
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of metal-coordinating comonomers on the coordination structure and binding in magnetic poly(ionic liquid)s†
Kayla
Foley
 a,
Lucas
Condes
b and
Keisha B.
Walters
*a
a,
Lucas
Condes
b and
Keisha B.
Walters
*a
aRalph E. Martin Department of Chemical Engineering, University of Arkansas, Fayetteville, AR 72701, USA. E-mail: keisha.walters@uark.edu
bSchool of Chemical, Biological and Materials Engineering, University of Oklahoma, Norman, OK 73019, USA
First published on 28th July 2023
Abstract
A poly(ionic liquid) (PIL), poly(acrylamide-co-diallyl dimethylammonium chloride), was systematically complexed with Co2+, Fe3+, and Co2+/Fe3+ (mixed) at different molar equivalencies to form a series of magnetic PILs (MPILs). These novel MPILs were utilized to examine molecular structure and binding between the polymer and metal species using comprehensive spectroscopic studies in both dry and liquid states. FTIR, X-ray photoelectron, UV-vis, and Raman spectroscopies showed evidence of metal coordination of both the iron and cobalt chloride species with the acrylamide comonomer. Using AC susceptibility measurements, the MPILs were found to have magnetic properties within the range of typical MPIL homopolymers with magnetic mass susceptibilities dependent on metal cation type and concentration. This work demonstrates MPILs can be tuned using the metal ion species and concentration, to increase the magnetic mass susceptibility and alter metal-ion coordination structure and binding with the PIL copolymer. Furthermore, the coordination structure of the transition metal halide complex was determined to be dependent on metal halide concentration in the polymer for the iron-based system, and it was shown that both metal cations (Fe3+ and Co2+) bond not only electrostatically with the PIL monomer but also predominantly with the acrylamide comonomer. In particular, metal–oxygen and metal–nitrogen bonding was observed with the amide group present in the acrylamide. Understanding and controlling the coordination structure and binding in PILs is significant as these materials allow for the formability and flexibility advantages of polymers to be combined with properties atypical for polymers, such as high ion conductivity, electrical and thermal stability, antibacterial properties, and magnetic responsiveness, in the case of MPILs.
Design, System, ApplicationThis work thoroughly examines the effects of a metal-coordinating comonomer in a magnetically responsive poly(ionic liquid) (MPIL) system on the paramagnetic complex formation, polymer–metal ion binding, and magnetic properties and response in both dry and wet states. A quaternary ammonium PIL was copolymerized with an acrylamide comonomer and systematically complexed at different molar equivalencies with Co2+, Fe3+, and Co2+/Fe3+ (mixed) chloride salts. While single-component MPIL systems bind anionic metal complexes (e.g., [FeCl4]−, [CoCl4]2−) to the polymer through ionic bonding, FTIR and XPS findings indicate that paramagnetic salts predominately bind to the MPIL copolymers through metal ion-coordination with the acrylamide groups. The paramagnetic complex structure is dependent on metal salt concentration as shown by UV-vis and Raman spectroscopy studies. AC susceptibility measurements reveal magnetic mass susceptibilities similar to single-component MPILs. Molecular structure and binding, such as the metal ion-polymer coordination, is demonstrated to impact the macroscale properties, and an understanding of co-material interactions supports a targeted and tailored design of complex multi-component systems such as MPIL random or block copolymers and blends. This work provides insight towards the design and development of functional magnetically responsive polymers for applications such as organic magnets, sensors, and magnetically responsive self-assembled nanomaterials. |
Introduction
Poly(ionic liquid)s, or PILs, are composed of polymerized ionic liquid monomers that form strong polyelectrolytes with permanent charge groups on the polymer pendant groups and exchangeable counterions. PILs exhibit high ion conductivity, electrical and thermal stability, antibacterial properties, and a variety of other favorable properties which are tunable through counterion exchange.1–3 Task-specific counterions may also be introduced to impart PILs with stimuli responsive properties to environmental conditions such as pH,4 light,5 electric potential,6 and magnetic fields.7Magnetic poly(ionic liquid)s (MPILs) are an emerging class of PILs with ionically bound paramagnetic counterions, frequently metal halides (i.e., [FeCl4]−, [CoCl4]2−), that imbue the polymers with magnetic responsiveness. Instead of a counterion exchange, MPILs are typically synthesized through coordination interactions between PIL halide counterions and added transition metal salts that electrostatically bind to the polyelectrolyte. Dobbelin et al. developed some of the first paramagnetic MPILs by complexing FeCl3 or FeBr3 salts with poly(vinyl alkyl imidazolium chloride) and poly(diallyl dimethylammonium chloride) type PILs to form MPILs with high spin paramagnetic iron complexes ([FeCl4]−, [FeBr3Cl]−, [FeBr4]−, [Fe2Cl7]−, [Fe3Cl10]−).7 These MPILs could be processed into powders or films and showed comparable or higher magnetic susceptibilities than their non-polymerized, small molecule counterparts, magnetic ionic liquid (MILs)8–10 and magnetic surfactant (Mag-surf)11,12 complexes.
In the past decade, the MPIL field has expanded to a variety of applications including catalysis,7 organic templates and metal ion sources for in situ metal nanoparticles synthesis,13 magnetic separations,14,15 antibacterial materials,16 optical and fluorescent quantum dots for biological labeling, and molecular magnet development.17,18 In many of these systems, MPILs have been incorporated into multicomponent systems, such as copolymers and composites. For instance, Bonnefond et al. developed magnetic latexes from the copolymerization of an MPIL with butyl acrylate and methyl methacrylate copolymers.19 Thermoset MPILs with weak ferromagnetic properties were formed by Carrasco et al. from a partially quaternized pyridine MPIL and acrylate block copolymers blended with epoxy. Cui et al. developed imidazolium-based superparamagnetic MPIL copolymers with FeCl4− anions that acted as quenchers for the fluorescent pyrene comonomer.20 MPILs and MILs have been used to form composites with carboxyl-functionalized graphene oxide, as well as other nanoparticles, that were magnetically separatable from aqueous solution.15,21
The co-materials in these multicomponent systems often contain functional groups (e.g., carboxylic acids, amides, amines, thiols) capable of interacting or coordinating with transition metal complexes which may in turn influence the magnetic properties of those complexes or how they are integrated into the MPIL system. However, very few studies have examined how the transition metal halide complex interacts with the polymer in multicomponent MPIL systems. While it was not a traditional MPIL, it is noteworthy to mention the work of Yang et al. where metal ion coordination (in this case, Ni2+ and Nd3+) in a imidazolium BF4− based PIL copolymerized with N-2-thiazolylmethacrylamide greatly influenced the magnetic behavior and properties of the polymer depending on metal cation type, random or block copolymerization, and coordination structure with the thiazole ring.22 Similar coordination behavior is expected for traditional MPILs with metal-coordinating comonomers or co-materials in multicomponent systems.
This work provides an in-depth analysis into the metal ion coordination structure and binding interactions in magnetically responsive multi-component PIL systems, an effort that, to the authors' knowledge, has not been previously presented in the literature. After systematic complexation of a PIL copolymer with different metal salts at different molar equivalencies, molecular structure and binding were characterized through a comprehensive spectroscopic study. In particular, poly(acrylamide-co-diallyl dimethylammonium chloride), a PIL copolymer with a quaternary ammonium PIL group and a comonomer capable of metal coordinating interactions, was selected as the halide PIL for this study, and this PIL was complexed with Co2+ and Fe3+ metal halide salts to form the MPIL copolymers. After synthesis, the chemical properties and molecular interactions of these copolymers were studied in both the liquid and dry states as a function of metal salt concentration and cation type using FTIR, X-ray photoelectron, UV-vis, and Raman spectroscopies. The magnetic properties of these MPIL copolymers were evaluated using AC susceptibility measurements. As magnetically responsive PIL functional materials show tremendous promise for technological innovation and novel applications, it is critical to understand how transition metal ions interact in MPIL multicomponent systems and subsequently impact the properties of these unique materials.
Materials and experimental methods
Materials
A 10 wt% solution of poly(acrylamide-co-diallyldimethylammonium chloride) ([Pam-co-PDADMA][Cl−]) in water was purchased from Sigma Aldrich. The copolymer contained 55% acrylamide (Pam) and 45% diallyldimethylammonium chloride (DADMA[Cl−]) by weight. CoCl2·6H2O was purchased from Sigma Aldrich, and FeCl3·6H2O was purchased from Alfa Aesar. All materials were used as received.Synthesis of magnetic copolymers
Magnetic copolymers were synthesized through complexation of the metal halide in aqueous solution with the copolymer. In a typical synthesis, 10 ml of the 10 wt% [Pam-co-PDADMA][Cl−] copolymer solution was added to a round bottom flask and further diluted with 5 mL of Millipore water. Aqueous solutions of FeCl3·6H2O, CoCl2·H2O, or ratioed mixtures of iron and cobalt chlorides were prepared in various concentrations to yield magnetic copolymers with 1 (3.09 mmol), 2 (6.19 mmol), or 3 (9.28 mmol) equivalents of the metal halide anions with respect to the PDADMA group. The metal halide solutions were then added dropwise to the copolymer solution under stirring (note gelling was observed after addition of 3–5 drops of FeCl3·6H2O solution, but the gel structure collapsed with continued addition of the metal halide solutions). For the cases where significant gelling occurred, the system was further diluted with water until completely solubilized. All solutions were stirred overnight at room temperature. The water was evaporated from the system, and the magnetic MPIL polymer product was placed in a vacuum oven (∼45 °C) for further drying before characterization.Spectroscopic characterizations
Attenuated total reflection (ATR) FTIR spectra of dried polymer were collected using a ThermoFisher Scientific Nicolet iS50R spectrometer with 4 cm−1 resolution and a minimum of 64 scans over the 4000–400 cm−1 spectral range. Raman spectroscopy was performed on a Renishaw inVia Raman microscope with a 785 nm red laser at a 1% power setting and 30 second scans for the 1200–200 cm−1 spectral range. Samples were prepared by drop casting thin films from aqueous solution onto microscope slides.Physical Electronics PHI VersaProbe 5000 station X-ray photoelectron spectrophotometer (XPS) was used to obtain XPS spectra of dry polymer films. A monochromated 25.0 W Al 1486.6 eV source was used for data collection. Survey scans and high-resolution scans were obtained at pass energies of 117.40 eV and 23.5 eV, respectively. Samples were prepared by drop casting MPIL aqueous solutions onto gold-coated silicon wafers. The peak maximum in the C1s spectra was pinned to 285 eV, corresponding to the aliphatic C–C/C–H C1s band, used to provide for charge correction. All data were analyzed using CasaXPS (version 2.3.23PR1.0) software. The C1s, N1s, O1s, and Cl2p high-resolution spectra were fit using Shirley backgrounds and Gaussian-Lorentzian mixed line shapes, typical for polymers;23 the experimental error for all high-resolution spectra was ±0.2 eV. The Fe 2p and Co 2p high-resolution spectra were fit using asymmetrical line shapes typical for transition metals.24,25 To avoid collecting photoelectrons from excess unbound metal chloride salts, the 3 eq. samples were not examined with XPS.
Solution samples were analyzed with a double beam Shimadzu UV-2450 ultraviolet-visible (UV-vis) spectrophotometer. UV-vis spectra were collected for each sample at room temperature at a scan rate of 500 nanometers per minute over a 190–750 nm spectral range with a 0.5 nm sampling interval and 2.0 nm slit width. All samples were measured in UV grade fused silica cuvettes with a path length of 10 mm (International Crystal Laboratories). UV-vis absorption spectra of solid films coated on microscope slides were also obtained with a Shimadzu Biospec 1601 spectrophotometer over 800–190 nm spectral range and a 1 nm sampling interval.
Magnetic characterizations
Magnetic mass susceptibilities were obtained with a Kappabridge MFK1 magnetic AC susceptibility meter. Solid sample flakes were added to 4 mL quartz cuvettes. The instrument was operated at a field strength of 450 A m−1 and at an AC field frequency of 976 Hz. The magnetic susceptibility reported is an average of 10 measurements. The diamagnetic contribution of the quartz cuvette was subtracted from the volume susceptibility and then normalized by mass, yielding the magnetic mass susceptibilities. A similar procedure was followed for liquid samples where 4 mL of copolymer solution was added to a quartz cuvette. The mass susceptibility measured from a cuvette filled with 4 mL of Millipore water was used to subtract the diamagnetic contributions from the water. Two back-to-back 0.5 T neodymium magnets with a 1 inch diameter and 0.75 inch thickness were used to examine the magnetic response of the copolymers. The combined magnetic field of the two magnets was calculated to be ∼0.6 T at the magnet surface.Results and discussion
Paramagnetic poly(ionic liquid) copolymers were synthesized by complexing FeCl3, CoCl2 or a ratioed mixture of the iron and cobalt metal halide salts with [Pam-co-PDADMA][Cl−] in aqueous solution to form the corresponding magnetic copolymer complexes [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][Co2+] (Scheme 1). During synthesis, the metal halide salts solutions were slowly added dropwise in concentrations to provide 1, 2, or 3 mole equivalents of the metal halide with respect to the DADMA monomer unit. During the synthesis, gelation initially occurred upon addition of the first ∼3–5 drops for the Fe3+ samples, likely due to the formation of metal coordination bonds between acrylamide units. The gel structure collapsed with continued metal halide solution addition. Following complexation and removal of the solvent, magnetic PIL copolymers were obtained. Similar copolymer systems with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio of mixed FeCl3 and CoCl2 salts were also prepared to form the [Pam-co-PDADMA][mixed Fe3+/Co2+] series. For example, [Pam-co-PDADMA][mixed Fe3+/Co2+ 1 eq.] consists of 0.5 mol equivalents of CoCl2 and 0.5 mol equivalents of FeCl3 for a total of 1 mole equivalent of metal salts added to the copolymer with respect to the PDADMA monomer unit. In total, nine magnetic copolymers were prepared with varying metal type and concentration. As observed in other MPIL systems, these copolymers displayed different colors based on the metal species and concentration as seen in Fig. 1A. The [Pam-co-PDADMA][Cl−] copolymer with no added metal content was a light pink color. The iron copolymers, [Pam-co-PDADMA][Fe3+], were colored dark brown (1 eq.), light brown (2 eq.), and orange-brown (3 eq.). All equivalents of the cobalt copolymer series, [Pam-co-PDADMA][Co2+], were a dark blue color. The [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymers were green (1 eq.), dark green (2 eq.), and green-brown (3 eq.). In general, the color of MPILs, and other metal-ion polymer complexes, can vary significantly depending on metal complex coordination structure (e.g., [FeCl4]−vs. [FeCl3Br]−), metal ion concentration, and the ligand or polymer chemical composition. Dobbelin et al. reported a yellow color for the poly(diallyl dimethylammonium [FeCl4]−) homopolymer and light brown for poly(vinyl butylimidazolium [FeCl4]−) homopolymer; the latter darkened upon formation of the [Fe2Cl7]− and [Fe3Cl10]− complexes.7 The iron copolymers in this work start with a dark brown color at the lowest iron concentration, which lightens to a light-brown color for the [Pam-co-PDADMA][Fe3+ 2 eq.] copolymer and darkens again upon an increase in iron content. The cobalt series copolymers display a darker blue color than the typical light blue colors reported for the MPILs poly((3-acrylamidopropyl)-trimethylammonium [CoCl4]−)13 and poly(vinyl dodecyl imidazolium [CoCl3Br]−).19 The green color of the mixed metals series derives from the combination of blue and yellow of the cobalt and iron systems, indicating the ability to tune polymer color by combining different metal salts. These color differences in the MPIL copolymers, particularly in those containing iron chloride salts, provide qualitative evidence of changes in metal–polymer complexation at different mole equivalencies. This observation was corroborated by corresponding changes in solubility.
:1 mole ratio of mixed FeCl3 and CoCl2 salts were also prepared to form the [Pam-co-PDADMA][mixed Fe3+/Co2+] series. For example, [Pam-co-PDADMA][mixed Fe3+/Co2+ 1 eq.] consists of 0.5 mol equivalents of CoCl2 and 0.5 mol equivalents of FeCl3 for a total of 1 mole equivalent of metal salts added to the copolymer with respect to the PDADMA monomer unit. In total, nine magnetic copolymers were prepared with varying metal type and concentration. As observed in other MPIL systems, these copolymers displayed different colors based on the metal species and concentration as seen in Fig. 1A. The [Pam-co-PDADMA][Cl−] copolymer with no added metal content was a light pink color. The iron copolymers, [Pam-co-PDADMA][Fe3+], were colored dark brown (1 eq.), light brown (2 eq.), and orange-brown (3 eq.). All equivalents of the cobalt copolymer series, [Pam-co-PDADMA][Co2+], were a dark blue color. The [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymers were green (1 eq.), dark green (2 eq.), and green-brown (3 eq.). In general, the color of MPILs, and other metal-ion polymer complexes, can vary significantly depending on metal complex coordination structure (e.g., [FeCl4]−vs. [FeCl3Br]−), metal ion concentration, and the ligand or polymer chemical composition. Dobbelin et al. reported a yellow color for the poly(diallyl dimethylammonium [FeCl4]−) homopolymer and light brown for poly(vinyl butylimidazolium [FeCl4]−) homopolymer; the latter darkened upon formation of the [Fe2Cl7]− and [Fe3Cl10]− complexes.7 The iron copolymers in this work start with a dark brown color at the lowest iron concentration, which lightens to a light-brown color for the [Pam-co-PDADMA][Fe3+ 2 eq.] copolymer and darkens again upon an increase in iron content. The cobalt series copolymers display a darker blue color than the typical light blue colors reported for the MPILs poly((3-acrylamidopropyl)-trimethylammonium [CoCl4]−)13 and poly(vinyl dodecyl imidazolium [CoCl3Br]−).19 The green color of the mixed metals series derives from the combination of blue and yellow of the cobalt and iron systems, indicating the ability to tune polymer color by combining different metal salts. These color differences in the MPIL copolymers, particularly in those containing iron chloride salts, provide qualitative evidence of changes in metal–polymer complexation at different mole equivalencies. This observation was corroborated by corresponding changes in solubility.
 | ||
| Scheme 1 Complexation of [Pam-co-PDADMA][Cl−] with metal halide salts to form magnetic [Pam-co-PDADMA][Mn+]. MClx refers to the metal halide salts FeCl3 or CoCl2. MClxn− denotes either [FeCl4]− or [CoCl4]2− anionic complexes. Mn+ refers to Fe3+ or Co2+ metal cations. | ||
 | ||
| Fig. 1 (A) Dried magnetic copolymers flakes displaying blue (cobalt), orange-brown (iron), and green (mixed) colors based on metal ion type and concentration. (B) Aqueous solutions at 7 mg mL−1 of the magnetic copolymers. The [Pam-co-PDADMA][Co2+] MPIL copolymers showed complete solubility at all equivalencies, while the [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] MPIL copolymers showed limited solubility and some gelation and suspension behaviors. | ||
Solubilities of the magnetic poly(ionic liquid) copolymers were examined in water (Fig. 1B). The neat (unmodified) Cl− based copolymer, [Pam-co-PDADMA][Cl−], was completely soluble in water at room temperature. Upon complexation with the cobalt metal halide salts, the copolymers (at all equivalents) were found to be completely soluble in water, and all resulted in pink solutions. In contrast, [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymer series were both found to display only partial solubility. The [Pam-co-PDADMA][Fe3+ 1 eq.] sample appeared to form some orange-brown hydrogel-like structures, while higher equivalents resulted in only pale-yellow suspensions that were stable for 2–3 h. The [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymer series displayed the formation of large particulates for all equivalents when placed in water. Hazell et al. produced PDADMA homopolymers complexed with iron halide salts that were completely soluble in water.15 This suggests here that the iron halide salts interact with the acrylamide units of the copolymer in solution during synthesis, forming coordinating crosslinked regions in the polymer, resulting in precipitation or gelation. Differences in the solubilities obtained for the iron based MPIL copolymers containing a metal coordinating comonomer compared to other similar homopolymer systems further suggest that polymer–metal ion binding are occurring at least partially through metal-ion coordination rather than purely electrostatic interactions in aqueous solution and possibly during the complexation reaction.
Multiple spectroscopic methods were used to study the chemical interactions and structure of the copolymers and transition metal complexes in both the dry and solution state. MPILs are formed through electrostatic binding with paramagnetic ion complexes, typically through tetrahedral transition metal halide anions ([FeCl4]−, [CoCl4]2−, etc.). However, the comonomer, acrylamide, is capable of forming metal coordination bonds through interaction with the carbonyl oxygen atom (O-binding) or the amide nitrogen atom (N-binding)26,27 as shown in Fig. 2. Acrylamide predominately binds with metal ions through the more basic carbonyl site, though N-binding is also possible depending on the acidity or hardness of the metal cation and the availability of adjacent anchoring chelation sites to stabilize the N-binding complex.26–29 Monomeric acrylamide coordinated with metal chloride salts has been found to form mainly 6-coordinated structures (e.g., [Co(AAm)4Cl2], [Fe(AAm)4Cl2], [Ni(AAm)4Cl2]) or associated cation-anion pair complexes (e.g., [Co(AAm)6]2+[CoCl4]2−),26,30 though steric constraints imposed by the polymeric backbone mainly alter these structures in the MPIL copolymers. As coordination with the acrylamide may influence the formation of the transition metal complex in the MPIL copolymers, the coordination structure of the transition metal species were first examined using Raman and UV-vis spectroscopies. Characterization using FTIR and XPS spectroscopies was then performed to investigate the interactions between the transition metal species and the MPIL copolymers.
 | ||
| Fig. 2 Possible transition metal ion binding sites for single-site (A) O-binding and its resonance structure, (B) N-binding, and (C) two-site O- and N-binding cases. (B) is adapted from Girma et al.26 | ||
Raman microscopy is typically performed on MPILs to confirm the formation of the tetrahedral anionic complex (i.e., [FeCl4]− or [CoCl4]2−). Fig. 3 shows the Raman spectra for the MPIL copolymer films in the dry state. The Fe–Cl str 334 cm−1 peak, typically considered characteristic for the [FeCl4]− and FeCl3 species, was observed in the [Pam-co-PDADMA][Fe3+] samples.7,14 For the solid samples, the [FeCl4]− can form [Fe2Cl7−] dimers as the metal halide concentration increases, resulting in Raman peaks at 315 and 420 cm−1.7,31 Here, this bridging complex peak was observed at ∼314 cm−1 for the [Pam-co-PDADMA][Fe3+ 2 eq.] and [Pam-co-PDADMA][Fe3+ 3 eq.] samples. The broad absorbance observed between 250 and 500 cm−1 prevents the observation of a distinct 420 cm−1 peak. In the cobalt-based samples, there is an intense broad peak from 250 to 500 cm−1 with a maximum at ∼358 cm−1 that is not observed for the [Pam-co-PDADMA][Cl−] MPIL spectra. This broad peak is likely composed of multiple peaks related to metal species complexes—including the characteristic tetrahedral cobalt halide complex peak expected at 270 cm−1.32–34 Specifically, this broad peak at low wavenumbers is likely due to metal bonding with oxygen and/or nitrogen (M–O or M–N). Freire et al. reported a broad band centered at ∼313 cm−1 for Cu–O bonding in formamide Cu2+ coordination complexes.35 Other examples of M–O and M–N stretching modes have been reported in the 600–200 cm−1 region.35–39 Therefore, the presence of this band is expected to be due to metal cation coordination with the acrylamide unit. Due to the intensity of the band in the cobalt and mixed series, it is possible that the tetrahedral [CoCl4]2− and [FeCl4]− complex modes are overlapping in this region. Yu et al. observed similar broad peak behavior in this region for small degrees of quaternization in polyethyleneimine polymers complexed with the [FeCl4]− anions,40 where the majority of the amine groups have lone electron pairs available for coordination bonding.
 | ||
| Fig. 3 Raman spectra of the MPIL copolymers from 1200 to 220 cm−1 are shown for [Pam-co-PDADMA][Cl−] (black), [Pam-co-PDADMA][Fe3+] (red), [Pam-co-PDADMA][Co2+] (blue), and [Pam-co-PDADMA][mixed Fe3+/Co2+] (green) at each mole equivalence. The solid arrows indicate the intense Fe–Cl stretch peak at 334 cm−1 for the FeCl3 or [FeCl4]− species and the 315 cm−1 shoulder characteristic of the [Fe2Cl7]− bridging complex. Dashed lines mark the peak center for the broad band assigned to coordination of the metal cation with oxygen or nitrogen groups (i.e., M–O or M–N) in acrylamide. | ||
The broad peak absorbance observed with Raman in all the MPIL copolymer samples indicates the presence of the acrylamide–metal species coordination. Further, while evidence is observed for the presence of Fe–Cl bonds and the possible presence of the [FeCl4]− and [Fe2Cl7]− for MPIL copolymers, particularly at higher iron halide mole equivalencies, the formation of the [CoCl4]− structure is less clear. UV-vis spectroscopy was therefore used to provide additional insight into the [FeCl4]− and [CoCl4]2− complexes.
UV-vis spectroscopy of the solid MPIL copolymer films was performed to provide further insight into the metal species coordination structure, and the results are displayed in Fig. 4(A–C). Second derivative analyses were performed to aid in peak assignments and are shown in Fig. 4(D–F). The [Pam-co-PDADMA][Cl−] copolymer (Fig. 4A and D, black line) had a major peak absorbance at 282 nm that is attributed to the acrylamide carbonyl band as the quaternary ammonium group does not contain a chromophore.
 | ||
| Fig. 4 UV-vis spectra and their second derivatives of the solid MPIL films are displayed in (A and D, blue) [Pam-co-PDADMA][Co2+], (B and E, red) [Pam-co-PDADMA][Fe3+], and (C and F, green) [Pam-co-PDADMA][mixed Fe3+/Co2+]. UV-vis spectra and second derivative plots for the base copolymer [Pam-co-PDADMA][Cl−] are also shown (A and D, black). Characteristic [CoCl4]2− and [FeCl4]− peaks are indicated by blue and red arrows, respectively, in the second derivative plots. The tetrahedral structure of the cobalt species is confirmed by the presence of the 628, 665, and 692 nm peaks (blue arrows). Characteristic [FeCl4]− complex is also confirmed for the 2 and 3 eq. [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymers (red arrows). | ||
In the visible region, three main bands are observed at ca. 628, 665, and 692 nm with two minor peaks at ∼610 and 640 nm for both the cobalt and mixed series films. The tetrahedral geometry of cobalt complexes typically display d–d transition bands at ∼625 nm and 665 nm while an octahedral coordination complex is designated by a band at ∼525 nm.41,42 The absence of the ∼525 nm band in the films indicates that the cobalt species only have a tetrahedral structure in these film samples. These three main bands and the general shape of the spectra are in close agreement with the ∼630, ∼665, and ∼690 nm reported for the [CoCl4]2−.43–46 This UV-vis data confirms the presence of the paramagnetic [CoCl4]2− anion in the MPIL films for all equivalents examined. However, other tetrahedral coordination structures (e.g., [CoCl2L2], [CoCl3L]−, [CoL4]2+[Cl]2−, where L is acrylamide ligand) cannot be dismissed. Similar absorbances in the 600–700 nm range have been previously assigned to tetrahedral coordination of CoCl2 with other ligands such as nitrate and diethyl sulfoxide.46–48 The similarity of the UV-vis spectra in the 500–800 nm range for all [Pam-co-PDADMA][Co2+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymers indicates that the cobalt complex structure forms similarly for all mole equivalences.
In the UV region, bands at ∼248 nm and 286 nm are also observed in the cobalt series films. These peaks are attributed to the acrylamide carbonyl and are slightly shifted compared to the [Pam-co-PDADMA][Cl−] copolymer, possibly indicating metal ion coordination with the amide group. In the iron and mixed series films, the first broad peak centered at ∼270 nm can be deconstructed into 2–3 bands in the 240–300 nm range (ca. 250, 270, and 286 nm) by second derivative analysis. These bands are similarly assigned to acrylamide carbonyl modes interacting with Fe3+ species. These results are in agreement with similar work reported by Sowwan and Dweik et al. for metal coordinated polyacrylamide with bands at 276 nm and 285 nm for Cu2+ and Ni2+ complexes, respectively.49,50
The [FeCl4]− complex characteristically shows intense absorbances at ca. 250, 315, and 360 nm.51–54 These peaks are evident in the 2 and 3 eq. [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] series films, but not in the 1 eq. sample, which corroborates the Raman results for 2 and 3 eq. [Pam-co-PDADMA][Fe3+]. Second derivative analysis of the second broad band centered at ∼370 nm in the [Pam-co-PDADMA][Fe3+ 1 eq.] sample shows a set of three peaks at 327, 354, and 395 nm, which are tentatively assigned to FeCl3 complexation with the acrylamide unit. While identification of the specific FeCl3–acrylamide coordination complex structures is beyond the scope of this work, these results demonstrate that changes in coordination structure occur as a function of concentration of iron chloride salts. At low concentrations there is first coordination of FeCl3 with acrylamide units of the polymer followed by formation of [FeCl4]− and [Fe2Cl7]− anions, which bind electrostatically with the quaternary ammonium PIL units, at higher metal salt concentrations as acrylamide binding sites have become occupied. Compared to the Co2+ cation, the trivalent nature of the Fe3+ cation likely leads to more rapid occupation of the acrylamide binding sites as metal halide concentration increases. Overall, it was observed that the acrylamide monomer unit influences the transition metal complex coordination structure in the dry iron-containing MPIL copolymer samples as metal content is increased. The findings are significant as they demonstrate that comonomer-metal coordination is occurring in these multi-component MPIL materials and provide additional understanding of the role of the metal species and concentration in this binding.
UV-vis spectroscopy of the copolymer solutions indicates significant hydration of the metal complexes for all systems (Fig. 5). Hydration of the Co2+ ions was evidenced by the presence of two peaks at ∼465 and 510 nm typical of the octahedral Co aqueous species45 as will be discussed further in the magnetic properties discussion below. The iron and mixed series showed broad absorbances between 300–500 nm, which are attributed to various Fe(OH)x and FeCl3 aq. species53,55 and resemble spectra of the FeCl3 and CoCl2 salt-only solutions (ESI,† Fig. S1). The acrylamide carbonyl is indicated by two peaks at ∼216 and ∼250 nm in [Pam-co-PDADMA][Cl−]. These peaks do not shift appreciably in the cobalt series films, indicating little interaction of the metal cation with the amide group in solution. Unfortunately, the iron species absorbances dominated in the iron and mixed copolymer solutions and the acrylamide carbonyl peaks could not be identified. However, it may be inferred from the gelation and solubility behavior exhibited by the [Pam-co-PDADMA][Fe3+] systems, partial coordination between the metal ions and the acrylamide units is taking place in the aqueous suspensions.
 | ||
| Fig. 5 UV-vis spectra of the copolymer solutions shows the predominant presence of hydrated Co(OH)6 and Fe(OH)x in aqueous solutions as indicated by the series of absorbances between 300–500 nm in the [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymers and by the characteristic octahedral cobalt species peaks at 465 nm and 510 nm in the [Pam-co-PDADMA][Co2+] copolymers. | ||
Fe3+ coordination in solution of the [Pam-co-PDADMA][Fe3+] copolymers during the synthesis influences the final iron coordination structure in the dry state as seen in the UV-vis spectra of the dry films. Conversely, the dissociation of the hydrated Co2+ species in solution and the similarity of the dry film UV-vis spectra observed for the [Pam-co-PDADMA][Co2+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymers at all molar equivalences suggests that acrylamide coordination with Co2+ species in solution does not play a significant role in the final cobalt species coordination structure.
ATR-FTIR spectroscopy was performed to examine binding interactions between the transition metal complexes and the MPIL copolymers. The unmodified commercial copolymer PIL–Cl− copolymer displayed the characteristic amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str) and amide II (NH2 wag) bands at 1662 cm−1 and 1615 cm−1, respectively (Fig. 6A and B, black trace). The amide III (C–N str and NH bending) modes are typically characterized by several bands in the 1430–1200 cm−1 region.56,57 Here, the band at ∼1417 cm−1 is assigned to the amide III (C–N str) mode, though there are several weaker bands in the 1400–1200 cm−1 region which may also contribute to the NH bending modes. The corresponding symmetrical primary amide N–H stretching mode is seen at ∼3200 cm−1. The asymmetrical N–H stretch is overlapping with the broad peak at 3370 cm−1 assigned to hydrogen bonding of the Cl− ion with the polymer and molecularly absorbed water. The peak at 1489 cm−1 is due to the bending modes of the quaternary ammonium methyl groups.58–61
O str) and amide II (NH2 wag) bands at 1662 cm−1 and 1615 cm−1, respectively (Fig. 6A and B, black trace). The amide III (C–N str and NH bending) modes are typically characterized by several bands in the 1430–1200 cm−1 region.56,57 Here, the band at ∼1417 cm−1 is assigned to the amide III (C–N str) mode, though there are several weaker bands in the 1400–1200 cm−1 region which may also contribute to the NH bending modes. The corresponding symmetrical primary amide N–H stretching mode is seen at ∼3200 cm−1. The asymmetrical N–H stretch is overlapping with the broad peak at 3370 cm−1 assigned to hydrogen bonding of the Cl− ion with the polymer and molecularly absorbed water. The peak at 1489 cm−1 is due to the bending modes of the quaternary ammonium methyl groups.58–61
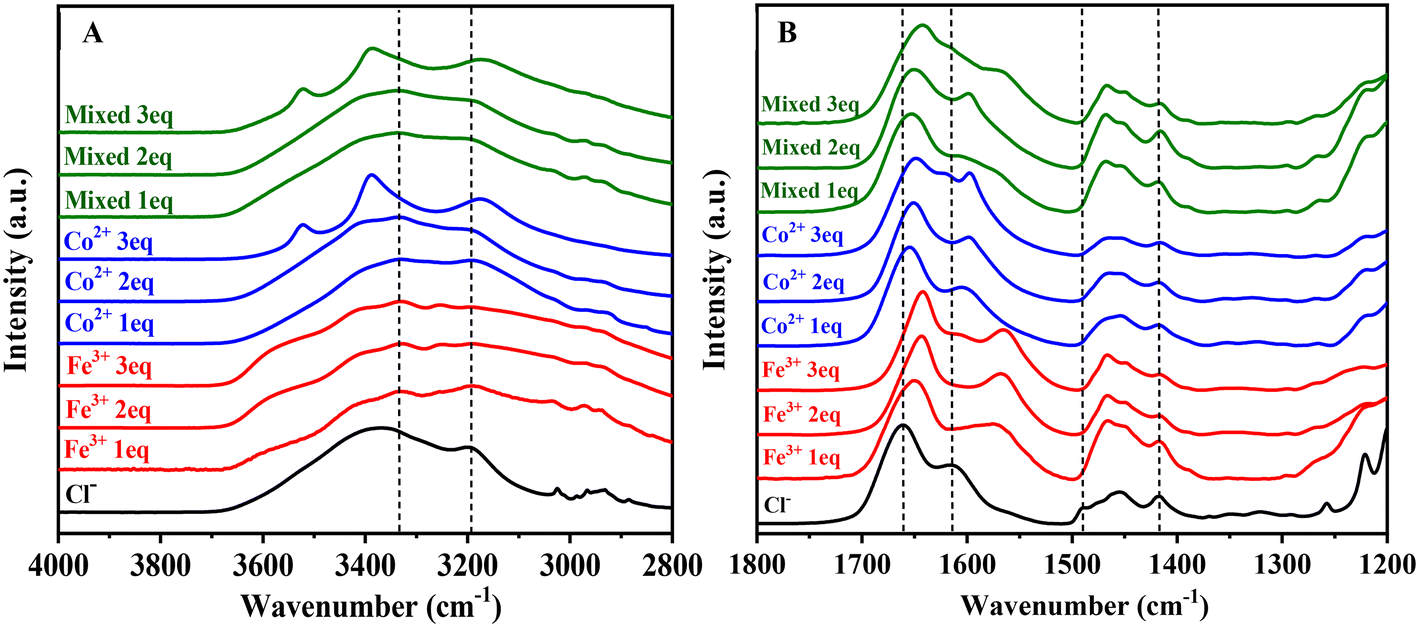 | ||
| Fig. 6 ATR-FTIR spectra of the MPIL copolymers from (A) 4000–2800 cm−1 and (B) 1800–1350 cm−1. At higher wavenumbers (A) the black dashed lines show the general location for the N–H asymmetrical and symmetrical stretches. In (B) the black dashed lines show the peak centers for the [Pam-co-PDADMA][Cl−] copolymer amide I (CO str), amide II (NH2 wag), the PDADMA methyl bending mode, and the amide III (CN str) (left to right). Significant shifts to lower wavenumbers are observed for both the amide I and amide II bands for each of the metal-copolymers, indicating transition metal species coordination with acrylamide unit, likely through both O- and N-bonding. Greater downshifts are observed for the [Pam-co-PDADMA][Fe3+] series compared to the [Pam-co-PDADMA][Co2+] and [Pam-co-PDADMA][mixed Fe3+/Co2+]. | ||
Fig. 6 and Table S1† show the absorbance shifts of the copolymers after metal complexation. Above 3000 cm−1, a slight decrease in frequency to ∼3190–3196 cm−1 in the symmetrical N–H stretch is observed (Fig. 6A). A defined band at ∼3330–3335 cm−1 emerges and is tentatively assigned to the N–H asymmetrical stretching mode. The decrease in the N–H stretching modes seen here is an indication of increased interactions of the amide hydrogen atoms in the intermolecular or intramolecular environment of the copolymers upon metal complexation, though not necessarily an indication of metal coordination.57 However, due to significant overlap with the hydrated metal complex –OH stretches, assignments and peak shifts in this region must be interpreted with caution. The spectra for the pure hexahydrate metals show peaks (ESI,† Fig. S2) at 3522, 3383, and 3163 cm−1 for CoCl2·6H2O and 3526, 3386, 3217, 3005 cm−1 for FeCl3·6H2O. These modes are clearly observed in the spectra for the 3 eq. metal complexed copolymers. For copolymers with lower metal content, these peaks are not clearly defined, though broadening is observed in the region.
All copolymer samples showed significant downshifts in both the amide I and amide II bands, indicating metal ion interactions with the polyacrylamide units (Fig. 6B). The cobalt series showed the smallest shifts with 6 to 14 cm−1 and 11 to 18 cm−1 downshifts for the amide CO str and NH2 wagging modes, respectively. The [Pam-co-PDADMA][Fe3+] series showed the greatest decreases, shifting downwards ∼12 to 19 cm−1 for the amide I band and 41–50 cm−1 for the amide II band. The CO bands for the [Pam-co-PDADMA][mixed Fe3+/Co2+] series showed intermediate downshifts to ∼1653, 1650, and 1642 cm−1 for the 1 eq., 2 eq., and 3 eq. samples, respectively. Assignment of the amide II band was more difficult for the [Pam-co-PDADMA][mixed Fe3+/Co2+] series due to a complex overlapping of bands in the amide II region. Peak deconvolution was performed for the [Pam-co-PDADMA][mixed Fe3+/Co2+ (1 eq.)] copolymer, yielding three fitted peaks at 1657, 1607, and 1572 cm−1 (Fig. S3†). The first peak is attributed to the amide carbonyl band, as determined above. The peaks at 1607 and 1572 cm−1 are attributed to the NH2 wagging mode due to cobalt and iron coordination interactions, respectively. Peak fittings for the [Pam-co-PDADMA][mixed Fe3+/Co2+ 2 eq.] and [Pam-co-PDADMA][mixed Fe3+/Co2+ 3 eq.] samples are shown in ESI,† Fig. S4 and S5. For the samples with 3 eq. equivalents of metal content, another band emerges at approximately 1618 cm−1 for the cobalt and 1608 cm−1 for the iron samples. These bands are attributed to the M–OH bending of uncoordinated metal halide salt in the copolymer. The ∼1417 cm−1 amide III band did not shift appreciably upon metal complexation, though several of the modes in the 1400–1200 cm−1 region displayed small shifts to higher wavenumbers.
Metal ion coordination with amides is typically indicated by shifts in the amide I and amide II bands.57,62 Similar redshifts were observed by Halevi et al. upon Ni2+ ion complexation with a poly(acrylamide) homopolymer,62 which were attributed to O-binding interactions. N-bonding of acrylamide may also occur depending on the stability of the ligand complex and cation acidity with possible acrylamide N-bonding modes of N-amide, N-amidate, and iminol groups as shown in Fig. 2. Deprotonation of the amide group results in the formation of either an n-amidate or an iminol group characterized by the absence or decrease in intensity of the amide II band.28,29,63 As the amide II band and N–H stretches are present in the MPIL copolymers, it is unlikely that significant amide tautomerization to iminol or deprotonation to n-amidate has occurred. Decreases in the CO frequencies potentially indicate O-binding interactions with both the Fe3+ and Co2+. Monodentate O-binding of amides results in a decrease in the amide I frequency but an increase in the amide II frequency due to weakening of the CO bond and subsequent stiffening of the C–N bond in the amide group due to resonance.38,57,64,65 However, both the amide I and amide II bands display shifts to lower wavenumbers in all cases, indicating binding with both the oxygen (Fig. 2A) and nitrogen (Fig. 2B) of the amide groups. It is clear coordination between the transition metal complex and the polymer acrylamide unit is evidenced for both the Fe3+ and Co2+ halide species at all equivalences with possible bonding at both the acrylamide oxygen and nitrogen atoms.
It should be noted that if hydrolysis of the amide group occurred with metal complexation, it would result in the formation of either carboxylic acids, denoted by a CO str in the 1740–1700 cm−1 region, or carboxylate groups, characterized by a CO asymmetrical str mode between 1695–1540 cm−1 and a symmetrical stretching mode between 1440–1335 cm−1.38 No modes are observed above 1700 cm−1, excluding carboxylic acid formation. Further, no new bands emerge in the 1440–1335 cm−1 region suggesting no carboxylate groups are formed either. Therefore, there is no evidence of hydrolysis of the acrylamide group occurring upon the addition of metal salts.
The quaternary ammonium methyl group bending mode at 1489 cm−1 disappears in the metal systems, but an increase in the peak intensity at ∼1465 cm−1 was observed. This suggests the peak downshifts and overlaps with the polymer backbone CH2/CH3 bending modes. However, due to this overlap, electrostatic interaction between the transition metal complexes and quaternary ammonium group cannot be confirmed. As many of the C–N infrared modes for the PDADMA group overlap with the acrylamide modes, XPS spectroscopy was performed to better understand the metal halide interactions with the polymer.
XPS studies were carried out to provide further understanding of the polymer–metal salt binding. Fig. 7 displays representative nitrogen, oxygen, carbon, and chloride high resolution spectra for the [Pam-co-PDADMA][Cl−] sample, and Tables S2 and S3 in the ESI† summarize the XPS data for all of the copolymer samples. [Pam-co-PDADMA][Cl−] showed the N1s peaks for the quaternary ammonium nitrogen at 402.4 eV and the acrylamide peak at 399.6 eV, which is in good agreement with the literature.66,67 Three peaks in the C1s spectra—285.0 eV, 286.1 eV, and 287.7 eV—are attributed to the aliphatic C–C/C–H bonds, quaternary ammonium ring C–N bonds, and the acrylamide carbonyl bond.23,66 The Cl 2p spectrum shows two chemical species as indicated by two sets of 2p3/2 and 2p½ peaks with binding energy separations of 1.6 eV. The major chemical species has a Cl 2p3/2 peak at 198.8 eV which is attributed to the chloride anion of the quaternary ammonium group. This value is approximately 2 eV higher than that seen for the poly(diallyl dimethyl ammonium chloride) homopolymer (∼196.9 eV) and pyrrolidinium based ionic liquids,68,69 which is likely due to the hydrogen bonding interactions with the acrylamide units. This is supported by the slightly lower binding energy for the Pam–NH2 N1s peak (399.6 eV) compared to 399.9 eV reported by Chen et al. in pure polyacrylamide.66 The minor component Cl 2p3/2 peak at 197.6 eV is assigned to residual NaCl salt in the source polymer. The main peak in the O1s spectrum at 531.3 eV is attributed to the acrylamide carbonyl band. The peaks at 536.1 eV and 538.0 eV are likely the Na KLL auger peaks. A small amount of sodium was observed in the survey spectra (Fig. S6†), likely due to residual salt in the polymer stock solution. The 532.5 eV peak was assigned to either molecularly absorbed water, which is typically observed from 532–533 eV,62,70,71 or partially exchanged -OH anions.69
 | ||
| Fig. 7 High resolution XPS spectra of the MPIL copolymers were collected over the (A) N1s, (B) O1s, (C) C1s, and (D) Cl 2p binding energy ranges. The red trace is the spectra for the original non-magnetic [Pam-co-PDADMA][Cl−] copolymer. The dotted black lines show peak shifts in the MPIL copolymers for 1 eq. and 2 eq. in comparison to the [Pam-co-PDADMA][Cl−] copolymer. Where appropriate, the N1s spectra was fitted with three peaks for the PDADMA quaternary ammonium, O-bonded Pam–NH2, and the N-bonded Pam–H2N⋯M groups; these peaks are marked for the [Pam-co-PDADMA][Co2+ 2 eq.] sample in (A). While slight shifts are observed in the N1s spectra for the PDADMA quaternary ammonium, the greater shifts observed for the O-bonded Pam–NH2 and the emergence of the N-bonded Pam–H2N⋯M band suggests more interaction of the transition metal complexes with the acrylamide group than the PIL quaternary ammonium. | ||
Adding the transition metal salts resulted in significant changes in several of the high-resolution spectra shown in Fig. 7. An initial examination of the N1s spectra (Fig. 7A) still shows two peaks for the PDADMA quaternary ammonium at higher binding energies and the Pam–NH2 nitrogen group at lower energies. Interestingly, a change in the area ratio of the (Pam–NH2/PDADMA) N1s peak is observed for all the metal complexed copolymers with the exception of the [Pam-co-PDADMA][Co2+ 1 eq.] sample. As significant hydrolysis was eliminated in the FTIR analysis, the N1s spectra were refitted to include a metal coordinated Pam–H2N+⋯M peak to account for the area ratio change and maintain the monomer balance of Pam and PDADMA in the copolymer. The resulting three peaks (ca. 402.5, 402.1, and ∼400 eV) were assigned to PDADMA quaternary ammonium peak, the Pam–H2N+⋯M, and Pam–NH2 peaks.
The PDADMA N1s peak shows slight increases in binding energy (∼0.1–0.2 eV) depending on metal type and mole equivalent. For instance, the quaternary ammonium N1s peak increases from 402.4 eV in the [Pam-co-PDADMA][Cl−] sample to 402.4 and 402.6 eV for the [Pam-co-PDADMA][Co2+] 1 eq. and 2 eq. samples, respectively (Table S2†). Both the [Pam-co-PDADMA][Fe3+] 1 and 2 eq. samples had a N1s binding energy of 402.5 eV. Similar binding energy increases have been seen in pyrrolidinium, imidazolium, and ammonium ionic liquids for different counterions.67,72,73 Counterion size, basicity, and coordinating ability influences the counterion's ability to transfer charge to the nitrogen cation, resulting in different binding energies for the quaternary ammonium nitrogen for different counterions. Generally, quaternary ammonium N 1s binding energy increases with different counterions in the order of Cl− < I− ∼ [CoCl4]2− < [BF4]− < [PF6]− < [Tf2N]− ∼ [FeCl4]−.73,74 However, these shifts are within the instrument resolution (±0.2 eV), so while an overall trend is evidenced, increases with specific metal cation type cannot be discerned here. Further, the small ∼0.1–0.2 eV binding energy shifts observed here are lower than those observed in other MIL systems. For example, Taylor et al. observed an increase of binding energy from 401.7 eV in an imidazolium based ionic liquid to 402.2 eV (Δ = 0.5 eV) and 401.9 eV (Δ = 0.2 eV) on addition of FeCl3 and CoCl2 salts, respectively.74 This suggests that the electrostatic environment of the PIL quaternary ammonium does not change significantly upon addition of the transition metal salts and that the main binding mechanism between the MPIL copolymers and the metals species is not due to electrostatic interactions with the PIL monomer unit.
Conversely, binding energy increases of ∼0.2–0.6 eV were observed for the original Pam–NH2 N1s peak (399.6 eV), with larger increases observed for the copolymers as a function of higher metal content (Table S2†). This peak is associated with the O-bonding case (Fig. 7A) where the metal cation coordinates with the carbonyl oxygen and the resonance shift results in a withdraw of electron density from the NH2 group. In the [Pam-co-PDADMA][Fe3+] copolymers, this peak is at 399.8 and 400.0 eV for 1 and 2 molar equivalences, respectively (Table S2†). Binding energies of 400.0 and 400.2 eV were observed for this peak in [Pam-co-PDADMA][Co2+ 1 eq.] and [Pam-co-PDADMA][Co2+ 2 eq.] copolymers. The [Pam-co-PDADMA][mixed 1 eq.] copolymer had a similar binding energy (399.8 eV) as the [Pam-co-PDADMA] [Fe3+ 1 eq.] copolymer.
The peak that emerges at ∼402 eV in several of the metal copolymer spectra is attributed to direct coordination of the metal cation with the acrylamide nitrogen Pam–H2N+⋯M (Fig. 7A). Zhang et al. assigned similar O- and N-bonded N1s peaks for CaCl2 coordinated polyacrylamide hydrogels.75 The associated Pam CO C1s (Fig. 7C) and O1s peak (Fig. 7B) also shift to higher binding energies between 288.1–288.8 eV and 531.9–532.3 eV respectively upon metals addition. These shifts agree well with binding energy increases observed in the literature for metal coordinated polyacrylamide.15,75 In the iron and mixed samples, an additional small peak at ∼530 eV emerges, which is typically associated with lattice metal–oxide bonds (Fe–O).71 In other FeCl3 based material systems, a similar peak at ∼530 eV has been attributed to the formation of FeClO due to storage of materials in ambient air conditions.76,77 Small amounts of Na were still observed in the survey spectra for several of the metal copolymers (Fig. S7–S11†), the peak at ∼536 eV is assigned to the Na KLL Auger peaks. A peak between 532.3 and 533.6 eV was observed at varying intensities in the metal copolymer O1s spectra and was assigned to absorbed water or hydroxyl groups.62,69–71 The XPS findings corroborate the FTIR spectroscopy results and show that the transition metal complexes predominately bind to the polymer by coordination interactions with the acrylamide unit through both oxygen and nitrogen bonding rather than by purely electrostatic binding with the quaternary ammonium group.
High resolution spectra for Fe 2p and Co 2p were also collected (Fig. 8A and B). High-spin coordination structures are characterized by observed spin–orbit splitting into 2p3/2 and 2p1/2 peaks for transition metals and are typically accompanied by shake-up satellite peaks.25,74,78 In all samples, these 2p3/2 and 2p1/2 multiplet splitting peaks and satellite peaks were observed in both the Fe 2p and Co 2p high resolution spectra. This indicates that the MPIL copolymers contain metal species with paramagnetic high spin-states as expected for magnetically responsive MPILs.
 | ||
| Fig. 8 XPS high resolution (A) Fe 2p and (B) Co 2p spectra show the 2p splitting and satellite peaks characteristic of the paramagnetic high spin iron and cobalt species in [Pam-co-PDADMA][Co2+], [Pam-co-PDADMA][Fe3+], and [Pam-co-PDADMA][mixed Fe3+/Co2+] MPIL copolymers. | ||
As high-resolution spectra for metals are often highly complex, component fitting was not attempted for the Fe 2p and Co 2p spectra. Rather, the peak maximums were taken to be the binding energy values for the predominate metal species (Table S3†). The intense Fe 2p3/2 peak is located at 710.9 eV, 711.4 eV, and 710.9 eV for the [Pam-co-PDADMA] [Fe3+ 1 eq.], [Pam-co-PDADMA] [Fe3+ 2 eq.], and [Pam-co-PDADMA] [mixed Fe3+/Co2+ 1 eq.] samples, respectively. The [Pam-co-PDADMA][Fe3+ 2 eq.] Fe 2p3/2 binding energy is similar to those reported for FeCl3 salt (711.5–712.0 eV),25 imidazolium-based FeCl4− ionic liquid (711.9 eV),74 and quaternary tetraethylammonium FeCl4− salt (711.2 eV).79 A similar trend is observed in the corresponding Cl 2p spectra where the main Cl 2p3/2 peak was found to be at 198.4 eV, 198.7 eV, and 198.4 eV for the [Pam-co-PDADMA] [Fe3+ 1 eq.], [Pam-co-PDADMA] [Fe3+ 2 eq.], and [Pam-co-PDADMA] [mixed Fe3+/Co2+ 1 eq.] samples, respectively. The lower Fe 2p3/2 binding energy of the [Pam-co-PDADMA] [Fe3+ (1 eq.)] and [Pam-co-PDADMA][mixed Fe3+/Co2+ 1 eq.] samples paired with corresponding lower Cl 2p binding energies indicate that overall the iron halide complex species are more electron rich than the [Pam-co-PDADMA][Fe3+ 2 eq.] sample (Fig. 7D, Table S3†). This may be due to a change in the charge distribution across the metal cation and Cl atom as electron density is gained from acrylamide coordination, which is also supported by the binding energy increases for the acrylamide O1s and N1s peaks and the downward shift in wavenumbers for the amide modes in the FTIR spectroscopy results. Further, the presence of the shake-up satellite peaks in the [Pam-co-PDADMA][Fe3+ 1 eq.] sample also suggests that the iron–acrylamide coordinated species are also in a paramagnetic high spin-state. In the [Pam-co-PDADMA][Fe3+ 2 eq.] sample, the Cl− concentration is higher from the increased concentration of metal halide, allowing for the formation of the [FeCl4]− anion as suggested in the UV-vis film results. For the iron and mixed cases, the Cl 2p spectra could not be adequately fit with one chemical state and maintain the 1.6 eV spin–orbit coupling splitting.80 An additional minor Cl 2p3/2 peak was also fitted at ∼200 eV in the iron and mixed copolymer samples, which is attributed to the [Fe2Cl7]− bridging complex as noted in the Raman results.81,82
The Co 2p3/2 and Cl 2p3/2 peaks for the [Pam-co-PDADMA][Co2+ 1 eq.], [Pam-co-PDADMA][Co2+ 2 eq.], and [Pam-co-PDADMA][mixed Fe3+/Co2+ 1 eq.] samples were located at 781.6 eV, 781.9 eV, and 781.3 eV and 198.4 eV, 198.8 eV, and 198.4 eV, respectively. These Co 2p3/2 binding energies are bounded by those reported for CoCl2 salt (782.1 eV)83 and the 780.6 eV reported for an imidazolium-based [CoCl4]2− ionic liquid.74 As was seen for the iron samples, similar lower binding energies in the 1 eq. samples compared to the 2 eq. sample are also noted for the Co 2p3/2 and Cl 2p3/2 peaks, indicating a more electron rich coordination environment for the Co2+ cation at lower metal mole equivalents in the polymer. This may also be related to changes in tetrahedral speciation from acrylamide coordinated Co2+ (e.g., [CoCl2L2], [CoCl3L]−, or [CoL4]2+[CoCl4]2−, where L is acrylamide ligand) to the formation of predominantly [CoCl4]2− anions at higher Cl− concentrations.
Magnetic studies were conducted on these [Pam-co-PDADMA][Fe3+], [Pam-co-PDADMA][Co2+], and [Pam-co-PDADMA][mixed Fe3+/Co2+] magnetic PIL copolymers. Samples were examined both in the dry (solid) state and as solutions (Co2+ copolymers) or suspensions (Fe3+ and Fe/Co-mixed copolymers). Table 1 and Fig. 9A show the magnetic mass susceptibilities for the dry polymer samples. Magnetic susceptibility, χm, is a measure of the paramagnetic copolymer's strength of attraction to a magnetic field. The magnetic mass susceptibilities increased linearly with increasing metal ion content in the MPILs copolymers. The iron copolymer series displayed mass susceptibilities that were consistently higher than those for the cobalt and Fe3+/Co2+-mixed copolymers, at a given metal halide equivalency. The dry [Pam-co-PDADMA][Fe3+] samples showed magnetic susceptibilities that increased from ∼25 × 10−6 to 44 × 10−6 emu g−1 as the metal equivalency was increased from 1 to 3. These values correspond well with the 29.3 × 10−6 and 35.3 × 10−6 emu g−1 values obtained by Dobbelin et al. for similar diallyl dimethylammonium-based PIL homopolymers containing FeCl4− and Fe2Cl7− counterions, respectively.7 The cobalt series displayed the lowest mass susceptibilities from this study, with values of 17.5 × 10−6, 29.4 × 10−6, and 34 × 10−6 emu g−1, for the 1, 2, and 3 equivalent samples, respectively. The [Pam-co-PDADMA][mixed Fe3+/Co2+] series showed apparent mixing rule behavior, with intermediate magnetic susceptibility values between those of iron and cobalt (ranging from 22 × 10−6 to 36.2 × 10−6 emu g−1). The dry copolymers were also responsive to a 0.62 T magnetic field produced by neodymium magnets, as shown for the [Pam-co-PDADMA][Co2+ 2 eq.] sample in the insert of Fig. 9A and ESI† Video V2. Similar responses were seen for all other samples (ESI† Videos V1–V9).
| Sample | Metal halidea wt% | Dry sample | Polymer solution or suspension |
|---|---|---|---|
| χ m (×10−6 emu g−1) | χ m (×10−6 emu g−1) | ||
| a Calculated based on added metal salt. | |||
| [Pam-co-PDADMA][Fe3+ 1 eq.] | 42.9% | 24.73 (± 0.03) | 23.5 (± 0.6) |
| [Pam-co-PDADMA][Fe3+ 2 eq.] | 60.1% | 37.22 (± 0.04) | 27.9 (± 0.5) |
| [Pam-co-PDADMA][Fe3+ 3 eq.] | 69.3% | 43.95 (± 0.05) | 35.9 (± 0.9) |
| [Pam-co-PDADMA][Co2+ 1 eq.] | 39.8% | 17.53 (± 0.03) | 20.8 (± 0.9) |
| [Pam-co-PDADMA][Co2+ 2 eq.] | 57.0% | 29.37 (± 0.03) | 21.6 (± 0.2) |
| [Pam-co-PDADMA][Co2+ 3 eq.] | 66.5% | 33.58 (± 0.04) | 23.3 (± 0.3) |
| [Pam-co-PDADMA][mixed Fe3+/Co2+ 1 eq.] | 41.4% | 21.99 (± 0.02) | 24.1 (± 0.4) |
| [Pam-co-PDADMA][mixed Fe3+/Co2+ 2 eq.] | 58.6% | 30.39 (± 0.06) | 30.9 (± 0.7) |
| [Pam-co-PDADMA][mixed Fe3+/Co2+ 3 eq.] | 68.0% | 36.17 (± 0.03) | 35.0 (± 0.5) |
 | ||
| Fig. 9 Magnetic mass susceptibilities of (A) the dry MPIL copolymers and (B) the aqueous solution (∼7 mg ml−1) as a function of molar equivalents of the metal halide(s). In both cases, χm correspondingly increased with increasing metal content, though the χm for the dry polymers were ∼1–10 × 10−6 emu g−1 higher than the MPIL copolymers in solution or suspension. The standard deviations in χm for the solid samples was on the order of 0.02–0.06 × 10−6 emu g−1 while the standard deviations for the liquid samples were slightly higher at 0.2–0.9 × 10−6 emu g−1. [Pam-co-PDADMA][Fe3+] copolymers generally had the highest χm followed by the [Pam-co-PDADMA][mixed Fe3+/Co2+] and the [Pam-co-PDADMA][Co2+] MPIL copolymers. The insert in (A) shows the dried [Pam-co-PDADMA][Co2+ 2 eq.] copolymer attracted to a 0.62 T magnet in the video frame capture of ESI† Video V2. All other dry MPIL copolymers also demonstrated magnetic responsiveness (see ESI† Videos V1–V9). | ||
MPIL systems have generally been found to exhibit magnetic susceptibilities ranging between 10 × 10−6 to 60 × 10−6 emu g−1 depending on the metal complex concentration, the type of transition metals, and the diamagnetic contributions of the organic polymer chemistries.7,14,84 MPIL systems with higher concentrations of the high spin paramagnetic MCl4n− species (where M = Fe3+, Co2+, etc.) have demonstrated higher susceptibilities7,14 as was observed here for the MPIL copolymers with higher metal mole equivalencies. MPILs and MILs containing Co2+ counterions generally have lower magnetic susceptibilities than Fe3+ based systems19,85 as was also observed here. The paramagnetism of a material is dependent on the total orbital and spin angular moment of unpaired electrons in the complex under an applied magnetic field. For the tetrahedral anions [MCl4]n−, the higher magnetic mass susceptibilities of [FeCl4]− species compared to [CoCl42−] species is due to the higher electron spin state of the [FeCl4−] (5/2 spin) compared to the [CoCl4]2− anion (3/2 spin). However, for different coordinating ligands, the geometry and distortion of the coordination complex, the ligand field strength (i.e., spectrochemical series), and the oxidation state of the transition metal ion may significantly influence the spin state and the net orbital angular momentum of unpaired electrons in the complex.86,87 Therefore, the formation of metal-ion complexes coordinated with the acrylamide comonomer and different concentrations of this complex compared to the high spin [FeCl4]− and [CoCl4]2− complexes may potentially influence the overall magnetic susceptibility of each MPIL copolymer. Due to instrument limitations, a full temperature study of magnetic susceptibility could not be performed to confirm paramagnetic behavior. However, assuming the MPILs are paramagnetic at room temperature with negligible magnetic correlations, the effective magnetic moment can be estimated by  where χ is the molar susceptibility and T is the temperature 298 K.88 The μeff per mole of FeCl3 was found to be 4.72, 4.89, and 4.95μB (Bohr magnetron) for the [Pam-co-PDADMA][Fe3+ 1 eq.], [Pam-co-PDADMA][Fe3+ 2 eq.], and [Pam-co-PDADMA][Fe3+ 3 eq.], respectively. These effective magnetic moments are lower than the expected 5.9μB for high spin Fe3+ (5/2 spin) and similar moments observed for single component MILs and MPILs containing [FeCl4]− counterions.9,19,88 The cobalt-based MPIL copolymers had effective magnetic moments of 3.69, 3.99, and 3.95μB per mole of CoCl2 with increasing cobalt halide concentration. These values are in closer agreement with the expected high spin Co2+ (3/2) spin of 3.88μB though still lower than effective moments typically observed for [CoCl4]2− based systems (4.2–5.2μB).19,88,89 These results suggest that the metal ion-acrylamide coordination does contribute to a reduction in the overall effective magnetic moment, and that coordination effects from comonomers or co-materials should be carefully considered in future design of multi-component MPIL systems. The authors are currently conducting further magnetometry studies as a function of metal chloride concentration, magnetic field strength, and temperature to verify paramagnetic behavior of these MPIL copolymers and evaluate the presence of magnetic correlations.
where χ is the molar susceptibility and T is the temperature 298 K.88 The μeff per mole of FeCl3 was found to be 4.72, 4.89, and 4.95μB (Bohr magnetron) for the [Pam-co-PDADMA][Fe3+ 1 eq.], [Pam-co-PDADMA][Fe3+ 2 eq.], and [Pam-co-PDADMA][Fe3+ 3 eq.], respectively. These effective magnetic moments are lower than the expected 5.9μB for high spin Fe3+ (5/2 spin) and similar moments observed for single component MILs and MPILs containing [FeCl4]− counterions.9,19,88 The cobalt-based MPIL copolymers had effective magnetic moments of 3.69, 3.99, and 3.95μB per mole of CoCl2 with increasing cobalt halide concentration. These values are in closer agreement with the expected high spin Co2+ (3/2) spin of 3.88μB though still lower than effective moments typically observed for [CoCl4]2− based systems (4.2–5.2μB).19,88,89 These results suggest that the metal ion-acrylamide coordination does contribute to a reduction in the overall effective magnetic moment, and that coordination effects from comonomers or co-materials should be carefully considered in future design of multi-component MPIL systems. The authors are currently conducting further magnetometry studies as a function of metal chloride concentration, magnetic field strength, and temperature to verify paramagnetic behavior of these MPIL copolymers and evaluate the presence of magnetic correlations.
The magnetic properties of the solution samples were also examined. Fully solvated polymer solutions, such as the [Pam-co-PDADMA][Co2+] series samples, did not respond to the neodymium magnet. This lack of magnetic response for the MPIL is due to some degree of dissociation of the magnetic metal complexes from the polymer and hydration of the Co2+ ion into a cationic [Co(H2O)6]2+ coordination complex as indicated by the pink color of these solutions (Fig. 1B).90 Precipitants from the partially insoluble iron and mixed series samples did visually respond to the magnet in water (ESI† Video V10), which has been observed for other MPIL particles in nonsolvents40,91,92 or MPILs complexed to insoluble particles.15 In similar small molecule MIL and Mag-surf systems, this magnetic response in an aqueous environment is predominately attributed to confining the paramagnetic ions into a hydrophobic domain (e.g. immiscible MILs or hydrophobic surfactant cores) or the Stern layer of an insoluble particle.21,93,94 Here, the observed magnetic responses for the [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] series MPILs are due to the confinement of the paramagnetic iron species within the insoluble polymer precipitants.
Magnetic mass susceptibilities of the copolymer solutions and suspensions were also measured (Fig. 9B). Generally, the susceptibilities of the MPILs in solution were approximately 1 to 10 × 10−6 emu g−1 lower in an aqueous environment than their dry counterparts, likely due to dissociation of some of the paramagnetic species into the aqueous phase. Both the [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymer series showed a linear increase in magnetic susceptibilities with metals content while the cobalt series did not significantly increase. These magnetic susceptibility data match the qualitative visual magnet responses. As stated before, the iron species in the [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][mixed Fe3+/Co2+] MPIL copolymers is being retained in the insoluble polymeric precipitants while the Co2+ species dissociate, at least in part, from the polymer and hydrate in solution. Interestingly, the [Pam-co-PDADMA][Co2+ 1 eq.] samples showed a slight increase to 20.8 × 10−6 emu g−1 in the liquid state from 17.5 × 10−6 emu g−1 in the dry state. One possible interpretation would be that, as the Co2+ species are dissociating from the polymer, the measured susceptibility for this sample is predominately from the high spin [Co(H2O)6]2+ species without the added diamagnetic contribution of the [Pam-co-PDADMA][Cl−] copolymer. However, even though the concentration of the cobalt species increases in the 2 and 3 eq. cases, the ions are dispersed in the liquid phase and do not interact or contribute to long-range ordering in solution. Whereas the iron species confined in the polymer matrix do interact allowing for long-range ordering at higher equivalents. These results indicate that the acrylamide metal coordinating comonomer also plays a role in the MPIL copolymer magnetic behavior in solutions or suspensions.
Conclusions
Poly(acrylamide-co-diallyl dimethylammonium chloride) is a poly(ionic liquid) (PIL) copolymer containing quaternary ammonium IL groups and comonomer groups capable of metal coordinating interactions. Complexation with Co2+ and Fe3+ metal halide salts was utilized to form magnetic PIL (MPIL) copolymers. The impact of metal salt cation type and concentration were examined. Color and solubility results showed metal–polymer complexation was occurring, but the complexation was changing as a function of metal type and molar equivalency. To verify the metal species-polymer binding interactions and the metal complex structure, the MPIL samples were examined with multiple spectroscopic methods.Binding interactions for these unique MPIL polymers were characterized in both the liquid and dry states using multiple molecular absorption spectroscopic techniques. Raman and UV-vis spectroscopy results confirmed the formation of the [FeCl4]− and [CoCl4]2− species in the dry MPIL copolymer particularly through the presence of the Fe–Cl str 314 cm−1 Raman mode and UV-vis 250, 315, and 360 nm absorbances for the [FeCl4]− anion and the ∼630, ∼665, and ∼690 nm UV-vis absorbances for [CoCl4]2−. However, these spectroscopies also indicated the presence of metal cation–acrylamide coordination species as well through a broad Raman band between 600–200 cm−1 typical for M–O and M–N stretching modes for both iron and cobalt-based MPILs. UV-vis further showed a change in the Fe3+ species coordination structure with increasing iron concentration from 1 to 2 mole equivalences, indicating the iron coordinates first with acrylamide before forming the anionic [FeCl4]− and [Fe2Cl7]− species denoted by characteristic peaks at ca. 250, 315, and 360 nm. UV-vis results of the MPILs in aqueous solution indicate the formation of octahedral Co2+ species through the emergences of the peaks at 465 and 510 nm and various Fe(OH)x and FeCl3 aq. species with broad absorbances between 300–500 nm. FTIR results confirm metal–acrylamide coordination for both metal cations through both O- and N-bonding with the amide group as indicated by significant downshifts in both the amide I (CO str) and amide II (NH2 wag) IR modes. Downshifts were found to be greater for the [Pam-co-PDADMA][Fe3+] copolymer (12–19 cm−1 amide I, 41–50 cm−1 amide II) compared to the [Pam-co-PDADMA][Co2+] copolymers (6–14 cm−1 amide I, 11–18 cm−1 amide II) with intermediate behavior for the [Pam-co-PDADMA][mixed Fe3+/Co2+] copolymer system. While downshifts in the quaternary ammonium methyl bending modes were also observed, XPS spectroscopy results indicated only small increases in binding energy (∼0.1–0.2 eV) for the corresponding ammonium N1s peak. Large increases in binding energy for the O-bonding Pam–NH2 N1s peak and the emergence of the N-bonding Pam–H2N+⋯M N1s reveal that the main binding mechanism between the copolymer and metal species is through acrylamide coordination.
Magnetic properties of the MPIL copolymers were confirmed using AC susceptibility measurements and showed magnetic mass susceptibilities comparable to other MPIL homopolymer and copolymer systems (∼17 to 44 × 10−6 emu g−1) in the dry state and were visually responsive to a 0.6 T magnet. While the cobalt-based MPILs were completely soluble in water and displayed no visual magnetic response, the partially insoluble iron-based MPILs were visually responsive to magnetic field and displayed magnetic mass susceptibilities between 23.5 and 35.9 × 10−6 emu g−1. Here, coordination with the acrylamide unit is believed to account for the partial insolubility and resulting magnetic response in aqueous based systems.
This work describes the first synthesis of [Pam-co-PDADMA][Fe3+] and [Pam-co-PDADMA][Co2+] MPIL copolymers reported in the literature. In addition, the authors are not aware of MPILs being characterized using XPS prior to this study. Most importantly this study demonstrated not only binding between the metal in the counterion and the amide group of the acrylamide monomer group, but it provided insight into the coordination structures of the transition metal halide complexes. Understanding transition metal ions interactions in MPIL systems will guide the design and selection criteria for other magnetically responsive PIL functional materials, particularly in multicomponent systems.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Andrew Rodriguez and Dr. Megan Madden (University of Oklahoma) for their aid in Raman data collection, as well as Dr. Matt Hamilton and Dr. Richard D. Elmore (University of Oklahoma) for their assistance with the magnetic mass susceptibility measurements.References
- A. Munoz-Bonilla and M. Fernandez-Garcia, Eur. Polym. J., 2018, 105, 135–149 CrossRef CAS
.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS
- J. Guo, L. Qiu, Z. Deng and F. Yan, Polym. Chem., 2013, 4, 1309–1312 RSC
- J. Yang, H. Wang, J. Wang, X. Guo and X. Pei, Langmuir, 2016, 32, 66–72 CrossRef CAS PubMed
- H. L. Ricks-Laskoski and A. W. Snow, J. Am. Chem. Soc., 2006, 128, 12402–12403 CrossRef CAS PubMed
- M. Dobbelin, V. Jovanovski, I. Llarena, L. J. C. Marfil, G. Cabanero, J. Rodriguez and D. Mecerreyes, Polym. Chem., 2011, 2, 1275–1278 RSC
- K. D. Clark, O. Nacham, J. A. Purslow, S. A. Pierson and J. L. Anderson, Anal. Chim. Acta, 2016, 934, 9–21 CrossRef CAS PubMed
- S. Hayashi, S. Saha and H. Hamaguchi, IEEE Trans. Magn., 2006, 42, 12–14 CAS
- A. Joseph, G. Żyła, V. I. Thomas, P. R. Nair, A. S. Padmanabhan and S. Mathew, J. Mol. Liq., 2016, 218, 319–331 CrossRef CAS
- P. Brown, T. Alan Hatton and J. Eastoe, Curr. Opin. Colloid Interface Sci., 2015, 20, 140–150 CrossRef CAS
- L. Wang, S. Dong and J. Hao, Curr. Opin. Colloid Interface Sci., 2018, 35, 81–90 CrossRef CAS
- N. Sahiner, S. Demir and S. Yildiz, Colloids Surf., A, 2014, 449, 87–95 CrossRef CAS
- I. Chikh Alard, J. Soubhye, G. Berger, M. Gelbcke, S. Spassov, K. Amighi, J. Goole and F. Meyer, Polym. Chem., 2017, 8, 2450–2456 RSC
- G. Hazell, M. Hinojosa-Navarro, T. M. McCoy, R. F. Tabor and J. Eastoe, J. Colloid Interface Sci., 2016, 464, 285–290 CrossRef CAS PubMed
- C. de la Fuente-Nunez, P. Brown, M. D. T. Torres, J. Cao and T. K. Lu, Colloid Interface Sci. Commun., 2018, 22, 11–13 CrossRef CAS
- X. Yu, Z. Xia, T. Zhao, X. Yuan and L. Ren, Macromolecules, 2021, 54, 4227–4235 CrossRef CAS
- X. Yu, X. Yuan, Y. Zhao and L. Ren, ACS Macro Lett., 2019, 8, 1504–1510 CrossRef CAS PubMed
- A. Bonnefond, M. Ibarra, D. Mecerreyes and J. R. Leiza, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1145–1152 CrossRef CAS
- J. Cui, S. Yang, J. Zhang, S. Zhao and Y. Yan, RSC Adv., 2012, 2, 12224–12230 RSC
- T. M. McCoy, P. Brown, J. Eastoe and R. F. Tabor, ACS Appl. Mater. Interfaces, 2015, 7, 2124–2133 CrossRef CAS PubMed
- J. Yang, W. Sun, W. Lin and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5123–5132 CrossRef CAS
- C. D. Easton, C. Kinnear, S. L. McArthur and T. R. Gengenbach, J. Vac. Sci. Technol., A, 2020, 38, 023207 CrossRef CAS
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Coord. Chem. Rev., 2005, 249, 1283–1293 CrossRef CAS
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS
- R. C. Dunbar, J. Martens, G. Berden and J. Oomens, Int. J. Mass Spectrom., 2018, 429, 198–205 CrossRef CAS
- R. C. Dunbar, N. C. Polfer, G. Berden and J. Oomens, Int. J. Mass Spectrom., 2012, 330-332, 71–77 CrossRef CAS
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Z. Anorg. Allg. Chem., 2005, 631, 2763–2769 CrossRef CAS
- T. Bäcker, O. Breunig, M. Valldor, K. Merz, V. Vasylyeva and A.-V. Mudring, Cryst. Growth Des., 2011, 11, 2564–2571 CrossRef
-
K. Nakamoto, in Handbook of Vibrational Spectroscopy, 2001, DOI:10.1002/0470027320.s4104
- R. P. Rathore, S. S. Khatri and T. Chakraborty, J. Raman Spectrosc., 1987, 18, 429–434 CrossRef CAS
- A. Wesełucha-Birczyńska and C. Paluszkiewicz, J. Mol. Struct., 2002, 614, 339–343 CrossRef
- A. I. Freire and W. A. Alves, Vib. Spectrosc., 2014, 73, 73–78 CrossRef CAS
- S. H. J. De Beukeleer and H. O. Desseyn, Spectrochim. Acta, Part A, 1994, 50, 2291–2309 CrossRef
- K. Murata and D. E. Irish, Spectrochim. Acta, Part A, 1988, 44, 739–743 CrossRef
-
G. Socrates, Infrared and Raman Characteristic Group Frequencies Tables and Charts, John Wiley & Sons, LTD, Baffins Lane, Chichester, West Sussex PO19 1UD, England, 3rd edn, 2001 Search PubMed
- S. K. Sharma, J. Chem. Phys., 1974, 60, 1368–1375 CrossRef CAS
- X. Yu, X. Yuan, Y. Zhao and L. Ren, RSC Adv., 2015, 5, 92207–92211 RSC
- R. Ma, Z. Liu, K. Takada, K. Fukuda, Y. Ebina, Y. Bando and T. Sasaki, Inorg. Chem., 2006, 45, 3964–3969 CrossRef CAS PubMed
- V. Sivo, G. D'Abrosca, L. Russo, R. Iacovino, P. V. Pedone, R. Fattorusso, C. Isernia and G. Malgieri, Bioinorg. Chem. Appl., 2017, 2017, 1527247 Search PubMed
- J. Bjerrum, A. S. Halonin, L. H. Skibsted, A. F. Andresen, J. T. Southern, K. R. Edlund, M. Eliasen, C. Herskind, T. Laursen and P. M. Pedersen, Acta Chem. Scand., 1975, 6, 326–332 CrossRef
- S. Chaouachi, S. Elleuch, B. Hamdi and R. Zouari, J. Mol. Struct., 2016, 1125, 149–161 CrossRef CAS
- H. Ma, C. Wan and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12754–12757 CrossRef PubMed
- M. B. Vraneš, S. M. Papović and S. B. Gadžurić, J. Solution Chem., 2019, 48, 1364–1377 CrossRef
- N. Banić, M. Vraneš, B. Abramović, J. Csanádi and S. Gadžurić, Dalton Trans., 2014, 43, 15515–15525 RSC
- S. A. Markarian, H. H. Ghazoyan, H. R. Sargsyan and G. A. Shahinyan, J. Solution Chem., 2019, 48, 1378–1392 CrossRef CAS
- H. Dweik, W. Sultan, M. Sowwan and S. Makharza, Int. J. Polym. Mater. Polym. Biomater., 2008, 57, 228–244 CrossRef CAS
- M. Sowwan, Int. J. Phys. Sci., 2011, 6, 6280–6285 CAS
- H. Aoshima, K. Satoh, T. Umemura and M. Kamigaito, Polym. Chem., 2013, 4, 3554–3562 RSC
- G. A. Gamlen and D. O. Jordan, J. Chem. Soc., 1953, 1435–1443, 10.1039/JR9530001435
- W. Liu, B. Etschmann, J. Brugger, L. Spiccia, G. Foran and B. McInnes, Chem. Geol., 2006, 231, 326–349 CrossRef CAS
- M. Torras, C. Moya, G. A. Pasquevich and A. Roig, Microchim. Acta, 2020, 187, 488 CrossRef CAS PubMed
- C. Loures, M. Alcantara, H. Filho, A. Teixeira, F. Silva, T. Paiva and G. Samananmud, Int. Rev. Chem. Eng., 2013, 5, 102–120 Search PubMed
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Z. Anorg. Allg. Chem., 2005, 631, 1843–1848 CrossRef CAS
- S. J. Kim, S. G. Yoon, I. Y. Kim and S. I. Kim, J. Appl. Polym. Sci., 2004, 91, 2876–2880 CrossRef CAS
- E. Pigorsch, Starch/Staerke, 2009, 61, 129–138 CrossRef CAS
- Y. Shin, W. H. Cheung, T. T. M. Ho, K. E. Bremmell and D. A. Beattie, Phys. Chem. Chem. Phys., 2014, 16, 22409–22417 RSC
- J. Zhang, J. Qiao, G. Jiang, L. Liu and Y. Liu, J. Power Sources, 2013, 240, 359–367 CrossRef CAS
- O. Halevi, J. Chen, G. Thangavel, S. A. Morris, T. Ben Uliel, Y. R. Tischler, P. S. Lee and S. Magdassi, RSC Adv., 2020, 10, 14812–14817 RSC
- B. M. Marsh, J. Zhou and E. Garand, RSC Adv., 2015, 5, 1790–1795 RSC
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Z. Anorg. Allg. Chem., 2005, 631, 1419–1422 CrossRef CAS
- T. F. Zafiropoulos, S. P. Perlepes, P. V. Ioannou, J. M. Tsangaris and A. G. Galinos, Z. Naturforsch. B, 1981, 36, 87–93 CrossRef
- S. Chen and H. Tanaka, J. Wood Sci., 1998, 44, 303–309 CrossRef CAS
- S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244–15255 RSC
- R. K. Blundell and P. Licence, Chem. Commun., 2014, 50, 12080–12083 RSC
- R. L. McLaren, G. R. Owen and D. J. Morgan, Results in Surfaces and Interfaces, 2022, 6, 100032 CrossRef
- E. Desimoni and B. Brunetti, Chemosensors, 2015, 3, 70–117 CrossRef CAS
- Y. J. Kim and C. R. Park, Inorg. Chem., 2002, 41, 6211–6216 CrossRef CAS PubMed
- R. K. Blundell and P. Licence, Phys. Chem. Chem. Phys., 2014, 16, 15278–15288 RSC
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H.-P. Steinrück, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed
- A. W. Taylor, S. Men, C. J. Clarke and P. Licence, RSC Adv., 2013, 3, 9436–9445 RSC
- H. Zhang, Z. Liu, J. Mai, N. Wang, H. Liu, J. Zhong and X. Mai, Adv. Sci., 2021, 8, 2100320 CrossRef CAS PubMed
- C. Madrona, M. Vila, F. E. Oropeza, V. A. de la Peña O'Shea and J. J. Vilatela, Carbon, 2021, 173, 311–321 CrossRef CAS
- Y. Wang, H. Zhang, Y. Zhu, Z. Dai, H. Bao, Y. Wei and W. Cai, Adv. Mater. Interfaces, 2016, 3, 1500801 CrossRef
- T. Ivanova, A. Naumkin, A. Sidorov, I. Eremenko and M. Kiskin, J. Electron Spectrosc. Relat. Phenom., 2007, 156–158, 200–203 CrossRef CAS
- M. V. Russo, G. Polzonetti, A. Furlani, A. Bearzotti, I. Fratoddi and P. Altamura, J. Vac. Sci. Technol., A, 1998, 16, 35–44 CrossRef CAS
-
J. F. Moulder and J. Chastain, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin-Elmer Corporation, 1992 Search PubMed
- N. Calisi, S. Martinuzzi, A. Giaccherini, C. S. Pomelli, L. Guazzelli and S. Caporali, J. Electron Spectrosc. Relat. Phenom., 2021, 247, 147034 CrossRef CAS
- M. D. Nguyen, L. V. Nguyen, E. H. Jeon, J. H. Kim, M. Cheong, H. S. Kim and J. S. Lee, J. Catal., 2008, 258, 5–13 CrossRef CAS
- D. G. Brown and U. Weser, Z. Naturforsch. B, 1979, 34, 1468–1470 CrossRef
- J. J. Malecha, J. R. Biller, B. Lama and D. L. Gin, ACS Macro Lett., 2020, 9, 140–145 CrossRef CAS PubMed
- R. E. Del Sesto, T. M. McCleskey, A. K. Burrell, G. A. Baker, J. D. Thompson, B. L. Scott, J. S. Wilkes and P. Williams, Chem. Commun., 2008, 447–449, 10.1039/B711189D
-
S. Blundell, Magnetism in Condensed Matter, Oxford University Press Inc., New York, 2001 Search PubMed
-
M. W. Duward Shriver, T. Overton, J. Rourke and F. Armstrong, Inorganic Chemistry, Oxford University Press, Great Britain, 6th edn, 2014 Search PubMed
- S. Mugiraneza and A. M. Hallas, Commun. Phys., 2022, 5, 95 CrossRef
- P. Brown, C. P. Butts, J. Eastoe, S. Glatzel, I. Grillo, S. H. Hall, S. Rogers and K. Trickett, Soft Matter, 2012, 8, 11609–11612 RSC
- A. Fortenberry, D. Reed, A. Smith and P. Scovazzo, Langmuir, 2019, 35, 11843–11849 CrossRef CAS PubMed
- Y. Ohara, Y. Kawata, A. Hyde, C. Phan, R. Takeda, Y. Takemura and S.-i. Yusa, Chem. Lett., 2017, 46, 1473–1475 CrossRef CAS
- T. Zhang, X. Yu, X. Yuan, Y. Zhao and L. Ren, Polymer, 2018, 135, 9–15 CrossRef CAS
- P. Beck, M. Liebi, J. Kohlbrecher, T. Ishikawa, H. Rüegger, H. Zepik, P. Fischer, P. Walde and E. Windhab, J. Phys. Chem. B, 2010, 114, 174–186 CrossRef CAS PubMed
- O. Nacham, K. D. Clark, H. Yu and J. L. Anderson, Chem. Mater., 2015, 27, 923–931 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3me00076a |
| This journal is © The Royal Society of Chemistry 2023 |