
A processable, scalable, and stable full-color ultralong afterglow system based on heteroatom-free hydrocarbon doped polymers†
Xiaoxin
Zheng
a,
Quanxiang
Han
a,
Qinglian
Lin
a,
Cuicui
Li
a,
Jinke
Jiang
a,
Qing
Guo
a,
Xin
Ye
a,
Wang Zhang
Yuan
 b,
Yang
Liu
*a and
Xutang
Tao
*a
b,
Yang
Liu
*a and
Xutang
Tao
*a
aState Key Laboratory of Crystal Materials, Institute of Crystal Materials, Shandong University, Jinan 250100, P. R. China. E-mail: liuyangicm@sdu.edu.cn; txt@sdu.edu.cn
bSchool of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Key Lab of Electrical Insulation and Thermal Aging, Shanghai Jiao Tong University, Shanghai, 200240, China
First published on 23rd October 2022
Abstract
Although room-temperature phosphorescence (RTP) organic materials are a widely-studied topic especially popular in recent decades, long-lived RTP able to fulfil broad time-resolved application requirements reliably, are still rare. Polymeric materials doped with phosphorescent chromophores generally feature high productivity and diverse applications, compared with their crystalline counterparts. This study proves that pure polycyclic aromatic hydrocarbons (PAHs) may even outperform chromophores containing hetero- or heavy-atoms. Full-color (blue, green, orange and red) polymer–PAHs with lifetimes >5000 ms under ambient conditions are constructed, which provide impressive values compared to the widely reported polymer-based RTP materials in the respective color regions. The polymer–PAHs could be fabricated on a large-scale using various methods (solution, melt and in situ polymerization), be processed into diverse forms (writing ink, fibers, films, and complex 3D architectures), and be used in a range of applications (anti-counterfeiting, information storage, and oxygen sensors). Plus their environmental (aqueous) stability makes the polymer–PAHs a promising option to expand the portfolio of organic RTPs.
New conceptsOne of the main methods in the study of organic room temperature phosphorescence (RTP) materials is using hetero- or heavy-atoms containing chromophores to promote sufficient intersystem crossing (ISC) processes. However, as the spin-orbit coupling (SOC) effect induced by hetero- or heavy-atoms promotes ISC transitions of both S1 to Tn and T1 to S0, an intrinsic dilemma of increasing phosphorescence efficiency versus decreasing lifetime exists. This study provides a non-traditional family of polymer-based RTP materials doped with polycyclic aromatic hydrocarbons (PAHs). The weak SOC of the PAHs favors a slow radiative ISC transitions from T1 to S0, where nonradiative transitions are effectively suppressed in the rigid matrix. The T1 energy levels of PAHs could be tuned effectively by increasing the conjugation degree. Blue, green, orange and red polymer–PAHs with lifetimes of >5000 ms under ambient conditions, were constructed; which are impressive values compared to the widely-reported polymer-based RTP materials in the respective color regions. Furthermore, the polymer–PAHs could be produced through various methods (solution, fibers, films, and complex 3D architectures). Applications as anti-counterfeiting ink, information storage film, and oxygen sensor were fulfilled to demonstrate the wide potential of the polymer–PAHs. |
Introduction
Due to the radiative transition of excitons from the triplet excited state (Tn) to the singlet ground state (S0), organic room temperature phosphorescence (RTP) materials can provide interesting optical properties compared with conventional phosphorescent materials: including a long luminescence lifetime, large Stokes shifts and high exciton utilization.1–7 Consequently RTPs are superior and have promising potential applications in data encryption,8–11 environmental sensing,12–14 information storage,15,16 bioimaging and diagnostics,17–19 color displays20–22 and organic light-emitting diodes (OLEDs),23–26etc.In general, the challenge to generate RTP for organic chromophores lies in the insufficient intersystem crossing (ISC) caused by weak spin-orbit coupling (SOC), ultrafast nonradiative decays, and oxygen quenching. A variety of strategies have been proposed to promote ISC processes by inclusion of heavy- and/or hetero-atoms20,27,28 and deuteration,29,30 and to suppress nonradiative relaxations using crystal engineering,31–35 host–guest doping,2,7,36 crosslinking,37 polymerization38–41 and many other methods. As a result, a growing number of organic RTP materials with long-lived lifetimes (>100 ms) under ambient conditions have been reported in recent years.11,15,42–51 However, to realize the practical use of organic RTP materials in the above-mentioned applications, all-round demands must be satisfied for the materials. That is, besides the key criteria for optical properties of an organic RTP material, e.g., phosphorescence lifetime (τPhos), phosphorescence quantum yield (ΦPhos), and afterglow time, the material should also be processable into different compounds, scalable, stable, cost-effective and color-tunable. In such a case, some effective strategies such as crystal engineering – where many crystalline examples have been endowed with long-lived τPhos and tunable phosphorescence32,52–55 – may lose their competitiveness. Indeed the confinement of crystal lattice protects radiative phosphorescence transitions from molecular vibration and oxygen quenching,31,32,56–59 and our group has grown inch-size RTP single-crystals;35 but poor reproducibility caused by the tricky growth conditions and weak processability caused by the fragile nature of organic crystals, really hamper their application.
On the other hand, polymer-based RTP materials, which normally bear amorphous morphology, have attracted increased attention due to their features in large-scale production and accessible processability. Quite a few polymer-based RTP materials have been prepared by chemical modification of traditional polymers and doping of organic chromophores into the polymer matrices.5,40,41,60 However, to create overall promising options – with both long-lived RTP lifetimes (e.g., the afterglow time persists longer than 10 s to ensure naked-eye visibility during the dynamic processes) and acceptable productivity with applicability – most polymer-based materials are still unsatisfactory. The contradiction is even more severe for long-wavelength phosphorescent materials, because most of the reported RTP strategies can only provide long-lived RTP materials with similar blue and green phosphorescent colors; and phosphorescent color tuning at a longer wavelength via molecular structure modification, with negligible sacrifice in lifetime, is a hard task in most cases. Therefore RTP polymers with multicolored and long-lived phosphorescence still rare.
Herein we introduce an effective strategy to obtain ultralong lifetime and color-tunable phosphorescence in polymer-based RTP materials by doping with a series of polycyclic aromatic hydrocarbons (PAHs). The PAH dopants, derived from coronene to truxene and hexabenzocoronene, are pure hydrocarbons bearing no heavy- or hetero-atoms. They are proved to be excellent long-lived afterglow phosphorescence emitters when being doped into polymethyl methacrylate (PMMA) and polycarbonate (PC) matrices, wherein their two-dimensional fused and rigid structures contribute to lower the vibrational and rotational nonradiative transition. Coronene and its deuteride may be the earliest discovered organic RTP materials; reported back in 1967 by Kropp and Dawson, they had ultralong lifetimes of 5580 ms and 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ms, respectively.29,61,62 Unfortunately, such an outstanding result even today, has not received the deserved attention, and no other PAHs have been found to possess lifetimes approximating that of coronene in the subsequent few decades.1–3,7,51,63 Alternatively, researchers have turn their attention to hetero- or heavy-atoms containing chromophores, which are currently mainstream in the study of RTP materials. However, because the SOC effect induced by hetero- or heavy-atoms promotes ISC transitions of both S1 to Tn and T1 to S0, an intrinsic dilemma between increasing phosphorescence efficiency and decreasing lifetime exists in these systems.44 In this context, as pure hydrocarbons without heavy- or hetero-atoms, the PAHs may have more chance to realize ultralong afterglow time in virtue of the sluggish transition of excitons from the triplet excited state to the singlet ground state.
000 ms, respectively.29,61,62 Unfortunately, such an outstanding result even today, has not received the deserved attention, and no other PAHs have been found to possess lifetimes approximating that of coronene in the subsequent few decades.1–3,7,51,63 Alternatively, researchers have turn their attention to hetero- or heavy-atoms containing chromophores, which are currently mainstream in the study of RTP materials. However, because the SOC effect induced by hetero- or heavy-atoms promotes ISC transitions of both S1 to Tn and T1 to S0, an intrinsic dilemma between increasing phosphorescence efficiency and decreasing lifetime exists in these systems.44 In this context, as pure hydrocarbons without heavy- or hetero-atoms, the PAHs may have more chance to realize ultralong afterglow time in virtue of the sluggish transition of excitons from the triplet excited state to the singlet ground state.
Therefore, distinct from the reported materials that achieve single morphology of multi-color ultra-long organic phosphorescence by adjusting the composition of crystals,54 CDs,50 MOFs,51etc., or co-doping multiple color dyes,51 we aim to expand the portfolio of PAHs beyond coronene by systematically developing a generic approach to construct multi-color high-performance organic RTP materials with various morphologies. Firstly, to broaden the color palette of the polymer–PAHs-based phosphorescence, another two PAHs – truxene and hexabenzocoronene – were adopted besides coronene (Fig. 1a). With the increasing conjugation degree, the T1 energy levels of truxene, coronene, and hexabenzocoronene are found to decrease accordingly, resulting in the RTP color of the doped PMMA and PC changing from blue to orange. Additionally, upon co-doping with a fluorescent dye, triplet-to-singlet energy transfer from the long-lived phosphorescent donor to the fluorescent acceptor further extended the afterglow wavelength to 650 nm. Moreover, all the blue, green and orange polymer–PAHs RTP materials possess ultralong lifetimes (>4000 ms) and afterglow times (>30 s), and the lifetime of the co-doped red RTP material is over 2800 ms with an afterglow time >20 s. To our knowledge, these results are impressive when compared to the ever-reported polymer-based RTP materials in blue, orange and red regions (Table S1 and Fig. S1, ESI†). Taking advantage of the facile high productivity and diverse products of polymer-based RTP materials, we show that they can be produced by various methods, e.g., solution, melt and in situ polymerization, and could be processed into various forms, e.g., writing ink, fiber, film, and bulk architecture. Dynamic anti-counterfeiting ink, information storage films and oxygen sensors are demonstrated, proving the heteroatom-free PAHs doped polymers to be long-lived, color-tunable, processable into different compounds, scalable, and stable RTP systems.
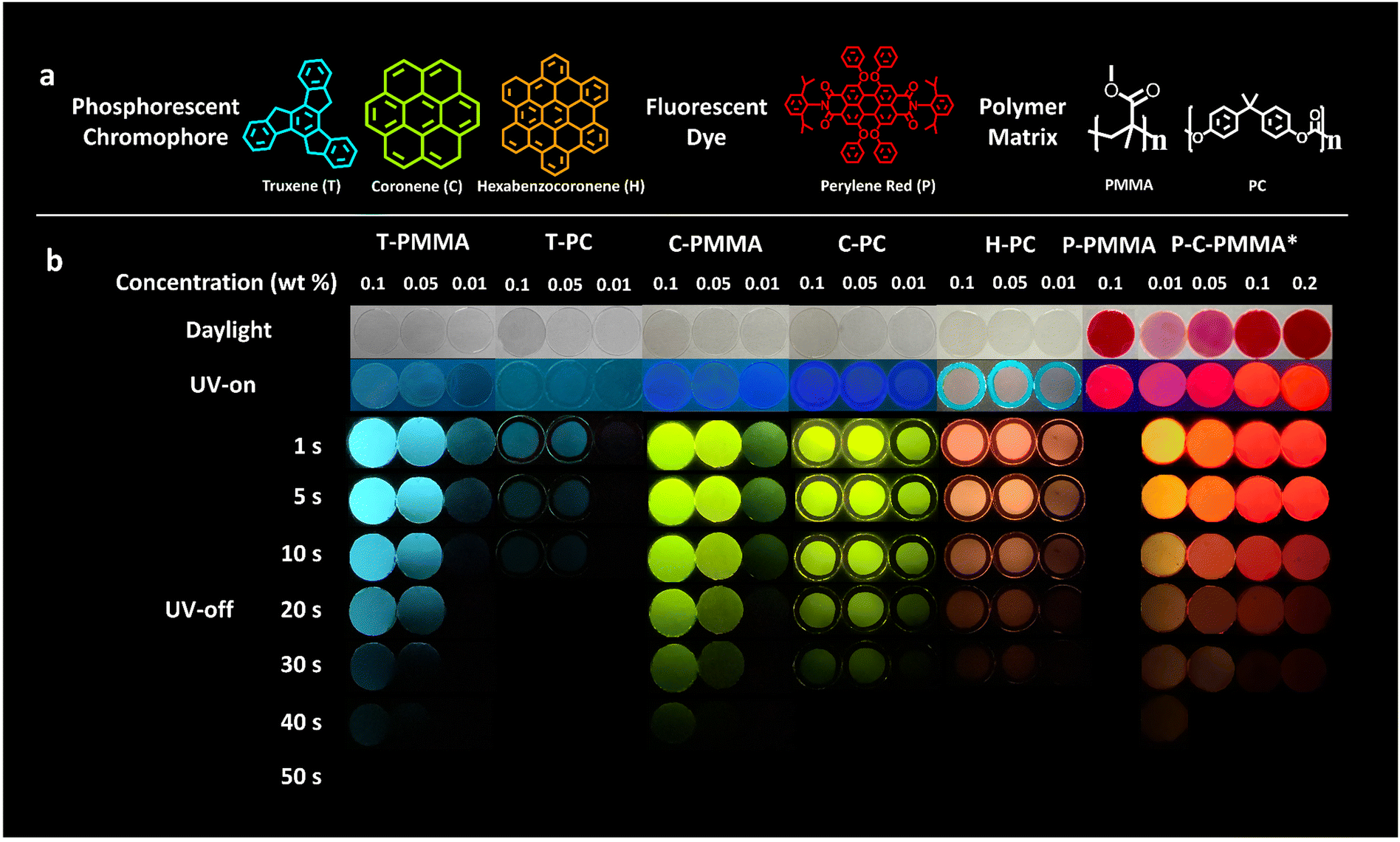 | ||
| Fig. 1 Molecular structures and RTP performance of the polymer–PAHs. (a) Molecular structures of the PAH dopants, fluorescent dye perylene red, and the polymer matrices PMMA and PC. (b) Photographs of the polymer–PAHs and fluorescent dye-doped and co-doped films with different doping concentrations taken in daylight, UV irradiation, and after turning off the UV irradiation for 0 to 50 s (* the concentration of coronene for P-C-PMMA is 0.1 wt%, and the concentration of perylene red for P-C-PMMA is 0.01, 0.05, 0.1, 0.2 wt%, respectively). | ||
Results and discussion
As depicted in Fig. 1a, the three kinds of PAH we employed to fabricate the color-tunable doped polymer–PAH RTP systems are truxene (T), coronene (C) and hexabenzocoronene (H). Coronene is commercially available, while truxene and hexabenzocoronene were synthesized in our lab according to the literature.64,65 Detailed synthetic procedures and characterizations including nuclear magnetic resonance (NMR), matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS), and elemental analysis to ensure the identity and purity of the target compounds are presented in the ESI.†PMMA and PC are selected as the amorphous matrices of the PAHs. Other polymers such as polyethylene glycol terephthalate (PET) and polystyrene (PS), were also employed as the dispersion matrices; but they were found to be incompatible with the selected PAH dopants, forming non-uniform and opaque films (Fig. S7, ESI†). The compatibility between the PAH dopants and PMMA and PC matrices was carefully explored to optimize the appropriate doping concentration using the solution mixing method (Fig. S8, ESI†). The results are shown in Fig. S9, S10 and Table S2 (ESI†). Truxene is soluble in PMMA when the concentration is not higher than 0.1 wt%; while coronene is soluble in PMMA even at a concentration of 0.5 wt%. Hexabenzocoronene is found to be almost immiscible with PMMA, which may be due to its large and rigid molecular structure. On the contrary, PC has better compatibility with the three PAHs; hexabenzocoronene is miscible in it at a concentration of 0.1 wt%. Thus, for a fair comparison, the doping concentrations of 0.1, 0.05, 0.01 wt% were chosen to investigate the photophysical properties of all the doped polymer–PAH systems. The compatibility of the fluorescent co-dopant, perylene red, in PMMA was also determined by the solution method, which showed a solubility of no less than 2.5 wt%. (Fig. S11, ESI†).
Fig. 1b shows the RTP properties of the polymer–PAH systems. Firstly, we could see the color-tunability realized by the inclusion of the different PAH dopants into the polymer matrices. The truxene-doped PMMA/PC (T-PMMA, T-PC), coronene-doped PMMA/PC (C-PMMA, C-PC), and hexabenzocoronene-doped PC (H-PC) are purple or blue under ultraviolet (UV) irradiation; while after removing the UV-excitation source, the truxene-doped polymers provide a blue afterglow emission, the coronene-doped polymers provide a green afterglow emission, and hexabenzocoronene-doped polymers provide an orange afterglow emission. The red afterglow RTP is constructed by co-doping C-PMMA with a fluorescent dye, perylene red (chemical structure is shown in Fig. 1a), in which (P-C-PMMA) Förster-resonance energy transfer (FRET) from the long-lived phosphorescent donor to the fluorescent acceptor further extend the afterglow wavelength to 650 nm. Fig. 2c shows the Commission International de l’Eclairage (CIE) coordinate diagram of these hybrid films calculated from their phosphorescence spectra. The coordinates of T-PMMA/PC (0.14, 0.28), C-PMMA/PC (0.36, 0.62), H-PC (0.61,0.39), and P-C-PMMA (0.68, 0.32) cover the primary colours (blue-green-red) triangle. Secondly, the afterglows are facilely visible under ambient conditions as the duration time is >30 s, except for T-PC. Movies S1–S6 (ESI†) record the afterglow decay processes of the five polymer–PAH systems with the same doping concentration (0.1, 0.05, and 0.01, wt%) as those in Fig. 1b. Such a long afterglow duration time would benefit the dynamic processes in the encryption and sensing applications. Thirdly, the doping concentrations affect the RTP performance, mainly on the intensity and duration time. Upon increasing the doping concentration from 0.01 wt% to 0.1 wt%, the afterglow luminance brightened and the duration time was elongated, indicating that these polymer–PAHs systems had optimal doping concentrations limited by the miscibility, where 0.1 wt% is optional. The RTP behavior of the coronene and perylene red co-doped P-C-PMMA with the perylene red concentrations from 0.01 to 0.2 wt% were also investigated. As shown in Fig. 1b, P-PMMA with no coronene doping showed red fluorescence under UV excitation; while no afterglow could be recorded after removing the excitation source, indicating that doping of the fluorescent dye into PMMA alone, cannot trigger RTP. When keeping the coronene doping concentration at 0.1 wt%, afterglow color changes from orange to red could be seen along with the doping concentration of perylene red increasing from 0.01 to 0.2 wt%. At an equal doping ratio of 0.1 wt% for both coronene and perylene red, the triplet-to-singlet energy transfer from coronene to perylene red may establish an equilibrium, but the fluorescence and afterglow from the former could not be monitored at this ratio. Here it should be noted that the polymer–PAH samples were prepared by sandwiching the drop-casting films between two pieces of quartz glass under UV-activation for 30 s to consume the triplet oxygen (Fig. S12, ESI†).66 The morphology of these doped films was characterized to be amorphous by powder X-ray diffraction (PXRD) (Fig. S13, ESI†), proving that the amorphous polymeric matrix is effective and robust to boost long-lived RTP of the planner and rigid PAHs.
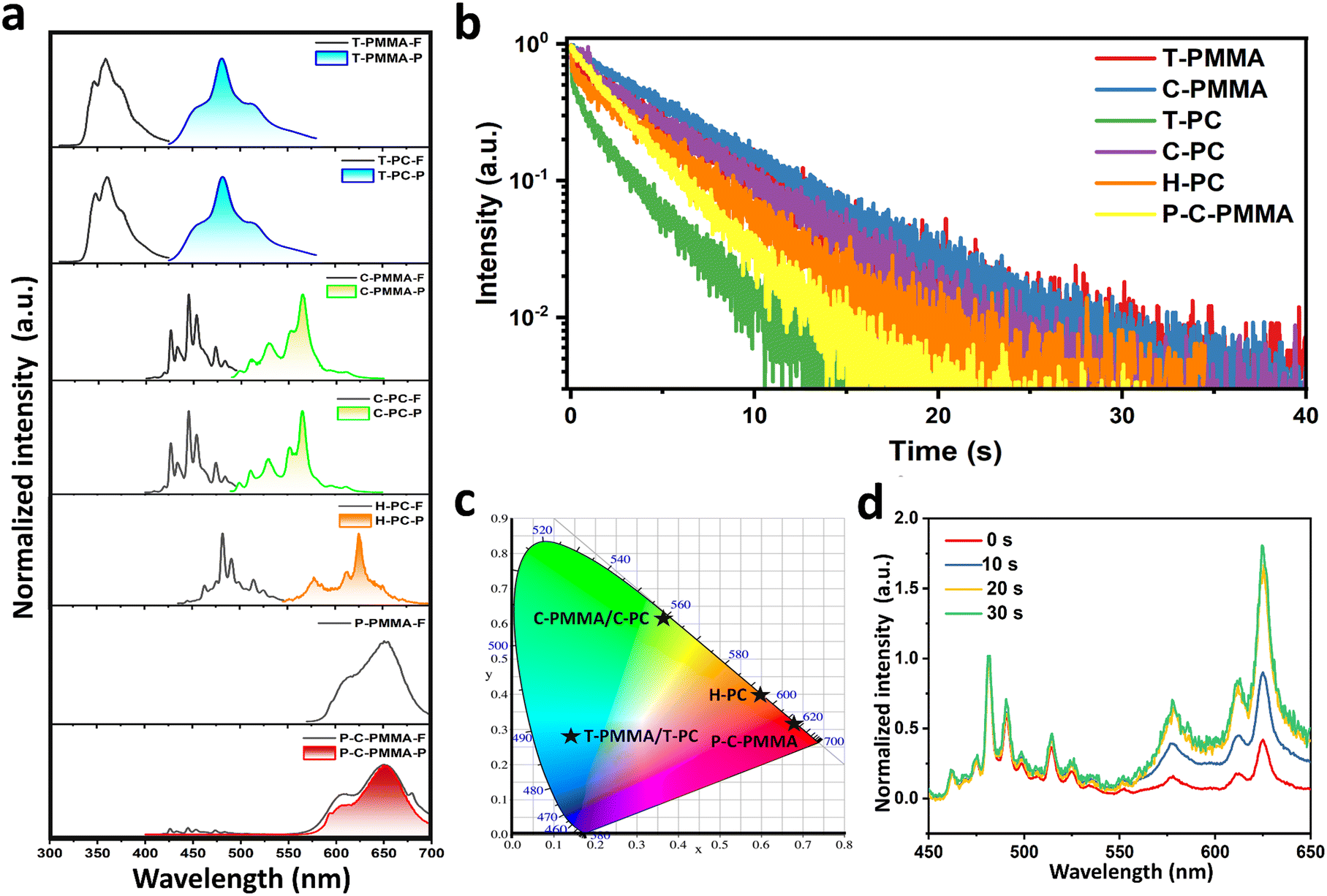 | ||
| Fig. 2 Photophysical properties of polymer–PAHs. (a) Normalized prompt (black) and delayed (colored) photoluminescence spectra (delay time = 10 ms) of polymer–PAHs. (b) Phosphorescence lifetime decay profiles of polymer–PAHs. (c) Commission International de l’Eclairage (CIE) color coordinates of polymer–PAHs. (d) The steady-state photoluminescence spectra of H-PC after different periods of UV-activation (356 nm). | ||
To comprehensively investigate the optical properties of polymer–PAHs, the spectrometric and time-related photophysical profiles of the doped systems with 0.1 wt% doping concentration were recorded under ambient conditions. Detailed photophysical information is shown in Fig. 2 and Table 1. Fig. 2a presents the steady-state (prompt) photoluminescence (PL) spectra (black line, marked with X-F) and the time-resolved (delayed) spectra (colored line, marked with X-P) of all the PMMA- and PC-based systems, including those of the perylene red co-doped system. Both truxene and coronene show nearly identical spectral profiles when doped into PMMA and PC matrices, with a prompt PL emission peak at ∼359 nm and a delayed phosphorescence peak at ∼480 nm for T-PMMA and T-PC, and a prompt PL emission peak at ∼445 nm and a delayed phosphorescence peak at ∼565 nm for C-PMMA and C-PC. The similarity of both the prompt and delayed spectra in different polymer matrices indicates that the fluorescence and phosphorescence of polymer–PAHs originate from the PAH dopants, whose emission wavelength is unaffected by the polymer matrices. The molecularly dispersing nature of the PAH molecules in the polymeric matrices are further confirmed by comparison with their fluorescence spectra in solutions. Truxene, coronene and hexabenzocoronene in dilute (10−5 mol L−1) CH2Cl2 solutions exhibit identical fluorescence spectra to those of the doped polymers (Fig. S14, ESI†). Due to the poor immiscibility of hexabenzocoronene in PMMA, we could only measure the H-PC to have a similar fluorescence spectrum as it is in solution, and an orange phosphorescence emission peaking at ∼625 nm.
| T-PMMA | T-PC | C-PMMA | C-PC | H-PC | P-PMMA | P-C-PMMA | |
|---|---|---|---|---|---|---|---|
| a λ ex: excitation wavelength. b τ fluo: fluorescence lifetime. c τ Phos: phosphorescence lifetime. d Φ total: total luminescence quantum efficiency. e Φ fluo: fluorescence quantum efficiency. f Φ Phos: phosphorescence quantum efficiency. | |||||||
| λ ex [nm] | 297 | 297 | 339 | 339 | 356 | 565 | 339 |
| τ fluo [ns] | 50.57@359 [nm] | 43.06@359 [nm] | 155.93@445 [nm] | 166.38@445 [nm] | 50.78@482 [nm] | 10.01@650 [nm] | — |
| τ Phos [ms] | 4687@480 [nm] | 2646@480 [nm] | 5501@565 [nm] | 4705@565 [nm] | 4007@625 [nm] | — | 2804@650 [nm] |
| Φ total | 12.59% | 6.81% | 16.64% | 9.53% | 16.68% | 92.61% | 52.14% |
| Φ fluo | 8.72% | 4.74% | 12.56% | 6.96% | 4.81% | 92.61% | — |
| Φ Phos | 3.87% | 2.07% | 4.08% | 2.56% | 11.87% | — | — |
As the red RTP material was realized through triplet-to-singlet energy transfers following a FRET mechanism (Fig. S15, ESI†),67–73 the excitation spectrum of perylene red doped PMMA (P-PMMA, 0.1 wt%) was examined (Fig. S16a, ESI†) and was found to overlap significantly with the phosphorescence spectrum of C-PMMA, satisfying the prerequisite of FRET. Furthermore, the high fluorescence quantum yield, 92.6% of P-PMMA (0.1 wt%) could facilitate the FRET conversion efficiency. P-C-PMMA and P-PMMA possess nearly identical steady-state PL spectra with a maximum peak at 650 nm attributed to perylene red, and this fluorescence spectrum also superposes the delayed PL spectrum of P-C-PMMA. In the resultant P-C-PMMA, neither the fluorescence at 445 nm nor the phosphorescence at 565 nm from coronene were observed, evidencing the nearly complete energy transfer of the triplet-to-singlet FRET. P-C-PMMA showed a long-lived afterglow emission peaking at 650 nm with lifetimes over 2804 ms (Fig. 2b), with a duration time over 20 s (Fig. 1b and Movie S6, ESI†). Accordingly, the triplet-to-singlet FRET efficiencies (ΦFRET) could be calculated to be 72.5% based on the equation ΦFRET = 1 − τ/τ0, where τ0 and τ are the RTP lifetimes of the energy donor (C-PMMA) before and after energy transfer (Table 1 and Fig. S16b, ESI†).67
The phosphorescence lifetime profiles of the polymer–PAHs are presented in Fig. 2b with the data listed in Table 1. We can see that all of the materials exhibit ultralong phosphorescence following a power law decay. Among them the PMMA-based materials generally hold longer lifetimes compared with those of the PC-based materials. The lifetimes of T-PMMA and C-PMMA are up to 4687 ms and 5501 ms, respectively; correspondingly, the lifetimes of the PC-based materials T-PC and C-PC are 2646 ms and 4705 ms, respectively. This indicates that although the emission wavelength is independent on the polymer matrices, the dynamic process of the excited state related to the lifetime and afterglow duration time is significantly affected by the surrounding matrices. The more rigid mechanical property of PMMA than that of PC is regarded to be responsible for this difference; where in PMMA the molecular vibrations and rotations cause nonradiative transitions, as well as oxygen quenching, which are more effectively suppressed than in PC.
Because the oxygen molecule has a unique triplet ground state, oxygen quenching usually plays a significant role in phosphorescence. Consumption of oxygen buried in the sample through a continuous period of UV irradiation is found to be effective at inducing RTP.68,74–78 In our work, the prepared samples before UV irradiation also showed obvious weak RTP emissions, especially on the peripheral parts of the films, as shown in Fig. S17 (ESI†); after UV irradiation for about 30 s, afterglow phosphorescence was recovered in all regions of the samples. This phenomenon is even noteworthy for H-PC, as its prompt emission colour changed from blue to orange after UV irradiation, becoming similar to that of the delayed emission (Fig. S17, ESI†). This process is confirmed by recording the prompt emission spectra after each 10 s UV irradiation, during which, the phosphorescence gradually increases in intensity to reach 1.8 times that of the fluorescence peak (Fig. 2d). This means that with consumption of oxygen the phosphorescence gradually takes over the fluorescence. For the UV-activated H-PC, its phosphorescence quantum efficiency is measured to be 11.87%, which is even higher than that of its fluorescence quantum efficiency (4.81%). This indicates hexabenzocoronene in PC may have exceptionally high efficiency intersystem crossing or stabilization of the triplet excited states, which has been rarely discovered previously. The evolution of the prompt photoluminescence spectra as a result of different periods of UV photoactivation for T-PMMA, C-PMMA, T-PC and C-PC are shown Fig. S18 (ESI†). To further confirm that the activation is due to inhibition of the adverse effects of oxygen, the phosphorescence of the H-PC film was monitored under nitrogen or oxygen purging. As shown in Fig. S19 (ESI†), under a nitrogen environment, the H-PC exhibited phosphorescence without UV activation: when oxygen is introduced, the phosphorescence of H-PC is quenched and so continuous UV irradiation cannot trigger its phosphorescence. These results prove the effect of oxygen under UV activation. Generally speaking, PMMA provides more compact protection of the embedded PAH molecules from oxygen quenching. As shown in Fig. S17 (ESI†), the UV-activated films of PC–PAHs lose their afterglow activity on their outer perimeter after 24 hours, resulting in an obviously dark annular region as oxygen permeates into the films from the perimeter. While such oxygen permeation induced phosphorescence quenching was not observed in the PMMA–PAHs films.
Thus through doping with PAHs with tunable T1 energy levels and FRET fluorescent dye PMMA, full-color afterglows with ultralong lifetimes (4687 ms for blue, 5501 ms for green and 2804 ms for red) were realized, which are impressive values compared to the currently reported polymer-based RTP materials in the respective color regions (Table S1 and Fig. S1, ESI†). And on the other hand, PC–PAHs whose prompt and afterglow emissions are sensitive to oxygen are potential oxygen indicators.
It is worth noting that the three PAHs – truxene, coronene, and hexabenzocoronene – exhibited RTP properties either in their solution (10−5 mol L−1 in CH2Cl2) or crystal forms under ambient conditions (Fig. S14 and S20, ESI†). Considering crystallization is regarded as one of the best strategies to boost RTP by: (i) suppression of molecular vibration and rotation decay pathways; (ii) obstructing oxygen out of the crystal lattice; and (iii) facilitating triplet excitons generalization through compact intermolecular coupling,31,32,56–59 single crystal structures of the three materials were determined to excavate such an exception by analyzing the molecular conformation and intermolecular interactions in crystals. As shown in Table S3 (ESI†) and Fig. 3a, all three crystals belong to a same space group, P21/c, wherein the molecules stack parallel so they are partially face to face. The π–π stacking distances (d) for truxene, coronene, and hexabenzocoronene are 3.499, 3.457, and 3.527 Å, respectively; and the angles between the molecular transition dipoles and the interconnecting axis (θ) are 43.254°, 42.398°, and 47.737°, respectively, manifesting J-aggregates according to the critical value of 54.7°.79 The formation of J-aggregates was further evidenced by the red-shift of the absorption and fluorescence peaks of the crystals relative to those of the corresponding solutions (Fig. S14 and S20, ESI†). The weakly coupled J-aggregates may afford insufficient intersystem crossing and inadequate stabilization of the triplet excited states,33,54,80–82 thus preventing RTP activity in the pure crystals. On the other hand, PAH molecules in polymer matrices exist as isolated individuals, as evidenced by the coincidence of both the absorption and fluorescence spectra of the polymer–PAHs with those of PAHs in dilute solutions (Fig. 2a, Fig. S14 and S21, ESI†). The point of the polymer matrix relative to the liquid solutions, lies in providing the isolated PAH chromophores with a rigid environment to sustain triplet exciton formation and to alleviate nonradiative decay.
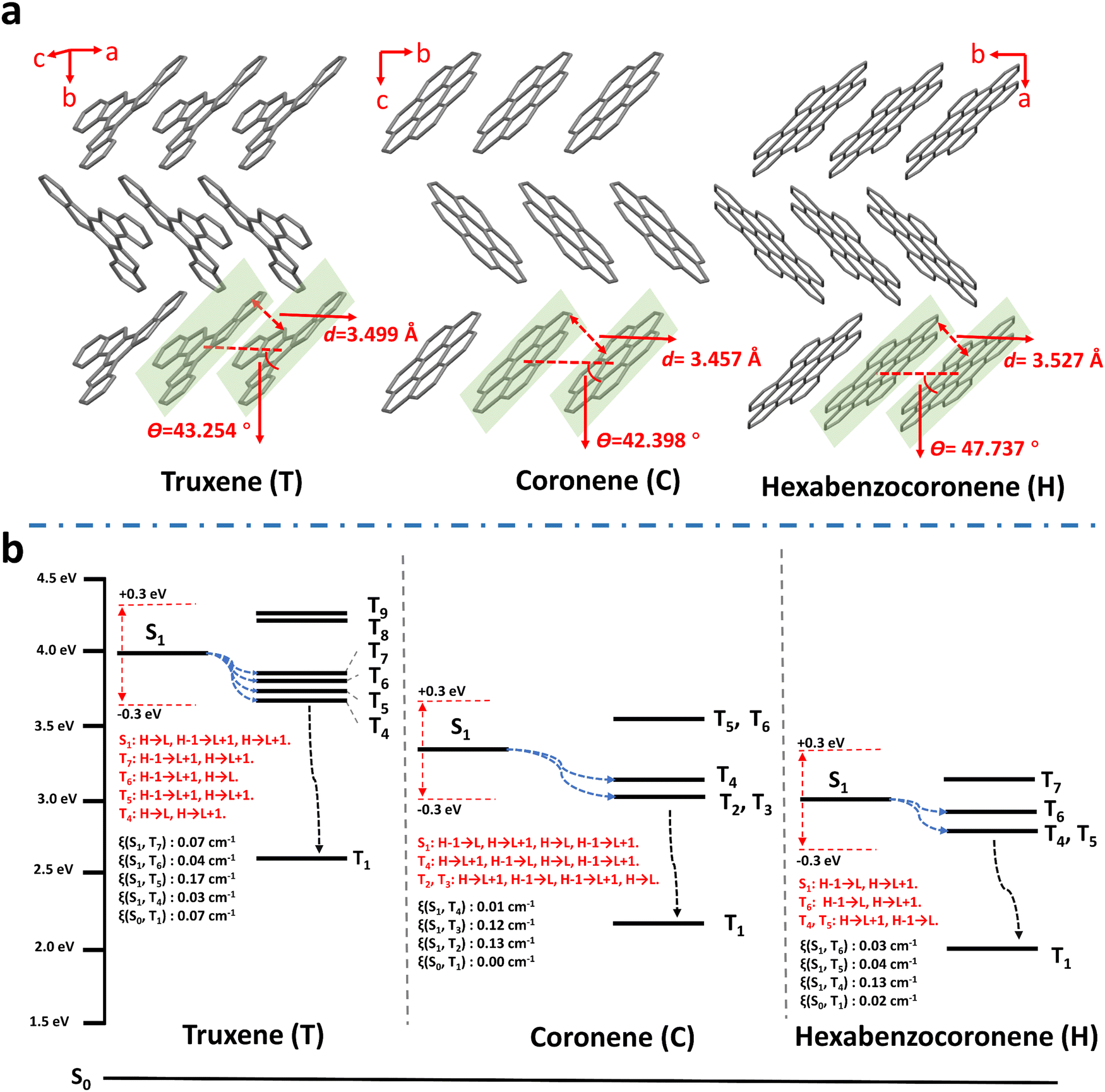 | ||
| Fig. 3 Single-crystal structures and energy level diagrams of isolated molecules of truxene, coronene and hexabenzocoronene. (a) Molecular packing structures of truxene, coronene, and hexabenzocoronene crystals. The π–π stacking distances (d) and the angles between the molecular transition dipoles and the interconnecting axis (θ) are labelled. (b) The excited-state energy level diagrams for isolated molecules of truxene, coronene and hexabenzocoronene. The blue dotted arrows represent the possible ISC channels, and the black dotted arrows represent the transition channels of Tn → T1. | ||
Based on the isolated single PAH molecular structure optimization using time-dependent density functional theory (TD-DFT), the excited energy levels of both singlet and triplet states, and the ISC transitions were calculated and analyzed15,44,54,83–85 (Fig. 3b and Tables S4–S6, ESI†). Firstly, along with increasing the conjugation degree from truxene and coronene, to hexabenzocoronene, their corresponding ET1 (energy level of the lowest triplet excited state) decreases from 2.621, 2.181, to 1.960 eV, being also in accordance with their respective experimental phosphorescent emission at 480, 565, and 625 nm, respectively. This on the one hand further confirms the phosphorescence of polymer–PAHs originating from T1–S0 radiation transitions of isolated PAH species, rather than the aggregates or radical ions, and on the other hand, proves the feasibility of RTP color tuning through dopant molecular structure modification. Secondly, the ISC transition from S1 to Tn is regarded as possible when the energy level of a triplet state (Tn) lies within the range ±0.3 eV to ES1 (the energy level of the lowest excited singlet S1 state), while with the same transition orbital compositions.54,81,82,86 The SOC constants (ξ) between the involved singlet and triplet states can be referred to estimate the possibility of ISC.87–89 As shown in Fig. 3b and Tables S4–S6 (ESI†), PAHs present plenty of ISC channels owing to their multi-resonance conjugated structure; however, the SOC constants of S1–Tn and S0–T1 are much smaller when compared with those of the reported molecules bearing heavy- or hetero-atoms.87–91 Honestly, we can still not conclude the full contributors leading to the prominent RTP activities of the polymer–PAHs. However, at least for the ISC transition of T1 → S0, a small SOC constant means a slow radiative transition process if the nonradiative transitions are effectively suppressed in the rigid matrix. This is consistent with El-Sayed's rule (Fig. S22, ESI†), in which the transition from 3(π, π*) to 1π2 (S0) would bring about a slow RTP process in the absence of 1(n, π*) and 3(n, π*) for the heavy- and hetero-atom free PAHs.2,92 The above experimental and theoretical results prove the polymer–PAHs are strong candidates for ultralong-lived and color-tunable RTP materials.63,70
Considering the limitation of computing power, the current calculations of SOC for the doped polymer systems are based on the optimized gaseous single-molecular structure, wherein the effects of the polymer matrix are not taken into account. Obviously the polymeric matrices play a crucial role in the RTP of polymer–PAHs, because pure PAHs in crystal form or liquid organic solvents, display no observable RTP phenomena. Thus, the SOC calculations neglecting the polymeric matrix may give results deviating from the actual situation. Here we suggest that the multi-resonance conjugated structures of the PAHs are involved in the mechanism. The natural transition orbital (NTO) calculations based on the excited electronic structures were performed. As depicted in Fig. S23–S25 (ESI†), truxene, coronene and hexabenzocoronene all show planar and rigid structures, leading to delocalized orbital distributions over the π-conjugated skeletons. Multi-resonance characters in an alternating pattern across the π-backbones of the hole and particle pairs in NTOs are observed. In the multi-resonant structures, according to reports,93–95 the partial overlap of the hole and the particle demonstrates the hybrid local and charge transfer character in the S1 state, which facilitates the ISC process from S1 to Tn. Meanwhile, for the T1 state, the hole and the particle both distribute on the same atoms. The overlap of the hole and the particle demonstrates a typical locally excited character, which makes the transition from T1 to S0 difficult, and leads to a smaller SOC value and phosphorescent radiative transition rate. This phenomenon is particularly evident in truxene, and can also be seen in coronene and hexabenzocoronene through atom-by-atom analysis.
To make full use of the long-lived RTP activity in multifarious scenarios, high productivity and diverse applications are prerequisites. Deriving from the intrinsic properties and fabrication techniques of polymers, as well as the low doping concentration of PAHs in the polymers, the polymer–PAHs could be produced through various methods such as solution, melt and in situ polymerization, and be processed into diverse forms, such as for writing inks, fibers, films, and bulk architectures. As shown in Fig. 4a, by directly loading the solutions of T-PMMA (blue), C-PMMA (green), and P-C-PMMA (red) with a certain concentration (200 mg ml−1 of PMMA) into inkwells, any words or figures can be expediently drawn on the paper or other substrates; after removing the UV excitation, the drawn words and figures exhibited vivid and high contrast blue, green and red afterglows sustained for >30 s (Movies S7–S9, ESI†). By spin-coating or drop-casting the solutions on a mould, RTP films could be prepared with a specific size. The films are flexible and could be tailored into diverse forms, e.g., pinwheel and bracelet with colorful afterglows (Fig. 4b and Movie S10, ESI†). Furthermore, casting or hot-pressing the melt of polymer–PAHs in proper moulds also produced large-area RTP films. Fig. 4b shows a ∼60 cm long tube composed of extruded RTP films (Fig. 4b and Fig. S26, ESI†), drawing from the melt we manufactured fibers of polymer–PAHs with diameters of several hundreds of micrometers tuned by the drawing speed. Fig. 4c shows that the full-color RTP fibers are flexible and could be bent into complex shapes (Movies S11 and S12, ESI†). Moreover, the RTP materials can also be processed through in situ chemical polymerizations of the PAH-doped monomers. As shown in Fig. 4d, three-dimensional figures – a deer with blue-afterglow horn, red-afterglow eyes, and green-afterglow body, and a red-afterglow fish – were fabricated by using in situ polymerization (Movie S13, ESI†). By assembly of in situ polymerized art called “FU” (means luck and fortune) and a bunch of fibers, we could create a pendant with multi-color RTP (Fig. 4e and Movie S14, ESI†), Fig. S26 (ESI†) gives some experimental details. In a word, the polymer–PAHs are indeed a versatile RTP system which is compatible with multiple manufacturing methods and various product modalities to accommodate wider application scenarios.
 | ||
| Fig. 4 Manufacturing polymer–PAHs into inks, films, fibers and 3D objects, through solution, melt and in situ polymerization methods. (a) Photograph of solution inks under daylight, (left) and the afterglow photographs of written words and figures after turning off the UV irradiation (right). (b) Photograph of the solution drop-casted films and a ∼60 cm long melt-extruded RTP tube under daylight, (left) and the afterglow photographs of the RTP films and the film-made pinwheel, bracelets, and tube after turning off the UV irradiation (right). (c) Photograph of the RTP fibers drawn from the melt under daylight (top left) and after turning off the UV irradiation (bottom left); and the afterglow photographs of complex shapes constructed by the fibers (right). (d) Photograph of in situ polymerized deer and fish under daylight (left) and the afterglow photographs after turning off the UV irradiation (right). (e) Photographs of a pendant composed of in situ polymerized artware “FU” and different color afterglow fibers under daylight (left) and after turning off the UV irradiation (right). | ||
It should be noted that for the blue-afterglow T-PMMA and green-afterglow C-PMMA, their ink solutions, inkblots and films are colorless and transparent under daylight. The transmission of T-PMMA and C-PMMA films (thickness ∼600 μm) was measured to be 91% across the visible region, similar to that of the pure PMMA film (Fig. S27, ESI†). On the contrary, P-C-PMMA has a significant absorption peak at 580 nm, corresponding to the red color under daylight. Accordingly, T-PMMA and C-PMMA could be used as invisible RTP materials; while P-C-PMMA is consistently red under daylight, UV irradiation, and after UV irradiation is removed. Thus, in light of the long-lived afterglows with response-dependent colors, the polymer–PAHs should be a good platform for developing time-resolved multi-level encryption and anti-counterfeiting.
As a proof-of-concept, we used inks of P-C-PMMA and P-PMMA to write the sentence “If winter comes, can spring be far behind?”. As shown in Fig. 5a, all the letters appear red under daylight and UV irradiation; while after removing UV irradiation, only the letters written by P-C-PMMA, “w e come n f r i.”, could be read because of their long-lived red RTP. The other letters written by P-PMMA disappeared when the UV irradiation was off, because they have no RTP effects (Movie S15, ESI†). By further introduction of another red dye with neither fluorescence nor phosphorescence emissions, a dual-responsive encryption system could be constructed. As demonstrated in Fig. 5b, the letters “RTP” were written with the non-emissive red dye, the ink of P-PMMA, and the ink of P-C-PMMA, respectively. Under daylight, the three letters “RTP” look the same in a red color; but under UV irradiation the fluorescence-active letters “TP” remain red emissive while the fluorescence-inactive “R” becomes invisible; after removing UV irradiation, only the letter “P” could be read because of its persistent RTP, thus realizing the secondary anti-counterfeiting (Movie S16, ESI†).
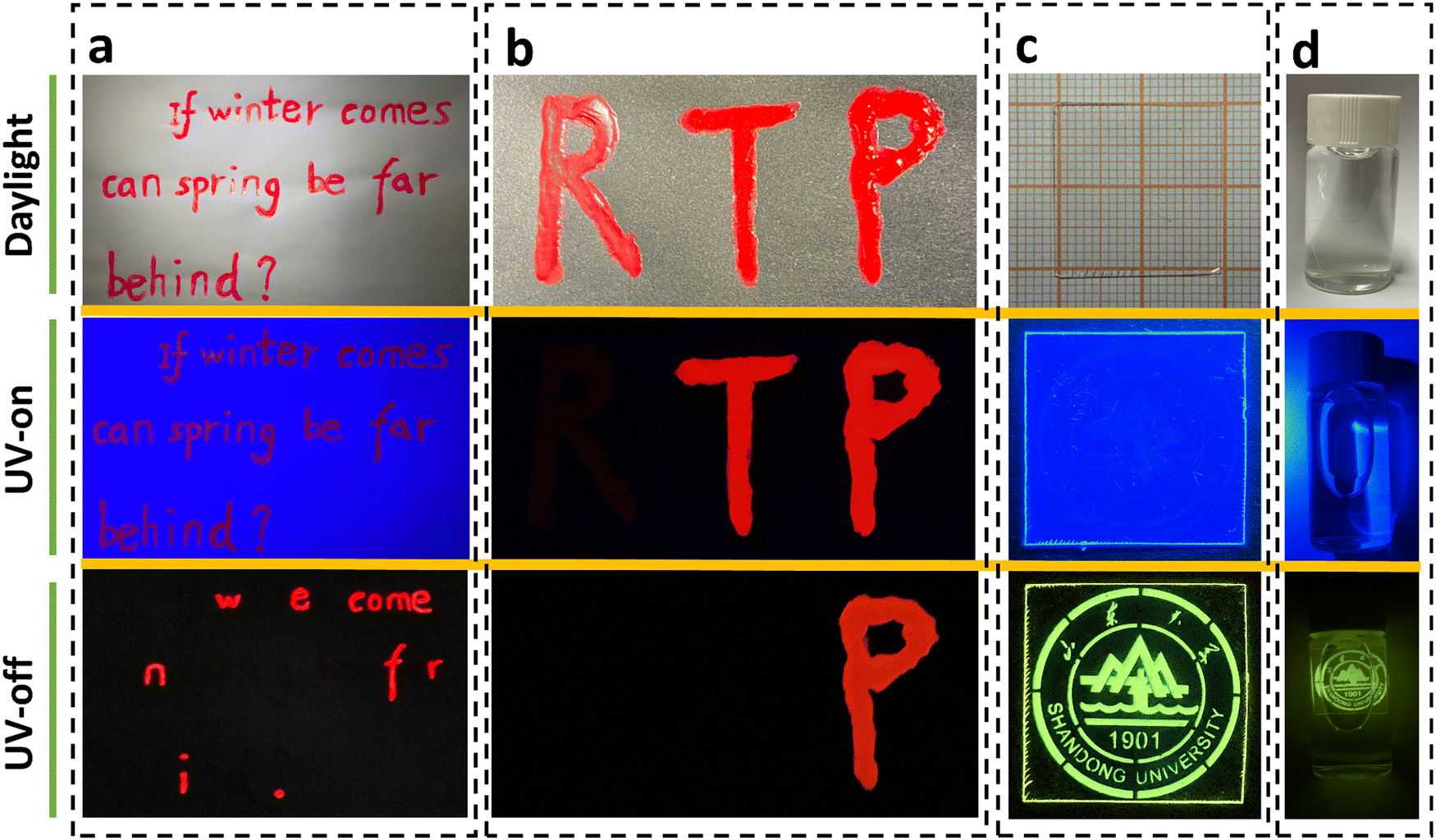 | ||
| Fig. 5 Information encryption and anti-counterfeiting demonstrations using polymer–PAH inks and films. (a) Photographs of a sentence written by P-PMMA and P-C-PMMA inks under daylight, UV irradiation and after turning off the UV irradiation. (b) Photographs of dual-responsive encryption letters written by non-emissive red dye, P-PMMA and P-C-PMMA. (c) Photographs of information written on a C-PMMA film under daylight, UV irradiation and after turning off the UV irradiation. (d) Photographs of information written on a C-PMMA film immersed in water. | ||
As mentioned above, the density of molecular oxygen plays a significant role in the triplet-involved phosphorescence. This enables us to dynamically and spatially regulate the RTP activities via adjusting the concentration of oxygen in the doped films. As depicted in Fig. S28 (ESI†), by filtered illumination of the C-PMMA film with continuous 254 nm UV light for ∼30 s, the images containing information of the filter, e.g., here an emblem of Shandong University, could be written on the film. The writing process is achieved by local removal of molecular oxygen on the spot illuminated by the UV. Fig. 5c shows a film after information writing, which was transparent under daylight and provides uniform blue emission under UV light. The encrypted information could only be captured after removal of the instantaneous excitation light, because the long persistent RTP pattern carries the corresponding information (Movie S17, ESI†). Furthermore, the written RTP image can be erased easily through 100 °C heating of the film; as the oxygen barrier permeability increases at elevated temperature. Thus, using filtered illumination and heat treatment, the photo-printed pattern can be conveniently changed, gifting the RTP films with optically accessible writing, reading, and erasing for information storage.
Moreover, the RTP property of the PAH doped PMMA exhibits excellent stability under atmospheric conditions or even in an aqueous environment. As shown in Fig. 5d, a C-PMMA film with local activation of the Shandong University emblem represents good RTP emission when soaked in water (Movie S18, ESI†). And thanks to the high phosphorescence brightness, the RTP emission in water could also be easily visualized in daylight (Fig. S29a, ESI†). Given the good water resistance of PMMA and the good chemical stability of PAHs, the phosphorescence of the written information remains perfectly readable after 3 days (Fig. S29b, ESI†).
Unlike PMMA, PC acting as a matrix provides relatively high oxygen permeability as aforementioned. The RTP effect of PC–PAHs, in consequence, is affected more heavily by the circumambient molecular oxygen, which makes them a good platform to design oxygen sensors. Considering that H-PC bears blue fluorescence and orange phosphorescence, such a high contrast in emission colors (Δ wavelength of ∼143 nm) makes a clear distinction between the oxygenated regions and oxygen-free regions in a H-PC film. As shown in Fig. 6a, because of the residual oxygen in the as-prepared H-PC film, most of the film displays blue fluorescence; while the phosphorescence is quenched in most areas with only a small area remaining orange. Along with the continuous irradiation of UV light, the residual molecular oxygen is gradually consumed, being accompanied by a noticeable color change of the H-PC film from dominant blue to dominant orange. After UV-activation, the H-PC film stored in air could be refilled with molecular oxygen from the periphery to the centre. The permeated oxygen quenched the RTP emission of H-PC film from the outside in, forming a growing annular non-RTP region.
