs. E. G. and G. R. are supported by the CNRS MITI ‘Défi Auto-Organisation’ grant. G. R. acknowledges the GdR ImaBio for support, and the ANR for funding (ANR-21-CE45-0028) L. C. acknowledges financial support from the Agence Nationale de la Recherche (ANR-15-CE16-0004-03) and ITMO Cancer AVIESAN within the framework of the Cancer Plan (18CPl21-00). A. B. acknowledges financial support from the Agence Nationale de la Recherche (ANR-17-CE18-0026). Confocal microscopy was performed at the Bordeaux Imaging Center, a service unit of the CNRS-INSERM and Bordeaux University, a member of the national infrastructure France BioImaging supported by the French National Research Agency (ANR-10-INBS-04).References
- S. Brasselet; P. Ferrand; A. Kress; X. Wang; H. Ranchon and A. Gasecka, Imaging Molecular Order in Cell Membranes by Polarization-Resolved Fluorescence Microscopy. In Fluorescent Methods to Study Biological Membranes, in Springer Series on Fluorescence, ed. Y. Mély and G. Duportail, Springer, Berlin, Heidelberg, 2013, pp. 311–337 DOI:10.1007/4243_2012_51.
- T.-H. Lin, Y. Huang, A. Y. G. Fuh and S.-T. Wu, Polarization Controllable Fresnel Lens Using Dye-Doped Liquid Crystals, Opt. Express, 2006, 14(6), 2359–2364, DOI:10.1364/OE.14.002359.
- R. W. MacQueen and T. W. Schmidt, Molecular Polarization Switching for Improved Light Coupling in Luminescent Solar Concentrators, J. Phys. Chem. Lett., 2013, 4(17), 2874–2879, DOI:10.1021/jz4013905.
- S. G. Lukishova, R. P. Knox, P. Freivald, A. McNamara, R. W. Boyd, C. R. S. Jr, A. W. Schmid and K. L. Marshall, Single-Photon Source for Quantum Information Based on Single Dye Molecule Fluorescence in Liquid Crystal Host, Mol. Cryst. Liq. Cryst., 2006, 454(1), 1/[403]–14/[416], DOI:10.1080/15421400600653977.
- T. Ha, T. Enderle, D. S. Chemla, P. R. Selvin and S. Weiss, Single Molecule Dynamics Studied by Polarization Modulation, Phys. Rev. Lett., 1996, 77(19), 3979–3982, DOI:10.1103/PhysRevLett.77.3979.
- J. N. Forkey, M. E. Quinlan, M. Alexander Shaw, J. E. T. Corrie and Y. E. Goldman, Three-Dimensional Structural Dynamics of Myosin V by Single-Molecule Fluorescence Polarization, Nature, 2003, 422(6930), 399–404, DOI:10.1038/nature01529.
- R. N. Marks, M. Muccini, E. Lunedi, R. H. Michel, M. Murgia, R. Zamboni, C. Taliani, G. Horowitz, F. Garnier, M. Hopmeier, M. Oestreich and R. F. Mahrt, Disorder Influenced Optical Properties of α-Sexithiophene Single Crystals and Thin Evaporated Films, Chem. Phys., 1998, 227(1), 49–56, DOI:10.1016/S0301-0104(97)00274-7.
- E. Betzig and R. J. Chichester, Single Molecules Observed by Near-Field Scanning Optical Microscopy, Science, 1993, 262(5138), 1422–1425, DOI:10.1126/science.262.5138.1422.
- G. S. Harms, M. Sonnleitner, G. J. Schütz, H. J. Gruber and T. Schmidt, Single-Molecule Anisotropy Imaging, Biophys. J., 1999, 77(5), 2864–2870, DOI:10.1016/S0006-3495(99)77118-3.
- S. Sengupta and F. Würthner, Chlorophyll J-Aggregates: From Bioinspired Dye Stacks to Nanotubes, Liquid Crystals, and Biosupramolecular Electronics, Acc. Chem. Res., 2013, 46(11), 2498–2512, DOI:10.1021/ar400017u.
- H. Manzano, I. Esnal, T. Marqués-Matesanz, J. Bañuelos, I. López-Arbeloa, M. J. Ortiz, L. Cerdán, A. Costela, I. García-Moreno and J. L. Chiara, Unprecedented J-Aggregated Dyes in Pure Organic Solvents, Adv. Funct. Mater., 2016, 26(16), 2756–2769, DOI:10.1002/adfm.201505051.
- S. Herbst, B. Soberats, P. Leowanawat, M. Stolte, M. Lehmann and F. Würthner, Self-Assembly of Multi-Stranded Perylene Dye J-Aggregates in Columnar Liquid-Crystalline Phases, Nat. Commun., 2018, 9(1), 2646, DOI:10.1038/s41467-018-05018-6.
- B. Heyne, Self-Assembly of Organic Dyes in Supramolecular Aggregates, Photochem. Photobiol. Sci., 2016, 15(9), 1103–1114, 10.1039/C6PP00221H.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Self-Assembled π-Stacks of Functional Dyes in Solution: Structural and Thermodynamic Features, Chem. Soc. Rev., 2009, 38(2), 564–584, 10.1039/B809359H.
- T. Yokoyama, S. Yokoyama, T. Kamikado, Y. Okuno and S. Mashiko, Selective Assembly on a Surface of Supramolecular Aggregates with Controlled Size and Shape, Nature, 2001, 413(6856), 619–621, DOI:10.1038/35098059.
- T. Song, J. Yu, Y. Cui, Y. Yang and G. Qian, Encapsulation of Dyes in Metal–Organic Frameworks and the Tunable Nonlinear Optical Properties, Dalton Trans., 2016, 45, 4218–4223 RSC.
- G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Host–Guest Antenna Materials, Angew. Chem., Int. Ed., 2003, 42(32), 3732–3758, DOI:10.1002/anie.200300570.
- C. Allard, L. Schué, F. Fossard, G. Recher, R. Nascimento, E. Flahaut, A. Loiseau, P. Desjardins, R. Martel and E. Gaufrès, Confinement of Dyes inside Boron Nitride Nanotubes: Photostable and Shifted Fluorescence down to the Near Infrared, Adv. Mater., 2020, 32(29), 2001429, DOI:10.1002/adma.202001429.
- J. Niskanen, I. Zhang, Y. Xue, D. Golberg, D. Maysinger and F. M. Winnik, Boron Nitride Nanotubes as Vehicles for Intracellular Delivery of Fluorescent Drugs and Probes, Nanomedicine, 2016, 11(5), 447–463, DOI:10.2217/nnm.15.214.
- Z. J. Jakubek, M. Chen, Y. Martinez Rubi, B. Simard and S. Zou, Conformational Order in Aggregated Rra-P3HT as an Indicator of Quality of Boron Nitride Nanotubes, J. Phys. Chem. Lett., 2020, 11(10), 4179–4185, DOI:10.1021/acs.jpclett.0c01023.
- Y. Martinez-Rubi, Z. J. Jakubek, M. B. Jakubinek, K. S. Kim, F. Cheng, M. Couillard, C. Kingston and B. Simard, Self-Assembly and Visualization of Poly(3-Hexyl-Thiophene) Chain Alignment along Boron Nitride Nanotubes, J. Phys. Chem. C, 2015, 119(47), 26605–26610, DOI:10.1021/acs.jpcc.5b09049.
- M. Smith, C60 Encapsulation inside Carbon Nanotubes, Nature, 1998, 396(6709), 323, DOI:10.1038/24519.
- L. Shi, P. Rohringer, K. Suenaga, Y. Niimi, J. Kotakoski, J. C. Meyer, H. Peterlik, M. Wanko, S. Cahangirov, A. Rubio, Z. J. Lapin, L. Novotny, P. Ayala and T. Pichler, Confined Linear Carbon Chains as a Route to Bulk Carbyne, Nat. Mater., 2016, 15(6), 634–639, DOI:10.1038/nmat4617.
- K. Yanagi, Y. Miyata and H. Kataura, Highly Stabilized β-Carotene in Carbon Nanotubes, Adv. Mater., 2006, 18(4), 437–441, DOI:10.1002/adma.200501839.
- E. Gaufrès, N. Y.-W. Tang, F. Lapointe, J. Cabana, M.-A. Nadon, N. Cottenye, F. Raymond, T. Szkopek and R. Martel, Giant Raman Scattering from J-Aggregated Dyes inside Carbon Nanotubes for Multispectral Imaging, Nat. Photonics, 2014, 8(1), 72–78, DOI:10.1038/nphoton.2013.309.
- M. A. Loi, J. Gao, F. Cordella, P. Blondeau, E. Menna, B. Bártová, C. Hébert, S. Lazar, G. A. Botton, M. Milko and C. Ambrosch-Draxl, Encapsulation of Conjugated Oligomers in Single-Walled Carbon Nanotubes: Towards Nanohybrids for Photonic Devices, Adv. Mater., 2010, 22(14), 1635–1639, DOI:10.1002/adma.200903527.
- C. Roquelet, F. Vialla, C. Diederichs, P. Roussignol, C. Delalande, E. Deleporte, J.-S. Lauret and C. Voisin, Local Field Effects in the Energy Transfer between a Chromophore and a Carbon Nanotube: A Single-Nanocompound Investigation, ACS Nano, 2012, 6(10), 8796–8802, DOI:10.1021/nn302566e.
- E. Gaufrès, N. Y.-W. Tang, A. Favron, C. Allard, F. Lapointe, V. Jourdain, S. Tahir, C.-N. Brosseau, R. Leonelli and R. Martel, Aggregation Control of α-Sexithiophene via Isothermal Encapsulation Inside Single-Walled Carbon Nanotubes, ACS Nano, 2016, 10(11), 10220–10226, DOI:10.1021/acsnano.6b05660.
- S. van Bezouw, D. H. Arias, R. Ihly, S. Cambré, A. J. Ferguson, J. Campo, J. C. Johnson, J. Defillet, W. Wenseleers and J. L. Blackburn, Diameter-Dependent Optical Absorption and Excitation Energy Transfer from Encapsulated Dye Molecules toward Single-Walled Carbon Nanotubes, ACS Nano, 2018, 12(7), 6881–6894, DOI:10.1021/acsnano.8b02213.
- C. Roquelet, D. Garrot, J. S. Lauret, C. Voisin, V. Alain-Rizzo, P. Roussignol, J. A. Delaire and E. Deleporte, Quantum Efficiency of Energy Transfer in Noncovalent Carbon Nanotube/Porphyrin Compounds, Appl. Phys. Lett., 2010, 97(14), 141918, DOI:10.1063/1.3496470.
- D. Garrot, B. Langlois, C. Roquelet, T. Michel, P. Roussignol, C. Delalande, E. Deleporte, J.-S. Lauret and C. Voisin, Time-Resolved Investigation of Excitation Energy Transfer in Carbon Nanotube–Porphyrin Compounds, J. Phys. Chem. C, 2011, 115(47), 23283–23292, DOI:10.1021/jp207267e.
- E. Gaufrès, S. Marcet, V. Aymong, N. Y.-W. Tang, A. Favron, F. Thouin, C. Allard, D. Rioux, N. Cottenye, M. Verhaegen and R. Martel, Hyperspectral Raman Imaging Using Bragg Tunable Filters of Graphene and Other Low-Dimensional Materials: Hyperspectral Raman Imaging Using Bragg Tunable Filters of Graphene and Other Low-Dimensional Materials, J. Raman Spectrosc., 2018, 49(1), 174–182, DOI:10.1002/jrs.5298.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd edn, Spinger, 1999 Search PubMed.
- S. Frisk, R. M. Ikeda, D. B. Chase and J. F. Rabolt, Determination of the Molecular Orientation of Poly(Propylene Terephthalate) Fibers Using Polarized Raman Spectroscopy: A Comparison of Methods, Appl. Spectrosc., 2004, 58(3), 279–286, DOI:10.1366/000370204322886618.
- L. Svenningsson, Y.-C. Lin, M. Karlsson, A. Martinelli and L. Nordstierna, Molecular Orientation Distribution of Regenerated Cellulose Fibers Investigated with Polarized Raman Spectroscopy, Macromolecules, 2019, 52(10), 3918–3924, DOI:10.1021/acs.macromol.9b00520.
- Y. Almadori, L. Alvarez, R. Le Parc, R. Aznar, F. Fossard, A. Loiseau, B. Jousselme, S. Campidelli, P. Hermet, A. Belhboub, A. Rahmani, T. Saito and J.-L. Bantignies, Chromophore Ordering by Confinement into Carbon Nanotubes, J. Phys. Chem. C, 2014, 118(33), 19462–19468, DOI:10.1021/jp505804d.
- L. Alvarez, Y. Almadori, A. Belhboub, R. L. Parc, R. Aznar, P. Dieudonné-George, A. Rahmani, P. Hermet, F. Fossard, A. Loiseau, B. Jousselme, S. Campidelli, T. Saito, G. Wang and J.-L. Bantignies, Supramolecular Organization of Pi-Conjugated Molecules Monitored by Single-Walled Carbon Nanotubes, J. Nat. Prod., 2015, 10(1), 012514, DOI:10.1117/1.JNP.10.012514.
- B. Klett, C. Cocchi, L. Pithan, S. Kowarik and C. Draxl, Polymorphism in α-Sexithiophene Crystals: Relative Stability and Transition Path, Phys. Chem. Chem. Phys., 2016, 18(21), 14603–14609, 10.1039/C6CP01405D.
- G. Horowitz, B. Bachet, A. Yassar, P. Lang, F. Demanze, J.-L. Fave and F. Garnier, Growth and Characterization of Sexithiophene Single Crystals, Chem. Mater., 1995, 7(7), 1337–1341, DOI:10.1021/cm00055a010.
- C. Lorch, R. Banerjee, C. Frank, J. Dieterle, A. Hinderhofer, A. Gerlach and F. Schreiber, Growth of Competing Crystal Phases of α-Sexithiophene Studied by Real-Time in Situ X-Ray Scattering, J. Phys. Chem. C, 2015, 119(1), 819–825, DOI:10.1021/jp510321k.
- A. Celik-Aktas, J. M. Zuo, J. F. Stubbins, C. Tang and Y. Bando, Structure and Chirality Distribution of Multiwalled Boron Nitride Nanotubes, Appl. Phys. Lett., 2005, 86(13), 133110, DOI:10.1063/1.1885177.
- R. Arenal, M. Kociak, A. Loiseau and D.-J. Miller, Determination of Chiral Indices of Individual Single- and Double-Walled Boron Nitride Nanotubes by Electron Diffraction, Appl. Phys. Lett., 2006, 89(7), 073104, DOI:10.1063/1.2335379.
- H. Shin, E. Yeverovich and K. S. Kim, Poly(4-Vinylpyridine) Adsorption on Boron Nitride Nanotubes and Hexagonal Boron Nitride: A Comparative Molecular Dynamics Study, J. Mater. Res., 2022, 37, 4483–4495, DOI:10.1557/s43578-022-00705-z.
- L. Jin, C. Bower and O. Zhou, Alignment of Carbon Nanotubes in a Polymer Matrix by Mechanical Stretching, Appl. Phys. Lett., 1998, 73(9), 1197–1199, DOI:10.1063/1.122125.
- S. Badaire, V. Pichot, C. Zakri, P. Poulin, P. Launois, J. Vavro, C. Guthy, M. Chen and J. E. Fischer, Correlation of Properties with Preferred Orientation in Coagulated and Stretch-Aligned Single-Wall Carbon Nanotubes, J. Appl. Phys., 2004, 96(12), 7509–7513, DOI:10.1063/1.1810640.
- J. Ollion, J. Cochennec, F. Loll, C. Escudé and T. Boudier, TANGO: A Generic Tool for High-Throughput 3D Image Analysis for Studying Nuclear Organization, Bioinformatics, 2013, 29(14), 1840–1841, DOI:10.1093/bioinformatics/btt276.
- D. Legland, I. Arganda-Carreras and P. Andrey, MorphoLibJ: Integrated Library and Plugins for Mathematical Morphology with ImageJ, Bioinformatics, 2016, 32(22), 3532–3534, DOI:10.1093/bioinformatics/btw413.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh01239a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |