
Novel indolyl 1,2,4-triazole derivatives as potential anti-proliferative agents: in silico studies, synthesis, and biological evaluation†
Sarah A.
Ghobish
 a,
Khaled O.
Mohamed
bc,
Nahla
Farag
*a and
Doaa B.
Farag
*a
a,
Khaled O.
Mohamed
bc,
Nahla
Farag
*a and
Doaa B.
Farag
*a
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Misr International University, Cairo, Egypt. E-mail: nahla.farag@miuegypt.edu.eg; doaa.boshra@miuegypt.edu.eg
bPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo, Egypt
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Sinai University (Arish branch), El Arish, Egypt
First published on 24th November 2023
Abstract
A new series of indolyl 1,2,4-triazole scaffolds was designed, synthesised, and biologically evaluated for their inhibitory activity against both CDK4 and CDK6. The results ranged from 0.049 μM to 3.031 μM on CDK4 and from 0.075 μM to 1.11 μM on CDK6 when compared to staurosporine, with IC50 values of 1.027 and 0.402 μM, respectively. Moreover, all compounds were tested for their cytotoxicity against two breast cancer cell lines, MCF-7 and MDA-MB-231. All of the synthesised compounds showed promising anti-proliferative activity, with two compounds Vf (IC50 = 2.91 and 1.914 μM, respectively) and Vg (IC50 = 0.891 and 3.479 μM, respectively) having potent cytotoxic activity in comparison to the reference staurosporine (IC50 = 3.144 and 4.385 μM, respectively). Vf and Vg were also found to significantly induce apoptosis to 45.33% and 37.26% (control = 1.91%) where Vf arrested the cell cycle at the S phase while Vg arrested the cycle at the G0/G1 phase. The binding mode and interactions of all compounds were studied and found to mimic those of the FDA approved CDK4/6 inhibitor palbociclib that was used as a reference throughout the study.
1. Introduction
Breast cancer is the world's most extensive and prevailing malignant tumour. Approximately 7.8 million women were diagnosed with breast cancer by the end of 2020 considering the 5 preceding years.1 The classic treatment methods of breast cancer such as hormonal therapy, chemotherapy, radio therapy and surgery are still being used as treatment options to this day.2 However, these are faced with great side effects and high risks, and the search for newer treatment techniques that are cheaper and more targeted is necessary.3–5Novel target enzymes such as the CDKs (cyclin dependent kinases) became of great interest and priority. There are 21 members in the CDK family, seven of which have a direct role in cell cycle progression.6 CDK1, CDK2, CDK4/6, CDK5, CDK7, CDK8, and CDK9 are believed to be good targets for drug discovery, but CDK4/6 in particular is hyperactivated in a variety of malignant human tumours, mainly breast cancer, due to the overexpression of a positive regulator called cyclin D.7 Evidently, cyclin D forms a complex with CDK4 at the G1 phase of the cell cycle, making CDK4 a key cell cycle regulator responsible for the progression from the G1 phase to the S phase.8,9 Experimental evidence suggested that inhibitors of this kinase can be promising as non-toxic and effective agents against breast cancer.10,11 Early generations of CDK inhibitors were mildly effective but also nonselective and inhibited many kinases, and that led to severe toxicities and a too narrow therapeutic window.12 Therefore, developing new potential inhibitors targeting CDK4/6 was the next step. Currently, CDK4/6 inhibitors are being used in the first line treatment of metastatic breast cancer; however, only three are FDA approved: palbociclib, ribociclib and abemaciclib (Fig. 1).13
 | ||
| Fig. 1 The rationale for the design of the new indolyl 1,2,4-triazole derivatives as CDK4/6 inhibitors. | ||
With the aim to expand the treatment possibilities against breast cancer, a new scaffold as CDK4/6 inhibitor was designed and evaluated with the aid of a new valid pharmacophore model based on CDK4/6 inhibitors of known reported activities. Carefully studying the clinically approved FDA drugs that are currently used against breast cancer (Fig. 2), the importance of the heterocyclic system was noticed and that immensely inspired the design of indolyl 1,2,4-triazole derivatives as a new scaffold targeting the CDK4/6 receptor. The choice of an indole/triazole moiety was supported by extensive research into heterocyclic compounds and their role in cancer therapy.14–16 Their pharmacophore was found to be built out of a head (the heterocyclic moiety) which is essential for binding in the hydrophobic pocket of the receptor, a linker that forms a necessary hydrogen bonding interaction at the hinge region and a tail that extends to the solvent region.17–22 We adopted this design in the new indole derivatives in which we also decided on a head, linker and a tail moiety.
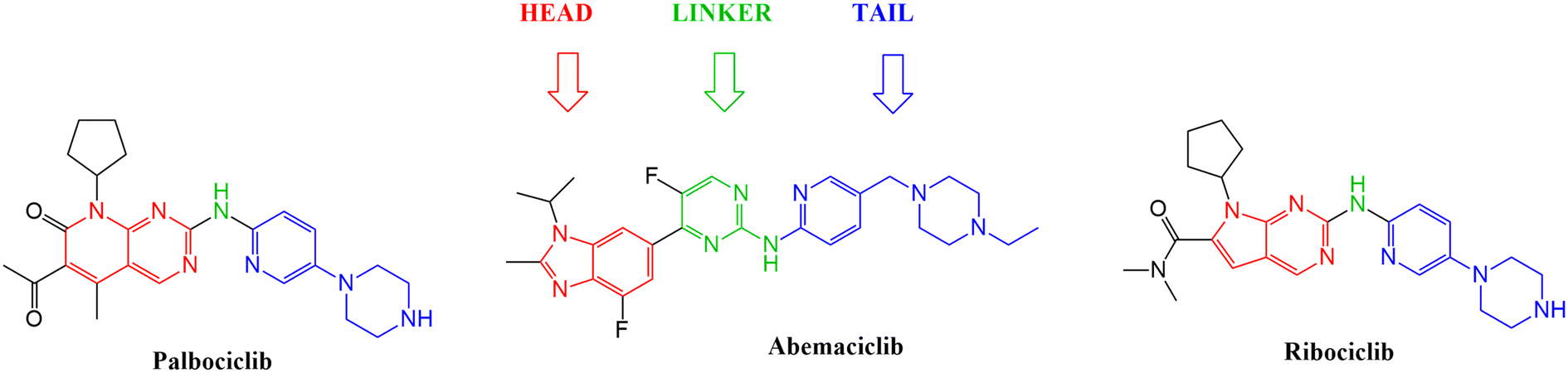 | ||
| Fig. 2 Structures of the three FDA approved CDK4/6 inhibitors highlighting the importance of the heterocyclic system, the linker and the tail moiety. | ||
The proposed compounds were mapped into the pharmacophore model, then docked into the target's binding site to confirm their mode of action and compare their interactions with CDK4/6 to those of the approved agents. Knowing that breast cancer cell (MCF-7 and MDA-MB-231) growth is dependent on CDK4, all of the synthesized derivatives were examined for their potential activity against MCF-7 and MDA-MB-231 breast cancer cell lines and their inhibitory effect towards both CDK4 and CDK6.23 The most potent compounds were further explored for their effect on cell cycle progression, apoptosis induction possibility and finally their cytotoxicity against normal human cells.
2. Molecular modelling studies
2.1. Advanced pharmacophore model generation
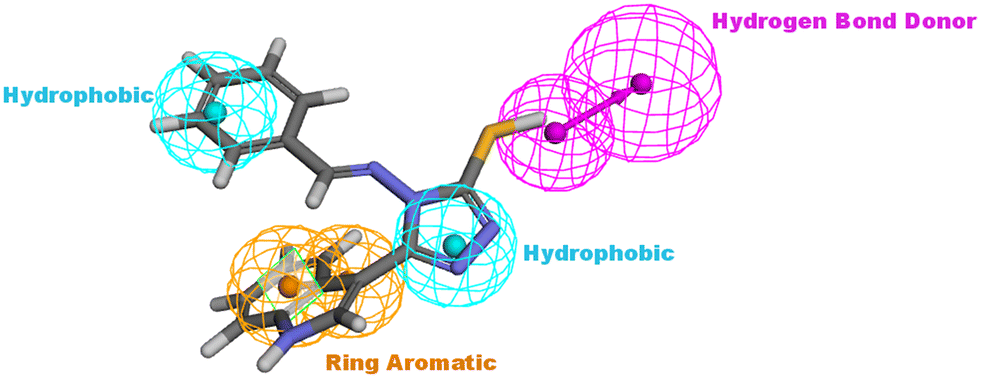
The design process of the new indolyl 1,2,4-triazole derivatives started by constructing a 3D QSAR pharmacophore model using Accelrys Discovery Studio V4.5. First, a data set of 41 pyrido[2,3-d]pyrimidin-7-one derivatives (CDK4/6 inhibitors) were collected from the literature with reported activities (IC50) ranging from 0.0007 to 3.3 μM.24 The ligands that were used for the modelling study were sketched in 3D using the Discovery Studio program and were prepared using the ligand preparation tool. The data set was randomly divided into 21 compounds as the training set (provided in the ESI†) and 20 compounds as a test set for model validation. The training set was then subjected to a feature mapping protocol defining the pharmacophore features, which turned out to be hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), hydrophobic moiety (HYP), positive ionizable (PI) groups and aromatic rings (AR). Ten models (hypothesis) were generated and validated for their statistical significance and accuracy of prediction based on the cost difference, RMS and correlation coefficient (Table 2). Hypothesis 1 proved to be the most valid model with the highest cost difference of 95.74, indicating statistical significance and prediction power of over 90%, RMS value of 1.35 and correlation coefficient of 0.92 giving confidence in the model's prediction of the activity of the newly designed compounds. The model had four features (hydrogen bond donor (HBD), 2 × hydrophobic (HYP) and ring aromatic (RA)) (Fig. 3 and Table 1).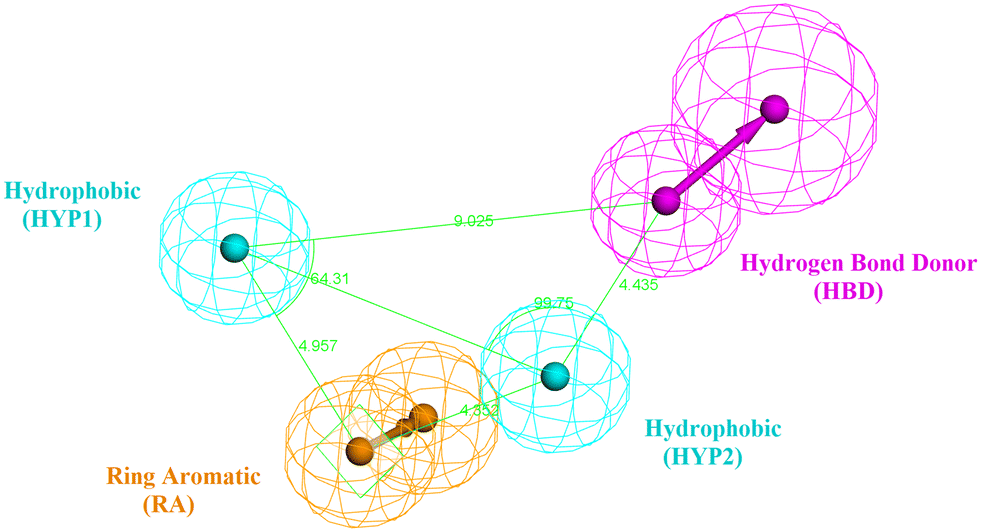 | ||
| Fig. 3 The best generated pharmacophore model illustrating the distances and angles between features. | ||
| Features | Distance (Å) |
|---|---|
| HYP1–HBD | 9.025 |
| HYP1–RA | 4.957 |
| RA–HYP2 | 4.352 |
| HYP2–HBD | 4.435 |
| HYP1–HYP2 | 7.145 |
| Angle (Å) | |
|---|---|
| RA–HYP1–HYP2 | 36.85 |
| HYP1–HYP2–HBD | 99.75 |
| RA–HYP2–HBD | 137.85 |
| Hypothesis | Cost difference | Maximum fit | Total cost | RMS | Correlation coefficient |
|---|---|---|---|---|---|
| 1 | 95.74 | 5.66 | 84.45 | 1.35 | 0.92 |
| 2 | 92.07 | 6.37 | 88.12 | 1.44 | 0.91 |
| 3 | 90.31 | 6.46 | 89.87 | 1.56 | 0.89 |
| 4 | 89.64 | 5.35 | 90.55 | 1.53 | 0.89 |
| 5 | 86.59 | 5.14 | 93.59 | 1.54 | 0.89 |
| 6 | 86.15 | 5.14 | 94.03 | 1.63 | 0.88 |
| 7 | 84.37 | 5.86 | 95.81 | 1.71 | 0.87 |
| 8 | 83.78 | 4.89 | 96.41 | 1.68 | 0.87 |
| 9 | 83.47 | 5.78 | 96.72 | 1.67 | 0.87 |
| 10 | 82.98 | 5 | 97.2 | 1.71 | 0.87 |
2.2. Ligand pharmacophore mapping of the FDA approved drugs as CDK4/6 inhibitors
The generated pharmacophore model was then utilized for the ligand pharmacophore mapping protocol using Accelrys Discovery Studio V4.5 software. To validate the model, the three FDA approved drugs, palbociclib, abemaciclib and ribociclib, were mapped into the generated model where the mapping of abemaciclib showed the fitting of the phenyl moiety of the benzimidazole into the ring aromatic (RA) feature. The isopropyl and the fluoro moiety of pyrimidine fitted into 2 hydrophobic (HYP) features and the NH linker between the pyridine and pyrimidine moieties fitted into the hydrogen bond donor (HBD) feature of the pharmacophore model with fit value = 4.19214 and estimated activity = 0.0572476 μM (Fig. 4).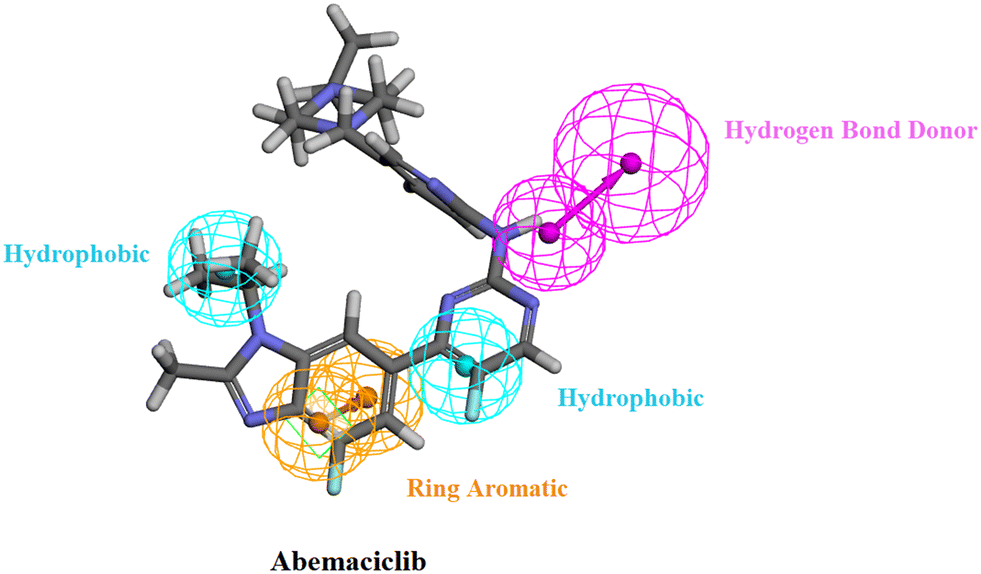 | ||
| Fig. 4 Abemaciclib mapped against the generated pharmacophore model with fit value = 4.19214 and estimated activity = 0.0572476 μM. | ||
2.3. Docking study
Searching the Protein Data Bank (PDB) revealed that the crystal structure of CDK4 with its inhibitor is not yet reported. According to relevant reports, there is a high degree of structural homology between CDK4 and CDK6.25,26 Thus, the crystal structure of CDK6 with its inhibitor palbociclib (PDB 5L2I) was downloaded from the PDB and used throughout the docking studies.27 Accelrys Discovery Studio V4.5 program was used to perform the docking of palbociclib into the target receptor CDK6. To validate the docking protocol, self-docking of the co-crystallised ligand (LQQ900) was performed, and the results were found to replicate those reported in the literature.20,21,26 The CDOCKER interaction energy of the reference crystallised ligand was −55.1849 kcal mol−1 and was found to form 4 hydrogen bonds mainly with the key amino acids of the ATP pocket of CDK6 (ASP163 and VAL101) and unconventional hydrogen bonds with amino acid residues of ASP104 and GLU99. The planar regions formed hydrophobic interactions in the hydrophobic pocket of amino acid residues ILE19, VAL27, ALA41, VAL77, PHE98, LEU152 and ALA162 as shown in Fig. 5. The self-docking validation process was further confirmed by the low RMSD of only 1.093 Å between the docked pose and the crystallised ligand (Fig. 6).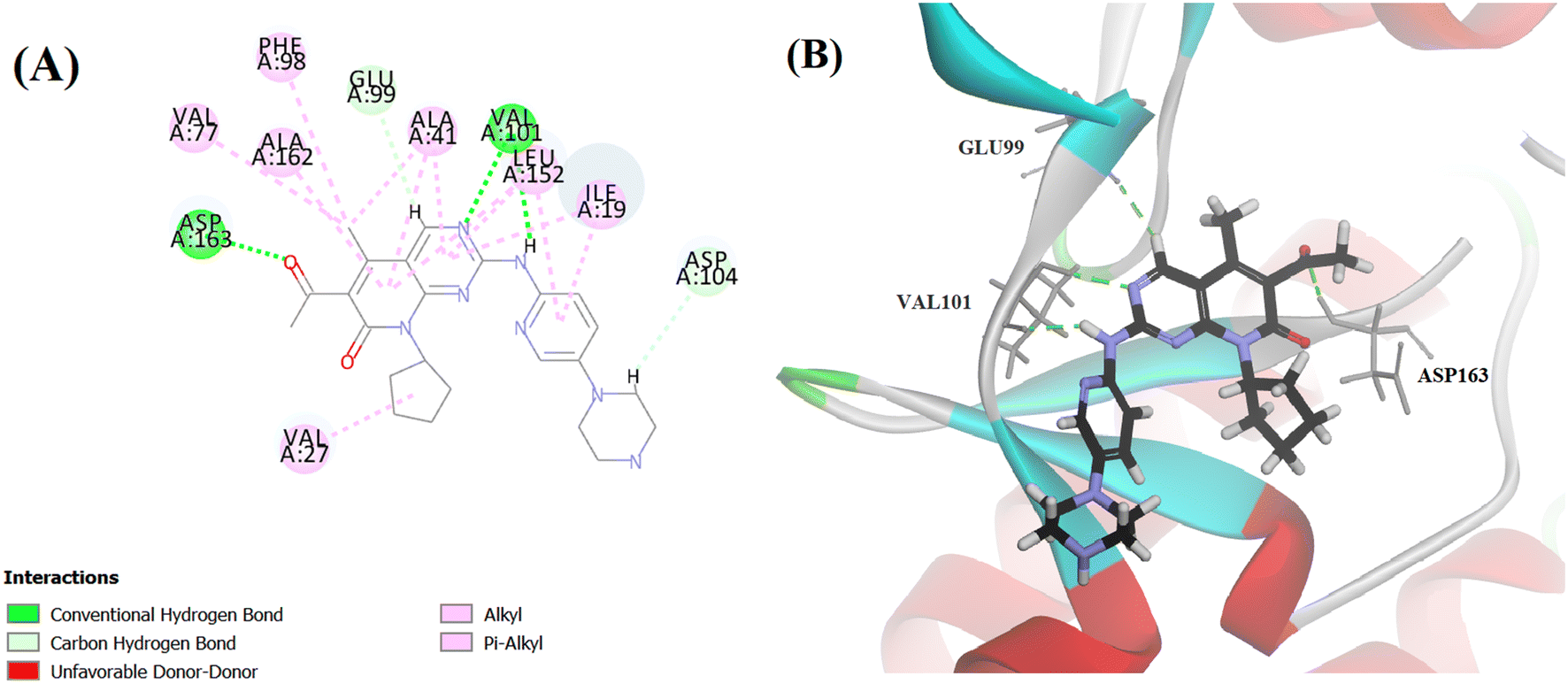 | ||
| Fig. 5 (A) 2D image of palbociclib displaying all interactions with the receptor CDK6. (B) 3D image displaying only the hydrogen bonds (represented by the green dotted lines) between palbociclib and amino acids GLU99, VAL101 and ASP163. | ||
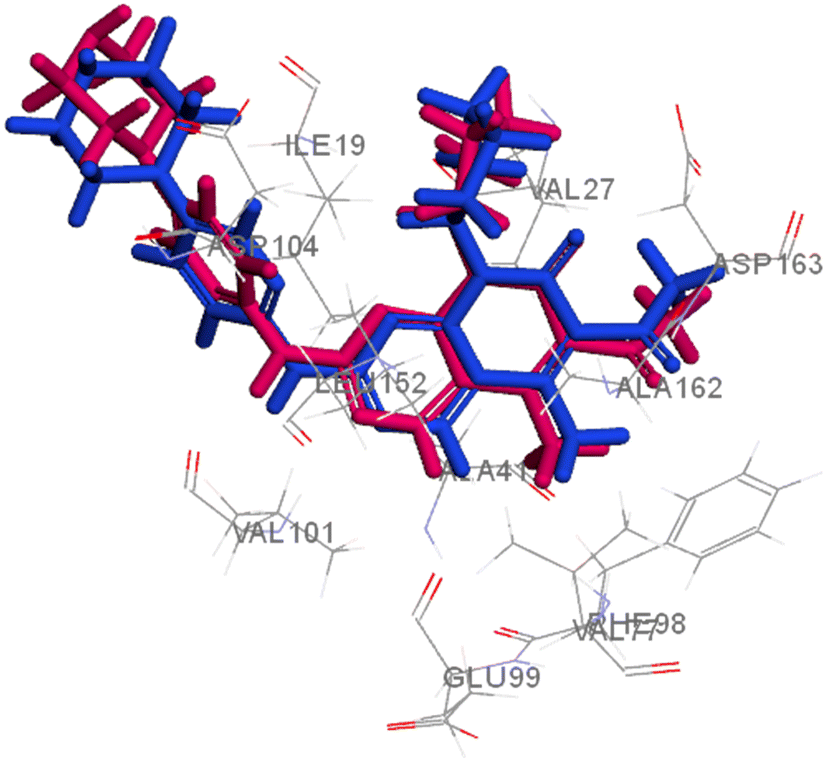 | ||
| Fig. 6 3D representation for the crystalised ligand (magenta) superimposed on the docked pose (blue) in the target receptor CDK6 binding site (RMSD = 1.093 Å). | ||
2.4. Design of the proposed derivatives
After studying the pharmacophore model and inspired by the FDA approved drugs' structural features, 4-(benzylideneamino)-5-(1H-indol-3-yl)-4H-1,2,4-triazol-3-thiol Va was designed in order to mimic the chemical and geometrical features necessary to target the CDK4/6 enzyme with potential cytotoxic activity. The pharmacophore and the docking study of the three FDA approved drugs highlighted the presence of the heterocyclic system which was mirrored in our design by an indole moiety. In addition, the linker that forms a hydrogen bond with the target receptor was mimicked in our design by the triazole part. Furthermore, a benzylideneamino moiety was added and considered as the tail that forms the hydrophobic interaction with the receptor at the solvent region (Fig. 7).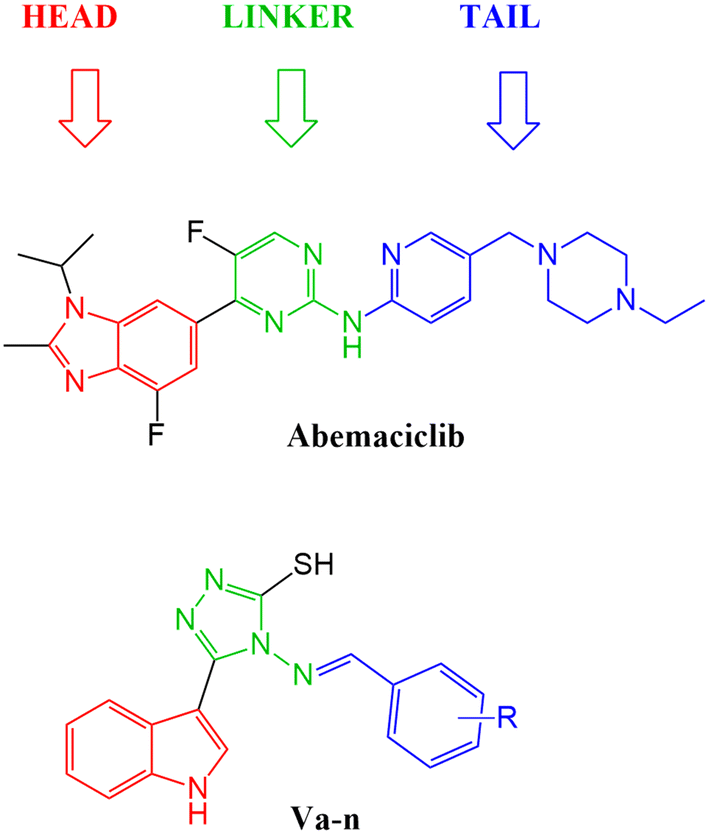 | ||
| Fig. 7 Illustration of abemaciclib's essential features that were mimicked in the design of the new indolyl 1,2,4-triazole derivatives Va–n. | ||
The proposed structure Va was successfully mapped into the generated 3D QSAR model in which it fitted into all the features, giving a comparably good fit value of 3.85023 and an estimated IC50 of 0.125798 μM (Fig. 8). Following that, Va was docked into the binding site of CDK6 (PDB code 5L2I). The docking results for Va showed a CDOCKER interaction energy score of −39.674 kcal mol−1. The binding mode of Va also agreed with that of the reference, forming the essential hydrogen bonds with ASP104 and GLN149 and forming hydrophobic interactions with the hydrophobic pocket's amino acid residues ALA41, VAL77, PHE98, LEU152 and ALA162 (Fig. 9).
 | ||
| Fig. 8 Illustration of Va mapped against the generated pharmacophore model with fit value = 3.75926 and estimated activity = 0.125798 μM. | ||
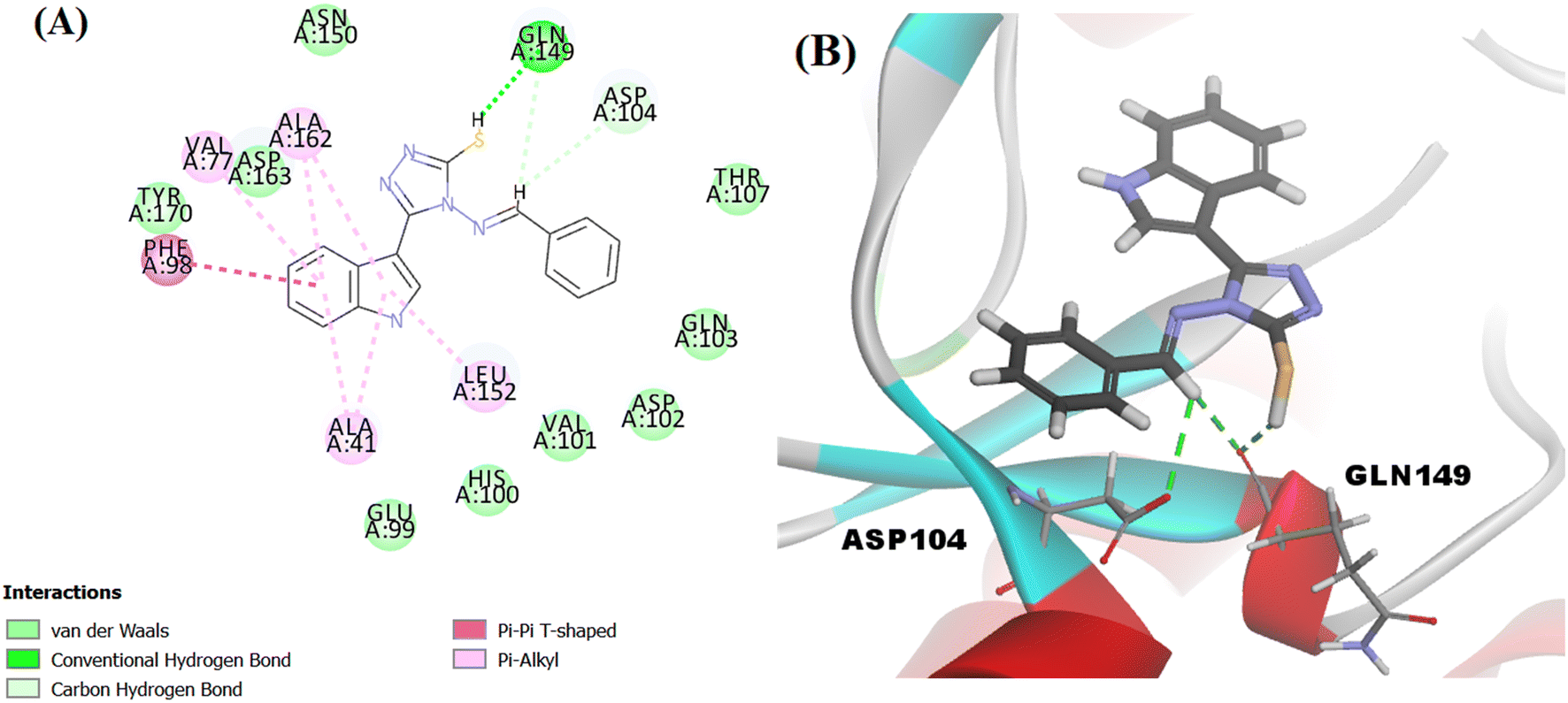 | ||
| Fig. 9 (A) 2D image of Va displaying all interactions with the receptor CDK6. (B) 3D image displaying only the hydrogen bonds (represented by the green dotted lines) between Va and amino acids ASP104 and GLN149. | ||
Based on the results of the previous mapping and docking studies, a series of 14 novel indolyl 1,2,4-triazole derivatives (Va–n) with various bio-isosteric substituents at the benzylidene moiety was designed, mapped into the generated 3D QSAR model and docked into the receptor's binding site. The obtained results of fit values and estimated IC50 were comparable to those of the FDA drugs and the docking of all the proposed compounds (Va–n) achieved competitive scores resembling a similar mode of action and binding patterns to those of the FDA approved drug (palbociclib) that was used as a reference. All of the obtained results from the molecular modelling study (collected and illustrated in Table 3) encouraged the synthesis of the proposed compounds and their biological evaluation on breast cancer cell lines, cell cycle progression, apoptosis induction and CDK4/6 inhibition.
| Compound | CDOCKER interaction energy (kcal mol−1) | Fit value | Estimated activity (μM) |
|---|---|---|---|
| Va | −39.674 | 3.75926 | 0.125798 |
| Vb | −43.3895 | 4.02751 | 0.109154 |
| Vc | −44.1266 | 5.59309 | 0.121975 |
| Vd | −40.5691 | 3.85111 | 0.129805 |
| Ve | −41.0798 | 4.07462 | 0.108881 |
| Vf | −43.5299 | 6.06381 | 0.111152 |
| Vg | −42.8016 | 6.98151 | 0.108747 |
| Vh | −45.1012 | 5.35718 | 0.111912 |
| Vi | −43.6843 | 8.97937 | 0.0787683 |
| Vj | −41.6258 | 4.40196 | 0.117547 |
| Vk | −42.4303 | 4.66412 | 0.116805 |
| Vl | −41.2136 | 3.9244 | 0.124244 |
| Vm | −47.331 | 6.46826 | 0.118933 |
| Vn | −42.0286 | 5.87994 | 0.127926 |
| Palbociclib | −55.1849 | 5.30676 | 0.00439687 |
| Abemaciclib | −53.1618 | 4.19214 | 0.0572476 |
| Ribociclib | −51.5638 | 5.34846 | 0.0039943 |
3. Results and discussion
3.1. Chemistry
The title compounds were synthesised as illustrated in Scheme 1. 1H-Indole-3-carbohydrazide II was obtained through the reaction of hydrazine hydrate with ethyl-1H-indole-3-carboxylate I.28,29 Reaction of 1H-indole-3-carbohydrazide II with CS2/KOH gave the corresponding potassium thiocarbamate salt 2-(1H-indole-3-carbonyl)hydrazinecarbodithioate III.29–31 The intermediate salt III was reacted with hydrazine hydrate to obtain the cyclized triazole 4-amino-3-(1H-indol-3-yl)-1H-1,2,4-triazole-5(4H)-thione IV, followed by the condensation of the amino group with different aromatic aldehydes in the presence of glacial acetic acid as a catalyst to afford the 4-(benzylideneamino)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol derivatives Va–n.28–35 The structures of all the derivatives were confirmed based on the investigation of their analytical and spectral data. All series showed IR signals at 3390–3201 cm−1 for the NH groups in addition to signals at 3097–3001 cm−1 for the aromatic CH groups and signals at 1658–1573 cm−1 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety. Also, the 1H NMR spectra of all the derivatives showed signals at δ = 11.27–11.76 ppm for the indole-NH group, peaks at δ = 11.73–14.05 ppm for the SH group and a range of aromatic protons at δ = 6.3–8.5 ppm. Furthermore, all the derivatives revealed chemical shifts at δ = 8.22–9.90 ppm corresponding to the NCH moiety, confirming its presence in the E stereochemical configuration.36,37 The structures of the compounds were also further confirmed by 13C NMR in which they shared the same important peaks at δ = 147.13–161.45 for the CN moiety as well as signals around δ = 146.88–166.68 for C5 of the triazole linker moiety.
N moiety. Also, the 1H NMR spectra of all the derivatives showed signals at δ = 11.27–11.76 ppm for the indole-NH group, peaks at δ = 11.73–14.05 ppm for the SH group and a range of aromatic protons at δ = 6.3–8.5 ppm. Furthermore, all the derivatives revealed chemical shifts at δ = 8.22–9.90 ppm corresponding to the NCH moiety, confirming its presence in the E stereochemical configuration.36,37 The structures of the compounds were also further confirmed by 13C NMR in which they shared the same important peaks at δ = 147.13–161.45 for the CN moiety as well as signals around δ = 146.88–166.68 for C5 of the triazole linker moiety.
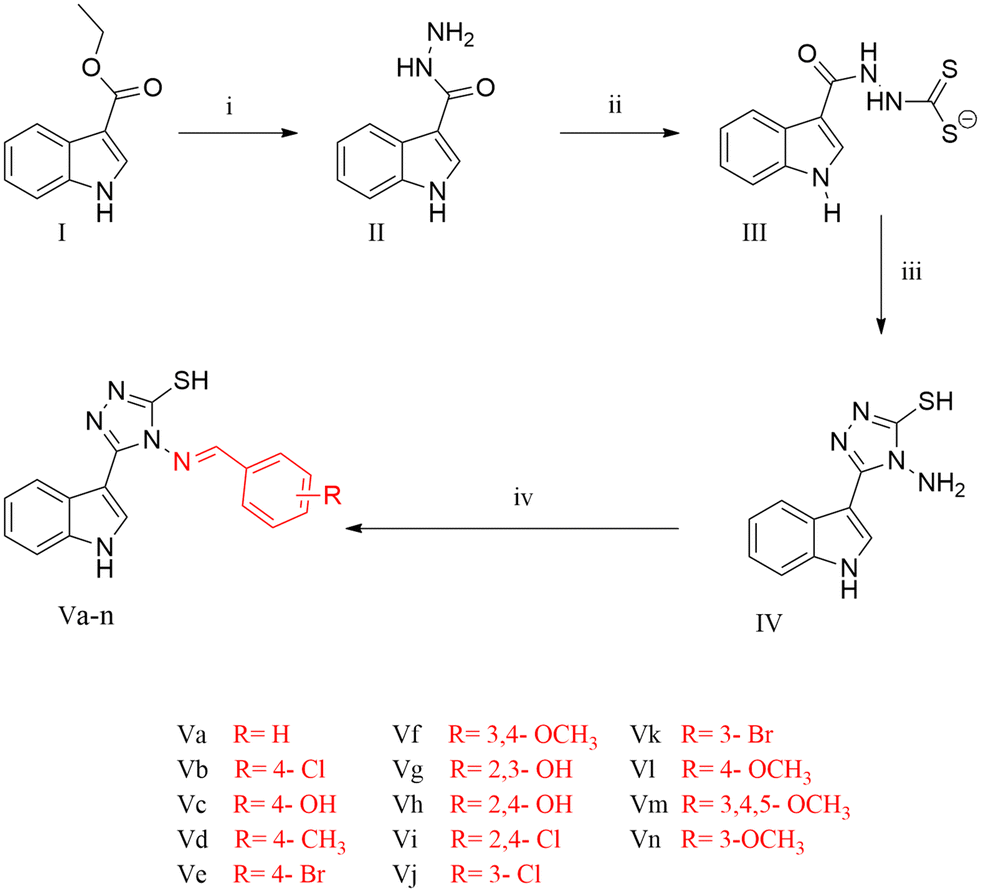 | ||
| Scheme 1 Synthesis of the targeted compounds (Va–n). Reagents and conditions: (i) hydrazine hydrate (80%), ethanol, reflux for 3 h; (ii) CS2, KOH, ethanol, stirred in room temperature for 12 h; (iii) hydrazine hydrate (80%), H2O, reflux for 1 h, followed by neutralization with conc. HCl; (iv) aromatic aldehydes, ethanol, glacial acetic acid, reflux for 5 h. | ||
3.2. Biological activity
| Compound | CDK4 IC50 (μM) | CDK6 IC50 (μM) |
|---|---|---|
| Va | 0.662 ± 0.039 | 0.577 ± 0.022 |
| Vb | 0.056 ± 0.003 | 0.235 ± 0.009 |
| Vc | 0.227 ± 0.013 | 0.817 ± 0.032 |
| Vd | 0.126 ± 0.007 | 0.075 ± 0.003 |
| Ve | 0.162 ± 0.009 | 0.462 ± 0.018 |
| Vf | 0.32 ± 0.016 | 0.198 ± 0.009 |
| Vg | 3.031 ± 0.154 | 0.77 ± 0.034 |
| Vh | 0.527 ± 0.031 | 0.095 ± 0.004 |
| Vi | 1.337 ± 0.078 | 0.271 ± 0.011 |
| Vj | 0.882 ± 0.052 | 1.11 ± 0.043 |
| Vk | 0.049 ± 0.003 | 0.146 ± 0.006 |
| Vl | 0.14 ± 0.008 | 0.574 ± 0.022 |
| Vm | 0.073 ± 0.004 | 0.2 ± 0.008 |
| Vn | 0.297 ± 0.017 | 1.108 ± 0.043 |
| Staurosporine | 1.027 ± 0.062 | 0.402 ± 0.018 |
| Abemaciclib | — | 0.026 ± 0.001 |
| Compound | R | IC50 (μM) | |
|---|---|---|---|
| MCF-7 | MDA-MB-231 | ||
| Va | H | 22.55 ± 1.11 | 13.98 ± 0.87 |
| Vb | 4-Cl | 10.77 ± 0.53 | 81.46 ± 5.04 |
| Vc | 4-OH | 12.79 ± 0.63 | 2.053 ± 0.13 |
| Vd | 4-CH3 | 36.4 ± 1.8 | 26.07 ± 1.61 |
| Ve | 4-Br | 9.241 ± 0.46 | 8.796 ± 0.54 |
| Vf | 3,4-OCH3 | 2.91 ± 0.14 | 1.914 ± 0.12 |
| Vg | 2,3-OH | 0.891 ± 0.04 | 3.479 ± 0.22 |
| Vh | 2,4-OH | 17.82 ± 0.88 | 28.18 ± 1.74 |
| Vi | 2,4-Cl | 27.9 ± 1.38 | 23.06 ± 1.43 |
| Vj | 3-Cl | 11.73 ± 0.58 | 2.799 ± 0.17 |
| Vk | 3-Br | 1.131 ± 0.06 | 5.398 ± 0.33 |
| Vl | 4-OCH3 | 7.677 ± 0.38 | 11.44 ± 0.71 |
| Vm | 3,4,5-OCH3 | 21.38 ± 1.06 | 14.75 ± 0.91 |
| Vn | 3-OCH3 | 4.636 ± 0.23 | 2.24 ± 0.14 |
| Staurosporine | — | 3.144 ± 0.16 | 4.385 ± 0.27 |
| Palbociclib | — | 6.21 ± 1.23 | 5.22 |
| Ribociclib | — | 48.66 ± 0.5 | 49.51 ± 0.76 |
| Abemaciclib | — | 7.29 ± 0.25 | 18.58 ± 1.07 |
 | ||
| Fig. 10 Structures of the most potent derivatives against both MCF-7 and MDA-MB-231 cell lines. | ||
| Compound | % G0–G1 | % S | % G2/M | Comment |
|---|---|---|---|---|
| Vf | 52.86 | 38.55 | 8.59 | Cell growth arrest@S |
| Vg | 66.92 | 25.07 | 8.01 | Cell growth arrest@G1 |
| Control | 60.07 | 29.72 | 10.21 | — |
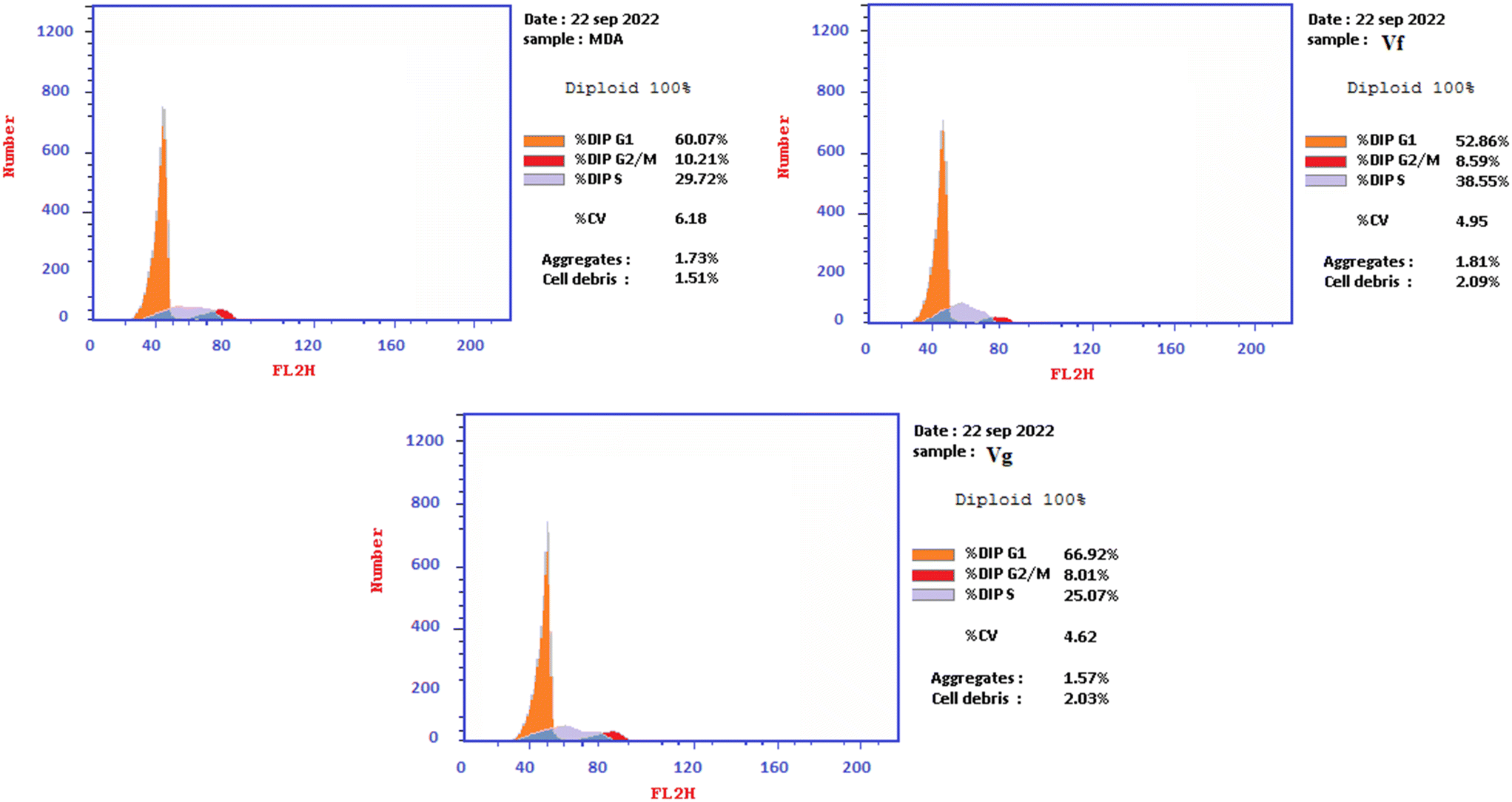 | ||
| Fig. 11 Illustration of the cell cycle analysis results of Vf and Vg on MDA-MB-231 cancer cell lines. | ||
| Compound | % Apoptosis | % Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| Vf | 45.33 | 28.06 | 12.7 | 4.57 |
| Vg | 37.26 | 11.61 | 15.85 | 9.8 |
| Control | 1.91 | 0.51 | 0.13 | 1.27 |
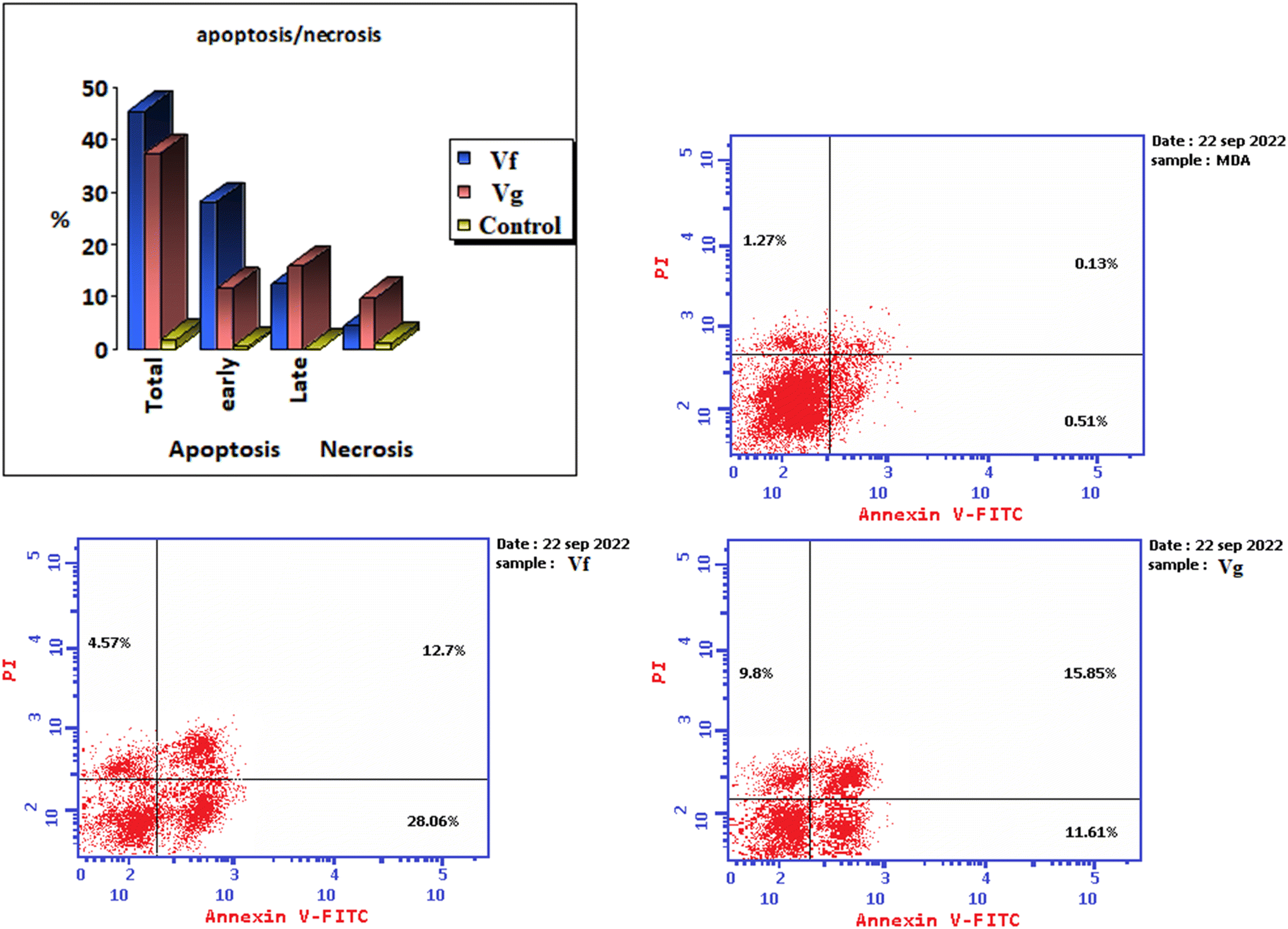 | ||
| Fig. 12 Illustration of the effect of the derivatives Vf and Vg on apoptotic induction in the MDA-MB-231 cancer cell line. | ||
| Compound | WI38 IC50 (μM) |
|---|---|
| Vf | 33.23 ± 1.39 |
| Vg | 28.6 ± 1.77 |
| Staurosporine | 22.45 ± 1.24 |
3.3. Molecular modelling studies of Vf and Vg
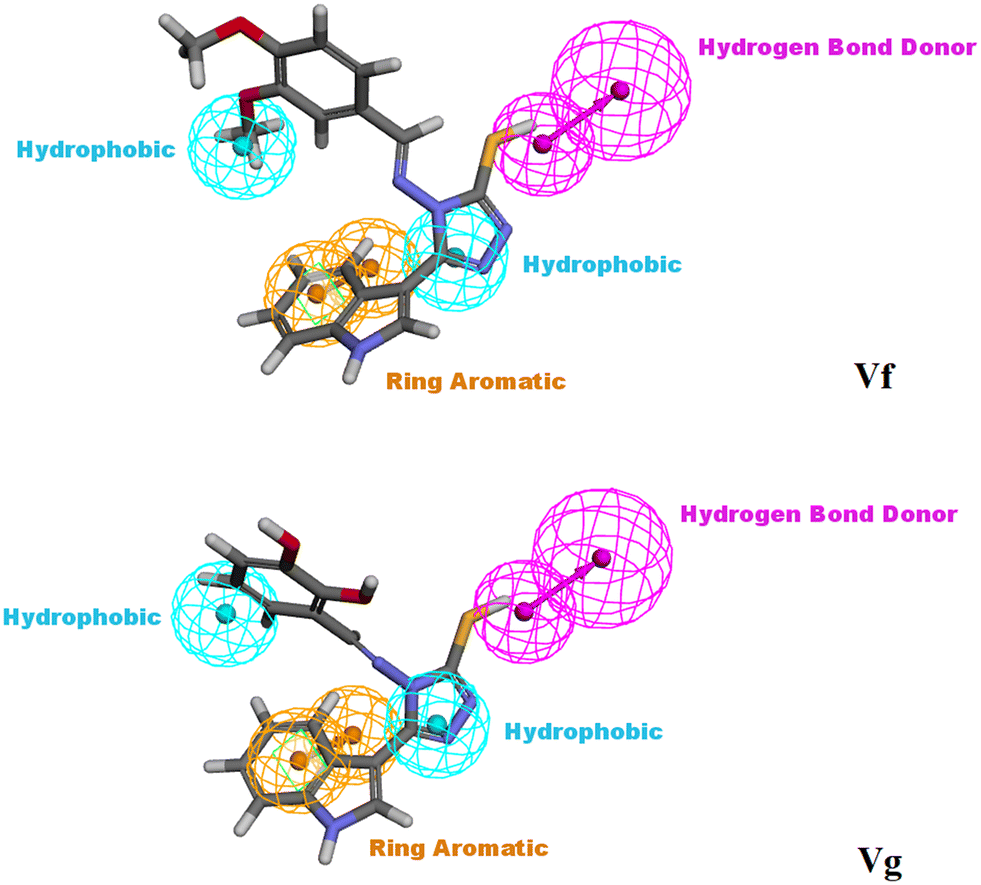 | ||
| Fig. 13 Vf and Vg mapped against the generated pharmacophore model (hypothesis 1), illustrating the main essential features required to act as CDK4/6 inhibitors. | ||
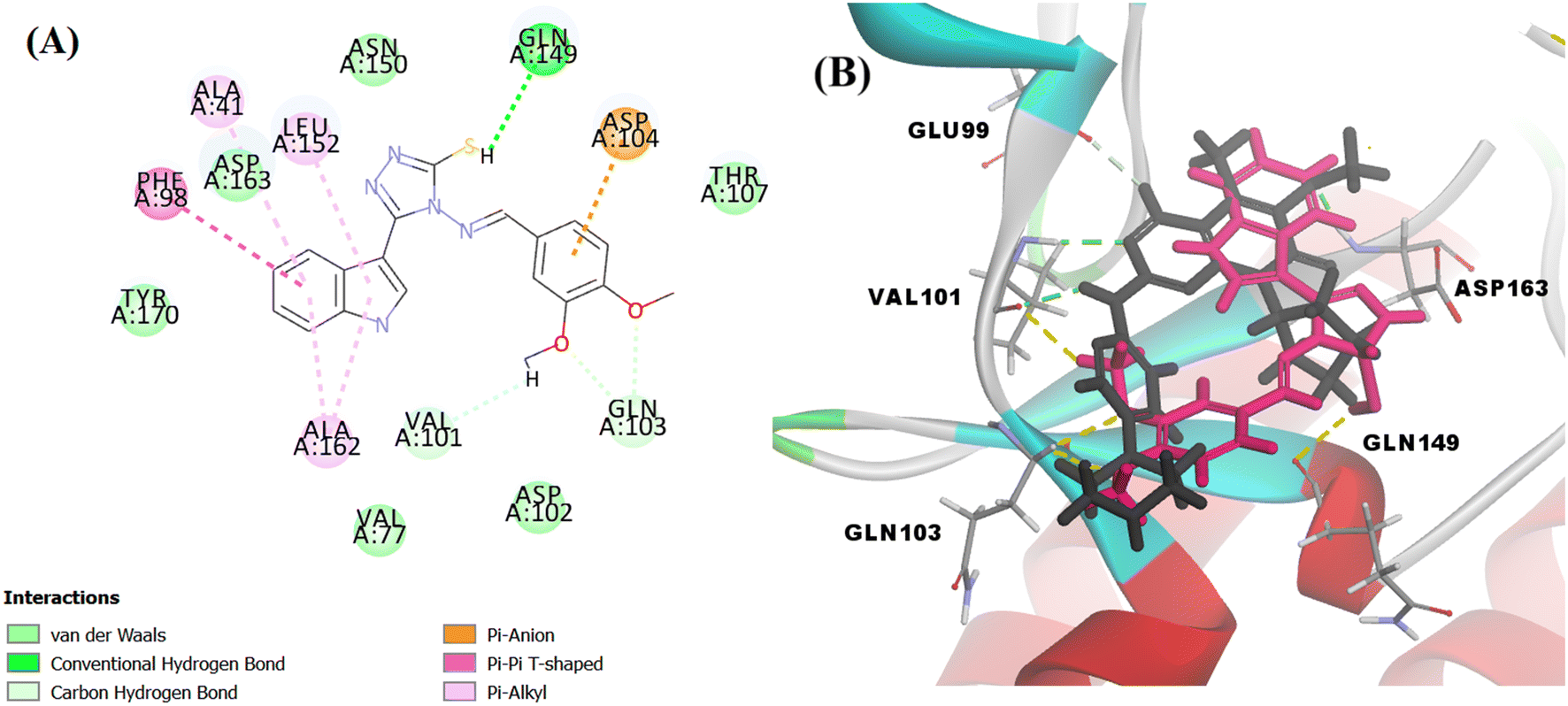 | ||
| Fig. 14 (A) 2D image of Vf displaying all interactions with the receptor CDK6. (B) overlapped 3D image displaying only the hydrogen bonds between the amino acid residues and palbociclib (represented by black colour and green dotted lines) and Vf (represented by magenta colour and yellow dotted lines). | ||
The docking score of Vg was −42.8016 kcal mol−1. Vg also binds in the ATP pocket and forms classical hydrogen bonds with the key amino acid VAL101 as well as ASP102 via the functional group SH and forms another hydrogen bond with GLN103 using the OH of the terminal aromatic ring. Furthermore, the indole part fitted into the hydrophobic pocket of the CDK6 active site and formed 9 hydrophobic bonds with the surrounding amino acids (ALA41, VAL77, PHE98, HIS100, LEU152 and ALA162) and the tail formed one electrostatic bond with the important amino acid ASP104, exactly the amino acids with which the reference palbociclib interacted, showing its suitability as an inhibitor for this target (Fig. 15).
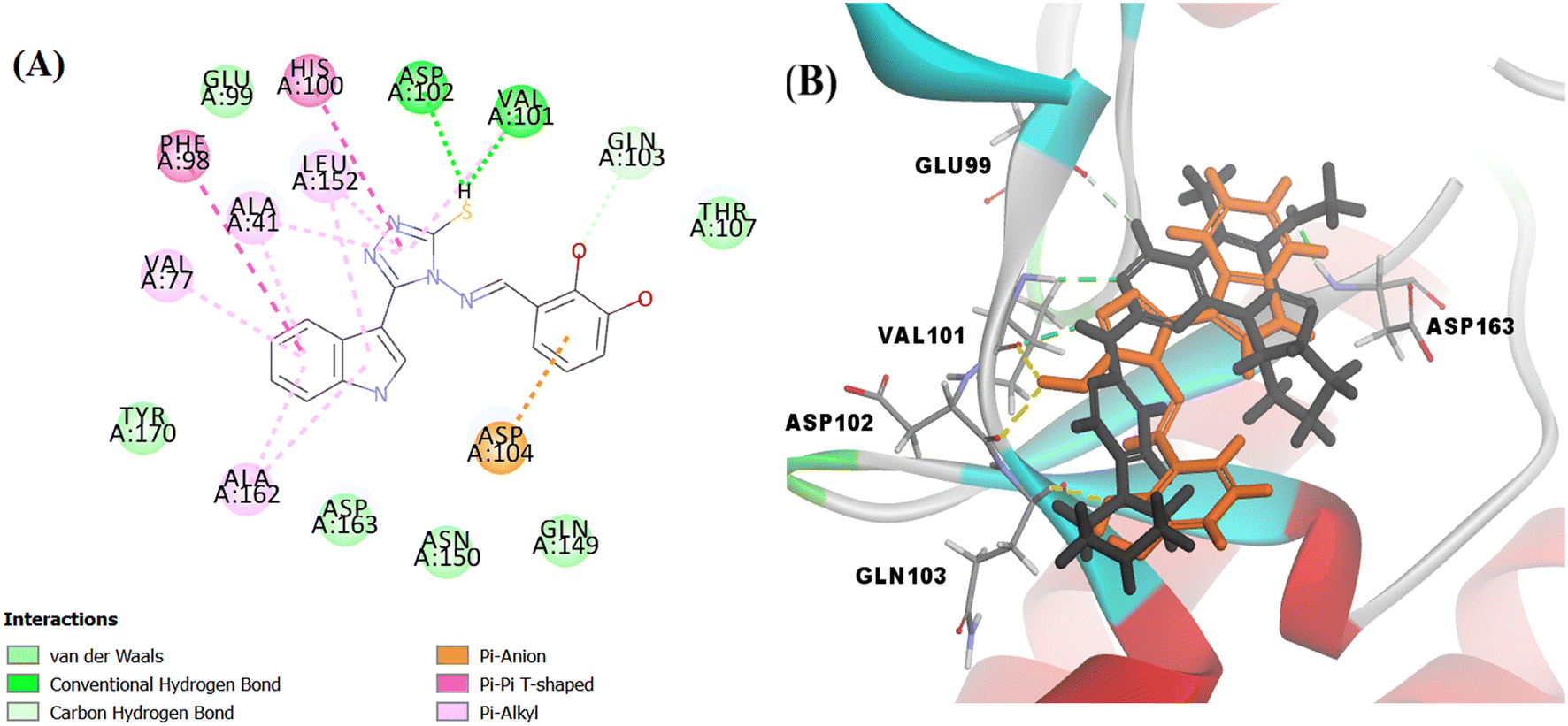 | ||
| Fig. 15 (A) 2D image of Vg displaying all interactions with the receptor CDK6. (B) overlapped 3D image displaying only the hydrogen bonds between the amino acid residues and palbociclib (represented by black colour and green dotted lines) and Vg (represented by orange colour and yellow dotted lines). | ||
3.4. Structure–activity relationship (SAR) study
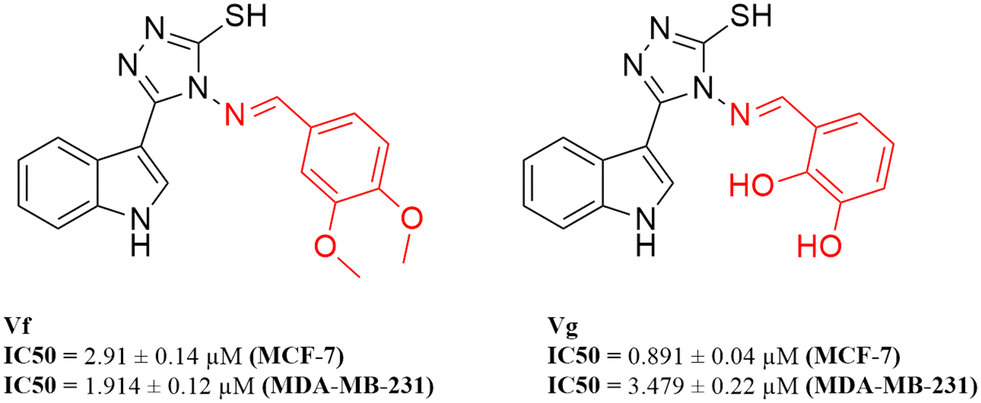
According to the results obtained from the cytotoxic investigation of the new indole compounds, a brief SAR study was extracted. Judging by the IC50 values of the fourteen compounds, it was established that compound Vf substituted with 2 electron donating groups (3,4-dimethoxy) was the best in the series with activity on both MCF-7 (IC50 = 2.91 μM) and MDA-MB-231 (IC50 = 1.914 μM) when compared with the reference staurosporine with IC50 values of 3.144 μM and 4.385 μM, respectively. Vg that was also di-substituted with electron donating groups (2,3-dihydroxy) expressed great results on both cell lines with IC50 values of 0.891 μM and 3.479 μM, respectively.Substitution at the meta position such as in compounds Vn, Vk and Vj was accompanied by good cytotoxic activity as well specially when that position was substituted with an electron donating group as in compound Vn (IC50 = 4.636 μM and 2.24 μM, respectively). The electron withdrawing group (3-Br) at the meta position in compound Vk showed interesting activity (IC50 = 1.131 μM and 5.398 μM, respectively) similarly to the electron withdrawing group (3-Cl) of compound Vj that also showed encouraging cytotoxic activity (IC50 = 11.73 μM and 2.799 μM, respectively) (Fig. 16).
 | ||
| Fig. 16 An illustration simplifying the SAR study. | ||
Despite the good results obtained from the substitution at the meta positions, the same groups when substituted at the para positions seemed to reduce the activity of the compounds as apprehended in Ve, Vl, Vb and Vd, leading to higher IC50 values on either MCF-7 or MDA-MB-231 to 9.241 μM and 8.796 μM, 7.677 μM and 11.44 μM, 10.77 μM and 81.46 μM and finally 36.4 μM and 26.07 μM, respectively. One exception to the para position relatively low results is compound Vc that has a substitution at the para position with a hydroxy group (4-hydroxy), accomplishing amazing cytotoxic activity particularly against the MDA-MB-231 cell line (IC50 = 2.053 μM) in contrast to the reference with half of that activity (IC50 = 4.385 μM) possibly due to the ability of its hydroxy group to form hydrogen bonds with the target receptor as perceived from the docking study.
On the other hand, compounds with di-substitutions at the ortho and para positions simultaneously were accompanied by low cytotoxic activity on both MCF-7 and MDA-MB-231 as shown in compounds Vh (IC50 = 17.82 μM and 28.18 μM, respectively) and Vi (IC50 = 27.9 μM and 23.06 μM, respectively). The tri-substitution on Vm (3,4,5-trimethoxy) proved less advantageous because of the bulkiness of the moiety, giving pretty low results (IC50 = 21.38 μM and 14.75 μM, respectively), and finally, the unsubstituted variant of the series Va showed very low results with IC50 of 22.55 μM and 13.98 μM, respectively.
4. Experimental
4.1. Molecular modelling study
4.2. Chemistry
4.2.5.1. (E)-4-(Benzylideneamino)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Va). Buff solid, yield 79%, M.P. 224–226 °C. IR (KBr, cm−1): 3309 (NH), 3055–3001 (arom. CH), 1654–1597 (C
N), 1577–1512 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.18–7.27 (m, 2H, ArH), 7.53–7.72 (m, 4H, ArH), 7.90 (d, 1H, J = 8.0 Hz, ArH), 8.02–8.03 (m, 2H, ArH), 8.17 (d, 1H, J = 8.0 Hz, ArH), 9.83 (s, 1H, NCH), 11.76 (s, 1H, indole-NH), 14.04 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 100.58 (C3 indole), 112.60 (C7 indole), 121.25 (C5 indole), 121.31 (C4 indole), 123.18 (C6 indole), 125.22 (C2 indole), 129.60 (C3, C5 benzene), 129.76 (C2, C6 benzene), 132.65 (C3a indole), 133.28 (C4 benzene), 135.02 (C1 benzene), 136.33 (C7a indole), 146.83 (C3 triazole), 161.26 (CN), 166.55 (C5 triazole). MS m/z (%): 320 ([M + H]+, 80), 136 (100). Anal. calcd for C17H13N5S (319): C, 63.93; H, 4.10; N, 21.93; S, 10.04; found: C, 64.17; H, 4.29; N, 22.20%.
4.2.5.2. (E)-4-(4-Chlorobenzylideneamino)-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Vb). Yellow solid, yield 82%, M.P. 273–275 °C. IR (KBr, cm−1): 3228 (NH), 3059–3032 (arom. CH), 1658–1597 (C
N), 1577–1504 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.15–7.27 (m, 2H, ArH), 7.49–7.54 (m, 2H, ArH), 7.66 (d, 1H, J = 8.0 Hz, ArH), 7.73 (d, 1H, J = 8.0 Hz, ArH), 8.00 (d, 1H, J = 8.0 Hz, ArH), 8.03 (d, 1H, J = 8.0 Hz, ArH), 8.15 (d, 1H, J = 8.0 Hz, ArH), 9.86 (s, 1H, NCH), 11.76 (s, 1H, indole-NH), 14.05 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 100.76 (C3 indole), 112.61 (C7 indole), 121.28 (C4 indole, C6 indole), 123.18 (C5 indole), 125.21 (C2 indole), 128.80 (C3a indole),129.92 (C3, C5 benzene), 130.93 (C2, C6 benzene), 131.54 (C1 benzene), 136.34 (C4 benzene), 137.96 (C7a indole), 146.84 (C3 triazole), 161.25 (CN), 165.21 (C5 triazole). MS m/z (%): 352 (M−, 100). Anal. calcd for C17H12ClN5S (353): C, 57.71; H, 3.42; Cl, 10.02; N, 19.79; S, 9.06; found: C, 57.98; H, 3.50; N, 20.03%.
4.2.5.3. (E)-4-({[3-(1H-Indol-3-yl)-5-mercapto-4H-1,2,4-triazol-4-yl]imino}methyl)phenol (Vc). Buff solid, yield 81%, M.P. 286–287 °C. IR (KBr, cm−1): 3305 (NH), 3001 (arom. CH), 1635 (C
N), 1597 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.86 (d, 2H, J = 8.0 Hz, ArH), 7.23 (t, 1H, J = 8.0 Hz, ArH), 7.52–7.59 (m, 2H, ArH), 7.88 (d, 2H, J = 8.0 Hz, ArH), 8.02 (d, 1H, J = 8.0 Hz, ArH), 8.19 (s, 1H, ArH), 8.29 (s, 1H, OH), 9.51 (s, 1H, NCH), 11.76 (s, 1H, indole-NH), 13.96 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 109.28 (C3 indole), 112.40 (C7 indole), 116.19 (C3, C5 benzene), 121.15 (C5 indole), 121.65 (C4 indole), 121.70 (C6 indole), 122.64 (C2, C3a indole), 126.25 (C1 benzene), 127.09 (C2 benzene), 128.92 (C6 benzene, C7a indole), 136.45 (C3 triazole), 145.73 (C4 benzene), 159.45 (C5 triazole, CN). Anal. calcd for C17H13N5OS (335): C, 60.88; H, 3.91; N, 20.88; O, 4.77; S, 9.56; found: C, 61.07; H, 4.15; N, 21.19%.
4.2.5.4. (E)-5-(1H-Indol-3-yl)-4-[(4-methylbenzylidene)amino]-4H-1,2,4-triazole-3-thiol (Vd). Brown solid, yield 67%, M.P. 264–265 °C. IR (KBr, cm−1): 3298 (NH), 3096–3055 (arom. CH), 2966 (aliph. CH), 1634–1616 (C
N), 1581–1512 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.42 (s, 3H, OCH3), 7.19–7.27 (m, 2H, ArH), 7.41 (d, 2H, J = 8.0 Hz, ArH), 7.52 (d, 1H, J = 8.0 Hz, ArH), 7.90 (d, 2H, J = 8.0 Hz, ArH), 8.00 (d, 1H, J = 8.0 Hz, ArH), 8.17 (d, 1H, J = 8.0 Hz, ArH), 9.73 (s, 1H, NCH), 11.75 (s, 1H, indole-NH), 14.00 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 21.77 (CH3), 100.90 (C3 indole), 112.59 (C7 indole), 121.23 (C5 indole), 121.32 (C4 indole), 123.17 (C6 indole), 125.21 (C2 indole), 128.62 (C3a indole), 129.29 (C3, C5 benzene), 129.95 (C1 benzene), 130.35 (C2, C6 benzene), 136.32 (C7a indole), 143.68 (C4 benzene), 146.78 (C3 triazole), 161.25 (CN), 166.68 (C5 triazole). MS m/z (%): 334 ([M + H]+, 100). Anal. calcd for C18H15N5S (333): C, 64.84; H, 4.53; N, 21.01; S, 9.62; found: C, 65.09; H, 4.60; N, 21.24%.
4.2.5.5. (E)-4-[(4-Bromobenzylidene)amino]-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Ve). Faint-yellow solid. Yield 55%, M.P. 281–282 °C. IR (KBr, cm−1): 3213 (NH), 3062–3047 (arom. CH), 1620–1593 (C
N), 1539 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.18–7.27 (m, 2H, ArH), 7.52 (d, 1H, J = 8.0 Hz, ArH), 7.81 (d, 2H, J = 8.0 Hz, ArH), 7.95–8.00 (m, 3H, ArH), 8.14 (d, 1H, J = 8.0 Hz, ArH), 9.85 (s, 1H, NCH), 11.75 (s, 1H, indole-NH), 14.04 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 100.73 (C3 indole), 112.61 (C7 indole), 121.26 (C5 indole), 123.18 (C4 indole), 125.19 (C6 indole), 126.99 (C4 benzene), 128.74 (C3a indole, C2 benzene), 131.07 (C2, C6 benzene), 131.87 (C1 benzene), 132.86 (C3, C5 benzene), 136.33 (C7a indole), 146.82 (C3 triazole), 161.23 (CN), 165.39 (C5 triazole). MS m/z (%): 399.8 ([M + H]+, 50), 279.9 (100). Anal. calcd for C17H12BrN5S (397): C, 51.27; H, 3.04; Br, 20.06; N, 17.58; S, 8.05; found: C, 51.56; H, 3.25; N, 17.85%.
4.2.5.6. (E)-4-[(3,4-Dimethoxybenzylidene)amino]-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Vf). Buff solid, yield 79%, M.P. 252–254 °C. IR (KBr, cm−1): 3329 (NH), 3078–3059 (arom. CH), 2954–2931 (aliph. CH), 1635–1600 (C
N), 1550–1512 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.88 (s, 6H, 2OCH3), 7.16–7.27 (m, 3H, ArH), 7.51 (d, 1H, J = 8.0 Hz, ArH), 7.61 (s, 1H, ArH), 8.01 (d, 1H, J = 8.0 Hz, ArH), 8.11 (d, 1H, J = 8.0 Hz, ArH), 8.43 (s, 1H, ArH), 9.60 (s, 1H, NCH), 11.75 (s, 1H, indole-NH), 13.68 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 56.09 (CH3), 101.04 (C3 indole), 110.02 (C5 benzene), 112.48 (C7 indole), 112.59 (C2 benzene), 121.03 (C5 indole), 121.26 (C4 indole), 122.92 (C6 indole), 123.15 (C6 benzene), 124.90 (C2 indole), 125.19 (C3a indole), 128.38 (C1 benzene), 136.19 (C7a indole), 147.67 (C3 benzene), 149.75 (C3 triazole), 153.42 (C4 benzene), 161.24 (CN), 165.68 (C5 triazole). MS m/z (%): 369.3 (M+, 84), 164 (100). Anal. calcd for C19H17N5O2S (379): C, 60.14; H, 4.52; N, 18.46; O, 8.43; S, 8.45; found: C, 60.41; H, 4.67; N, 18.75%.
4.2.5.7. (E)-3-({[3-(1H-Indol-3-yl)-5-mercapto-4H-1,2,4-triazol-4-yl]imino}methyl)benzene-1,2-diol (Vg). Yellow solid, yield 54%, M.P. 264–266 °C. IR (KBr, cm−1): 3460–3367 (OH), 3332–3201 (NH), 3055–3043 (arom. CH), 1631 (C
N), 1539 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.73 (t, 1H, J = 8.0 Hz, ArH), 6.87 (d, 1H, J = 8.0 Hz, ArH), 6.96 (s, 1H, OH), 7.17–7.24 (m, 2H, ArH), 7.51 (d, 1H, J = 8.0 Hz, ArH), 8.24 (d, 2H, J = 8.0 Hz, ArH), 8.52 (s, 1H, ArH), 9.19 (s, 1H, NCH), 11.46 (s, 1H, OH), 11.74 (s, 1H, indole-NH), 11.81 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 108.68 (C3 indole), 112.58 (C7 indole), 117.42 (C4 benzene), 119.57 (C5 indole, C1 benzene), 120.45 (C4 indole), 121.39 (C6 indole), 121.50 (C5 benzene), 122.87 (C6 benzene), 126.89 (C2 indole), 129.33 (C3a indole), 136.64 (C7a indole), 146.05 (C2 benzene, C3 triazole), 146.29 (C3 benzene), 146.87 (C5 triazole), 161.25 (CN). Anal. calcd for C17H13N5O2S (351): C, 58.11; H, 3.73; N, 19.93; O, 9.11; S, 9.13; found: C, 58.38; H, 3.89; N, 20.14%.
4.2.5.8. (E)-4-({[3-(1H-Indol-3-yl)-5-mercapto-4H-1,2,4-triazol-4-yl]imino}methyl)benzene-1,3-diol (Vh). Faint-yellow solid, yield 82%, M.P. 302–305 °C. IR (KBr, cm−1): 3483–3421 (OH), 3317 (NH), 3055–3012 (arom. CH), 1631–1612 (C
N), 1573–1519 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.34–6.38 (m, 2H, ArH), 7.15–7.22 (m, 2H, ArH), 7.28 (s, 1H, OH), 7.48 (d, 1H, J = 8.0 Hz, ArH), 8.18 (d, 2H, J = 8.0 Hz, ArH), 8.41 (s, 1H, ArH), 9.90 (s, 1H, NCH), 11.51 (s, 1H, OH), 11.68 (s, 1H, indole-NH), 11.75 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 103.16 (C3 benzene), 107.99 (C3 indole), 108.82 (C5 benzene), 111.34 (C1 benzene), 112.49 (C7 indole), 121.25 (C5 indole, C4 indole), 121.48 (C6 indole), 122.75 (C2 indole), 126.84 (C3a indole), 128.92 (C6 benzene), 131.58 (C7a indole), 136.58 (C3 triazole), 147.14 (CN), 159.70 (C2 benzene), 160.68 (C4 benzene, C5 triazole). MS m/z (%): 351 (M+, 10), 136 (100). Anal. calcd for C17H13N5O2S (351): C, 58.11; H, 3.73; N, 19.93; O, 9.11; S, 9.13; found: C, 58.34; H, 3.85; N, 20.18%.
4.2.5.9. (E)-4-[(2,4-Dichlorobenzylidene)amino]-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Vi). Yellow solid, yield 89%, M.P. 249–251 °C. IR (KBr, cm−1): 3383 (NH), 3089–3008 (arom. CH), 1635–1602 (C
N), 1585–1550 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.15–7.25 (m, 2H, ArH), 7.49–7.54 (m, 2H, ArH), 7.72 (s, 1H, ArH), 8.02 (d, 1H, J = 8.0 Hz, ArH), 8.11 (d, 1H, J = 8.0 Hz, ArH), 8.42 (s, 1H, ArH), 8.66 (s, 1H, NCH), 11.69 (s, 1H, indole-NH), 13.68 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 101.04 (C3 indole), 112.49 (C7 indole), 121.27 (C5 indole), 121.37 (C4 indole), 121.60 (C6 indole), 122.92 (C2 indole), 125.19 (C3a indole), 128.37 (C5 benzene), 128.44 (C4 benzene), 129.81 (C6 benzene), 131.64 (C3 benzene), 133.88 (C2 benzene), 134.91 (C1 benzene), 136.19 (C7a indole), 136.48 (C3 triazole), 147.67 (CN), 165.68 (C5 triazole). Anal. calcd for C17H11Cl2N5S (387): C, 52.59; H, 2.86; Cl, 18.26; N, 18.04; S, 8.26; found: C, 52.67; H, 3.04; N, 18.23%.
4.2.5.10. (E)-4-[(3-Chlorobenzylidene)amino]-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Vj). Buff solid, yield 34%, M.P. 230–232 °C. IR (KBr, cm−1): 3251 (NH), 3066–3008 (arom. CH), 1631 (C
N), 1527 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.16–7.23 (m, 2H, ArH), 7.46–7.51 (m, 3H, ArH), 7.66 (d, 1H, J = 8.0 Hz, ArH), 7.77 (s, 1H, ArH), 8.23 (d, 1H, J = 8.0 Hz, ArH), 8.33 (s, 2H, ArH, NCH), 11.61 (s, 1H, indole-NH), 11.84 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 101.06 (C3 indole), 112.48 (C7 indole), 121.30 (C5 indole), 121.63 (C4 indole), 122.76 (C6 indole), 125.94 (C2 indole), 126.39 (C3a indole), 129.57 (C2 benzene), 131.15 (C6 benzene), 134.14 (C5 benzene), 136.47 (C4 benzene), 137.57 (C3 benzene), 143.15 (C1 benzene), 146.90 (C7a indole), 147.67 (C3 triazole), 161.28 (CN), 165.67 (C5 triazole). MS m/z (%): 346.8 (M+, 100). Anal. calcd for C17H12ClN5S (353): C, 57.71; H, 3.42; Cl, 10.02; N, 19.79; S, 9.06; found: C, 57.98; H, 3.60; N, 20.05%.
4.2.5.11. (E)-4-[(3-Bromobenzylidene)amino]-5-(1H-indol-3-yl)-4H-1,2,4-triazole-3-thiol (Vk). Faint-yellow solid, yield 51%, M.P. 232–235 °C. IR (KBr, cm−1): 3248 (NH), 3066–3005 (arom. CH), 1631 (C
N), 1523 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.16–7.23 (m, 2H, ArH), 7.38 (t, 1H, J = 8.0 Hz, ArH), 7.49 (d, 1H, J = 8.0 Hz, ArH), 7.58 (d, 1H, J = 8.0 Hz, ArH), 7.70 (d, 1H, J = 8.0 Hz, ArH), 7.92 (s, 1H, ArH), 8.23 (d, 1H, J = 8.0 Hz, ArH), 8.30 (s, 2H, ArH, NCH), 11.56 (s, 1H, indole-NH), 11.81 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 101.07 (C3 indole), 112.48 (C7 indole), 121.32 (C5 indole), 122.68 (C4 indole), 122.78 (C6 indole), 122.92 (C3 benzene), 125.21 (C2 indole), 126.34 (C3a indole), 128.38 (C6 benzene), 129.27 (C5 benzene), 131.41 (C2 benzene), 132.47 (C4 benzene), 136.21 (C1 benzene), 137.78 (C7a indole), 143.01 (C3 triazole), 147.68 (CN), 165.68 (C5 triazole). Anal. calcd for C17H12BrN5S (398): C, 51.27; H, 3.04; Br, 20.06; N, 17.58; S, 8.05; found: C, 51.49; H, 3.21; N, 17.80%.
4.2.5.12. (E)-5-(1H-Indol-3-yl)-4-[(4-methoxybenzylidene)amino]-4H-1,2,4-triazole-3-thiol (Vl). Buff solid, yield 65%, M.P. 249–251 °C. IR (KBr, cm−1): 3390 (NH), 3097–3001 (arom. CH), 2966–2912 (aliph. CH), 1602–1597 (C
N), 1566–1512 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.88 (s, 3H, OCH3), 7.15–7.27 (m, 4H, ArH), 7.52 (d, 1H, J = 8.0 Hz, ArH), 7.96–8.00 (m, 3H, ArH), 8.16 (d, 1H, J = 8.0 Hz, ArH), 9.60 (s, 1H, NCH), 11.73 (s, 1H, indole-NH), 13.96 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 56.06 (CH3), 100.95 (C3 indole), 112.58 (C7 indole), 115.27 (C3, C5 benzene), 121.22 (C5 indole), 121.32 (C4 indole), 123.17 (C6 indole), 125.05 (C1 benzene), 125.20 (C2 indole), 128.54 (C3a indole), 131.30 (C2, C6 benzene), 136.31 (C7a indole), 146.70 (C3 triazole), 161.26 (CN), 163.50 (C5 triazole), 166.76 (C4 benzene). Anal. calcd for C18H15N5OS (349): C, 61.87; H, 4.33; N, 20.04; O, 4.58; S, 9.18; found: C, 61.74; H, 4.60; N, 20.28%.
4.2.5.13. (E)-5-(1H-Indol-3-yl)-4-[(3,4,5-trimethoxybenzylidene)amino]-4H-1,2,4-triazole-3-thiol (Vm). Faint-yellow solid, yield 73%, M.P. 229–230 °C. IR (KBr, cm−1): 3352 (NH), 3043–3005 (arom. CH), 2974–2924 (aliph. CH), 1602–1577 (C
N), 1504 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.72 (s, 3H, OCH3), 3.85 (s, 6H, 2OCH3), 7.02 (s, 2H, ArH), 7.14–7.25 (m, 2H, ArH), 7.35 (s, 1H, ArH), 7.48 (d, 1H, J = 8.0 Hz, ArH), 8.15 (d, 1H, J = 8.0 Hz, ArH), 8.22 (s, 1H, NCH), 11.41 (s, 1H, indole-NH), 11.77 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 56.37 (CH3 of C3, C5 benzene), 60.59 (CH3 of C4 benzene), 104.39 (C2, C6 benzene), 106.56 (C3 indole), 112.43 (C7 indole), 121.21 (C5 indole), 122.68 (C4 indole), 123.15 (C6 indole), 125.20 (C2 indole), 128.01 (C3a indole), 130.85 (C1 benzene), 139.25 (C7a indole), 141.87 (C4 benzene), 146.83 (C3 triazole), 153.68 (C3, C5 benzene), 161.20 (CN), 165.91 (C5 triazole). MS m/z (%): 411.1 ([M + H]+, 48). Anal. calcd for C20H19N5O3S (409): C, 58.67; H, 4.68; N, 17.10; O, 11.72; S, 7.83; found: C, 58.90; H, 4.85; N, 17.36%.
4.2.5.14. (E)-5-(1H-Indol-3-yl)-4-[(3-methoxybenzylidene)amino]-4H-1,2,4-triazole-3-thiol (Vn). Yellow solid, yield 32%, M.P. 219–221 °C. IR (KBr, cm−1): 3290 (NH), 3062–3001 (arom. CH), 2958–2935 (aliph. CH), 1597–1573 (C
N), 1508 (CC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.86 (s, 3H, OCH3), 7.19–7.28 (m, 3H, ArH), 7.49–7.59 (m, 4H, ArH), 8.05 (d, 1H, J = 8.0 Hz, ArH), 8.20 (s, 1H, ArH), 9.86 (s, 1H, NCH), 11.78 (s, 1H, indole-NH), 14.06 (s, 1H, SH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 55.82 (CH3 of C3 benzene), 100.87 (C3 indole), 112.61 (C7 indole), 113.26 (C2 benzene), 119.29 (C4 benzene), 121.26 (C5 indole), 121.35 (C4 indole), 122.09 (C6 benzene), 123.18 (C6 indole), 125.25 (C2 indole), 128.76 (C3a indole), 130.89 (C5 benzene), 134.06 (C1 benzene), 136.33 (C7a indole), 146.90 (C3 triazole), 160.20 (CN), 161.25 (C5 triazole), 165.88 (C3 benzene). MS m/z (%): 349.9 ([M + H]+, 40), 135.9 (100). Anal. calcd for C18H15N5OS (349): C, 61.87; H, 4.33; N, 20.04; O, 4.58; S, 9.18; found: C, 61.73; H, 4.51; N, 20.28%.
4.3. Biological evaluation
All of the biological evaluation assays were conducted in the Egyptian company for the production of vaccines, sera and drugs labs (VACSERA, Giza, Egypt). All cell lines used in this study are from the American Type Culture Collection (ATCC). The cell lines used were MCF-7, MDA-MB-231 (human epithelial cells of mammary gland tissue with adenocarcinoma) and WI-38 (fibroblasts from human lung tissue). All the newly obtained compounds Va–n were examined for their potential cytotoxic activity against two breast cancer cell lines (MCF-7 and MDA-MB-231) of the ATCC. The MTT method was used where cells in the log phase of growth were employed. Firstly, an amount equal to 10% of the culture medium volume was used from the reconstituted MTT and cultures were returned to the incubator for 2–4 hours judging by the cell type and density. After the incubation, MTT solubilization solution [M-8910] was added to dissolve the remaining formazan crystals. Finally, the absorbance at a wavelength of 570 nm was measured spectroscopically.42–46For CDK6 assay, the enzyme, substrate, ATP and inhibitors were diluted in kinase buffer. After that, 1 μL of inhibitor + 2 μL of enzyme + 2 μL of substrate/ATP mix were added to each well and incubated at room temperature for 60 minutes. The luminescence reagent (5 μL) was added and incubated once more at room temperature for 40 minutes. 10 μL of kinase detection reagent was added and incubated for another 30 minutes and eventually the luminescence was recorded.53
5. Conclusion
The current research introduces a new series of indolyl 1,2,4-triazole derivatives as potent anti-tumour agents against breast cancer with promising activity against CDK4/6. Initially, a valid pharmacophore model was generated using CDK4/6 inhibitors with known IC50 values, inspiring and aiding the design of the new compounds. The novel designed compounds revealed high fit values and were very competitive to that of palbociclib (a CDK4/6 inhibitor) which was used as a reference. The docking protocol was performed on the CDK6 binding site while comparing the results to the crystalised ligand palbociclib. All the newly designed and synthesised compounds Va–n were tested for their inhibitory activity against both CDK4 and CDK6 and the results ranged from 0.049 μM to 3.031 μM on CDK4, while on CDK6 the values ranged from 0.075 μM to 1.11 μM when compared to staurosporine with IC50 values of 1.027 and 0.402 μM, respectively. Also, their cytotoxic abilities against two breast cancer cell lines MCF-7 (IC50 = 0.891–36.4 μM) and MDA-MB-231 (IC50 = 1.91–81.46 μM) were tested and showed promising anti-proliferative activities towards both. The bio-isosteric replacement at the tail terminal moiety with electron donating groups proved to be the most potent, while substitution at the para position displayed low activity. Triple substitution on the other hand seemed to be the least effective maybe due to the bulkiness of the compound. Two compounds, Vf and Vg, showed the most potent cytotoxic performance against both MCF-7 and MDA-MB-231 and thus were further investigated for cell cycle analysis, apoptosis induction and cytotoxicity against normal human cells. Vf was able to arrest the cell cycle at the S phase while Vg arrested the cycle at the G0/G1 phase and both compounds induced apoptosis significantly. Collectively, all results of the investigations that were carried out in this study suggested promising ability for the new compounds to work as anti-tumour agents in breast cancer and introduced an encouraging activity for this class of compounds on CDK4/6, although these compounds require further investigation as more variations of this scaffold could prove to be better. However, it is highly possible for these compounds to also act on other kinases or even other target enzymes, leaving this study open for more research in the future.This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. J. Bray, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2021, 71(3), 209–249 CrossRef PubMed.
- N. J. Bundred, Prognostic and predictive factors in breast cancer, Cancer Treat. Rev., 2001, 27(3), 137–142 CrossRef CAS PubMed.
- T. Al-Warhi, A. M. El Kerdawy, N. Aljaeed, O. E. Ismael, R. R. Ayyad, W. M. Eldehna, H. A. Abdel-Aziz and G. H. J. Al-Ansary, Synthesis, biological evaluation and in silico studies of certain oxindole–indole conjugates as anticancer CDK inhibitors, Molecules, 2020, 25(9), 2031 CrossRef CAS PubMed.
- E. Davies and S. J. Hiscox, New therapeutic approaches in breast cancer, Maturitas, 2011, 68(2), 121–128 CrossRef CAS PubMed.
- J. Singh Sidhu, R. Singla and V. J. Jaitak, Indole derivatives as anticancer agents for breast cancer therapy: a review, Anti-Cancer Agents Med. Chem., 2016, 16(2), 160–173 CrossRef PubMed.
- C. Sanchez-Martinez, L. M. Gelbert, M. J. Lallena and A. J. de Dios, Cyclin dependent kinase (CDK) inhibitors as anticancer drugs, Bioorg. Med. Chem. Lett., 2015, 25(17), 3420–3435 CrossRef CAS PubMed.
- S. J. Baker and E. P. J. C. r. Reddy, CDK4: a key player in the cell cycle, development, and cancer, Genes Cancer, 2012, 3(11–12), 658–669 CrossRef CAS PubMed.
- J. Bloom and F. R. J. Cross, Multiple levels of cyclin specificity in cell-cycle control, Nat. Rev. Mol. Cell Biol., 2007, 8(2), 149–160 CrossRef CAS PubMed.
- M. Ingham and G. K. J. Schwartz, Cell-cycle therapeutics come of age, J. Clin. Oncol., 2017, 35(25), 2949 CrossRef CAS PubMed.
- H. K. Reddy, R. V. Mettus, S. G. Rane, X. Grana, J. Litvin and E. P. J. Reddy, Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis, Cancer Res., 2005, 65(22), 10174–10178 CrossRef CAS PubMed.
- T. Tsutsui, B. Hesabi, D. S. Moons, P. P. Pandolfi, K. S. Hansel, A. Koff and H. J. Kiyokawa, Targeted disruption of CDK4 delays cell cycle entry with enhanced p27Kip1 activity, Mol. Cell. Biol., 1999, 19(10), 7011–7019 CrossRef CAS PubMed.
- J. Cicenas, K. Kalyan, A. Sorokinas, A. Jatulyte, D. Valiunas, A. Kaupinis and M. J. Valius, Highlights of the latest advances in research on CDK inhibitors, Cancers, 2014, 6(4), 2224–2242 CrossRef PubMed.
- T. K. Eggersmann, T. Degenhardt, O. Gluz, R. Wuerstlein and N. J. Harbeck, CDK4/6 inhibitors expand the therapeutic options in breast cancer: palbociclib, ribociclib and abemaciclib, BioDrugs, 2019, 33(2), 125–135 CrossRef CAS PubMed.
- L. Chen, T.-Y. Ji, X.-S. Huo, Z.-Y. Zeng, W.-X. Ye, C.-C. Dai, Y.-Q. Zhang, W.-W. You and P.-L. Zhao, Rational design, synthesis and biological evaluation of novel 2-(substituted amino)-[1, 2, 4] triazolo [1, 5-a] pyrimidines as novel tubulin polymerization inhibitors, Eur. J. Med. Chem., 2022, 244, 114864 CrossRef CAS PubMed.
- X.-S. Huo, X.-E. Jian, J. Ou-Yang, L. Chen, F. Yang, D.-X. Lv, W.-W. You, J.-J. Rao and P.-L. Zhao, Discovery of highly potent tubulin polymerization inhibitors: Design, synthesis, and structure-activity relationships of novel 2, 7-diaryl-[1, 2, 4] triazolo [1, 5-a] pyrimidines, Eur. J. Med. Chem., 2021, 220, 113449 CrossRef CAS PubMed.
- T. Ji, X. Jian, L. Chen, W. Zeng, X. Huo, M. Li, P. Chen, Y. Zhang, W. You and P. Zhao, Discovery of novel 6-p-tolyl-3-(3, 4, 5-trimethoxybenzyl)-7H-[1, 2, 4] triazolo [3, 4-b][1, 3, 4] thiadiazine derivative as a potent tubulin inhibitor with promising in vivo antitumor activity, Eur. J. Med. Chem., 2023, 256, 115437 CrossRef CAS PubMed.
- A. Ammazzalorso, M. Agamennone, B. De Filippis and M. J. M. Fantacuzzi, Development of CDK4/6 inhibitors: a five years update, Molecules, 2021, 26(5), 1488 CrossRef CAS PubMed.
- H. He, Q. Liu, L. Chen, J. Wang, Y. Yuan, H. Li, X. Qian, Z. Zhao and Z. J. Chen, Design, synthesis and biological evaluation of pteridine-7 (8H)-one derivatives as potent and selective CDK4/6 inhibitors, Bioorg. Med. Chem. Lett., 2022, 128991 CrossRef CAS PubMed.
- M. Poratti and G. J. Marzaro, Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib, Eur. J. Med. Chem., 2019, 172, 143–153 CrossRef CAS PubMed.
- S. Tadesse, M. Yu, L. B. Mekonnen, F. Lam, S. Islam, K. Tomusange, M. H. Rahaman, B. Noll, S. K. Basnet and T. J. Teo, Highly potent, selective, and orally bioavailable 4-thiazol-N-(pyridin-2-yl) pyrimidin-2-amine cyclin-dependent kinases 4 and 6 inhibitors as anticancer drug candidates: design, synthesis, and evaluation, J. Med. Chem., 2017, 60(5), 1892–1915 CrossRef CAS PubMed.
- D. Triki and D. Fourches, Palbociclib and Ribociclib analogs targeting CDK4 and CDK6 for breast cancer treatment: A molecular modeling study, ChemRxiv, 2020, preprint, Cambridge, Cambridge Open Engage, DOI:10.26434/chemrxiv.12902504.v1.
- P. Wang, J. Huang, K. Wang and Y. J. Gu, New palbociclib analogues modified at the terminal piperazine ring and their anticancer activities, Eur. J. Med. Chem., 2016, 122, 546–556 CrossRef CAS PubMed.
- R. Lamb, S. Lehn, L. Rogerson, R. B. Clarke and G. J. Landberg, Cell cycle regulators cyclin D1 and CDK4/6 have estrogen receptor-dependent divergent functions in breast cancer migration and stem cell-like activity, Cell Cycle, 2013, 12(15), 2384–2394 CrossRef CAS PubMed.
- M. Barvian, D. H. Boschelli, J. Cossrow, E. Dobrusin, A. Fattaey, A. Fritsch, D. Fry, P. Harvey, P. Keller and M. J. Garrett, Pyrido [2, 3-d] pyrimidin-7-one inhibitors of cyclin-dependent kinases, J. Med. Chem., 2000, 43(24), 4606–4616 CrossRef CAS PubMed.
- T. A. Chohan, J.-J. Chen, H.-Y. Qian, Y.-L. Pan and J.-Z. J. Chen, Molecular modeling studies to characterize N-phenylpyrimidin-2-amine selectivity for CDK2 and CDK4 through 3D-QSAR and molecular dynamics simulations, Mol. BioSyst., 2016, 12(4), 1250–1268 RSC.
- H. Zhao, X. Hu, K. Cao, Y. Zhang, K. Zhao, C. Tang and B. J. Feng, Synthesis and SAR of 4, 5-dihydro-1H-pyrazolo [4, 3-h] quinazoline derivatives as potent and selective CDK4/6 inhibitors, Eur. J. Med. Chem., 2018, 157, 935–945 CrossRef CAS PubMed.
- P. Chen, N. V. Lee, W. Hu, M. Xu, R. A. Ferre, H. Lam, S. Bergqvist, J. Solowiej, W. Diehl and Y.-A. J. He, Spectrum and Degree of CDK Drug Interactions Predicts Clinical PerformanceSpectrum and Degree of CDK Drug Engagement, Mol. Cancer Ther., 2016, 15(10), 2273–2281 CrossRef CAS PubMed.
- L. Gavara, L. Sevaille, F. De Luca, P. Mercuri, C. Bebrone, G. Feller, A. Legru, G. Cerboni, S. Tanfoni and D. J. Baud, 4-Amino-1, 2, 4-triazole-3-thione-derived Schiff bases as metallo-β-lactamase inhibitors, Eur. J. Med. Chem., 2020, 208, 112720 CrossRef CAS PubMed.
- Z. Shi and Z. J. Zhao, Microwave irradiation synthesis of novel indole triazole Schiff base fluorescent probe for Al3+ ion, Inorg. Chim. Acta, 2019, 498, 119135 CrossRef CAS.
- S. A. Al-Hussain, T. A. Farghaly, M. E. Zaki, H. G. Abdulwahab, N. T. Al-Qurashi and Z. A. J. B. C. Muhammad, Discovery of novel indolyl-1, 2, 4-triazole hybrids as potent vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors with potential anti-renal cancer activity, Bioorg. Chem., 2020, 105, 104330 CrossRef CAS PubMed.
- R. Hamdy, A. T. Jones, M. El-Sadek, A. M. Hamoda, S. B. Shakartalla, Z. M. Al Shareef, S. S. Soliman and A. D. J. Westwell, New bioactive fused triazolothiadiazoles as Bcl-2-targeted anticancer agents, Int. J. Mol. Sci., 2021, 22(22), 12272 CrossRef CAS PubMed.
- A. T. Boraei, A. A. Sarhan, S. Yousuf and A. J. Barakat, Synthesis of a New Series of Nitrogen/Sulfur Heterocycles by Linking Four Rings: Indole; 1, 2, 4-Triazole; Pyridazine; and Quinoxaline, Molecules, 2020, 25(3), 450 CrossRef PubMed.
- M. Darestani-Farahani, F. Faridbod and M. R. J. Ganjali, A sensitive fluorometric DNA nanobiosensor based on a new fluorophore for tumor suppressor gene detection, Talanta, 2018, 190, 140–146 CrossRef CAS PubMed.
- S. M. Gomha and S. M. J. Riyadh, Synthesis under microwave irradiation of [1, 2, 4] triazolo [3, 4-b][1, 3, 4] thiadiazoles and other diazoles bearing indole moieties and their antimicrobial evaluation, Molecules, 2011, 16(10), 8244–8256 CrossRef CAS PubMed.
- M. S. Nafie and A. T. J. Boraei, Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer, Bioorg. Chem., 2022, 122, 105708 CrossRef CAS PubMed.
- A. Alsaygh, J. Al-Humaidi and I. Al-Najjar, Synthesis of some new pyridine-2-yl-benzylidene-imines, Int. J. Org. Chem., 2014, 2014, 116–121 CrossRef.
- M. Bakkar, M. Monshi, I. Warad, M. Siddiqui and A. J. Bahajaj, 1H 13C NMR investigation of E/Z-isomerization around CN bond in the trans-alkene-Pt (II) imine complexes of some ketimines and aldimines, J. Saudi Chem. Soc., 2010, 14(2), 165–174 CrossRef CAS.
- M. M. Ahmed, F. Fatima, A. Alali, M. A. Kalam, K. Alhazzani, S. Bhatia, S. Alshehri and M. M. Ghoneim, Ribociclib-Loaded Ethylcellulose-Based Nanosponges: Formulation, Physicochemical Characterization, and Cytotoxic Potential against Breast Cancer, Adsorpt. Sci. Technol., 2022, 2022, 1922263 Search PubMed.
- M. K. Anwer, F. Fatima, M. M. Ahmed, M. F. Aldawsari, A. S. Alali, M. A. Kalam, A. Alshamsan, M. Alkholief, A. Malik and A. J. Az, Abemaciclib-loaded ethylcellulose based nanosponges for sustained cytotoxicity against MCF-7 and MDA-MB-231 human breast cancer cells lines, Saudi Pharm. J., 2022, 726–734 CrossRef CAS PubMed.
- Q. Li, X.-E. Jian, Y.-F. Ma, L. Chen, X.-S. Huo, Y. Wang, R.-X. He, W.-W. You and P.-L. J. Zhao, Synthesis and antiproliferative evaluation of novel 8-cyclopentyl-7, 8-dihydropteridin-6 (5H)-one derivatives as potential anticancer agents, Bioorg. Med. Chem. Lett., 2021, 31, 127684 CrossRef CAS PubMed.
- A. Polisety, G. Misra, J. Rajawat, A. Katiyar, H. Singh and A. N. J. Bhatt, Therapeutic natural compounds Enzastaurin and Palbociclib inhibit MASTL kinase activity preventing breast cancer cell proliferation, Med. Oncol., 2022, 39(5), 1–9 Search PubMed.
- J. Carmichael, W. G. DeGraff, A. F. Gazdar, J. D. Minna and J. B. J. Mitchell, Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing, Cancer Res., 1987, 47(4), 936–942 CAS.
- F. Denizot and R. J. Lang, Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability, J. Immunol. Methods, 1986, 89(2), 271–277 CrossRef CAS PubMed.
- J. M. Edmondson, L. S. Armstrong and A. O. J. Martinez, A rapid and simple MTT-based spectrophotometric assay for determining drug sensitivity in monolayer cultures, J. Tissue Cult. Methods, 1988, 11(1), 15–17 CrossRef CAS.
- D. M. Morgan, Tetrazolium (MTT) assay for cellular viability and activity, in Polyamine protocols, Springer, 1998, pp. 179–184 Search PubMed.
- D. T. Vistica, P. Skehan, D. Scudiero, A. Monks, A. Pittman and M. R. J. Boyd, Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production, Cancer Res., 1991, 51(10), 2515–2520 CAS.
- J. Gray, F. Dolbeare, M. Pallavicini, W. Beisker and F. Waldman, Related Studies in Physics, Cell cycle analysis using flow cytometry, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1986, 49(2), 237–255 CrossRef CAS PubMed.
- C. Jayat and M. H. J. Ratinaud, Cell cycle analysis by flow cytometry: principles and applications, Biol. Cell, 1993, 78(1–2), 15–25 CrossRef CAS PubMed.
- R. J. Nunez, DNA measurement and cell cycle analysis by flow cytometry, Curr. Issues Mol. Biol., 2001, 3(3), 67–70 CAS.
- P. Pozarowski and Z. Darzynkiewicz, Analysis of cell cycle by flow cytometry, in Checkpoint controls and cancer, Springer, 2004, pp. 301–311 Search PubMed.
- E. Miller, Apoptosis measurement by annexin v staining, in Cancer cell culture, Springer, 2004, pp. 191–202 Search PubMed.
- Y. Shounan, X. Feng and P. J. J. O'Connell, Apoptosis detection by annexin V binding: a novel method for the quantitation of cell-mediated cytotoxicity, J. Immunol. Methods, 1998, 217(1–2), 61–70 CrossRef CAS PubMed.
- M. O. Sarhan, S. S. Abd El-Karim, M. M. Anwar, R. H. Gouda, W. A. Zaghary and M. A. J. Khedr, Discovery of New Coumarin-Based Lead with Potential Anticancer, CDK4 Inhibition and Selective Radiotheranostic Effect: Synthesis, 2D & 3D QSAR, Molecular Dynamics, In Vitro Cytotoxicity, Radioiodination, and Biodistribution Studies, Molecules, 2021, 26(8), 2273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00524k |
| This journal is © The Royal Society of Chemistry 2024 |