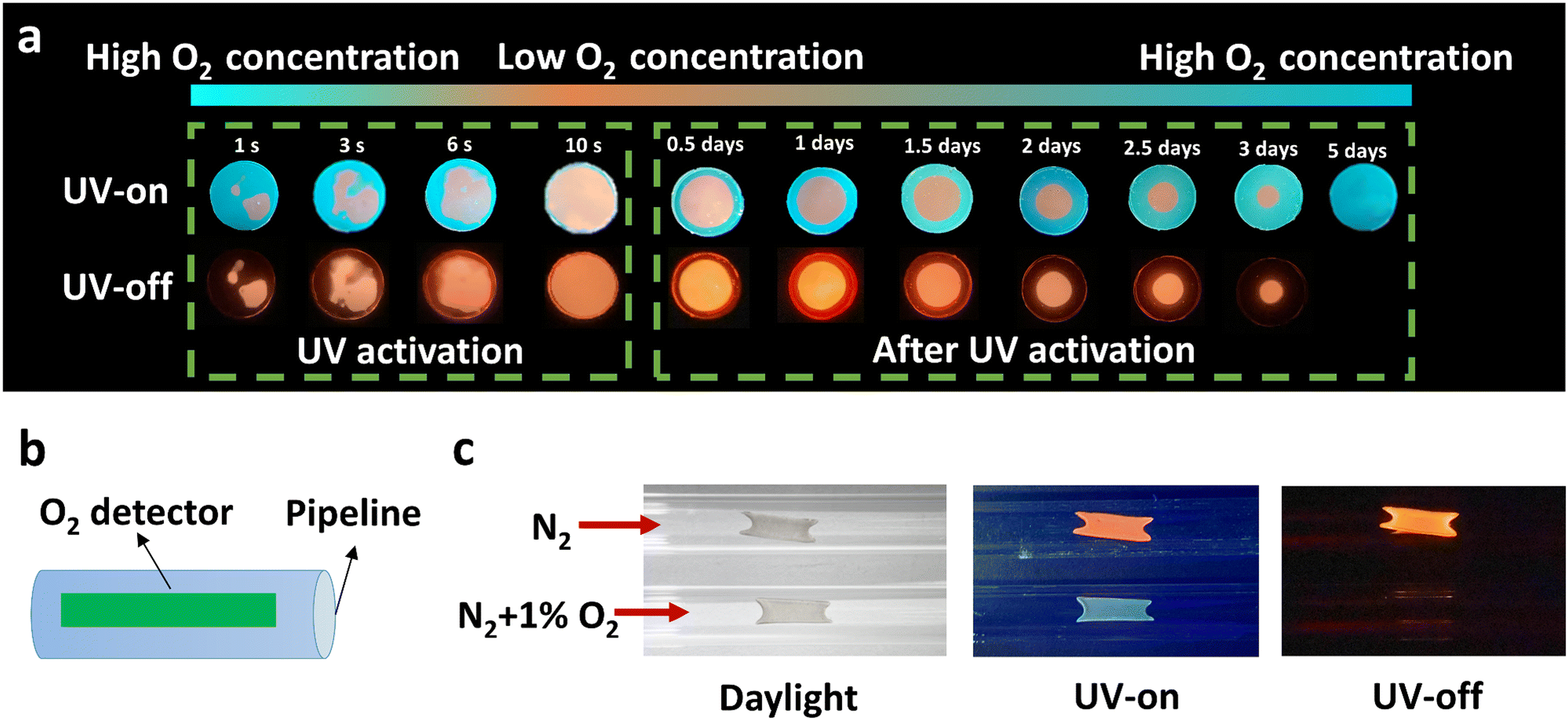 | ||
| Fig. 6 Molecular oxygen monitoring demonstration using H-PC. (a) Prompt (UV-on) and afterglow (UV-off) images show the change of H-PC films under UV activation, caused by the consumption of residual oxygen; and after UV activation, caused by oxygen permeation. (b) Schematic illustration of an H-PC oxygen monitor coated on the inner wall of a nitrogen pipeline. (c) Photographs of the H-PC oxygen monitor exposed to pure and 1% (v/v) O2 containing N2 under daylight, UV irradiation, and after turning off the UV irradiation. | ||
Based on this phenomenon, an on-line oxygen sensor is demonstrated, as shown in Fig. 6b and c. An H-PC film was coated on the inner wall of a nitrogen pipeline as the oxygen detector. After three hours piping of pure nitrogen gas, the H-PC detector showed orange RTP emission; while when the nitrogen gas contains 1% (v/v) oxygen, the H-PC detector showed no RTP effect with only blue steady-state emission (Fig. 6c). Fig. S30 (ESI†) shows the process of the gas mixture of N2 and O2 (1%) quenching the ultralong organic phosphorescence. The accumulative oxygen-quenching effect of RTP renders it more suitable to be utilized as a cumulated oxygen monitor.
Conclusions
In conclusion, by using a selected series of hetero- or heavy-atom free PAHs as dopants and PMMA or PC as polymeric matrices, a family of polymer–PAH RTP materials were constructed. The T1 energy levels of PAHs could be tuned effectively from blue to orange by increasing the conjugation degree; and upon co-doping with a FRET fluorescent dye, the RTP wavelength can be further extended to 650 nm, realizing full-color afterglows. Due to the weak SOC of the pure hydrocarbon PAHs, a slow radiative ISC transition from T1 to S0 leads to the blue, green, orange and red RTP possessing long-lived phosphorescence with lifetimes of >5000 ms under ambient conditions, which are impressive values compared to the commonly-reported polymer-based RTP materials in the respective color regions. Most importantly, the polymer–PAHs could be produced through various ways (solution, fibers, films, and complex 3D architectures). Application as anti-counterfeiting ink, information storage films, and as oxygen sensors were demonstrated showing the wide potential of the polymer–PAHs. This study provides a family of all-round players with long-lived RTP lifetimes, high brightness afterglow, color-tunability, high productivity and diverse applications, as well as environmental (aqueous) stability – which we believe will facilitate practical applications of the widely-studied RTP materials.Author contributions
X. T. and Y. L. conceived the project. X. Z. synthesized all of the materials and performed all the measurements. Q. H., Q. L., C. L., J. J., Q. G., X. Y., and W. Z. Y. discussed and revised the manuscript. All of the authors approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (Grants 51973106, 52273185, 52102006, and 51932004), the National Key Research and Development Program of China (Grants 2018YFB0406502), the 111 Project 2.0 in China (Grant PB2018013), and the Natural Science Foundation of Shandong Province (Grant ZR2021QE091, ZR202105230005). X. Y. is thankful for the China Postdoctoral Science Foundation (2021M701973). Y. L. is thankful for the support from the Distinguished Young Scholars of Shandong Province (ZR2019JQ03), and the Shandong University multidisciplinary research and innovation team of young scholars.Notes and references
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Small, 2022, 18, 2104073 CrossRef CAS.
- A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- M.-M. Fang, J. Yang and Z. Li, Chin. J. Polym. Sci., 2019, 37, 383–393 CrossRef CAS.
- Q. Li, Y. Tang, W. Hu and Z. Li, Small, 2018, 14, 1801560 CrossRef.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- X. Dou, T. Zhu, Z. Wang, W. Sun, Y. Lai, K. Sui, Y. Tan, Y. Zhang and W. Z. Yuan, Adv. Mater., 2020, 32, 2004768 CrossRef CAS.
- H. Liu, W. Ye, Y. Mu, H. Ma, A. Lv, S. Han, H. Shi, J. Li, Z. An, G. Wang and W. Huang, Adv. Mater., 2021, e2107612 Search PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS.
- J. Zhang, S. Xu, Z. Wang, P. Xue, W. Wang, L. Zhang, Y. Shi, W. Huang and R. Chen, Angew. Chem., Int. Ed., 2021, 60, 17094–17101 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CrossRef CAS.
- C. Qian, Z. Ma, X. Fu, X. Zhang, Z. Li, H. Jin, M. Chen, H. Jiang, X. Jia and Z. Ma, Adv. Mater., 2022, 34, 2200544, DOI:10.1002/adma.202200544.
- M. Gmelch, H. Thomas, F. Fries and S. Reineke, Sci. Adv., 2019, 5, eaau7310 CrossRef PubMed.
- Y. Fan, S. Liu, M. Wu, L. Xiao, Y. Fan, M. Han, K. Chang, Y. Zhang, X. Zhen, Q. Li and Z. Li, Adv. Mater., 2022, 34, 2201280, DOI:10.1002/adma.202201280.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS.
- C. Chen, H. Gao, H. Ou, R. T. K. Kwok, Y. Tang, D. Zheng and D. Ding, J. Am. Chem. Soc., 2022, 144, 3429–3441 CrossRef CAS PubMed.
- Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS.
- Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 139, 6376–6381 CrossRef CAS.
- S. Hirata and M. Vacha, Adv. Opt. Mater., 2017, 5, 1600996 CrossRef.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Brase, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- G. Zhan, Z. Liu, Z. Bian and C. Huang, Front. Chem., 2019, 7, 305 CrossRef CAS.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef.
- H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Adv. Opt. Mater., 2016, 4, 1015–1021 CrossRef CAS.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29 Search PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- S. Cui, T. Zhu, L. Zhang, X. Ye, Q. Han, C. Ge, Q. Guo, X. Zheng, Q. Lin, C. Li, J. Jiang, W. Yuan, Y. Liu and X. Tao, Adv. Opt. Mater., 2022, 10, 2102355 CrossRef CAS.
- N. Notsuka, R. Kabe, K. Goushi and C. Adachi, Adv. Funct. Mater., 2017, 27, 1703902 CrossRef.
- X. Lin, Q. Xu and X. Ma, Adv. Opt. Mater., 2021, 10, 2101646 CrossRef.
- Y. Zhu, Y. Guan, Y. Niu, P. Wang, R. Chen, Y. Wang, P. Wang and H. L. Xie, Adv. Opt. Mater., 2021, 9, 2100782 CrossRef CAS.
- B. Zhao, S. Yang, X. Yong and J. Deng, ACS Appl. Mater. Interfaces, 2021, 13, 59320–59328 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28 Search PubMed.
- H. Gao and X. Ma, Aggregate, 2021, 2 Search PubMed.
- Z. Y. Zhang, W. W. Xu, W. S. Xu, J. Niu, X. H. Sun and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 18748–18754 CrossRef CAS PubMed.
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 186 CrossRef CAS.
- H. Zhu, I. Badia-Dominguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. Ruiz Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS PubMed.
- S. Cai, Z. Sun, H. Wang, X. Yao, H. Ma, W. Jia, S. Wang, Z. Li, H. Shi, Z. An, Y. Ishida, T. Aida and W. Huang, J. Am. Chem. Soc., 2021, 143, 16256–16263 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS.
- L. Gu, H. Shi, M. Gu, K. Ling, H. Ma, S. Cai, L. Song, C. Ma, H. Li, G. Xing, X. Hang, J. Li, Y. Gao, W. Yao, Z. Shuai, Z. An, X. Liu and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 8425–8431 CrossRef CAS PubMed.
- J. Chen, X. Chen, L. Cao, H. Deng, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2022, 61, e202200343 CAS.
- H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Adv. Funct. Mater., 2019, 29 Search PubMed.
- C. Lin, Y. Zhuang, W. Li, T. L. Zhou and R. J. Xie, Nanoscale, 2019, 11, 6584–6590 RSC.
- J. Liu, Y. Zhuang, L. Wang, T. Zhou, N. Hirosaki and R. J. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 1802–1809 CrossRef CAS PubMed.
- F. Li, C. Qian, J. Lu, Y. Ma, K. Y. Zhang, S. Liu and Q. Zhao, Adv. Opt. Mater., 2021, 10, 2101773 CrossRef.
- Z. Wang, H. Yuan, Y. Zhang, D. Wang, J. Ju and Y. Tan, J. Mater. Sci. Technol., 2022, 101, 264–284 CrossRef.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- M. Singh, K. Liu, S. Qu, H. Ma, H. Shi, Z. An and W. Huang, Adv. Opt. Mater., 2021, 9, 2002197 CrossRef CAS.
- E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977–9981 CrossRef CAS PubMed.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29 Search PubMed.
- W. Jia, Q. Wang, H. Shi, Z. An and W. Huang, Chemistry, 2020, 26, 4437–4448 CrossRef CAS.
- J. X. Wang, Y. G. Fang, C. X. Li, L. Y. Niu, W. H. Fang, G. Cui and Q. Z. Yang, Angew. Chem., Int. Ed., 2020, 59, 10032–10036 CrossRef CAS.
- J. Wang, X. Y. Lou, Y. Wang, J. Tang and Y. W. Yang, Macromol. Rapid Commun., 2021, 42, e2100021 CrossRef.
- J. L. Kropp and W. R. Dawson, J. Phys. Chem., 1967, 71, 4499–4506 CrossRef CAS.
- M. Wu, X. Wang, Y. Pan, J. Li, X. Li, Y. Sun, Y. Zou, H. Zhang and K. Zhang, J. Phys. Chem. C, 2021, 125, 26986–26998 CrossRef CAS.
- I. Bhattacharjee, K. Hayashi and S. Hirata, JACS Au, 2021, 1, 945–954 CrossRef CAS PubMed.
- R. Sharma, M. B. Thomas, R. Misra and F. D'Souza, Angew. Chem., Int. Ed., 2019, 58, 4350–4355 CrossRef CAS.
- J. Cao, Y. M. Liu, X. Jing, J. Yin, J. Li, B. Xu, Y. Z. Tan and N. Zheng, J. Am. Chem. Soc., 2015, 137, 10914–10917 CrossRef CAS PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
- D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Nat. Commun., 2022, 13, 347 CrossRef CAS.
- H. Peng, G. Xie, Y. Cao, L. Zhang, X. Yan, X. Zhang, S. Miao, Y. Tao, H. Li and C. Zheng, Sci. Adv., 2022, 8, eabk2925 CrossRef CAS PubMed.
- J. Du, L. Sheng, Y. Xu, Q. Chen, C. Gu, M. Li and S. X. Zhang, Adv. Mater., 2021, 33, 2008055 CrossRef CAS.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS.
- S. Kuila, S. Garain, S. Bandi and S. J. George, Adv. Funct. Mater., 2020, 30 Search PubMed.
- A. Kirch, M. Gmelch and S. Reineke, J. Phys. Chem. Lett., 2019, 10, 310–315 CrossRef CAS PubMed.
- Y. Yang, J. Wang, D. Li, J. Yang, M. Fang and Z. Li, Adv. Mater., 2021, 33, 2104002 CrossRef CAS PubMed.
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31 Search PubMed.
- X. Yao, J. Wang, D. Jiao, Z. Huang, O. Mhirsi, F. Lossada, L. Chen, B. Haehnle, A. J. C. Kuehne, X. Ma, H. Tian and A. Walther, Adv. Mater., 2021, 33, 2005973 CrossRef CAS.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef.
- Y. Liu, X. Huang, Z. Niu, D. Wang, H. Gou, Q. Liao, K. Xi, Z. An and X. Jia, Chem. Sci., 2021, 12, 8199–8206 RSC.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- H. Li, H. Li, W. Wang, Y. Tao, S. Wang, Q. Yang, Y. Jiang, C. Zheng, W. Huang and R. Chen, Angew. Chem., Int. Ed., 2020, 59, 4756–4762 CrossRef CAS.
- S. Li, L. Fu, X. Xiao, H. Geng, Q. Liao, Y. Liao and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 18059–18064 CrossRef CAS PubMed.
- J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS.
- M. Petersilka, U. Gossmann and E. Gross, Phys. Rev. Lett., 1996, 76, 1212 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li, Z. Cai, Y. Zhao and C. Yang, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed.
- Y. Yang, Y. Liang, Y. Zheng, J. A. Li, S. Wu, H. Zhang, T. Huang, S. Luo, C. Liu, G. Shi, F. Sun, Z. Chi and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202201820 CAS.
- B. Roy, I. Maisuls, J. Zhang, F. C. Niemeyer, F. Rizzo, C. Wölper, C. G. Daniliuc, B. Z. Tang, C. Strassert and J. Voskuhl, Angew. Chem., Int. Ed., 2021, 61, e202111805 Search PubMed.
- C. Dong, X. Wang, W. Gong, W. Ma, M. Zhang, J. Li, Y. Zhang, Z. Zhou, Z. Yang, S. Qu, Q. Wang, Z. Zhao, G. Yang, A. Lv, H. Ma, Q. Chen, H. Shi, Y. M. Yang and Z. An, Angew. Chem., Int. Ed., 2021, 60, 27195–27200 CrossRef CAS PubMed.
- K. Zheng, X. Yang, F. Ni, Z. Chen, C. Zhong and C. Yang, Chem. Eng. J., 2021, 408 Search PubMed.
- Y. Tian, J. Yang, Z. Liu, M. Gao, X. Li, W. Che, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2021, 60, 20259–20263 CrossRef CAS.
- T. Zhu, T. Yang, Q. Zhang and W. Z. Yuan, Nat. Commun., 2022, 13, 2658 CrossRef CAS PubMed.
- J. Zhang, P. Alam, S. Zhang, H. Shen, L. Hu, H. H. Y. Sung, I. D. Williams, J. Sun, J. W. Y. Lam, H. Zhang and B. Z. Tang, Nat. Commun., 2022, 13, 3492 CrossRef CAS PubMed.
- M. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Nat. Commun., 2022, 13, 347 CrossRef CAS PubMed.
- L. Deng, Z. Ma, J. Zhou, L. Chen, J. Wang, X. Qiao, D. Hu, D. Ma, J. Peng and Y. Ma, Chem. Eng. J., 2022, 137834 CrossRef CAS.
- Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li and Z. Cai, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: synthesis and characterization, experimental details. CCDC 2201069 and 2201070. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2mh00998f |
| This journal is © The Royal Society of Chemistry 2023 |