
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface polymerisation and self-assembly of DPP-based molecular wires†
Michael
Clarke
a,
Abigail
Bellamy-Carter
 ab,
Ferdinando
Malagreca
c,
Jack
Hart
a,
Stephen P.
Argent
c,
James N.
O'Shea
a,
David B.
Amabilino
d and
Alex
Saywell
*a
ab,
Ferdinando
Malagreca
c,
Jack
Hart
a,
Stephen P.
Argent
c,
James N.
O'Shea
a,
David B.
Amabilino
d and
Alex
Saywell
*a
aSchool of Physics & Astronomy, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: alex.saywell@nottingham.ac.uk
bSchool of Liberal Arts and Natural Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
cSchool of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK
dInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus Universitari de Bellaterra, 08193 Cerdanyola del Vallès, Spain
First published on 16th February 2023
Abstract
The incorporation of organic semiconducting materials within solid-state electronic devices provides a potential route to highly efficient photovoltaics, transistors, and light emitting diodes. Key to the realisation of such devices is efficient intramolecular charge transport within molecular species, as well as intermolecular/interdomain transport, which necessitates highly ordered supramolecular domains. The on-surface synthesis of polymeric organic materials (incorporating donor and/or acceptor moieties) is one pathway towards the production of highly ordered molecular domains. Here we study the formation of a polymer based upon a diketopyrrolopyrrole (DPP) monomer unit, possessing aryl-halide groups to facilitate on-surface covalent coupling and functionalised with alkyl chains which drive the self-assembly of both the monomer material prior to reaction and the domains of polymeric material following on-surface synthesis. The self-assembled structure of close-packed domains of the monomer units, and the ordered polymers, are investigated and characterised using scanning tunnelling microscopy and X-ray photoelectron spectroscopy.
Design, System, ApplicationThe alignment of photo sensitive polymers within optoelectronic devices is expected to play a significant role with respect to device operation and efficiency. Understanding the role played by molecular level interactions in driving the ordering of polymers is an important step towards fabrication of devices based upon the directed self-assembly of molecular wires. Here, a molecule containing an electron deficient π-functional unit was designed to incorporate a leaving group promoting on-surface synthesis along a specific direction, governed by the substitution pattern of the reactive parts, and driving the formation of 1D polymer chains. Lateral units orthogonal to the polymerization reaction arrange the molecular precursors in an ordered, chiral, assembly. The construction of the polymers on the metal surface promises to facilitate the preparation of functional interfaces for optoelectronic materials, and the order-directing interactions may help produce high quality films with a low number of defects. These characteristics would be attractive for the preparation of thermally stable and well-ordered films. |
Introduction
An underlying concept of molecular electronics is that the bespoke properties of specific molecular species (e.g. electronic, magnetic, chemical, and optical functionalities) can be incorporated into solid-state devices. Diketopyrrolopyrrole (DPP) is a chemical moiety which may act as both an electron acceptor and donor within organic electronic-devices, and it has been shown to exhibit a high charge carrier mobility when utilised in extended polymer structures,1,2 and has been utilised within organic field effect transistors (OFET) and other nanoelectronic devices.3 Charge transport within such systems is affected by the local arrangement, and orientation, of molecules and their domains; hence, the controlled formation of ordered structures is of interest. Additionally, the chirality of the molecular sub-units within ordered structures is also known to influence electronic properties of the materials,4,5 and hence methodologies for controlling these structural arrangements, and techniques which provide molecular level characterisation, are an important facet within the development of molecular-based devices.The on-surface self-assembly of molecular structures, where non-covalent interactions give rise to local ordering,6 is a promising approach. Scanning probe microscopy techniques (such as scanning tunnelling microscopy, STM, and atomic force microscopy, AFM) have frequently been employed to study such systems as they provide single-/sub-molecular resolution of surface confined molecules. The ordered assembly of domains may be driven by a wide range of intermolecular interactions7 (e.g. hydrogen-bonding, metal–organic, van der Waals) and the self-assembly of DPP based species has been achieved via functionalisation with alkyl side chains; driving the formation of ordered domains confined to a surface.8,9
Importantly, molecular properties can be changed and enhanced upon polymerisation, and an appropriately designed monomer with specific functional groups may be polymerised into ordered nano or microstructures (with the on-surface synthesis of graphene nano ribbons being an important demonstration of this approach10). A range of on-surface synthesis protocols that result in monomer units being covalently coupled under appropriate reaction conditions have been explored (e.g. Ullmann-type reactions11 amongst others12–18) and precursor units possessing acceptor/donor functionality have been covalently coupled to produce individual molecular wires characterised by STM methodologies.19,20
Here we show the on-surface synthesises of polymers formed from a functionalised DPP-monomer on an Au(111) surface under ultra-high vacuum (UHV); as characterised by scanning tunnelling microscopy (STM). Structural characterisation of molecular and polymeric species at different stages of growth within the reaction allow us to identify the role of molecular chirality and molecular interactions, driven by alkyl chain and bromine functionalisation of the DPP core, within the observed ordered structures. Specifically, we show how the alkyl chains give alignment and order to the DPP polymers and how the thermal stability of these groups is key in maintaining this long-range order.
Results
The on-surface synthesis of polymeric DPP structures requires a suitably functionalised DPP core. We employ DPP species functionalised with aryl halide and alkyl chain groups (3,6-bis(4-bromophenyl)-2,5-dihexyl-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione – Br2Hex2DPP, see ESI† for details of synthesis); chemical structure of Br2Hex2DPP shown in Fig. 1a with dimensions indicated in Fig. 1b(i). The nitrogen atoms of the DPP core are alkylated with hexyl chains, which have previously been shown to facilitate ordered on-surface self-assembly,8,9 and aryl bromide groups provide access to Ullmann-type covalent coupling reactions on surfaces.11,15,16 It is worth noting that the self-assembly of precursor units may have significant impact upon the reaction pathway of coupling processes21 and that this approach, amongst others22 (including alignment to surface features23,24 and reactions within molecular pores25,26) offers a paradigm for controlling on-surface synthesis. The functionalisation of the DPP core with hexyl chains is selected as a route to modulate the balance of molecule–molecule interactions driven by interactions between the aromatic DPP cores and those driven by alkyl chain interactions. Within three-dimensional structures formed from DPP variants functionalisation with short (e.g. butyl), or no, alkyl chains the self-assembly is driven by interactions between the DPP cores. The inclusion of longer chains (e.g. octyl) results in structures dominated by lamella-type packing of interdigitating alkyl chains.27 Such a motif is frequently observed within surface confined two-dimensional self-assembly.28 Here we have obtained, by inclusion of hexyl chains, a balance between interacting DPP cores and between alkyl chains such that the self-assembled structure contains rows of DPP cores (where proximity may allow Ullmann-type coupling) and local ordering of the rows is facilitated by the interdigitated alkyl chains. | ||
| Fig. 1 (a) Scheme of the Ullmann-type coupling reaction expected from heating Br2Hex2DPP on Au(111). (b) (i) Predicted dimensions of a single Br2Hex2DPP as modelled via molecular mechanics geometry optimisation (see ESI†). (ii) and (iii) Show the M and P chiral arrangements of the molecule as induced by surface interaction. (c) Scheme showing two possible chiral arrangements of monomers within a polymer. This adsorption-induced chirality could lead to non-linear/kinked polymers [hexyl chains removed for clarity]. (d) STM topograph showing self-assembled structures formed by Br2Hex2DPP (bias = 2 V, current = 20 pA). Accompanying this is a zoom of a 2D-FFT filtered image of the lattice, with lattice dimensions, a and b, indicated alongside overlay of proposed molecular structure (a = 1.1 ± 0.1 nm and b = 1.4 ± 0.1 nm). (e) Shows the molecular lattice as modelled by molecular mechanics geometry optimisation (a = 1.2 ± 0.1 nm and b = 1.5 ± 0.1 nm). | ||
A scheme of the on-surface reaction for Br2Hex2DPP on Au(111) is shown in Fig. 1a. The pro-chiral nature29,30 of Br2Hex2DPP, with respect to adsorption on the surface leads to two enantiomeric chiral species (labelled M and P, as shown in Fig. 1b(ii) and (iii)), and hence on-surface coupling may give rise to M–M, P–P, or equivalent M–P and P–M products (see Fig. 1c) which will influence the geometry of the resultant polymers.
A monolayer coverage of Br2Hex2DPP on Au(111) was prepared via thermal deposition under UHV conditions and subsequently imaged using STM. Fig. 1d shows the observed self-assembled structures where a majority and minority phase are observed (minority phase indicated with a white arrow). Within the majority phase, two principal directions are identified (green/blue arrows), with the ‘a’ axis of the molecular lattice running approximately parallel to the 〈1 1 −2〉 directions of the Au(111) surface. The measured dimensions of the unit cell, a and b, are found to be a = 1.1 ± 0.1 nm, b = 1.4 ± 0.1 nm; which is in excellent agreement with the proposed model (shown in Fig. 1e – derivation presented in ESI†), with a = 1.2 ± 0.1 nm and b = 1.5 ± 0.1 nm. The proposed model is a geometry optimised structure based upon the experimentally determined lattice measurement and a consideration of inter and intra-molecular steric effects (see ESI†). Our proposed arrangement suggests that the alkyl chains play a role in ordering the observed structures (van der Waals interactions between parallel alkyl chains are known to be a driving force in self-assembled structures7–9 – indicated by blue dashed lines in Fig. 1e). The self-assembled structure proposed is homo-chiral (either all M or P species present), but the resolution of the STM data precludes identification of molecular chirality within individual domains. However, a consideration of homo- and hetero-chiral domains allows us to preclude the presence of hetero-chiral molecular structures due to steric effects (see ESI†). Following deposition, a racemic mixture of the adsorption-induced chiral enantiomers is assumed with co-existing homochiral domains being formed during self-assembly. This would suggest that homochiral interactions are an important driver of the self-assembled structures, and that the monolayer crystal formed can be considered a conglomerate (overall racemic mixture preserved with distinct homochiral domains present).29
Another feature of the proposed self-assembled structure shown in Fig. 1e is the potential for halogen bonding between bromine and oxygen atoms attached to the DPP core (dashed red line). Indeed, this interaction is seen in the solid state for this compound (see ESI†). Halogen bonding is highly directional, and may influence molecular self-assembly via the anisotropic electron distribution around covalently bonded halogens, allowing the strongly electronegative halogen species to interact with another electronegative group by virtue of a small electrophilic region which forms on the halogen atom at the opposite side to the covalent bond.31,32
To explore the progress of the on-surface polymerisation reaction, the sample was sequentially heated; to 100 °C, 200 °C, 250 °C and 350 °C. The reaction products following heating were characterised by STM, (see Fig. 2a–c). Previous studies of aryl-bromides on the Au(111) surface indicate that scission of the C–Br is likely to occur in the range 100–250 °C.33–42 Our results indicate that initial polymerisation occurs at a temperature of 100 °C (Fig. 2a); short (1–30 nm length) chains are observed to form distinct ordered domains of varying size. These polymer domains are typically of a smaller size than those seen in the close-packed islands observed before annealing (cf.Fig. 1d). Further discussion of the polymerisation details on the molecular level are provide below. Initially we focus on the large-scale morphologies observed as a function of temperature.
 | ||
| Fig. 2 STM images of the polymer structures formed following annealing the surface to; (a) 100 °C (bias = 2 V, current = 40 pA); (b) 200 °C (bias = 2 V, current = 20 pA); and (c) 350 °C (bias = 2 V, current = 20 pA). (d) Angular orientation of polymer chains in (a). (e) Angular orientation of polymer chains in (b). (f) XPS data showing the Br 3p region following deposition of Br2Hex2DPP on Au(111) (orange line, top) and subsequent annealing of the surface to 100 °C (blue line, bottom). | ||
Following annealing at 100 °C, the polymer domains are observed to run along several directions, with the polymers within each domain being highly aligned. The angular distribution of polymer alignment, relative to the fast-scan direction (x-axis), for the chains in Fig. 2a is shown in Fig. 2d. While there is a high degree of polymer alignment within individual domains, when several images are compared there is no evidence to support a unique relationship between the polymer orientation and the major crystallographic directions of the underlying Au(111) substrate (although our data does not categorically preclude a more subtle interaction with the surface crystallography or the herringbone reconstruction – see details in the ESI†). We propose that the local ordering within the domains, post-anneal, is driven by the initial ordering of the unreacted Br2Hex2DPP species within close-packed domains, with the proximity of the aryl-bromide groups facilitating covalent coupling.
Similar analysis is performed for the polymer structures obtained after annealing to 200 °C (STM topography shown in Fig. 2b, with angular distribution shown in Fig. 2e). In contrast to the range of polymer orientations observed within a single STM image after annealing at 100 °C, additional annealing has resulted in larger domains, where the polymer chains are locally aligned; for example, polymer chains running from the bottom left to the top right of the STM image (shown in Fig. 2b) maintain local parallel ordering, but the chain direction varies across the image. We suggest that during the anneal sufficient thermal energy is provided to allow the short polymers to diffuse, producing longer chains which often align with surface structures (such as parallel to step-edges) to maximise the polymer length. Further annealing at 350 °C (Fig. 2c) results in a loss of long range ordering of the polymer structure, with local ordering of neighbouring chains (parallel alignment) observed, but with frequent intersection of polymer chains being present. The elevated temperature here may facilitate cross-linking reactions between DPP-polymers as a result of C–H activation in the phenyl rings in the biphenyl units of neighbouring chains. Since there are several positions that may be activated, the inter-chain reactions are expected to lead to a loss of order, as seen for other aromatic systems.18 The lack of any regular structure supports this hypothesis of non-specific C–C linking with loss of hydrogen.
Our interpretation, that the change in structure is due to an on-surface coupling reaction, is supported by X-ray photoelectron spectroscopy (XPS) measurements of the Br 3p region (Fig. 2f) for the as deposited (orange) and annealed to 100 °C (blue) samples. Due to spin–orbit splitting two peaks are expected for each Br environment (1/2 and 3/2). Comparison of the expected binding energies of C–Br and Au–Br species (red and green lines in Fig. 2f) with our experimental data indicates that prior to annealing, the Br–C bond within Br2Hex2DPP is intact.42 Following annealing to 100 °C no significant peaks are observed, indicating that the environment of the Br has changed, which we attribute to the cleavage of C–Br bonds. Although we observe no peaks in the Br 3p region following annealing, it is likely that the presence of two distinct chemical environments, the expected spin–orbit splitting, and the potential for the desorption of Br species at these temperatures40 will combine to obfuscate any peaks within the noise level of our measurements. As such, our XPS data supports the cleavage of the C–Br but does not necessarily support the complete removal of Br from the surface at 100 °C (note: C1s shows no significant changes – see ESI†). In line with several STM studies of Ullmann-type coupling reactions involving bromine functionalised species, we do not observe evidence within our STM data for Br atoms at the Au substrate following C–Br bond scission23,43 (NB related studies involving iodine materials frequently show the presence of iodine atoms25,44).
We now focus on molecular level details of the polymer structures formed after annealing. Fig. 3a shows an overview of a domain of the linearly aligned polymer chains formed by heating to 100 °C. There is a clear uniform directionality for chains within the region, alongside discontinuities along the length of the chain (indicated by red arrows). The presence of kinks in predominantly straight chains is likely caused by the adsorption-induced chirality of the Br2Hex2DPP discussed in Fig. 1c: we find that the geometry of these kink sites match our chiral models (see ESI†). The prevalence of kink sites amongst the 100 °C structures was measured to be 1 in 27 (56 kinks identified within a region of 1530 monomer units), implying a largely defect free and enantiopure polymer structures. The homochiral nature, (M)n or (P)n, is confirmed via a comparison with the racemic, P–(M–P)n–M, structure (Fig. S10†). Alignment of the homochiral polymer structure provides good agreement with respect to the bright/high regions within the topographic STM images (assigned to the aromatic species within the polymer chains), whereas the racemic polymer structure does not spatially align with the STM data. Domains of homochiral polymers are observed to occur separately to one another (see Fig. 2a), potentially driven by the homochirality of the large self-assembled domains of the unreacted monomer prior to annealing. This may give rise to localised regions with a high proportion of monomers of a single enantiomer that would lead to the observed homochiral reaction products. An overlay of Br2Hex2DPP monomers onto the structures formed after annealing at 100 °C structures is shown in Fig. 3b. Here, monomers, dimers, and longer polymers are identified (position of a line profile indicated with a red line – data in Fig. 3e). Fig. 3c demonstrates how the arrangement of the hexyl chain interdigitation (green lines) determines the chain separation (discussed below – see Fig. 4). Similar to the self-assembled structures prior to annealing, our STM derived model of the chain structure indicates that the linear polymer chains are homochiral. The resolution of our STM data does not allow the chirality (M or P) of the homochiral polymer chains to be determined from a consideration of the chirality of individual monomer units. However, the kinks may be identified as a specific chiral structure, allowing the specific chirality of the covalently coupled linear chains either side of the kink to be identified.
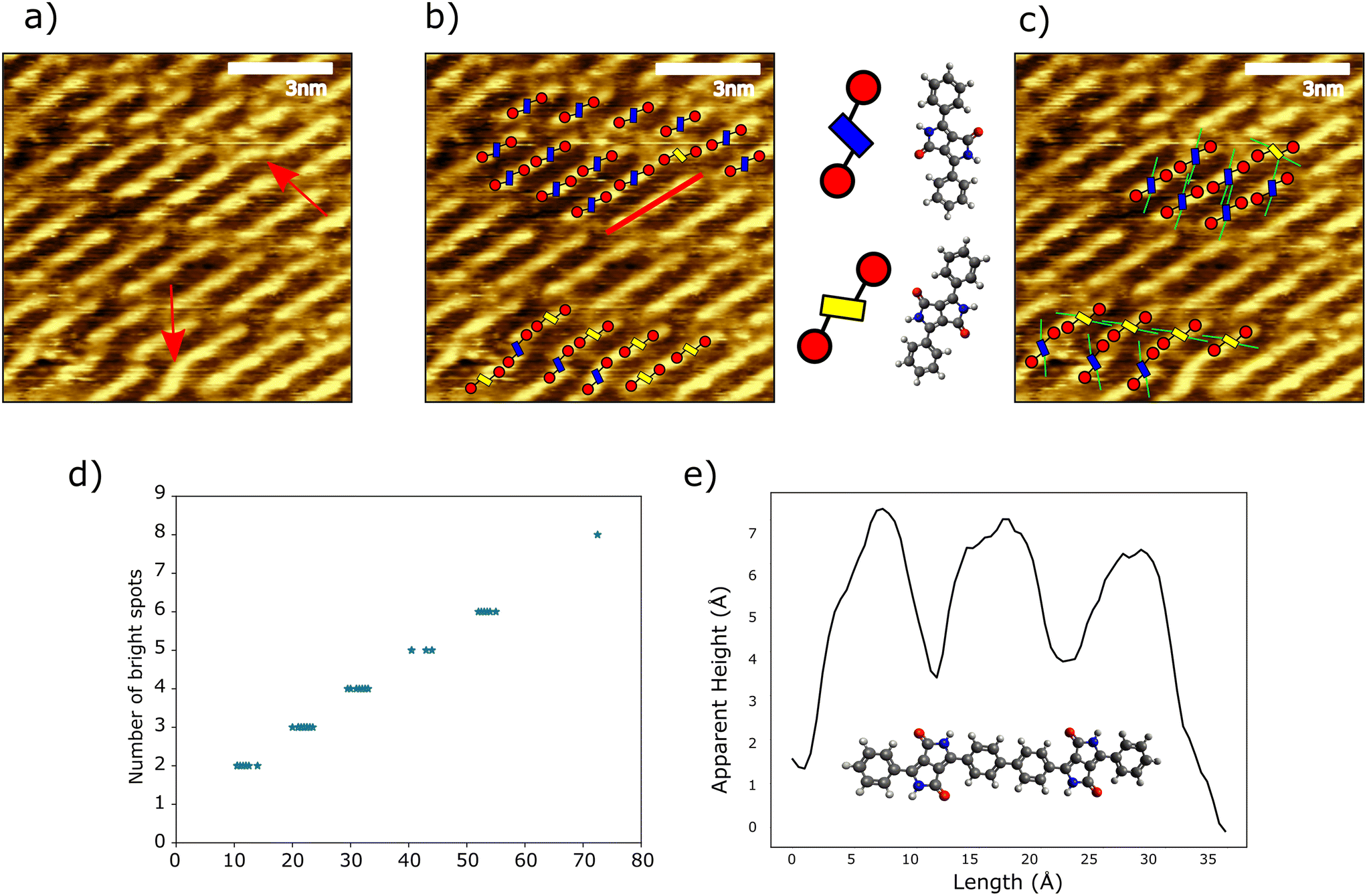 | ||
| Fig. 3 a) STM image (bias = 2 V, current = 40 pA) of an area of polymer chains formed by heating to 100 °C, kinks within polymers indicated by red arrows. b) Molecular model overlaid onto the image of polymer chains, with the blue and yellow DPP cores highlighting the two interface enantiomers. c) Simplified model demonstrating the role of alkyl chain interactions in influencing the shape of the domains. d) Plot of the number of visible bright spots within a chain against length of polymer chain. e) Line profile along a polymer chain showing the correspondence between peak positions and molecular structure (line profile position shown by red line in Fig. 3b). | ||
 | ||
| Fig. 4 a) Histogram showing the distribution of polymer chain lengths following annealing at 100 °C and 250 °C. b) Histogram of chain spacing between polymer rows following annealing at 100 °C and 250 °C. c–e) Models of different hexyl chain configurations, and the corresponding expected chain spacing. | ||
The line profile in Fig. 3e shows variations in apparent height along the length of the polymer, resulting in ‘brighter’ regions (the location the line profile was taken is shown with a red line in high resolution image Fig. 3b). The frequency of bright spots is observed to increase linearly with the chain length (see Fig. 3d). The separation between the first and last bright features in chains with three bright features is measured to be 2.18 nm ± 0.02 nm, in good agreement with the expected separation of the outer aryl rings within a covalently bonded DPP dimer (see line profile – Fig. 3e); indicating that under these imaging parameters the DPP units within the polymers appear ‘dark’. As can be seen in Fig. 3d, each additional monomer to the chain adds one additional bright spot (with pairs of phenyl rings forming one indistinguishable peak).
A comparison of the polymers formed after annealing to 100 °C and 250 °C reveals details of additional reactions at higher temperatures and provides information on the thermal stability of the polymers. Fig. 4a shows the distribution of the measured chain lengths following annealing at 100 °C and 250 °C, with an average length of 4.14 nm for the chains at 100 °C. The comparatively shorter chain lengths (for the lower temperature anneal) are potentially driven by the initial arrangement of the close-packed structure of the as-deposited monomer material. These close-packed structures bring the reactive Br-aryl groups into close proximity, with only minimal rearrangement being required to facilitate the required bonding geometry. In this respect, the van der Waals interactions between hexyl chains, and halogen bonding, drive both the order of the close-packed structure and the aligned polymers. Heating further, to 250 °C, results in a broadening of the distribution of the measured polymer lengths as well as an increase in the average length to 9.14 nm (as shown in Fig. 4a). This observation indicates that the Ullmann-type polymerisation had not progressed to completion following the initial anneal to 100 °C (with the small domains of polymers prohibiting longer chain growth due to local misalignment of the reactive end groups – from the STM data it is not possible to determine if these chains retain Br atoms at their termination).
The role of the hexyl chains in the alignment of the polymer structures is further elucidated by considering the separation between neighbouring polymers. Fig. 4b shows the distribution of chain separations following annealing. Following the anneal at 100 °C, the separation of the chains is broadly uniform, being generally around 0.9–1.0 nm (which is in agreement with a hexyl chain configuration for the polymers with the alkyl chains orientated in the rotamer (2-2) position– see Fig. 4d and S9†). This specific chain conformation results in the expected inter chain separation and could be the cause of the visible diagonal staggering between smaller chains in the more heterogeneous domains, as shown in Fig. 3c. Heating further, to 250 °C, results in a much wider spread of separations (0.7–1.5 nm) with a significant reduction in the minimum separation between polymer chains (to around 0.7–0.8 nm). These smaller dimensions are consistent with a process whereby hexyl chains begin to break off at higher temperatures, with a separation of 0.7 nm being consistent with two parallel polymers with no hexyl chains present (Fig. 4c). The additional space generated by the loss of alkyl chains may facilitate a conformational change between the two accessible rotamers. Fig. 4e shows how an alternative alkyl chain conformation could be expected to stabilise a chain separation of around 1.3 nm. Here the chains are in the rotamer (1-1) orientation, which increases interchain separation compared to the rotamer (2-2) orientation. The combination of the loss of alkyl chains and conformation change is proposed to stabilise the structures shown in Fig. 4c and e, and is in agreement with the distribution of inter-chain separations shown in Fig. 4b.
Conclusion
In conclusion, we have demonstrated that the on-surface self-assembly and polymerisation of functional DPP species is significantly influenced by specific functional groups. Here, alkyl chains are observed to influence both the close-packing of the reactant molecules and the ordering of the polymeric product. Similarly, the bromine moieties (selected to facilitate on-surface synthesis) also influence the stability of the close-packed precursor units, which in turn is likely to affect the reaction pathway and ultimate alignment of the polymeric product. We demonstrate that the alkyl chains play a key role in the alignment of the DPP polymers and show how the thermal stability of these groups is key in maintaining the long-range order of the polymers. In addition, we have shown that pro-chiral nature of the DPP-based monomers results in homochiral self-assembled structures of the unreacted monomers and the polymer reaction product. It is expected that the alignment of photo sensitive polymers is key to maximising the efficiency of optoelectronic devices, and hence the role of molecular level interactions driving the ordering of these polymers is an important step towards development of such devices.Experimental methods
STM
Au on mica samples (Georg Albert PVD GmbH) were cleaned via cycles of Ar ion sputtering (20 minutes at 0.75 keV, 8.5 × 10−6 mbar) and annealing (573 K for 20 minutes). Sample cleanliness was determined via scanning tunnelling microscopy (STM) prior to deposition of Br2Hex2DPP. Thermal deposition was conducted using a Knudsen-type evaporation cell (K-Cell) at a temperature of 190 °C for a period of 2 hours at a pressure of 1 × 10−8 mbar. STM data was acquired using an Omicron STM-1 system, with Nanonis control electronics, operating at room temperature under ultra-high vacuum (UHV) conditions: base pressure <2 × 10−9 mbar. Imaging was performed using electrochemically etched tungsten tips, functionalised with gold during on-surface tip preparation (bias is applied to the sample). Sample annealing is performed via resistive heating of a silicon wafer mounted behind the Au/mica sample: temperature estimates are based upon thermocouple readings from the sample receptor which were calibrated to the sample temperature via a combination of pyrometer measurements of the Si wafer and a thermocouple affixed to the sample plate. Samples were annealed to 100 °C, 200 °C, 250 °C and 350 °C with an estimated error of ±50 °C.XPS
X-ray photoelectron spectroscopy (XPS) was acquired using a SPECS DeviSim near ambient pressure XPS (NAP-XPS) instrument operating in ultra-high vacuum (UHV) mode at a pressure <1 × 10−9 mbar. Spectra were measured using a Phoibos 150 NAP hemispherical analyzer with 20 eV pass energy and monochromatic Al Kα X-rays (1486.7 eV). The samples were transported between the STM and XPS sites using a vacuum suitcase at a pressure <1 × 10−10 mbar.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. J. H., M. C., A. B.-C., and J. O'S. acquired the XPS data; M. C. and A. B.-C. acquired STM data; F. M. synthesised the Br2Hex2DPP material; M. C. and A. S. analysed the data; conceptualization, A. S. and D. B. A.; funding acquisition, A. S. and D. B. A.; supervision, A. S. and D. B. A.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. C. acknowledges support via the MI&A Doctoral Training Programme at the University of Nottingham (EPSRC). F. M. thanks the LDMI Doctoral Training Programme (EPSRC) and the University of Nottingham's Propulsion Futures Beacon of Excellence for funding. A. S. would like to acknowledge support via a Royal Society University Research Fellowship. D. B. A. acknowledges support from Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR), Grant 2021SGR01085.References
- Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684–1710 RSC.
- Y. Zang, S. Ray, E. D. Fung, A. Borges, M. H. Garner, M. L. Steigerwald, G. C. Solomon, S. Patil and L. Venkataraman, J. Am. Chem. Soc., 2018, 140, 13167–13170 CrossRef CAS PubMed.
- C. Fu, P. J. Beldon and D. F. Perepichka, Chem. Mater., 2017, 29, 2979–2987 CrossRef CAS.
- S. J. Krivickas, C. Hashimoto, J. Yoshida, A. Ueda, K. Takahashi, J. D. Wallis and H. Mori, Beilstein J. Org. Chem., 2015, 11, 1561–1569 CrossRef PubMed.
- C. Réthoré, N. Avarvari, E. Canadell, P. Auban-Senzier and M. Fourmigué, J. Am. Chem. Soc., 2005, 127, 5748–5749 CrossRef PubMed.
- J. V. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375–407 CrossRef CAS PubMed.
- S. De Feyter and F. C. De Schryver, Chem. Soc. Rev., 2003, 32, 139–150 RSC.
- A. Honda, Y. Tamaki and K. Miyamura, Bull. Chem. Soc. Jpn., 2015, 88, 969–975 CrossRef CAS.
- C. Fu, F. Bélanger-Gariépy and D. F. Perepichka, CrystEngComm, 2016, 18, 4285–4289 RSC.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- L. Grill and S. Hecht, Nat. Chem., 2020, 12, 115–130 CrossRef CAS PubMed.
- T. Wang and J. Zhu, Surf. Sci. Rep., 2019, 74, 97–140 CrossRef CAS.
- L. Verstraete and S. De Feyter, Chem. Soc. Rev., 2021, 50, 5884–5897 RSC.
- X. Li, H. Ge, R. Xue, M. Wu and L. Chi, JACS Au, 2022, 2, 58–65 CrossRef CAS PubMed.
- L. Dong, P. N. Liu and N. Lin, Acc. Chem. Res., 2015, 48, 2765–2774 CrossRef CAS PubMed.
- Q. Fan, J. M. Gottfried and J. Zhu, Acc. Chem. Res., 2015, 48, 2484–2494 CrossRef CAS PubMed.
- M. In't Veld, P. Iavicoli, S. Haq, D. B. Amabilino and R. Raval, Chem. Commun., 2008, 1536–1538 RSC.
- S. Haq, F. Hanke, J. Sharp, M. Persson, D. B. Amabilino and R. Raval, ACS Nano, 2014, 8, 8856–8870 CrossRef CAS PubMed.
- C. Nacci, F. Ample and D. Bleger, et al. , Nat. Commun., 2015, 6, 7397 CrossRef PubMed.
- D. Skidin, T. Erdmann, S. Nikipar, F. Eisenhut, J. Krüger, F. Günther, S. Gemming, A. Kiriy, B. Voit, D. A. Ryndyk, C. Joachim, F. Moresco and G. Cuniberti, Nanoscale, 2018, 10, 17131–17139 RSC.
- X. Meng, L. Liu, F. García, B. Álvarez, D. Pérez, H. Y. Gao, D. Pena and H. Fuchs, J. Phys. Chem. C, 2018, 122, 6230–6235 CrossRef CAS.
- S. Clair and D. G. De Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- A. Saywell, J. Schwarz, S. Hecht and L. Grill, Angew. Chem., 2012, 124, 5186–5190 CrossRef.
- C. J. Judd, S. L. Haddow, N. R. Champness and A. Saywell, Sci. Rep., 2017, 7, 1–7 CrossRef CAS PubMed.
- C. J. Judd, N. R. Champness and A. Saywell, Chem. – Eur. J., 2018, 24, 56–61 CrossRef CAS PubMed.
- C. J. Judd, D. V. Kondratuk, H. L. Anderson and A. Saywell, Sci. Rep., 2019, 9, 1–8 CrossRef CAS PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94–102 CrossRef CAS PubMed.
- D. B. Amabilino, et al. , J. Phys.: Condens. Matter, 2008, 20, 184003 CrossRef.
- K. H. Ernst, Phys. Status Solidi B, 2012, 249, 2057–2088 CrossRef CAS.
- R. Raval, Chem. Soc. Rev., 2009, 38, 707–721 RSC.
- J. Teyssandier, K. S. Mali and S. De Feyter, ChemistryOpen, 2020, 9, 225–241 CrossRef CAS PubMed.
- J. Lawrence, G. C. Sosso, L. Đorđević, H. Pinfold, D. Bonifazi and G. Costantini, Nat. Commun., 2020, 11, 7–13 CrossRef PubMed.
- A. Batra, D. Cvetko, G. Kladnik, O. Adak, C. Cardoso, A. Ferretti, D. Prezzi, E. Molinari, A. Morgante and L. Venkataraman, Chem. Sci., 2014, 5, 4419–4423 RSC.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215–220 CrossRef CAS PubMed.
- K. A. Simonov, N. A. Vinogradov, A. S. Vinogradov, A. V. Generalov, E. M. Zagrebina, N. Mårtensson, A. A. Cafolla, T. Carpy, J. P. Cunniffe and A. B. Preobrajenski, J. Phys. Chem. C, 2014, 118, 12532–12540 CrossRef CAS.
- T. Faury, S. Clair, M. Abel, F. Dumur, D. Gigmes and L. Porte, J. Phys. Chem. C, 2012, 116, 4819–4823 CrossRef CAS.
- M. Koch, M. Gille, A. Viertel, S. Hecht and L. Grill, Surf. Sci., 2014, 627, 70–74 CrossRef CAS.
- D. G. De Oteyza, A. García-Lekue, M. Vilas-Varela, N. Merino-Díez, E. Carbonell-Sanromà, M. Corso, G. Vasseur, C. Rogero, E. Guitián, J. I. Pascual, J. E. Ortega, Y. Wakayama and D. Peña, ACS Nano, 2016, 10, 9000–9008 CrossRef CAS PubMed.
- M. Di Giovannantonio, Q. Chen, J. I. Urgel, P. Ruffieux, C. A. Pignedoli, K. Müllen, A. Narita and R. Fasel, J. Am. Chem. Soc., 2020, 142, 12925–12929 CrossRef CAS PubMed.
- T. A. Pham, F. Song, M. T. Nguyen, Z. Li, F. Studener and M. Stöhr, Chem. – Eur. J., 2016, 22, 5937–5944 CrossRef CAS PubMed.
- A. Basagni, F. Sedona, C. A. Pignedoli, M. Cattelan, L. Nicolas, M. Casarin and M. Sambi, J. Am. Chem. Soc., 2015, 137, 1802–1808 CrossRef CAS PubMed.
- G. Galeotti, F. De Marchi, T. Taerum, L. V. Besteiro, M. El Garah, J. Lipton-Duffin, M. Ebrahimi, D. F. Perepichka and F. Rosei, Chem. Sci., 2019, 10, 5167–5175 RSC.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, J. Phys. Chem. C, 2014, 118, 1719–1728 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic materials and methods; prochirality and rotomerisation; liquid STM; example line profiles; XPS; chain separation measurements; molecular modelling; tip changes during STM; details on crosslinking and image resolution. CCDC 2208888. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2me00232a |
| This journal is © The Royal Society of Chemistry 2023 |