
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid molecules of protoflavones and spirooxindole derivatives with selective cytotoxicity against triple-negative breast cancer cells†
Gábor
Girst‡
 a,
Elizabeth A.
Lopes‡
b,
Lídia M.
Gonçalves
b,
Margarida
Espadinha
b,
Norbert
Kúsz
a,
Hui-Chun
Wang
c,
Maria M. M.
Santos
*b and
Attila
Hunyadi
*a
a,
Elizabeth A.
Lopes‡
b,
Lídia M.
Gonçalves
b,
Margarida
Espadinha
b,
Norbert
Kúsz
a,
Hui-Chun
Wang
c,
Maria M. M.
Santos
*b and
Attila
Hunyadi
*a
aInstitute of Pharmacognosy, Interdisciplinary Excellence Centre, Faculty of Pharmacy, University of Szeged, Eötvös str. 6, H-6720, Szeged, Hungary. E-mail: hunyadi.attila@szte.hu
bResearch Institute for Medicines, Faculty of Pharmacy, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal. E-mail: mariasantos@ff.ulisboa.pt
cGraduate Institute of Natural Products, Kaohsiung Medical University, Shih-Chuan 1st Rd. 100, Kaohsiung 807, Taiwan
First published on 1st August 2023
Abstract
The combination of compounds with complementary bioactivities into hybrid molecules is an emerging concept in drug discovery. In this study, we aimed to synthesize new hybrid compounds based on p53-MDM2/X protein–protein interaction spiropyrazoline oxindole-based inhibitors and ataxia telangiectasia and Rad3-related (ATR) protoflavone-based inhibitors through copper(I) catalysed azide–alkyne cycloaddition. Five new hybrids were prepared along with three representative reference fragments. The compounds were tested against human breast cancer cell lines MCF-7 (hormone-dependent, wild-type p53) and MDA-MB-231 (triple-negative, mutant p53). Most of the new hybrids were more cytotoxic than their reference fragments and several showed 2–4 times selective toxicity against MDA-MB-231 cells. Relevant pharmacological benefit gained from the hybrid coupling was further confirmed by virtual combination index calculations using the Chou method. Compound 13 modulated doxorubicin-induced DNA damage response through inhibiting the ATR-dependent activation of Chk-1, while increasing the activation of Chk-2. Our results suggest that the new hybrids may serve as new leads against triple negative breast cancer.
Introduction
Breast cancer is the most common cancer worldwide, with more than 2.26 million new cases in women in 2020.1 Triple-negative breast cancer (TNBC) accounts for about 10–15% of all breast cancers.2 It is aggressive and has a poor prognosis. Compared to other breast cancer subtypes, TNBC is more resistant and more likely to recur after treatment.3 Hence, there is an urgent need for the development of novel chemotherapeutics against TNBC.The p53 protein is a tumour suppressor that is activated by several cellular stressors, including DNA-damaging agents and hypoxia. Breast cancers with mutated p53 have poor prognosis.4 For those reasons, p53 is considered a highly relevant antitumor therapeutic target in anticancer treatment. Two main approaches have been developed to activate p53: 1) in cancers with wild-type (wt) p53, inhibition of the two main p53 negative regulators (MDM2 and MDMX); 2) in cancers with mutated p53, restoration of wt tumour suppressor function of mutant p53.5,6 Recently, spiropyrazoline oxindoles (Fig. 1), like compound 1 (Fig. 2) were identified as dual p53-MDM2/X protein–protein interaction inhibitors with inhibitory activity in the nM range, representing valuable leads for the development of more effective p53 activators.7
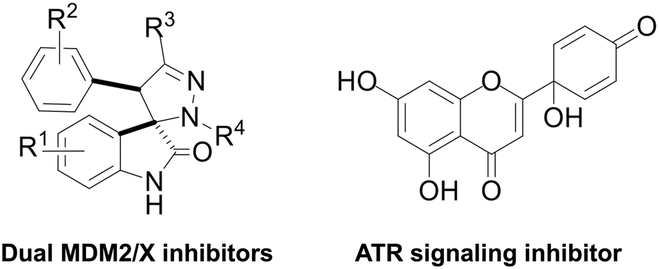 | ||
| Fig. 1 Chemical structure of spiropyrazoline oxindoles (dual MDM2/X inhibitors) and protoapigenone (PA, ATR signaling inhibitor). | ||
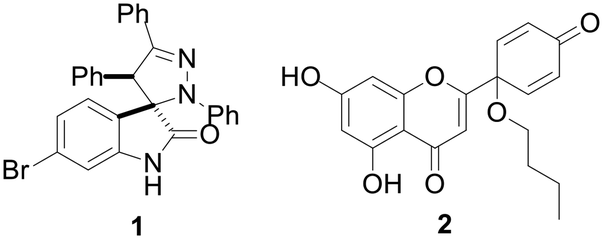 | ||
| Fig. 2 Chemical structure of spiropyrazole oxindole 1 and protoapigenone 1′-O-butyl ether 2. | ||
The ataxia telangiectasia and Rad3-related protein (ATR) is a replication stress-response kinase that also plays a central role in DNA damage response. This kinase is considered a ‘hot-topic’ selective antitumor target, and several promising clinical studies are currently ongoing to obtain the first ATR inhibitor drug.8,9 Among other potential therapeutic benefits, ATR inhibitors were shown to selectively kill p53-deficient cancer cells,10,11 which also makes such agents potential candidates to complement p53-targeted therapeutic approaches.
Protoapigenone (PA, Fig. 1), the protoflavone analogue of apigenin, was previously identified to inhibit the ATR-dependent phosphorylation of checkpoint kinase-1 (Chk1).12 Its 1′-O-butyl ether derivative 2 (Fig. 2) exerts a slightly stronger cytotoxic activity against human breast cancer cell lines and, unlike PA, has a two-fold selective toxicity against the TNBC cell line MDA-MB-231 compared with MCF-7.13 Moreover, 1′-O-coupled protoflavone–chalcone hybrids, exploiting the effect of simultaneous inhibition of ATR signaling (attributed to the protoflavone fragment) and induction of oxidative stress (attributed to both fragments), exerted a greatly improved antitumor activity, indicating that 1′-O-alkyl substitution of protoflavones does not eliminate their effect on ATR-dependent signaling.14
In the current study, we prepared novel hybrid compounds with two different pharmacophores with complementary mechanisms of action. Specifically, we prepared hybrid compounds of protoflavone and spiropyrazoline oxindole derivatives as a multitarget approach and evaluated their antiproliferative activity in breast cancer cell lines MCF-7 and MDA-MB-231.
Results and discussion
Chemistry
The spiropyrazoline oxindole 1 was prepared as previously described.15 Briefly, compound 1 was obtained in 94% yield by reacting 6-bromo-(E)-3-benzylideneindolin-2-one and (Z)-N-phenylbenzohydrazonoyl chloride (Scheme 1).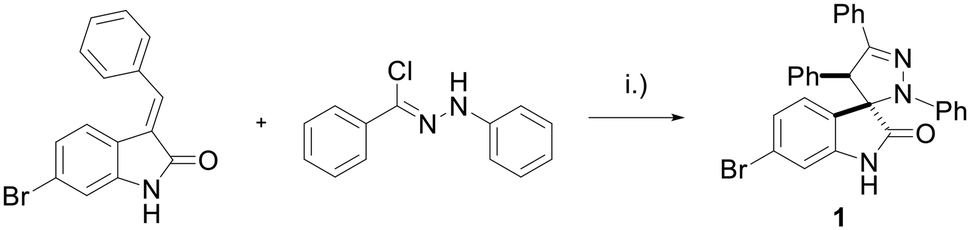 | ||
| Scheme 1 Synthesis of spiropyrazoline oxindole 1. Reaction conditions: (i) NEt3, CH2Cl2, r.t., 5 h, 94%. | ||
Linkers a and b were prepared from the nucleophilic substitution of the corresponding bromoalcohols with sodium azide to afford azidoalcohols, followed by mesylation of the hydroxyl group. Linker c was prepared by mesylation of azido-PEG3-alcohol (Scheme 2). The reaction of spiropyrazoline oxindole 1 with the three linkers led to compounds 3a–c in good yields containing an azide handle for further modification (Scheme 3).
 | ||
| Scheme 2 Synthesis of azide linkers. Reaction conditions: (i) NaN3, H2O, 80 °C, 16 h, 91%; (ii) MsCl, NEt3, CH2Cl2, r.t., 16 h, 91–100%. | ||
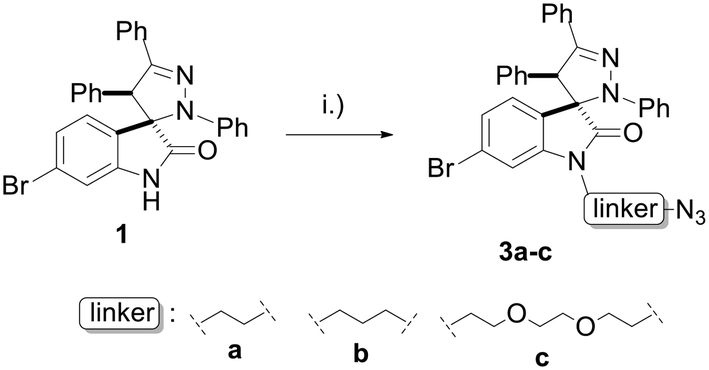 | ||
| Scheme 3 Synthesis of spiropyrazoline oxindoles 3a–c. Reaction conditions: (i) K2CO3, MsO-linker-N3, DMF, 50 °C – r.t., 16 h, 65–85%. | ||
Protoflavonoids 2 and 4 were prepared from apigenin, following a previously reported procedure.13 Protoapigenone 1′-O-butyl ether 2 was then functionalized using methyl iodide and propargyl bromide to obtain 7-O-alkyl derivatives 6 and 7, respectively. The 1′-methoxy derivative 4 and the 7-methoxy derivative 6 were prepared and used as reference fragments for the hybrids coupled at positions C-1′ and C-7, respectively (Scheme 4). The choice of these compounds as controls, instead of our original lead PA, is based on their i) improved chemical stability, and ii) retained effect on ATR-dependent signaling. These make them more relevant reference compounds than the non-substituted PA for a comparative evaluation of antitumor activity of the designed hybrids.
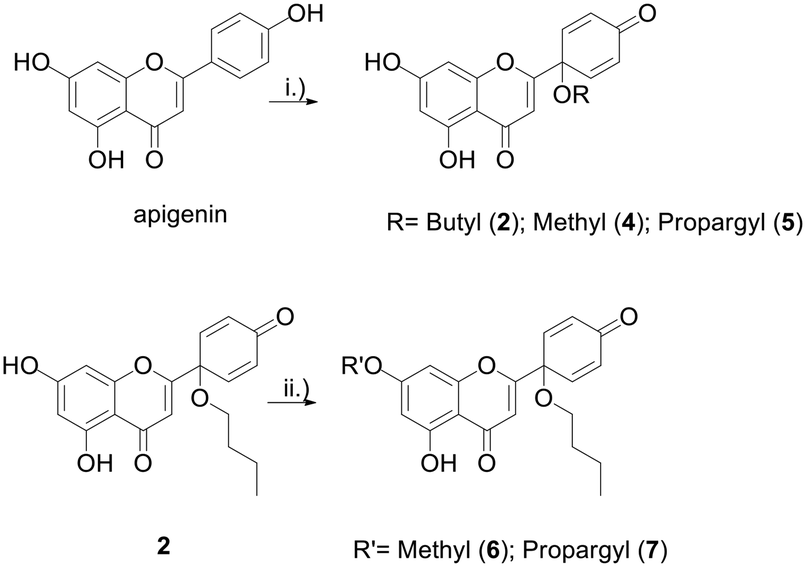 | ||
Scheme 4 Synthesis of protoflavone propargyl derivatives 2 and 4–7. Reaction conditions: (i) PIFA, CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ROH (9:1), 80 °C, 1 h, 30–43%; (ii) K2CO3, MeI/propargylBr-, DMF, rt, 3 h, 72–79%. :ROH (9:1), 80 °C, 1 h, 30–43%; (ii) K2CO3, MeI/propargylBr-, DMF, rt, 3 h, 72–79%. | ||
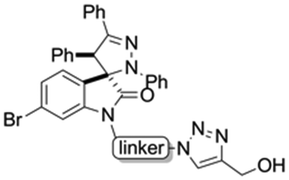
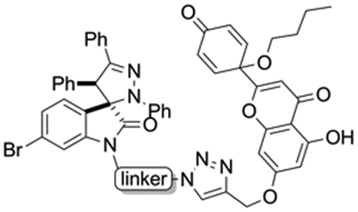
Hybrids 9–13 were prepared from spirooxindoles 3a–c with protoflavone derivatives 5 or 7via copper(I) catalyzed azide–alkyne cycloaddition reaction. Reference compounds 8a–c were also synthesised to evaluate the effect of the N′-substitution of the oxindole moiety as well as the effect of the size and nature of the linker. For that, the same protocol was used for the synthesis of the reference compounds 8a–c (Scheme 5), using propargyl alcohol instead of protoflavones 5 and 7. Structures of the hybrid compounds (9–13) and their respective controls (8a–c) are compiled in Table 1.
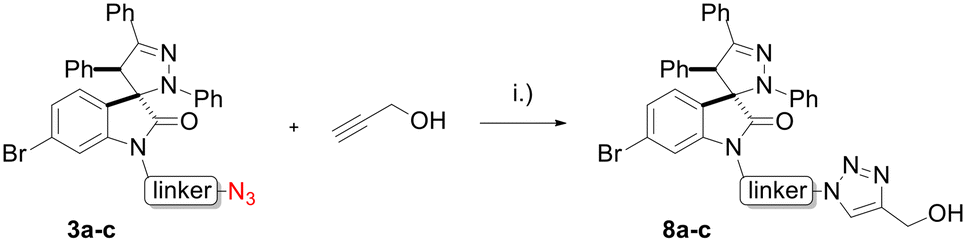 | ||
| Scheme 5 Synthesis of reference compounds 8a–c. Reaction conditions: (i): CuSO4·5H2O, sodium ascorbate, n-BuOH:H2O (1:1), r.t., 12–16 h, 44–53%. | ||
| Structure | Linker | Triazole compound |
|---|---|---|
|
a | 8a |
| b | 8b | |
| c | 8c | |
|
b | 9 |
| c | 10 | |
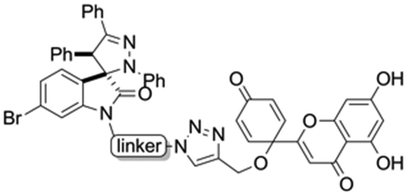
|
a | 11 |
| b | 12 | |
| c | 13 |
Bioactivity
The hybrid compounds (9–13) and their relevant fragments were tested for their cytotoxicity on MCF-7 (wild-type p53, estrogen-dependent), and MDA-MB-231 (mutant p53, TNBC) cancer cell lines. The results are compiled in Table 2.| Comp. | IC50 (μM) | ST | IC50 (μM) | |
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | MCF 10A | ||
| a Ambiguous fitting, confidence interval cannot be calculated due to the high slope of the regression curve, the values given are the experimental dilutions below and above the IC50; n.a.: not available; n.d.: not determined. | ||||
| 1 | 11.8 [10.0–14.0] | 19.2 [18.1–20.4] | 0.6 | >40 |
| 4 | 14.2 [12.2–16.7] | 10.5 [9.29–11.8] | 1.4 | n.d. |
| 6 | 11.9 [9.72–14.6] | 7.55 [6.62–8.61] | 1.6 | n.d. |
| 8a | 11.9 [10.9–13.0] | ∼9.30 [5.00–10.0]a | ∼1 | >40 |
| 8b | >40 | >40 | n.a. | n.d. |
| 8c | 16.4 [15.1–17.8] | 25.9 [25.2–26.7] | 0.6 | >40 |
| 9 | >40 | 10.6 [9.62–11.60] | >3.8 | >40 |
| 10 | >40 | >40 | n.a. | n.d. |
| 11 | 2.92 [2.68–3.18] | 1.08 [0.96–1.22] | 2.6 | 4.56 [3.87–5.38] |
| 12 | 4.08 [3.56–4.67] | 2.03 [1.78–2.32] | 2.0 | 5.32 [4.79–5.92] |
| 13 | 3.18 [2.77–3.66] | ∼1.18 [0.625–1.25]a | ∼2.7 | 6.64 [5.80–7.60] |
As the most important finding, hybrids 11–13, coupled at the C-1′ of the protoflavone fragment, exerted potent cytotoxic activity at low micromolar concentrations against both breast cancer cell lines, and two-times or higher selectivity towards the TNBC cells. This greatly exceeds the potency of all control fragments (1, 4, and 6) as well as that of their linker-containing derivatives (8a–c). On the other hand, hybrids 9 and 10, coupled at the C-7 of the protoflavone, were in general much less active. Still, it may be of interest that compound 9 showed the highest selectivity towards MDA-MB-231 cells among the tested compounds, despite its relatively moderate activity.
The control compounds containing spiropyrazoline oxindole 1, linker and triazole either retained (8a and 8c) or lost (8b) the antiproliferative activity in MCF-7 cells compared with compounds 1, 4, and 6. However, in MDA-MB-231 cells, control 8a showed increased potency compared to spiropyrazoline oxindole 1, while the antiproliferative effect of other reference compounds was lost.
The antiproliferative effects of the active compounds containing the triazole linker were also tested against the non-cancerous MCF 10A mammary epithelial cell line. The results are shown in Table 2. Comparing the IC50 values of the hybrids in the normal vs. the MDA-MB-231 cell line, compounds 11–13 all have quite good tumour selectivity and 13 is the best with a value of ca. 5.5. Concerning the normal vs. MCF-7 cell line selectivity, compound 13 is clearly the best again, exerting about two times stronger activity on the tumour cells.
To further elaborate on the relative efficacy of hybrids 11–13 compared to that of their fragments, additional calculations were performed based on the same raw cell viability data as a mathematical approach. This was performed by calculating for each hybrid compound a theoretical interaction between its corresponding fragments. A virtual combination index (VCI) was used similar to our previous work on protoflavone–chalchone hybrids.14 Briefly, relevant fragments' cell viability data were taken as control datasets, and the hybrid was considered as a 1:1 mixture of its fragments. Then, the Chou method was used to calculate combination index. Considering that the bioactivity of covalently linked hybrids was evaluated this way and not a mixture of separate compounds, the results were expressed as VCI values and are shown in Table 3.
:1 combinations of cell viability data obtained for fragments A (1) and B (4 or 6). VCI values are shown at 50%, 75%, and 90% of inhibition; 0 < VCI < 1, VCI = 1, and VCI > 1 represent synergism, additivity, and antagonism, respectively. Dm, m, and r represent the antilog of the x-intercept (activity), slope (shape of the dose-effect curve), and linear correlation coefficient (conformity of the data) of the median effect plot, respectively16
| Hybrid | Combo of | MCF-7 | MDA-MB-231 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VCI values at | VCIavg | D m | m | r | VCI values at | VCIavg | D m | m | r | ||||||
| ED50 | ED75 | ED90 | ED50 | ED75 | ED90 | ||||||||||
| a CI values cannot be calculated; compound 9 has an IC50 > 40 μM on MCF-7 cells, therefore it is considered inactive in this cell line. | |||||||||||||||
| 9 | 1 and 6 | n.a.a | 1.73 | 1.73 | 1.81 | 1.77 | 10.012 | 1.999 | 0.997 | ||||||
| 11 | 1 and 4 | 0.50 | 0.69 | 0.95 | 0.79 | 2.951 | 1.065 | 0.953 | 0.19 | 0.13 | 0.09 | 0.12 | 1.450 | 5.467 | 0.917 |
| 12 | 1 and 4 | 0.26 | 0.21 | 0.16 | 0.19 | 1.550 | 2.338 | 0.918 | 0.33 | 0.25 | 0.20 | 0.24 | 2.592 | 3.626 | 0.890 |
| 13 | 1 and 4 | 0.69 | 0.95 | 1.29 | 1.08 | 4.115 | 1.081 | 0.968 | 0.16 | 0.12 | 0.11 | 0.12 | 1.235 | 3.116 | 0.970 |
VCI values for hybrid compounds 11 and 13 showed much stronger synergism against the TNBC MDA-MB-231 cell line than against MCF-7 cells. This shows that coupling their fragments into hybrid molecules increased the anti-TNBC selectivity compared to their both respective fragments. This was not observed for compound 12, even though this compound also has approximately two-fold TNBC MDA-MD-231 cell line selectivity (see Table 2). This suggests that coupling the fragments into hybrid molecules similarly increased efficacy against both cell lines, i.e., a relevant selectivity increase compared to that of the fragments did not occur in the case of compound 12.
Compound 13 was selected to study its effect on the doxorubicin induced DNA-damage response in MCF-7 cell line. The serine–threonine checkpoint kinases Chk1 and Chk2 have an important role in the DNA damage response, being activated by ATR and ataxia telangiectasia mutated kinase (ATM), respectively. Compound 13 exerted a significant inhibition of the ATR-mediated phosphorylation of Chk1, while it increased the phosphorylation of Chk2 (Fig. 3). The Chk1 inhibitory property suggests that this hybrid can potentially target specific cancer cells with high levels of endogenous DNA damage.9 The increase in Chk2 activation suggests that it could induce DNA double-strand breaks and, consequentially, activate the ATM-Chk2 pathway. This pharmacodynamic behaviour is most similar to that of graviquinone, a p-coumaric acid methyl ester-derived protoflavone analogue, and markedly differs from that of PA that does not influence the ATM-dependent DNA damage response.17
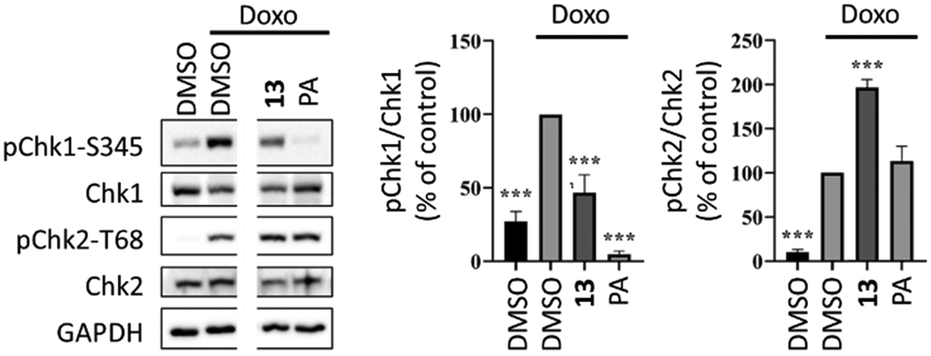 | ||
| Fig. 3 Activity of compound 13 on doxorubicin-induced DNA damage response in MCF-7 cells. Cells were pretreated with 10 μM of 13 or 5 μM of protoapigenone (PA) for 30 minutes and the phosphorylation of Chk1 and Chk2 was detected at 6 hours after exposure to 1 μM doxorubicin; n = 3; ***: p < 0.0001 by one-way ANOVA, Dunnett's multiple comparisons test. Original, non-cut images of the gel bands are available as ESI.† | ||
Materials and methods
General information
Reagents were purchased from Sigma (Merck KGaA, Darmstadt, Germany), Fluorochem Ltd (UK) and Alfa Aesar (Thermo Fisher Scientific, USA) without further purification. Solvents were obtained from Macron Fine Chemicals (Avantor Performance Materials, Center Valley, PA, USA), Honeywell (Germany), PanReac AppliChem (Spain) and Fluorochem Ltd (UK). Apigenin was obtained from Biosynth (Biosynth s.r.o, Bratislava, Slovakia). Hereinafter, solvent compositions are always given in volumetric ratios.Flash purification of compounds 2 and 4–7 were carried out on a CombiFlash RF+ Lumen (TELEDYN Isco, Lincoln, Ne, USA) instrument equipped with ELSD and UV detector with disposable 40 g RediSep Gold columns. Compounds 9–10 were purified using an Agilent 1100 series HPLC pump (Waters Co. Milford, MA, USA) connected to a Jasco UV-2075 detector (Jasco Co., Tokyo, Japan) with a Kinetex Gemini C18 (5 μm, 110 Å, 250 × 10 mm) semipreparative column as the stationary phase and an isocratic mixture of acetonitrile and water (74:26 and 90:10, respectively) as eluent at a flow rate of 3 mL min−1. Compounds 11–13 were purified by LaPrep VWR system with P110 pump and UV detector P314 set at 250 nm. A Luna® 100 C18 (5 μm, 100 Å, 250 × 4.6 mm) reverse phase column was used as stationary phase and acetonitrile:H2O (70:30) was used as mobile phase. The elution flow rate was set to 2 mL min−1 for 3 min and increased to 7 mL min−1 for 57 min.
All compounds were identified by one- and two-dimensional NMR techniques and mass spectrometry. The NMR measurements were recorded on a Bruker Avance NEO 500 MHz spectrometer equipped with a Prodigy BBO 5 mm CryoProbe or on a Bruker 300 Ultra-Shield (300 MHz) spectrometer.
Melting points were measured using an RK Tech melting point apparatus with microscope (compounds 7, 9 and 10) or a Kofler camera Bock monoscope M (compounds 8a–8c and 11–13).
Synthesis of protoflavone derivatives (2, 4–7) from apigenin
1′-O-Protoflavones 2, 4, 5 were synthesised based on our previously reported method.13 Briefly, apigenin (3.7 mmol, 1 equiv.) was dissolved in 100 mL of a 9:1 ratio mixture of acetonitrile and methanol, n-butanol, or propargyl alcohol as appropriate for the targeted ether group, and [bis(trifluoroacetoxy)iodo]benzene (PIFA) (7.4 mmol, 2 equiv.) was added. After an hour stirring at 80 °C, the solvent was evaporated, and the reaction mixture was purified by flash chromatography. For the purification of compounds 2 and 5, an isocratic mixture of n-hexane:ethyl acetate:acetone (80:15:5) was used as eluent. Compound 4 was purified using a gradient of n-hexane (A) and ethyl acetate (B), going from 15% to 25% B in an hour.
The protoflavone 7-O-ethers 6 and 7 were synthesised from protoapigenone 1′-O-butyl ether 2. The starting material (0.28 mmol, 1.0 equiv.) was dissolved in dry DMF (3.57 mL mmol−13), followed by addition of potassium carbonate (0.37 mmol, 1.2 equiv.). Methyl iodide (0.37 mmol, 1.2 equiv.) or propargyl bromide (0.37 mmol, 1.2 equiv.) was added for the synthesis of compounds 6 or 7, respectively. After 3 hours stirring at room temperature, the reaction mixture was purified by flash chromatography using a gradient of n-hexane (A) and ethyl acetate (B), going from 10% B to 15% B in 30 minutes, to afford compounds 6 (74.8 mg, 72% yield) and 7 (87.6 mg, 79% yield). Compound 6 was previously synthesized by our group using a different method.18
Synthesis of linkers a–c
To a solution of 2-bromoethanol (14.11 mmol, 1 equiv.) or 3-bromo-1-propanol (2.59 mmol, 1 equiv.) in water (1.25 mL mmol−1 of alcohol), sodium azide (3 equiv.) was added. The reaction was stirred overnight at 80 °C. The product was extracted twice with diethyl ether, the organic layers were combined, dried over anhydrous MgSO4 and the solvent was evaporated to afford the azidoalcohols that were used in the next step without further purification.To a solution of the respective azidoalcohol (1 equiv.) in dry dichloromethane (0.36 mL mmol−1 of azidoalcohol), triethylamine (1 equiv.) was added. After cooling the mixture at 0 °C, a solution of methanesulfonyl chloride (1 equiv.) in CH2Cl2 (0.17 mL mmol−1 of reactant) was added, and stirred at room temperature overnight, under nitrogen atmosphere. The reaction mixture was diluted with water and the product was extracted with CH2Cl2. The organic layer was washed with water, dried over anhydrous Na2SO4, and concentrated in vacuo to afford the mesylated azido linkers a (1839.2 mg, 91% yield), b (464.1 mg, 100% yield) and c (455.3 mg, 100% yield).
Synthesis of spiropyrazoline oxindole 1
Spiropyrazoline oxindole derivative 1 was synthesised as reported previously.15 Briefly, (Z)-N-phenylbenzohydrazonoyl chloride (2.51 mmol, 2.0 equiv.) was dissolved in CH2Cl2 (12 mL, 10 mL mmol−1) previously dried with CaH2. 6-Bromo-(E)-3-benzylideneindolin-2-one (1.22 mmol, 1.0 equiv.) was added, and the solution was stirred for 15 min. Then, triethylamine (2.51 mmol, 2.0 equiv.) was added dropwise. The reaction was stirred overnight at room temperature under nitrogen atmosphere. The reaction was quenched with cold water and the organic layer was washed with brine twice. The resulting solution was dried with anhydrous MgSO4 and the solvent was evaporated. The crude was purified by flash chromatography using ethyl acetate:n-hexane (1:3) to afford 6-bromo-2′,4′,5′-triphenyl-2′,4′-dihydrospiro[indoline-3,3′-pyrazol]-2-one (1) (565.8 mg, 94% yield).
Synthesis of spiropyrazoline oxindoles 3a–c
The spiropyrazoline oxindole 1 (0.20 mmol, 1.0 equiv.) and potassium carbonate (0.60 mmol, 3.0 equiv.) were dissolved in anhydrous DMF (1 mL) and stirred at room temperature for 30 min under nitrogen atmosphere. The corresponding mesylate azido linker a–c (0.20 mmol, 1.0 equiv.) was added to the previous solution and stirred overnight at 50 °C. The reaction was cooled down, and diethyl ether was added. The organic phase was washed with water, dried with MgSO4 and the solvent was evaporated. The crude mixture was purified by flash chromatography using ethyl acetate:n-hexane (3:2, 3a; 4:1, 3b; or 1:1, 3c).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH), 3.74 (p, J = 2.7 Hz, 2H, NCH2), 3.46 (q, J = 5.2 Hz, 2H, CH2N3); 13C NMR (75 MHz, CDCl3) δ 175.9 (N–CO), 149.5 (CN), 144.3 (Cq), 143.6 (Cq), 134.4 (Cq), 131.5 (Cq), 129.3 (2CH), 129.1 (CH), 129.0 (2CH1), 129.0 (2CH2), 128.5 (2CH), 128.4 (CH), 127.9 (CH), 127.0 (2CH), 125.7 (CH), 124.2 (Cq), 123.6 (Cq), 121.9 (CH), 116.3 (2CH), 112.3 (CH), 76.6 (Cspiro), 62.6 (NC–
C–CH), 3.74 (p, J = 2.7 Hz, 2H, NCH2), 3.46 (q, J = 5.2 Hz, 2H, CH2N3); 13C NMR (75 MHz, CDCl3) δ 175.9 (N–CO), 149.5 (CN), 144.3 (Cq), 143.6 (Cq), 134.4 (Cq), 131.5 (Cq), 129.3 (2CH), 129.1 (CH), 129.0 (2CH1), 129.0 (2CH2), 128.5 (2CH), 128.4 (CH), 127.9 (CH), 127.0 (2CH), 125.7 (CH), 124.2 (Cq), 123.6 (Cq), 121.9 (CH), 116.3 (2CH), 112.3 (CH), 76.6 (Cspiro), 62.6 (NC–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H), 49.1 (CH2N3), 39.9 (NCH2).
C–CH), 3.72 (p, J = 6.8 Hz, 1H, NCH2), 3.52 (dt, J = 14.3, 6.2 Hz, 1H, N–CH2), 2.96 (p, J = 6.1 Hz, 2H, CH2N3), 1.65 (p, J = 6.8 Hz, 2H, C–CH2–C); 13C NMR (75 MHz, CDCl3) δ 175.5 (N–CO), 150.2 (CN), 144.6 (Cq), 143.8 (Cq), 134.4 (Cq), 131.5 (Cq), 129.4 (2CH), 129.3 (CH), 129.0 (2CH1), 129.0 (2CH2), 128.5 (2CH), 128.4 (CH), 128.1 (CH), 127.0 (2CH), 125.5 (CH), 124.4 (Cq), 123.7 (Cq), 122.5 (CH), 117.4 (2CH), 112.0 (CH), 77.1 (Cspiro), 62.2 (NC–H), 48.5 (–CH2–N3), 37.5 (NCH2–), 26.8 (C–H2–C).
C–CH), 3.88–3.72 (m, 2H, N–CH2), 3.63–3.46 (m, 8H, OCH2), 3.23 (t, J = 4.9 Hz, 2H, CH2N3); 13C NMR (75 MHz, CDCl3) δ 175.9 (N–CO), 149.2 (CN), 144.3 (Cq), 134.5 (Cq), 131.6 (Cq), 129.3 (2CH), 129.0 (2CH), 128.9 (CH), 128.8 (2CH), 128.5 (2CH), 128.2 (CH), 127.5 (CH), 126.9 (2CH), 125.2 (CH), 124.1 (Cq), 123.1 (Cq), 121.5 (CH), 116.0 (2CH), 113.3 (CH), 76.5 (Cspiro), 71.0 (NCH2H2O), 70.8 (H2OCH2CH2N3), 70.4 (H2CH2–N3), 68.9 (NCH2CH2OH2), 62.7 (NC–H), 50.8 (H2–N3), 40.5 (N–H2).
H), 49.1 (CH2N3), 39.9 (NCH2).
C–CH), 3.72 (p, J = 6.8 Hz, 1H, NCH2), 3.52 (dt, J = 14.3, 6.2 Hz, 1H, N–CH2), 2.96 (p, J = 6.1 Hz, 2H, CH2N3), 1.65 (p, J = 6.8 Hz, 2H, C–CH2–C); 13C NMR (75 MHz, CDCl3) δ 175.5 (N–CO), 150.2 (CN), 144.6 (Cq), 143.8 (Cq), 134.4 (Cq), 131.5 (Cq), 129.4 (2CH), 129.3 (CH), 129.0 (2CH1), 129.0 (2CH2), 128.5 (2CH), 128.4 (CH), 128.1 (CH), 127.0 (2CH), 125.5 (CH), 124.4 (Cq), 123.7 (Cq), 122.5 (CH), 117.4 (2CH), 112.0 (CH), 77.1 (Cspiro), 62.2 (NC–H), 48.5 (–CH2–N3), 37.5 (NCH2–), 26.8 (C–H2–C).
C–CH), 3.88–3.72 (m, 2H, N–CH2), 3.63–3.46 (m, 8H, OCH2), 3.23 (t, J = 4.9 Hz, 2H, CH2N3); 13C NMR (75 MHz, CDCl3) δ 175.9 (N–CO), 149.2 (CN), 144.3 (Cq), 134.5 (Cq), 131.6 (Cq), 129.3 (2CH), 129.0 (2CH), 128.9 (CH), 128.8 (2CH), 128.5 (2CH), 128.2 (CH), 127.5 (CH), 126.9 (2CH), 125.2 (CH), 124.1 (Cq), 123.1 (Cq), 121.5 (CH), 116.0 (2CH), 113.3 (CH), 76.5 (Cspiro), 71.0 (NCH2H2O), 70.8 (H2OCH2CH2N3), 70.4 (H2CH2–N3), 68.9 (NCH2CH2OH2), 62.7 (NC–H), 50.8 (H2–N3), 40.5 (N–H2).
Synthesis of reference compounds 8a–c
Propargyl alcohol (10.0 mmol, 1 equiv.) and spiropyrazoline oxindoles 3a–c (10.0 mmol, 1 equiv.) were dissolved in 1 mL of n-ButOH:water (1:1). To the stirred solution, CuSO4·5H2O (2.0 mmol, 0.2 equiv.) and sodium ascorbate (10.0 mmol, 1.0 equiv.) were added. After 12 hours at room temperature, the reaction mixture was dissolved in water (20 mL) and extracted with ethyl acetate five times. The organic layers were combined, dried over anhydrous MgSO4, and concentrated in vacuo. Compounds 8a–c were purified by LaPrep VWR system with P110 pump and UV detector P314 set at 250 nm. A Luna® 100 C18 (5 μm, 100 Å, 250 × 4.6 mm) reverse phase column was used as stationary phase and acetonitrile:H2O (70:30) as mobile phase. The elution flow rate was set to 2 mL min−1 for 3 min and increased to 7 mL min−1 for 57 min.
C), 7.68–7.61 (m, 2H, ArH), 7.42–7.32 (m, 4H, ArH), 7.26–7.17 (m, 3H, ArH), 7.14–7.06 (m, 4H, ArH), 6.97 (m, 1H, ArH), 6.84–6.67 (m, 5H, ArH), 6.21 (d, J = 8.0 Hz, 1H, ArH), 5.37 (s, 1H, NC–CH), 5.15 (t, J = 5.5 Hz, 1H, OH), 4.53 (d, J = 5.1 Hz, 2H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2OH), 4.33–4.25 (m, 2H, N–CH2), 3.76 (t, J = 5.4 Hz, 2H, N–N–CH2); 13C NMR (75 MHz, DMSO-d6) δ 174.5 (N–CO), 149.2 (CN), 148.7 (NN–C), 143.7 (Cq), 142.9 (Cq), 134.4 (Cq), 131.0 (Cq), 129.0 (4CH), 128.6 (3CH), 128.0 (3CH), 126.8 (CH), 126.7 (2CH), 123.4 (CH), 123.3 (Cq), 122.9 (N–HC), 122.7 (Cq), 120.6 (CH), 114.6 (2CH), 112.6 (CH), 74.9 (Cspiro), 63.2 (NC–H), 55.7 (CH2–OH), 49.1 (N–H2–C), 40.3 (CH2–N–N). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 619, found: 619.
C), 7.69–7.64 (m, 2H, ArH), 7.43 (d, J = 1.7 Hz, 1H, ArH), 7.39–7.30 (m, 3H, ArH), 7.24–7.17 (m, 3H, ArH), 7.15–7.08 (m, 2H, ArH), 7.04–6.98 (m, 2H, ArH), 6.84–6.72 (m, 4H, ArH), 6.22 (d, J = 8.0 Hz, 1H, ArH), 5.45 (s, 1H, NC–CH), 5.16 (t, J = 5.6 Hz, 1H, OH), 4.53 (d, J = 5.5 Hz, 2H, C2OH), 4.32 (t, J = 7.0 Hz, 2H, N–CH2), 3.83–3.71 (m, 2H, N–N–CH2), 2.21–2.04 (m, 2H, C–CH2–C); 13C NMR (75 MHz, DMSO-d6) δ 174.8 (N–CO), 149.8 (CN), 148.0 (NN–C), 143.9 (Cq), 143.5 (Cq), 134.5 (Cq), 131.0 (Cq), 129.0 (4CH), 128.6 (3CH1), 128.0 (3CH), 127.2 (CH), 126.6 (2CH), 124.7 (CH), 123.8 (Cq), 122.9 (N–HC), 121.0 (CH), 114.9 (2CH), 112.6 (CH), 75.4 (Cspiro), 60.8 (NC–H), 55.1 (CH2–OH), 46.8 (N–H2–C), 37.2 (CH2–N–N), 27.7 (C–CH2–C). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 633, found: 633.
C), 7.70–7.61 (m, 2H, ArH), 7.42 (d, J = 1.7 Hz, 1H, ArH), 7.39–7.29 (m, 3H, ArH), 7.18 (dd, J = 5.1, 1.9 Hz, 3H, ArH), 7.14–7.06 (m, 2H, ArH), 6.98 (dt, J = 6.5, 2.2 Hz, 2H, ArH), 6.82–6.73 (m, 4H, ArH), 6.22 (d, J = 8.0 Hz, 1H, ArH), 5.38 (s, 1H, NC–CH), 5.16 (t, J = 5.6 Hz, 1H, OH), 4.51 (d, J = 5.1 Hz, 2H, C2OH), 4.44 (td, J = 4.9, 4.6, 1.3 Hz, 2H, N–N–CH2), 4.00–3.90 (m, 2H, N–CH2), 3.76 (t, J = 5.3 Hz, 2H, C
2OH), 4.33–4.25 (m, 2H, N–CH2), 3.76 (t, J = 5.4 Hz, 2H, N–N–CH2); 13C NMR (75 MHz, DMSO-d6) δ 174.5 (N–CO), 149.2 (CN), 148.7 (NN–C), 143.7 (Cq), 142.9 (Cq), 134.4 (Cq), 131.0 (Cq), 129.0 (4CH), 128.6 (3CH), 128.0 (3CH), 126.8 (CH), 126.7 (2CH), 123.4 (CH), 123.3 (Cq), 122.9 (N–HC), 122.7 (Cq), 120.6 (CH), 114.6 (2CH), 112.6 (CH), 74.9 (Cspiro), 63.2 (NC–H), 55.7 (CH2–OH), 49.1 (N–H2–C), 40.3 (CH2–N–N). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 619, found: 619.
C), 7.69–7.64 (m, 2H, ArH), 7.43 (d, J = 1.7 Hz, 1H, ArH), 7.39–7.30 (m, 3H, ArH), 7.24–7.17 (m, 3H, ArH), 7.15–7.08 (m, 2H, ArH), 7.04–6.98 (m, 2H, ArH), 6.84–6.72 (m, 4H, ArH), 6.22 (d, J = 8.0 Hz, 1H, ArH), 5.45 (s, 1H, NC–CH), 5.16 (t, J = 5.6 Hz, 1H, OH), 4.53 (d, J = 5.5 Hz, 2H, C2OH), 4.32 (t, J = 7.0 Hz, 2H, N–CH2), 3.83–3.71 (m, 2H, N–N–CH2), 2.21–2.04 (m, 2H, C–CH2–C); 13C NMR (75 MHz, DMSO-d6) δ 174.8 (N–CO), 149.8 (CN), 148.0 (NN–C), 143.9 (Cq), 143.5 (Cq), 134.5 (Cq), 131.0 (Cq), 129.0 (4CH), 128.6 (3CH1), 128.0 (3CH), 127.2 (CH), 126.6 (2CH), 124.7 (CH), 123.8 (Cq), 122.9 (N–HC), 121.0 (CH), 114.9 (2CH), 112.6 (CH), 75.4 (Cspiro), 60.8 (NC–H), 55.1 (CH2–OH), 46.8 (N–H2–C), 37.2 (CH2–N–N), 27.7 (C–CH2–C). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 633, found: 633.
C), 7.70–7.61 (m, 2H, ArH), 7.42 (d, J = 1.7 Hz, 1H, ArH), 7.39–7.29 (m, 3H, ArH), 7.18 (dd, J = 5.1, 1.9 Hz, 3H, ArH), 7.14–7.06 (m, 2H, ArH), 6.98 (dt, J = 6.5, 2.2 Hz, 2H, ArH), 6.82–6.73 (m, 4H, ArH), 6.22 (d, J = 8.0 Hz, 1H, ArH), 5.38 (s, 1H, NC–CH), 5.16 (t, J = 5.6 Hz, 1H, OH), 4.51 (d, J = 5.1 Hz, 2H, C2OH), 4.44 (td, J = 4.9, 4.6, 1.3 Hz, 2H, N–N–CH2), 4.00–3.90 (m, 2H, N–CH2), 3.76 (t, J = 5.3 Hz, 2H, C![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) CH2N–N), 3.69–3.55 (m, 2H, NCH2C2), 3.55–3.44 (m, 4H, OCH2); 13C NMR (75 MHz, DMSO-d6) δ 175.1 (N–CO), 149.2 (NC), 147.9 (NN–C), 144.1 (Cq), 143.5 (Cq), 134.5 (Cq), 131.1 (Cq), 129.0 (4CH), 128.6 (3CH), 128.0 (3CH), 126.8 (CH), 126.5 (2CH), 124.4 (CH), 123.4 (Cq), 123.0 (N–HC), 122.4 (Cq), 120.7 (CH), 114.5 (2CH), 113.3 (CH), 75.1 (Cspiro), 69.8 (OCH2), 69.6 (OCH2), 69.0 (H2CH2N–N), 67.6 (NCH2H2O), 61.3 (NC–H), 53.9 (CH2OH), 49.2 (CH2–N–N), 40.4 (NH2CH2O). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 707, found: 707.
CH2N–N), 3.69–3.55 (m, 2H, NCH2C2), 3.55–3.44 (m, 4H, OCH2); 13C NMR (75 MHz, DMSO-d6) δ 175.1 (N–CO), 149.2 (NC), 147.9 (NN–C), 144.1 (Cq), 143.5 (Cq), 134.5 (Cq), 131.1 (Cq), 129.0 (4CH), 128.6 (3CH), 128.0 (3CH), 126.8 (CH), 126.5 (2CH), 124.4 (CH), 123.4 (Cq), 123.0 (N–HC), 122.4 (Cq), 120.7 (CH), 114.5 (2CH), 113.3 (CH), 75.1 (Cspiro), 69.8 (OCH2), 69.6 (OCH2), 69.0 (H2CH2N–N), 67.6 (NCH2H2O), 61.3 (NC–H), 53.9 (CH2OH), 49.2 (CH2–N–N), 40.4 (NH2CH2O). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 707, found: 707.
Synthesis of hybrid compounds 9–13
The alkyne containing protoflavones 5 or 7 (1 equiv.) and spiropyrazoline oxindoles 3a–c (1 equiv.) were dissolved in 1 mL of n-ButOH:water (1:1). To the stirred solution, CuSO4·5H2O (0.2 equiv.) and sodium ascorbate (1.0 equiv.) were added. After 12 hours at room temperature, the reaction mixture was dissolved in water (20 mL) and extracted with ethyl acetate five times. The organic layers were combined, dried over anhydrous MgSO4, and concentrated in vacuo. The hybrids were purified by RP-HPLC.
C), 7.63–7.55 (m, 2H, ArH), 7.37 (d, J = 1.8 Hz, 1H, ArH), 7.34–7.17 (m, 5H, ArH), 7.14–6.99 (m, 7H, ArH), 6.83–6.68 (m, 5H, ArH), 6.53 (ddd, J = 9.9, 5.5, 1.8 Hz, 2H, CH), 6.43 (s, 1H, ArH), 6.17 (t, J = 2.8 Hz, 1H, ArH), 6.08 (d, J = 8.1 Hz, 2H, CH), 4.98 (s, 1H, NC–CH), 4.79–4.68 (m, 2H, C2OH), 4.55 (d, J = 2.7 Hz, 2H, N–CH2), 4.37–4.24 (m, 1H, N–N–CH2), 4.22–4.10 (m, 1H, N–N–CH2); 13C NMR (75 MHz, DMSO-d6) δ 184.2 (O–CC–O), 181.5 (C–CC–O), 174.5 (N–CO), 164.7 (Cq), 161.4 (Cq), 157.3 (Cq), 149.6 (CN), 145.4 (CH), 143.9 (Cq), 143.7 (Cq), 143.0 (N–NN–C), 134.4 (Cq), 132.4 (CH), 131.0 (Cq), 129.0 (CH), 128.7 (CH), 128.6 (CH), 128.1 (CH), 126.8 (CH), 126.3 (CH), 125.3 (CH), 124.8 (Cq), 123.3 (Cq), 122.7 (Cq), 120.8 (Cq), 114.7 (CH), 112.4 (CH), 107.4 (CH), 104.0 (CH), 99.3 (CH), 93.9 (CH), 75.0 (Cspiro), 74.3 (Cq), 60.5 (NC–H), 58.5 (CH2O),47.0 (N–CH2), 40.4 (CH2–N–N). MS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 887, found: 887.
C), 7.69–7.62 (m, 2H, ArH), 7.42 (d, J = 1.8 Hz, 1H, ArH), 7.36–7.29 (m, 4H, ArH), 7.24–7.06 (m, 8H, ArH, CH), 7.04–6.97 (m, 2H, ArH), 6.82 (dd, J = 8.0, 1.6 Hz, 1H, ArH), 6.79–6.71 (m, 2H, ArH), 6.56 (dt, J = 11.6, 5.1, 3.3 Hz, 2H, CH), 6.49 (s, 1H, Ar), 6.22 (d, J = 8.0 Hz, 1H, ArH), 6.18 (q, J = 2.1 Hz, 2H, CH), 5.43 (s, 1H, NC–CH), 4.64 (s, 2H, C2O), 4.34 (t, J = 7.1 Hz, 2H, N–CH2), 3.88–3.69 (m, 2H, N–N–CH2), 2.24–2.05 (m, 2H, C–CH2–C); 13C NMR (75 MHz, DMSO-d6) δ 184.2 (O–CC–O), 181.3 (C–CC–O), 174.8 (N–CO), 164.2 (Cq), 161.4 (Cq), 157.3 (Cq), 149.8 (CN), 145.4 (CH), 143.9 (Cq), 143.5 (Cq), 143.3 (N–NN–C), 134.5 (Cq), 132.2 (CH), 131.0 (Cq), 129.0 (CH), 128.6 (CH), 128.5 (CH), 128.0 (CH), 126.8 (CH), 126.6 (CH), 124.7 (CH), 124.5 (Cq), 123.8 (Cq), 122.9 (Cq), 121.0 (Cq), 114.9 (CH), 112.5 (CH), 107.4 (CH), 103.7 (CH), 99.4 (CH), 94.0 (CH), 75.4 (Cspiro), 74.2 (Cq), 60.8 (NC–H), 58.6 (CH2O), 37.0 (CH2–N–N), 27.5 (C–H2–C). HRMS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 901.19799, found: 901.19941.
C), 7.66–7.60 (m, 2H, ArH), 7.40 (d, J = 1.8 Hz, 1H, ArH), 7.37–7.28 (m, 4H, ArH), 7.20–7.13 (m, 4H, ArH), 7.13–7.04 (m, 4H, ArH, CH), 6.99–6.93 (m, 2H, ArH), 6.82–6.73 (m, 4H, ArH), 6.57–6.45 (m, 3H, CH), 6.24–6.16 (m, 3H, ArH, CH), 5.35 (s, 1H, NC–CH), 4.58 (s, 2H, CH2O), 4.49–4.40 (m, 2H, N–NCH2), 3.98–3.88 (m, 2H, N–CH2), 3.77 (t, J = 5.4 Hz, 2H, N–N–CH2H2), 3.68–3.55 (m, 1H, N–CH2H2), 3.54–3.45 (m, J = 2.6 Hz, 4H, OCH2); 13C NMR (75 MHz, DMSO-d6) δ 184.2 (O–CC–O), 181.4 (C–CC–O), 175.1 (N–CO), 164.3 (Cq), 161.4 (Cq), 157.3 (Cq), 149.1 (CN), 145.4 (CH), 144.0 (Cq), 143.5 (Cq), 143.1 (N–NN–C), 134.4 (Cq), 132.2 (CH), 131.1 (Cq), 128.9 (CH), 128.6 (CH), 128.2 (CH), 127.9 (CH), 126.5 (CH), 125.3 (CH), 124.8 (Cq), 123.4 (Cq), 122.4 (Cq), 114.5 (CH), 113.3 (CH), 107.4 (CH), 103.9 (CH), 99.3 (CH), 93.9 (CH), 75.1 (Cspiro), 74.2 (Cq), 69.8 (OCH2), 69.5 (OCH2), 68.8 (H2CH2N–N), 67.6 (NCH2H2O), 61.3 (NC–H), 58.6 (N–C–H2O), 49.5 (CH2–N–N), 40.4 (NH2CH2O). HRMS (ESI): C56H51BrN6O9 calculated m/z [M + H+]: 975.23477, found: 975.23609.
Antiproliferative MTT assay
The day before experiments, cells obtained from the American Type Culture Collection MCF-7 (ATCC HTB-22™) and MDA-MB-231 (ATCC HTB-26™) were seeded at 2 × 104 cells per well in 96 well tissue culture plates, in 100 μL of RPMI 1640 culture medium supplemented with 10% foetal bovine serum, 100 units of penicillin G (sodium salt), 100 mg of streptomycin sulphate and 2 mM L-glutamine. MCF 10A (ATCC CRL-10317™) cells were seeded at the same cell density in 100 μL in DMEM/F12 medium, containing 5% horse serum, 100 U mL−1 penicillin, 0.1 mg mL−1 streptomycin, 0.01 mg mL−1 insulin, 0.5 μg mL−1 hydrocortisone, 100 ng mL−1 cholera toxin, and 20 ng mL−1 human epidermal growth factor.Tested compounds were dissolved in dimethyl sulfoxide (DMSO), serially diluted in the culture medium, and added to the cells. After 48 h, cell media was removed and replaced with fresh medium, the MTT dye solution was added to each well and after 3 h of incubation the media was removed, and intracellular formazan crystals were solubilized and extracted with DMSO. After 15 min at room temperature, absorbance was measured at 570 nm in a microplate reader spectrophotometer (FLUOstar Omega, BMG Labtech, Germany), and the percentage of viable cells was determined for each compound concentration as described previously.15
The IC50 values were calculated by the variable slope log(inhibitor) vs. normalized response nonlinear regression model of GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA), and data presented as the estimated value (95% confidence intervals).
Virtual combination index determination
Virtual combination indices were calculated according to a previously published method.14 Briefly, a purely mathematical approach was taken to allow a comparison of the cytotoxic activity of each hybrid with that of its two relevant fragments, and this was expressed as a virtual combination index (VCI). This was calculated by the Chou method16 by re-analysing the raw data of the MTT assays that gave the IC50 values shown in Table 2. Each hybrid was considered as a 1:1 ratio mixture of its two corresponding fragments, and combination index values were computed by the CalcuSyn software (version 2.1, Biosoft). In this context, VCI close to 1 refers to similar, VCI <1 to a stronger, and VCI >1 to a weaker effect of a hybrid than what would be expected based on a simple addition of the effects of its fragments.14
DNA damage response (DDR) evaluation
To test the compound with ATR inhibitory activity, we produced DNA damage by doxorubicin, and ATM/ATR-mediated Chk1 and Chk2 phosphorylation represented DDR was detected by Western blot assay. Briefly, cells were pre-treated with 13 or PA for 30 min, then co-treating with doxorubicin for an additional 6 hours. Harvested proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Specific primary antibodies of phospho-Chk1, phospho-Chk2, Chk1, Chk2, and the internal control GAPDH were individually recognized on the membrane. HRP substrate reacted with HRP-conjugated secondary antibodies on the membrane will elicit chemiluminescence, which reflects the relative protein expression levels and can be captured by the Chemiluminescence Imaging System.12Conclusions
Altogether, we can conclude that the hybrid compounds 11–13 are promising new leads against breast cancer, and that in the case of compounds 11 and 13 the anti-TNBC selectivity also exceeds that expected based on the pharmacodynamic properties of their corresponding fragments. Inhibition of the ATR-mediated DNA damage response by compound 13 further emphasizes the drug development potential of this hybrid compound towards an effective and safe therapeutic agent against cancers with mutated p53 including most triple negative cancers.Author contributions
Conceptualisation, M. M. M. S. and A. H.; methodology, G. G. and E. A. L.; investigation, G. G., E. A. L., M. E., L. M. G., N. K. and H. C. W.; resources, M. M. M. S. and A. H.; data curation, A. H.; writing—original draft, G. G., E. A. L., M. M. M. S. and A. H.; writing—review and editing, M. M. M. S. and A. H.; supervision, M. M. M. S. and A. H.; funding acquisition, M. M. M. S. and A. H. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the bilateral action FCT/NKFIH 2019/2020 with references 2018-2.1.15-TÉT-PT-2018-00016 (NKFIH, Hungary) and 5183/2019 (FCT, Portugal). We also acknowledge the funding of National Research, Development and Innovation Office, Hungary through NKFIH; K-134704, TKP2021-EGA-32 and ÚNKP-22-4-SZTE-164 by the Ministry of Innovation and Technology of Hungary, and FCT (Fundação para a Ciência e a Tecnologia, I.P.) through iMed.ULisboa (UID/DTP/04138/2020), project PTDC/QUI-QOR/1304/2020, principal investigator grant CEECIND/03143/2017 (L. M. Gonçalves) and PhD fellowship SFRH/BD/137544/2018 (E. A. Lopes). The NMR spectrometers are part of the National NMR Network (PTNMR) and are partially supported by Infrastructure Project No 022161 (co-financed by FEDER through COMPETE 2020, POCI and PORL and FCT through PIDDAC). Financial support from FCT and Portugal 2020 to the Portuguese Mass Spectrometry Network (Rede Nacional de Espectrometria de Massa – RNEM; LISBOA-01-0145-FEDER-402-022125) is also acknowledged.References
- W. International, Breast cancer statistics, https://www.wcrf.org/cancer-trends/breast-cancer-statistics/, (accessed 23/01/2023, 2023) Search PubMed.
- A. C. Society, Triple-negative Breast Cancer, https://www.cancer.org/cancer/breast-cancer/about/types-of-breast-cancer/triple-negative.html, (accessed 23/01/2023, 2023) Search PubMed.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS PubMed.
- M. Olivier, M. Hollstein and P. Hainaut, Cold Spring Harbor Perspect. Biol., 2010, 2, a001008 CrossRef PubMed.
- E. A. Lopes, S. Gomes, L. Saraiva and M. M. Santos, Curr. Med. Chem., 2019, 26, 7323–7336 CrossRef CAS PubMed.
- A. Aguilar and S. Wang, Pharmaceuticals, 2023, 16, 24 CrossRef CAS PubMed.
- M. Espadinha, E. A. Lopes, V. Marques, J. D. Amaral, D. J. V. A. dos Santos, M. Mori, S. Daniele, R. Piccarducci, E. Zappelli, C. Martini, C. M. P. Rodrigues and M. M. M. Santos, Eur. J. Med. Chem., 2022, 241, 114637 CrossRef CAS PubMed.
- E. Lecona and O. Fernandez-Capetillo, Nat. Rev. Cancer, 2018, 18, 586–595 CrossRef CAS PubMed.
- A. Bradbury, S. Hall, N. Curtin and Y. Drew, Pharmacol. Ther., 2020, 207, 107450 CrossRef CAS PubMed.
- P. M. Reaper, M. R. Griffiths, J. M. Long, J.-D. Charrier, S. MacCormick, P. A. Charlton, J. M. C. Golec and J. R. Pollard, Nat. Chem. Biol., 2011, 7, 428–430 CrossRef CAS PubMed.
- M. Kwok, N. Davies, A. Agathanggelou, E. Smith, C. Oldreive, E. Petermann, G. Stewart, J. Brown, A. Lau, G. Pratt, H. Parry, M. Taylor, P. Moss, P. Hillmen and T. Stankovic, Blood, 2016, 127, 582–595 CrossRef CAS PubMed.
- H. C. Wang, A. Y. Lee, W. C. Chou, C. C. Wu, C. N. Tseng, K. Y. Liu, W. L. Lin, F. R. Chang, D. W. Chuang, A. Hunyadi and Y. C. Wu, Mol. Cancer Ther., 2012, 11, 1443–1453 CrossRef CAS PubMed.
- A. Hunyadi, D. W. Chuang, B. Danko, M. Y. Chiang, C. L. Lee, H. C. Wang, C. C. Wu, F. R. Chang and Y. C. Wu, PLoS One, 2011, 6, e23922 CrossRef CAS PubMed.
- A. D. Latif, T. Jernei, A. Podolski-Renić, C. Y. Kuo, M. Vágvölgyi, G. Girst, I. Zupkó, S. Develi, E. Ulukaya, H. C. Wang, M. Pešić, A. Csámpai and A. Hunyadi, Antioxidants, 2020, 9, 519 CrossRef CAS PubMed.
- Â. Monteiro, L. M. Gonçalves and M. M. M. Santos, Eur. J. Med. Chem., 2014, 79, 266–272 CrossRef PubMed.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- L. Fási, F. Di Meo, C.-Y. Kuo, S. Stojkovic Buric, A. Martins, N. Kúsz, Z. Béni, M. Dékány, G. T. Balogh, M. Pesic, H.-C. Wang, P. Trouillas and A. Hunyadi, J. Med. Chem., 2019, 62, 1657–1668 CrossRef PubMed.
- B. Danko, A. Martins, D. W. Chuang, H. C. Wang, L. Amaral, J. Molnár, F. R. Chang, Y. C. Wu and A. Hunyadi, Anticancer Res., 2012, 32, 2863–2869 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00251a |
| ‡ The first two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |