
Catalytic routes towards polystyrene recycling

Abstract
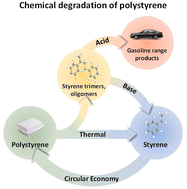
Polystyrene (PS) is one of the most popular plastics due to its versatility, which renders it useful for a large variety of applications, including laboratory equipment, insulation and food packaging. However, its recycling is still a challenge, as both mechanical and chemical (thermal) recycling strategies are often cost-prohibitive in comparison to current disposal methods. Therefore, catalytic depolymerization of PS represents the best alternative to overcome these economical drawbacks, since the presence of a catalyst can improve product selectivity for chemical recycling and upcycling of PS. This minireview focuses on the catalytic processes for the production of styrene and other valuable aromatics from PS waste, and it aims to lay the ground for PS recyclability and long-term sustainable PS production.
- This article is part of the themed collections: Polymer Upcycling, Emerging horizons in polymer applications and Recent Review Articles
 Please wait while we load your content...
Please wait while we load your content...

