DOI:
10.1039/D2MH01218A
(Review Article)
Mater. Horiz., 2023,
10, 698-721
Copper-based catalysts for the electrochemical reduction of carbon dioxide: progress and future prospects
Received
30th September 2022
, Accepted 19th December 2022
First published on 20th December 2022
Abstract
There is an urgent need for the development of high performance electrocatalysts for the CO2 reduction reaction (CO2RR) to address environmental issues such as global warming and achieve carbon neutral energy systems. In recent years, Cu-based electrocatalysts have attracted significant attention in this regard. The present review introduces fundamental aspects of the electrocatalytic CO2RR process together with a systematic examination of recent developments in Cu-based electrocatalysts for the electroreduction of CO2 to various high-value multicarbon products. Current challenges and future trends in the development of advanced Cu-based CO2RR electrocatalysts providing high activity and selectivity are also discussed.
1. Introduction
Fossil fuels currently play an increasingly crucial role in global economic growth, technological progress and industry.1,2 However, rapid worldwide economic development and population growth have led to the excessive utilization of conventional nonrenewable fossil fuels such as coal, petroleum and natural gas. This scenario has, in turn, significantly increased the concentration of carbon dioxide (CO2) in the atmosphere, causing a series of global disasters and environmental problems such as global warming, desertification, ocean acidification, extreme weather and catastrophic floods.3–7 Therefore, there is an urgent need to develop reliable and efficient methods of converting CO2 into value-added chemicals and/or fuels with the goal of solving the above-mentioned crisis and achieving a sustainable carbon neutral society.
Reducing the level of CO2 in the atmosphere while also converting CO2 into valuable chemicals and/or fuels as a means of realizing carbon neutral energy conversion could possibly be achieved by the capture, sequestration and utilization of this compound.5,8–10 However, it is well known that CO2 is a thermodynamically stable molecule with strong covalent bonds such that it is not readily converted into other compounds. The electrochemical CO2 reduction reaction (CO2RR) is a promising and environmentally friendly approach to converting CO2 into valuable fuels and/or chemicals under ambient conditions. This process is therefore a potential solution to issues related to future energy shortages and could provide sustainable carbon-neutral energy conversion.2,5,11–14 Although CO2RR technologies have been studied since the early 19th century, it was not until 1985 that Hori et al. reported that CO2 could be converted into methane (CH4) as the major product along with various other compounds, including carbon monoxide (CO), formate (HCOO−) and hydrocarbons on a variety of metal electrodes.15 Since then, many studies have examined the development of advanced CO2RR catalysts so as to increase the efficiency of the CO2RR.
To date, many catalytic materials have been proposed as CO2RR electrocatalysts on the basis of possessing high electrical conductivity and good intrinsic catalytic activity while being readily available. The carbon-based catalysts (e.g., N-doped carbon, B- and N-co-doped carbon, Ru(II) polypyridyl carbene, graphene-based materials etc.) have shown the catalytic ability to electrochemically reduce CO2 to a variety of hydrocarbons and oxides, but they usually suffering from low current densities, large overpotential, poor stability, as well as difficult to prepare at scale.16,17 Among the potential metal-based catalytic materials candidates, Fe, Mn, Zn, Au, Ag, Pd and Ga have been demonstrated to be highly efficient CO2RR catalysts for the production of CO while Sn, Bi, Sb, In, Pb, Hg, Ti and Cd have been primarily used to generate liquid products such as formic acid (HCOOH) or HCOO−. As an example, a wide variety of carbon-based compounds can be obtained using Cu-based electrocatalysts. These include C1 products (e.g., CO, HCOOH, methanol (CH3OH) and CH4), C2 products (e.g., ethylene (C2H4), acetaldehyde (CH3CHO), acetate (CH3COO−) and ethanol (C2H5OH)) and C2+ products (e.g., acetic acid (CH3COOH), acetone (CH3COCH3) and n-propanol (C3H7OH)). Compared with C1 products, the C2 and C2+ products have higher energy densities and are more valuable and so currently play important roles in the energy supply and chemical industries.10,18–27 However, Cu catalysts commonly suffer from high overpotentials and poor selectivity for the CO2RR. In addition, the CO2RR is a multi-component reaction process capable of generating up to 16 different products, such that it is difficult to use on an industrial scale.28 Another issue identified by many studies is that the rate of the Cu-catalyzed CO2RR rapidly decreases within several hours.10,29 Therefore, it is imperative to develop advanced Cu-based electrocatalysts exhibiting high selectivity, improved activity and excellent stability while providing the desired reduction products.
In recent years, there has been considerable research with the aim of designing efficient Cu-based heterogeneous materials as CO2RR electrocatalysts. These materials have comprised monometallic Cu, Cu-based oxides and other Cu-based compounds, Cu-based bimetallic systems, single/dual Cu atoms and Cu-based metal-organic frameworks (MOFs) among others.
The present review examines the performance of Cu-based catalysts along with the associated reaction mechanisms. The focus is on introducing fundamental aspects of the electrocatalytic CO2RR after which the latest developments in the design of Cu-based electrocatalysts for the electroreduction of CO2 to high-value multi-carbon products are systematically summarized. Lastly, unsolved challenges and anticipated future progress related to the development of advanced Cu-based CO2RR electrocatalysts with high activity and selectivity are examined. It is the hope of the authors that this review will provide a comprehensive overview of recent developments in the study of Cu-based CO2RR electrocatalysts.
2. Fundamental aspects of the electrocatalytic CO2RR
2.1 Performance evaluation parameters, electrochemical cell configurations and electrolytes
2.1.1 Performance evaluation parameters.
The parameters that are commonly used to compare and evaluate the catalytic performances of CO2RR systems include the overpotential (η), current density (j), partial current density (jpartial), faradaic efficiency (FE), turnover number (TON) and turnover frequency (TOF). The parameter η is the potential difference between the measured and theoretical potentials required to drive the CO2RR and so a catalyst with superior CO2RR activity will typically exhibits a low η with respect to the generation of certain products. The value of η is calculated aswhere E is the measured electrode potential and Eeq is the standard potential for the formation of the product. The parameter j is the total current (i) per unit area (A) of the cathode electrode and is calculated asThe value of j indicates the overall rate of the CO2RR and so this variable is an important aspect of evaluating the electrocatalytic activity of a catalyst. The parameter jpartial for a given product can be obtained from the relationshipHere, FE is calculated according to Faraday's Law and equals the ratio of the charge consumed by the formation of the product to the total charge (Q). This term is determined aswhere z is the number of electrons transferred, n is the amount of product in moles and F is Faraday's constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1). FE is used to describe the selectivity of the catalyst during the electrocatalytic reduction of CO2. The TON is the yield of a product acquired using a unit amount of the catalyst while the TOF is the yield of a product generated using a unit amount of the catalyst over a unit time period. Both the TON and TOF are used to describe the electrocatalytic activity of the catalyst.
485 C mol−1). FE is used to describe the selectivity of the catalyst during the electrocatalytic reduction of CO2. The TON is the yield of a product acquired using a unit amount of the catalyst while the TOF is the yield of a product generated using a unit amount of the catalyst over a unit time period. Both the TON and TOF are used to describe the electrocatalytic activity of the catalyst.
2.1.2 Electrochemical cell configuration.
The electrochemical cell structure is an important factor affecting the CO2 reduction process and will determine the FE, current density and stability. Since Hori first reported the electrochemical reduction of CO2 in the 1980s,15 different electrochemical cells have been developed. At present, reactor vessels can be primarily divided into H-type, flow, solid-oxide electrolysis and differential electrochemical mass spectrometry cells (Fig. 1). It is worth noting that the CO2RR is still in the laboratory research stage and the catalytic current density based on mass transport limitations is currently the primary factor preventing the commercial application of this technology. Therefore, further research is needed with regard to the design of more efficient electrochemical cells.
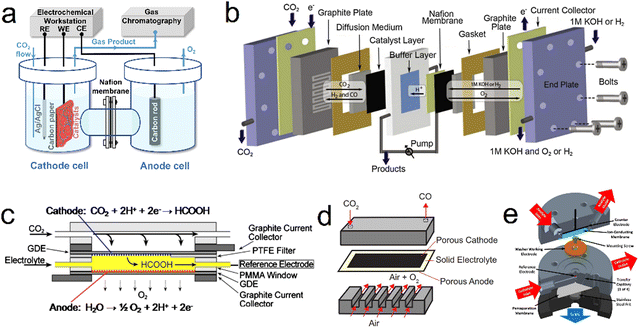 |
| | Fig. 1 Diagrams showing various electrochemical cell concepts. (a) H-Type. (b) PEM flow. (c) Microfluidic. (d) Solid-oxide electrolysis. (e) DEMS cells. Reproduction with permission from ref. 31, Copyright 2022, Chinese Chemical Society and Reproduction with permission from ref. 34, Copyright 2013, The Electrochemical Society and Reproduction with permission from ref. 35, Copyright 2013, The Electrochemical Society and Reproduction with permission from ref. 74, Copyright 2018, Cell Press and Reproduction with permission from ref. 37, Copyright 2022, American Chemical Society. | |
To date, commercially available H-type cells (Fig. 1a) are still the most common laboratory reactors for the CO2RR. In these units, the working and reference electrodes are placed in the cathode compartment while the counter electrode is situated in the anode compartment.30,31 The two compartments are usually connected by a circular channel and separated by an ion exchange membrane to prevent the oxidation of reduction products during the CO2RR. Throughout the CO2RR process, gaseous CO2 continuously flows into the cathodic compartment through a conduit while the gas phase products are sampled and transferred to a gas chromatograph to determine the composition of the product mixture. It should be noted that the electrochemical cell must be airtight and the gas flow rate is typically controlled by a mass flow meter at the inlet or outlet of the cell. In addition, liquid products are collected from the electrolyzed electrolyte and evaluated by nuclear magnetic resonance spectrometry or liquid chromatography-mass spectrometry. However, it should be noted that the concentration of each liquid product has to be above the detection limit of the analytical technique (either high performance liquid chromatography or liquid chromatography-mass spectrometry), which may not always be the case because the FE associated with most such products is very low. Therefore, longer electrolysis durations and smaller compartment volumes are recommended for the determination of such compounds in trials involving an H-type cell. In addition, although the H-type cell is widely used in the study of the CO2RR, the relatively large distance between the counter and working electrodes, the low solubility of CO2 in the electrolyte and the inherent mass transfer limitations in this system tend to produce low catalytic current densities (j < 100 mA cm−2), which greatly limits practical applications.
Various flow cell designs have been developed to increase the catalytic current density that can be obtained from the CO2RR. In a typical flow cell, CO2 is continuously supplied to the cathode using a gas diffusion electrode (GDE) or a system involving the external circulation of an electrolyte in which CO2 is dissolved.32,33 In contrast to an H-type cell, the reduction of CO2 reduction at a GDE can remove the effect of mass transfer limitations and provide higher CO2 concentrations on the catalyst surface, thus enabling higher catalytic current densities in such flow cells. The polymer electrolyte membrane (PEM) concept (Fig. 1b) is currently the most widely used type of CO2 flow cell and has led to significant progress in the study of the CO2RR.34 The configuration of such units is almost the same as those of proton exchange membrane fuel cells and typically comprises a membrane electrode assembly, cathode/anode current collectors and cathode/anode flow plates. Because this cell configuration contains only working and counter electrodes, without a reference electrode, adjustment of the current rather than the potential is often used to control the reaction, such that it can be challenging to differentiate anode and cathode degradation effects. The membrane electrode assembly, which is the most important component of such cells, consists of a cathode and anode, a GDE and a PEM. The electrodes are positioned close to one another such that the cell resistance is decreased. In a PEM flow cell, the catalysts are typically deposited on a carbon-based gas diffusion layer (made of either carbon paper or carbon cloth) to prolong the contact time with CO2 while also providing a high surface area. Fig. 1c presents a diagram of a typical sandwich-structured microfluidic flow cell.35 This reactor consists of two GDEs separated by a Nafion-117 membrane. During the CO2RR, the electrolyte is injected into the cell at a specific flow rate selected to allow online collection of the reaction products for analysis. Importantly, the performance of such devices can be effectively improved by adjusting both cell parameters and reaction conditions, including the system pressure distribution, electrolyte flow rate, chamber/channel size and electrode structure.
In addition to electrochemical cells operating at room temperature such as those described above, solid oxide electrolysis cells (SOECs) can be used for CO2 reduction at high temperatures (>873 K) (Fig. 1d). These devices have recently become of interest because they provide advantages such as improved reaction kinetics and reduced internal resistance, thereby achieving higher reaction efficiencies without using noble metal catalysts. A typical SOEC has three primary parts: a cathode for CO2 reduction, an anode for oxygen evolution and a solid electrolyte for ion transport at temperature range from 300 °C to 1500 °C. In this cell configuration, the main product of the CO2RR is CO, although coke and CH4 are also produced. Despite the narrow range of products obtainable from such systems, the product selectivity and catalytic performance of SOECs are generally superior to those of low temperature systems.36
Differential electrochemical mass spectrometry (DEMS) can continuously separate and collect electrochemical reaction products in real time based on pervaporation technology, followed by rapid analysis of these products (with an analysis time on the order of 1 s). In 2015, Clark and coworkers designed a novel DEMS cell (Fig. 1e) and demonstrated the applicability of this unit to the analysis of the electrochemical reduction of CO2 on polycrystalline Cu.37 In this DEMS cell a parallel working and counter electrode are separated by an ion-conducting membrane to ensure a uniform distribution of potential on the electrode surface and prevent unwanted parasitic reactions. During the CO2RR, a CO2-saturated electrolyte is pumped into the cell at a constant flow rate to supplement the CO2 consumed by electroreduction while simultaneously providing efficient mass transfer to the cathode. The catholyte volume between the working electrode and pervaporation membrane is minimized to ensure that the delay time between product generation and detection is greatly shortened, with a delay on the order of 2 s. Products are delivered to a collection chamber and then efficiently assessed by mass spectrometry.
2.1.3 Electrolytes.
The electrolyte acts as a conductive medium during the electrolysis process and the specific cations and anions in this medium can significantly affect the CO2RR performance. The electrolytes employed for the CO2RR can be divided into three categories: aqueous solutions, organic solvents and ionic liquids. The most common inorganic electrolytes used in aqueous solutions are sodium bicarbonate (NaHCO3) and potassium bicarbonate (KHCO3), which act as both proton donors and pH buffers. The addition of halide anions to the electrolyte has been found to have a positive effect on the CO2RR, as demonstrated by reduced overpotentials and enhanced reaction rates.38,39 In addition, the sizes of the cations and anions in the electrolyte can also affect CO2RR performance.40–43 Except bicarbonate, potassium hydroxide (KOH) electrolyte is also commonly used in CO2RR. KOH electrolyte with high pH value is conducive to the electroreduction of CO2, which may be due to the fact that anions are more easily adsorbed on the electrode surface or the anode potential is reduced in KOH electrolyte. Because CO2 is more soluble in organic solvents than in aqueous solutions, electrochemical CO2 reduction occurs more readily in organic solvents. The most commonly used organic solvents for the CO2RR are propylene carbonate, dimethyl sulfoxide, N,N-dimethylformamide, acetonitrile and methanol. The liquid products produced by these solvents typically include formic acid, oxalic acid, glyoxylate and glycolate. Among these potential solvents, methanol is superior because of its relatively low toxicity and minimal cost together with its ability to provide high yields.44
Ionic liquids are a class of molten salts having very low melting points. These compounds can also be employed in CO2RR systems because they tend to exhibit wide electrochemical windows, good electrical conductivity, high CO2 solubility and essentially nil vapor pressure.45 Ionic liquids are generally thought to serve as co-catalysts in addition to solvents because they can reduce the generation of the CO2 radical anion intermediate (CO2˙−), thus lowering the overpotential associated with CO2 reduction.46,47 Among the many ionic liquids, 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIM-BF4) is widely used because the moderate binding energy of EMIM with CO2 promotes CO2 reduction.48
Modifying the electrolyte can also affect the electronic structure of the catalyst while also stabilizing Cu+ ions during the reaction, representing another means of enhancing the CO2RR. As an example, Yang et al. reported that the controlled surface reconstruction of commercial polycrystalline Cu could be readily accomplished using ethylenediamine tetramethylenephosphonic acid as an electrolyte additive, leading to a substantial improvement in the extent of CO2 electroreduction to CH4.49
2.2 Reaction mechanism
The CO2RR in an aqueous electrolyte involves proton coupled electron transfer. This is a complicated process comprising multiple single-step reactions and so a variety of reduction products are obtained. The mechanism by which such products are generated can be examined based on the specific carbon products that are formed. The most commonly reported C1 products are CO, CH4, HCOOH/HCOO− and CH3OH. The formation of CO requires that adsorbed *CO2 (where * indicates an active site) on the catalyst is initially reduced to generate *CO2˙−. Following this, the *CO2˙− is converted to *COOH. In the case that this species then undergoes hydrogenation, HCOOH/HCOO− will be formed, representing pathway 1.50 However, if dehydration occurs, gaseous CO is obtained, representing pathway 2.51 The associated reaction pathways are
| *HCOOH → HCOOH(l) + * (pathway 1) |
and
| *COOH + H+ + e− → *CO + H2O |
| *CO → CO (g) + * (pathway 2). |
If the *CO generated by pathway 2 does not desorb from the catalyst, it will be further reduced to *CHO, *CH2O and *CH3O with the eventual formation of CH4, representing pathway 3.51 This sequence can be summarized as
| *COOH + H+ + e− → *CO + H2O |
| *CH3O + H+ + e−→ CH4(g) + *O |
| *OH + H+ + e− → H2O + * (pathway 3). |
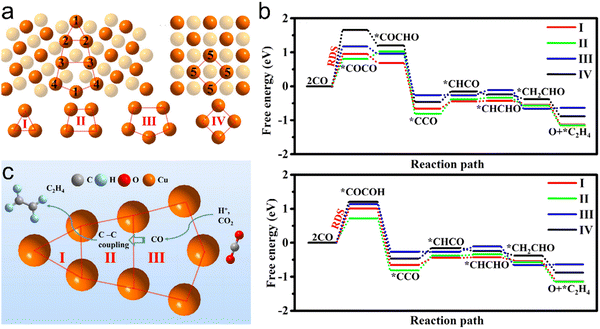
According to Suzuki et al., the rate-limiting step in the formation of CH4 is *CO → *CHO.52 It should also be noted that CH3OH is a side product of the CH4 formation process.50 The C2 products from this process are typically C2H4 and C2H5OH and early research established that *CO is a key intermediate in the formation of these two products.53 If a dimerization reaction based on PCET between two *CO takes place on the catalyst, the resulting *C2O2H intermediate will lead to the formation of C2 products. In the following step, the outgoing H+ can bond with the α-carbon on *C2O2H to produce C2H4 as the end product via pathway 4, which can be summarized as
| *C2O2H + H+ + e− → *COCHOH |
| *COCHOH + 2H+ + 2e− → *OCH2CHOH |
| *OCH2CHOH + H+ + e− → *CHCH2O + H2O |
| *CHCH2O + H+ + e− → *CH2CH2O |
| *CH2CH2O → C2H4(g) + *O (pathway 4). |
Another proposed mechanism of C2H4 production from CO2RR has also been reported by Janik and Asthagiri (pathway 5):54
| *COCOH + H+ + e− → *CCO + H2O |
| *CCO + 2H+ + 2e− → *CHCHO |
| *CHCHO + H+ + e− → *CH2CHO |
| *CH2CHO + H+ + e− → C2H4(g) + *O (pathway 5) |
If there is no loss of *O after the generation of *CH2CHO, the reaction will continue as follows (pathway 6):
| *CH2CHO + H+ + e− → *CH2CH2O |
| *CH2CH2O + H+ + e− → C2H4(g) + *O (pathway 6) |
If there is no loss of *O after *CH2CH2O production, the reaction will continue as follows (pathway 7):
| *CH2CH2O + H+ + e− → *CH3CH2O |
| *CH3CH2O + H+ + e− → C2H4(g) + *O + H2O (pathway 7) |
In addition, Goddard and coworkers found that when the *COCOH intermediate is produced, C2H4 can be produced through another path (pathway 8):55
| *COCOH + H+ + e− → *COHCOH |
| *COHCOH + 2H+ + 2e− → *CHCOH + H2O |
| *CHCOH + H+ + e− → *CCH + H2O |
| *CCH + 2H+ + 2e− → *CHCH2 |
| *CHCH2 + H+ + e− → C2H4(g) (pathway 8) |
After the formation of *C2O2H intermediate, if the proton bonds with the β-carbon of *C2O2H, the final product is C2H5OH via the reaction sequence:
| *C2O2H + H+ + e− → *C2O + H2O |
| *CHCHO + H+ + e− → *CH2CHO |
| *CH2CHO + H+ + e− → *CH3CHO |
| *CH3CHO + H+ + e− → *CH3CH2O |
| *CH3CH2O + H+ + e− → *CH3CH2OH |
| *CH3CH2OH → CH3CH2OH(l) + * (pathway 9). |
Another proposed mechanism of CH3CH2OH generation is reported as follows (pathway 10):55
| *COCOH + H+ + e− → *COHCOH |
| *COHCOH + 2H+ + 2e− → *CHCOH + H2O |
| *CHCOH + H+ + e− → *CHCHOH |
| *CHCHOH + H+ + e− → *CH2CHOH |
| *CH2*CHOH + H+ + e− → *CH3CHOH |
| *CH3CHOH + H+ + e− → CH3CH2OH(l) (pathway 10) |
Bell and Head-Gordon found that CH3CH2OH can be produced via another pathway (pathway 11):56
| *COCHO + H+ + e− → C2H2O2 (glyoxal) |
| C2H2O2 + 3H+ + 3e− → *CH2CHO |
| *CH2CHO + H+ + e− → *CH3CHO |
| *CH3CHO + H+ + e− → *CH2CH2OH |
| *CH2CH2OH + H+ + e− → CH3CH2OH(l) (pathway 11) |
CH3COOH is another possible C2 product but, as a consequence of the associated energy barriers and multiple proton-electron transformations, only a few electrocatalysts are capable of efficiently reducing CO2 to CH3COOH.57–59
The reaction sequence for this mechanism is
| *COOH + H+ + e− → *CO + H2O |
| *CH3OH + H+ + e− → *CH3 + H2O |
| *CH3COO + H+ + e− → *CH3COOH (pathway 12). |
For the CH3COOH formation route, *CH3 on the active site is a critical intermediate. If dimerization of *CH3 is occurred instead of the reaction with *CO, ethane will be formed.2,60
The formation of C3 products such as propanol61 and acetone62 is also sometimes observed during the CO2RR process although the associated mechanism remains unclear and the FE values for these products are less than 25%. It is assumed that the insertion of C1 species into stabilizing C2 intermediates is an important step in the generation of C3 products such as n-propanol.63 Highly efficient catalysts are needed to improve the selectivity and yields of C3 products.
Theoretical investigations (e.g., Density Function Theory (DFT) calculations) could provide vital support to the experimental results and predicting the product selectivity as well as the mechanistic insights in the case of electrocatalysts.64–68 Garza et al. propose a reaction mechanism for the reduction of CO2 to C2 products over a copper electrode using DFT calculations combined with experimental findings.56 Additionally, theoretical modeling can also accelerate the rational design of catalysts compared to traditional experimental studies. Scaling relationships and some activity descriptors including the d-band center for the transition-metal surface and the valence state of metals have been successfully established,69–71 which correlate the simple parameters of catalysts with complex catalytic performance, lead to develop excellent electrocatalysts. Recently, Wang et al. develop a simple universal descriptor 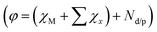 to assess the catalytic performance of 2D material-supported dual-atom catalysts (DACs@2D) for electrochemical reduction based on first-principles calculations, the conservation of the orbital symmetry, and feature engineering via the machine-learning method.72 This descriptor is closely correlated with inherent atomic properties such as electro-negativity (χ), electron type and number (Nd/p). On this basis, CuCr/g-C3N4 for CH4 and CuSn/N-BN for HCOOH with extremely low onset potentials of −0.24 and −0.11 V have been identified.
to assess the catalytic performance of 2D material-supported dual-atom catalysts (DACs@2D) for electrochemical reduction based on first-principles calculations, the conservation of the orbital symmetry, and feature engineering via the machine-learning method.72 This descriptor is closely correlated with inherent atomic properties such as electro-negativity (χ), electron type and number (Nd/p). On this basis, CuCr/g-C3N4 for CH4 and CuSn/N-BN for HCOOH with extremely low onset potentials of −0.24 and −0.11 V have been identified.
2.3 Practical applications
Many products obtained from the CO2RR (such as CO, HCOO−, C2H4, ethanol and propanol) have important usage in modern industrial processes. Consequently, the practical applications of the CO2RR could have significant economic benefits. To thoroughly evaluate the industrial cost of practical products from CO2RR, the involved technologies such as CO2RR process, catalyst preparation, electrolyzer design, and the separation, purification and storage of product have to be taken into consideration. Due to the current CO2RR technology remains far from the level of maturity required for industrialization, we can screen out promising products from CO2RR process for practical application at the present stage. FE is widely considered as the primary standard to assess the practical application potential of CO2RR products.73 An outstanding FE means high selectivity of a specific product, which can significantly decrease the cost of product separation and purification. For a high FE product, its economic benefits can be calculated from the difference between the market price of raw materials and the electricity cost of per mol product.73 As an example, assuming a cost of electricity on an industrial scale of $0.05 kW h−1, the price of ethanol obtained from the electroreduction of CO2 would be on the order of $0.32 L−1,74 which is much less than the fuel ethanol price of $1.51 L−1 in 2022. According to the product selectivity and economic benefits of various CO2RR products, CO, HCOOH, ethanol, and ethene show high potential of industrial application. At the moment, the electroreduction of CO2 to CO is the most promising technology for practical applications and has been demonstrated at the pilot plant level. Haldor Topsoe built the world's first CO2-to-CO plant based on solid oxide electrolysis cells in 2017, capable of producing from 10 to 100 N m3 of gaseous CO per hour with a purity of 99.5%.75 In addition, in 2020, Schmid et al. set up an aqueous CO2-to-CO electrolyzer system with a power rating of approximately 300 W to pursue industrialization and achieved a CO production efficiency above 90% with an operating lifetime of over 1500 h.76 Based on reported CO2-to-CO plant or pilot plant test,58–61 the involved technical indicator for industrial application should be current density ≥500 mA cm−2, FE ≥ 90%, CO concentration in product gas ≥30%, purity of purified CO ≥ 99.5%, cell size in a square-meter range, and stability ≥1000 h. The industrial scale production of other CO2RR products will require significant progress before industrialization but the discovery of more efficient catalysts, electrolytes and electrolyzers could lead to the practical synthesis of these compounds via the CO2RR.
3. Cu-Based electrocatalysts for the CO2RR
3.1 Monometallic Cu
The synthesis of single-carbon products such as CO and CH4, as well as higher value-added C2+ hydrocarbons, alcohols and oxygenates, has attracted much attention. In particular, techno-economic analyses of possible CO2RR products have shown that C2+ products such as C2H4 and ethanol provide a reasonable balance between value and preparation difficulty. Although the synthesis of such compounds shows great potential, the energy conversion and chemical transformation efficiencies of the CO2RR are presently limited by the lack of efficient electrocatalysts. In addition, because of the unique value for the binding energy between the *CO intermediate and Cu, this is the only metal capable of catalyzing C–C coupling to form C2+ products at reasonable rates.77 Unfortunately, polycrystalline Cu is unselective and can generate up to 16 different products, which is extremely inconvenient with regard to the implementation of this process on an industrial scale.78 This section reviews the factors that can potentially affect the catalytic performance of copper. The effects of the Cu structure, including surface crystalline facets and grain boundaries, are initially examined, followed by an assessment of the viability of defect engineering and geometric structure regulation.
3.1.1 Facets.
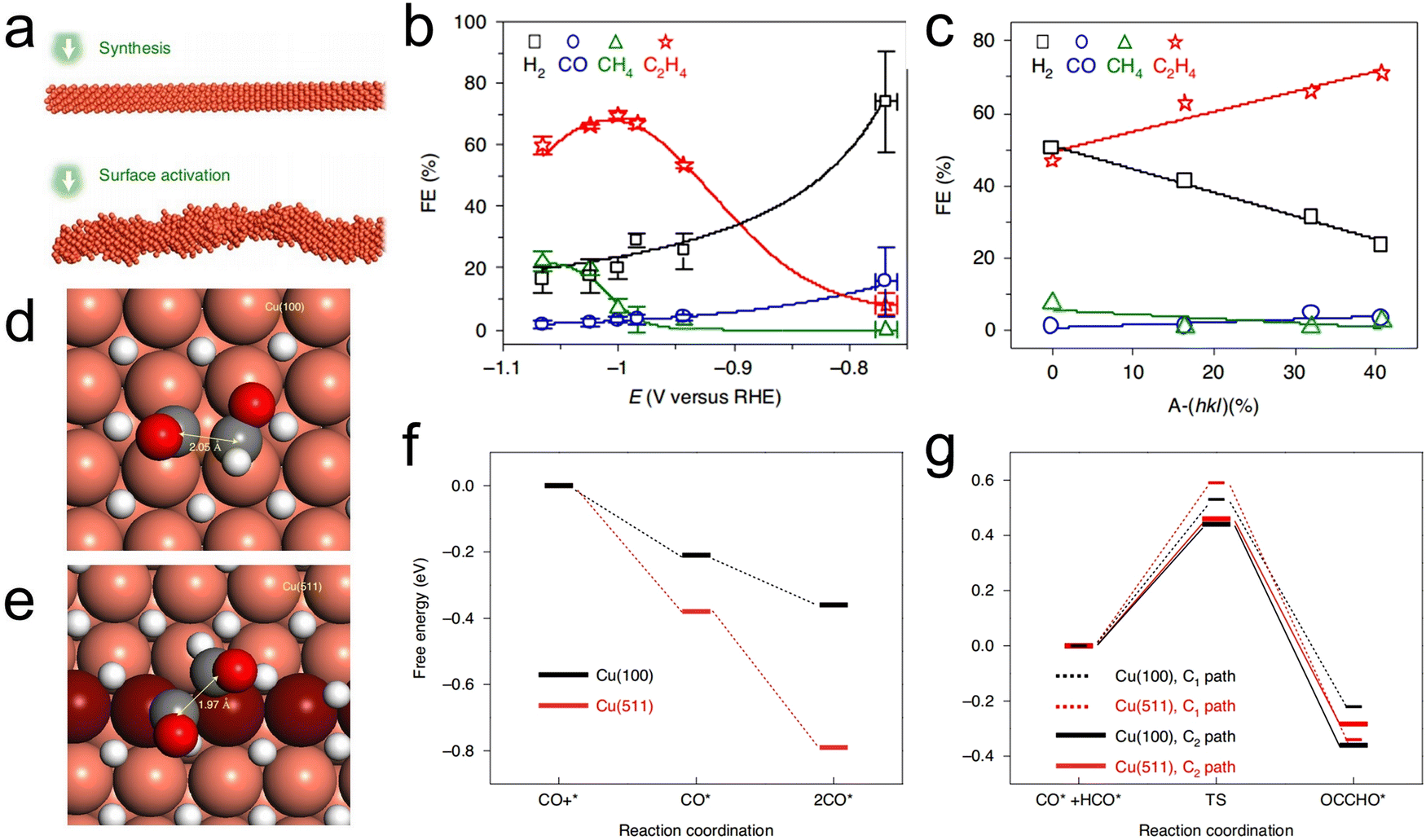
Polished Cu polycrystalline foils having primarily exposed (111) facets have been shown to catalyze C–C coupling for the formation of C2+ products. However, the associated FE values are often much lower than those for the generation of C1 products such as HCOOH, CO and CH4.28 Analyses of the structure-property relationships of single crystals have established that the selectivity of a Cu catalyst is determined by the degree of crystallinity of the Cu. Early work by Hori comprised the systematic assessment of the performance of 19 single-crystal Cu surfaces at different applied potentials and demonstrated the structure-sensitive nature of the CO2RR.79 Specifically, the reduction of CO2 at Cu(100) facets affords C2H4 instead of CH4 whereas oxygenated hydrocarbons such as acetaldehyde, ethanol and acetic acid are the primary compounds obtained from Cu(110).54,79 Wang et al. studied the effects of particular Cu facets on the initial C–C coupling steps during CO2 reduction using DFT calculations and suggested that both Cu(100) and stepped facets favor C2+ product formation compared with Cu(111).80 Wang's group also developed a metal ion cycling method to synthesize single crystalline Cu2O nanocubes having primarily Cu2O(100) facets. These oxide nanocubes could be subsequently reduced to polycrystalline Cu nanocubes with preferentially exposed Cu(100) facets for efficient C–C coupling. Sargent et al. proposed a strategy based on the in situ electrodeposition of Cu under CO2 reduction conditions that preferentially exposed and maintained Cu(100) facets and therefore favored the formation of C2+ products.81 More recently, Gong et al. described the effect of the facets of Cu crystals derived from Cu(OH)2, CuO or Cu2O as precursors on the CO2RR.82 The Cu catalysts obtained from Cu(OH)2 had relatively high densities of exposed Cu(110) and Cu(100) steps assembled into Cu(210) and Cu(310) facets. These materials also exhibited improved activity during the CO2RR to generate C2+ products based on the promotion of CO adsorption and C–C coupling. Consequently, superior selectivity for C2+ products, a high FE of 87% and a large partial current density of 217 mA cm−2 for C2+ products have been achieved at a voltage of only −0.54 V vs. a reversible hydrogen electrode (RHE; the reference for all potentials in this article unless otherwise specified) in a flow-cell electrolyzer in alkaline aqueous solutions.
Compared with Cu(100) facets, the high-index Cu(511), Cu(711), Cu(911), Cu(11,1,1), Cu(310), Cu(510), Cu(610) and Cu(810) facets provide even higher C2H4-to-CH4 ratios with greater overall C2+ selectivity.83–85 These high-index facets can be visualized as combinations of terraces and steps on low-index facets that maximize the contribution of active sites. Work with such materials provides a means of assessing the synergistic effects between different facets and eventually improving the selectivity beyond theoretically predicted limitations.85 Huang et al. reported the preparation of activated Cu nanowires (A-CuNWs) with highly active stepped surfaces (assigned to A-(hkl)) through an in situ electrochemical activation process (Fig. 2a). These materials were found to contain increasing proportions of A-(hkl) after prolonged activation durations, suggesting that the {100} and {110} facets expressed on the synthesized CuNWs surfaces were gradually transformed into the higher-energy A-(hkl) surface structures during the electrochemical activation process. As the proportion of the stepped A-(hkl) surfaces was gradually increased from 0 to 40.68%, the FE for C2H4 production underwent a corresponding increase from 47.04% to 71.19% (Fig. 2b and c). These A-CuNWs also demonstrated exceptionally high stability over a time span of approximately 200 h.84 DFT calculations demonstrated that the thermodynamically favored Cu(511) plane [3(100) × (111)] stepped surface provided a higher energy barrier for the C1 path and also slowed the hydrogen evolution reaction (HER) such that the selectivity for C2H4 was greatly increased (Fig. 2d–g). Guo et al. very recently investigated the C–C coupling process on Cu stepped surfaces based on ab initio molecular dynamics (AIMD) and density functional theory (DFT) calculations and established the nature of CO–CO adsorption sites, indicating that the high selectivity for C2+ products could ascribe to the high-index facet.86
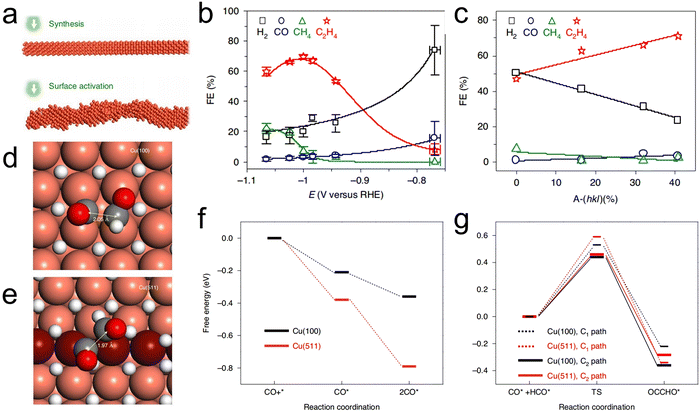 |
| | Fig. 2 (a) A diagram showing the preparation of Cu NWs with surface activation step, (b) the FE of A-CuNW at various potential, (c) the correlations between FE values and A-(hkl) over the approximate range of −0.99 to −1.00V (vs. RHE). Transition states determined for the C2 pathway on (d) Cu(100) and (e) Cu(511). (f) CO and 2CO adsorption energies (ΔGads) on Cu(100) and Cu(511) and (g) C1 and C2 pathways on Cu(100) and Cu(511). Reproduction with permission from ref. 84, Copyright 2020, Springer Nature. | |
3.1.2 Grain boundary engineering.
Grain boundary (GB) engineering is another important strategy for enhancing the catalytic activity during the CO2RR. In 2012, Li et al. reported the application of Cu electrodes prepared by the electrochemical reduction of oxidized Cu foil (OD-Cu) for CO2 reduction.87 An OD-Cu-500 (annealed at 500 °C) specimen was found to provide peak FE values of approximately 45% and 38% at approximately −0.3 and −0.5 V with regard to CO and HCOO− production, respectively. At overpotentials higher than −0.5 V, both FEs decreased dramatically and were essentially nil at −1.0 V. Simultaneously, the selectivity for hydrocarbon products slowly increased, although only C2H4 and ethane were obtained, both with FE values of less than 10%. As a consequence of such studies, there has been much interest in determining the mechanisms responsible for the enhanced reduction currents and FE values associated with nanostructured Cu electrodes.
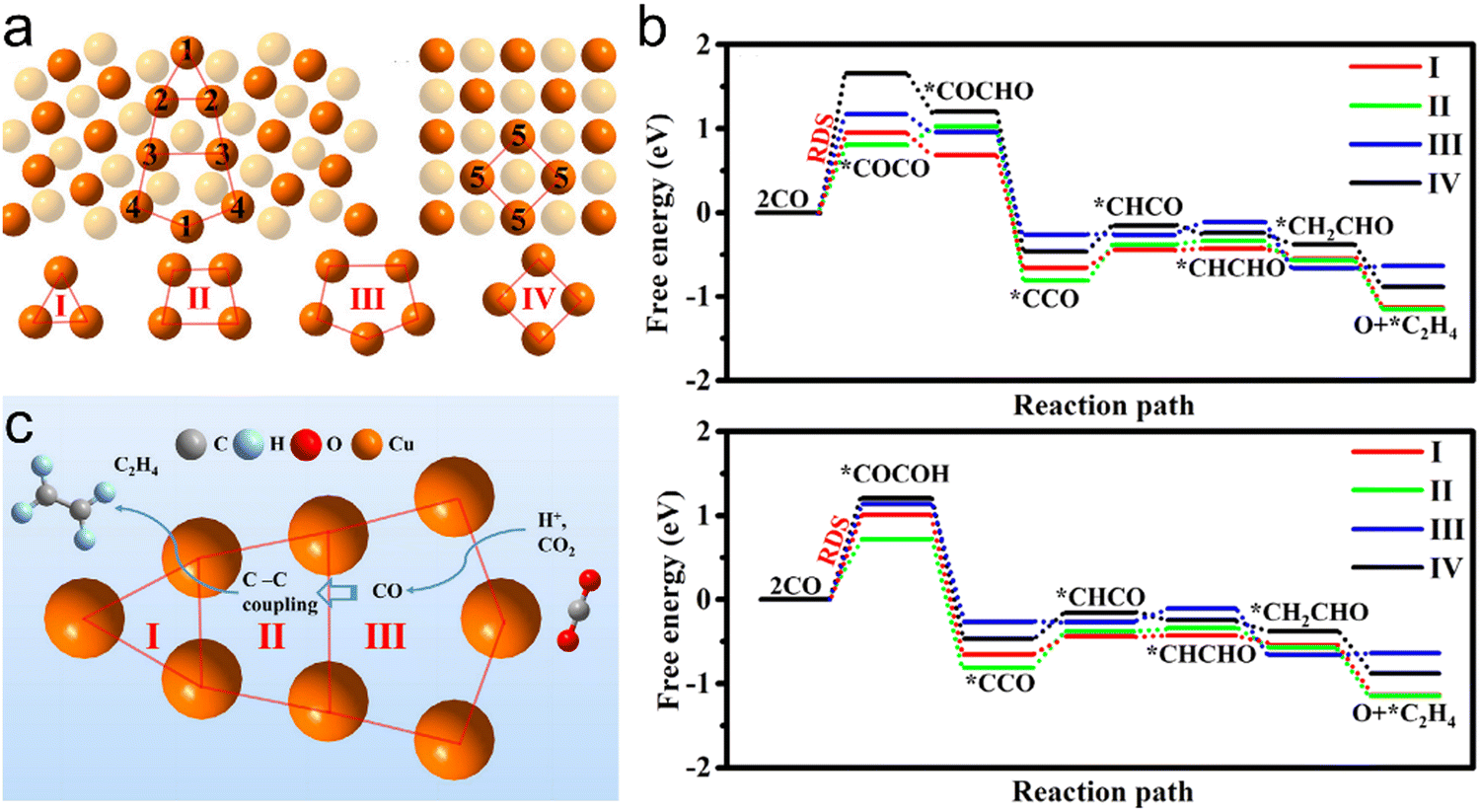
Subsequent studies determined that OD-Cu electrodes possess abundant GBs and identified a linear correlation between GB density and CO2RR performance.88,89 Raciti and Wang reported systematic investigations of high density Cu nanowires applied to the CO2RR.90,91 Electrocatalytic experiments showed that these materials were highly active during the electrochemical reduction of CO2 and selectively generated CO at low overpotentials (more positive than −0.5 V) but C2 species (ethane, C2H4 and ethanol) at more negative potentials. Interestingly, positive and negative correlations were observed between catalytic performance and high-angle and coherent GBs, respectively. Gong et al. generated realistic OD-Cu surface models by simulating the “oxide-derived” process based on molecular dynamics with global neural network potential coupled as well as density functional theory calculations and experimental verification.92 This work identified three square-shaped sites that were likely to be responsible for C–C coupling. Among these, planar-square and convex-square sites associated with Σ3 GBs were found to be responsible for C2H4 production while step-square sites (i.e. n(111) × (100)) favored the synthesis of alcohols.
Liu et al. established that dual catalytic pathways on adjacent active motifs of Cu GBs were responsible for enhanced C2+ production using first principles calculations.93Fig. 3a presents GB models with Cu(100) facets built according to the coincidence site lattice theory. In these models, the d-band center of GB sites is closer to the Fermi level than that of Cu(100) facets and the Cu atoms at GBs have shorter bond lengths and stronger bonding with *CO. This would be expected to enhance the adsorption of *CO at GBs. The pathways for the two different C–C coupling modes to produce C2H4 (Fig. 3b) indicate that the CO2RR at GBs does indeed proceed via a tandem reaction mechanism (Fig. 3c). This work provided an improved understanding of the CO2RR at GB surfaces and suggested a new approach to overcoming the limitations of the structure-performance relationship of single sites on Cu(100) facets.
 |
| | Fig. 3 (a) Cu atom structures of R5{021}/[100]GB and (100) facets, (b) the reaction pathways from CO2 to C2H4 based on (top) C–C coupling via *COCO as an intermediate and (bottom) C–C coupling via *COCOH as an intermediate. (c) Diagram of the cascade reaction of C2H4 between different sites on a GB surface. Reproduction with permission from ref. 93, Copyright 2022, Elsevier. | |
3.1.3 Defect engineering.
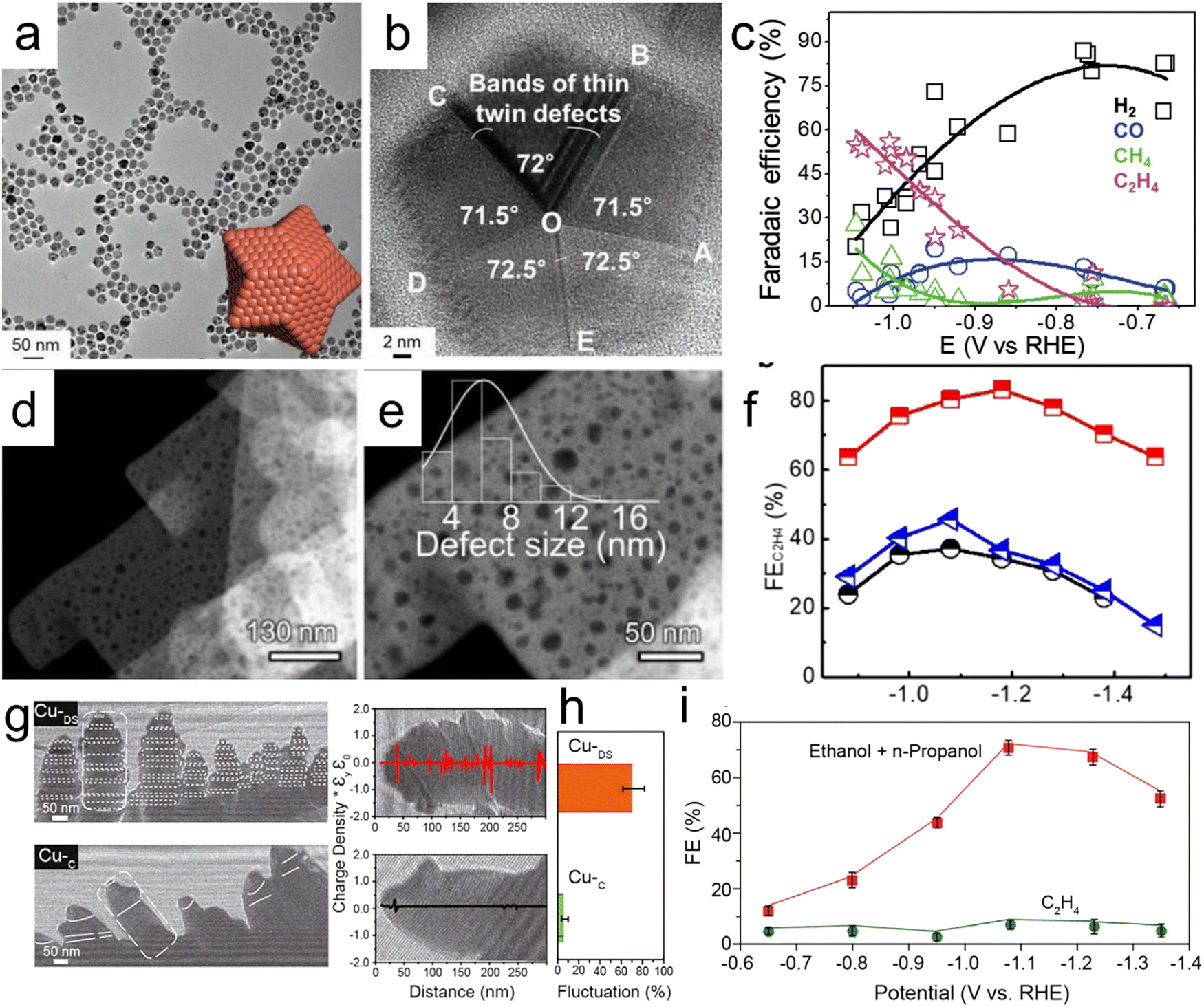
Defect engineering based on twin boundaries, stacking faults and stepped sites can be used to optimize the adsorption, enrichment and confinement of reaction intermediates and hydroxyl ions on electrocatalysts to improve electrocatalytic performance.94–96 Choi et al. reported the high-yield synthesis of star-shaped decahedron Cu nanocrystals having high densities of twin boundaries and stacking faults (Fig. 4a and b).94 The presence of these crystal defects evidently increases the *CO binding energy but significantly decrease the energy barrier to the formation of *CHO, a common intermediate for both CH4 and C2H4 generation. Consequently, multiple-twinned nanocrystals displayed a low overpotential for CH4 synthesis, such that the onset potential was lowered by 0.149 V relative to the value obtained using polycrystalline commercial Cu nanoparticles. In addition, high selectivity for C2H4 production was observed, with an FE of 52.43% at −0.993 V vs. RHE (Fig. 4c). Zhang et al. designed Cu nanosheets having nano-scaled defects with sizes of 2–14 nm for the electrochemical production of C2H4 from carbon dioxide.95 As shown in Fig. 4d and the enlarged HAADF-STEM image in Fig. 4e, each nanosheet contained numerous pits. On the basis of the high density of atomic defects that concentrated crucial adsorbates (*CO, *OCCO and OH−) required for C–C coupling, nanodefective Cu nanosheets provided a record C2H4 FE of 83.2% and a high current density of −60 mA cm−2 at −1.18 V vs. RHE (Fig. 4f).
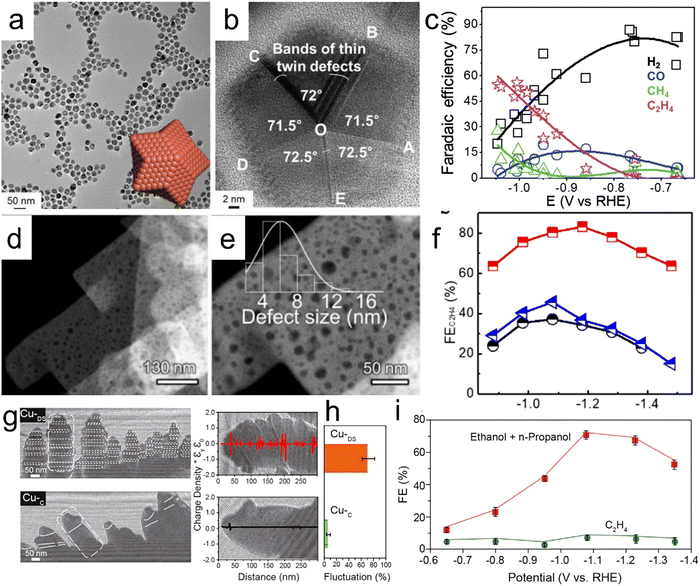 |
| | Fig. 4 (a) Low-magnification TEM image of SD-Cu NPs (the inset shows a theoretical atomic structure), (b) an HRTEM image of SD-Cu NPs demonstrating fivefold twin symmetry, (c) FE values of SD-Cu NPs as functions of E, (d and e) HAADF-STEM images of a n-Cu NS sample (the inset in (e) shows the size distribution of the nano-defects on the material), (f) C2H4 FE values at various applied potentials for n-CuNS, CuNS and CuNP, (g) electron holographs of Cu-DS and Cu-C, (h) charge density signal fluctuations and (i) FE values for Cu-DS and Cu-C under different potentials. Reproduction with permission from ref. 94, Copyright 2019, Wiley-VCH and Reproduction with permission from ref. 95, Copyright 2020, American Chemical Society and Wiley-VCH and Reproduction with permission from ref. 96, Copyright 2021, Cell Press. | |
Interestingly, CO-rich environments have been used to construct Cu catalysts with stepped sites that result in high surface coverages of *CO intermediates and bridge-bound *CO adsorption. These effects, in turn, trigger CO2 reduction pathways that form alcohols rather than C2H4. A FE value of 70% and high current densities over 100 mA cm−2 during the synthesis of C2+ alcohols in an H-cell system were obtained on this basis (Fig. 4g–i).96
3.1.4 Geometric structure regulation.
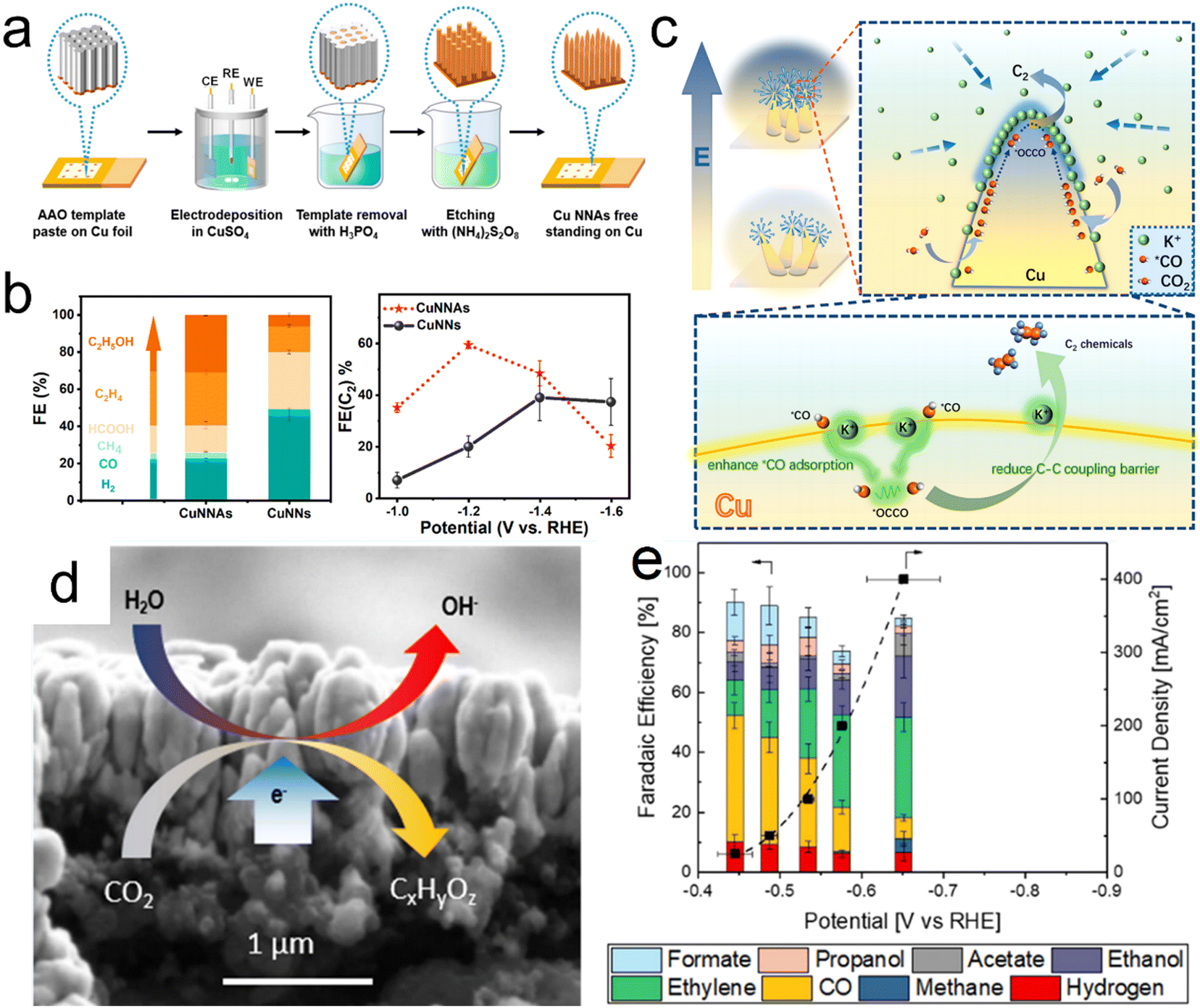
The efficiency with which multi-carbon products are obtained using Cu-based catalysts can be improved by optimizing *CO adsorption and reducing the energy barrier for C–C coupling. Liu et al. reported that a strong local electric field can be obtained by regulating the arrangement of Cu nanoneedle arrays (CuNNAs).97 According to finite element simulations and the results of DFT calculations, CuNNAs with vertically ordered tip arrangements should exhibit a stronger tip local electric field compared with randomly distributed nanoneedles (CuNNs), leading to localized K+ accumulation and stronger *CO adsorption, thus reducing the C–C coupling energy barrier. On the basis of these theoretical predictions, the same group prepared vertically ordered CuNNAs and randomly disordered CuNNs on Cu foil by template-assisted electrodeposition (Fig. 5a). CO2 reduction performance tests indicated that the CuNNAs provided an FE of 59% for multi-carbon products at −1.2 V vs RHE compared with 20% for the CuNNs (Fig. 5b). The very high localized electric fields produced by the ordered CuNN arrays evidently promoted the accumulation of K+ ions and this effect enhanced both *CO adsorption and C–C coupling (Fig. 5c). Biener et al. demonstrated that Cu catalysts synthesized by electron beam (EB) exhibit excellent current densities, selectivities and energy efficiencies. This superior performance can be ascribed to the faceted surface morphologies and narrow Cu/gas diffusion layer interfaces of such materials, which increase their hydrophobicity (Fig. 5d and e).98
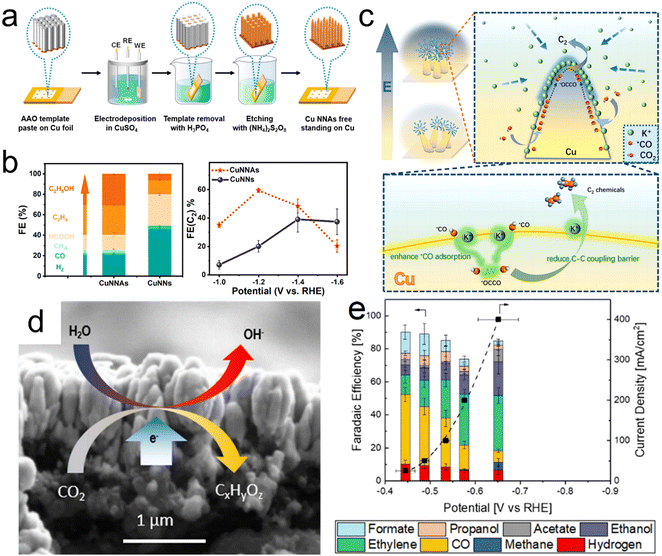 |
| | Fig. 5 (a) Diagram showing the synthesis of CuNNAs, (b) product distributions obtained from CuNNAs and CuNNs at −1.2 V vs. RHE together with FE(C2+) values at different potentials, (c) diagram showing C2+ formation on the single tip of a CuNNA. (d and e) Electron beam image of a Cu catalyst and corresponding FE values at different potentials. Reproduction with permission from ref. 97 and 98, Copyright 2022, American Chemical Society. | |
