
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Democratizing the rapid screening of protein expression for materials development†
Melody A.
Morris
 ,
Rogério A.
Bataglioli
,
Danielle J.
Mai
,
Yun Jung
Yang
,
Justin M.
Paloni
,
Carolyn E.
Mills
,
Zachary D.
Schmitz
,
Erika A.
Ding
,
Allison C.
Huske
and
Bradley D.
Olsen
*
,
Rogério A.
Bataglioli
,
Danielle J.
Mai
,
Yun Jung
Yang
,
Justin M.
Paloni
,
Carolyn E.
Mills
,
Zachary D.
Schmitz
,
Erika A.
Ding
,
Allison C.
Huske
and
Bradley D.
Olsen
*
Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: bdolsen@mit.edu
First published on 28th October 2022
Abstract
The function, structure, and mechanical properties of protein materials make them well-suited for a range of applications such as biosensors and biomaterials. Unlike in traditional polymer synthesis, their sequences are defined and, in the case of recombinant proteins, dictated by the chosen DNA sequence. As DNA synthesis has rapidly progressed over the past twenty years, the limiting bottleneck in protein materials development is the empirical optimization of protein expression. Herein, a low-cost, automated, high-throughput, combinatorial protein expression platform is developed to test permutations of DNA vectors and Escherichia coli (E. coli) strains in a 96-well plate format. Growth and expression are monitored with optical density at 600 nm (OD600) to measure growth, Bradford assays to establish the total protein concentration, and dot blot assays to determine the concentration of the protein of interest. With an eye toward accessibility for researchers without suites of biosynthetic equipment, automated camera-based assays are validated for the OD600 assay, via turbidimetry, and the Bradford assay, via colorimetry. High-yield expression conditions can be determined within a week. Notably, in several cases, previously un-expressible proteins are expressed successfully in viable yields. Collectively, an efficient approach to overcoming long-running synthesis challenges in protein materials development is established, which will expedite materials innovation.
Design, System, ApplicationProtein materials provide a deep design space, particularly in the area of sequence-defined macromolecules, but rapid materials design cycles have not yet been realized. As synthetic biology has rapidly progressed, the bottleneck in the exploration of this area has been the ability to rapidly express a protein due to almost entirely empirical guidelines for selecting cell machinery. Herein, a combinatorial, low-cost, automated strategy is employed to quantitatively screen for viable plasmid and strain combinations for a panel of protein materials. In comparison to methods with initial investments in the hundreds of thousands of dollars, the developed platform requires initial capital investments under 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 USD, with each protein costing approximately 600 dollars in materials and consumables. Additionally, inexpensive camera-based methods were verified to replace costly plate-reader-based assays. All automation and analysis codes are fully available, and the platform is highly modular to encourage immediate implementation by the community. Together, this platform promotes the rapid generation of a library of protein expression data, starting with over one thousand data points, to move toward data-driven optimization methods and more rapid protein materials design cycles. 000 USD, with each protein costing approximately 600 dollars in materials and consumables. Additionally, inexpensive camera-based methods were verified to replace costly plate-reader-based assays. All automation and analysis codes are fully available, and the platform is highly modular to encourage immediate implementation by the community. Together, this platform promotes the rapid generation of a library of protein expression data, starting with over one thousand data points, to move toward data-driven optimization methods and more rapid protein materials design cycles.
|
Introduction
With their unique combination of binding, enzymatic, and structural properties, protein materials have tremendous promise for a variety of biomaterials applications, including biosensors and industrial catalysts.1–6 To develop materials for these types of applications, it is essential to achieve the necessary mechanical properties,1,7,8 maintain function and stability of folded proteins,9,10 and manipulate the nanoscale orientation and morphology of the material.11 Each of these properties is affected by the protein material's sequence, molar mass, and processing conditions.12 Inspired by the Materials Genome Initiative,13,14 rapid, high-throughput materials development cycles are necessary to synthesize, discover, and optimize properties to compete with existing materials such as polymers and catalysts.As synthetic biology has advanced rapidly, DNA synthesis cost and time have decreased exponentially,15 positioning protein expression as the key bottleneck in materials innovation.16 Expression optimization of the protein of interest requires a largely empirical optimization process, with automated screening tools accessible but at extremely high cost.17 A variety of expression hosts, including bacteria, yeast, and mammalian cells as well as others, are possible for recombinant protein production.18 The choice of expression host can affect the final recombinant protein's glycosylation, post-translational modifications, yield, and ease of purification; thus, it is essential to match a protein's required usage to an appropriate host system.19 For many protein materials, Escherichia coli (E. coli) is the preferred host for recombinant protein expression due to its fast and high expression, inexpensive culture, and ease of genetic manipulation.20 However, because of E. coli's tightly coupled transcription and translation, many proteins do not properly fold or are insoluble in non-optimized conditions. In particular, desirable sequences for protein materials include many challenging characteristics for soluble protein expression, including repetitive sequences,21,22 rare codons,23–25 large protein sizes,26,27 hydrophobicity,22,28,29 toxicity,30 and disulfide bonds.31–33 Currently, methodologies to enhance solubility include decreased expression temperatures,34 engineered cell strains,16,25,35 and solubility-enhancement tags,36 such as glutathione-S-transferase (GST) or small ubiquitin-like modifier (SUMO).37,38 It is noted that not all proteins can be made in E. coli, such as proteins with certain post-translational modifications or glycosylations, so there is a wealth of opportunities for additional studies to probe other expression hosts. To leverage the wealth of achievable sequences, it is essential to establish an accessible, low-cost, combinatorial screening tool to identify high yield protein expression conditions suitable for protein materials in as little time as possible.16
Although there exist general guidelines for matching proteins to appropriate cell strains and DNA plasmids, there is currently no widely accessible framework for determining optimal conditions.16,39 In 2001, Knaust and Nordlund reported non-automated high-throughput screening of two constructs in deep-well plates,40 which was transformed to automated screenings shortly after.41–43 As synthetic biology has advanced in the last twenty years, new promoter systems, cell strains, and solubility tags have been developed, but most automation efforts have focused on novel interfaces between protocol development and liquid-handling robots.44–46 Although commercial automated systems exist, they are typically beyond the reach of academic groups and even many small businesses.47 Moreover, a framework that can compile and generate a large database of protein expression conditions for data-driven approaches, such as machine learning, is lacking; typically only successful expression conditions are published in the scientific literature, which makes it difficult to establish design rules for expression. Thus, there remains a gap in high-throughput protein expression screenings to accelerate protein materials development.
Herein, a high-throughput, combinatorial E. coli expression platform has been developed using a low-cost liquid handling robot and open-source software and tested on 17 constructs of interest to demonstrate its wide versatility, as demonstrated in Fig. 1. Specifically, the genes of interest were inserted into a small library of different DNA plasmids commonly used for biomaterial expression, which include a variety of inducible promoter systems, and further transformed into different cell strains to form a combinatorial expression library that can be tested in well-plate format. Cell growth was monitored by tracking the optical density at 600 nm (OD600), and a protocol with a simple automated camera was developed and validated such that the platform can be operated without a spectrophotometer. Post-expression, the yield of total protein, via Bradford assay, and of the protein of interest, via dot blot, were quantified to identify promising cell-plasmid combinations that can be further optimized with changes to media, temperature, and time. High yield expression conditions can now reliably be found within a week, and conditions for previously un-expressible proteins have been identified in several cases. Initial startup costs are under US $15000, with each protein's expression optimization totaling just over US $600, making this process significantly more accessible than previous expression optimization schemes with similar throughput levels. Compiled data is stored in a database-format to enable further data-driven approaches to optimizing the expression of protein materials moving forward. This work establishes an efficient approach to overcoming long-running synthesis challenges in protein materials development at a 100-fold lower capital cost than commercial systems, expediting innovation in this space.
 | ||
| Fig. 1 Combinatorial protein materials expression design. After a protein is designed, it is subcloned into a panel of six vectors using a common protocol for the whole panel. Once cloning is validated, an automated protocol transforms the vector panel into eleven different E. coli strains, which are directly carried forward into protein expression (monitored by OD600) and harvest. After clarifying the lysate, the total protein concentration and concentration of the protein of interest are quantified via Bradford assay and dot blot, respectively. | ||
Materials and methods
DNA cloning and preparation
Seventeen genes of interest were chosen, detailed in the ESI,† section A. Eight (047A, 047B, Catcher, CC43, PPxY, Tag, ZE, and ZR) were designed and purchased from GenScript as BamHI-NdeI-DNA sequence of interest-SpeI-XhoI-HindIII in pET-15b. An additional four (mCherry, hNup50, hNup62, hNup98) were purchased from GenScript as BamHI-NdeI-DNA sequence of interest-XhoI-HindIII-BglII in pUC57. The ELP series E10, E20, E40, and E80 were designed and cloned as detailed previously,48 with a final design of NdeI-NheI-DNA sequence of interest-SpeI-HindIII-BamHI in pET-15b. P4 was designed and cloned as detailed previously,49 with a final design of BamHI-NheI-DNA sequence of P4-SpeI-HindIII. Gene designs flanked by NdeI on the 5′- and XhoI on the 3′-end were directly subcloned into the custom-designed pGEX-4T-(1H) vector (complete sequence in ESI,† section B). Gene designs lacking these restriction sites were subcloned via restriction digest cloning with other restriction sites into vectors that did contain these flanking sites and subsequently cloned into pGEX-4T-1(H). To subclone into the remaining vectors, the following pairs of restriction sites were used: pET-15b (NdeI/XhoI), pET-22b(+) (NdeI/XhoI), pQE-9 (BamHI/HindIII), pQE-60 (BamHI/BglII), pGEX-4T-(1H) (NdeI/XhoI), and pET-SUMO (BamHI/XhoI). All sequences were confirmed via Sanger sequencing (Genewiz, USA).Competent cell preparation
BL21, T7 Express, T7 Express lysY, and T7 Express lysY/Iq were purchased from New England Biolabs, USA. Rosetta 2™ (DE3) was purchased from Millipore-Sigma, USA. BL21(DE3), BL21*(DE3), Tuner(DE3), C41(DE3), C43(DE3), NiCo21(DE3), and SG13009 were prepared from existing lab stocks. The Zymo Mix & Go! Kit was used to prepare large stocks of all competent cells, and cells were aliquoted in 430 μL aliquots and stored at −80 °C. Competency was tested with 0.05 ng μL−1 pUC19, and only cells with transformation efficiencies >106 transformants μg−1 were used (competent cell efficiencies are reported in ESI,† section K).Protein expression and cell lysis
All expressions were manipulated with an OpenTrons OT-2 pipetting robot in 96 shallow- and deep-well plates. Shallow well plates were 330 μL, clear, sterile, flat-bottom, untreated polystyrene plates. Deep-well plates were 2 mL, sterilized, square-shaped, cone-bottom polypropylene plates (PlateOne #1896-2110). Combinations of each plasmid/competent cell were transformed in a 96 shallow-well plate according to the designed plate layout using 45 μL of cells and 45 μL DNA at a concentration of 5 ng μL−1. After sitting on a cold plate for 30 minutes, 60 μL of cell/DNA solution was added to 200 μL of SOC broth in a deep well plate with glass beads and incubated for 60 min at 300 rpm at 37 °C (VWR 89232-904). Successful transformants were selected by subculturing 60 μL of the transformation into 600 μL of LB supplemented with the appropriate antibiotic for each vector/strain combination (100 μg mL−1 for ampicillin, 50 μg mL−1 for kanamycin, and 34 μg mL−1 for chloramphenicol) at 37 °C for 20 h in a ThermoScientific MaxQ 4000 Refrigerated Shaker. OD600 measurements were taken to determine the transformation success rate by transferring 200 μL of culture into a 96 shallow well plate and measuring on a plate reader (Tecan Infinite® 200 PRO). Protein expression was performed in 96 deep-well plates containing one glass bead per well to increase mixing. Two replicate plates were prepared for each expression experiment. 900 μL of LB supplemented with the appropriate antibiotic was inoculated with 30 μL of the overnight culture, and these cultures were grown at 37 C for 2.5 h with orbital shaking at 300 rpm in a VWR 1585 Orbital Shaking Incubator and a ThermoScientific MaxQ 4000 Refrigerated Shaker. OD600 measurements were taken to monitor the optical density at induction by transferring 200 μL of culture into a 96 shallow well plate and measuring on a plate reader. After the initial 2.5 h growth, expression was induced with 1 mM IPTG, and cultures were allowed to grow for an additional 20 h at 25 °C with orbital shaking at 300 rpm. OD600 of the cultures at harvest was measured by transferring 200 μL of culture into a 96 shallow well plate and measuring absorbance on a plate reader. The cells were harvested via centrifugation (3488 × g, 20 °C), and the supernatant was removed via multichannel pipetting. Plates containing cell pellets were frozen overnight at −20 °C; subsequently, pellets were resuspended in 200 μL of MENT lysis buffer (3 mM MgCl2, 1 mM ethylenediaminetetraacetic acid (EDTA), 100 mM NaCl, 10 mM trizma, 0.5 mg mL−1 lysozyme, 0.1 mg mL−1 DNase I, pH 7.5) and incubated at 37 °C for one hour to initiate lysis. The lysate was transferred to 96 shallow-well plates and subjected to two additional freeze–thaw cycles before being clarified by centrifugation (4816 × g, 4 °C). Clarified lysates were stored in a −20 °C freezer until assays were run.Bradford protocol
For each expression plate, 200 μL of Quick Start™ Bradford 1X Dye Reagent (Bio-Rad, USA) was added to each well of a 96 shallow-well plate (250 μL, flat bottom, untreated), and the absorbance at 595 nm was measured via plate reader. Bovine serum albumin (BSA) was dissolved in MENT buffer at 1 mg mL−1, 0.25 mg mL−1, 0.0625 mg mL−1, and 0 mg mL−1 for use as standards. Using the OT-2 robot, 20 μL of clarified lysate was added to each well and mixed 5 times by automated pipetting. BSA controls were added to wells E12–H12 in place of the negative controls on the initial plate. After all wells were filled, the plate was allowed to develop for 3 min, flamed to remove any bubbles, and measured at 595 nm on the plate reader.Dot blot protocol
Previously-expressed and purified 6×His-tag-containing P4 was used as a control and diluted to 1 mg mL−1, 0.25 mg mL−1, 0.0625 mg mL−1, and 0 mg mL−1 in MENT buffer.50 Using the OT-2 robot, 10 μL of clarified lysate was added to each well of a 200 μL PCR plate; P4 controls were added to wells E12–H12 in place of the negative controls. Plates were sealed with aluminum sealing film and stored overnight. The PCR plates were heated to 95 °C for 5 min in a Bio-Rad T100 Thermal Cycler (Bio-Rad, USA) before cooling to 4 °C. Plates were kept at 4 °C for at least 10 min and up to 1 h before 3 μL of each solution was transferred to a 0.45 μm nitrocellulose membrane cut to 7.5 cm × 12.5 cm (Bio-Rad, USA) with a filter paper backing. Membranes were allowed to dry for at least 5 min, at which time liquid spots were no longer visible. The blotting procedure followed existing chromogenic methods with anti-tetra-His mouse antibody (Qiagen, USA) used as the primary antibody and anti-mouse IgG-alkaline phosphatase (Sigma, USA) used as the secondary antibody.51 Blots were imaged with a ChemiDoc XRS+ system (BioRad, USA) and processed using the ImageJ Background Subtraction and Gel Analysis tool.Results and discussion
Platform development
The combinatorial design of this platform is built upon the 96-well plate format, allowing a library of 66 trials plus controls per plate for each gene of interest. Vectors were varied down the rows, and strains were varied across the columns. To ensure reproducibility and accuracy, thirty wells were reserved for positive and negative controls, as shown in the plate design in Fig. 2. Row G of the plate contained an empty pUC19 vector as a positive control for transformation and cell growth. Row H contained only 100 mM CaCl2 buffer to serve as a negative control for the transformation to ensure antibiotic resistance. These two transformation controls are necessary for each cell strain. In the twelfth column, the first four wells served as positive controls with protein (mCherry in A12 and B12 and P4, a disordered structural protein that has been widely expressed in the Olsen group,49,50,52 in C12 and D12)/vector/cell combinations that are known to successfully express;49,53 the final four wells acted as media and antibiotic-only negative controls. No edge effects were noted due to a multicomponent shaking insert to ensure even orbital shaking across the entire plate. The entire plate was rerun if either the negative or positive expression controls failed.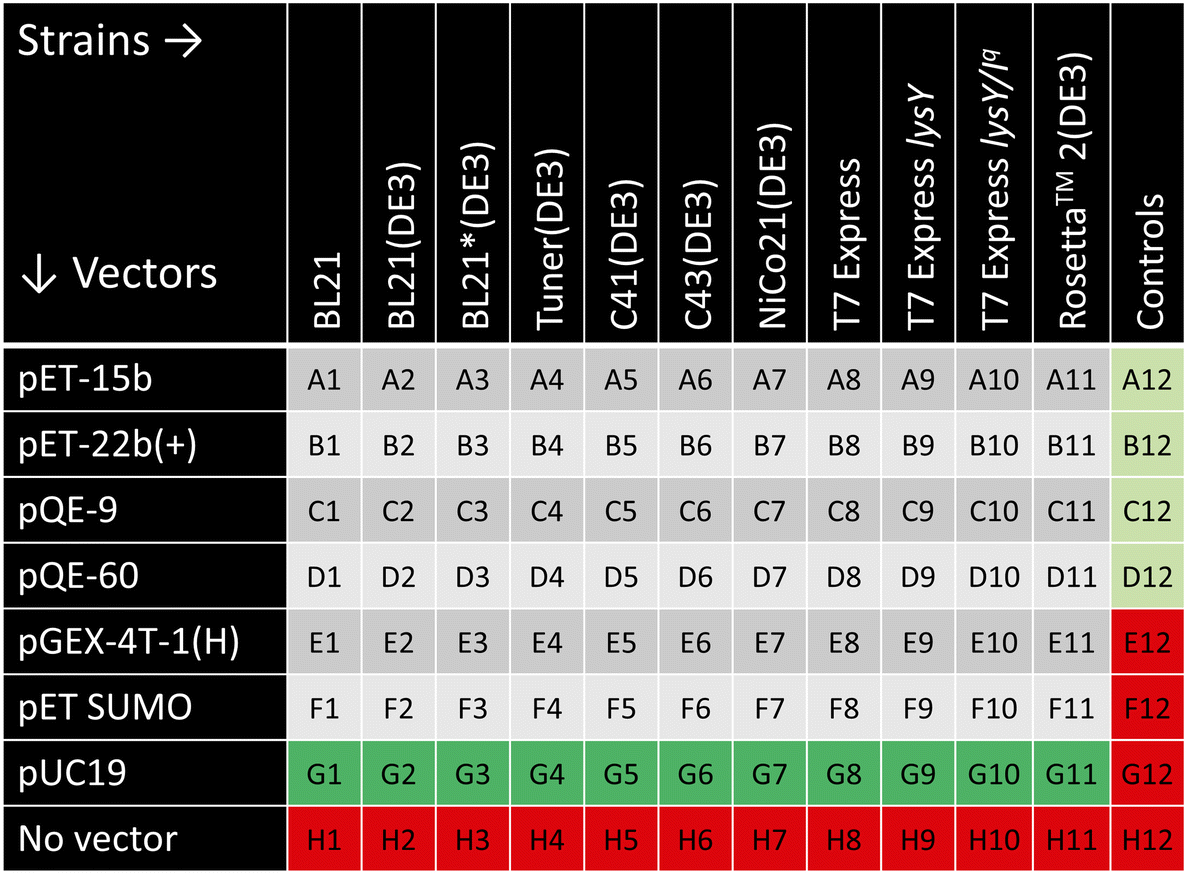 | ||
| Fig. 2 Plate layout for combinatorial protein expression platform including controls. Light and dark gray wells represent the test conditions, red wells designate negative controls, light green well denote positive expression controls, and dark green wells correspond to pUC19 transformation controls. | ||
| Vector | Origin of replication | Promoter | Antibiotic resistance | Tag(s) | Preferred sites for subcloning | Supplier | Cat. no. |
|---|---|---|---|---|---|---|---|
| pET-15b | pBR322 | T7-lac | Ampicillin | N-term 6×His | NdeI + XhoI | Novagen | 69661-3 |
| pET-22b(+) | pBR322 | T7-lac | Ampicillin | C-term 6×His | NdeI + XhoI | Novagen | 69744-3 |
| pQE-9 | ColE1 | T5-lac | Ampicillin | N-term 6×His | BamHI + HindIII | Qiagen | 32915 |
| pQE-60 | ColE1 | T5-lac | Ampicillin | C-term 6×His | BamHI + BglII | Qiagen | 32903 |
| N-term GST | |||||||
| pGEX-4T-1(H) | pBR322 | tac | Ampicillin | BamHI + XhoI | Custom | ||
| C-term 6×His | |||||||
| N-term SUMO | |||||||
| pET SUMO | pBR322 | T7-lac | Kanamycin | BamHI + XhoI | Custom | ||
| N-term 6×His |
| Strain | Features | Vendor | Cat. no. |
|---|---|---|---|
| BL21 | General purpose; negative control for T7-lac plasmids | NEB | C2530H |
| BL21 (DE3) | General purpose | Thermo Scientific | EC0114 |
| BL21* (DE3) | Enhanced mRNA stability | Thermo Scientific | C601003 |
| Tuner (DE3) | Homogeneous IPTG concentration | Novagen | 70623-3 |
| C41 (DE3) | Enhanced toxic protein tolerance | Sigma-Aldrich | CMC0017 |
| C43 (DE3) | Enhanced toxic protein tolerance | Sigma-Aldrich | CMC0019 |
| NiCo21 (DE3) | Reduced metal affinity chromatography contaminants | NEB | C2529H |
| T7 Express | NEB derivative of BL21(DE3) | NEB | C2566H |
| T7 Express lysY | Reduced basal expression (lysY expresses T7 lysozyme) | NEB | C3010I |
| T7 Express lysY/Iq | Lowest basal expression (T7 lysozyme + lacIq) | NEB | C3013I |
| Rosetta™ 2 (DE3) | Additional plasmid for rare codon expression | Novagen | 71400-3 |
The cell panel evaluates a variety of features related to regulation of protein expression levels, tolerance to toxic proteins, and sensitivity to plasmid copy number. BL21 (DE3) and T7 Express serve as general purpose derivatives of BL21 that provide baseline expression levels for each construct. BL21 Star™ (DE3) promotes mRNA stability, which is advantageous for expression of low copy-number plasmids.57 Tuner™ (DE3) promotes uniform IPTG uptake in a cell culture, allowing further tuning of concentration-dependent induction.58 OverExpress™ C41 (DE3) and C43 (DE3) strains include mutations that prevent cell death in response to toxic recombinant proteins.59 NiCo21 (DE3) is engineered to minimize basal E. coli proteins that contaminate immobilized metal affinity chromatography steps used in downstream purification.60 T7 Express lysY inhibits T7 RNA polymerase and reduces basal levels of potentially toxic recombinant proteins before expression is induced.61 T7 Express lysY/Iq further tightens the control of expression by producing a lac repressor.62 The final strain of interest is Rosetta™ 2 (DE3), which carries a chloramphenicol-resistant plasmid that supplies cell machinery for rare codon expression (arginine: AGG, AGA, CGG; isoleucine: AUA, leucine: CUA, proline: CCC, and glycine: GGA).58 Rosetta™ 2 (DE3) was specifically chosen because artificially engineered protein polymers such as elastin-like polypeptides are commonly enriched in these rare amino acids.
mCherry, a pink fluorescent protein, was used to validate the protocols. Across the three Selection plates, 93% of the viable cultures grew in at least one of the plates, 93% grew in at least two plates, and 92% grew in all three replicates (Fig. 3a). Selection plates were grown for 20 h at 37 °C and 300 rpm to produce a saturated culture, which was then subcultured 1:100 (v:v) into fresh antibiotic-containing media, termed growth plates. These cultures were grown for 2.5 hours, to reach log phase growth, at which point the OD600 was measured (Fig. 3b). Most of the cultures (68% of transformed cultures) reach the desired OD600 range (0.6–1.0 in LB media) at this point, though there are populations that are under- or overgrown, which could affect final protein yield. All cultures were induced at 1 mM IPTG and allowed to grow at 25 °C for an additional 20 h, reaching OD600 values spanning 1.5–4.0 (Fig. 3c). Cultures were harvested as described in the Materials and methods.64
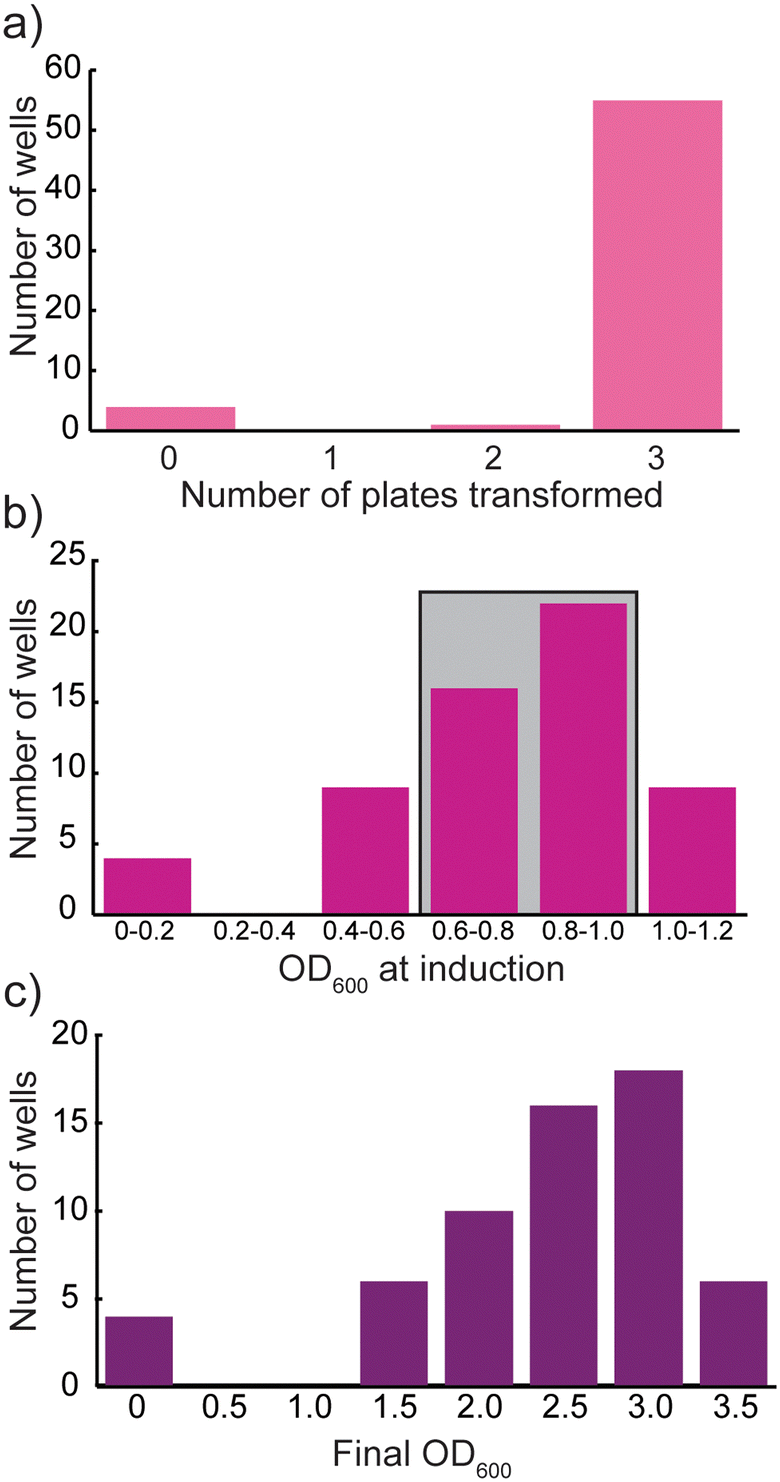 | ||
| Fig. 3 a) Histogram of number of transformed mCherry cultures for each vector/strain combination (total of 60 combinations), b) histogram of average OD600 for each vector/strain combination for mCherry cultures at induction (2.5 h after subculture), with cultures in the log phase boxed in grey c) histogram of average OD600 values for each vector/strain combination at harvest, 20 h post-induction, binned into 0.5-unit increments. BL21* (DE3) samples are not included in these plots. The four samples that did not successfully transform in panel (a) are the same wells in panels (b) and (c) with low OD600 values. | ||
 | (1) |
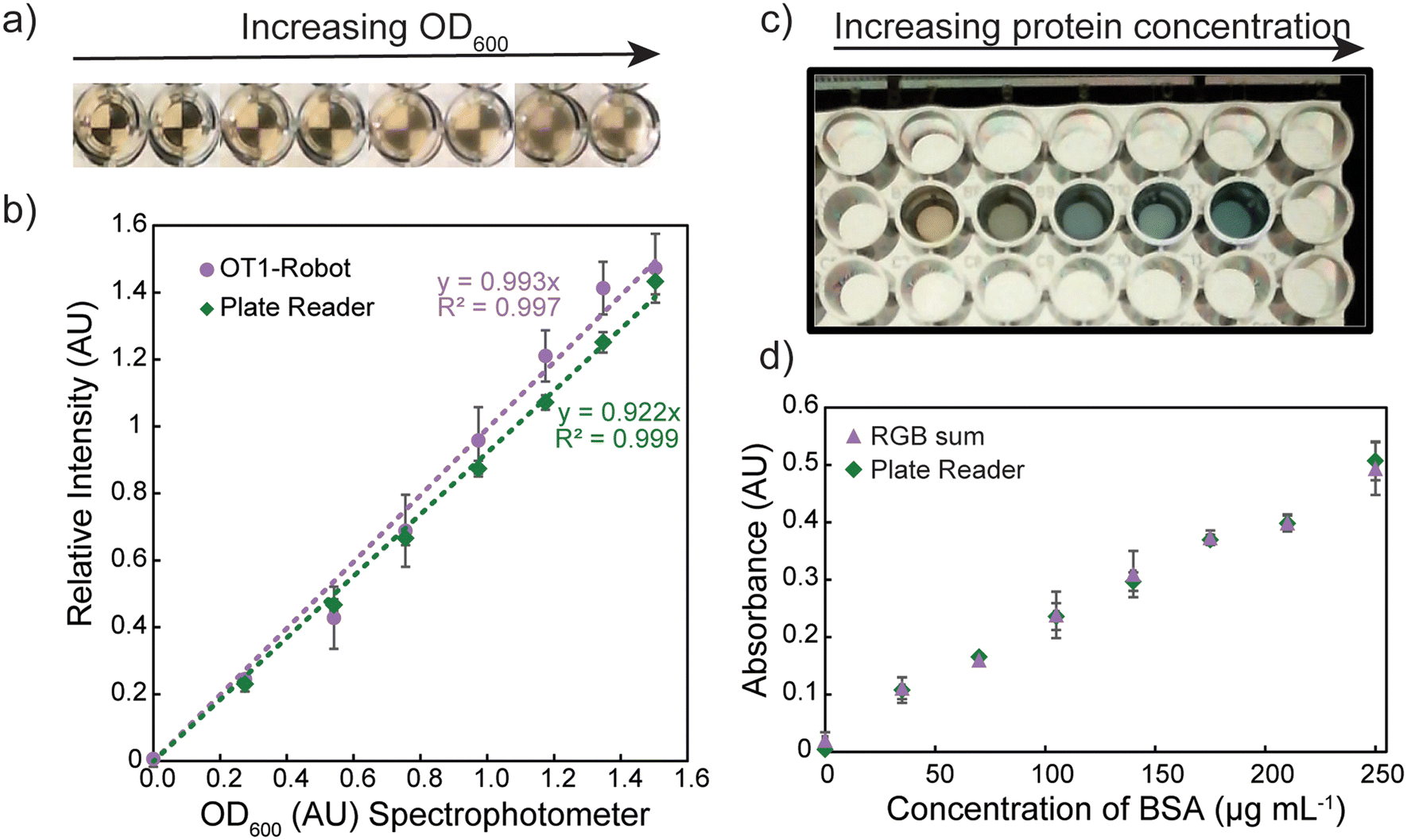 | ||
| Fig. 4 a) Photograph of wells measured via turbidometry-based automated OD600 measurements; b) OD600 measured on a traditional spectrophotometer (1 cm path length) vs. intensity measured by OT-1 robot (purple) and plate reader (green), corrected for path length; c) photograph of Bradford assay well with variable amounts of proteins; d) Bradford assay absorbance as a function of bovine serum albumin (BSA) concentration measured via least squares regression of photography-based assay from weighted red-green-blue (RGB) sums (purple) and by plate reader (green). Error bars represent the standard error across three replicates. | ||
The relative contrast was subtracted from unity to obtain an absorbance value (termed relative intensity), which was correlated to OD600 obtained with a traditional spectrophotometer to apply a linear correction (Fig. 4b). OD600 values below 0.2 and above 1.4 fell out of the linear regime and were reported as “<0.2” and “>1.4”; this limitation did not tremendously affect the platform's performance as highly concentrated cell cultures could be diluted before measurement, and the values close to the lower limit are not important for protein expression. OD600 values were taken at three points in the protocol: 20 h after transformation when cells were seeded into new growth plate, 2.5 h after seeding (before induction), and 20 h after induction.
After expression and clarification of the lysate, the total protein concentration was evaluated with a Bradford assay, as detailed in the Materials and methods. The Bradford assay uses a Coomassie Brilliant Blue G-250 dye in aqueous solution that has a maximum wavelength at 465 nm, which visually appears a yellow-tan color.65 If a protein that has basic and aromatic side chains is added, the absorption maximum shifts to 595 nm within two minutes to a solution that visually appears blue as shown in Fig. 4c.65 This color change has been harnessed to develop a protocol to quantify the protein concentration based on a robot-controlled camera instead of a spectrophotometer to measure absorbance. The camera was used to image each of the wells individually (filled with 200 μL of Bradford reagent) of a clear-bottomed 96-shallow well plate backlit by a tablet with a white screen. Cell lysate (20 μL) was added to each well, and the plate was reimaged after 3 min to allow for complete development of the dye. The red, green, and blue channels were separated with a facile Python code and the absorbance for each channel was calculated in eqn (2).
 | (2) |
The plot of these values is for a control set with bovine serum albumin (BSA) (ESI†). As expected, the absorbance of the blue channel decreases while the absorbance of the red channel increases in the visual shift from tan to blue. To correlate these values, a least-squares fit weighted by the standard deviation of the camera absorbance values to data obtained on the plate reader with a 595 nm absorbance was calculated (Fig. 4d). There is good agreement between the weighted sum of the absorbances and the plate reader, illustrating that this camera-based assay is sufficiently accurate for assessing the overall protein content in cell lysates. It is noted that the Bradford assay is nonlinear at higher protein concentrations, but this non-linearity is an effect of the chemistry of the assay and can be elucidated in the colorimetric assay just as it is by absorbance spectroscopy.
 | ||
| Fig. 5 a) Scatterplot of final OD600vs. concentration of mCherry as quantified by dot blot, sorted by vector; b) scatterplot of final OD600vs. concentration of mCherry as quantified by dot blot, sorted by cell strain; c) visualization of dot blots of mCherry. Dot area and color are normalized against the highest average concentration of 6×His-tagged protein; d) concentration obtained via dot blot vs. measured absorbance at 586 nm (A586). A best fit line is included to guide the eye. Outliers are circled in red and green for ease of discussion. Final concentration is based on 200 μL of lysate, generated from 570 μL of culture. Error bars reflect the standard error across transformed replicates. | ||
To further validate this protocol, the absorbance at 586 nm (A586), which is the maximum absorbance for mCherry, was assessed for all clarified lysate plates (Fig. 5d). Most of the samples fall on a single line, though there are some notable outliers. Falling significantly above the line, circled in red, implies that there is a protein in the lysate that does not show strong signal in the dot blot but absorbs at 586 nm. Although there are several possible explanations for this behavior, it is likely that these examples are mCherry truncation products in which the 6×His tag was either never synthesized or was degraded before the dot blot was run (see Fig. S89 in the ESI† for SDS-PAGE gels of the clarified lysates). Alternatively, some of these proteins could have a population of exceptionally well-folded protein that promote a high value of A586. Products below the line, circled in green, had strong dot blot signal but weaker absorbance, which could be a result of improper folding of the β-barrel or a limit of detection in the case of the highest concentration sample. Interestingly, all of the circled outliers are in the pET-22b(+) vector, which contains a C-terminal 6×His-tag and suggests that the green-circled points are not indicative of truncation products. In this context, any of the combinations with a high dot blot concentration would perform reasonably with sufficient optimization of temperature, media, and time, which indicates that the dot blot is a good metric for candidate selection.
Protein expression platform verification
| Protein | Class | Molar mass (kDa) | pIa | Rareb codon% | Vector | Cell strain | Yield (mg L−1) |
|---|---|---|---|---|---|---|---|
| a Calculated from ref. 66. b Determined using the eight codons calculated in Zhang et al.67 | |||||||
| 047A | ELP–GP pentablocks | 65.7 | 4.55 | 1.78 | pGEX-4T-1(H) | C41 (DE3) | 19.6 |
| 047B | ELP–GP pentablocks | 30.5 | 8.87 | 1.75 | pET-22b(+) | C41 (DE3) | 62.3 |
| Catcher | ELP–GP pentablocks | 62.6 | 4.73 | 1.85 | pET-15b | BL21 | 36.6 |
| CC43 | ELP–GP pentablocks | 37.5 | 8.12 | 2.98 | pET-SUMO | Rosetta 2™ (DE3) | 7.2 |
| PPxY | ELP–GP pentablocks | 29.5 | 6.35 | 3.55 | pET-22b(+) | Rosetta 2™ (DE3) | 311.6 |
| Tag | ELP–GP pentablocks | 29.7 | 9.57 | 3.53 | pQE-9 | T7 Express lysY/Iq | 10.1 |
| ZE | ELP–GP pentablocks | 40.1 | 4.66 | 2.79 | pET-22b(+) | C41 (DE3) | 39.98 |
| ZR | ELP–GP pentablocks | 40.8 | 11.74 | 2.79 | pET-SUMO | T7 Express lysY | 14.6 |
| E10 | Y-containing ELP | 13.6 | 7.80 | 2.78 | pET-SUMO | Rosetta 2™ (DE3) | 33.5 |
| E20 | Y-containing ELP | 24.7 | 7.71 | 3.01 | pET-SUMO | Rosetta 2™ (DE3) | 33.3 |
| E40 | Y-containing ELP | 46.9 | 7.60 | 3.14 | pET-SUMO | Tuner (DE3) | 2.2 |
| E80 | Y-containing ELP | 91.3 | 7.46 | 3.21 | pET-15b | Tuner (DE3) | 4.8 |
| hNup50 | hNup | 50.6 | 6.38 | 0 | pET-22b(+) | Rosetta 2™ (DE3) | 104.2 |
| hNup62 | hNup | 53.7 | 5.12 | 0 | pET-SUMO | Rosetta 2™ (DE3) | 3.4 |
| hNup98 | hNup | 96.4 | 6.92 | 0 | pGEX-4T-1(H) | Rosetta 2™ (DE3) | 1.8 |
| mCherry | Fluorescent protein | 31.2 | 6.02 | 4.68 | pET-22b(+) | Tuner (DE3) | 337.3 |
| P4 | Disordered protein | 62.3 | 4.16 | 0 | pET-22b(+) | C41 (DE3) | 21.9 |
DNA and amino acid sequences for all constructs are detailed in section A of the ESI,† and all constructs were cloned into the vector panel detailed above. Once cloned, each panel (thus, one combinatorial expression of a single gene of interest in 60 plasmid/strain combinations) was completed within 7 working days to ensure that the protocol can be a component of an envisioned one-month protocol from purchased gene to purified protein model to enable rapid protein materials synthesis. Across all panels, 83% of wells were successfully transformed and 91% of all vector/cell combinations were successfully tested with at least one replicate; of the most effective examples, 3 panels probed all possible combinations.
Considering the mCherry dot blot concentration vs. A586 results discussed in the dot blot verification, the dot blot concentrations were used as the determining metric of optimal vector/strain combination. As a benchmark, yields (mass of protein per volume of culture) above 15 mg L−1 are defined to be a reasonable protein expression condition for a recombinant protein of interest; of the 17 tested constructs, 10 achieved that metric with at least one vector/strain combination (Fig. 6a). Of these 10, 4 proteins showed yields over 45 mg L−1, which could be expressed without requiring significant (or any) additional optimization. Outside of these particularly high performers, the platform requires a second round of screening to optimize variables such as media formulation, IPTG concentration, expression time or temperature, and oxygenation, so these values are likely a lower limit of the expected yields of these protein materials. It is envisioned that the specifics of this secondary optimization would be protein-dependent, but likely would use 60 mL cultures and monitor growth post-induction to determine high yield conditions. The metric of 15 mg L−1 is chosen because it is expected that a 5 L fermenter-based culture could express at least 100 mg of protein for advanced materials testing after an optimization process that increases yield by at least 33%, as has been seen previously with optimizations of IPTG concentration, post-induction temperature, and post-induction time.68 In addition to the high yields, an additional 3 constructs achieved yields between 5 and 15 mg L−1, which, though less desirable, are likely able to be optimized to reach reasonable expression levels. Of the four constructs (E40, E80, hNup62, and hNup98) with very low (<5 mg L−1) yields, E40 did show faint dots on the dot blot, which could be used as a starting point for testing alternate expression systems, such as cold shock expression vectors like pCOLD,69 because it is expected that E40 exhibits molecular-weight-dependent lower-critical solution temperature (LCST) behavior. For these low-expressing proteins, panels containing alternate vectors that include different solubility tags or promoter systems will be required to enable robust expression. To extend this platform to functional globular proteins, such as enzymes, different vectors with tighter regulation or alternative tags can be incorporated into the vector panel. Additionally, specific colorimetric assays could be developed for each protein, and the combined activity and titer can be used to select the best expression conditions. Generally, as shown in Fig. 6b, this process is robust across a wide span of pI and Mn, though high molar mass proteins have difficulty, as expected. Nonetheless, with a relatively high success rate and fast return of results, this platform has been validated as a vector/strain screening tool.
 | ||
| Fig. 6 a) Histogram of maximum yield of protein of interest as determined by the dot blot. Colors of the bars reflect the yield sorting criteria discussed in the text: very high (dark green), high (green), acceptable (yellow) and low (red). b) Scatter plot of molar mass vs. pI for the protein panel, with colors reflecting the protein yield. | ||
With the development of a high-throughput platform to scan protein expression conditions, data-driven approaches can be used to begin to establish guidance for machinery used in expression. These initial results suggest a demand for collection of data on wider varieties of protein materials to span the physical and chemical space and to adequately describe the complexity of E. coli protein expression. Although simple correlations were attempted to be established between physical properties, the intricacy of the protein expression system requires more advanced analytics and large, unbiased data sets to achieve enhanced understanding of the problem. Along this same line, translation of categorical descriptors, such as vector, cell strain, and gene sequence, into features for machine learning and other data-driven statistical approaches will continue to require refinement. With these challenges in mind, it is important to note that the establishment of experimental techniques that are able to supply organized databases is one of the key technological obstacles toward achieving these goals. Alone, this work has generated over one thousand unique data points (60 vector/strain combinations for 17 proteins of interest = 1020 test conditions) toward this grand challenge with the ability to continue to produce 66 combinations per week by a single worker, largely dictated by the time necessary for E. coli growth as well as the outlined replication strategy (a Gantt chart for the process is included in the ESI,† section I). These data are stored in a database structure, keeping track of the protein, vector, strain, and data collected for all replicates (OD600 values, overall protein concentration, and concentration of protein of interest), which can be readily assembled using an automated MATLAB script that is available in the ESI,† section F. The throughput could be further enhanced by modifying the replication strategy and/or building out more incubators and robots to enable more runs to be run in parallel; theoretically, with unlimited incubator space, a run could commence every 4 hours, allowing up to 12 runs (792 combinations) per week. In comparison to many other existing high-throughput approaches, this system reflects the upstream batch production very closely. Most current high-throughput approaches generate new targets via synthetic biology70,71 or screen large libraries,72 which would be highly complementary to this new approach. By making this system widely available to the protein materials community with limited initial investment (a complete cost table for the platform is available in the ESI,† section J), this system represents a first step toward rapid protein materials design cycles.
Conclusions
Using a low-cost liquid handling robot and open-source software, a modular, high-throughput platform for E. coli vector and strain selection was developed and validated to optimize the expression of protein materials. A simple, robust cloning strategy was used to clone genes of interest into a small library of DNA vectors commonly used for biomaterial expression, including a suite of inducible promoter systems and solubility tags. Once cloned, the genes were transformed into eleven different E. coli strains to form a combinatorial expression library that was assessed in a well-plate format. Protocols using a simple automated camera were developed to measure the OD600 and verified such that the platform can be operated without a spectrophotometer. Post-expression, the yield of total protein, via Bradford assay, and of the protein of interest, via dot blot, were quantified to identify promising strain-plasmid combinations that can be further optimized with changes to media, temperature, and time. This expression optimization protocol was validated first with mCherry and then extended to a panel of 17 protein materials. Of these, expression yields >15 mg L−1 were attained for 10 of the proteins. Reasonable expression conditions can now reliably be found an order of magnitude faster, and conditions for previously un-expressible proteins have been elucidated in several cases. Compiled data is stored in a database-format to enable further data-driven approaches to optimizing the expression of protein materials moving forward. Together, this work established an efficient and modular approach to overcoming protein materials synthesis challenges, which will expedite continued development and innovation in this growing space.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under contracts CBET-1705923 and DMR-1253306. R. A. B. was supported by São Paulo Research Foundation (FAPESP) grants #2016/10193-9 and #2017/24668-1. We thank Dr. Helen Yao for helpful discussions and supply of purified His-tagged P4.References
- S. Bechtle, S. F. Ang and G. A. Schneider, Biomaterials, 2010, 31, 6378–6385 CrossRef CAS PubMed.
- R. L. DiMarco and S. C. Heilshorn, Adv. Mater., 2012, 24, 3923–3940 CrossRef CAS.
- M. K. Gupta, D. T. Wagner and M. C. Jewett, MRS Bull., 2020, 45, 999–1004 CrossRef CAS.
- Y. J. Yang, A. L. Holmberg and B. D. Olsen, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 549–575 CrossRef CAS.
- J. M. Paloni, X. H. Dong and B. D. Olsen, ACS Sens., 2019, 4, 2869–2878 CrossRef CAS.
- M. Richter, C. Schulenburg, D. Jankowska, T. Heck and G. Faccio, Mater. Today, 2015, 18, 459–467 CrossRef CAS.
- J. Wu, P. Li, C. Dong, H. Jiang, X. Bin, X. Gao, M. Qin, W. Wang, C. Bin and Y. Cao, Nat. Commun., 2018, 9, 620 CrossRef PubMed.
- B. D. Olsen, AIChE J., 2013, 59, 3558–3568 CrossRef CAS.
- H. V. Sureka, A. C. Obermeyer, R. J. Flores and B. D. Olsen, ACS Appl. Mater. Interfaces, 2019, 11, 32354–32365 CrossRef CAS.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- D. Chang, A. Huang and B. D. Olsen, Macromol. Rapid Commun., 2017, 38, 1600449 CrossRef.
- Y. Wang, P. Katyal and J. K. Montclare, Adv. Healthcare Mater., 2019, 8, 1801374 CrossRef PubMed.
- J. J. de Pablo, B. Jones, C. L. Kovacs, V. Ozolins and A. P. Ramirez, Curr. Opin. Solid State Mater. Sci., 2014, 18, 99–117 CrossRef.
- J. J. de Pablo, N. E. Jackson, M. A. Webb, L.-Q. Chen, J. E. Moore, D. Morgan, R. Jacobs, T. Pollock, D. G. Schlom, E. S. Toberer, J. Analytis, I. Dabo, D. M. DeLongchamp, G. A. Fiete, G. M. Grason, G. Hautier, Y. Mo, K. Rajan, E. J. Reed, E. Rodriguez, V. Stevanovic, J. Suntivich, K. Thornton and J.-C. Zhao, npj Comput. Mater., 2019, 5, 41 CrossRef.
- S. Kosuri and G. M. Church, Nat. Methods, 2014, 11, 499–507 CrossRef CAS.
- D. M. Francis and R. Page, Curr. Protoc. Protein Sci., 2010, 61, 5.24.1-25.24.29 Search PubMed.
- F. Kong, L. Yuan, Y. F. Zheng and W. Chen, J. Lab. Autom., 2012, 17, 169–185 CrossRef CAS PubMed.
- N. K. Tripathi and A. Shrivastava, Front. Bioeng. Biotechnol., 2019, 7, 420 CrossRef PubMed.
- S. K. Gupta and P. Shukla, Appl. Microbiol. Biotechnol., 2018, 102, 10457–10468 CrossRef CAS.
- J. Yin, G. Li, X. Ren and G. Herrler, J. Biotechnol., 2007, 127, 335–347 CrossRef CAS PubMed.
- N. C. Tang and A. Chilkoti, Nat. Mater., 2016, 15, 419–424 CrossRef CAS PubMed.
- M. A. DePristo, M. M. Zilversmit and D. L. Hartl, Gene, 2006, 378, 19–30 CrossRef CAS.
- P. M. Sharp and W.-H. Li, Nucleic Acids Res., 1987, 15, 1281–1295 CrossRef CAS PubMed.
- L. R. Cruz-Vera, M. A. Magos-Castro, E. Zamora-Romo and G. Guarneros, Nucleic Acids Res., 2004, 32, 4462–4468 CrossRef CAS.
- N. A. Burgess-Brown, S. Sharma, F. Sobott, C. Loenarz, U. Oppermann and O. Gileadi, Protein Expression Purif., 2008, 59, 94–102 CrossRef CAS.
- C.-S. Goh, N. Lan, S. M. Douglas, B. Wu, N. Echols, A. Smith, D. Milburn, G. T. Montelione, H. Zhao and M. Gerstein, J. Mol. Biol., 2004, 336, 115–130 CrossRef CAS.
- S. Gräslund, P. Nordlund, J. Weigelt, B. M. Hallberg, J. Bray, O. Gileadi, S. Knapp, U. Oppermann, C. Arrowsmith, R. Hui, J. Ming, S. Dhe-Paganon, H.-W. Park, A. Savchenko, A. Yee, A. Edwards, R. Vincentelli, C. Cambillau, R. Kim, S.-H. Kim, Z. Rao, Y. Shi, T. C. Terwilliger, C.-Y. Kim, L.-W. Hung, G. S. Waldo, Y. Peleg, S. Albeck, T. Unger, O. Dym, J. Prilusky, J. L. Sussman, R. C. Stevens, S. A. Lesley, I. A. Wilson, A. Joachimiak, F. Collart, I. Dementieva, M. I. Donnelly, W. H. Eschenfeldt, Y. Kim, L. Stols, R. Wu, M. Zhou, S. K. Burley, J. S. Emtage, J. M. Sauder, D. Thompson, K. Bain, J. Luz, T. Gheyi, F. Zhang, S. Atwell, S. C. Almo, J. B. Bonanno, A. Fiser, S. Swaminathan, F. W. Studier, M. R. Chance, A. Sali, T. B. Acton, R. Xiao, L. Zhao, L. C. Ma, J. F. Hunt, L. Tong, K. Cunningham, M. Inouye, S. Anderson, H. Janjua, R. Shastry, C. K. Ho, D. Wang, H. Wang, M. Jiang, G. T. Montelione, D. I. Stuart, R. J. Owens, S. Daenke, A. Schütz, U. Heinemann, S. Yokoyama, K. Büssow and K. C. Gunsalus, Nat. Methods, 2008, 5, 135–146 CrossRef.
- S. Costa, A. Almeida, A. Castro and L. Domingues, Front. Microbiol., 2014, 5, 63 Search PubMed.
- M. R. Dyson, S. P. Shadbolt, K. J. Vincent, R. L. Perera and J. McCafferty, BMC Biotechnol., 2004, 4, 32 CrossRef.
- F. Saïda, M. Uzan, B. Odaert and F. Bontems, Curr. Protein Pept. Sci., 2006, 7, 47–56 CrossRef.
- E. J. Stewart, F. Åslund and J. Beckwith, EMBO J., 1998, 17, 5543–5550 CrossRef CAS.
- J. Lefebvre, G. Boileau and P. Manjunath, Mol. Hum. Reprod., 2008, 15, 105–114 CrossRef PubMed.
- Y. Xu, A. Yasin, R. Tang, J. M. Scharer, M. Moo-Young and C. P. Chou, Appl. Microbiol. Biotechnol., 2008, 81, 79–87 CrossRef CAS PubMed.
- C. P. Chou, Appl. Microbiol. Biotechnol., 2007, 76, 521–532 CrossRef CAS PubMed.
- S. Gottesman, in Methods in Enzymology, Academic Press, 1990, ch. 11, vol. 185, pp. 119–129 Search PubMed.
- W. Peti and R. Page, Protein Expression Purif., 2007, 51, 1–10 CrossRef CAS.
- R. N. Armstrong, Chem. Res. Toxicol., 1997, 10, 2–18 Search PubMed.
- M. P. Malakhov, M. R. Mattern, O. A. Malakhova, M. Drinker, S. D. Weeks and T. R. Butt, J. Struct. Funct. Genomics, 2004, 5, 75–86 Search PubMed.
- G. L. Rosano, E. S. Morales and E. A. Ceccarelli, Protein Sci., 2019, 28, 1412–1422 CrossRef CAS.
- R. K. C. Knaust and P. Nordlund, Anal. Biochem., 2001, 297, 79–85 CrossRef CAS PubMed.
- H. Nguyen, B. Martinez, N. Oganesyan and R. Kim, J. Struct. Funct. Genomics, 2004, 5, 23–27 CrossRef CAS PubMed.
- R. Vincentelli, S. Canaan, J. Offant, C. Cambillau and C. Bignon, Anal. Biochem., 2005, 346, 77–84 CrossRef CAS.
- R. Vincentelli, A. Cimino, A. Geerlof, A. Kubo, Y. Satou and C. Cambillau, Methods, 2011, 55, 65–72 CrossRef CAS.
- V. Gupta, J. Irimia, I. Pau and A. Rodríguez-Patón, ACS Synth. Biol., 2017, 6, 1230–1232 CrossRef CAS.
- E. J. Chory, D. W. Gretton, E. A. DeBenedictis and K. M. Esvelt, Mol. Syst. Biol., 2021, 17, e9942 CrossRef PubMed.
- J. Konczal and C. H. Gray, Protein Expression Purif., 2017, 133, 160–169 CrossRef CAS.
- N. J. Saez, H. Nozach, M. Blemont and R. Vincentelli, J. Visualized Exp., 2014, e51464, DOI:10.3791/51464.
- B. M. Seifried, J. Cao and B. D. Olsen, Bioconjugate Chem., 2018, 29, 1876–1884 CrossRef CAS.
- M. J. Glassman, J. Chan and B. D. Olsen, Adv. Funct. Mater., 2013, 23, 1182–1193 CrossRef CAS.
- A. Rao, H. Yao and B. D. Olsen, Phys. Rev. Res., 2020, 2, 043369 CrossRef CAS.
- QIAexpress® Detection and Assay Handbook, 2015, https://www.qiagen.com/us/resources/resourcedetail?id=a8d20bf6-b88c-436b-a7be-de66b5dd70fd&lang=en Search PubMed.
- M. Kim, W. G. Chen, B. S. Souza and B. D. Olsen, Mol. Syst. Des. Eng., 2017, 2, 149–158 RSC.
- A. Huang, H. Yao and B. D. Olsen, Soft Matter, 2019, 15, 7350–7359 RSC.
- E. A. Woestenenk, M. Hammarström, S. van den Berg, T. Härd and H. Berglund, J. Struct. Funct. Genomics, 2004, 5, 217–229 CrossRef CAS PubMed.
- P. Daegelen, F. W. Studier, R. E. Lenski, S. Cure and J. F. Kim, J. Mol. Biol., 2009, 394, 634–643 CrossRef CAS PubMed.
- G. L. Rosano and E. A. Ceccarelli, Front. Microbiol., 2014, 5, 341 Search PubMed.
- L. Briand, G. Marcion, A. Kriznik, J. M. Heydel, Y. Artur, C. Garrido, R. Seigneuric and F. Neiers, Sci. Rep., 2016, 6, 33037 CrossRef CAS.
- D. Hartinger, S. Heinl, H. E. Schwartz, R. Grabherr, G. Schatzmayr, D. Haltrich and W.-D. Moll, Microb. Cell Fact., 2010, 9, 62 CrossRef PubMed.
- B. Miroux and J. E. Walker, J. Mol. Biol., 1996, 260, 289–298 CrossRef CAS PubMed.
- S. Y. Teow, S. A. Mualif, T. C. Omar, C. Y. Wei, N. M. Yusoff and S. A. Ali, BMC Biotechnol., 2013, 13, 107 CrossRef.
- I. Sermadiras, J. Revell, J. E. Linley, A. Sandercock and P. Ravn, PLoS One, 2013, 8, e83202 CrossRef PubMed.
- J. Granhøj, H. Dimke and P. Svenningsen, Sci. Rep., 2019, 9, 4118 CrossRef.
- J. A. Jessee and F. R. Bloom, US Pat., 4981797, 1988 Search PubMed.
- C. E. Mills, E. Ding and B. Olsen, Ind. Eng. Chem. Res., 2019, 58, 11698–11709 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- Expasy and Swiss Institute of Bioinformatics, Compute pI/Mw, https://web.expasy.org/compute_pi/, (accessed August 2021).
- S. Zhang, G. Zubay and E. Goldman, Gene, 1991, 105, 61–72 CrossRef CAS.
- M. Gutiérrez-González, C. Farías, S. Tello, D. Pérez-Etcheverry, A. Romero, R. Zúñiga, C. H. Ribeiro, C. Lorenzo-Ferreiro and M. C. Molina, Sci. Rep., 2019, 9, 16850 CrossRef PubMed.
- G. Qing, L.-C. Ma, A. Khorchid, G. V. T. Swapna, T. K. Mal, M. M. Takayama, B. Xia, S. Phadtare, H. Ke, T. Acton, G. T. Montelione, M. Ikura and M. Inouye, Nat. Biotechnol., 2004, 22, 877–882 CrossRef CAS.
- N. Tenhaef, R. Stella, J. Frunzke and S. Noack, ACS Synth. Biol., 2021, 10, 589–599 CrossRef CAS PubMed.
- K. Iwai, M. Wehrs, M. Garber, J. Sustarich, L. Washburn, Z. Costello, P. W. Kim, D. Ando, W. R. Gaillard, N. J. Hillson, P. D. Adams, A. Mukhopadhyay, H. Garcia Martin and A. K. Singh, Microsyst. Nanoeng., 2022, 8, 31 CrossRef CAS PubMed.
- N. Furtmann, M. Schneider, N. Spindler, B. Steinmann, Z. Li, I. Focken, J. Meyer, D. Dimova, K. Kroll, W. D. Leuschner, A. Debeaumont, M. Mathieu, C. Lange, W. Dittrich, J. Kruip, T. Schmidt and J. Birkenfeld, mAbs, 2021, 13, 1955433 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00150k |
| This journal is © The Royal Society of Chemistry 2023 |