
Conjugated polymer blends for faster organic mixed conductors†
Micah
Barker
a,
Tommaso
Nicolini
 a,
Yasmina Al
Yaman
a,
Damien
Thuau
b,
Olga
Siscan
a,
Sasikumar
Ramachandran
a,
Eric
Cloutet
a,
Cyril
Brochon
a,
Lee J.
Richter
c,
Olivier J.
Dautel
d,
Georges
Hadziioannou
*a and
Natalie
Stingelin
*ae
a,
Yasmina Al
Yaman
a,
Damien
Thuau
b,
Olga
Siscan
a,
Sasikumar
Ramachandran
a,
Eric
Cloutet
a,
Cyril
Brochon
a,
Lee J.
Richter
c,
Olivier J.
Dautel
d,
Georges
Hadziioannou
*a and
Natalie
Stingelin
*ae
aUniversité de Bordeaux, CNRS Bordeaux INP/ENSCBP, Laboratoire de Chimie des Polyméres Organiques, UMR 5629, Allée Geoffroy Saint-Hilaire, 33615, Pessac Cedex, France. E-mail: natalie.stingelin@gatech.edu; georges.hadziioannou@u-bordeaux.fr
bUniversité de Bordeaux, CNRS Bordeaux INP/ENSCBP Laboratoire de l’Intégration du Matériau au Système UMR 5218, 16 Avenue Pey Berland, 33607, Pessac Cedex, France
cMaterial Measurement Laboratory, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA
dInstitut Charles Gerhardt Montpellier, UMR 5253 CNRS-UM-ENSCM. Campus CNRS-Bâtiment Balard, 1919, route de Mende, 34293, Montpellier Cedex 05, France
eSchool of Materials Science & Engineering and School of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
First published on 14th November 2022
Abstract
A model mixed-conducting polymer, blended with an amphiphilic block-copolymer, is shown to yield systems with drastically enhanced electro-chemical doping kinetics, leading to faster electrochemical transistors with a high transduction. Importantly, this approach is robust and reproducible, and should be readily adaptable to other mixed conductors without the need for exhaustive chemical modification.
New conceptsSemiconducting polymers attract great interest for bioelectronics applications due to their soft nature and mixed ionic–electronic conduction capabilities. So far, most efforts focused on enhancing mixed-conducting functionalities by introducing polar side chains. Here, we show that blending offers a powerful, general approach to improve mixed conduction. We used a block copolymer of poly(3-hexylthiophene) and poly(ethylene oxide) (P3HT-b-PEO) as additive for a relatively poorly performing model material based on a random copolymer between 3-hexylthiophene and 3-(6-hydroxy)hexylthiophene, (P(3HT-co-3HHT)), to unambigously demonstrate the benefit of our strategy. Blends and neat P(3HT-co-3HHT) show similar transduction performance when implemented in organic electrochemical transistors (OECT)s, yet, intriguingly, blends display drastically reduced drain-current hysteresis because of faster electrochemical doping; i.e., blending introduces ion-transporting pathways without negatively affecting the semiconductor's electronic conductivity. This is desired for electrochemical transducer operation and is rendered possible via use of the amphiphilic block copolymer that imparts the required hydrophilicity to the active layer and promotes partial miscibility between active-layer components without the need of stabilizing the films by cross-linking. Additionally, a notable threshold-voltage stability across gate-potential sweep rates and a low impedance is found, thanks to the electrolyte/redox-polymer compatibilization due to the presence of the additive, rendering these blends promising for numerous applications, including electrochemical biosensing. |
Introduction
The growth of the organic bioelectronics field over the past two decades has been rapid, driven by the potential of semiconducting polymers to impact future medical technology platforms, neuromorphic sensor systems, batteries, and beyond.1 Among the most beneficial features of the versatile materials class of ‘plastic’ semiconductors are their unique, tunable electrochemical characteristics and the possibility to introduce mixed ionic and electronic conducting properties that, combined, lead to bulk doping and/or de-doping in the presence of electrolytes and may be exploited in applications where transduction between biochemical ionic fluxes and electronics is required. Specifically, electrochemical oxidation of the polymers’ conjugated backbone will modulate their electrical conductivity and can produce large volumetric capacitances (C*) – effects that, combined, can result in high signal amplification when integrated in devices such as biosensors.2,31 However, critical processes leading to mixed conduction32 rely on ion transport across polymer/analyte interfaces as well as through the bulk of the polymer structure, both of which can limit the response speed of devices such as organic electrochemical transistors (OECTs).3One common strategy to support ion transport in polymeric semiconductors33 involves chemically engineering the polarity of the materials’ side chains by introducing, e.g., organic acid or ionic groups, and/or using oligo(ethylene glycol) side chains,4–8 which all lead to mixed-conduction, bulk electrochemical doping, and high volumetric capacitances. To date, use of oligo(ethylene glycol) side chains has resulted in the highest augmentation of C*, enabling a high steady-state transduction performance in OECTs (cf. ref. 8). The choice of backbone chemistry, in addition, offers access to depletion, accumulation, or ambipolar modes of OECT operation, thereby, providing a plethora of options for materials design and having resulted in a steady growth of the polymer mixed conductor library.4–8
There are, however, limitations when using a single-component material for OECT applications. For example, because of the required polar side-chain functionalization, the polymers often become water soluble, resulting in the need for cross-linking the material post film deposition and/or for the use of slowly diffusing bulky counter-ions to be compatible with applications in aqueous media.5 Significant water uptake—passive or during operation—can bring about irreversible morphological changes in the material's solid-state structure, often with drastic consequences on their electronic function and device stability,9 in many cases combined with a pronounced sensitivity to humidity.9,10 Furthermore, bulky side groups can lead to torsional backbone disorder which can detrimentally affect electronic charge-carrier mobility.11,12 The interplay of these effects is typically not predictable; accordingly, a series of polymers generally needs to be synthesized, typically exploiting co-polymerization and variation of the side-chain substitution pattern, to understand and fine-tune materials performance.13–15
Alternatively, multicomponent systems may be used to introduce specific functionalities via blending together different materials, without the need for complex chemical designs of the active semiconducting polymer itself.16,17 For instance, in the organic thin-film transistor field, addition of the bulk commodity polymer, polyethylene, to a polymer semiconductor has resulted in structures of increased mechanical robustness, enhanced environmental stability, smaller gate-bias stress and, in many cases, improved electron and hole transport, even at very high content of the insulating ‘additive’.16,18,19 Transport was also maintained in the bulk,20 allowing fabrication of organic solar cells using a donor polymer, acceptor fullerene derivative and polyethylene ternary blend.21 Here, we show that a similar approach can be employed for enhancing the electronic/ionic mixed conduction properties of polymeric semiconductors. In contrast to thin-film transistors, however, where highly apolar additives need to be employed that are inactive and are incompatible with the semiconductor,18 for OECTs, where bulk doping/charge transport is required, an amphiphilic additive needs to be used that can simultaneously enhance the overall hydrophilicity and ionic conductivity of the system, while keeping the active layer water-insoluble without requiring a cross-linking agent and without affecting the molecular packing of the semiconductor, critical for electronic transport.
Results and discussion
We selected as model mixed conductor a random copolymer based on the archetypal polymer semiconductor poly(3-hexylthiophene) (P3HT) and resulting from the copolymerization of 3-hexylthiophene (3HT) and 3-(6-hydroxy)hexylthiophene (3HHT), i.e. P(3HT-co-3HHT) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mol ratio of 3HT:3HHT; number-average molecular mass, Mn = 30 kg mol−1, dispersity Đ = 1.9). We chose a material with this particular conjugated backbone because these polymers are known to have somewhat limited hole mobilities with respect to other state of the art organic mixed conductors24 thus can serve as an excellent model system to demonstrate the efficacy – and generality – of blending to enhance ion transport in OECT active layers. We like to also note that for this mixed conductor, it was previously shown that the hydroxyhexyl (–C6H12OH) side chains in the 3HHT moieties can enhance its electrochemical response in aqueous media,22,23 while not affecting the molecular order and the electronic/ionic properties of solution-cast films even after OECT device operation.
:50 mol ratio of 3HT:3HHT; number-average molecular mass, Mn = 30 kg mol−1, dispersity Đ = 1.9). We chose a material with this particular conjugated backbone because these polymers are known to have somewhat limited hole mobilities with respect to other state of the art organic mixed conductors24 thus can serve as an excellent model system to demonstrate the efficacy – and generality – of blending to enhance ion transport in OECT active layers. We like to also note that for this mixed conductor, it was previously shown that the hydroxyhexyl (–C6H12OH) side chains in the 3HHT moieties can enhance its electrochemical response in aqueous media,22,23 while not affecting the molecular order and the electronic/ionic properties of solution-cast films even after OECT device operation.
To produce a multicomponent system of desired function, we blended the P(3HT-co-3HHT) with a block-copolymer of P3HT and ion-conducting poly(ethylene oxide) (P3HT-b-PEO; Mn,P3HT = 5 kg mol−1, Mn,PEO = 20 kg mol−1). We used a block copolymer rather than a PEO homopolymer to limit phase separation when combined with the more apolar P(3HT-co-3HHT). An additional benefit of using a block-co-polymer is that this component still provides sites within the P3HT block that can be electrochemically oxidized, thus, potentially minimizing impact on the volumetric capacitance of the system. The chemical structures of the polymers are shown in the insets of Fig. 1a.
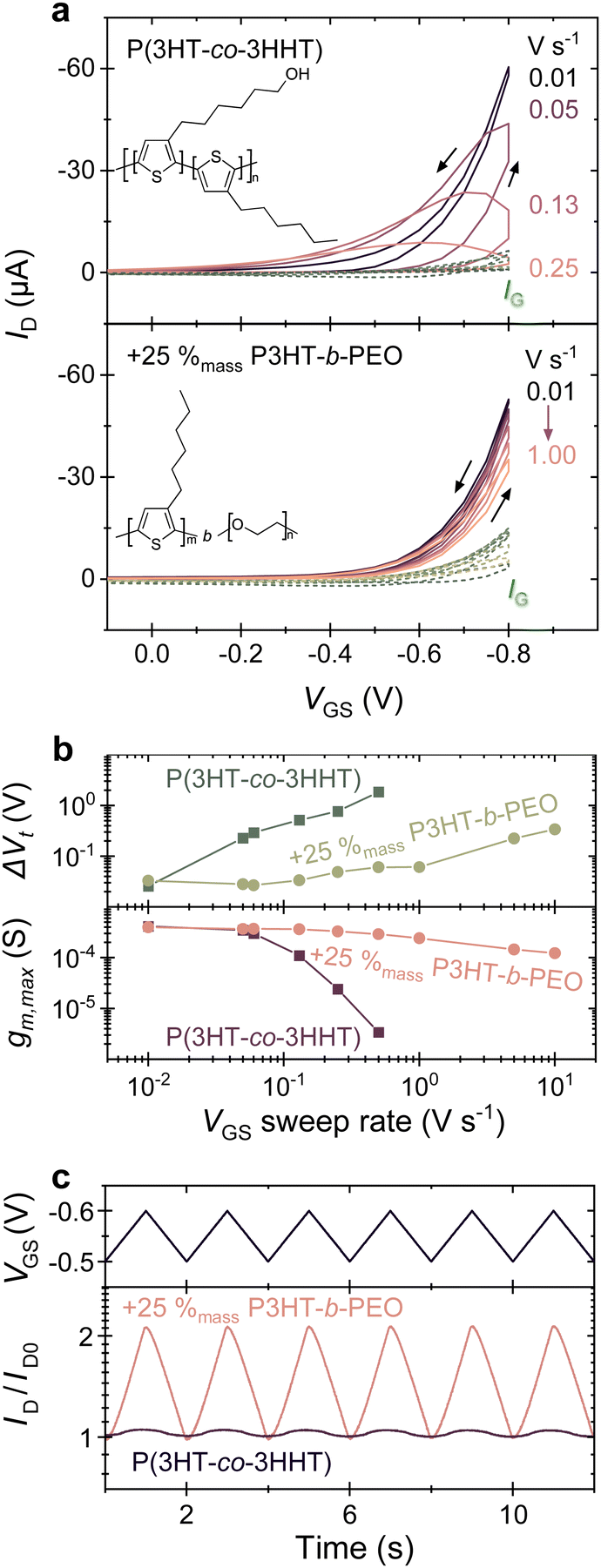 | ||
| Fig. 1 (a) OECT transfer curves of P(3HT-co-3HHT) (top) and its blend with P3HT-b-PEO (bottom) measured by cyclic application of gate–source voltage (VGS) at indicated sweep rates and constant drain–source voltage (VDS = −0.6 V). Blends show less hysteresis in current traces. The chemical structures of the components are shown in the inset. (b) Threshold voltage difference between forward and reverse scans (ΔVt) (top) and maximum transconductance during forward scans (gm,max) (bottom) extracted from data in (a). (c) OECT transient drain currents in response to the indicated VGS waveform (measured at VDS = −0.4 V). Results obtained from devices of similar channel dimensions (width W = 100 μm, length L = 10 μm, and thickness d = 110 nm) using 0.1 mol L−1 KCl aqueous electrolyte. | ||
Blend films could be readily prepared via spin-coating from a co-solvent—a 1:1 mixture (by volume fraction) of tetrahydrofuran (THF) and N-methylpyrrolidone (NMP)—using a concentration of 15 mg mL−1, followed by drying at 55 °C for (3 to 8) min and subsequent exposure to dynamic vacuum overnight to extract excess NMP. Reassuringly, no large scale phase separation was observed in optical microscopy, although distinct features for P(3HT-co-3HHT) and the PEO-block in P3HT-b-PEO were observed both in grazing-incidence wide-angle X-ray scattering (GIWAXS) and differential scanning calorimetry (DSC), see Fig. S3 to S5 (ESI†). [Note, no melting endotherm was found for the P3HT-blocks in neat P3HT-b-PEO suggesting that they are of relatively low molecular order]. Moreover, the water contact angle notably decreases upon addition of P3HT-b-PEO to P(3HT-co-3HHT) (Fig. S6, ESI†), from which we infer that the block-copolymer increases at least the surface polarity of the system.
Films of thicknesses of (80 to 200) nm were used to fabricate OECTs, following procedures detailed in the ESI.† The OECTs were operated in an aqueous solution of 0.1 mol L−1 KCl, and the drain current (ID) response to cyclic application of a gate–source potential (VGS) from 0.2 to −0.8 V in −0.05 V increments was measured at various gate–source voltage sweep rates and at a constant drain–source voltage (VDS) of −0.6 V. The resulting transfer characteristics (IDvs. VGS) for P(3HT-co-3HHT) and a blend of P(3HT-co-3HHT) with 25% mass ratio (hereafter %mass) of P3HT-b-PEO are displayed in Fig. 1a. Schematics of the device architecture used, transfer curve of the blend device with 25%mass P3HT-b-PEO, with ID plotted on a logarithmic scale, and data for other blend compositions, (10, 33, 40, and 45) %mass P3HT-b-PEO, are shown in Fig. S7 (ESI†). Corresponding output characteristics for the neat P(3HT-co-3HHT) and the 75:25 (by mass) P(3HT-co-3HHT):P3HT-b-PEO blends are displayed in Fig. S8 (ESI†).
Immediately apparent is that OECTs comprising the neat P(3HT-co-3HHT) as channel material display a pronounced hysteresis, especially at faster operation. Gate–source voltage sweep rates as low as 0.01 V s−1 were needed to minimize the observed hysteresis and attain reasonable drain currents (≈60 μA) for a transistor with a channel width, W, of 100 μm, a channel length, L, of 10 μm, and an active layer thickness, d, of 110 nm. In strong contrast, in 75:25 P(3HT-co-3HHT):P3HT-b-PEO blends, a minimal hysteresis is observed even at sweep rates up to 1 V s−1, while reaching drain currents of (30 to 60) μA, leading to a current modulation of 100 to 500 (Fig. S7, ESI†).
The different behavior of the blend vs. the neat P(3HT-co-3HHT) can be better illustrated by plotting the extrapolated threshold voltage shifts between the forward and reverse scans (ΔVt), see Fig. 1b, top panel (further details are given in Fig. S7, ESI†). At a gate–source voltage sweep rate of 0.01 V s−1, the lowest rate used in this work, ΔVt extracted for both systems are comparable, ≈(20 to 30) mV. However, increasing the sweep rate to 0.1 V s−1 leads to a notable increase of ΔVt by an order of magnitude for neat P(3HT-co-3HHT), reaching ≥1 V at sweep rates of 0.1 V s−1 and higher (see also Fig. S10, ESI†). In devices of the 75:25 blend, conversely, a low ΔVt of (20 to 50) mV was maintained even when the OECT was operated at sweep rates of 1 V s−1. Remarkably, even at 10 V s−1, the increase of ΔVt in the blends is smaller than that observed in devices of the neat material when operated at a rate of 1 V s−1, i.e., one order of magnitude slower. Data for a broader range of blend compositions are shown in Fig. S11 (ESI†).
A similar observation can be made for the transconductance, gm, an important OECT figure-of-merit that is directly related to the hole mobility (μ), C*, and device relevant parameters and dimensions including Vt, VGS, W, L and d, via the following equation:2
| gm = μC*(|Vt − VGS|)(Wd/L) |
As Fig. 1b, bottom panel, shows, at slow gate–source voltage sweeps, the maximum transconductance, gm,max, is comparable for the neat and blended systems, but gm,max distinctly diverges when higher sweep rates are used, with gm,max rapidly decreasing for neat P(3HT-co-3HHT) for rates >10−1 V s−1, while for the 75:25 blend it stayed relatively constant (around 0.4 mS) over the entire gate–source voltage sweep rate range tested, (0.1 to 10) V s−1. [Note: Fig. S9, ESI† illustrates how gm,max and ΔVt were deduced].
The consistency in performance of blend devices is further highlighted by comparing devices of a range of geometries (i.e., different channel dimensions; Fig. 2a and b), where we consistently observe for the 75:25 P(3HT-co-3HHT):P3HT-b-PEO a transconductance, gm, that scales with OECT dimensions, typically given as |(Vt − VGS)| (W·dL−1),24 starkly contrasting with the neat copolymer, for which a large dispersion in gm is found across gate–source voltage sweep rates, especially at low |(Vt − VGS)| (W·dL−1) values.
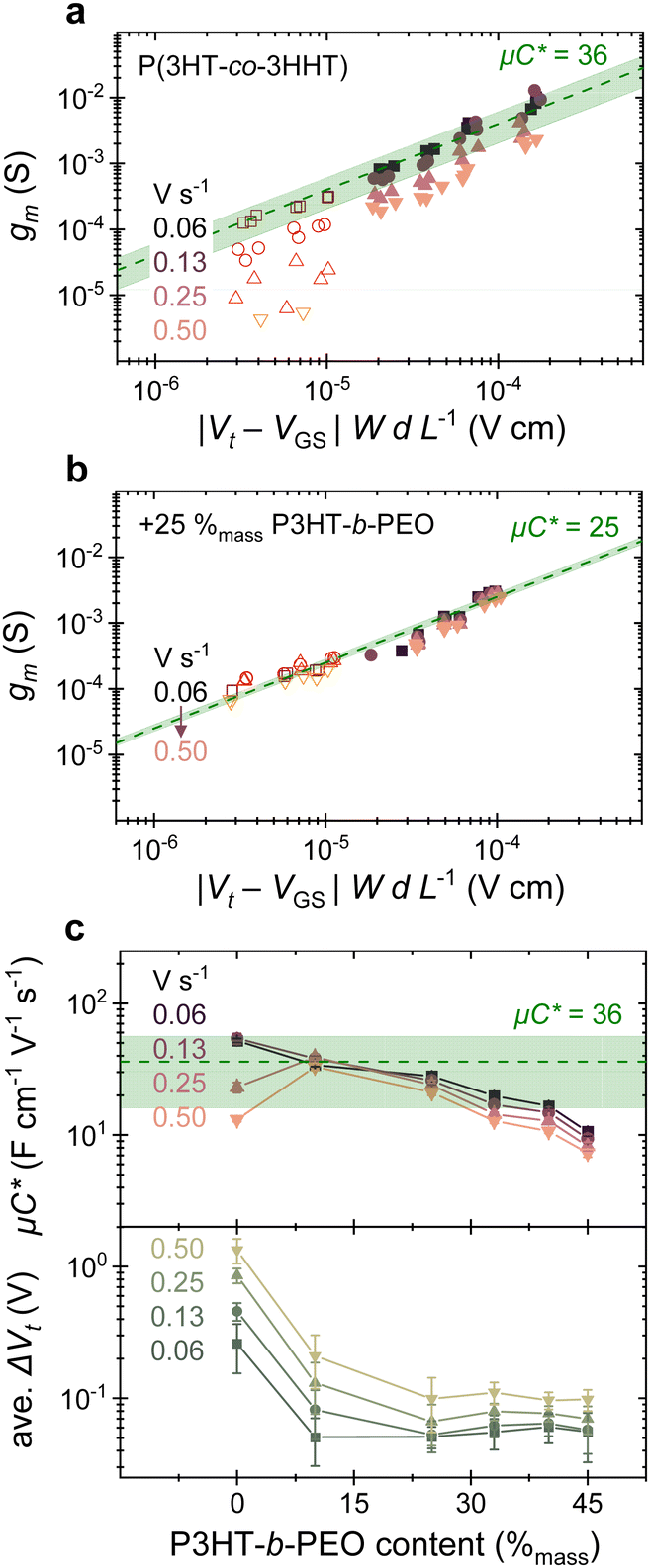 | ||
| Fig. 2 OECT transconductance (gm) plotted against the product of gate–source bias and channel dimensions (|Vt − VGS| WdL−1) for several devices with (a) P(3HT-co-3HHT) and (b) 25%mass P3HT-b-PEO as channel material. Data from devices with micro-scale source and drain electrodes patterned by photolithography are shown with a red gradient (open symbols), while those of large-scale devices are shown with a pink gradient (solid symbols). Data for other blend compositions are shown in Fig. S12 (ESI†). (c) Summary of μC* (top) and average Gate threshold voltage difference (ave. ΔVt) (bottom) extracted for various blend compositions at indicated scan rates. Error bars represent sample standard deviation of at least 12 devices. Green dashed line and shaded regions indicate average μC* in units of F cm−1 V−1 s−1and its standard deviation, respectively, measured across sweep rates for the neat P(3HT-co-3HHT) in (a) and (c), and for the 25%mass blend in (b). | ||
From the slope of gmvs. |(Vt − VGS)| (W·dL−1), we can deduce μ·C*, indicated in Fig. 2a and b with a dashed line. Intriguingly, μ·C* for the 75:25 blend (≈25 F cm−1 V−1 s−1 across sweep rates; ±3 F cm−1 V−1 s−1 standard deviation) converges with the range of μ·C* that can be deduced for the neat P(3HT-co-3HHT) across sweep rates investigated here (≈36 F cm−1 V−1 s−1; ±20 F cm−1 V−1 s−1 standard deviation). This observation suggests that introduction of the block-copolymer at this content (25%mass P3HT-b-PEO) does not strongly affect μ·C*, which implies that either μ or C* increases while C* or μ is decreasing, or μ·C* stays constant.
The importance of reaching maximum transconductance over a wider range of times scales for, e.g., the blend with 25%mass P3HT-b-PEO with respect to the neat material becomes clear from the transient measurements displayed in Fig. 1c. The triangular waveform applied at the gate is transduced to a significant ID-modulation only for the blend comprising 25%mass P3HT-b-PEO. Increasing the block-copolymer fraction above 25%mass, does however reduce μ·C*, especially at contents of ≥40%mass P3HT-b-PEO. Accordingly, blend compositions can be identified where a compromise is reached where ΔVt is sufficiently low while μ·C* remains relatively high. Comparing the top panel with the bottom panel of Fig. 2c, this is reached for a block-copolymer content between (20 to 40) %mass (see also Fig. S12, ESI†).
In order to gain a more mechanistic understanding of μ·C* upon blending, we analyzed various blends with electrochemical impedance spectroscopy (EIS) and compared these with the neat P(3HT-co-3HHT). The impedance magnitude (|Z|) and the corresponding Nyquist (Zimvs. Zre) plots are displayed in Fig. 3a and b, respectively. A few observations can be immediately made. In the high (>103 Hz) and low (<1 Hz) frequency range, the behavior with respect to |Z| of blends and the neat co-polymer are comparable, while addition of P3HT-b-PEO to P(3HT-co-3HHT) clearly depresses |Z| in the mid-frequency regime, (1 to 103) Hz.
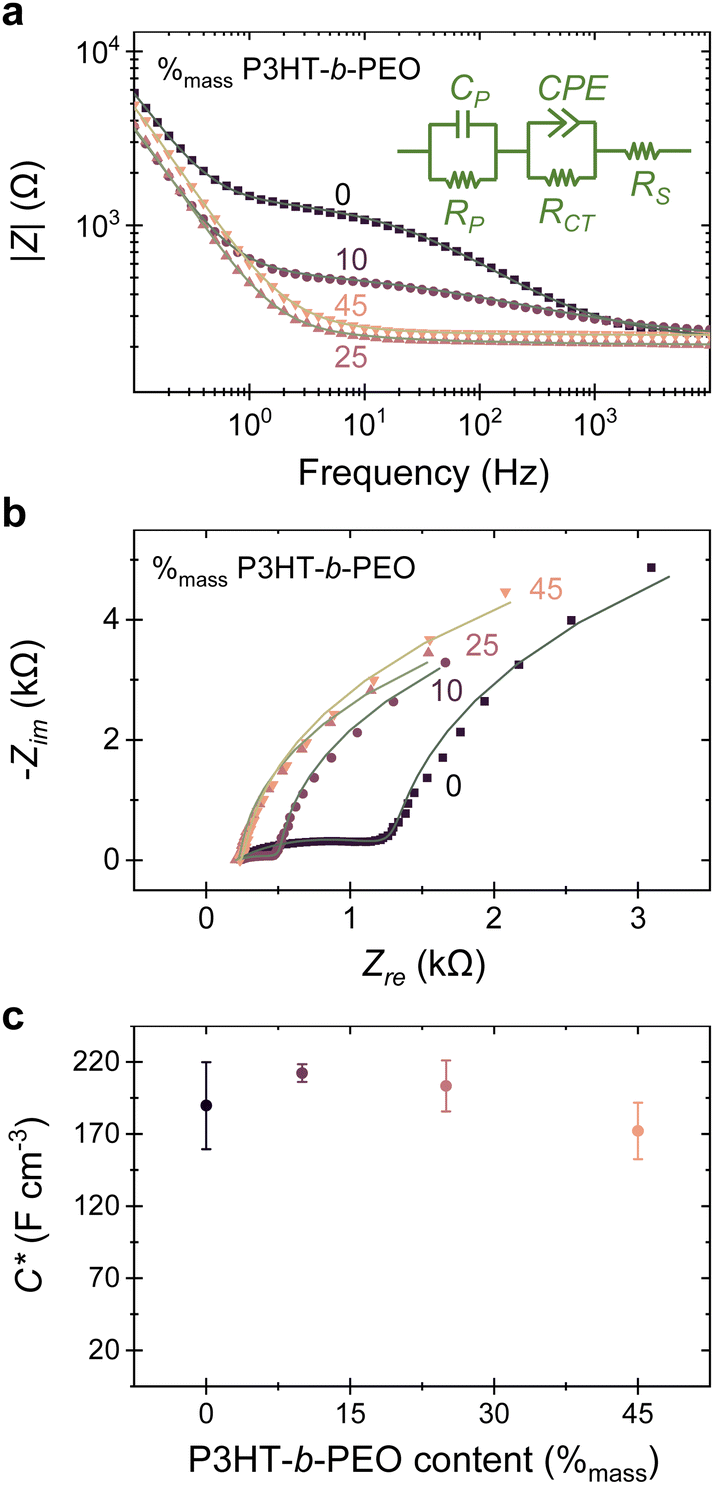 | ||
| Fig. 3 (a) Bode plots (|Z| vs. f) and (b) Nyquist plots (Zimvs. Zre) measured for P(3HT-co-3HHT) and its blends with P3HT-b-PEO. Experimental data (scatter plots) could be modelled reasonably well with the equivalent circuit shown in (a) (solid lines). [Note: a constant phase element (CPE) was used instead of an ideal capacitor element to better fit the experiment data.] (c) Volumetric capacitance (C*) calculated using CP extracted from model fits to experimental data for films of various thickness and blend compositions, showing similar values. Error bars represent standard deviation of at least 3 samples. | ||
This behavior can be modelled with a second-order Voigt-type equivalent circuit (Fig. 3a, inset), typically used in simplified models of Li-ion battery and supercapacitor cells25,26 and consisting of three elements: a resistance (RS) resulting from the solution/electrolyte resistance and dominating the high frequency regime; a capacitor (CP) in parallel with a resistor (RP) that we ascribe to the low frequency range and can be assigned to the capacitance and resistance of the polymer film (neat or blended); and a constant phase element (CPE) in parallel with a resistor (RCT) that we used to describe the mid-frequency behavior, which we attribute to inhomogeneous double layer capacitive charging27 and ion injection occurring at the polymer/electrolyte interface. Since |Z| decreases in this mid-frequency regime, indicating a lowering of ion injection resistance upon addition of the block-copolymer, we conclude that the PEO-block assists in ‘blurring’ the interfaces between the polymer layer and the electrolyte. This is likely due to the increased polarity of such blends, as deduced from the decrease of the water contact angles from 96° for the neat P(3HT-co-3HHT) to 72° for the blended film with 45 %mass P3HT-b-PEO (Fig. S6, ESI†), combined possibly with an enhanced ion transport due to the addition of the polar PEO moiety. This behavior is illustrated in the Nyquist plots, where blending minimizes the depressed semi-circle feature, converging at around 45%mass P3HT-b-PEO content. Parameters extracted for the various blends are listed in Table S1 (ESI†), and a plot at low impedance values is shown in Fig. S13 (ESI†).
Importantly, because the equivalent circuit that we used for modelling the impedance spectra is approaching that of a simple Randle cell of [RS (CP || RP)], average C* can be deduced for the various systems from the capacitance values CP in the low-frequency regime (the capacitor component in parallel with RP) for films of a range of thicknesses (Table S1, ESI†). The results are summarized in Fig. 3c. Relatively small variations in C* (≈190 F cm−3; ±20 F cm−3 standard deviation; measured at a DC offset of 0.75 V) are extracted for neat P(3HT-co-3HHT) and its blends with P3HT-b-PEO independent of block-copolymer content. Considering that μ·C* is of the order of 101 F cm−1 V−1 s−1, we estimate a hole mobility of the order of ≈10−1 cm2 V−1 s−1, which is in accordance with literature on such side-chain-functionalized thiophene-based mixed conducting materials.23,28
The finding that μ·C* is essentially constant over a large blend composition range is somewhat surprising, but may give some insights on the blends’ phase behavior, considering that C* describes the strength of the ionic–electronic coupling in a material and is related to the charge-carrier density.2,3,24,29 Specifically, the fact that C* is unaffected by the addition of the block-copolymer suggests that addition of the PEO-block does not reduce the amount of charge carriers that can be induced upon electrochemical doping.
We infer from this finding that in the blends similar amounts of 3HT- and 3HHT-segments are available that can be oxidized as in the neat P(3HT-co-3HHT). Since the crystalline regions in P(3HT-co-3HHT) seem not to drastically swell upon exposure to humidity (see Fig. S14, ESI†) and thus, likely, exposure to an electrolyte, we speculate that the amorphous regions in the thin-film structures are regulating the injection and transport of ions during application of a bias on the polymer film. Since these ions are necessary to balance the positive charge of polarons in the electroactive material, we attribute to the amorphous fractions a key role in determining the kinetics and electrochemical doping efficiency. In this context, it is important to note that blending leads to a certain degree of vitrification of both components. We infer this from DSC measurements, extracting the enthalpy values for the P(3HT-co-3HHT)/P3HT and PEO-block melting endotherms at ≈245 °C and ≈50 °C, i.e. ΔH3HT/3HHT and ΔHPEO respectively (Fig. S5, ESI,† bottom panel). The solid lines show the enthalpy values for the two moieties (3HT/3HHT vs. PEO) while the corresponding dashed lines describe the “ideal” values (the expected enthalpies in case the materials would fully phase separate and no interactions between the components occur); i.e., a scenario where both P(3HT-co-3HHT) and P3HT-b-PEO can crystallize essentially as unhindered as in the neat form. Enthalpy values below this ideal line as observed here, therefore, indicate that blending suppresses crystallization of the specific moiety/block, leading to a higher molecular disorder in the blend as compared to the neat films. A positive deviation, vice versa, would suggest that blending of the materials promotes the crystallization of one or both components.
Since addition of P3HT-b-PEO to P(3HT-co-3HHT) results in a larger fraction of amorphous phase, we conclude that blending does not reduce the amounts of segments available that can be oxidized but, rather, increases them. This effect counter-acts the reduction in overall active material through introduction of the PEO block. Hence, a compromise can be found where the polarity of a system is increased, and ion transport be enhanced, without negatively impacting C*. This results in drastically faster response times when such blends are implemented in OECTs, promising devices of higher sensitivity and signal amplification, as illustrated by the OECT time constants (τ) extracted from the transient drain currents in response to an applied gate–source bias (Fig. S15, ESI†). While τ for the neat material is around 8 s, for the 75:25 blends a time constant of 80 ms is found. Consequently, blend devices exhibit significantly larger output current amplitudes than those of the neat material upon application of a moderate gate–source bias wave-period signal (triangle wave, period T = 2 s), as shown in Fig. 1c.
Spectroelectrochemical data of the blends cast on indium tin oxide (ITO) electrodes support the view that the observed transient OECT behavior can be attributed to faster electrochemical doping of the semiconducting layer upon addition of P3HT-b-PEO (Fig. 4; details are given in the ESI†). In the neutral state, blends of different P3HT-b-PEO content of (0 to 45) %mass show essentially identical absorption with maxima at 560 nm and only slight differences in the 0–1/0–0 vibronic peak ratios (Fig. S16, ESI†).10 Application of a potential at the polymer/ITO electrode (0.75 V vs. an Ag/AgCl wire electrode in 0.1 mol L−1 KCl, chosen based on the cyclic voltammetry data shown in Fig. S17, ESI†) results in a decrease of this ground-state absorption band for all blend compositions, accompanied by an increase of a broad polaron absorption30 at longer wavelengths (Fig. S18, ESI†). This process is typically reversible if a 0 V bias is applied between the polymer film and the Ag/AgCl wire electrode in a subsequent step. Furthermore, no significant degradation of the doping kinetics nor the extent of doping was observed up to 10 doping/de-doping cycles for the blend with 25%mass P3HT-b-PEO as compared to the neat material (Fig. S19, ESI†).
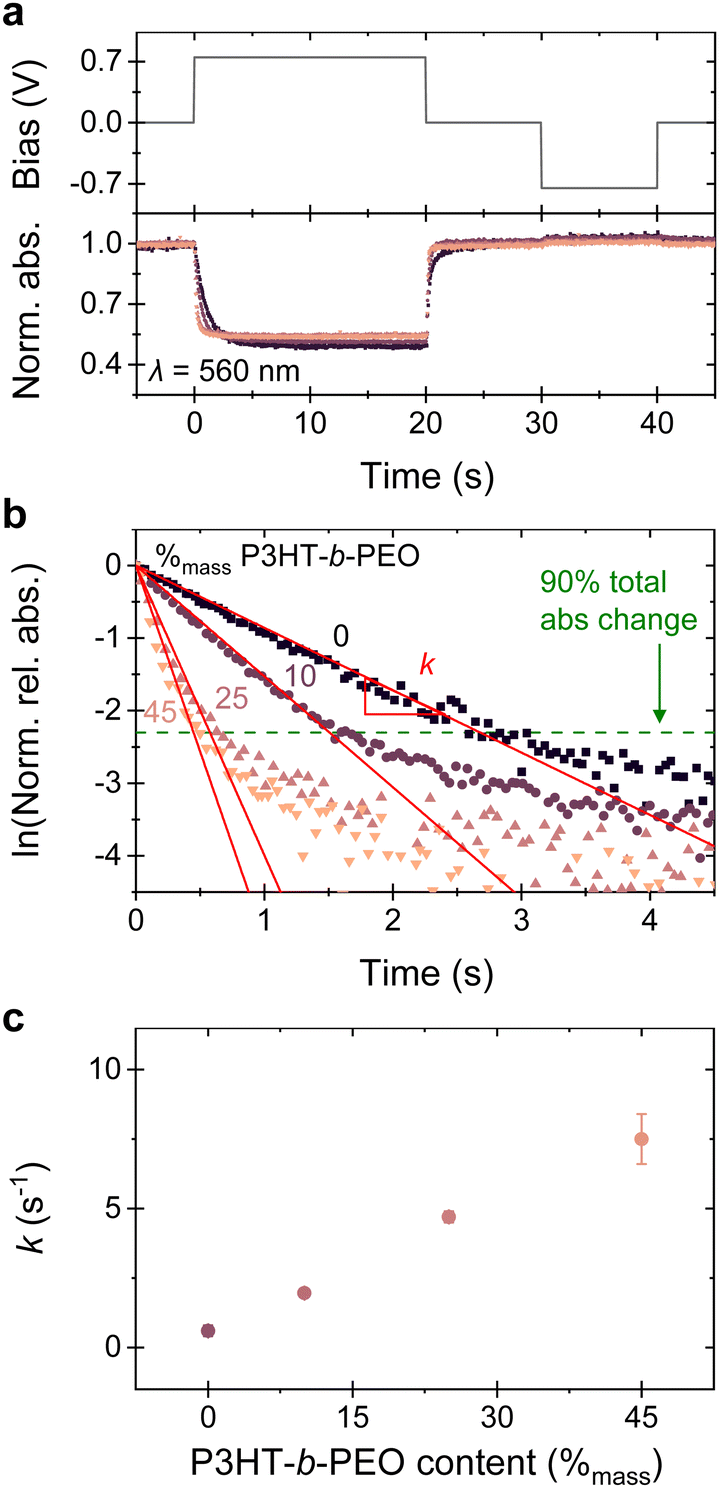 | ||
| Fig. 4 (a) Absorbance changes during application of potential on a polymer/ITO electrode (0.75 V vs. Ag/AgCl wire) in 0.1 mol L-1 KCl, showing similar intensity decrease for blends with (0 to 45) %mass P3HT-b-PEO. (b) Logarithmic plot of relative absorbance over time, showing faster decrease with higher P3HT-b-PEO content. (c) Rate constants (k) extracted from (b) plotted against corresponding blend composition. Results obtained from films of similar thickness (d = 110 ± 10 nm). Error bars represent standard deviation of at least 3 measurements. | ||
The dynamics of the electrochemical doping/de-doping can be visualized by plotting the normalized absorbance as function of time and applied bias22,30 (Fig. 4a) for the different blends/neat P(3HT-co-3HHT). An evident, faster response is observed for the blends (pink to purple traces) compared to the neat P(3HT-co-3HHT) (black trace), with blends of higher P3HT-b-PEO content (light pink traces) displaying the fastest response. Moreover, only small differences between the normalized absorption at saturation (at 0.75 V vs. an Ag/AgCl wire) are found between the blends and the neat P(3HT-co-3HHT), emphasizing that they can be electrochemically equally doped; i.e., equal amounts of charges are introduced, in agreement with the similar C* values we extract for all systems.
Closer inspection of the optical response within the first few seconds of applying a potential and using the normalized relative absorbance (Fig. 4b; further details are given in Fig. S18, ESI†) allows us to extract the electrochemical doping rate constants (k) from exponential decay fits of the data, assuming that the change in absorbance is directly proportional to the decrease of neutral 3HT or 3HHT segments and follows first order kinetics. A single-phase exponential decay function fits adequately with the experimental data until about 90% of the total absorbance change (green dotted line in Fig. 4b and Fig. S18, ESI†), after which the signal slowly saturates. Plotting the extracted rate constants, k, it is immediately apparent that increasing the P3HT-b-PEO content in the blends results in drastically higher k (Fig. 4c), further highlighting that introduction of the block-copolymer to the P(3HT-co-3HHT) assists ion transport in the system. Additionally, k is thickness dependent (Fig. S18d–f, ESI†), from which we conclude that the electrochemical doping/de-doping is a bulk process.
Conclusions
Our work shows that blending can be an effective strategy to enhance the transient performance of polymeric mixed conductors without the need for significant chemical modification. Adding P3HT-b-PEO to the mixed conducting P(3HT-co-3HHT) supports faster electrochemical doping of the system in aqueous media, enhancing in turn the response speed of OECT devices. Tellingly, our approach works even for relatively non-polar semiconductors, such as the prototypical P3HT. The electro-chemical spectroscopy and OECT data shown in Fig. S20 and S21 (ESI†) illustrate that blends of 75:25 P3HT:P3HT-b-PEO operate rather cleanly in an aqueous electrolyte, in stark contrast to neat P3HT. Indeed, no amplification of the source–drain current is observed for neat P3HT OECTs, while in blends, clear transistor behavior is recorded.
We attribute the beneficial effect of blending with the block-copolymer to an increase in hydrophilicity of blend thin-film structures introduced by the presence of the PEO moieties. This seems to assist in enhancing the compatibility between the active blend film and the aqueous electrolyte, effectively ‘blurring’/'softening’ this interface, thereby supporting more efficient ion uptake. In addition, the PEO-block may assist bulk ion transport as well. Intriguingly, despite ‘diluting’ the system with an electronically inert component (the PEO-blocks), the bulk capacitance C* related to the charge density of the electrochemically doped polymer,28 is relatively little impacted by blending. This means that blending does not significantly alter the hole concentration that can be induced by doping. Since μ·C* is comparable across blend composition, we propose that the overall electronic property of P(3HT-co-3HHT) are largely unaffected by blending. As importantly, the time scale range, over which the source–drain current is amplified in OECTs based on blends, often in a significant fashion, is notably extended (see Fig. 1b and c), opening applications of such OECTs for, e.g., monitoring biological signals of specific time scales.
To achieve the fast ion transport, leading to a relatively negligible ΔVt and a rather stable gm independent of gate–source bias sweep rate, it seems important to use a block-copolymer rather than a PEO homopolymer. The P3HT-block helps limiting large-scale phase separation and, indeed, seems to induce partial vitrification of the active P(3HT-co-3HHT). This vitrification may be the reason that a sufficiently large fraction of P(3HT-co-3HHT) segments are available for the electrochemical doping process. This point of view is emphasized when analyzing blends of P(3HT-co-3HHT) with PEO. Contrary to the P(3HT-co-3HHT):P3HT-b-PEO systems, blends between P(3HT-co-3HHT) and the PEO homopolymer feature enthalpies for the P(3HT-co-3HHT)/P3HT and PEO-melting endotherms that decrease in a linear fashion with PEO content (Fig. S5, ESI†), indicating that both components can relatively freely crystallize, unhindered by the addition of the other species, as is typically observed fully phase-separated systems.
Unambiguously, our blending approach can be further exploited. For instance, vitrification and compatibilization can be manipulated by the respective block length of the ‘additive’. Namely, increasing the P3HT-block size could further promote vitrification at the blurred interface between mixed conductor and ‘additive’, allowing deeper doping of the mixed-conductor. Moreover, length of the blocks of the additive, especially of the PEO-moiety, can be used to manipulate the overall mechanical properties of the system. Varying the absolute and relative size of each block, especially with respect to that of the redox active polymer, will open a versatile tool box for blend control. In addition, semiconducting blocks of higher charge-carrier mobilities may be used; or blocks that can be more readily oxidized than P3HT-moieties, rendering blending such a powerful strategy. Obviously, similar principles will also apply for systems that support electron transport, expanding the possibilities to design materials systems from the outset towards specific, desired property set.
Author contributions
M. B. performed most of the experiments in this work including electrochemical spectroscopy, OECT fabrication and impedance measurements. Experiments on P3HT were conducted by Y. A. Y.; while T. N. and D. T. provided assistance in the experiments. O. J. D. performed the synthesis of the random copolymer, and O. S. and S. R. performed the one of the block-copolymer under the guidance of C. B. and E. C.; L. J. R. and T. B. provided structural characterization. M. B. and T. N. prepared the manuscript. N. S. and G. H. designed the research and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. B. is grateful for the financial support provided by the French ministry, MESRI. N. S. acknowledges funding via the IdEX Bordeaux program (ANR-10-IDEX-03-02) and O.D. support from Chimie Balard Cirimat Carnot Institute through the ANR program N° 16 CARN 0008-01. Y. A. Y., E. C., C. B., O. J. D., G. H. and N. S. acknowledge funding by ANR project “MAPLLE” (ANR-20-CE06-0002). This research used the CMS BM11 beamline at the National Synchrotron Light Source II, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory NY, USA, under Contract No. DE-SC0012704. M. B. is grateful for access to the EquipEx ELORPrintTec facility (ANR-10-EQPX-28-01).References
- S. G. Higgins, A. Lo Fiego, I. Patrick, A. Creamer and M. M. Stevens, Organic Bioelectronics: Using Highly Conjugated Polymers to Interface with Biomolecules, Cells, and Tissues in the Human Body, Adv. Mater. Technol., 2020, 5(11), 2000384, DOI:10.1002/admt.202000384.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J.-M. Badier, C. Bernard and G. G. Malliaras, High-Performance Transistors for Bioelectronics through Tuning of Channel Thickness, Sci. Adv., 2015, 1(4), e1400251, DOI:10.1126/sciadv.1400251.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Organic Mixed Ionic–electronic Conductors, Nat. Mater., 2020, 19(1), 13–26, DOI:10.1038/s41563-019-0435-z.
- A. Laiho, L. Herlogsson, R. Forchheimer, X. Crispin and M. Berggren, Controlling the Dimensionality of Charge Transport in Organic Thin-Film Transistors, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(37), 15069–15073, DOI:10.1073/pnas.1107063108.
- E. Zeglio, J. Eriksson, R. Gabrielsson, N. Solin and O. Inganäs, Highly Stable Conjugated Polyelectrolytes for Water-Based Hybrid Mode Electrochemical Transistors, Adv. Mater., 2017, 29(19), 1605787, DOI:10.1002/adma.201605787.
- S. Inal, J. Rivnay, P. Leleux, M. Ferro, M. Ramuz, J. C. Brendel, M. M. Schmidt, M. Thelakkat and G. G. Malliaras, A High Transconductance Accumulation Mode Electrochemical Transistor, Adv. Mater., 2014, 26(44), 7450–7455, DOI:10.1002/adma.201403150.
- A. Giovannitti, D.-T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Controlling the Mode of Operation of Organic Transistors through Side-Chain Engineering, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(43), 12017–12022, DOI:10.1073/pnas.1608780113.
- A. Giovannitti, C. B. Nielsen, D. T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, N-Type Organic Electrochemical Transistors with Stability in Water, Nat. Commun., 2016, 7(1–9), 13066, DOI:10.1038/ncomms13066.
- A. A. Szumska, I. P. Maria, L. Q. Flagg, A. Savva, J. Surgailis, B. D. Paulsen, D. Moia, X. Chen, S. Griggs, J. T. Mefford, R. B. Rashid, A. Marks, S. Inal, D. S. Ginger, A. Giovannitti and J. Nelson, Reversible Electrochemical Charging of N-Type Conjugated Polymer Electrodes in Aqueous Electrolytes, J. Am. Chem. Soc., 2021, 143(36), 14795–14805, DOI:10.1021/jacs.1c06713.
- M. J. Dyson, E. Lariou, J. Martin, R. Li, H. Erothu, G. Wantz, P. D. Topham, O. J. Dautel, S. C. Hayes, P. N. Stavrinou and N. Stingelin, Managing Local Order in Conjugated Polymer Blends via Polarity Contrast, Chem. Mater., 2019, 31(17), 6540–6547, DOI:10.1021/acs.chemmater.8b05259.
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia, I. Nasrallah, I. Romanov, K. Broch, I. McCulloch, D. Emin, Y. Olivier, J. Cornil, D. Beljonne and H. Sirringhaus, Approaching Disorder-Free Transport in High-Mobility Conjugated Polymers, Nature, 2014, 515(7527), 384–388, DOI:10.1038/nature13854.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, A General Relationship between Disorder, Aggregation and Charge Transport in Conjugated Polymers, Nat. Mater., 2013, 12(11), 1038–1044, DOI:10.1038/nmat3722.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Side Chain Redistribution as a Strategy to Boost Organic Electrochemical Transistor Performance and Stability, Adv. Mater., 2020, 32(37), 2002748, DOI:10.1002/adma.202002748.
- P. Schmode, D. Ohayon, P. M. Reichstein, A. Savva, S. Inal and M. Thelakkat, High-Performance Organic Electrochemical Transistors Based on Conjugated Polyelectrolyte Copolymers, Chem. Mater., 2019, 31(14), 5286–5295, DOI:10.1021/acs.chemmater.9b01722.
- C.-K. Mai, T. Arai, X. Liu, S. L. Fronk, G. M. Su, R. A. Segalman, M. L. Chabinyc and G. C. Bazan, Electrical Properties of Doped Conjugated Polyelectrolytes with Modulated Density of the Ionic Functionalities, Chem. Commun., 2015, 51(99), 17607–17610, 10.1039/C5CC06690E.
- A. D. Scaccabarozzi and N. Stingelin, Semiconducting:Insulating Polymer Blends for Optoelectronic Applications—a Review of Recent Advances, J. Mater. Chem. A, 2014, 2(28), 10818–10824, 10.1039/C4TA01065E.
- S. Yamamoto and G. G. Malliaras, Controlling the Neuromorphic Behavior of Organic Electrochemical Transistors by Blending Mixed and Ion Conductors, ACS Appl. Electron. Mater., 2020, 2(7), 2224–2228, DOI:10.1021/acsaelm.0c00203.
- A. D. Scaccabarozzi, J. I. Basham, L. Yu, P. Westacott, W. Zhang, A. Amassian, I. McCulloch, M. Caironi, D. J. Gundlach and N. Stingelin, High-Density Polyethylene—an Inert Additive with Stabilizing Effects on Organic Field-Effect Transistors, J. Mater. Chem. C, 2020, 8(43), 15406–15415, 10.1039/D0TC03173A.
- S. Goffri, C. Müller, N. Stingelin-Stutzmann, D. W. Breiby, C. P. Radano, J. W. Andreasen, R. Thompson, R. A. J. Janssen, M. M. Nielsen, P. Smith and H. Sirringhaus, Multicomponent Semiconducting Polymer Systems with Low Crystallization- Induced Percolation Threshold, Nat. Mater., 2006, 5(12), 950–956, DOI:10.1038/nmat1779.
- A. Kumar, M. A. Baklar, K. Scott, T. Kreouzis and N. Stingelin-Stutzmann, Efficient, Stable Bulk Charge Transport in Crystalline/Crystalline Semiconductor-Insulatblends, Adv. Mater., 2009, 21(44), 4447–4451, DOI:10.1002/adma.200900717.
- T. A. M. Ferenczi, C. Müller, D. D. C. Bradley, P. Smith, J. Nelson and N. Stingelin, Organic Semiconductor:Insulator Polymer Ternary Blends for Photovoltaics, Adv. Mater., 2011, 23(35), 4093–4097, DOI:10.1002/adma.201102100.
- C. M. Pacheco-Moreno, M. Schreck, A. D. Scaccabarozzi, P. Bourgun, G. Wantz, M. M. Stevens, O. J. Dautel and N. Stingelin, The Importance of Materials Design to Make Ions Flow: Toward Novel Materials Platforms for Bioelectronics Applications, Adv. Mater., 2017, 29(4), 1604446, DOI:10.1002/adma.201604446.
- T. Nicolini, J. Surgailis, A. Savva, A. D. Scaccabarozzi, R. Nakar, D. Thuau, G. Wantz, L. J. Richter, O. Dautel, G. Hadziioannou and N. Stingelin, A Low-Swelling Polymeric Mixed Conductor Operating in Aqueous Electrolytes, Adv. Mater., 2021, 33(2), 2005723, DOI:10.1002/adma.202005723.
- S. Inal, G. G. Malliaras and J. Rivnay, Benchmarking Organic Mixed Conductors for Transistors, Nat. Commun., 2017, 8(1), 1767, DOI:10.1038/s41467-017-01812-w.
- N. Meddings, M. Heinrich, F. Overney, J.-S. Lee, V. Ruiz, E. Napolitano, S. Seitz, G. Hinds, R. Raccichini, M. Gaberšček and J. Park, Application of Electrochemical Impedance Spectroscopy to Commercial Li-Ion Cells: A Review, J. Power Sources, 2020, 480, 228742, DOI:10.1016/j.jpowsour.2020.228742.
- C. Zou, L. Zhang, X. Hu, Z. Wang, T. Wik and M. Pecht, A Review of Fractional-Order Techniques Applied to Lithium-Ion Batteries, Lead-Acid Batteries, and Supercapacitors, J. Power Sources, 2018, 390(March), 286–296, DOI:10.1016/j.jpowsour.2018.04.033.
- S. Amand, M. Musiani, M. E. Orazem, N. Pébère, B. Tribollet and V. Vivier, Constant-Phase-Element Behavior Caused by Inhomogeneous Water Uptake in Anti-Corrosion Coatings, Electrochim. Acta, 2013, 87, 693–700, DOI:10.1016/j.electacta.2012.09.061.
- L. Q. Flagg, C. G. Bischak, J. W. Onorato, R. B. Rashid, C. K. Luscombe and D. S. Ginger, Polymer Crystallinity Controls Water Uptake in Glycol Side-Chain Polymer Organic Electrochemical Transistors, J. Am. Chem. Soc., 2019, 141(10), 4345–4354, DOI:10.1021/jacs.8b12640.
- C. M. Proctor, J. Rivnay and G. G. Malliaras, Understanding Volumetric Capacitance in Conducting Polymers, J. Polym. Sci., Part B: Polym. Phys., 2016, 54(15), 1433–1436, DOI:10.1002/polb.24038.
- C. Enengl, S. Enengl, S. Pluczyk, M. Havlicek, M. Lapkowski, H. Neugebauer and E. Ehrenfreund, Doping-Induced Absorption Bands in P3HT: Polarons and Bipolarons, Chem. Phys. Chem., 2016, 17(23), 3836–3844, DOI:10.1002/cphc.201600961.
- A. Koklu, S. Wustoni, K. Guo, R. Silva, L. Salvigni, A. Hama, E. Diaz-Galicia, M. Moser, A. Marks, I. McCulloch, R. Grünberg, S. T. Arold and S. Inal, Convection Driven Ultrarapid Protein Detection via Nanobody-Functionalized Organic Electrochemical Transistors, Adv. Mater., 2022, 34(35), 2202972, DOI:10.1002/adma.202202972.
- E. M. Thomas, P. H. Nguyen, S. D. Jones, M. L. Chabinyc and R. A. Segalman, Electronic, Ionic, and Mixed Conduction in Polymeric Systems, Annu. Rev. Mater. Res., 2021, 51(1), 1–20, DOI:10.1146/annurev-matsci-080619-110405.
- J. H. Kim, S. Kim, G. Kim and M. Yoon, Designing Polymeric Mixed Conductors and Their Application to Electrochemical-Transistor-Based Biosensors, Macromol. Biosci., 2020, 20, 2000211, DOI:10.1002/mabi.202000211.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2mh00861k |
| This journal is © The Royal Society of Chemistry 2023 |