3.1.5 Cu clusters and amorphous nanoparticles.
Atomically-precise Cu clusters with high surface/volume ratios and more active sites have attracted attention with regard to the CO2RR process.99–101 Dong et al. reported that Cu79 clusters (where the subscript indicates the number of Cu atoms in the cluster) provide a lower energy barrier for CO2 reduction to CO than the values for bulk Cu (111) and (211) surfaces.99 Similarly, Cu20 clusters show superior selectivity during the CO2RR with an improved ability to prevent the HER. The Cu atoms at the vertices and edges on Cu20 cluster surfaces are potential active sites for CO2 reduction to HCOOH.100 Very recently, Zang et al. developed structurally precise Cu8 cluster isomers having different core structures (either cubes or ditetrahedra). The ditetrahedron-shaped Cu8 clusters exhibited a high FEHCOOH of approximately 92% at −1.0 V.101
Amorphous NPs possess low-coordinated surface atoms with dangling bonds that can serve as reactive sites for catalysis.102,103 Yan et al. achieved the first-ever tunable synthesis of either amorphous or crystalline Cu nanoparticles (having sizes on the order of 3 nm) in a similar reaction environment and explored their catalytic performance during the CO2RR.103 The amorphous Cu NPs exhibited superior CO2RR performance, achieving a total FE for liquid fuels of 59% at −1.4 V, with HCOOH and C2H6O accounting for 37% and 22%, respectively, of the total products. The superior performance of amorphous Cu can be ascribed to the larger electrochemical active surface area (ECSA) of this material along with enhanced CO2 adsorption. The reactive sites provided by regularly arranged atoms with short-range order are capable of binding and stabilizing the *CO intermediate and so facilitate the production of liquid fuels. This work suggested new techniques for improving the electroreduction of CO2 based on the use of amorphous metal catalysts.
3.2 Cu-Based oxides
The oxidation state of the Cu in a catalyst can affect the activity and selectivity of the material during the CO2RR by promoting reactant activation, regulating the adsorption of intermediates and facilitating the C–C coupling step.104 Using DFT calculations, Goddard and co-workers showed that the synergy between Cu+ and Cu0 can promote CO2 activation.105 Specifically, the C atom of CO absorbed at a Cu+ site is positively charged whereas the C atom at a Cu0 site will be negatively charged because of the back donation effect. Sargent and co-workers tuned the Cu oxidation state from −0.1 to +0.3 and found that the CO adsorption energy increased monotonically with increases in the oxidation number.106 DFT calculations by the same group established that the presence of Cu+ species favors dimerization.107
Over the past several years, several efficient Cu-based oxide catalysts have been developed that effectively inhibit the HER while improving both selectivity and reactivity during the CO2RR. The oxidation state of the metal can affect the intrinsic performance of such catalysts by affecting structure and other properties, including spin state, work function, active sites and energy band structure.104 Kanan et al. showed that the pre-oxidation of Cu can greatly increase its ability to promote C2+ formation.87 Yu et al. also found that oxygen in OD-Cu catalysts plays a critical role in CO adsorption and dimerization.108 DFT calculations indicated that oxygen on the Cu surface promotes both CO adsorption and dimerization and so enhances the C–C coupling reaction. Following this work, various OD-Cu catalysts were developed by generating Cu structures using an anodic treatment109 or O2 plasma and these materials exhibited outstanding CO2RR activity.110
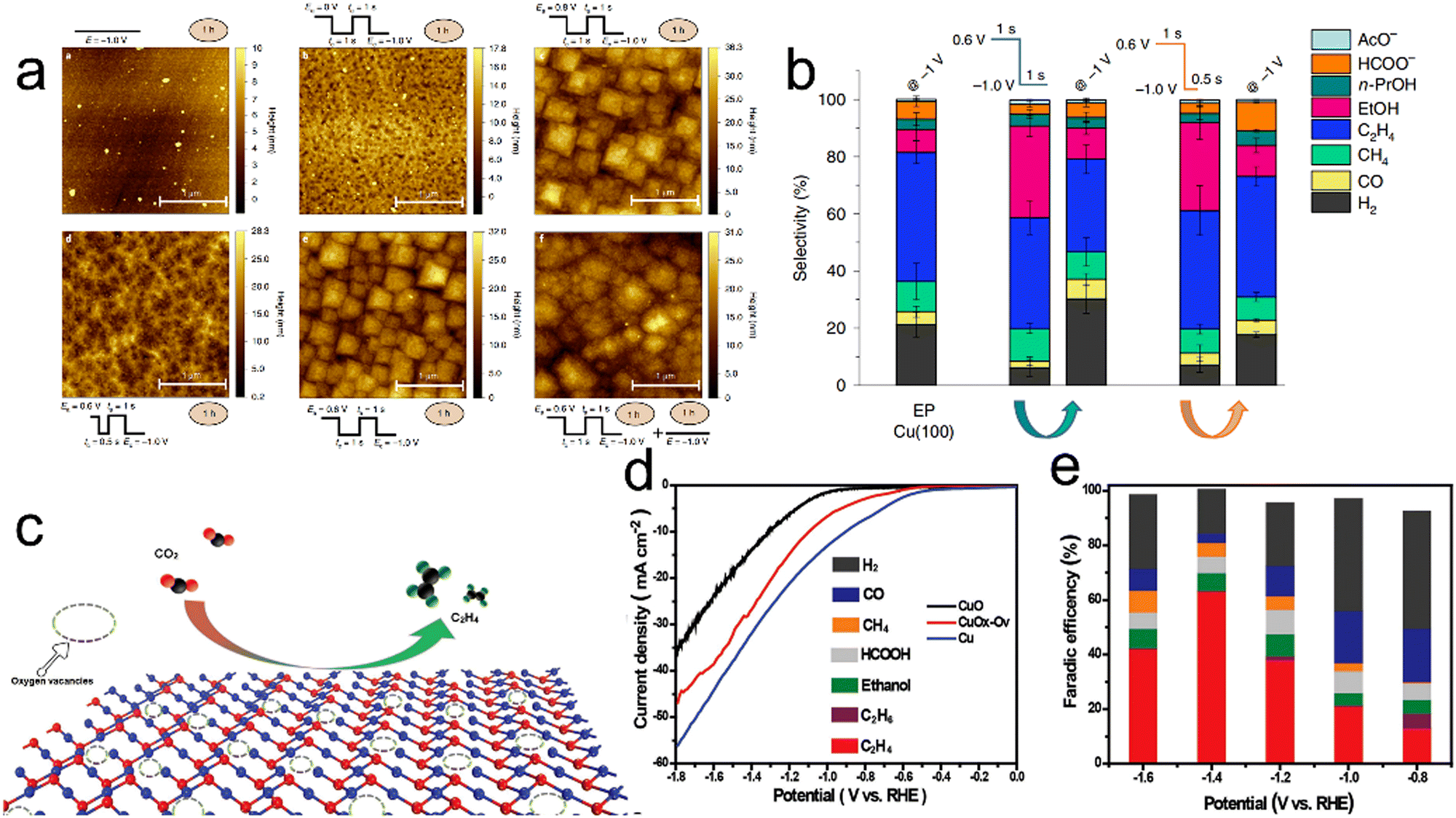
It should be noted that tuning the relative proportions of Cu0 and Cu+ species is also a useful approach to obtaining better CO2RR performance. Wu and co-workers synthesized three Cu electrodes with adjustable oxidation states of either Cu+ or Cu0 based on electrochemical deposition.111 A Cu electrode subjected to cyclic voltammetry (CV) was found to contain both Cu0 and Cu+ species that synergistically catalyzed CO2 reduction to generate C2H4 as a consequence of the dimerization of CO. This electrode exhibited a 40% FE for C2H4 at −1 V versus RHE. As shown in Fig. 6a, the relative proportions of Cu0 and Cu+ present during the CO2RR can be tuned by regulating the anodic pulse potential (Ea) and anodic pulse time (ta).112 Using Ea = 0.6 V versus RHE and ta = 1 s, Cu2O species were formed on a Cu surface at a proportion of 16% and remained at a proportion of 7% throughout the CO2RR. under these pulsed conditions, the Cu catalyst was able to convert CO2 to C2+ products with an FE of 76% at −1.0 V vs. RHE (Fig. 6b). Oxygen vacancies have also been demonstrated to act as catalytic sites capable of promoting CO2 activation and C1 adsorption to generate C2+ products. In a prior study, a Cu oxide with surface oxygen vacancies created by electrochemical reduction showed a FE of approximately 63% for the conversion of CO2 to C2H4 (Fig. 6d and e).113
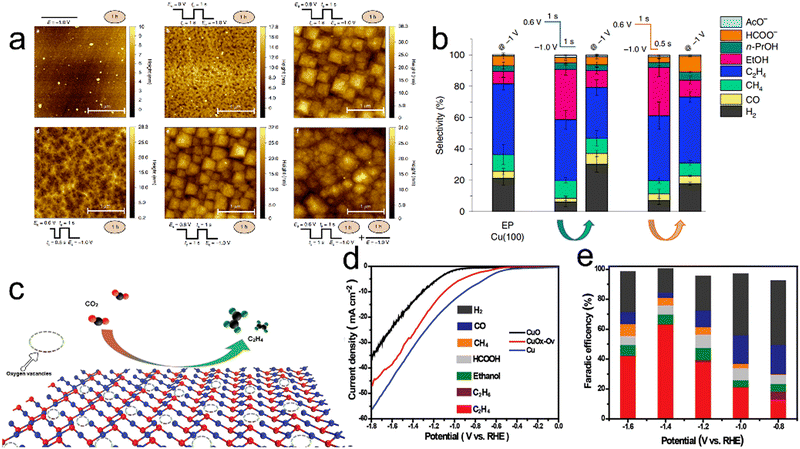 |
| | Fig. 6 (a) Atomic force microscopy images of a Cu(100) electrode after different surface treatments and reaction settings, (b) product selectivity of the aforementioned surfaces under potentiostatic (−1.0V) or pulsed conditions (product selectivity for tc = 0.5s is also included for comparison). The selectivity data reported here are averages of at least three different measurements and the error is given as the standard deviation. (c) Diagram showing the electrocatalytic reduction of CO2 to C2H4 on a Vo-rich CuOx–Vo surface, (d) LSV curves for CuOx–Vo, CuO and Cu in a CO2-saturated 0.1 M KHCO3 electrolyte and (e) FE values of CuOx–Vo, at various potentials in CO2-saturated 0.1 M KHCO3. Reproduction with permission from ref. 112, Copyright 2020, Springer Nature and Reproduction with permission from ref. 113, Copyright 2019, Wiley-VCH. | |
The above results indicate that the presence of Cu+ promotes the formation of C2+ products although it is not yet clear if the active oxide phase is present at the catalyst surface under the harsh CO2RR conditions. Strasser et al. studied the synthesis of a (001)-oriented CuO nanosheet CO2RR catalyst.114 Operando X-ray absorption spectroscopy (XAS) studies confirmed the chemical reduction of CuO and concomitant formation of disordered and coordinatively under-saturated Cu0 over a duration of approximately 2 h using reductive CO2RR conditions. These undercoordinated sites were thought to be responsible for the high C2+ production rates over this material. Using an electrochemical flow cell that allowed for in situ X-ray absorption spectroscopy (GIXAS) and X-ray diffraction (GIXRD) with improved CO2 mass transfer, Drisdell et al. successfully demonstrated that surface Cu oxide was reduced to metallic Cu prior to the onset of the CO2RR and that metallic Cu was the only detectable phase during the reaction.115 Very recently, Cui et al. combined in situ Raman spectroscopy, secondary ion mass spectrometry and isotope-labelling to demonstrate a “seesaw-effect” between the cathodic reduction and reoxidation induced by OH˙ radicals and determine the chemical state and proportion of Cuδ+ species during the CO2RR.116 It is evident that the evolution and preservation of Cuδ+ species during the CO2RR should be studied in more detail in future work.
To date, various strategies have been developed to maintain the Cu valence state during the CO2RR, including the use of heteroatoms,106 interface engineering117,118 and coordination polymers.119 Sargent and co-workers introduced boron as a dopant to tune the ratio of Cuδ+ to Cu0 active sites and achieved an FE of approximately 80% for C2 products at −1.1 V vs. RHE.106 In addition, Yan et al. reported that the Cu oxidization state can be stabilized by introducing strong electronic interactions to suppress electron accumulation around the Cu+ sites.118 Yan's group proposed a model catalyst based on depositing Cu2O nanoparticles onto hexagonal boron nitride (h-BN) nanosheets, and this material showed a 1.62-fold larger C2H4/CO ratio compared with bare Cu2O. Theoretical calculations indicated that electrophilic h-BN received some electron density from Cu2O and that this phenomenon strengthened the Cu–O bonds and stabilized the Cu+ species in the catalyst.
3.3 Cu-Based compounds
Cu-containing compounds such as sulfides, phosphides and selenides contain modified active Cu sites and so often exhibit outstanding CO2RR catalysis.63,120–125 Hod et al. developed a Cu2S catalyst for the CO2RR using a cation change method (Fig. 7a).121 The presence of S heteroatoms in this material changed the electronic structure of adjacent Cu sites, resulting in the presence of oxidized Cu+ sites based on the higher electronegativity of S. These positively charged Cu sites were able to bind CO2 to relatively electronegative oxygen, leading to the formation of *OCHO serving as the primary intermediate for the production of HCOO−. The sulfur-modified Cu catalyst provided HCOOH in 0.1 M NaHCO3 with an FE as high as 87.3% and record-high activity at –0.9 V vs. RHE (Fig. 7b). Sargent and coworkers reported the use of DFT calculations to show that the application of modified Cu2S cores having Cu surface vacancies could cause the C2+ reaction pathway to transition from the formation of C2H4 to the generation of multi-carbon alcohols.122 This same group synthesized a Cu2S–Cu–V (where V denotes a vacancy) nanoparticle structure based on the controllable introduction of vacancies on a Cu surface shell having a Cu sulfide core, leading to FE values for C3H7OH and C2H5OH of 8 ± 0.7% and 15 ± 1% with partial current densities of 2.5 ± 0.1 and 4.8 ± 0.1 mA cm−2 at –0.95 V vs. RHE, respectively.
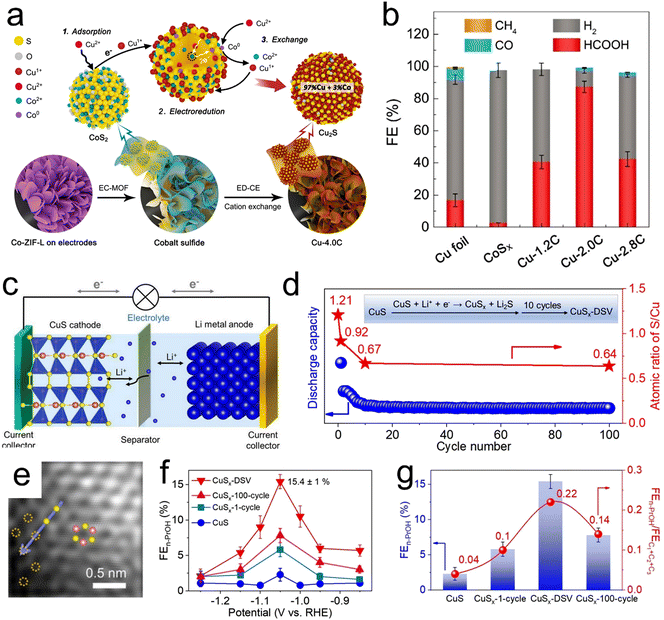 |
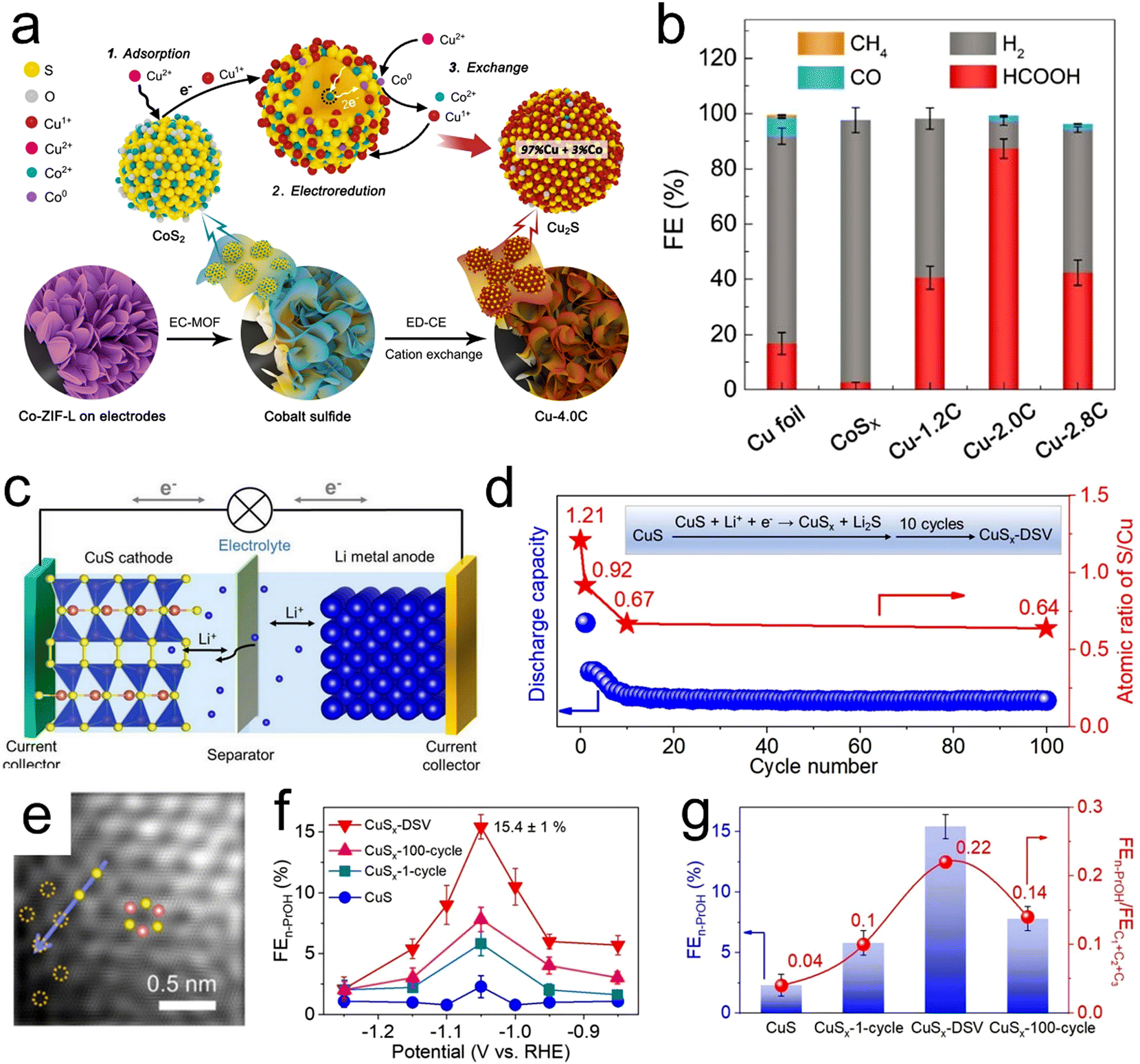
| | Fig. 7 (a) Diagram of the experimental paths and mechanisms for electrochemically-driven cation exchange, (b) FE values for CO2RR products using Cu foil, CoSx, Cu-1.2C, Cu-2.0C and Cu-2.8C at −0.9 V (vs. RHE). (c) Diagram of a lithium-ion battery assembled with CuS (cathode) and Li metal (anode), (d) discharge capacities of CuS (blue dots) and the S/Cu atomic ratios (red stars) with respect to the cycle number at a constant current of 0.044 mA cm−2 in the voltage range of 0.01–3 V. Sulfur atoms in the CuS lattice were selectively removed to form Li2S, resulting in CuSx-DSV. (e) HAADF-STEM image of CuSx-DSV and (f and g) FE and FEn-PrOH values together with FEn-PrOH/FEC1+C2+C3 ratios for n-propanol on four catalysts (CuS, CuSX-1-cycle, CuSX-DSV and CuSX-100-cycle) at different applied potentials. Reproduction with permission from ref. 121, Copyright 2020, Wiley-VCH and Reproduction with permission from ref. 63, Copyright 2021, Springer Nature. | |
In addition, CuS has been reported to electrochemically reduce CO2 to provide HCOOH (FE = approximately 60%)123 or CH4 (FE = approximately 73%) as the major product.124 The selectivity of this material can be tuned based on its morphology and the local electronic structures of Cu atoms around sulfur atoms. Recently, Zheng et al. synthesized CuS with double sulfur vacancies using an electrochemical lithium tuning strategy (Fig. 7c–e). This material enabled the stabilization of *CO and *OCCO dimers and CO–OCCO coupling to form the key *C2+ intermediate for n-propanol.63 As shown in Fig. 7f and g, the FE for n-propanol production was increased to 15.4% in H-cells and the partial current density for n-propanol production was further increased to 9.9 mA cm−2 in flow cells. This value was comparable to the performance reported for prior optimized electrochemical CO2RR systems.
Transition metal selenides have also been widely applied as electrocatalysts owing to their low cost and unique physicochemical properties. Han et al. provided the first report that Cu selenides exhibit outstanding performance during the conversion of CO2 to methanol with a high FE of 77.6% and a current density of 41.5 mA cm−2 at a low overpotential of 285 mV.126 Hu et al. found that the bimetallic compound CuInSe2 had Au-like catalytic properties with good CO2RR and poor HER activity and could serve as an electrocatalyst for the highly selective CO2RR to give CO with much greater efficiency than monometallic selenides.127 Se vacancies evidently promoted the delocalization of electrons in this material and further optimized the CO2RR to give an FE for CO production of 91% in aqueous solutions.
Dismukes et al. reported the synthesis, surface structure, electronic structure and catalytic activity of highly crystalline single-phase Cu3P (on which [00I] facets were primarily exposed) when applied to the electrochemical reduction of CO2 to formic acid.125 It was suggested that the Cu+ oxidation state was insufficient to achieve high CO2RR activity and that close multi-Cu sites were essential to produce C2 or larger products. The formation of a surface hydride at isolated *H-CuP3 sites was thought to provide catalytic sites forming both H2 and HCOO− while the long Cu–Cu bonds inhibited the formation of C–C coupling products.
Most recently, Qiao et al. have reported reliable ampere-level CO2-to-C2+ electrolysis on heteroatom engineering on Cu catalysts.128 Among various Cu-based compounds with heteroatom (N, P, S, O), N-engineered Cu (N–Cu) catalyst exhibits the best CO2-to-C2+ productivity, it achieves a C2+ partial current density of −909 mA cm−2 at −1.15 V, outperforms most reported Cu-based catalysts. Combined with in situ spectroscopy and density functional theory studies, indicating that the high adsorption strength of *CO on N–Cu results from the depressed HER and promoted *CO adsorption on both bridge and atop sites of Cu, which greatly reduces the energy barrier for C–C coupling.
3.4 Cu-Based bimetallic materials
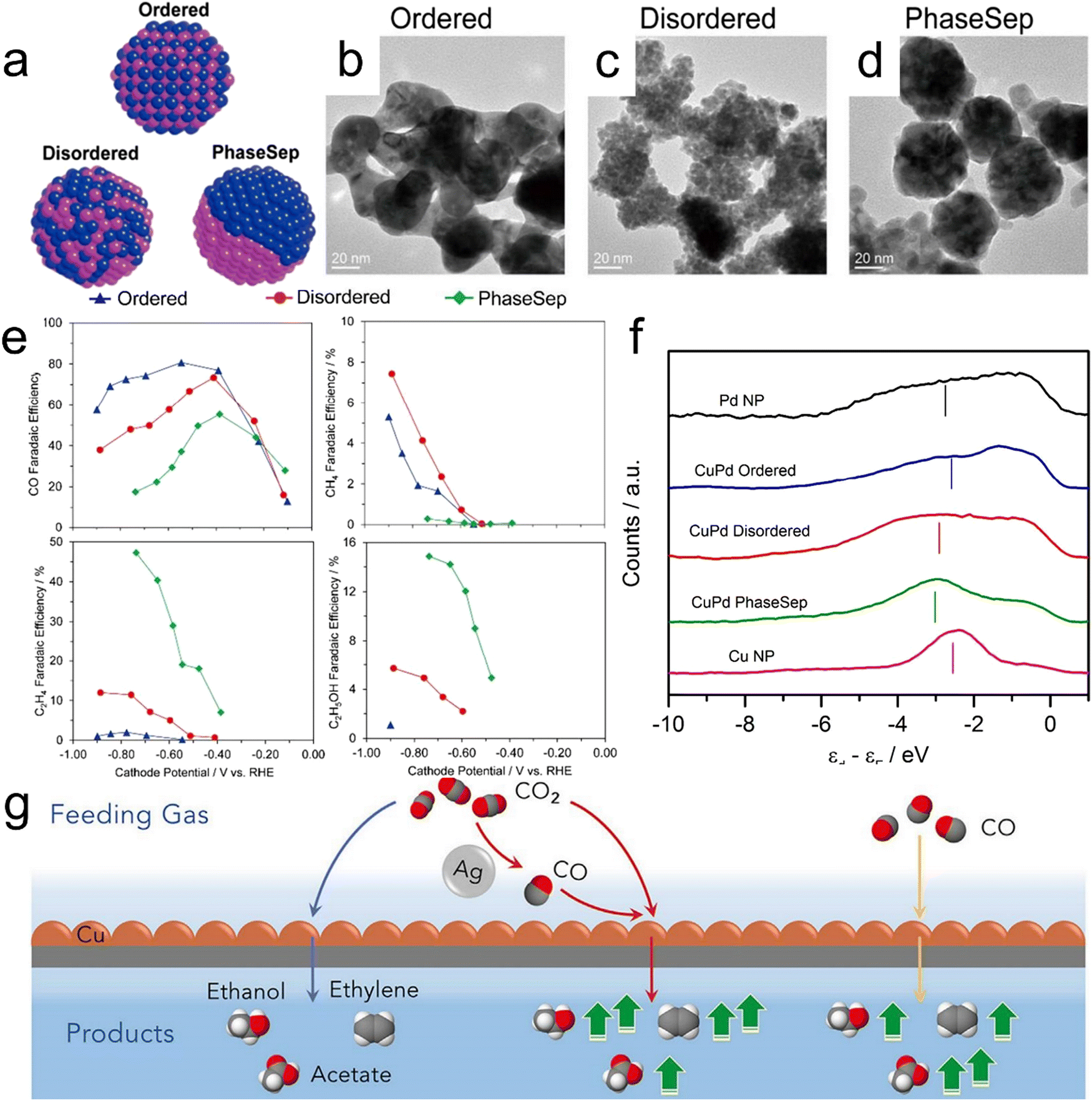
Cu-Based bimetallic compounds have emerged as another class of CO2RR electrocatalysts. The presence of a secondary metal near Cu can promote the efficiency of CO spillover and induce localized increases in interatomic distances. In addition, the ability to adjust the binding energies of adsorbates and intermediates can allow the formation of C2+ liquid products.129 Cu-based bimetallic catalysts such as Cu–Pd, Cu–Au, Cu–Ag, Cu–Co and Cu–Zn alloys have been developed and demonstrated to show promise activity for the CO2RR.130–134 In prior work, Kenis et al. synthesized bimetallic Cu–Pd catalysts with different elemental mixing patterns, such as ordered, disordered and phase-separated. Diagrams of these materials and corresponding high-resolution TEM images are presented in Fig. 8a–d.130 A specimen having neighboring Cu atoms (that is, a phase-separated material) favored the production of C2+ products while that featuring an alternating Cu–Pd arrangement (a disordered structure) promoted the formation of CH4 (Fig. 8e). Fig. 8f confirms that phase-separated Cu–Pd will have the lowest d-band center while Cu nanoparticles will have the highest. Even so, phase separated Cu–Pd and Cu nanoparticles show similar catalytic selectivity and activity, indicating that geometric/structural effects play a more important role in catalytic selectivity and activity than electronic effects.
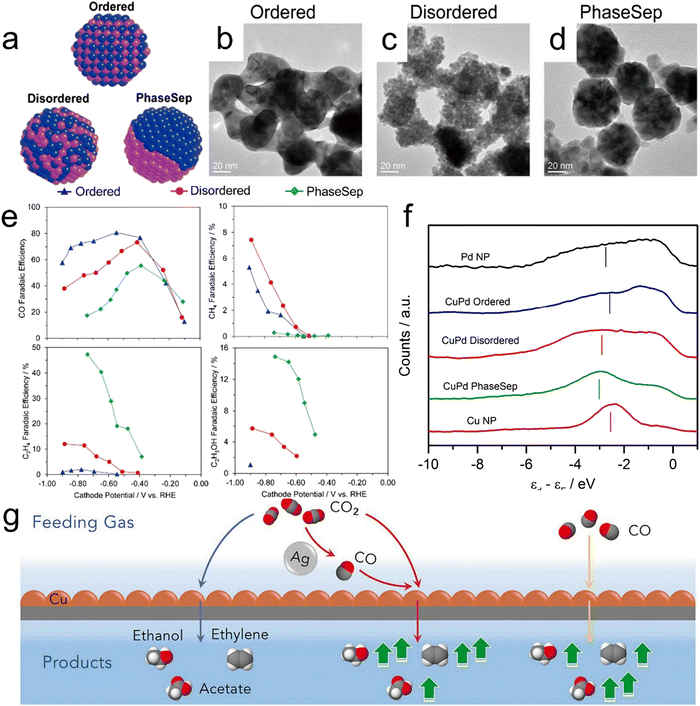 |
| | Fig. 8 (a) Diagrams of CuPd nanoalloys with different structures, (b–d) high-resolution TEM images of CuPd nanoalloys, (e) faradaic efficiencies for CO, CH4, C2H4 and C2H5OH obtained using bimetallic Cu–Pd catalysts with different mixing patterns, (f) surface valence band photoemission spectra of CuPd nanoalloys relative to the Fermi level. (g) A diagram summarizing the catalytic systems provided by the local environments produced by a Cu–Ag tandem catalyst (red) and the standard CORR (yellow) or CO2RR (blue). Reproduction with permission from ref. 130, Copyright 2017, American Chemical Society and Reproduction with permission from ref. 135, Copyright 2020, Cell Press. | |
Cuenya et al. explored the effect of metal composition and particle size on activity during the CO2RR over well-defined CuCo NPs.132 The FE and partial current density data confirmed that small amounts of Co enhanced the activity of these NPs while an increase in particle size favored the CO2RR over the HER. Under CO2RR conditions, the NPs underwent the surface segregation of Cu together with possible sintering, leading to a contraction of the Cu–Cu interatomic distance that weakened the binding energy between the surface and the key intermediates *COOH, *CO and *H. These structural and compositional changes have also been found to occur in CuZn nanoparticles. In such materials, metallic Cu in close proximity to ZnO leads to the production of CH4 during the initial stage or the reaction, after which the progressive reduction of ZnO occurs under CO2RR conditions. The simultaneous enhancement of Cu–Zn interactions and formation of a brass alloy later change the selectivity to exclusively generate CO and H2.133
In addition to Pd, Cu can also be doped with Co, Zn and Ag for the efficient conversion of CO2 to C2+ products. A Cu–Ag tandem catalyst has been found to enhance the C2+ production rate by promoting CO2 reduction to CO on Ag and subsequent carbon coupling on Cu (Fig. 8g).135 With the addition of Ag, the C2+ partial current over the Cu surface increased from 37 to 160 mA cm−2 at −0.70 V vs. RHE in 1 M KOH, indicating that the localized CO-enriched environment generated by Ag promoted C2+ formation on Cu. Very recently, He et al. used E-beam evaporation to synthesize a series of CuAg films with uniform distributions and controllable stoichiometries. A series of Cu1−xAgx (x = 0.05–0.2) alloys were found to suppress the formation of HCOOH, thus increasing the ratio of C2 liquid products (such as ethanol and acetate) to the C1 liquid product (HCOOH).136 Moreover, an Au/Cu bimetallic catalyst was reported to exhibit improved activity and selectivity for C2+ alcohols at ambient temperature and pressure.137 A tandem catalysis mechanism was proposed by Jaramillo et al. based on a combination of electrochemical testing and mass transport modelling. In this mechanism, CO2 reduction on gold nanoparticles generates a high local concentration of CO on the neighboring Cu surface and this CO is then reduced to alcohols such as ethanol and n-propanol under locally alkaline conditions.
Various sp-block non-noble metals such as In,138 Sn139 and Sb140 have previously been identified as highly selective electrocatalysts for CO2 reduction. Alloying these non-noble metals with Cu to form Cu–In,141,142 Cu–Sb143 and Cu–Sn144,145 has been found to promote CO formation while inhibiting the HER. As an example, Sun et al. demonstrated that Cu2Sb-decorated Cu nanowire arrays on Cu foil could serve as highly active and selective electrocatalysts for the conversion of CO2 to CO.143 Trials in CO2-saturated 0.1 M KHCO3 achieved a high FE of 86.5% for CO at –0.90 V vs. RHE with an onset potential observed for CO evolution of −0.50 V vs. RHE and complete suppression of HCOO− formation. The unique spike-like microstructure obtained by alloying Cu with Sn atoms can also tune the electronic structure of the catalyst and so balance the adsorption and protonation of the *CO2 intermediate while increasing the local electric field to raise the CO2 concentration.145 Porous Cu6.26Sn5 exhibits superior selectivity for HCOO− with an FE of 97.8 ± 2.4%.
3.5 Single/dual Cu atom catalysts
Metal-based single atomic catalysts (SACs) and dual atom catalysts combine the merits of both heterogeneous and homogeneous catalysts with unique geometric and electronic characteristics. The advantages of these materials include low coordination of metal atoms, high atomic utilization and strong metal–support interaction,146 all of which provide outstanding performance during the CO2RR.147–149 In this section, Cu SACs based on different support materials (such as oxides, nano-sized carbons and MXenes) as well as Cu-containing dual-atom catalysts with applications to electrochemical CO2 conversion are considered.
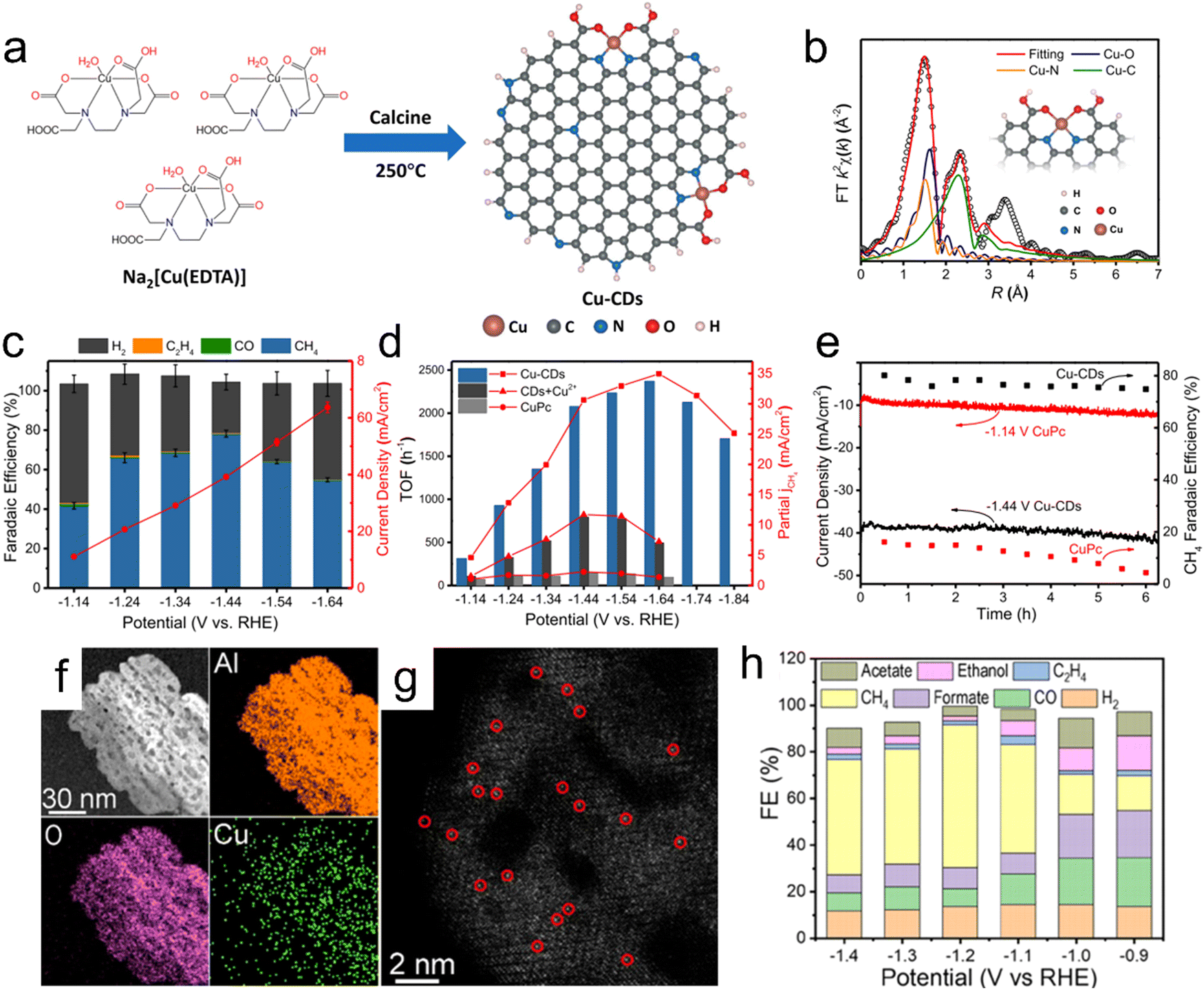
The different coordination environments around isolated Cu atoms and the substrate play an important role in determining the products obtained from the CO2RR. He et al. proposed a facile strategy for the large-scale synthesis of single-atom Cu-decorated through-hole carbon nanofibers (CuSAs/TCNFs).150 In this work, metal atoms embedded in the self-supporting through-hole structure of the material were reduced to generate abundant single atoms of Cu capable of effectively catalyzing the CO2RR, leading to a high partial current density of −93 mA cm−2 for C1 products. This catalyst also remained stable for more than 50 h in an aqueous solution. According to DFT calculations, single Cu atoms provide a relatively high binding energy for the *CO intermediate, allowing this intermediate to be further reduced to products such as methanol rather than being released as CO. Zheng et al. demonstrated a facile approach to tuning active Cu sites for CO2 electroreduction to form different hydrocarbons based on pyrolyzing MOF precursors at different temperatures.7 The presence of nitrogen in these materials enabled good dispersion and attachment of atomic Cu species on nitrogen-doped carbon frameworks with Cu–Nx configurations. DFT calculation results indicated that these catalysts could produce C2H4via the binding of two CO intermediates on adjacent Cu–N2 sites. In addition, the isolated Cu–N4, neighboring Cu–N4 and isolated Cu–N2 sites would all be expected to promote the formation of CH4. Pyrrolic-N4 sites were determined to provide the free energy required for the formation of the *COOH intermediate and for C–C coupling more readily than pyridinic-N4 sites.151 Recently, Zhu et al. reported the first synthesis of a carbon-dots-based SAC containing unique CuN2O2 sites and indicated that this material showed a remarkably high FE of 78% and selectivity of 99% for CO2RR products during the electrochemical conversion of CO2 to CH4 with a current density of 40 mA cm−2 in aqueous electrolytes (Fig. 9a–e).152 Theoretical calculations also established that the high selectivity and activity at CuN2O2 active sites could be attributed to the optimally increased energy barriers to CH4 and H2 formation as well as to the fine-tuned electronic structure of the active Cu sites.
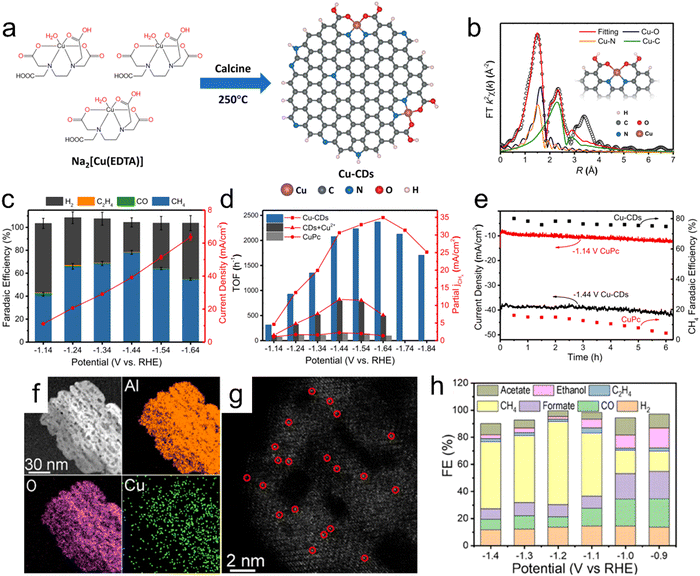 |
| | Fig. 9 (a) Diagram showing the low-temperature calcination of Cu-CD catalysts, (b) EXAFS fitting curves for Cu-CDs in R space based on the backscattering paths of Cu–N, Cu–O and Cu–C (the inset in (b) shows the structure of Cu sites in Cu-CDs), (c) dependence of FE (left y-axis) and current density (based on geometric surface area, right y-axis) of Cu-CDs on potential, (d) partial CH4 current density plots and TOFs of Cu-CDs, CDs + Cu2+ and CuPc at different applied potentials, (e) stability test of Cu-CDs and CuPc at their highest CO2RR FE potentials. (f and g) EDS elemental mapping images and aberration-corrected HAADF-STEM image (atomically-dispersed Cu is highlighted in red circles) of the Cu/p-Al2O3 SAC and (h) FE values for various products using the Cu/p-Al2O3 SAC. Reproduction with permission from ref. 152, Copyright 2021, Springer Nature and Reproduction with permission from ref. 157, Copyright 2021, American Chemical Society. | |
In addition to carbon-based materials, C3N4, metal oxides and MXenes have also been utilized as substrate materials for Cu SACs.153–156 As an example, Zheng et al. developed a Cu-doped CeO2 electrocatalyst with highly effective catalytic sites for the electroreduction of single CO2 molecules to CH4.153 The strong interaction between CeO2 and Cu in this material promoted the formation of single-atom dispersed Cu species as well as the surrounded multiple oxygen vacancies, and these effects were the primary cause of the excellent CH4 selectivity shown by this catalyst. In this work, Cu–CeO2-4% nanorods provided a CH4 FE of approximately 58% at –1.8 V vs. RHE. This study also demonstrated the rational design of highly dispersed metal catalytic centers at the single atom level with the aim of promoting the CO2RR. Li et al. found that the loading of Cu SAs onto Al2O3 and Cr2O3 (acting as Lewis acids) significantly improved the rate of CO2 reduction to CH4 (Fig. 9f–h). A Cu/Al2O3 SAC exhibited a high selectivity of 62% towards CH4 with a corresponding current density of 153.0 mA cm−2 at −1.2 V vs. RHE.157 This work provided useful techniques for tailoring the electronic structure of Cu single atoms for the highly efficient CO2RR.
MXenes having OH, O and F surface terminations have received much attention owing to the excellent electrical conductivity, chemical stability and abundant active catalytic sites of these materials.158,159 As a result, MXenes have been widely employed as substrates for SACs. Sun et al. reported that Cu single atoms anchored on Ti3C2Tx MXene nanosheets can act as effective and robust catalysts for electrochemical CO reduction, achieving an ultrahigh selectivity of 98% for the formation of multi-carbon products.154 As an example, a high FE of 71% for C2H4 was obtained at −0.7 V vs. RHE. Theoretical simulations suggested that atomically dispersed Cu–O3 sites favor the C–C coupling of CO molecules to generate the key *CO–-CHO intermediate and also lower the energy barrier associated with the potential rate-determining step. Yang et al. subsequently reported an efficient approach to producing single Cu atoms immobilized on MXene that exhibited a high FE value of 59.1% for CH3OH and showed good electrocatalytic stability.155 Single-atom Cu with an unsaturated electronic structure (Cuδ+, 0 < δ < 2) provided a low energy barrier for the rate-determining step (the conversion of *HCOOH to the absorbed *CHO intermediate) that was responsible for the efficient electrocatalytic reduction of CO2 to CH3OH.
The construction of paired atom structures to form dual atom Cu catalysts has also been proposed as a potentially useful means of modifying the catalytic behavior of atomic sites and increasing electrocatalytic performance.149 Based on the position of the atoms, these dual atom catalysts are classified as either isolated or binuclear. Chen et al. demonstrated the first fabrication of a novel Ni–Cu atomic pair configuration with binuclear dual-atom sites with the aim of obtaining improved CO2RR performance.160 The incorporation of Cu in this system positively shifted the Ni 3d orbital energy to the Fermi level and thus accelerated the rate-determining step (*COOH formation). During the electrocatalytic CO2RR, it is difficult to obtain a low overpotential because of scaling effects, by which increased *CO adsorption is always accompanied by stronger binding of *CHO (or *COH).69 Wang et al. reported that this scaling relationship could be avoided to obtain efficient CO2 electrochemical reduction by employing heteronuclear transition metal dimers embedded in a monolayer of C2N as dual active centers.161 In such systems, the binding energies of key reduction intermediates are completely decoupled so that the overpotential limitation no longer exists. CuCr/C2N and CuMn/C2N have exhibited the best performance among these materials to date, with very low limiting potentials (−0.37 and −0.32 V, respectively) for CH4 production.
3.6 Cu-Based MOFs
MOFs have large specific surface areas and highly dispersed unsaturated metal centers that can be used as electrocatalysts. The catalytic performance of an MOF can also be tailored by tuning its structure, including the types of ligands and metals, degree of porosity and pore sizes and size distribution.162 The first research regarding the electrocatalytic reduction of CO2 utilizing a Cu-MOF was conducted in 2012.163 In this work, Hinogami et al. utilized Cu rubenate (CR) MOFs on carbon paper for the selective reduction of CO2 to formic acid in an aqueous solution of KHCO3 with a 30% current efficiency. Compared with a normal Cu electrode, the CR-MOF was much more efficient and selective during the reduction. This higher selectivity was attributed to the lower electron densities at the metal centers and weaker adsorption of the reactant CO2 on the MOF surface. Subsequently, MOFs were extensively studied as catalysts for the synthesis of C1 products (CH4, CO, HCOOH and CH3OH) and C2+ products (C2H4, EtOH, C2H6 and CH3COOH).164–167
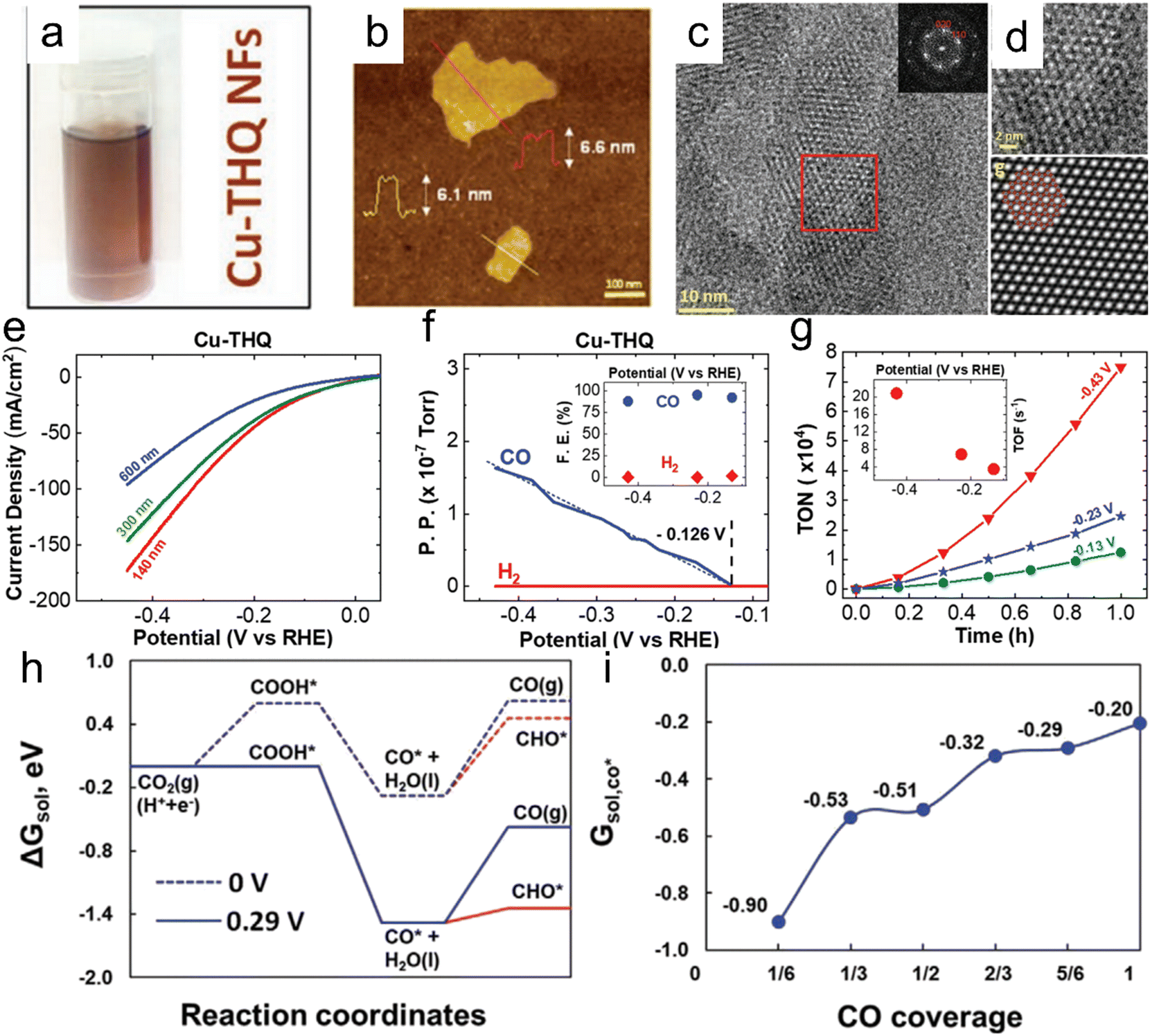
Majidi et al. prepared a two-dimensional (2D) Cu-based conductive MOF (Cu tetrahydroxyquinone (Cu–THQ)) having excellent catalytic activity (Fig. 10).164 In this material, reduced Cu (Cu+) was reversibly converted to Cu2+ after the CO2RR reaction. Cu–THQ nanoflakes with an average lateral size of 140 nm exhibited a negligible overpotential of 16 mV for activation, a high current density of approximately 173 mA cm−2 at −0.45 V vs. RHE, an average FE of approximately 91% for CO production and a remarkable TOF as high as approximately 20.82 s−1. Yang et al. synthesized a so-called Cu-ade MOF comprising Cu2+ ions coordinated with adeninato/carboxylato ligands and employed this material to promote electrocatalytic CO2 conversion to C2H4. A maximum FE of 45.0% was achieved at −1.4 V vs. RHE in a CO2-purged 0.1 mol L−1 KHCO3 electrolyte.165 Recently, Wang et al. reported a novel CO2RR catalyst consisting of CuO nanoparticles with sizes ranging from 1.4 to 3.3 nm anchored on Cu-MOF nanosheets that was obtained through a one-step facile solvothermal method. The electrocatalytic performance was significantly promoted by the interface between the CuO and Cu-MOF and the accessible metallic moieties and unique 2D structure of the Cu-MOF enhanced the adsorption and activation of CO2 molecules.166 Notably, compared with state-of-the-art 2D PcCu–Cu–O, the overpotential required to obtain a similar FE for C2H4 formation (50.0%) over the CuO/Cu-MOF composite was substantially lowered by 100 mV.168 Liu et al. prepared a 2D conductive MOF using a nitrogen-rich tricycloquinazoline (TQ)-based multitopic catechol ligand to coordinate Cu2+ and Ni2+ ions and form 2D graphene-like porous sheets (M3(HHTQ)2) (M = Cu, Ni; HHTQ = 2,3,7,8,12,13-hexahydroxytricycloquinazoline).169 A Cu3(HHTQ)2 specimen exhibited superior catalytic activity during the CO2RR with CH3OH as the sole product and an FE of 53.6% at a small overpotential of −0.4 V.
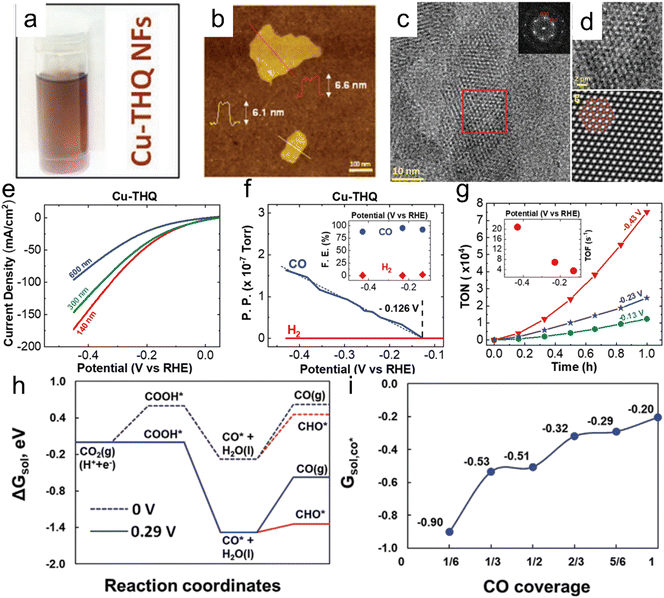 |
| | Fig. 10 (a) Cu–THQ NFs dispersed in IPA, (b) a representative AFM image of Cu–THQ NFs, (c) HRTEM images of Cu–THQ NFs along the [001] direction showing elliptical pore packing, (d) an enlarged view of the HRTEM image in the red box shown in panel (c) and corresponding lattice-averaged and symmetry-imposed image, (e) LSV results obtained during the CO2RR in a solution of 1 M choline chloride and 1 M KOH using Cu–THQ NFs having different particle sizes, (f) DEMS analysis results for CO and H2 production during an LSV experiment with Cu–THQ NFs, (g) TON values for CO production during 1 h chronoamperometry experiments at controlled potentials (the inset shows TOFs at these potentials after 1 h), (h) free energies including solvation corrections for CO production on Cu–THQ at 0 V vs. RHE (dotted blue lines) and at an overpotential, η, of 0.29 V (solid blue lines) and (i) adsorption free energies of CO at different coverages on Cu–THQ. Reproduction with permission from ref. 164, Copyright 2021, Wiley-VCH. | |
Carbonization and oxidation of Cu based MOF also have been proved to be an effective approach to improve the activity and selectivity of CO2RR, which can achieve high current density due to the presence of carbon matrix in addition to the intrinsic characteristics of MOFs remaining.170–172 Liang et al. prepared Cu-based catalyst derived from Cu-MOF, which remained porous morphology of Cu-MOF and achieved the conversion of CO2 to C2+ products.172 The optimized catalyst exhibits a 51% FE for C2H4 and a 70% FE for C2+ products, with 20 h operational stability in an H-cell configuration, and a partial ethylene current density of 150 mA cm−2 in a flow-cell configuration. The formation of bimetallic catalysts could change the electronic structure, and decrease the energy barriers for CO2 activation. Yang et al. synthesized rod-like CuBi bimetallic catalysts by carbonization and oxidation of CuBi-MOF precursors, showing an allured high FE formate of 100% at −0.77 V and excellent durability.170 The Bi2CuO4 in the interface of catalyst greatly enhanced the activity and selectivity of the bimetallic CuBi catalysts. Recently, Xue et al. prepared a Cu/Bi bi-metal catalyst derived from MOFs by a hydrothermal synthesis combining with calcination under N2 atmosphere, which shows cylindrical morphology composed of bi-metallic nanoparticles.171 The Cu/Bi bi-metallic system lowered the activate energy barrier of CO2 and shows a strong adsorption capability for the CO2–intermediate. The optimized Cu1–Bi/Bi2O3@C exhibits excellent selectivity toward HCOOH with FE of 93% at −0.94 V.
4. Conclusion and perspectives
This review introduced standard criteria used to evaluate the performance of CO2RR systems and assessed recent advances in the innovation and development of Cu-based CO2RR electrocatalysts. These materials have included monometallic Cu, Cu-based oxides and other Cu-based compounds, Cu-based bimetallics, single/dual Cu atoms and Cu-based MOFs. The results of prior studies and the performance data for various electrocatalysts were summarized in Table 1. It was noted that certain electrocatalysts have shown excellent catalytic activity and selectivity as well as suitable levels of stability during the CO2RR. Although much has been achieved in this regard, many challenges remain to be overcome and many areas must still be explored. The following are some of the most important advancements, challenges and potential research associated with this field of study.
Table 1 A summary of reported performance data for Cu-based CO2RR electrocatalysts
| Catalyst |
Products |
Electrolyte |
Performance |
Ref. |
| Cu(OH)2-D/Cu |
C2+ |
CO2-Saturated 0.1 M NaHCO3 |
FEs of ∼58% for C2H4 and ∼87% for C2+ hydrocarbons and alcohols with the C2+ partial current density of ∼217 mA cm−2 only at −0.54 V |
82
|
| (100)-Rich Cu |
C2H4, CH3COO, C2H5OH, n-C3H7OH |
CO2-Saturated 0.1 M NaHCO3 |
C2+ FEs of 88% and partial current density of 320 mA cm−2 at −0.63 V |
81
|
| Activated Cu nanowires |
C2H4 |
CO2-Saturated 0.1 M KHCO3 |
C2H4 FEs of ∼77% at −1.01 ± 0.01 V |
84
|
| OD-Cu-500 |
CO, HCOO− |
CO2-Saturated 0.1 M NaHCO3 |
CO and HCOO− FEs of ∼45% at −0.3 V |
87
|
| Cu nanowires |
CO |
CO2-Saturated 0.1 M NaHCO3 |
CO FEs of ∼60% and current density of 1 mA cm−2 at an overpotential of 0.3 V |
90
|
| Star decahedron Cu NPs |
C2H4 |
CO2-Saturated 0.1 M KHCO3 |
C2H4 FEs of 52.43% at −0.993 V |
94
|
| Cu nanosheets with nano-scaled defects |
C2H4 |
CO2-Saturated 0.1 M K2SO4 |
C2H4 FEs of 83.2% at −1.2 V |
95
|
| Defect-site-rich Cu catalyst |
C2+ alcohols |
CO2-Saturated 0.1 M KHCO3 |
C2+ alcohols FEs of 70% |
96
|
| Cu nanoneedle arrays |
C2+ |
CO2-Saturated 0.1 M KHCO3 |
C2+ FEs of 59% at −1.2 V (vs. RHE) |
97
|
| Electron beam (EB) Cu catalyst |
C2H4 |
15 sccm CO2 and 1 M KOH |
C2H4 FEs of 39% and C2+ FE of 70% at −0.65 V vs. RHE |
98
|
| Ditetrahedron-shaped Cu8 |
HCOOH |
CO2-Saturated 0.5 M KHCO3 |
HCOOH FE of ∼92% at −1.0 V vs. RHE |
101
|
| Amorphous Cu NPs |
HCOOH, C2H6O |
CO2-Saturated 0.1 M KHCO3 |
C2 FE of 59% at −1.4 V with formic acid (HCOOH) and ethanol (C2H6O) account for 37% and 22%. |
103
|
| CV-treated Cu electrode |
C2H4 |
CO2-Saturated 0.1 M KHCO3 |
C2H4 FE of 40% at −1 V versus RHE |
111
|
| Anodic treated Cu |
C2+ |
CO2-Saturated 0.1 M KHCO3 |
C2+ FE of 76% at −1.0 V versus RHE. |
112
|
| Cu oxide with surface oxygen vacancies |
C2H4 |
CO2-Saturated 0.1 M KHCO3 |
C2H4 FE of ∼63% at −1.4 versus RHE |
113
|
| Cu2O-BN |
CO, C2H4 |
CO2-Saturated 0.5 M KHCO3 |
CO FE of ∼14% at −1.2 V versus RHE, C2H4 FE of ∼16% at −1.4 V versus RHE |
118
|
| Cu2S |
Formate |
CO2-Saturated 0.1 M NaHCO3 aqueous solution |
Formate FE of 87% and partial current density over 19 mA cm−2 at −0.9 V vs. RHE |
121
|
| Cu2S–Cu–V NPs |
C3H7OH, C2H5OH |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
FE for C3H7OH and C2H5OH reaches 8 ± 0.7% and 15 ± 1% with a partial current density of 2.5 ± 0.1 and 4.8 ± 0.1 mA cm−2 at –0.95 V vs. RHE |
122
|
| CuS with double sulfur vacancies |
n-Propanol |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
n-Propanol FE of 15.4 ± 1% at −1.05 V vs. RHE |
63
|
| CuS |
HCOOH |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
HCOOH FE of ∼60% |
123
|
| CuS |
CH4 |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
CH4 FE of ∼73% |
124
|
| Cu2−xSe |
Methanol |
CO2-Saturated [Bmim]PF6-CH3CN-H2O |
Methanol FE of 77.6% and current density of 41.5 mA cm−2 at a low overpotential of 285 mV |
126
|
| Se-defective CuInSe2 |
CO |
CO2-Saturated 0.5 M KHCO3 aqueous solution |
CO FE of 91% at −0.7 V vs. RHE |
127
|
| Cu3P NS/Cu |
Formate |
CO2-Saturated 0.1 M KHCO3 |
FE of 90% at a low overpotential of 65 mV |
125
|
| Cu3N |
C2+ |
1 M KOH |
FE of 73.7% under −1100 mA cm−2, C2+ partial current density of −909 mA cm−2 at −1.15 V vs. RHE |
128
|
| CuPd |
CO, C2 |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
The ordered CuPd exhibits the highest CO FE of ∼80% |
130
|
| The separated CuPd exhibits the highest FE (up to 63%) for C2 such as C2H4 and C2H5OH |
| Cu100−xCox NPs |
HCOOH and CO |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
Cu90Co10 at constant NP size of ∼5.2 nm show HCOOH FE of ∼10% and CO FE of 7% at −1.1 vs. RHE |
132
|
| CuZn NPs |
CO, CH4 |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
CH4 FE of ∼70% (Zn contents from 10–50), CO FE of ∼40% (Zn contents from 70–100) |
133
|
| Cu–Ag Tandem catalysts |
C2H4, C2H5OH, CH3COO− |
1 M KOH |
C2+ partial current over a Cu surface increases from 37 to 160 mA cm−2 |
135
|
| At 0.70 V vs. RHE |
| Gold NPs on Cu foil |
Ethanol and n-propanol |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
Alcohol production is observed at over 265 mV more positive potentials on the Au/Cu catalyst compared with Cu |
137
|
| In1.5Cu0.5 NPs |
HCOOH |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
HCOOH FE of 90% at −1.2 V vs. RHE |
141
|
| Cu–CuI composite catalyst |
C2+ |
1 M KOH |
C2+ partial current density of 591 mA cm−2 at −1.0 V vs. RHE |
142
|
| Cu2Sb decorated Cu nanowire arrays |
CO |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
CO FE of 86.5% at −0.90 V vs. RHE |
143
|
| Cu–Sn bimetallic catalyst |
CO |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
CO FE of 90% and a current density of −1.0 mA cm−2 at −0.6 V vs. RHE |
144
|
| Porous Cu6.26Sn5 |
Formate |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
Formate FE of 97.8 ± 2.4% at −1.08 V vs. RHE |
145
|
| Cu–SACs/TCNFs (through-hole carbon nanofibers) |
CH3OH, CO |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
FE of 44% for CH3OH and 56% for CO at the potentials of 0.9 vs. RHE |
150
|
| Cu–SACs/nitrogen-doped carbon |
CH4, C2H4 |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
Cu–N2 with C2H4 FE of 24.8% at 1.4 V, Cu–N4 with CH4 FE of 38.6% at 1.6 V. |
7
|
| Cu–SA/NPC |
CH3COCH3 |
CO2-Saturated electrolyte |
FE of 36.7% and production rate of 336.1 μg h−1 at 0.36 V. |
151
|
| Carbon-dots-based SAC with unique CuN2O2 sites |
CH4 |
CO2-Saturated 0.5 M KHCO3 solution |
CH4 FE of 78% and selectivity of 99% with current density of 40 mA cm−2 at 1.64 V. |
152
|
| Cu–C3N4 |
C2H5OH, C2H4, and C2H6 |
0.1 M KHCO3 solution |
— |
156
|
| Cu–CeO2-4% nanorods |
CH4 |
CO2-Saturated 0.1 M KHCO3 aqueous solution |
CH4 FE of ∼58% at –1.8 V vs. RHE |
153
|
| Cu/p-Al2O3 SAC |
CH4 |
— |
CH4 FE of 62% at −1.2 V with current density of 153.0 mA cm−2. |
157
|
| Cu SA/MXene |
CH3OH |
CO2-Saturated in the electrolyte |
CH3OH FE of 59.1% |
155
|
| Ni–Cu dual atom catalysts |
CO |
CO2-Saturated 0.5 M KHCO3 solution |
TOF of 20695 h−1 and CO FE of 97.7% at −0.6 V |
160
|
| HKUST-1 |
CH3OH, CH3CH2OH |
CO2-Saturated 0.5 M KHCO3 |
Methanol FE of 5.6% and ethanol FE of 10.3% at −1.0 vs. Ag/AgCl |
175
|
| Cu/C |
CH3OH, CH3CH2OH |
CO2-Saturated 0.1 M KHCO3 |
Methanol FE of 43.2%, ethanol FE of 34.8% at −0.1 V vs. RHE |
176
|
| Cu/Bi-MOFs |
CH3OH, CH3CH2OH |
CO2-Saturated 0.5 M KHCO3 |
Methanol FE of 18.2%, ethanol FE of 28.3% at −0.37 vs. RHE |
177
|
| CuPc |
CH4 |
CO2-Saturated 0.5 M KHCO3 |
CH4 FE of 66% at −1.06 vs. RHE |
178
|
| Cu-MOF-74/Cu NPs |
CH4 |
CO2-Saturated 0.1 M KHCO3 |
CH4 FE of >50% at −1.3 vs. RHE |
179
|
(1) CO2RR performance should be improved by controlling various reaction conditions, such as the electrolyte that is used, as well as by modification of the Cu catalyst surface and the application of an external electric field or electric–thermal coupling. CO2 conversion to value-added C2+ chemicals could be enhanced with the aid of localized electric-thermal field synergy as a means of improving the activity and selectivity of the CO2RR.173
(2) Compared with that of other transition metals, the selectivity of Cu-based nanomaterials for specific CO2RR products is very low. Optimization of these materials is therefore required, particularly in the case of the newly-developed Cu-based amorphous and high entropy alloys.
(3) Single/dual atom Cu catalysts can promote the CO2RR to form different products. Effective approaches to improving the overall selectivity for deep reduction products are necessary, such as the rational design of the structure of single-atom Cu sites and the construction of atom pairs. Other techniques have included the selection of substrate materials having high surface areas and strong coordination sites. Remaining challenges include difficulties in precisely controlling multiple active centers, low intrinsic activities of catalysts, poor loading capacities and low yields of C2+ products (the majority of SACs provide CO or HCOOH as the major CO2RR product). The reversible transformation of Cu-SACs to Cu clusters during the CO2 reduction process has been established based on operando XAS analyses.174 Therefore, strategies to improve the stability of Cu-SACs must be devised.
(4) In the case of Cu-based MOFs, the effects of various substituents on organic ligands and metal centers on the CO2RR activities of these materials should be further investigated. Other potential improvements include the design of conducive ligand structures to facilitate electron transfer and the use of nano-sized 2D structures and bimetallic catalysts to further improve the selectivity and efficiency of CO2 conversion.
(5) A thorough understanding of changes in the catalyst and of reaction intermediates would be helpful to the rational design of more effective Cu-based CO2RR catalysts. Such work will require an improved understanding of the CO2RR mechanism based on a combination of in situ/operando characterization techniques such as in situ spectroscopy and TEM with theoretical calculations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to acknowledge funding from the National Natural Science Foundation of China (52201037), Research Projects of Sichuan Province (2022NSFSC1965, 2022JDRC0085).
References
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981–6000 RSC.
- C. Xiao and J. Zhang, ACS Nano, 2021, 15, 7975–8000 CrossRef CAS PubMed.
- V. A. Semenov, Nat. Climate Change, 2012, 2, 315–316 CrossRef.
- Q. Schiermeier, Nature, 2011, 470, 316 CrossRef CAS.
- K. Xiang, F. Shen, Y. Fu, L. Wu, Z. Wang, H. Yi, X. Liu, P. Wang, M. Liu, Z. Lin and H. Liu, Environ. Sci.: Nano, 2022, 9, 911–953 RSC.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- L. Ye, Y. Ying, D. Sun, Z. Zhang, L. Fei, Z. Wen, J. Qiao and H. Huang, Angew. Chem., Int. Ed., 2020, 59, 3244–3251 CrossRef CAS.
- C. Liu, J. Gong, Z. Gao, L. Xiao, G. Wang, J. Lu and L. Zhuang, Sci. China: Chem., 2021, 64, 1660–1678 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- E. B. Creel and B. D. McCloskey, Nat. Catal., 2018, 1, 6–7 CrossRef CAS.
- F. Li, D. R. MacFarlane and J. Zhang, Nanoscale, 2018, 10, 6235–6260 RSC.
- M. M. Ayyub and C. N. R. Rao, Mater. Horiz., 2021, 8, 2420–2443 RSC.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 1695–1698 CrossRef CAS.
- Y. Du, X. Meng, Z. Wang, X. Zhao and J. Qiu, Acta Phys. Chim. Sin., 2021, 0, 2101009 Search PubMed.
- J. Chen, T. Wang, Z. Li, B. Yang, Q. Zhang, L. Lei, P. Feng and Y. Hou, Nano Res., 2021, 14, 3188–3207 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- Q. Zhu, J. Ma, X. Kang, X. Sun, H. Liu, J. Hu, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 9012–9016 CrossRef CAS PubMed.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8(35), 1801536 CrossRef.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie, A. S. Jalilov, Z. Li, H. Li, B. I. Yakobson, Q. Wu, E. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8(19), 1703487 CrossRef.
- H. Chen, J. Chen, J. Si, Y. Hou, Q. Zheng, B. Yang, Z. Li, L. Gao, L. Lei, Z. Wen and X. Feng, Chem. Sci., 2020, 11, 3952–3958 RSC.
- Y. Fu, T. Wang, W. Zheng, C. Lei, B. Yang, J. Chen, Z. Li, L. Lei, C. Yuan and Y. Hou, ACS Appl. Mater. Interfaces, 2020, 12, 16178–16185 CrossRef CAS.
- F. Wei, T. Wang, X. Jiang, Y. Ai, A. Cui, J. Cui, J. Fu, J. Cheng, L. Lei, Y. Hou and S. Liu, Adv. Funct. Mater., 2020, 30(39), 2002092 CrossRef CAS.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS PubMed.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2020, 30(4), 1907658 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef CAS PubMed.
- C. Zhao and J. Wang, Chem. Eng. J., 2016, 293, 161–170 CrossRef CAS.
- Y. Wang, X.-P. Zhang, H. Lei, K. Guo, G. Xu, L. Xie, X. Li, W. Zhang, U.-P. Apfel and R. Cao, CCS Chem., 2022, 0, 1–9 Search PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- J. Wu, F. G. Risalvato, P. P. Sharma, P. J. Pellechia, F.-S. Ke and X.-D. Zhou, J. Electrochem. Soc., 2013, 160, F953–F957 CrossRef CAS.
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, B109 CrossRef CAS.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 CrossRef CAS.
- C. Zou, C. Xi, D. Wu, J. Mao, M. Liu, H. Liu, C. Dong and X.-W. Du, Small, 2019, 15, 1902582 CrossRef CAS.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- S. Kaneco, K. Iiba, S.-K. Suzuki, K. Ohta and T. Mizuno, J. Phys. Chem. B, 1999, 103, 7456–7460 CrossRef CAS.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 403–408 RSC.
- N. Hollingsworth, S. F. R. Taylor, M. T. Galante, J. Jacquemin, C. Longo, K. B. Holt, N. H. de Leeuw and C. Hardacre, Angew. Chem., Int. Ed., 2015, 54, 14164–14168 CrossRef CAS PubMed.
- V. Vedharathinam, Z. Qi, C. Horwood, B. Bourcier, M. Stadermann, J. Biener and M. Biener, ACS Catal., 2019, 9, 10605–10611 CrossRef CAS.
- B. Braunschweig, P. Mukherjee, J. L. Haan and D. D. Dlott, J. Electroanal. Chem., 2017, 800, 144–150 CrossRef CAS.
- Z. Han, D. Han, Z. Chen, J. Gao, G. Jiang, X. Wang, S. Lyu, Y. Guo, C. Geng, L. Yin, Z. Weng and Q.-H. Yang, Nat. Commun., 2022, 13, 3158 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- H. Xiao, T. Cheng and W. A. Goddard, 3rd, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- B. Zha, C. Li and J. Li, J. Catal., 2020, 382, 69–76 CrossRef CAS.
- Q. Qian, J. Zhang, M. Cui and B. Han, Nat. Commun., 2016, 7, 11481 CrossRef.
- X. F. Qiu, J. R. Huang, C. Yu, Z. H. Zhao, H. L. Zhu, Z. Ke, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202206470 CAS.
- A. D. Handoko, K. W. Chan and B. S. Yeo, ACS Energy Lett., 2017, 2, 2103–2109 CrossRef CAS.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 16459–16464 CrossRef CAS.
- M.-G. Kim, Y. Choi, E. Park, C.-H. Cheon, N.-K. Kim, B. K. Min and W. Kim, ACS Appl. Energy Mater., 2020, 3, 11516–11522 CrossRef CAS.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T.-K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS.
- C. Liu, T. Wang, D. Hao, Q. Li, S. Li and C. Sun, J. Mater. Sci. Technol., 2022, 110, 96–102 CrossRef.
- N. Li, X. Chen, W.-J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS.
- R. Zhang, Y. Zhang, L. Liu, X. Li, Y. Tang, Y. Ni, C. Sun and H. Zhang, Appl. Surf. Sci., 2022, 582, 152472 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- Y. Ji, J. K. Nørskov and K. Chan, J. Phys. Chem. C, 2019, 123, 4256–4261 CrossRef CAS.
- C. Shi, H. A. Hansen, A. C. Lausche and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 4720–4727 RSC.
- S. Zheng, C. Zuo, X. Liang, S. Li and F. Pan, J. Energy Chem., 2021, 56, 444–448 CrossRef CAS.
- H. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 115114 CrossRef.
- C. Ren, S. Lu, Y. Wu, Y. Ouyang, Y. Zhang, Q. Li, C. Ling and J. Wang, J. Am. Chem. Soc., 2022, 144, 12874–12883 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- R. Küngas, P. Blennow, T. Heiredal-Clausen, T. Holt, J. Rass-Hansen, S. Primdahl and J. B. Hansen, ECS Trans., 2017, 78, 2879–2884 CrossRef.
- R. Krause, D. Reinisch, C. Reller, H. Eckert, D. Hartmann, D. Taroata, K. Wiesner-Fleischer, A. Bulan, A. Lueken and G. Schmid, Chem. Ing. Tech., 2020, 92, 53–61 CrossRef CAS.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- Y. H. Wang, Z. Y. Wang, C. T. Dinh, J. Li, A. Ozden, M. G. Kibria, A. Seifitokaldani, C. S. Tan, C. M. Gabardo, M. C. Luo, H. Zhou, F. W. Li, Y. Lum, C. McCallum, Y. Xu, M. X. Liu, A. Proppe, A. Johnston, P. Todorovic, T. T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- D. Zhong, Z. J. Zhao, Q. Zhao, D. Cheng, B. Liu, G. Zhang, W. Deng, H. Dong, L. Zhang, J. Li, J. Li and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 4879–4885 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ACS Catal., 2019, 9, 7894–7899 CrossRef CAS.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- L. Zaza, K. Rossi and R. Buonsanti, ACS Energy Lett., 2022, 7, 1284–1291 CrossRef CAS.
- M. Song, Z. Jiao, W. Jing, Y. Liu and L. Guo, J. Phys. Chem. Lett., 2022, 13, 4434–4440 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef CAS PubMed.
- L. Cao, D. Raciti, C. Li, K. J. T. Livi, P. F. Rottmann, K. J. Hemker, T. Mueller and C. Wang, ACS Catal., 2017, 7, 8578–8587 CrossRef CAS.
- D. Cheng, Z. J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- T. Luo, K. Liu, J. Fu, S. Chen, H. Li, J. Hu and M. Liu, J. Energy Chem., 2022, 70, 219–223 CrossRef CAS.
- C. Choi, T. Cheng, M. Flores Espinosa, H. Fei, X. Duan, W. A. Goddard III and Y. Huang, Adv. Mater., 2019, 31, 1805405 CrossRef.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- Y. Zhou, Y. Liang, J. Fu, K. Liu, Q. Chen, X. Wang, H. Li, L. Zhu, J. Hu, H. Pan, M. Miyauchi, L. Jiang, E. Cortés and M. Liu, Nano Lett., 2022, 22, 1963–1970 CrossRef PubMed.
- E. Jeng, Z. Qi, A. R. Kashi, S. Hunegnaw, Z. Huo, J. S. Miller, L. B. Bayu Aji, B. H. Ko, H. Shin, S. Ma, K. P. Kuhl, F. Jiao and J. Biener, ACS Appl. Mater. Interfaces, 2022, 14, 7731–7740 CrossRef CAS.
- H. Dong, Y. Li and D.-E. Jiang, J. Phys. Chem. C, 2018, 122, 11392–11398 CrossRef CAS.
- S. Zhang, L. Chen, X. S. Luan and H. Li, Chem. Phys., 2022, 557, 111487 CrossRef CAS.
- L. J. Liu, Z. Y. Wang, Z. Y. Wang, R. Wang, S. Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2022, 134(35), e202205626 Search PubMed.
- S. J. Li, D. Bao, M. M. Shi, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29(33), 1700001 CrossRef PubMed.
- Y.-X. Duan, F.-L. Meng, K.-H. Liu, S.-S. Yi, S.-J. Li, J.-M. Yan and Q. Jiang, Adv. Mater., 2018, 30, 1706194 CrossRef.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci., 2017, 114, 6685–6688 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS.
- Y. Kwon, Y. Lum, E. L. Clark, J. W. Ager and A. T. Bell, ChemElectroChem, 2016, 3, 1012–1019 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- T.-C. Chou, C.-C. Chang, H.-L. Yu, W.-Y. Yu, C.-L. Dong, J.-J. Velasco-Vélez, C.-H. Chuang, L.-C. Chen, J.-F. Lee, J.-M. Chen and H.-L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- Z. X. Gu, N. Yang, P. Han, M. Kuang, B. B. Mei, Z. Jiang, J. Zhong, L. Li and G. F. Zheng, Small Methods, 2019, 3, 1800449 CrossRef.
- X. Wang, K. Klingan, M. Klingenhof, T. Moller, J. Ferreira de Araujo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS.
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Aviles Acosta, J. W. Beeman, Y. Ye, J. Yano, A. Mehta, R. C. Davis, T. F. Jaramillo, C. Hahn and W. S. Drisdell, J. Am. Chem. Soc., 2021, 143, 588–592 CrossRef CAS PubMed.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- J. Li, A. Ozden, M. Y. Wan, Y. F. Hu, F. W. Li, Y. H. Wang, R. R. Zamani, D. Ren, Z. Y. Wang, Y. Xu, D. H. Nam, J. Wicks, B. Chen, X. Wang, M. C. Luo, M. Graetzel, F. L. Che, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2808 CrossRef CAS PubMed.
- Y. Zhou, Y. Yao, R. Zhao, X. Wang, Z. Fu, D. Wang, H. Wang, L. Zhao, W. Ni, Z. Yang and Y.-M. Yan, Angew. Chem., Int. Ed., 2022, e202205832 CAS.
- N. Sakamoto, Y. F. Nishimura, T. Nonaka, M. Ohashi, N. Ishida, K. Kitazumi, Y. Kato, K. Sekizawa, T. Morikawa and T. Arai, ACS Catal., 2020, 10, 10412–10419 CrossRef CAS.
- Z. Sun, Y. Hu, D. Zhou, M. Sun, S. Wang and W. Chen, ACS Energy Lett., 2021, 6, 3992–4022 CrossRef CAS.
- W. He, I. Liberman, I. Rozenberg, R. Ifraemov and I. Hod, Angew. Chem., Int. Ed., 2020, 59, 8262–8269 CrossRef CAS.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407–4412 CrossRef CAS.
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, J. Mater. Chem. A, 2017, 5, 20239–20243 RSC.
- A. B. Laursen, K. U. D. Calvinho, T. A. Goetjen, K. M. K. Yap, S. Hwang, H. Yang, E. Garfunkel and G. C. Dismukes, Electrochim. Acta, 2021, 391, 138889 CrossRef CAS.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS.
- J. Wang, X. Zheng, G. Wang, Y. Cao, W. Ding, J. Zhang, H. Wu, J. Ding, H. Hu, X. Han, T. Ma, Y. Deng and W. Hu, Adv. Mater., 2022, 34, 2106354 CrossRef CAS.
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CrossRef CAS.
- A. Herzog, A. Bergmann, H. S. Jeon, J. Timoshenko, S. Kuhl, C. Rettenmaier, M. Lopez Luna, F. T. Haase and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2021, 60, 7426–7435 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS.
- M. Bernal, A. Bagger, F. Scholten, I. Sinev, A. Bergmann, M. Ahmadi, J. Rossmeisl and B. R. Cuenya, Nano Energy, 2018, 53, 27–36 CrossRef CAS.
- H. S. Jeon, J. Timoshenko, F. Scholten, I. Sinev, A. Herzog, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2019, 141, 19879–19887 CrossRef CAS PubMed.
- D. Higgins, A. T. Landers, Y. Ji, S. Nitopi, C. G. Morales-Guio, L. Wang, K. Chan, C. Hahn and T. F. Jaramillo, ACS Energy Lett., 2018, 3, 2947–2955 CrossRef CAS.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- Y. Xu, C. Li, Y. Xiao, C. Wu, Y. Li, Y. Li, J. Han, Q. Liu and J. He, ACS Appl. Mater. Interfaces, 2022, 14, 11567–11574 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- W. Luo, W. Xie, M. Li, J. Zhang and A. Züttel, J. Mater. Chem. A, 2019, 7, 4505–4515 RSC.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS.
- F. Li, M. Xue, J. Li, X. Ma, L. Chen, X. Zhang, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14718–14722 CrossRef CAS PubMed.
- B. Wei, Y. Xiong, Z. Zhang, J. Hao, L. Li and W. Shi, Appl. Catal., B, 2021, 283, 119646 CrossRef CAS.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS.
- S. Mou, Y. Li, L. Yue, J. Liang, Y. Luo, Q. Liu, T. Li, S. Lu, A. M. Asiri, X. Xiong, D. Ma and X. Sun, Nano Res., 2021, 14, 2831–2836 CrossRef CAS.
- S. Sarfraz, A. T. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- D. Li, L. Huang, Y. Tian, T. Liu, L. Zhen and Y. Feng, Appl. Catal., B, 2021, 292, 120119 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- M. Wang, M. Li, Y. Liu, C. Zhang and Y. Pan, Nano Res., 2022, 15, 4925–4941 CrossRef CAS.
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Chin. J. Catal., 2022, 43, 1547–1597 CrossRef CAS.
- B. Mohanty, S. Basu and B. K. Jena, J. Energy Chem., 2022, 70, 444–471 CrossRef CAS.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS.
- Y. Cai, J. Fu, Y. Zhou, Y. C. Chang, Q. Min, J. J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 586 CrossRef CAS.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- H. Bao, Y. Qiu, X. Peng, J. A. Wang, Y. Mi, S. Zhao, X. Liu, Y. Liu, R. Cao, L. Zhuo, J. Ren, J. Sun, J. Luo and X. Sun, Nat. Commun., 2021, 12, 238 CrossRef CAS.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15, 4927–4936 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y.-Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS.
- J.-C. Lei, X. Zhang and Z. Zhou, Front. Phys., 2015, 10, 276–286 CrossRef.
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
- Z. Liang, C. Qu, W. Guo, R. Zou and Q. Xu, Adv. Mater., 2018, 30, 1702891 CrossRef.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, ECS Electrochem. Lett., 2012, 1, H17 CrossRef CAS.
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou, P. Zapol, J. Cabana, L. A. Curtiss and A. Salehi-Khojin, Adv. Mater., 2021, 33, 2004393 CrossRef CAS PubMed.
- F. Yang, A. Chen, P. L. Deng, Y. Zhou, Z. Shahid, H. Liu and B. Y. Xia, Chem. Sci., 2019, 10, 7975–7981 RSC.
- L. Wang, X. Li, L. Hao, S. Hong, A. W. Robertson and Z. Sun, Chin. J. Catal., 2022, 43, 1049–1057 CrossRef CAS.
- I. U. Din, M. Usman, S. Khan, A. Helal, M. A. Alotaibi, A. I. Alharthi and G. Centi, J. CO2 Util., 2021, 43, 101361 CrossRef CAS.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- Z. Yang, H. Wang, X. Fei, W. Wang, Y. Zhao, X. Wang, X. Tan, Q. Zhao, H. Wang, J. Zhu, L. Zhou, H. Ning and M. Wu, Appl. Catal., B, 2021, 298, 120571 CrossRef CAS.
- Y. Xue, C. Li, X. Zhou, Z. Kuang, W. Zhao, Q. Zhang and H. Chen, ChemElectroChem, 2022, 9, e202101648 CAS.
- K. Yao, Y. Xia, J. Li, N. Wang, J. Han, C. Gao, M. Han, G. Shen, Y. Liu, A. Seifitokaldani, X. Sun and H. Liang, J. Mater. Chem. A, 2020, 8, 11117–11123 RSC.
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortés, S. A. Maier and M. Liu, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castaño and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig and V. S. Batista, Nat. Commun., 2018, 9, 1–9 CrossRef CAS.
- M. K. Kim, H. J. Kim, H. Lim, Y. Kwon and H. M. Jeong, Electrochim. Acta, 2019, 306, 28–34 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Xuguang
An
ab,
Qian
Liu
b,
Lisi
Xie
b,
Jing
Zhang
ab,
Qinye
Li
cd,
Weitang
Yao
ab,
Xuguang
An
ab,
Qian
Liu
b,
Lisi
Xie
b,
Jing
Zhang
ab,
Qinye
Li
cd,
Weitang
Yao
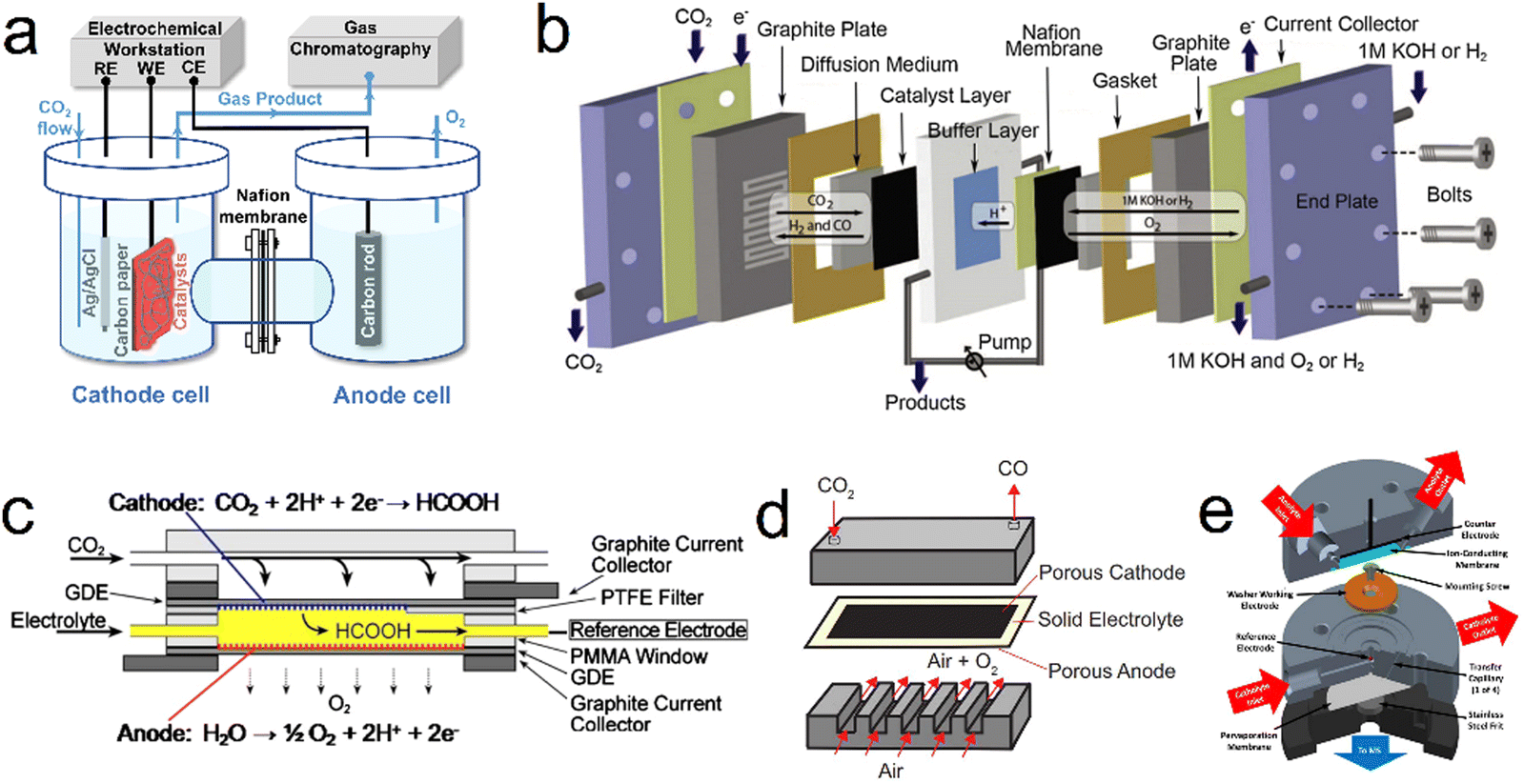
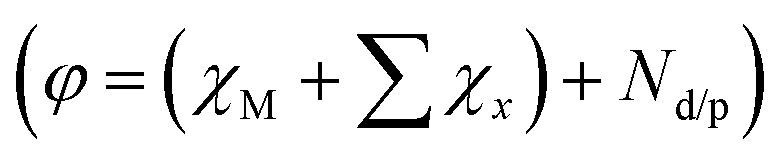 to assess the catalytic performance of 2D material-supported dual-atom catalysts (DACs@2D) for electrochemical reduction based on first-principles calculations, the conservation of the orbital symmetry, and feature engineering via the machine-learning method.72 This descriptor is closely correlated with inherent atomic properties such as electro-negativity (χ), electron type and number (Nd/p). On this basis, CuCr/g-C3N4 for CH4 and CuSn/N-BN for HCOOH with extremely low onset potentials of −0.24 and −0.11 V have been identified.
to assess the catalytic performance of 2D material-supported dual-atom catalysts (DACs@2D) for electrochemical reduction based on first-principles calculations, the conservation of the orbital symmetry, and feature engineering via the machine-learning method.72 This descriptor is closely correlated with inherent atomic properties such as electro-negativity (χ), electron type and number (Nd/p). On this basis, CuCr/g-C3N4 for CH4 and CuSn/N-BN for HCOOH with extremely low onset potentials of −0.24 and −0.11 V have been identified.