
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesoscale artificial synthesis of conjugated microporous polymers†
Catherine
Mollart
 *,
Bartosz
Ciborowski
and
Abbie
Trewin
*
*,
Bartosz
Ciborowski
and
Abbie
Trewin
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: c.mollart@lancaster.ac.uk; a.trewin@lancaster.ac.uk
First published on 12th October 2023
Abstract
This work reports the mesoscale artificial synthesis of a conjugated microporous polymer, CMP-1, using a hybrid coarse-grained methodology. Whilst using a coarse grain approach does give a lower density and surface area when compared to the all-atom equivalent, this allowed a simulation cell volume scale-up of up to 64 times, and an overall speed-up factor of 44% when compared to the all-atom equivalent.
Design, System, ApplicationConjugated microporous polymers (CMPs) are used within a range of applications, including as anodes for ion storage batteries, supercapacitors, photocatalysts, and as solid-state electrolyte materials. They demonstrate high stability, flexibility of synthesis, and therefore offer great potential. However, CMPs are amorphous, making it difficult to rationalise the structure through traditional characterisation techniques alone. Simulation is therefore an extremely important tool. Previously, we have shown an ‘Artificial Synthesis’ method using an in-house developed code called Ambuild. Ambuild simulates the full catalytic synthetic mechanism to generate fully atomistic models, a unique capacity and currently the only way to probe properties. However, this approach is limited to length scales that are too small to fully incorporate larger scale effects, including phase separation of the reaction mix, that strongly influence the resulting properties. Here, we show that we can use the same ‘Artificial Synthesis’ approach but incorporate a hybrid coarse grained methodology that enables simulation of the mesoscale structure, a scale that has not been achieved before for these complex polymer systems. The resulting mesoscale structures show new insights and point towards new directions for understanding, which will ultimately result in the design and discovery of new materials with exceptional properties. |
Microporous materials, such as zeolites,1 metal–organic frameworks (MOFs),2,3 covalent organic frameworks (COFs),4–6 and microporous organic polymers (MOPs)5,7–10 have been explored for a variety of applications, such as energy storage,11,12 gas uptake,13 heterogeneous catalysis14 and molecular separations.2,5,15 However, despite the ultra-high surface areas of the crystalline MOFs and COFs due to their defined pore structures,3 some suffer from a lack of stability on exposure to ‘real-world’ conditions that would be used, for example, in a rechargeable battery. The amorphous MOPs, however, do not show these same limitations in stability, with examples of MOP materials that can be boiled in acid with no loss to porosity reported in the literature.16 This paves the way for the rational design of new MOP materials for applications such as those given above.
The design of amorphous materials is challenging due to the lack of information offered by traditional characterisation techniques such as powder X-ray diffraction (XRD) due to the inherent random nature of these polymer frameworks.17 Additionally, due to the large degree of crosslinking observed within these polymer networks, many are insoluble, rendering solution-state nuclear magnetic resonance (NMR) spectroscopy very difficult to obtain.17
This makes modelling approaches vital to understanding the atomic structure of these amorphous structures. However, as has been previously reported by Mollart et al.,18 it is not simply possible to model the idealised end structure of a particular polymer, rather, each step in the synthetic protocol, including the roles of solvent and catalyst, must be considered during the artificial synthesis procedure (Fig. 1).18
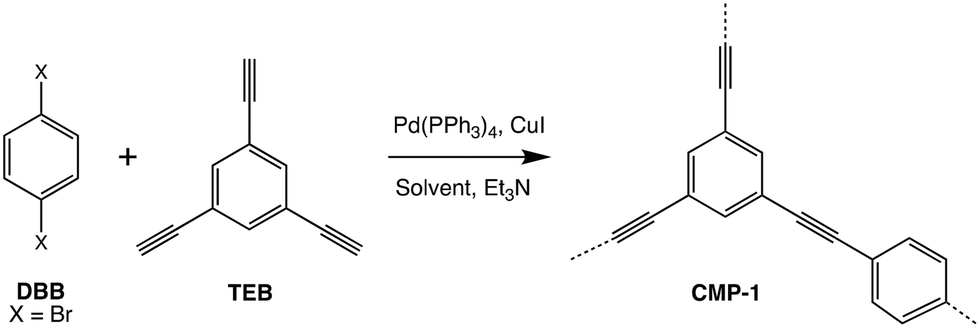 | ||
| Fig. 1 The reaction scheme used to synthesise CMP-1 from the reaction of 1,4-dibromobenzene (DBB) with 1,3,5-triethynylbenzene (TEB) in the presence of a Pd(PPh3)4 catalyst (cat), CuI co-catalyst, solvent, and triethylamine (TEA). | ||
When modelling amorphous structures, it is necessary to generate as many repeats of the structure as possible. This ensures an increased sampling of the polymer, which reduces the likelihood of oversimplifying the structure by considering it to be homogenous, when, in fact, it is known that MOPs do phase separate into fused spherical particles.16,19,20 In addition, generating multiple repeat structures ensures the reliability of porosity properties obtained, such as the surface area and micropore volume.16 It is also important to model as large a structure as possible, both to observe larger-scale effects such as gelation, and to prevent inducing artificial crystallinity in the material.
Previous work reported by Thomas et al. described the artificial synthesis of the first reported conjugated microporous polymer (CMP), CMP-1.16 CMP-1 in particular was chosen as an ideal first test case to model as this has an abundance of experimental data to compare simulation results to.19–21 As the name suggests, CMPs comprise a fully π-conjugated polymer backbone and a three-dimensional arrangement of micropores throughout the framework. CMP-1 is made via the Pd(PPh3)4-catalysed Sonogashira–Hagihara reaction of 1,4-dibromobenzene (DBB) and 1,3,5-triethynylbenzene (TEB), in the presence of a CuI co-catalyst, triethylamine (TEA) and a solvent such as toluene (the original solvent used to prepare CMPs) or N,N-dimethylformamide (DMF, which has since become the ‘standard’ solvent used in many CMP syntheses due to the enhanced porosity properties observed when this is the reaction solvent employed).19–21 The reaction scheme and catalytic cycle used to prepare CMP-1 are given in Fig. S1 and S2.† To summarise the Sonogashira–Hagihara reaction mechanism, the first step is oxidative addition of 1,4-dibromobenzene monomer to the palladium catalyst. Following this, a transmetallation step occurs, whereby the product of the transmetallation side cycle (copper-terminated 1,3,5-triethynylbenzene), adds to the catalyst in exchange for the bromine ligand added as part of the oxidative addition. Then, an isomerisation step occurs to place the vinyl halide and copper(I) acetylide ligands cis to each other, before the new oligomer leaves via reductive elimination.16
The artificial synthesis approach used by Thomas et al., which included a comprehensive study of the reaction ratios, was able to accurately describe the porosity properties seen for CMP-1 by modelling the full synthetic conditions, including the catalytic mechanism and desolvation processes, but was limited by the simulation size that could be reasonably generated, meaning that it was only possible to model the micropore region of 0–20 Å.16
Coarse graining is a technique designed to reduce the number of atoms within the system, where a group of atoms may be replaced by a single grain of similar shape and chemistry.22–24 It is often employed in the simulation of large protein and peptide structures to enable a larger simulation size to be modelled within the same timescale in comparison to the equivalent all-atom system.22 Increasing the system size modelled allows the possibility to study larger-scale effects, such as gelation and phase separation, and reduces the likelihood of artificial crystallinity being induced within the system.
In this work, we therefore discuss the results obtained from modelling CMP-1 using the same methodology described by Thomas et al.,16 but also incorporating a hybrid coarse-grain (CG) approach to the modelling process to increase the system size by a factor of up to 64 times the all-atom (AA) system.
Methodology
The structural models discussed in this work were generated using the Ambuild code, a Python-based structure generation tool that exploits GPU architecture to increase simulation speed.16 Ambuild integrates with the GPU-based HOOMD-blue,25,26 which was used as our geometry optimisation and molecular dynamics engine throughout, and Poreblazer, used for the porosity analysis and described in detail elsewhere.27,28 Unless otherwise stated in the ESI,† all building blocks were generated using the polymer-consistent forcefield (PCFF),29 which is an appropriate forcefield to model organic polymers such as CMP-1.Detailed information about the Ambuild modelling process is available in ESI† section S2, but to summarise, we firstly generate a cubic simulation cell (50 Å × 50 Å × 50 Å to compare to the original paper16 unless otherwise stated), to which we randomly seed stoichiometric quantities of monomers and catalyst, along with TEA and solvent. Following the seed step, we run a loop containing geometry optimisation, NVT (constant number of molecules, cell volume, and temperature) molecular dynamics, and Ambuild zip steps, whereby the initial bond length and bond angle criteria within which two building blocks must be to form a bond are changed, and any new bonds formed, followed by a geometry optimisation of the resulting structure. Throughout the molecular dynamics steps, the integration timestep was set to 0.0001 ps, the number of molecular dynamics cycles was set to one million, and the HOOMD-blue temperature factor set to 55.0.25,26 The average simulated MD time was 16.4 ns. We then desolvate our structures using the ‘Strategy 6’ approach described by Thomas et al.16
Grain mapping
When designing coarse grains to replicate an AA system, an inherent error is present simply due to the approximation made that individual atoms may be replaced by grains. It is therefore essential that the grains chosen can replicate the geometry and chemistry of the atoms as closely as possible to minimise this error.22–24 With that in mind, we decided to use the phenyl-based coarse grains reported by DeVane et al. as a starting point for our modelling,22 as this allowed us to replace the phenyl rings in the DBB and TEB monomers and Pd(PPh3)4 catalyst with the four of the so-called BER grains designed by DeVane et al.22 To ensure the correct connectivity of the Ambuild product, we retained the end group and cap atoms of each building block (C–Br for DBB, C–Cu for TEB and Pd–H for the catalyst, respectively), giving us hybrid-CG building blocks (Fig. 2 and S3–S5†).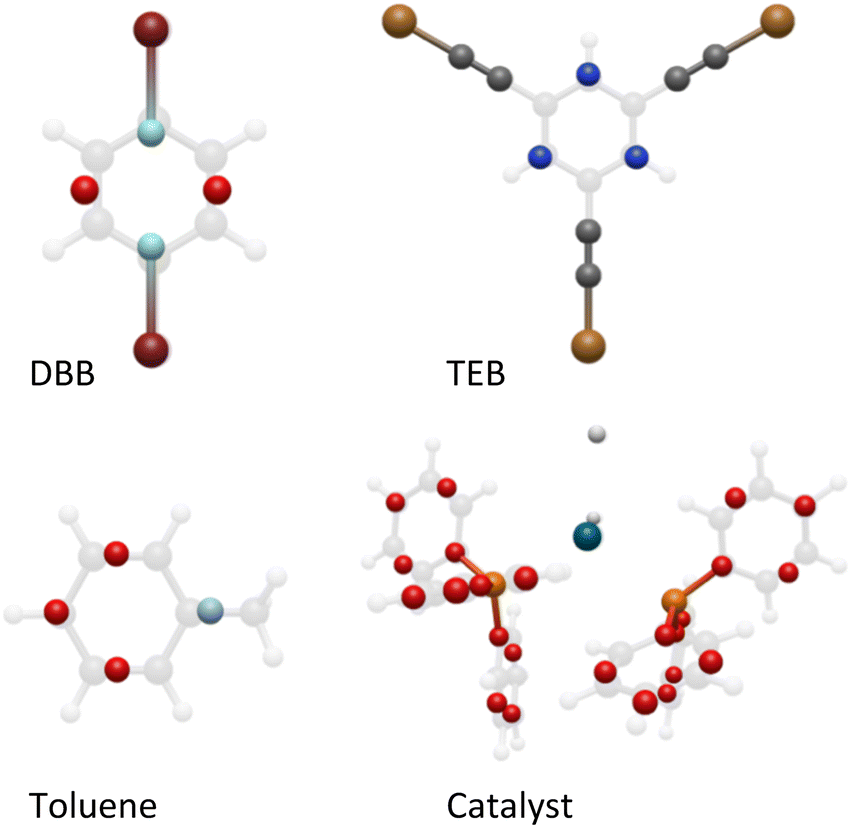 | ||
| Fig. 2 The all-atom (faded) and coarse-grained (bold) blocks used in the artificial synthesis and based on the building block of DeVane et al.22 Key: BER grain – red, dBER grain (DBB) – pale blue, Br – burgundy, tBER grain (TEB) – blue, Cu – gold, C – grey, XYR grain (toluene) – pale blue, H – white, Pd (catalyst) – blue, P – orange. | ||
DeVane and co-workers also designed a coarse-grained model of toluene where each toluene molecule is replaced by four grains (Fig. 2 and S6†).22 As the solvent does not take part in bonding, this can be represented by the fully coarse-grained model. When considering that the solvent makes up the largest percentage of the building blocks within our system, followed by the monomer building blocks, this drastically reduces the number of atoms within our system.
As the phenyl rings in the catalyst do not participate in bonding, it was possible to describe these using four BER grains per phenyl ring, which maintain the geometry and chemistry of the catalyst molecule. However, the BER grains do not match the all-atom potentials well for the DBB and TEB blocks. To optimise the CG fitting so that the Lennard-Jones (LJ) potentials of the hybrid CG monomer building blocks better matched the AA, we designed two new grain types for DBB and TEB, called dBER and tBER, respectively (Fig. S7 and S8†). This allowed us to differentiate between the two building block types, giving us a much better match to the AA equivalents. The optimised hybrid CG fitting plots for DBB and TEB are given in Fig. S9 and S10,† and full details of the building block fitting process are given in ESI† section S4.
Once the potential fitting optimisation was complete, we compared the reaction profiles of six AA CMP-1 network generation processes to six equivalent hybrid CG networks over a small number of steps, where each simulation type was identical in all apart from the nature of the building blocks (hybrid CG or AA). When comparing the bond formation observed in the two simulation types, we can see that the hybrid CG models show very similar behaviour to the AA models, especially when the consideration is made that CMP-1 is an amorphous material, and as such will show varying behaviour from one simulation repeat to the next, even when employing identical reaction conditions.16
Comparison of networks
We simulated the full CMP-1 network formation and desolvation processes by generating four repeat models of our hybrid CG system, and four repeat models of the AA system, both using 130 molecules of toluene as the solvent. We chose to use 130 solvent molecules as this gave a level of solvation corresponding to relatively high surface areas in the work reported by Thomas et al.,16 and we chose to use toluene, rather than DMF, which has become one of the ‘default’ solvents in CMP synthesis, as our models do not currently allow us to coarse-grain DMF. A relatively small sized cell of 50 Å × 50 Å × 50 Å was used so that we can easily compare the different approaches.For the hybrid CG systems, we firstly fit the CG pair potentials to ensure that the pair LJ energy per particle in the hybrid CG systems are as close as possible to the AA systems. This was done by plotting the Lennard-Jones energy per particle during the MD within the desolvation procedure as a function of timestep for the AA system and for the CG systems with epsilon values of 1× the AA epsilon, 0.9× the AA, 0.8× the AA, and 0.7× the AA. The plots were then compared independently for toluene (Fig. S16†) and DMF (Fig. S17†). When this fitting was completed, we found that the CG pair potentials that best matched the AA systems in both toluene and DMF solvents had epsilon values that were 0.8× the AA pair potentials, as these corresponded to the smallest difference between the CG Lennard-Jones energy per particle and the AA equivalent at each timestep (Fig. S16 and S17, Table S5†).
When comparing the maximum block size in the cell against the reaction coordinate for the AA systems (Fig. S18†), it is possible to distinguish the various steps in the CMP-1 network formation mechanism, first proposed by Laybourn et al.,20 where there is an initial increase in the maximum block mass as the monomers react together to form small oligomers. As the reaction continues, the block mass increases as these small oligomers continue to react and eventually precipitate out of solution to form a gel-like phase. The large oligomers then fuse together to form a number of insoluble CMP-1 clusters, at which point the maximum block mass plateaus. As the reaction proceeds, it is also possible to observe a steady decrease in the number of building blocks within the cell, which is again, expected behaviour during the polymerisation. When comparing the block analysis for the AA systems to the hybrid CG systems with fitted LJ pair potentials (Fig. S19†), we observe the same general trend, but the steps appear less defined for the hybrid CG systems. This may be due to the reduced number of atoms in the hybrid CG systems, however, due to the amorphous nature of these materials, definitive conclusions cannot be made as to why this difference arises.
Porosity analysis
To assess the influence of solvent on the porosity of the systems, we also generated a set of models using 130 molecules of DMF as the solvent using both the AA and hybrid CG techniques, with the DMF molecules modelled AA in both cases. These can also be compared to the porosity of the atomistic models reported previously by Thomas et al.,16 which we consider here as a benchmark. The resulting data is given in Table S6.† The hybrid CG models with fitted LJ pair potentials match the atomistic very well, with an almost identical averaged micropore volume, and a surface area that is well within the large error associated with this characterisation technique for microporous materials like CMP-1.30,31 This suggests that our methodology is appropriate to model these materials, however, we need to consider the relationship between the all-atom and hybrid coarse-grain surface areas, in order to calculate an approximate atomistic surface area for a model that is too large to model using the AA approach, which is the ultimate goal of this modelling approach.To do this, we generated a range of small fragments consisting of two-ten monomer building blocks using both the AA and hybrid CG techniques (Table S8†), then back-mapped the atom locations of the hybrid CG models to match the AA atom locations (Fig. S22–S50†). Following this, we calculated the porosity properties of the AA and back-mapped hybrid CG models for each polymer fragment (Table S9†). We then plotted the average AA network accessible surface area for each polymer fragment size (grouped by number of building blocks in the fragment) against the averaged back-mapped hybrid CG network accessible surface area for the same fragment size (Fig. S51†). This gives us a relationship between the two that could be applied to calculate an approximate surface area for the AA equivalent of a simulated hybrid CG model.
Fig. 4 shows the average time per Poreblazer calculation for the all-atom and back-mapped hybrid CG small fragment models.27 It is clear that there is an advantage to coarse graining the system, even at the small system sizes, with a reduction in the calculation time required per system.
Scale-up
Finally, we test larger systems using our hybrid CG approach. To ensure that as much of the system could be coarse-grained as possible, the models were artificially synthesised using toluene, which, unlike DMF, can be coarse-grained using our current approach, therefore allowing us to artificially synthesise a larger-scale model. We generated short timescale runs at cell lengths beginning at the original cell length of 50 Å and increasing in increments of 25 Å up to a final cell length of 200 Å (using an integration timestep of 0.000005 ps to match the scaled-up artificial synthesis described in ESI† section S11, Table S10, Fig. S52). We also generated these short timescale runs using the AA approach so that we could compare how the time per output step changed on coarse graining the system, and we found that whilst the time to simulate 1 ps of molecular dynamics followed an exponential curve as the cell size increased, the largest model generated (cell length 200 Å, volume scale-up of 64× the original cell length) demonstrated a molecular dynamics percentage speed-up of 35% to simulate 1 ps of MD when simulated using the hybrid CG approach in comparison to the AA (Fig. 3 and S53†).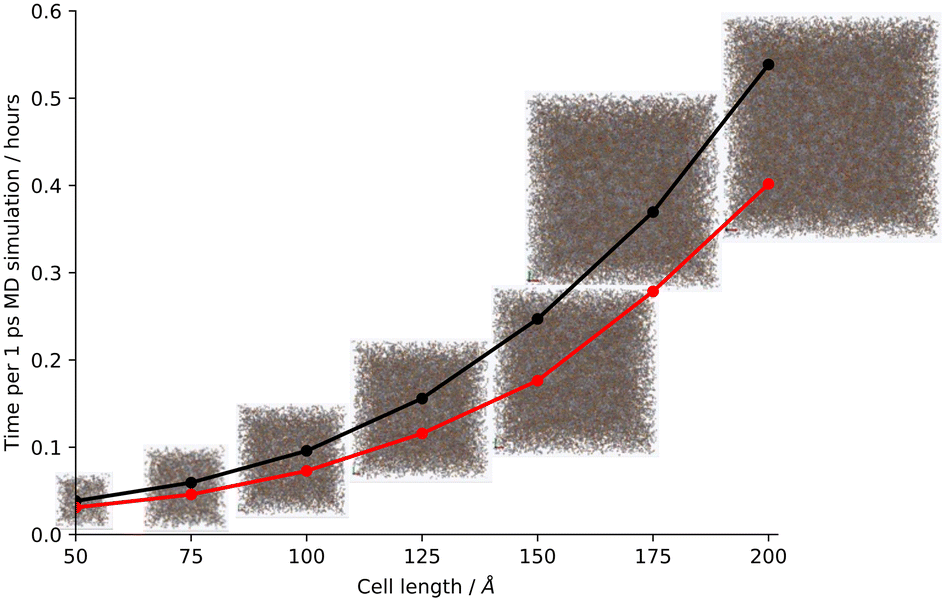 | ||
| Fig. 3 Plot of the unit cell length against the average time to simulate 1 ps MD per step for steps 1–2 of the CMP-1 network formation process. Key: all-atom – black, hybrid coarse-grain – red. Showing the respective models used for each cell length true to scale with each other. | ||
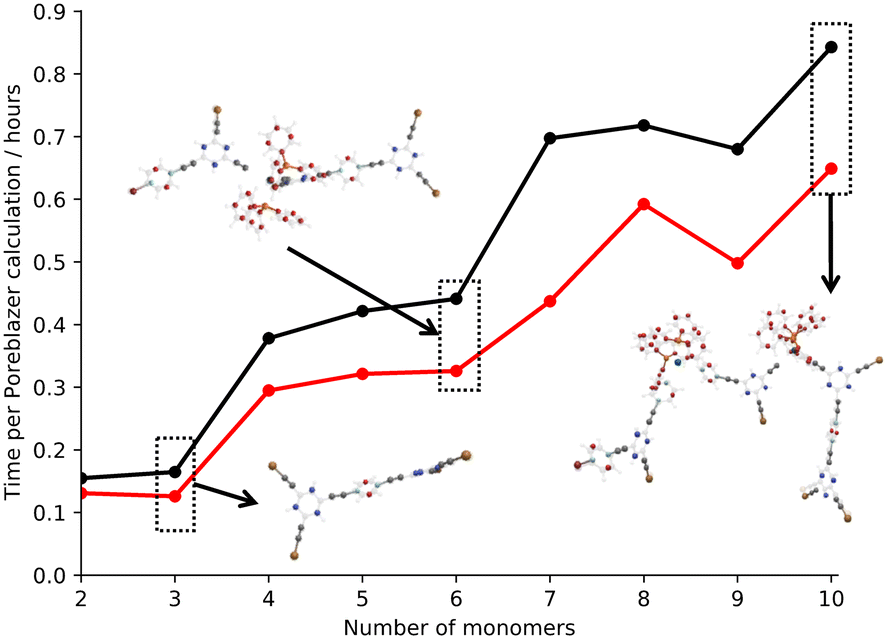 | ||
| Fig. 4 Plot of the number of monomers within the small fragment systems against the average time per Poreblazer calculation.27 Key: all-atom – black, hybrid coarse-grain – red. A selection of all-atom (faded) and back-mapped coarse-grain structures (bold) are also included for clarity. | ||
We then simulated the full artificial synthesis procedure for our largest model (cell length 200 Å). Full details about the requirements needed to modify the artificial synthesis of the scaled-up model compared to the original models generated at a cell length of 50 Å are given in ESI† section S11, but in summary, a larger quantity of geometry optimisation and molecular dynamics were required, and tighter restrictions of the bond formation process were necessary. The porosity properties of this scaled-up, mesoporous model compared to those of the original models simulated show that the scaled-up model was almost twice the density of the original models, leading to a far lower network-accessible surface area and micropore volume (Table S11†).
Previously we have considered different desolvation strategies designed to mimic the effect of diffusion-limiting pore diameters to the removal of solvent, building blocks, and oligomers. Here we used strategy 6 for desolvation, which comprises of removing both the solvent and the building blocks from the entire cell. This has the result of leaving large regions of void which can potentially collapse during the desolvation and work up process leading to a denser structure. It may be that these large models require a more sophisticated approach to desolvation and work up to accurately model the diffusion limiting effect that the pores have on the desolvation process.
It has been previously hypothesised by Thomas et al. that CMP materials such as CMP-1 are comprised of spherical polymer particles, containing a very dense, non-porous central core, made up of building blocks that would rapidly react in the absence of solvent, that is surrounded by regions of gradually decreasing density and increasing porosity on moving closer to the outside of the particle, towards the solvent phase.16 Computationally, this was modelled by considering different quantities of solvation within the polymer, where models artificially synthesised using very small quantities of solvent represented the central core region, whilst the edge regions were modelled with larger quantities of solvent present during the synthesis.16 The decreased porosity and increased density of the scaled-up models compared to the original may also be indicative of the scaled-up model being a closer match to the regions of denser, less porous polymer close to the central core region of the spherical CMP-1 polymer clusters, whilst the smaller original models more closely represent the less dense, more porous edges of the polymer particles, however predicting the relative size of each distinct region of this amorphous polymer is beyond the scope of this work.
Conclusions
In summary, this work reports the mesoscale artificial synthesis of CMP-1 using a hybrid coarse-grained methodology based on the phenyl-based grains reported by DeVane et al.22 and the methodology reported by Thomas et al.16 This enabled a simulation cell scale-up of 64 times, and an overall speed-up factor of 44% when compared to the all-atom equivalent. This suggests that our methodology could be used to generate mesoscale structural models that can demonstrate larger-scale effects such as phase separation and gelation, therefore reducing the number of models required to reliably compare to real-world materials.Author contributions
All authors contributed to conceptualisation and data analysis. Catherine Mollart: data collection and draft preparation. Bartosz Ciborowski: data collection. Abbie Trewin: draft preparation, editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Lancaster University High-End Computing facility.Notes and references
- M. Moshoeshoe, M. S. Nadiye-Tabbiruka and V. Obuseng, Am. J. Mater. Sci., 2017, 7, 196–221 Search PubMed.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 86 CrossRef.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- H. Wang, L. Huang and D. Cao, in Porous Polymers: Design, Synthesis and Applications, ed. S. Qiu and T. Ben, The Royal Society of Chemistry, 2015, pp. 121–150, 10.1039/9781782622260-00121.
- A. Herrera, A. Riaño, R. Moreno, B. Caso, Z. D. Pardo, I. Fernández, E. Sáez, D. Molero, A. Sánchez-Vázquez and R. Martínez-Alvarez, J. Org. Chem., 2014, 79, 7012–7024 CrossRef CAS PubMed.
- A. Liu, C. Mollart, A. Trewin, X. Fan and C. H. Lau, ChemSusChem, 2023, 16, e202300019 CrossRef CAS PubMed.
- S. Qiu and T. Ben, in Porous Polymers: Design, Synthesis and Applications, ed. S. Qiu and T. Ben, The Royal Society of Chemistry, 2015, pp. 95–120 Search PubMed.
- J. M. H. Thomas and A. Trewin, J. Phys. Chem. C, 2014, 118, 19712–19722 CrossRef CAS.
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS.
- Z. Zhao, S. Das, G. Xing, P. Fayon, P. Heasman, M. Jay, S. Bailey, C. Lambert, H. Yamada, T. Wakihara, A. Trewin, T. Ben, S. Qiu and V. Valtchev, Angew. Chem., Int. Ed., 2018, 57, 11952–11956 CrossRef CAS PubMed.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J. M. H. Thomas, C. Mollart, L. Turner, P. Heasman, P. Fayon and A. Trewin, J. Phys. Chem. B, 2020, 124, 7318–7326 CrossRef CAS PubMed.
- R. Dawson and A. Trewin, in Porous Polymers: Design, Synthesis and Applications, ed. S. Qiu and T. Ben, The Royal Society of Chemistry, Cambridge, 2015, ch. 7, vol. 17, pp. 155–185 Search PubMed.
- C. Mollart and A. Trewin, Phys. Chem. Chem. Phys., 2020, 22, 21642–21645 RSC.
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS.
- A. Laybourn, R. Dawson, R. Clowes, T. Hasell, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2014, 5, 6325–6333 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- R. DeVane, M. L. Klein, C.-c. Chiu, S. O. Nielsen, W. Shinoda and P. B. Moore, J. Phys. Chem. B, 2010, 114, 6386–6393 CrossRef CAS PubMed.
- S. J. Marrink, A. H. de Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S.-J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS PubMed.
- J. A. Anderson, C. D. Lorenz and A. Travesset, J. Comput. Phys., 2008, 227, 5342–5359 CrossRef.
- J. Glaser, T. D. Nguyen, J. A. Anderson, P. Lui, F. Spiga, J. A. Millan, D. C. Morse and S. C. Glotzer, Comput. Phys. Commun., 2015, 192, 97–107 CrossRef CAS.
- L. Sarkisov, R. Bueno-Perez, M. Sutharson and D. Fairen-Jimenez, Chem. Mater., 2020, 32, 9849–9867 CrossRef CAS.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- H. Sun, Macromolecules, 1995, 28, 701–712 CrossRef CAS.
- M. F. De Lange, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2014, 200, 199–215 CrossRef CAS.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X.-H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K.-i. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3me00130j |
| This journal is © The Royal Society of Chemistry 2023 |