
Recent advances of phenotypic screening strategies in the application of anti-influenza virus drug discovery
Huinan
Jia†
a,
Lide
Hu†
a,
Jiwei
Zhang
a,
Xing
Huang
a,
Yuanmin
Jiang
a,
Guanyu
Dong
a,
Chuanfeng
Liu
*ab,
Xinyong
Liu
 *a,
Meehyein
Kim
*c and
Peng
Zhan
*a
*a,
Meehyein
Kim
*c and
Peng
Zhan
*a
aDepartment of Medicinal Chemistry, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, 44 West Culture Road, 250012, Jinan, Shandong, P.R. China. E-mail: liuchuanfeng214@163.com; xinyongl@sdu.edu.cn; zhanpeng1982@sdu.edu.cn
bSuzhou Research Institute of Shandong University, Room 607, Building B of NUSP, No. 388 Ruoshui Road, SIP, Suzhou, Jiangsu 215123, P.R. China
cInfectious Diseases Therapeutic Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon 34114, Korea. E-mail: mkim@krict.re.kr
First published on 9th November 2023
Abstract
Seasonal and pandemic influenza virus infections not only pose a serious threat to human health but also cause tremendous economic losses and social burdens. However, due to the inherent high variability of influenza virus RNA genomes, the existing anti-influenza virus drugs have been frequently faced with the clinical issue of emerging drug-resistant mutants. Therefore, there is an urgent need to develop efficient and broad-spectrum antiviral agents against wild-type and drug-resistant mutant strains. Phenotypic screening has been widely employed as a reliable strategy to evaluate antiviral efficacy of novel agents independent of their modes of action, either directly targeting viral proteins or regulating cellular factors involved in the virus life cycle. Here, from the point of view of medicinal chemistry, we review the research progress of phenotypic screening strategies by focusing direct acting antivirals against influenza virus. It could provide scientific insights into discovery of a distinctive class of therapeutic candidates that ensure high efficiency but low cytotoxicity, and address issues from circulation of drug-resistant influenza viruses in the future.
1. Introduction
Influenza is a contagious respiratory disease caused by influenza virus. This virus belongs to the Orthomyxoviridae family with eight-segmented, single-stranded negative-sense RNA genomes and can be classified into four different types (type A, B, C and D).1–3 Among them, influenza A virus (IAV), which infects poultry and mammals including humans, has been identified as the main pathogen responsible for influenza pandemics. Every year, the number of seasonal influenza (IAV and influenza B virus) infections worldwide is as high as 1 billion, resulting in 3 million to 5 million severe cases and 290![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 650000 deaths.4–7 Therefore, it is still an important task to prevent and treat influenza viral infections.
000 to 650000 deaths.4–7 Therefore, it is still an important task to prevent and treat influenza viral infections.
With the in-depth exploration of influenza virus, the virus life cycle can be divided into following six steps (Fig. 1).8,9
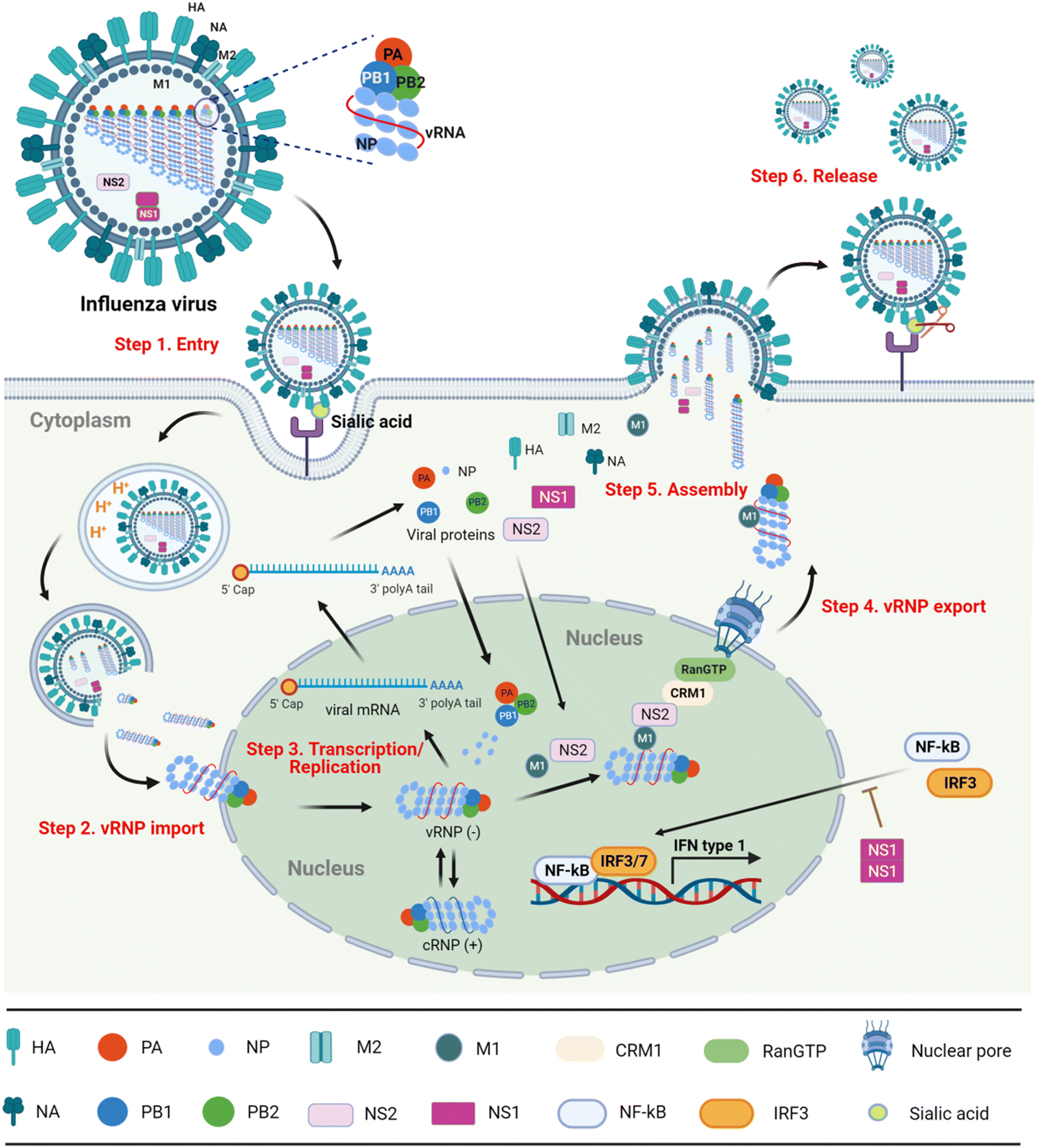 | ||
| Fig. 1 The life cycle of influenza A virus (the image was created in https://BioRender.com). | ||
Step 1) entry into the host cell
The hemagglutinin (HA) on the surface of the virus particles recognizes and binds to the sialic acid (SA) receptors on the surface of host cells. After the receptor-mediated endocytosis, the acidic environment of the endosome induces the conformational change of HA that promotes virus–endosome membrane fusion and opens the M2 ion channel. Finally, the viral genome is released into the cytoplasm in the form of nucleosomes named viral ribonucleoproteins (vRNPs).10Step 2) importing vRNPs into the nucleus
The viral proteins vRNPs are composed of nucleoproteins (NPs), polymerase acidic (PA) protein, polymerase basic protein 1 (PB1) and polymerase basic protein 2 (PB2). Migration of vRNPs into the nucleus relies on the nuclear localization signals (NLSs) existing within NPs.11,12Step 3) transcription and replication of viral genomes
Since the influenza virus itself cannot synthesize the 5′ cap primers necessary for mRNA transcription, it needs to capture the 5′ cap structure of the host cellular mRNA, namely ‘cap-snatching’. In this process, the cap binding region of PB2 first recognizes the 5′ cap structure of the cellular precursor mRNA (pre-mRNA) randomly, and then endonuclease PA cleaves at the 3′ end of a guanine base of 10–13 nucleotides from the cap, generating 5′ cap primers. The resulting 5′-capped oligonucleotide fragment triggers viral mRNA transcription under the catalytic activity of RNA-dependent RNA polymerase (RdRp) PB1. In parallel, the replication intermediate complementary RNA (cRNA) synthesized by viral RdRp can be used as a template for amplifying the copies of negative-sense vRNAs that are coated with NPs for constructing vRNPs again.13–15Step 4) export of vRNPs from the nucleus
Newly synthesized vRNPs are exported from the nucleus with the assistance of NP-bound M1.16,17 In detail, as M1 also interacts with NS2 (nonstructural protein 2), named nuclear export protein (NEP), the vRNP–M1–NS2 complex is able to penetrate the nuclear pore in host chromosome maintenance protein 1 (CRM1) in a GTP-loaded Ran-dependent manner.Step 5) assembly and budding
After the vRNPs exit the nucleus, the assembled complex with M1 is further transported to the cell membrane, on which HA, neuraminidase (NA) and M2 are spiked to form progeny viral particles. The assembled viral particles bud from the apical side of polarized cells.18Step 6) release
Finally, the terminal SA residue between HA and the cell receptor is cleaved by NA, and the progeny virions are released from the infected cells.19In summary, blocking any step of the virus life cycle can efficiently inhibit influenza virus replication. Therefore, the viral structural and non-structural proteins have been primarily targeted for the design of antiviral drugs.
In order to effectively prevent the replication process of influenza virus, pharmacologists have developed diverse influenza virus inhibitors to various targets. Currently approved anti-influenza virus drugs (Fig. 2) mainly include M2 ion channel blockers: amantadine (1) and rimantadine (2), NA inhibitors: zanamivir (3), oseltamivir phosphate (4), peramivir (5) and laninamivir octanoate (6), and a PA inhibitor: baloxavir marboxil (7).20–22
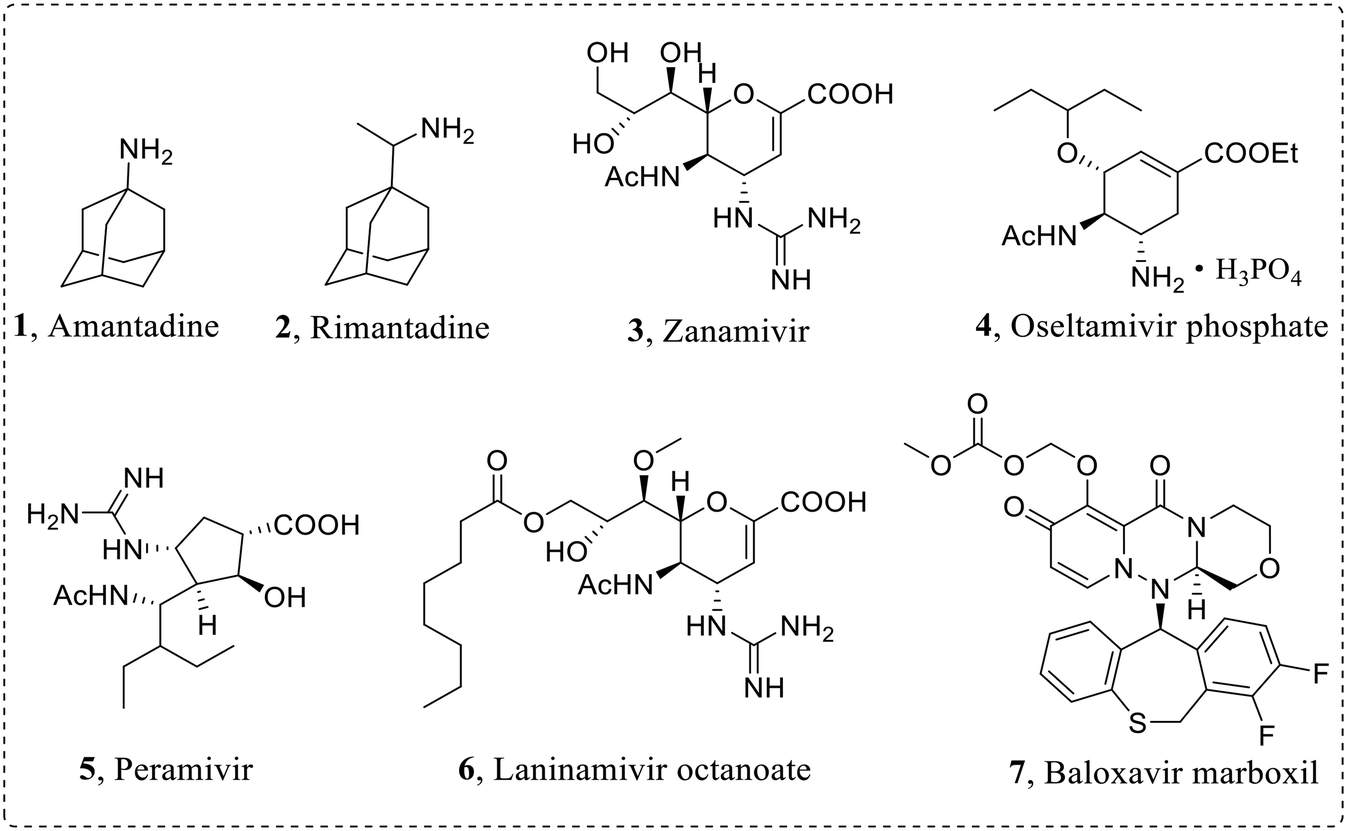 | ||
| Fig. 2 Structures of approved anti-influenza virus drugs. | ||
In the early stage, the existing antivirals have achieved remarkable results in the prevention and treatment of influenza. However, due to the highly error-prone properties of viral RdRp, drug-resistant viruses with reduced drug sensitivity emerge consistently, and their ability of human-to-human transmission threatens again public health. For example, the emergence of M2-V27A mutant strains leads to drug resistance of M2 ion channel inhibitors.23,24 In addition, M2 ion channel blockers have central nervous system adverse reactions, and the Centers for Disease Control and Prevention (CDC) no longer recommends the use of such drugs for the treatment of IAV.23 The first-line anti-influenza virus drug oseltamivir is frequently limited in clinical application due to the emergence of drug-resistant strains, such as N1-H274Y mutant.25,26 Moreover, as the first marketed PA inhibitor, baloxavir has the strongest anti-influenza A and B virus activity, but there are also drug-resistant mutants such as I38T mutant.27,28 At the same time, the mutation of influenza virus also leads to cross-resistance to different anti-influenza virus drugs.27 Therefore, it is urgent to develop anti-drug resistant influenza virus inhibitors with new skeletons and distinctive mechanisms.
Phenotypic screening strategies play an important role in the discovery of innovative drugs with discriminated mechanisms or to first-in-class targets.29–31 Originally, they have been based on evaluation of therapeutic efficacy of drug candidates in animal disease models by observing the phenotypic changes such as body weight, survival, mobility or feeding. According to the development of cellular and molecular biology, this concept has been applied to the minimized system of organic metabolism such as cells, proteins, and even genes, which allow more elaborate pharmacological studies.32,33
Importantly, compared with traditional target-based drug screening, the advantage of the phenotypic screening strategy is to expand the space of drug targets and new mechanisms of action, and to screen out drugs with low molecular weight.34,35 Target-based drug design plays a dominant role in drug discovery strategies. However, 90.6% of the small molecule drugs approved for marketing were found by phenotype-based screening strategies. In addition, data show that the number of approved first-in-class small-molecule drugs identified through phenotypic screening each year exceeds the number found through molecular target-based drug design methods,36 and this strategy has been extensively applied to the research and development of antitumor drugs,37 anti-inflammatory drugs,38 antimalarial drugs39,40 and antibacterial drugs.41,42
The conventional approach to drug phenotypic screening involves the use of animal disease models to observe the effects of drugs. However, this method is not suitable for high-throughput screening of small molecule sample banks. Therefore, it is primarily used for evaluating approved drugs or later-stage drug candidates. With the advancement of technology, it has become feasible to use minute components such as cells, proteins, and genes that constitute organisms. Among these components, the utilization of cell-based phenotypic screening is widespread due to its inherent advantages, including a reduced sample size, decreased screening costs, and simplified assessment of drug efficacy.33 In recent years, cell-based phenotypic screening approach has also been widely used in the development of antiviral agents against influenza virus. Therefore, from the perspective of medicinal chemistry, we summarized its progress in the cell culture-based evaluation of antiviral efficacy with selected representative research examples. On the basis of the influenza virus life cycle, the antiviral agents validated in virus-infected cell culture systems are divided into the following categories: influenza glycoprotein inhibitors, polymerase inhibitors, NP inhibitors, non-structural protein 1 (NS1) inhibitors, other inhibitors and host-targeted inhibitors.
2. Influenza virus glycoprotein inhibitors
2.1 NA inhibitors
Wang et al.43 screened active compounds against influenza virus (H5N1) from twenty-three phloroglucinols isolated from Dryopteris crassirhizoma. Firstly, molecular docking was performed and found that compounds dryocrassin ABBA (8) and filixic acid ABA (9, Fig. 3) could dock into the active sites of NA. Furthermore, compounds 8 and 9 exhibited inhibitory effects on NA with IC50 values of 18.59 μmol L−1 and 29.57 μmol L−1, respectively in NA inhibition assay. In addition, compound 8 displayed inhibitory activity against H5N1 with an EC50 value of 16.55 μmol L−1 but no cytotoxicity in MDCK cells (CC50 > 400 μmol L−1). In short, the study unveiled the potential utilization of Dryopteris crassirhizoma in the exploration of anti-influenza drugs.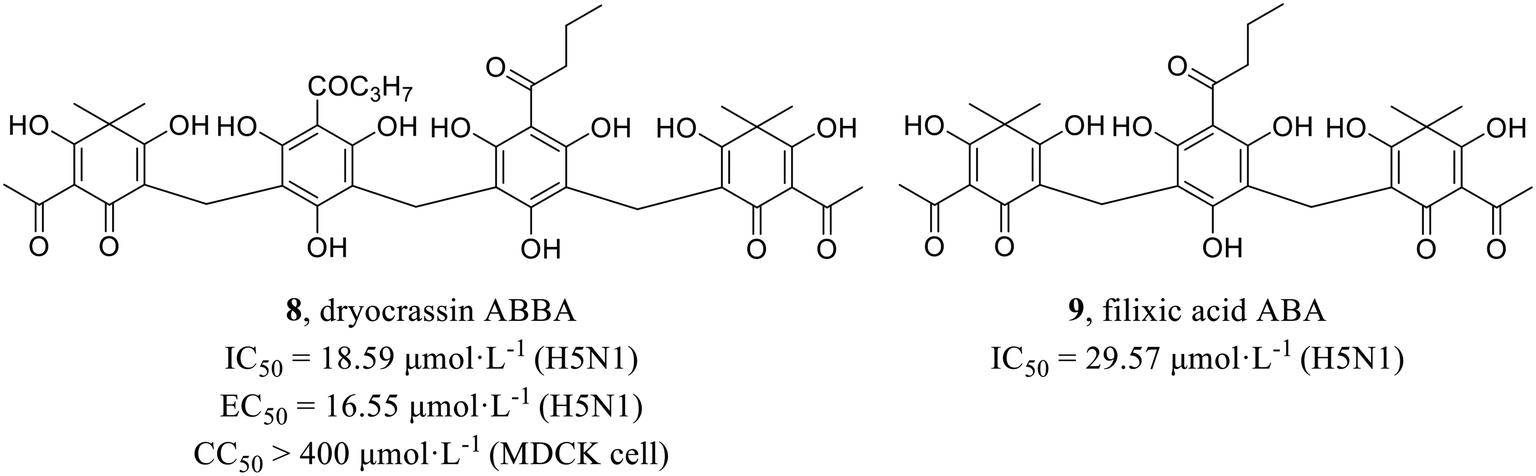 | ||
| Fig. 3 Structures of phloroglucinol derivatives 8 and 9. | ||
Xiao et al.44 isolated phytochemicals from the aqueous extract of olive leaves and tested their antiviral activities. Among them, phenolic glycoside 10 (EC50 = 11.63–29.15 μmol L−1) showed better anti-H1N1 activities than favipiravir (EC50 = 37.42–85.77 μmol L−1) with low cytotoxicity in MDCK cells (10, CC50 = 964.5 μmol L−1; favipiravir, CC50 > 1000 μmol L−1). Then, HA inhibitory assay and NA inhibitory assay were carried out to seek for the target protein. The results showed that compound 10 exhibited NA inhibitory activity. Interestingly, compound 11 (Fig. 4) could suppress both HA and NA at the same time, although its antiviral activities at the cellular level (EC50 = 24.61–30.65 μmol L−1) was less potent than 10. Moreover, molecular docking showed that both 10 and 11 could form multiple hydrogen bonds with amine of Arg152 of NA. This study provided a favorable support for the application of olives in the field of antiviral drug development.
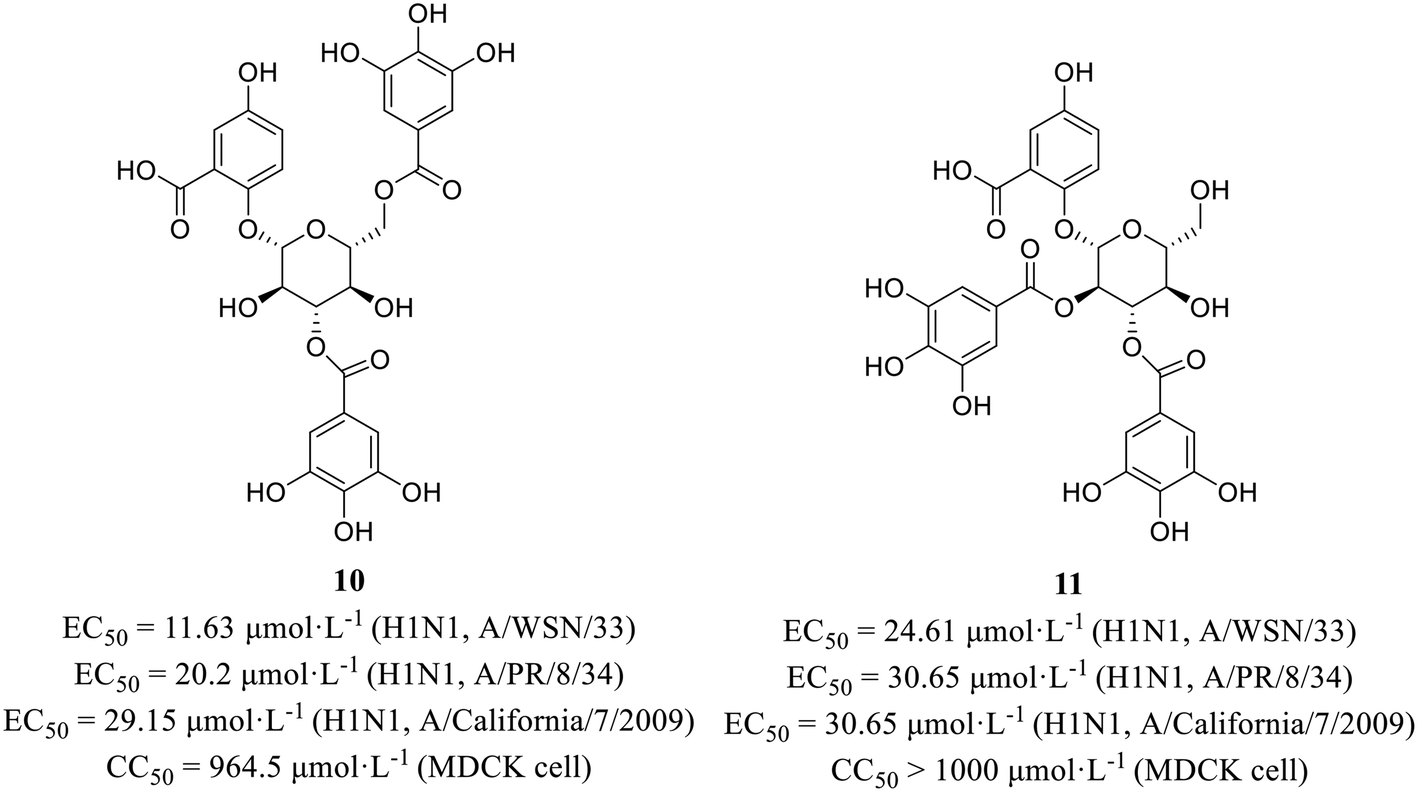 | ||
| Fig. 4 Structures of phenol glycoside derivatives 10 and 11. | ||
2.2 HA inhibitors
In 2010, Kao et al.45 screened from a chemical library containing more than 50000 compounds using cell-based influenza A infection assays and obtained 950 compounds which showed cytoprotective activity. Based on previous work of this group, Lai et al.46 performed a secondary cell-level activity test and identified two compounds FA-583 (12, H1N1, EC50 = 0.0698 μmol L−1; H5N1, EC50 = 0.08 μmol L−1) and FA-617 (13, H1N1, EC50 = 0.007 μmol L−1; H5N1, EC50 = 0.0868 μmol L−1, Fig. 5), which showed potent antiviral activities against different virus subtypes with low cytotoxicity (12, CC50 = 73 μmol L−1; 13, CC50 > 100 μmol L−1). Time-of-compound-addition assay and a pseudotyped virus assay showed that they inhibited the early stage of viral infection. And they also found that these compounds carried mutations near the B-loop of the hemagglutinin 2 (HA2) protein. Further mechanism research showed that they inhibited viruses by interfering with the structural changes of HA for membrane fusion at acidic pH.
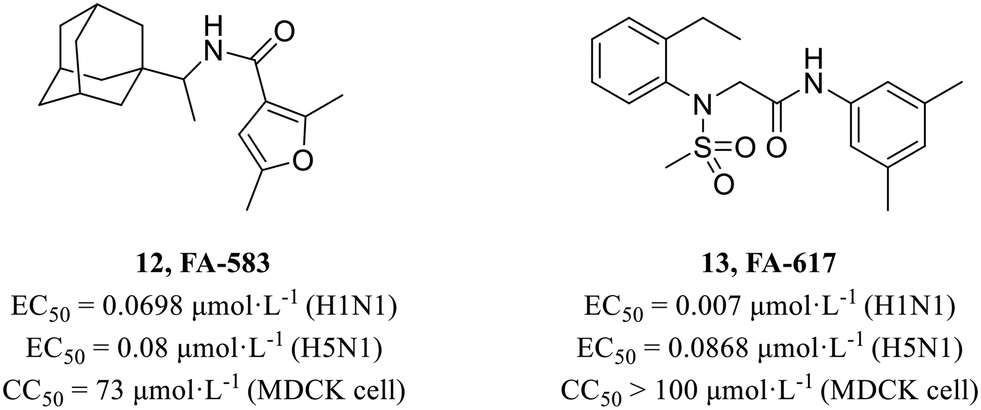 | ||
| Fig. 5 Structures of compounds 12 and 13. | ||
In 2016, Weisshaar et al.47 adopted a high-throughput screening protocol for double myxoviruses, which combined a fully replicated IAV-WSN strain with a respiratory syncytial virus reporter strain to screen a library containing over 142000 compounds. In the primary screening, three structurally distinct scaffolds (Fig. 6) inhibiting influenza H1N1 (EC50 = 0.17–0.38 μmol L−1) with low toxicity in MDCK cells (CC50 > 100 μmol L−1) were selected for further evaluation. Consequently, time-of-addition, minigenome, and viral entry studies were carried out and indicated that these compounds blocked HA2-mediated membrane fusion. Biolayer interferometry and resistance profiling further confirmed the target. In summary, their study identified some novel and potent chemical scaffolds, while also outlining diverse routes of IAV escape from entry inhibition.
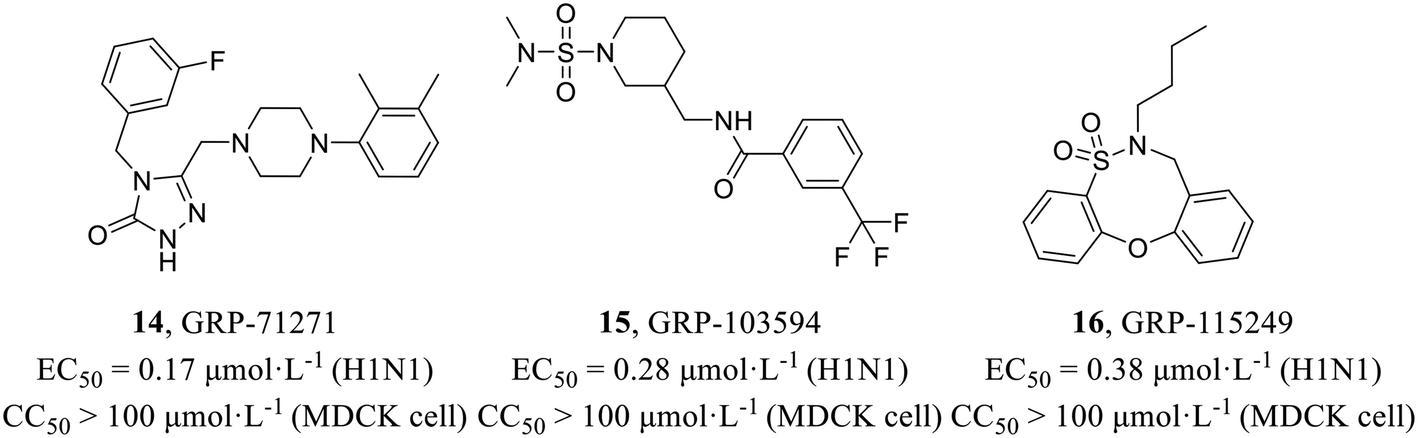 | ||
| Fig. 6 Structures of compounds 14–16. | ||
3. Polymerase inhibitors
3.1 PA inhibitors
In 2012, Yamada et al.48 found a putative PA inhibitor THC19 (17, Fig. 7) via computational screening from about 290000 compounds followed by plaque reduction assay. Time-of-addition experiments proved that it acted on the early stage of the viral life cycle and minigenome assay was conducted to further verify its PA-dependent inhibitory effect. Moreover, it demonstrated antiviral activities against various strains (H1N1, EC50 = 30.7–35.6 μmol L−1; H3N2, EC50 = 44.7 μmol L−1; H5N1, EC50 = 39.8 μmol L−1) and no cytotoxicity in MDCK cells (CC50 > 200 μmol L−1), which could be a starting material to explore the mechanisms of PA endonuclease activity.
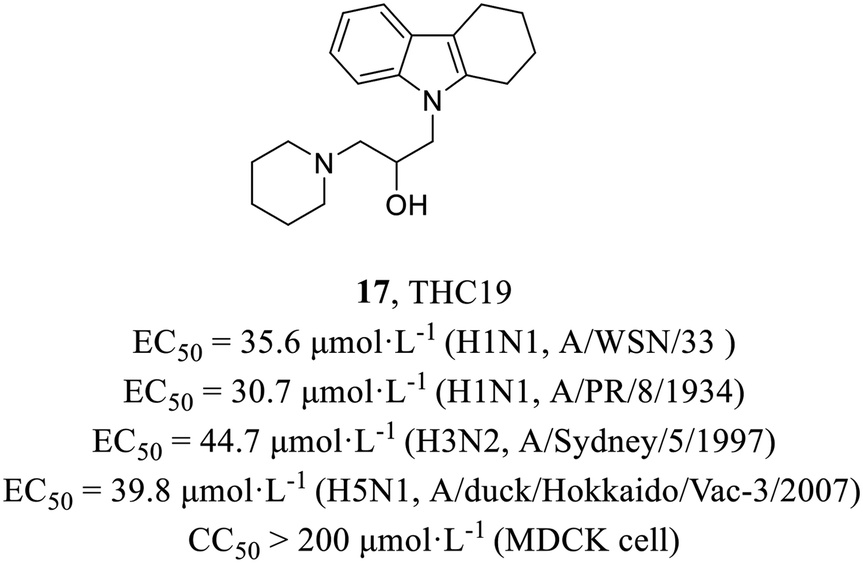 | ||
| Fig. 7 The structure of compound 17. | ||
In 2022, Zhao et al.49 found compound APL-16-5 (18, Fig. 8) by screening from microbial metabolites which exhibited inhibitory activities against IAV in HEK293T cells and influenza B virus in MDCK cells with EC50 values of 0.28 μmol L−1 and 1.22 μmol L−1, respectively. Subsequently, it was found that 18 inhibited influenza virus by diminishing reducing viral RNA levels. Further antiviral mechanism research with compound 18 showed that it induced ubiquitination of PA by engaging E3 ligase TRIM25, and then degraded PA by recruiting them to the proteasome. This mode of action was consistent with proteolysis targeting chimeras (PROTACs), which used the cellular ubiquitin–proteasome machinery to chemically induce the degradation of target proteins. As a key subunit of the influenza virus RdRp complex, the degradation of PA disrupted the synthesis of viral mRNA and consequently inhibited the replication of influenza virus. The research provided the possibility of finding new PROTACs from microbial metabolites due to their structural diversities and biological activities.
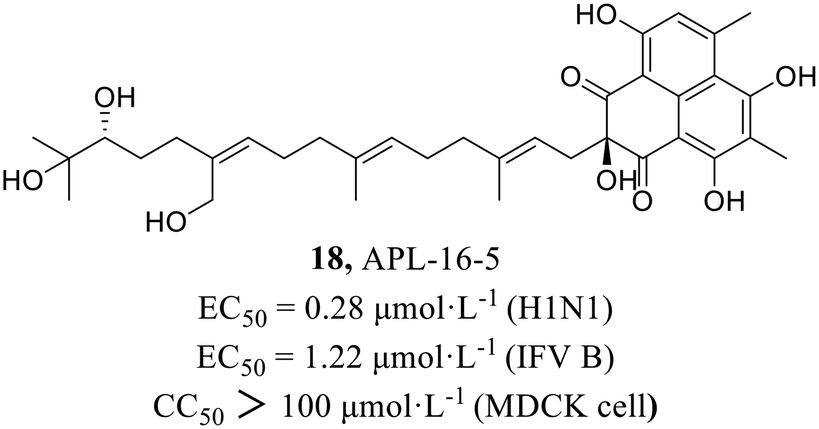 | ||
| Fig. 8 The structure of compound 18. | ||
3.2 PB2 inhibitors
In 2014, Clark et al.50,51 found a class of azaindole-based compounds from 1000 compounds which exerted distinct cell protective activity at 10 μmol L−1 concentration by using cytopathic effect (CPE) assay. A competition binding fluorescence polarization (FP) assay was used to measure the binding affinity of compounds to the cap binding domain of PB2. Among them, the Kd value of compound 19 binding to PB2 reached 0.1 μmol L−1 and its X-ray crystal structure (PDB code: 4NCM, Fig. 9) bound to the PB2 cap-binding domain was resolved, whose azaindole formed hydrogen bonds with the side chains of Glu361 and Lys376 and was between the aromatic side chains of His357 and Phe404, while the pyrimidine ring formed π–π stacking with Phe323. Structure-guided modification pointing at its pyrimidine ring and azaindole ring was carried out and then cyclohexylcarboxylic acid analogue 20 (pimodivir, VX-787) was discovered, which was bound to PB2 with a Kd value of less than 0.003 μmol L−1, more than 30 times higher than that of compound 19. Moreover, pimodivir exhibited anti-IAV activity with EC50 values ranging from 0.15 to 2.8 nmol L−1 and excellent pharmacokinetic properties in rats (AUC = 0.5 μg h mL−1, F = 26%). Unfortunately, the research and development of VX-787 was terminated due to drug metabolism and drug resistance in clinical phase III trials.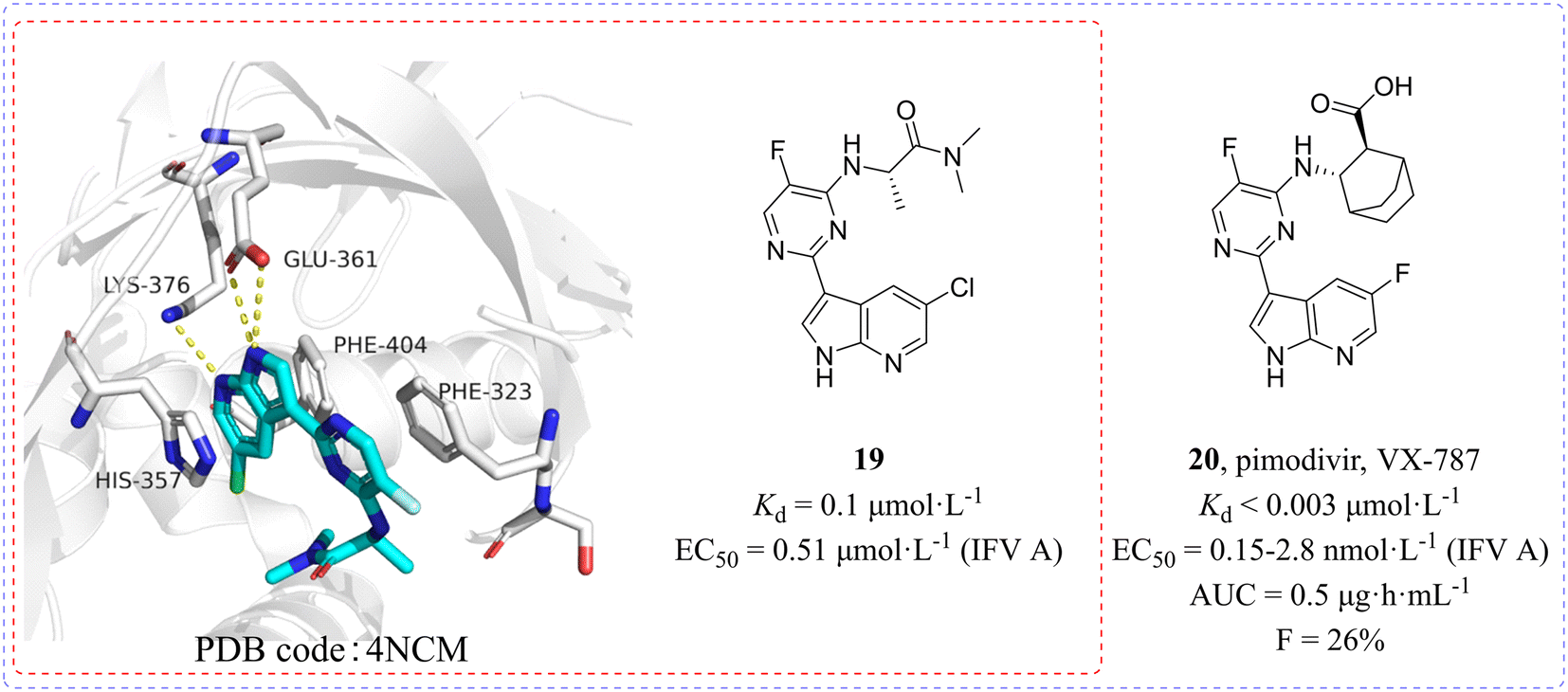 | ||
| Fig. 9 Structures of compounds 19 and 20 and the complex structure of compound 19 bound to the PB2 cap-binding domain. | ||
4. NP inhibitors
In 2010, Su et al.52 established a screening procedure to identify new anti-influenza inhibitors from 1200000 compounds. Compound 3061 (21, Fig. 10) as the most potent inhibitor could inhibit the replication of multiple influenza virus strains (H1N1, EC50 = 0.07–4.2 μmol L−1; H3N2, EC50 = 0.7 μmol L−1; H5N1, EC50 = 0.32 μmol L−1) in MDCK cells. Molecular mechanism studies have found that 3061 (21) targeted the influenza NP with an IC50 value of 0.1 μmol L−1 against reconstituted RdRp but led to resistance with the Y52H mutation on the NP (IC50 > 30 μmol L−1). In addition, they found another compound, 367 (22), targeting PB1, which showed an inhibitory effect on RdRp enzymatic activity (IC50 = 0.3–1 μmol L−1), and the mutant recombinant virus with H456P substitution in PB1 as well as the corresponding mutant protein expressed in the cell-based viral polymerase assay system had elevated resistance to 22 (IC50 > 100 μmol L−1). In summary, the screening yielded two distinct classes of influenza virus inhibitors, thereby establishing a solid groundwork for the development of novel anti-influenza drugs.
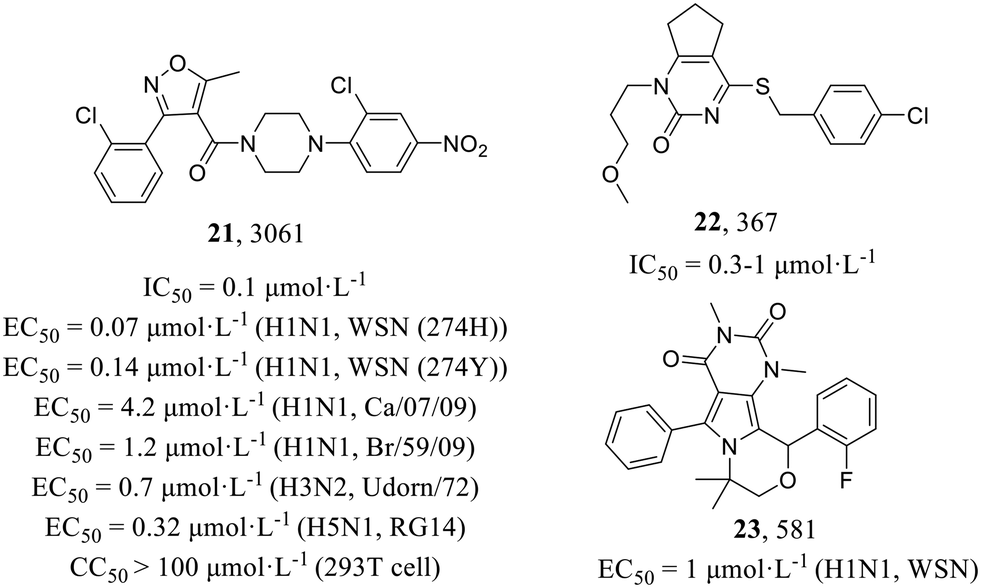 | ||
| Fig. 10 Structures of compounds 21–23. | ||
Compound 581 (23, Fig. 10) was identified as a strong anti-influenza pyrimido-pyrrolo-quinoxalinedione (PPQ) analogue (H1N1, EC50 = 1 μmol L−1) by Su et al. in 2010.52 Lin et al.53 further characterized its antiviral mechanism consequently and suggested that 23 possessed similar anti-influenza effects to 21, not only inhibiting viral protein synthesis at the early stage of H1N1 infection but also affecting vRNP nuclear export and its cytoplasmic aggregation at a later time. They discovered that the recombinant influenza virus carrying the S377G NP mutation was resistant to 23 in anti-influenza and viral RNA polymerase assays and lacked 23-mediated RNP nuclear retention and cytoplasmic aggregation activity, but not resistant to NP inhibitor 21. Similarly, the 21-resistant IAV was not resistant to 23, suggesting that 23 targeted a different site from 21.
He et al.54 examined the antiviral activities of 15 amaryllidaceae alkaloids isolated from the bulbs of L. radiata against IAV H5N1, and AA1 (24, Fig. 11) was found to be significantly active against H5N1 (EC50 < 0.46 μmol L−1) with low cytotoxicity in MDCK cells (CC50 = 20.9 μmol L−1). A series of mechanism experiments suggested that 24 could not affect the function of viral membrane proteins, viral invasion process and viral RNP activity. However, intracellular NP localization explained that 24 inhibited the nuclear export of the vRNP complex at the single-cycle or multi-cycle replication stage. The compound exhibited potential as an inhibitor of IAV, but further investigation is required to elucidate its precise mechanism of action for development.
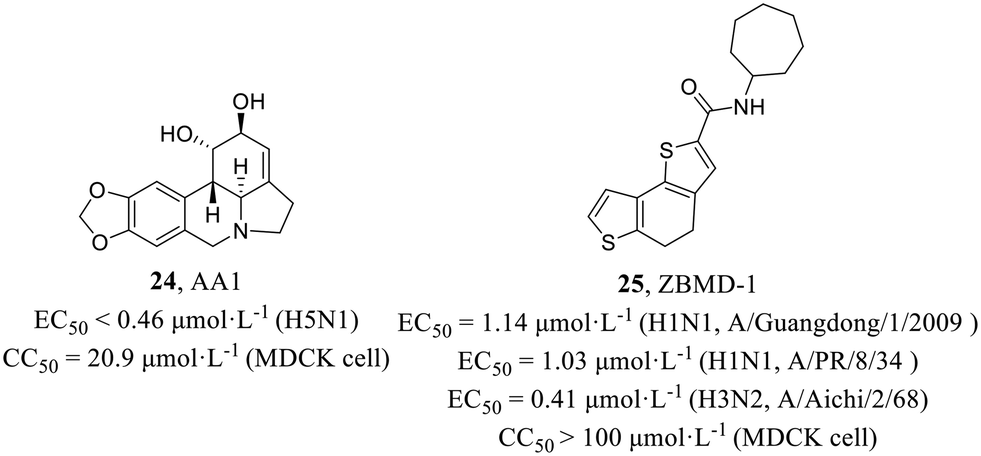 | ||
| Fig. 11 Structures of compounds 24 and 25. | ||
Huang et al.55 screened from a library of 20000 compounds by cell-based IAV infection assays, presenting a new anti-influenza compound, ZBMD-1 (25, Fig. 11), which inhibited the replication of H1N1 and H3N2 with EC50 values of 1.03–1.14 μmol L−1 and 0.41 μmol L−1, respectively and had low cytotoxicity in MDCK, A549 and 293T cells (CC50 > 100 μmol L−1). At the same time, 25 protected the mice challenged with lethal doses of A/PR/8/1934 (H1N1) virus and effectively reduced pathological changes in lung tissues without inducing toxic effects in vivo. Moreover, 25 inhibited the polymerase activity and specifically blocked the nuclear export of NPs through binding to the nuclear export signal 3 (NES3) domain within NPs and inhibiting its oligomerization. Taken together, this screening identified a compound exhibiting potent inhibition of IAV replication both in vitro and in vivo, thereby establishing its potential as a promising candidate for an anti-influenza virus drug.
In 2022, Wang et al.56 employed an in situ-generated directing group strategy and a macrocyclization strategy to efficiently synthesize diverse complex macrolactams, thereby creating a screening library comprising various macrocycles. On the basis of the phenotypic screening principle, several oxime-containing macrolactams (Fig. 12) were found to show strong anti-H1N1 activity (26, EC50 = 2.3 μmol L−1; 27, EC50 = 0.57 μmol L−1) with no obvious cytotoxic effects (26, CC50 = 210.6 μmol L−1; 27, CC50 > 100 μmol L−1). Furthermore, the CPE inhibition assay and immunofluorescence studies clarified that these compounds exerted antiviral effects by targeting nucleoproteins. All in all, the method they developed facilitated the rapid construction of compound libraries, laying the foundation for phenotypic screening in the quest for effective anti-influenza virus drugs.
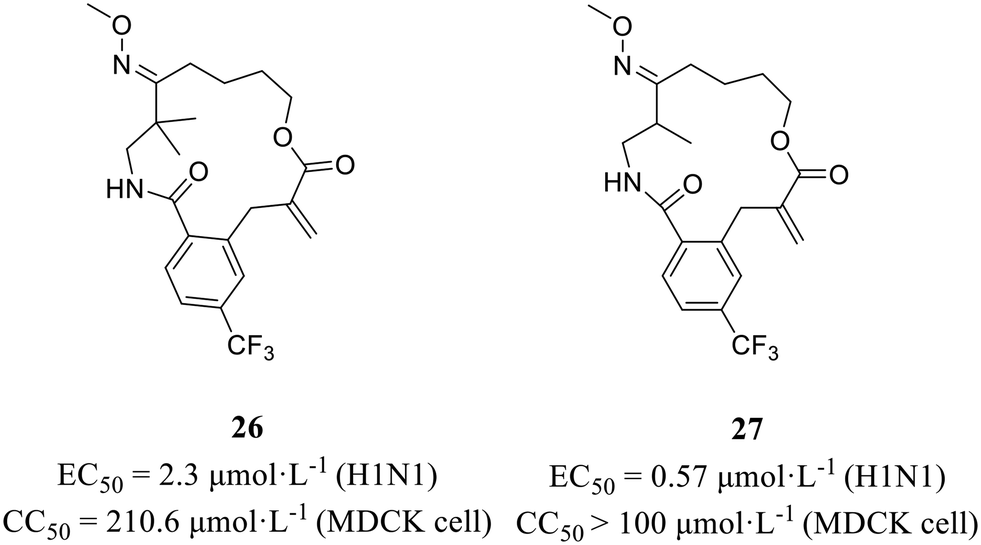 | ||
| Fig. 12 Structures of compounds 26 and 27. | ||
Recently, Li et al.57 utilized a phenotypic screening system based on an influenza luciferase reporter virus to screen an internal library comprising 110 structurally distinct compounds and discovered ligustrazine and chalcone derivative A9 (28, Fig. 13), exhibiting broad spectrum antiviral activities (H1N1, EC50 = 2.74 μmol L−1; H3N2, EC50 = 3.44 μmol L−1; influenza B viruses, EC50 = 2.41–3.13 μmol L−1), including oseltamivir-resistant strain A (H1N1) pdm09 (EC50 = 1.04 μmol L−1). The efficacy of A9 in inhibiting the nuclear export of vRNPs was demonstrated through a series of mechanistic experiments. Moreover, escape mutant analyses and surface plasmon resonance (SPR) experiments (Kd = 6.06 μmol L−1) provided evidence that A9 specifically targeted the NP. In addition, A9 could markedly reduce the viral load in the lung tissues of mice at 30 mg kg−1 day−1 and showed antiviral activity in vivo. Taken together, this was the first report of the antiviral activity of ligustrazine and chalcone derivatives against influenza virus, which provided a potential lead compound for the development of anti-influenza drugs.
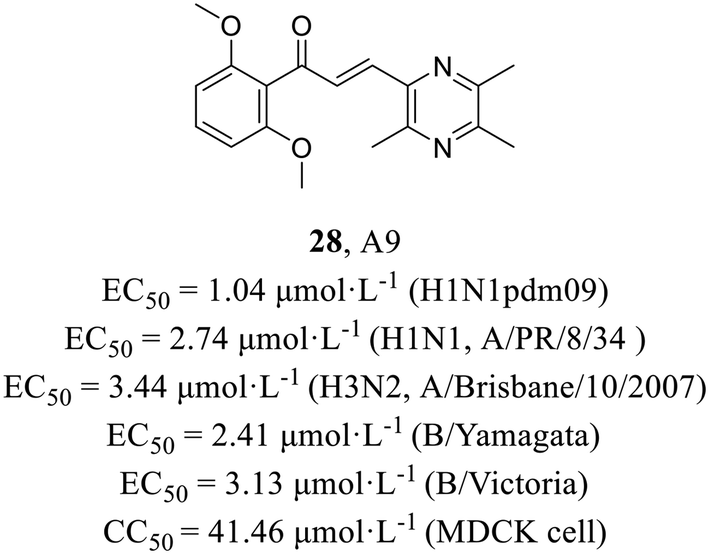 | ||
| Fig. 13 The structure of ligustrazine and chalcone derivative 28. | ||
5. NS1 inhibitors
NS1 is an immunosuppressive molecule that antagonizes type I interferon signaling, playing a critical role in influenza virus evasion of host innate immune response (Fig. 1). More recently, Marsili et al.58 conducted a cell-based screening assay expressing IFN-β promoter-driven luciferase to discover inhibitors targeting NS1 from a commercial library of 84 molecules. Finally, compounds 29 and 30 (Fig. 14) were selected based on their potent anti-NS1 activity. These compounds exhibited significant inhibitory effects against H1N1 with EC50 values of 51.6 μmol L−1 and 46.4 μmol L−1, respectively, while demonstrating no cytotoxicity (29, CC50 > 400 μmol L−1; 30, CC50 >150 μmol L−1). At the same time, obvious 2.7- and 3.2-fold increases in IFN-β mRNA accumulation were observed with 29 and 30, respectively, proving that the two compounds inhibited viral replication by restoring IFN-β expression in human lung epithelial A549 cells.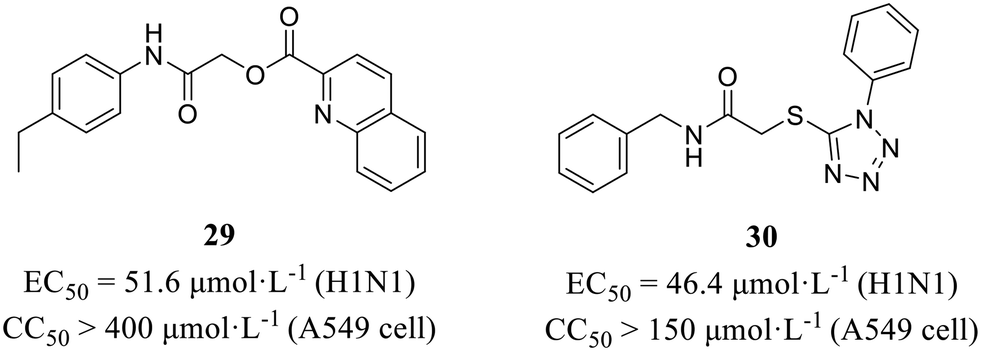 | ||
| Fig. 14 Structures of compounds 29 and 30. | ||
6. Other inhibitors based on phenotypic screening
Maddry et al.59 adopted a cell-based HTS assay to screen 100000 compounds from the NIH Molecular Libraries Screening Center Network (MLSCN) compound library at 50 μM against H5N1 virus and identified 5 active compounds (SI > 3). Further activity evaluation showed that benzoquinazolinone SR#22700 (31, Fig. 15) had inhibitory activity on H1N1 and H5N1 with EC50 values of 0.098 μmol L−1 and 0.046 μmol L−1, respectively and high selectivity (H1N1, SI > 510; H5N1, SI > 1087). The mechanism studies revealed that 31 distinctly reduced the M1 RNA levels as compared to the no-drug MDCK cells infected with influenza viruses.
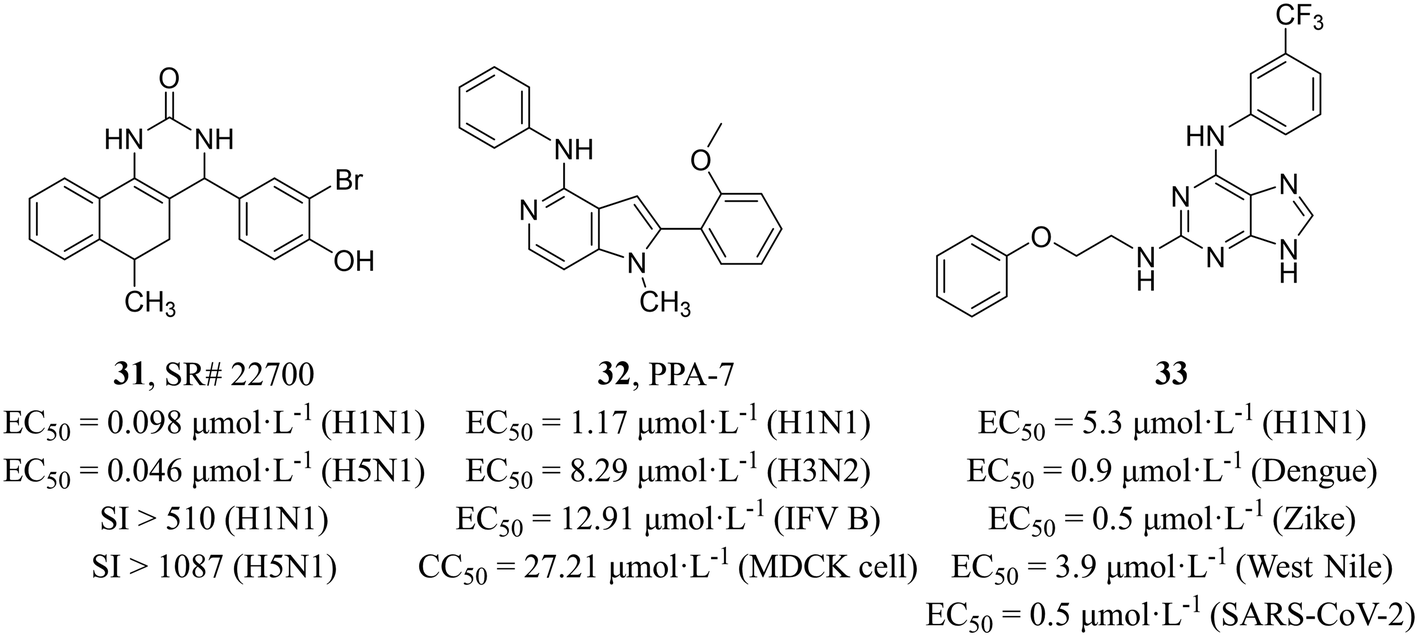 | ||
| Fig. 15 Structures of compounds 31–33. | ||
Chang et al.60 screened compounds with inhibitory activity against IAV from 4000 small molecules of different structural types sourced from the BioFocus library by using GFP-expressing H1N1 virus (PR8), and pyrrolopyridinamine (PPA) derivative PPA-7 (32, Fig. 15) was found to have broad-spectrum activity for IAV and influenza B virus (H1N1, EC50 = 1.17 μmol L−1; H3N2, EC50 = 8.29 μmol L−1; influenza B virus, EC50 = 12.91 μmol L−1) and low cytotoxicity in MDCK cells (CC50 = 27.21 μmol L−1). Compound 32 was verified to block virus entry by the time-of-addition assay. Furthermore, image-based assays were performed to analyze its antiviral mechanism and found that 32 disrupted the virus uncoating and vRNP nuclear import during the influenza virus entry process. Unfortunately, the precise target of 32 during influenza virus invasion remained unclear and required further investigation.
Vicenti et al.61 reported a class of 2,6-diaminopurine broad-spectrum antiviral drugs from phenotypic screening on cell cultures and a system-oriented optimization. Among them, compound 33 (Fig. 15) exhibited great potency against IAV and other vector-borne flaviviruses, such as dengue, Zika and West Nile viruses (EC50 = 0.5–5.3 μmol L−1). At the same time, they verified the activity against various influenza strains (the human H1N1 (PR8), the pandemic 2009 H1N1 and the avian Ulster H7N1) by using time-of-addition assay. In addition, compound 33 also showed excellent anti-SARS-CoV-2 activity in pulmonary Calu-3 cells (EC50 = 0.5 μmol L−1) with high selectivity (SI = 240). All in all, the compound exhibited a diverse range of antiviral activities and held potential for further development as a broad-spectrum antiviral drug.
7. Host-targeted inhibitor
Host-targeted compounds are less sensitive to drug resistance resulting from viral mutations and have the potential for exhibiting broad-spectrum antiviral activity. O'Hanlon et al.62 screened from 9000000 compounds using cell-based assay and finally identified compound M85 (34, Fig. 16) with potent activities against both IAV (EC50 = 2.05 μmol L−1) and influenza B virus (EC50 = 0.4 μmol L−1) and low cytotoxicity in MDCK cells and A549 cells (CC50 > 50 μmol L−1). In addition, it also demonstrated broad-spectrum antiviral activities against various other viruses (H5N1, EC50 = 1.77 μmol L−1; H3N2, EC50 = 1.22 μmol L−1; hepatitis C virus, EC50 = 0.76 μmol L−1; human rhinovirus 14, EC50 = 0.05 μmol L−1). A series of mechanistic experiments proved that 34 targeted host kinases, epidermal growth factor receptor (EGFR, IC50 = 79 nmol L−1) and phosphoinositide 3 class II β (PIK3C2β, IC50 = 430 nmol L−1) and blocks endocytosis of viruses. In addition, 34 showed a strong synergistic effect with oseltamivir both in vitro and in vivo. To summarize, this research represented the initial documentation of PIK3C2β as a crucial host factor facilitating influenza virus entry, and paved the way for potential combination therapy in the treatment of influenza.
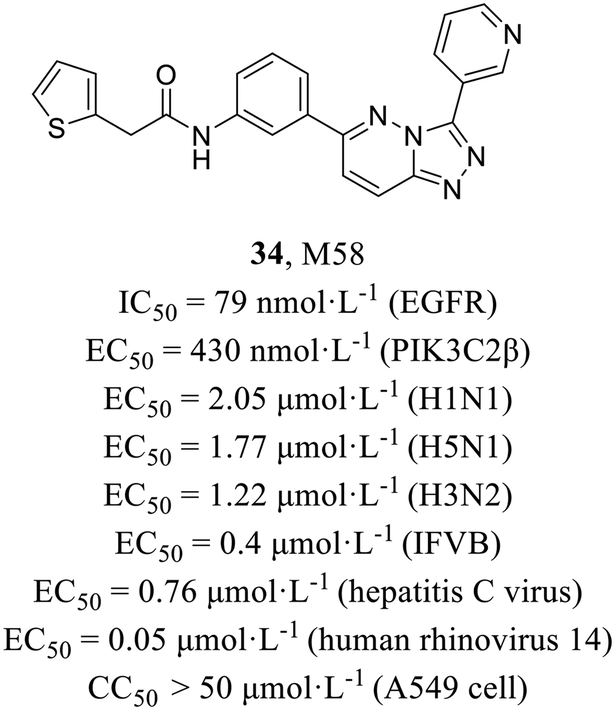 | ||
| Fig. 16 The structure of compound 34. | ||
8. Conclusions
At present, influenza viruses still pose a continuous threat to public health. This virus is prone to variation, resulting in the emergence of reassortant strains or drug-resistant variants, which makes the influenza vaccine lag and cause antiviral resistance. Therefore, it is necessary to develop high-efficiency, low-toxicity, and anti-drug resistant influenza virus inhibitors.In order to overcome drug resistance, researchers have developed many drug design strategies, such as covalent binding to catalytic sites,63 multi-site binding within a single target molecule,64 PROTAC technology65 and phenotypic screening.66 These drug design options have their own advantages in overcoming drug resistance. Among them, the phenotypic screening approach can support drug discovery and lead compounds to a first-in-class target, which are unique advantages particularly in overcoming drug resistance. Therefore, here we summarize its applications in the discovery of antiviral drugs at different steps with the understanding of the life cycle of influenza virus.
Drug discovery based on phenotypic screening to drug targets is directly related to the biological disease model and can preemptively exclude compounds that do not penetrate the cell membrane. Most of all, different kinds of compounds can be selected at one time to multiple targets and with multiple mechanisms. These targets are likely to be entirely novel, so there is strong potential for overcoming emergence of drug-resistant variants. However, because the target molecules are unknown, it is necessary to elucidate their action mechanisms through additional molecular virological studies or characterization of proteins binding to the compounds. Technologies that have been discovered so far include labeling-based chemoproteomics, chemoproteomic-enabled phenotypic screening libraries, label-free methods, and artificial intelligence (AI), which still need further development. And finally, target validation is also indispensable after finding putative target molecules.30,67,68
Author contributions
The writing of the first draft was initiated by Huinan Jia and Lide Hu. Jiwei Zhang, Xing Huang, Yuanmin Jiang and Guanyu Dong put forward constructive suggestions. Chuanfeng Liu, Xinyong Liu, Meehyein Kim and Peng Zhan edited the manuscript. Meehyein Kim and Peng Zhan reviewed and approved the final manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors have received financial support from the National Natural Science Foundation of China (NSFC No. 82173677 and 82211530493), the Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31), the Natural Science Foundation of Jiangsu Province (SBK2023041680) and the China Postdoctoral Science Foundation (2022M711938). This study was also supported by the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (HI22C2067).References
- E. C. Hutchinson, Trends Microbiol., 2018, 26, 809–810 CrossRef CAS.
- C. Adlhoch, M. Sneiderman, O. Martinuka, A. Melidou, N. Bundle, J. Fielding, S. J. Olsen, P. Penttinen, L. Pastore and R. Pebody, Eurosurveillance, 2021, 26, 2100077 CAS.
- J. To and J. Torres, Cell, 2019, 8, 654 CrossRef CAS PubMed.
- T. Flerlage, D. F. Boyd, V. Meliopoulos, P. G. Thomas and S. Schultz-Cherry, Nat. Rev. Microbiol., 2021, 19, 425–441 CrossRef CAS.
- W. N. Harrington, C. M. Kackos and R. J. Webby, Exp. Mol. Med., 2021, 53, 737–749 CrossRef CAS.
- N. J. Roberts, Jr. and L. R. Krilov, Viruses, 2022, 14, 883 CrossRef PubMed.
- P. Yazici Özkaya, E. E. Turanli, H. Metin, A. Aydın Uysal, C. Çiçek and B. Karapinar, J. Med. Virol., 2022, 94, 575–581 CrossRef.
- T. Samji, Yale J. Biol. Med., 2009, 82, 153–159 CAS.
- R. Du, Q. Cui, Z. Chen, X. Zhao, X. Lin and L. Rong, Virol. Sin., 2023, 38, 1–8 CrossRef CAS PubMed.
- L. Hou, Y. Zhang, H. Ju, S. Cherukupalli, R. Jia, J. Zhang, B. Huang, A. Loregian, X. Liu and P. Zhan, Acta Pharm. Sin. B, 2022, 12, 1805–1824 CrossRef CAS.
- X. Liu, J. Liang, Y. Yu, X. Han, L. Yu, F. Chen, Z. Xu, Q. Chen, M. Jin, C. Dong, H. B. Zhou, K. Lan and S. Wu, J. Med. Chem., 2022, 65, 3814–3832 CrossRef CAS.
- A. P. Walker and E. Fodor, Trends Microbiol., 2019, 27, 398–407 CrossRef CAS.
- A. J. Te Velthuis and E. Fodor, Nat. Rev. Microbiol., 2016, 14, 479–493 CrossRef CAS.
- Z. Zhou, T. Liu, J. Zhang, P. Zhan and X. Liu, Drug Discovery Today, 2018, 23, 503–518 CrossRef CAS.
- F. T. Vreede and E. Fodor, Virulence, 2010, 1, 436–439 CrossRef PubMed.
- C. T. Höfer, F. Jolmes, I. Haralampiev, M. Veit and A. Herrmann, Cell. Microbiol., 2017, 19, e12679 CrossRef PubMed.
- K. Ma, A. M. Roy and G. R. Whittaker, Virology, 2001, 282, 215–220 CrossRef CAS.
- S. S. Lakdawala, E. Fodor and K. Subbarao, Annu. Rev. Virol., 2016, 3, 411–427 CrossRef CAS PubMed.
- A. Jagadesh, A. A. Salam, P. P. Mudgal and G. Arunkumar, Arch. Virol., 2016, 161, 2087–2094 CrossRef CAS.
- H. Ju, J. Zhang, B. Huang, D. Kang, B. Huang, X. Liu and P. Zhan, J. Med. Chem., 2017, 60, 3533–3551 CrossRef CAS.
- R. Voelker, JAMA, 2021, 325, 20 Search PubMed.
- R. Jia, J. Zhang, J. Zhang, C. Bertagnin, A. Bonomini, L. Guizzo, Z. Gao, X. Ji, Z. Li, C. Liu, H. Ju, X. Ma, A. Loregian, B. Huang, P. Zhan and X. Liu, Molecules, 2022, 27, 6426 CrossRef CAS PubMed.
- Y. Hu, R. Musharrafieh, C. Ma, J. Zhang, D. F. Smee, W. F. DeGrado and J. Wang, Antiviral Res., 2017, 140, 45–54 CrossRef CAS.
- F. G. Hayden, S. J. Sperber, R. B. Belshe, R. D. Clover, A. J. Hay and S. Pyke, Antimicrob. Agents Chemother., 1991, 35, 1741–1747 CrossRef CAS.
- N. Lee and A. C. Hurt, Curr. Opin. Infect. Dis., 2018, 31, 520–526 CrossRef CAS PubMed.
- E. Takashita, S. Fujisaki, M. Shirakura, K. Nakamura, N. Kishida, T. Kuwahara, Y. Shimazu, T. Shimomura, S. Watanabe and T. Odagiri, Eurosurveillance, 2016, 21, 1560–7917 CrossRef.
- F. G. Hayden, N. Sugaya, N. Hirotsu, N. Lee, M. D. de Jong, A. C. Hurt, T. Ishida, H. Sekino, K. Yamada, S. Portsmouth, K. Kawaguchi, T. Shishido, M. Arai, K. Tsuchiya, T. Uehara and A. Watanabe, N. Engl. J. Med., 2018, 379, 913–923 CrossRef CAS PubMed.
- J. H. Park, B. Kim, K. J. C. Antigua, J. H. Jeong, C. I. Kim, W. S. Choi, S. Oh, C. H. Kim, E. G. Kim, Y. K. Choi, Y. H. Baek and M. S. Song, Antiviral Res., 2021, 193, 105126 CrossRef CAS PubMed.
- J. G. Moffat, F. Vincent, J. A. Lee, J. Eder and M. Prunotto, Nat. Rev. Drug Discovery, 2017, 16, 531–543 CrossRef CAS.
- L. P. Conway, W. Li and C. G. Parker, Cell Chem. Biol., 2021, 28, 371–393 CrossRef CAS PubMed.
- E. Chatelain and J. R. Ioset, Expert Opin. Drug Discovery, 2018, 13, 141–153 CrossRef CAS.
- F. Vincent, A. Nueda, J. Lee, M. Schenone, M. Prunotto and M. Mercola, Nat. Rev. Drug Discovery, 2022, 21, 899–914 CrossRef CAS.
- W. Zheng, N. Thorne and J. C. McKew, Drug Discovery Today, 2013, 18, 1067–1073 CrossRef CAS.
- D. C. Swinney, Clin. Pharmacol. Ther., 2013, 93, 299–301 CrossRef CAS.
- I. H. Gilbert, J. Med. Chem., 2013, 56, 7719–7726 CrossRef CAS PubMed.
- A. Sadri, J. Med. Chem., 2023, 66, 12651–12677 CrossRef CAS PubMed.
- O. D. Staples, J. J. Hollick, J. Campbell, M. Higgins, A. R. McCarthy, V. Appleyard, K. E. Murray, L. Baker, A. Thompson, S. Ronseaux, A. M. Slawin, D. P. Lane, N. J. Westwood and S. Lain, Cell Cycle, 2008, 7, 3417–3427 CrossRef CAS PubMed.
- D. M. DuBreuil, X. Lai, K. Zhu, G. Chahyadinata, C. Perner, B. M. Chiang, A. Battenberg, C. L. Sokol and B. J. Wainger, Mol. Pain, 2023, 19, 17448069221148351 CrossRef.
- M. L. Hovlid and E. A. Winzeler, Trends Parasitol., 2016, 32, 697–707 CrossRef CAS PubMed.
- R. Kaur, L. A. Sloan, A. D. Blanchard, J. L. Smith, I. Churcher, G. J. Wayne and S. B. Ludbrook, J. Biomol. Screening, 2013, 18, 1223–1233 CrossRef PubMed.
- W. G. Cochrane, P. R. Fitzgerald and B. M. Paegel, ACS Chem. Biol., 2021, 16, 2752–2756 CrossRef CAS PubMed.
- D. D. C. Silva, R. F. Rampelotto, V. V. Lorenzoni, S. O. D. Santos, J. Damer, M. Hörner and R. Hörner, Rev. Soc. Bras. Med. Trop., 2017, 50, 173–178 CrossRef PubMed.
- J. Wang, Y. T. Yan, S. Z. Fu, B. Peng, L. L. Bao, Y. L. Zhang, J. H. Hu, Z. P. Zeng, D. H. Geng and Z. P. Gao, Molecules, 2017, 22, 431 CrossRef PubMed.
- M. Xiao, B. Lim-Ho Kong, Y. Zhang, Y. Yang, J. Lu, H. K. Lee, F. Cao and P. C. Shaw, J. Ethnopharmacol., 2022, 292, 115175 CrossRef CAS.
- R. Y. Kao, D. Yang, L. S. Lau, W. H. Tsui, L. Hu, J. Dai, M. P. Chan, C. M. Chan, P. Wang, B. J. Zheng, J. Sun, J. D. Huang, J. Madar, G. Chen, H. Chen, Y. Guan and K. Y. Yuen, Nat. Biotechnol., 2010, 28, 600–605 CrossRef CAS PubMed.
- K. K. Lai, N. N. Cheung, F. Yang, J. Dai, L. Liu, Z. Chen, K. H. Sze, H. Chen, K. Y. Yuen and R. Y. Kao, J. Virol., 2015, 90, 2690–2701 CrossRef.
- M. Weisshaar, R. Cox, Z. Morehouse, S. Kumar Kyasa, D. Yan, P. Oberacker, S. Mao, J. E. Golden, A. C. Lowen, M. G. Natchus and R. K. Plemper, J. Virol., 2016, 90, 7368–7387 CrossRef CAS PubMed.
- K. Yamada, H. Koyama, K. Hagiwara, A. Ueda, Y. Sasaki, S. N. Kanesashi, R. Ueno, H. K. Nakamura, K. Kuwata, K. Shimizu, M. Suzuki and Y. Aida, Microbes Infect., 2012, 14, 740–747 CrossRef CAS PubMed.
- J. Zhao, J. Wang, X. Pang, Z. Liu, Q. Li, D. Yi, Y. Zhang, X. Fang, T. Zhang, R. Zhou, T. Zhang, Z. Guo, W. Liu, X. Li, C. Liang, T. Deng, F. Guo, L. Yu and S. Cen, Nat. Commun., 2022, 13, 2079 CrossRef CAS PubMed.
- S. Pautus, P. Sehr, J. Lewis, A. Fortuné, A. Wolkerstorfer, O. Szolar, D. Guilligay, T. Lunardi, J. L. Décout and S. Cusack, J. Med. Chem., 2013, 56, 8915–8930 CrossRef CAS.
- M. P. Clark, M. W. Ledeboer, I. Davies, R. A. Byrn, S. M. Jones, E. Perola, A. Tsai, M. Jacobs, K. Nti-Addae, U. K. Bandarage, M. J. Boyd, R. S. Bethiel, J. J. Court, H. Deng, J. P. Duffy, W. A. Dorsch, L. J. Farmer, H. Gao, W. Gu, K. Jackson, D. H. Jacobs, J. M. Kennedy, B. Ledford, J. Liang, F. Maltais, M. Murcko, T. Wang, M. W. Wannamaker, H. B. Bennett, J. R. Leeman, C. McNeil, W. P. Taylor, C. Memmott, M. Jiang, R. Rijnbrand, C. Bral, U. Germann, A. Nezami, Y. Zhang, F. G. Salituro, Y. L. Bennani and P. S. Charifson, J. Med. Chem., 2014, 57, 6668–6678 CrossRef CAS PubMed.
- C. Y. Su, T. J. Cheng, M. I. Lin, S. Y. Wang, W. I. Huang, S. Y. Lin-Chu, Y. H. Chen, C. Y. Wu, M. M. Lai, W. C. Cheng, Y. T. Wu, M. D. Tsai, Y. S. Cheng and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19151–19156 CrossRef CAS PubMed.
- M. I. Lin, B. H. Su, C. H. Lee, S. T. Wang, W. C. Wu, P. Dangate, S. Y. Wang, W. I. Huang, T. J. Cheng, O. A. Lin, Y. S. Cheng, Y. J. Tseng and C. M. Sun, Eur. J. Med. Chem., 2015, 102, 477–486 CrossRef CAS PubMed.
- J. He, W. B. Qi, L. Wang, J. Tian, P. R. Jiao, G. Q. Liu, W. C. Ye and M. Liao, Influenza Other Respir. Viruses, 2013, 7, 922–931 CrossRef CAS PubMed.
- F. Huang, J. Chen, J. Zhang, L. Tan, G. Lu, Y. Luo, T. Pan, J. Liang, Q. Li, B. Luo, H. Zhang and G. Lu, J. Cell. Mol. Med., 2018, 22, 1826–1839 CrossRef CAS PubMed.
- H. Wang, Z. Li, X. Chen, J. J. Wong, T. Bi, X. Tong, Z. Xu, M. Zhen, Y. Wan, L. Tang, B. Liu, X. Zong, D. Xu, J. Zuo, L. Yang, W. Huang, K. N. Houk and W. Yang, Chem, 2023, 9, 607–623 CAS.
- P. Li, H. Ju, Y. Zhang, J. G. Achi, D. Kang, J. Zou, R. Du, Q. Cui, X. Liu, L. Rong and P. Zhan, J. Med. Virol., 2023, 95, e28968 CrossRef CAS PubMed.
- G. Marsili, C. Acchioni, A. L. Remoli, D. Amatore, R. Sgarbanti, M. De Angelis, R. Orsatti, M. Acchioni, A. Astolfi, N. Iraci, S. Puzelli, M. Facchini, E. Perrotti, V. Cecchetti, S. Sabatini, F. Superti, M. Agamennone, M. L. Barreca, J. Hiscott, L. Nencioni and M. Sgarbanti, Int. J. Mol. Sci., 2023, 24, 10495 CrossRef CAS PubMed.
- J. A. Maddry, X. Chen, C. B. Jonsson, S. Ananthan, J. Hobrath, D. F. Smee, J. W. Noah, D. Noah, X. Xu, F. Jia, C. Maddox, M. I. Sosa, E. L. White and W. E. Severson, J. Biomol. Screening, 2011, 16, 73–81 CrossRef CAS PubMed.
- S. Y. Chang, D. J. Cruz, Y. Ko and J. Y. Min, Biochem. Biophys. Res. Commun., 2016, 478, 1594–1601 CrossRef CAS PubMed.
- I. Vicenti, M. G. Martina, A. Boccuto, M. De Angelis, G. Giavarini, F. Dragoni, S. Marchi, C. M. Trombetta, E. Crespan, G. Maga, C. Eydoux, E. Decroly, E. Montomoli, L. Nencioni, M. Zazzi and M. Radi, Eur. J. Med. Chem., 2021, 224, 113683 CrossRef CAS PubMed.
- R. O'Hanlon, V. H. Leyva-Grado, M. Sourisseau, M. J. Evans and M. L. Shaw, ACS Infect. Dis., 2019, 5, 1779–1793 CrossRef.
- I. Deni, B. H. Stokes, K. E. Ward, K. J. Fairhurst, C. F. A. Pasaje, T. Yeo, S. Akbar, H. Park, R. Muir, D. S. Bick, W. Zhan, H. Zhang, Y. J. Liu, C. L. Ng, L. A. Kirkman, J. Almaliti, A. E. Gould, M. Duffey, A. J. O'Donoghue, A. C. Uhlemann, J. C. Niles, P. C. A. da Fonseca, W. H. Gerwick, G. Lin, M. Bogyo and D. A. Fidock, Cell Chem. Biol., 2023, 30, 470–485.e6 CrossRef CAS PubMed.
- R. Jia, J. Zhang, W. Ai, X. Ding, S. Desta, L. Sun, Z. Sun, X. Ma, Z. Li, D. Wang, B. Huang, P. Zhan and X. Liu, Eur. J. Med. Chem., 2019, 178, 64–80 CrossRef CAS PubMed.
- G. T. Lee, N. Nagaya, J. Desantis, K. Madura, H. E. Sabaawy, W. J. Kim, R. J. Vaz, G. Cruciani and I. Y. Kim, Mol. Cancer Ther., 2021, 20, 490–499 CrossRef CAS PubMed.
- Y. Han, J. Shi, Z. Xu, Y. Zhang, X. Cao, J. Yu, J. Li and S. Xu, Front. Pharmacol., 2022, 13, 802168 CrossRef CAS PubMed.
- H. Park, J. Ha and S. B. Park, Curr. Opin. Chem. Biol., 2019, 50, 66–72 CrossRef CAS PubMed.
- X. Liu, H. A. Baarsma, C. H. Thiam, C. Montrone, B. Brauner, G. Fobo, J. S. Heier, S. Duscha, M. Königshoff, V. Angeli, A. Ruepp and M. Campillos, Cell Chem. Biol., 2016, 23, 1302–1313 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |