
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, electrochemistry and anti-trypanosomatid hit/lead identification of nitrofuranylazines†
Maryna
Saayman
a,
Christina
Kannigadu
a,
Janine
Aucamp
a,
Helena D.
Janse van Rensburg
a,
Cassiem
Joseph
 b,
Andrew J.
Swarts
b and
David D.
N'Da
*a
b,
Andrew J.
Swarts
b and
David D.
N'Da
*a
aCentre of Excellence for Pharmaceutical Sciences, North-West University, Potchefstroom 2520, South Africa. E-mail: David.Nda@nwu.ac.za; Fax: +27 18 299 4243; Tel: +27 18 299 2256
bMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg-Braamfontein 2050, South Africa
First published on 16th August 2023
Abstract
Chagas disease and leishmaniasis are vector-borne infectious diseases affecting both humans and animals. These neglected tropical diseases can be fatal if not treated. Hundreds to thousands of new Chagas disease and leishmaniasis cases are being reported by the WHO every year, and currently available treatments are insufficient. Severe adverse effects, impractical administrations and increased pathogen resistance against current clinical treatments underscore a serious need for the development of new drugs to curb these ailments. In search for such drugs, we investigated a series of nitrofuran-based azine derivatives. Herein, we report the design, synthesis, electrochemistry, and biological activity of these derivatives against promastigotes and amastigotes of Leishmania major, and L. donovani strains, as well as epimastigotes and trypomastigotes of Trypanosoma cruzi. Two leishmanicidal early leads and one trypanosomacidal hit with submicromolar activity were uncovered and stand for further in vivo investigation in the search for new antitrypanosomatid drugs. Future objective will focus on the identification of involved biological targets with the parasites.
Introduction
Neglected tropical diseases (NTDs) consist of several conditions that are prevalent in tropical regions,1 and can negatively impact individuals through stigmatisation, social rejection, disabilities, discrimination, and financial strain.2 Leishmaniasis and Chagas disease are both NTDs caused by Leishmania and Trypanosoma parasites, respectively. These parasites are taxonomically related protozoa that belong to the Trypanosomatidae family (order Kinetoplastida) and accordingly have similar biochemical and structural features.3Leishmaniasis is transmitted to humans during hematophagy of infected female Phlebotomus sandflies.4 There are over twenty Leishmania species that can affect humans and three clinical forms in which leishmaniasis can occur.5 These forms are cutaneous (CL), which can be identified by lesions on the skin; mucocutaneous (MCL), a differentiated form with lesions on the mouth and skin; and visceral leishmaniasis (VL), identified by skin lesions and a systemic infection that results in an immune deficiency.6 CL mainly occur in the Americas, Middle East, Mediterranean basin, and Central Asia, while VL is more prevalent in Brazil, India, and East Africa, and MCL in Brazil, Peru, and Ethiopia.7 In 2021, there were 221![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 731 new CL8 and 11450 new VL9 cases reported by the World Health Organization (WHO); however, it is estimated that only 25 to 45% of VL cases are being reported, therefore making the annual occurrence 50000 to 90000 cases.7 VL is fatal when left untreated, while MCL can also be fatal when secondary complications occur.10
731 new CL8 and 11450 new VL9 cases reported by the World Health Organization (WHO); however, it is estimated that only 25 to 45% of VL cases are being reported, therefore making the annual occurrence 50000 to 90000 cases.7 VL is fatal when left untreated, while MCL can also be fatal when secondary complications occur.10
Chagas disease (American trypanosomiasis), on the other hand, is mainly transmitted to humans through contact with the urine and/or faeces of blood-sucking triatomine insects infected with T. cruzi parasites.11 It can also be transmitted through blood transfusions, congenital transmission, organ transplants, and laboratory accidents.12 This disease presents in two clinical phases, acute and chronic. The acute phase, lasting approximately 2 to 4 months, is usually asymptomatic. The chronic phase, which can last decades after infection, can either be asymptomatic or symptomatic.13 Symptoms of active chronic infection include chronic gastrointestinal disease and/or heart disease with low, fluctuating parasitaemia levels.14 Approximately 6 to 7 million individuals worldwide are infected with the T. cruzi parasite, with an annual occurrence of about 30000 new cases, 12000 deaths, and 8600 infants infected during gestation by the Pan American Health Organization (PAHO), a specialised agency of the United Nations (UN) in charge of international health cooperation in the Americas.15 Chagas disease is mostly found in endemic Latin American countries.11
There are no vaccines for protection against both infections; however, chemotherapeutic drugs are available for the treatment of leishmaniasis and Chagas disease. Leishmaniasis treatment consists of pentavalent antimonies (sodium stibogluconate and meglumine antimonate), amphotericin B, paromomycin, miltefosine, pentamidine and the azoles (ketoconazole, itraconazole and fluconazole).16–18 These drugs have several limitations, such as impractical administration, toxicity, steep cost, and parasite resistance.19 The fight against the latter is an ongoing battle due to the misuse of drugs, financial constraints, and the unavailability of new drugs.20
The treatment for Chagas disease consists only of benznidazole or nifurtimox.21 If administered soon after infection occurs, both medicines are nearly 100% effective in curing the disease. However, the efficacy of the treatment decreases as the disease progresses.22
The use of clinical nitrofurans (cNFs) to treat various infectious diseases has been well established over recent years.23,24 The diverse biological activities of nitrofurans have been attributed to their nitro group and its ability to undergo one-electron transfer reduction, thereby forming reactive oxygen species (ROS) upon Fenton reactions with enzymes.23,25 Additionally, cNFs (Fig. 1) also have a hydrazone moiety (blue in Fig. 1) that possesses anti-infective activity. The hydrazone moiety also confers diverse biological and pharmacological properties, such as anticancer,26 antimicrobial,27 antitrypanosomal,28 antimycobacterial,29 anticonvulsive,30 anti-inflammatory31 and analgesic properties.32 Altogether, these pharmacological features enhance the stand of cNFs as potentially repurposable drugs for the treatment of infectious diseases such as leishmaniasis and Chagas disease.
 | ||
| Fig. 1 Clinical nitrofuran drugs. | ||
Research literature indicates that azines consisting of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC functional unit (Fig. 2) possess bactericidal and antiparasitic properties.33 These compounds are highly basic and form salts very easily, thus they are endowed with increased water solubility and become equivalent to hydrazones in the protonated state. They are also thermally and chemically more stable than their hydrazone tautomers.33,34 The azine bridge acts as electronic effect communicator between the substituents on its terminal imine carbon atoms.35
N–NC functional unit (Fig. 2) possess bactericidal and antiparasitic properties.33 These compounds are highly basic and form salts very easily, thus they are endowed with increased water solubility and become equivalent to hydrazones in the protonated state. They are also thermally and chemically more stable than their hydrazone tautomers.33,34 The azine bridge acts as electronic effect communicator between the substituents on its terminal imine carbon atoms.35
 | ||
| Fig. 2 Structures of biologically active azines with: (a) trypanocidal, (b) antimalarial, (c) and (d) antibacterial agents. | ||
Moreover, the use of aromatic rings in drug design is a well-known strategy as they offer numerous unique and strong interaction modes with target proteins, which include classical arene–arene (π-stacking) interactions, as well as arene–H bonding (edge-to-face interactions), and other interactions, such as sulphur–arene interactions.36 Thus, the linking of nitrofuran, azines and aromatic rings may result in compounds that are more biologically active and stable.
Based on these previous findings, nitrofuran-based azine derivatives and their antiprotozoan activities were examined in vitro. We herein present the synthesis and the biological activities of these nitrofuranylazines.
Results and discussion
Chemistry
Nitrofuran-based azine derivatives (1a–9a, 1b–9b) were synthesised via a two-step process depicted in Scheme 1, starting from commercially available 5-nitro-2-furaldehyde (NFA) or 5-nitrothiophene-2-carboxaldehyde (NTA). Firstly, the hydrazone intermediates 1–9 were prepared by reacting various substituted aldehydes with excess hydrazine hydrate (6-fold equivalent), which was needed to prevent the early formation of unwanted symmetrical arylazines and were isolated after recrystallisation from ethyl acetate. This was then followed by a Schiff base reaction of intermediates with NFA or NTA to afford the targeted nitrofuran-based azine derivatives (1a–9a, 1b–9b) in poor to excellent yields of 13–98% after purification by recrystallisation from ethyl acetate or column chromatography. The poor yields were attributed mainly the poor rate of reaction and purification process. For example, the synthesis of 5a, 8a and 7b was unsuccessful in the acidic medium, hence a basic medium with the addition of potassium carbonate was used. Yet, after 48 hours, considerable amounts of unreacted reagents were still present in the reaction mixture. No optimisation was attempted, which may be a future endeavour especially if these poorly yielded azines happen to be among the most biologically active of compounds of this investigation. | ||
| Scheme 1 Two-step synthesis of target nitrofuranylazines. Reagents and conditions: i. hydrazine hydrate (6 equiv.), TEA (1.5 equiv.), EtOH (10 mL), rt, 12 h; ii. NFA/NTA/FA (1 equiv.), EtOH (10 mL), rt, 12 h. | ||
The aldazines were divided into two chalcogen series, nitrofuran (oxygen chalcogen, series A) and nitrothiophene (sulphur chalcogen, series B).
Furthermore, to validate nitrofuran as pharmacophore of the azines, furfural was reacted with selected hydrazone intermediates, namely 7 and 9, which resulted in azines 11 and 12, omitting the nitro group. The characterisation spectra of all synthesised compounds are provided as ESI† (Appendix A).
The formation of the hydrazone intermediates (1–9) was illustrated via1H NMR through the disappearance of the characteristic aldehyde proton at ca. δ 10.0 ppm and the appearance of a previously absent singlet at δ 7.65 ppm, indicative of the imine proton of the newly formed hydrazone bond CHNH–NH2. This was further corroborated by the appearance of the characteristic singlet at ca. δ 6.60, representative of the terminal primary amine protons NH2. The disappearance of this NH2 peak from the spectra of 1a–9a and 1b–9b confirmed the success of the Schiff base reaction. This was further verified by the appearance of two separate vinylic protons H-1′ and H-6 of the azine bond as characteristic singlets at ca. δ 8.79 and 8.65 ppm.
The fluorine-containing azines (i.e., 2a, 2b) exhibited proton-fluorine coupling, and it was found that the protons of interest, H-3′ and H-4′, underwent mostly ortho couplings with JH–F ∼ 8–9 Hz. Through careful examination of all the 1H spectra, it was determined that all the protons of each azine were accounted for.
The 13C spectra of the aldazines showed the two hydrazone moiety peaks of C-1′ at δ 162.7 and C-6 at δ 149.8. Carbon–fluorine coupling (JC–F) also occurred in compounds 2a and 2b which exhibited J values of ca.1JC-5′–F = 250 Hz, 2JC 3′–F = 9.0 Hz, 3JC-3′–F = 3.0 Hz and 4JC-2′–F = 22.0 Hz.
IR spectroscopy further confirmed the success of the Schiff base reactions through the appearance of characteristic stretching absorptions: CN (1600–1560 cm−1) and N–O (1550–1500 and 1350–1300 cm−1). The HRMS using atmospheric pressure chemical ionisation (APCI) source confirmed the integrity of the synthesised aldazines as in all cases, the molecular ions corroborated the proposed structures.
Of note, azines may occur in three configurational isomers, i.e. (E,E), (E,Z), and (Z,Z).37,38 Photochemical isomerisation of CN bonds can result in the production of isomers (E,Z) and (Z,Z) from (E,E), which is thermodynamically the most stable form.39 A sequence of four atoms (CN–NC) determines the configuration of aromatic azines. X-ray diffraction (XRD) studies indicate that mostly all of reported aromatic azines exhibit the preferred (E,E) configuration where large groups linked to the imine carbons (CN–) are trans to the N–N bond, due to reduced repulsive interactions with the lone pair of the N atoms.40 The current azines were not subjected to XRD analysis, and to the best of knowledge, they are novel. However, XRD studies of ferrocenylazines bearing the 5-nitrofuran and 5-nitrothiophene scaffolds have previously been conducted by Gómez et al.35,41 and confirmed the (E,E) configuration of these aromatic azines. In this configuration, the 1H chemical shifts (δ) of the azine bridge protons were found to be in the 8.38–8.36 ppm region, wherein those of protons H-6 and H-1′ of the current azines also fell. Accordingly, the (E,E)-configuration was presumed for all the aldazines in this study.
In silico physicochemical and pharmacokinetic properties
The physicochemical and pharmacokinetic properties of the synthesised compounds and standard nitrofuran drugs were determined using the SwissADME web tool and presented in ESI† (Appendix B).The logPo/w value of a molecule is the partition coefficient between octanol (lipophilic phase) and water (hydrophilic phase).42 logPo/w is used to examine important biological properties for drug action, which include lipid solubility, distribution in tissue, binding to receptors, cellular uptake, metabolism, and drug bioavailability.43 According to Lipinski et al.,44 logPo/w values are targeted in the 1–5 range, with 1–3 being ideal for orally deliverable drugs. All the synthesised compounds presented with a logPo/w of 1–5, whereas the parent drugs, NFA and NTA, and reference antibiotics, NFX, FZD, NFZ and NFT, fell below 1 due to their higher hydrophilic affinities. This in theory implies an overall improvement in oral bioavailability.
Furthermore, all synthesised azines complied with Lipinski's rules and had physicochemical properties well within the target ranges.44 Lipophilicity, although a crucial physiochemical parameter, is not the only determining characteristic of good drug design, as evident by other successful drugs that do not fall within the target range.45 Therefore, other drug parameters, such as topological polar surface area (TPSA), were predicted for these compounds. TPSA is the surface area of a molecule that emerges from nitrogen or oxygen atoms, as well as hydrogen atoms that are attached to oxygen or nitrogen atoms.46 TPSA shows the correlation with passive molecular transport through membranes, which allows the prediction of gastrointestinal (GI) absorption, Caco-2 monolayer permeability, and penetration of the blood–brain barrier. All the derivatives, apart from 9b, were predicted to be highly absorbed in the gastrointestinal (GI) tract through passive diffusion and were, therefore, expected to be druglike by nature (ESI† table). This also indicates that the possible mode of delivery for these azine analogues would be oral administration, except for 9b, which will possibly be intravenous or intramuscular administration.
Electrochemistry
The biological activity of nitroheterocyclic compounds results from their capacity to undergo oxidation–reduction process by one electron-transfer and their selective toxicity is determined by reduction to the biologically active form in the absence of oxygen.47–49 Hence, electrochemical testing was used to assess the redox properties of the synthesised aldazines. Cyclic voltammetric experiments determined their half redox potentials (E1/2) at room temperature using a scan rate of 100 mV s−1 and solution of 0.5 mM in anhydrous acetonitrile (MeCN), with 0.1 M [tBuN][PF6] as the background electrode. The redox potentials were measured using glassy carbon, Pt wire and Ag/AgCl as the working, counter, and reference electrodes, respectively. The data are summarised in Table 1.| Compd. | Nitroaromatic redox group | Redox potentials | ||||
|---|---|---|---|---|---|---|
| E pa (V) | E pc (V) | E 1/2 (V) | ΔEpe (V) | i pa/ipcf | ||
| NFA: 5-nitro-2-furaldehyde; NTA: 5-nitro-2-thiophenecarboxaldehyde; NTX: nifurtimox; NF: nitrofuran; NT: nitrothiophene; Ph: phenyl; nd: not determined.a Measured in acetonitrile at a scan rate of 100 mV s−1 (V vs. Ag/AgCl).b Anodic wave potential.c Cathodic wave potential.d Half wave potential (E1/2 = (Epa + Epc)/2).e Wave potential separation (Ep = Epa − Epc).f Ratio of anodic to cathodic current.g Historical data.35 | ||||||
| NFA | NO2–NF | −0.74 | −1.02 | −0.88 | 0.28 | 0.967 |
| 1a | NO2–NF | −0.76 | −1.00 | −0.88 | 0.24 | 1.016 |
| 2a | NO2–NF | −0.78 | −1.06 | −0.92 | 0.28 | 0.990 |
| 3a | NO2–NF | −0.80 | −1.08 | −0.94 | 0.28 | 0.967 |
| 4a | NO2–NF | −0.80 | −1.06 | −0.93 | 0.26 | 0.891 |
| 5a | NO2–NF | −0.82 | −1.06 | −0.94 | 0.24 | 1.007 |
| 6a | NO2–NF | −0.68 | −1.02 | −0.85 | 0.34 | 0.965 |
| 7a | NO2–NF | −0.78 | −1.04 | −0.91 | 0.26 | 1.008 |
| 8a | NO2–NF | −0.82 | −1.08 | −0.95 | 0.26 | 0.943 |
| 9a | NO2–NF & NO2–Ph | −0.84 | −1.06 | −0.95 | 0.22 | 0.963 |
| NTA | NO2–NT | −0.86 | −1.06 | −0.96 | 0.20 | 0.755 |
| 1b | NO2–NT | −0.84 | −1.06 | −0.95 | 0.22 | 1.032 |
| 2b | NO2–NT | −0.78 | −1.00 | −0.89 | 0.22 | 1.022 |
| 3b | NO2–NT | −0.74 | −0.96 | −0.85 | 0.22 | 0.889 |
| 4b | NO2–NT | −0.80 | −1.04 | −0.92 | 0.24 | 0.961 |
| 5b | NO2–NT | −0.78 | −1.02 | −0.90 | 0.24 | 1.000 |
| 6b | NO2–NT | −0.80 | −1.04 | −0.92 | 0.24 | 0.990 |
| 7b | NO2–NT | nd | nd | nd | nd | nd |
| 8b | NO2–NT | −0.82 | −1.06 | −0.94 | 0.24 | 1.002 |
| 9b | NO2–NT & NO2–Ph | −0.82 | −1.06 | −0.94 | 0.24 | 0.951 |
| NTX | NO2–NF | nd | nd | −0.88g | nd | nd |
The voltammograms of all azines displayed reversible waves with E1/2 values in the −0.96 to −0.85 V range, as shown in Fig. 3 for azine 7a, when scanned over the range of −1.7 to +1.0 V.
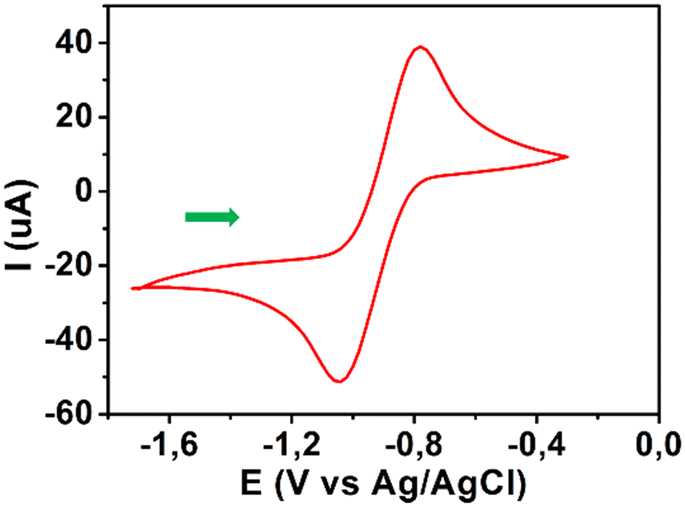 | ||
| Fig. 3 Cyclic voltammogram of 7a (0.5 mM) in 0.1 M tBuNPF6/MeCN recorded at a scan rate of 100 mV s−1. | ||
Comparing the reduction waves (Epc) and ipa/ipc ratios, all azines 1a–9a and 1b–9b had values of −1.0 to −1.08 V, and ca. 1, respectively, which are indicative of a reversible-controlled one-electron transfer process that was attributed to the reduction of the Ar–NO2 to form a stable anion radical (i.e., Ar–NO2˙−) as previously reported.35 However, all azines possessed negative E1/2 values which may also suggest that their nitroaromatic groups are less readily reduced. The E1/2 value of nitrofuran clinical trypanocidal drug, NTX, is −0.88 V, as reported by Gómez et al.35 Compounds NFA, 1a and 2b presented with E1/2 values comparable to that of NTX, whereas 6a and 3b were only slightly lower (−0.85 V). Since Leishmania and Trypanosoma are taxonomic kinetoplastid parasites, these aldazines and nifurtimox may display comparable trypanocidal and leishmanicidal potencies, which may be more pronounced for NFA and 1a as these two compounds have the same E1/2 value of −0.88 V as NTX. The remaining majority had higher half-potentials (E1/2 ≥ 0.9 V), which could suggest a tendency to be less reduced than NTX. As the general biological activity and selective toxicity of nitroheterocyclic compounds depend on their capacity to undergo oxidation–reduction via electron-transfer and anaerobic reduction, respectively, this may indicate reduced activity compared to NTX.
Further scrutiny of the data revealed that, in reference to NFA, the reduction potential (E1/2) of its derived azines, 2a–9a, show a slight cathodic shift which suggests that these azines are more readily reduced (though marginally) than the parent NFA due to the incorporation of more electron-donating groups. This assertion is corroborated by aldazines 5a, 6a, 7a and 8a that contain the electron-donating groups (EDGs), methyl (Me), MeO (methoxy in para-position), benzyloxy (BnO) and hydroxy (OH), respectively on the phenyl (Ph) ring while the remaining azines, 2a, 3a and 9a harbour electron-withdrawing groups (EWGs).
On the contrary, in reference to NTA, the reduction potential (E1/2) of the sulphur chalcogen azines 1b–9b showed no discernible differences except for 2b and 3b. This could be attributed to both azines bearing EWGs, chlorine and bromine, respectively, while for the remaining azines of the subseries, the electronic nature seemed relatively the same which resulted in their comparable E1/2 values.
Fig. 4 shows comparisons of molecular weight, logP and electronic effect with electrochemical data of the azines with no obvious correlations found.
 | ||
| Fig. 4 Comparison of electrochemistry data with electronic effect, (a) molecular weight and (b) in silico logP data. EWGs are indicated in green, whereas EDGs were indicated in orange and the neutral group was indicated in grey. | ||
Pharmacology
The Leishmania parasite presents in two different morphological forms throughout its life cycle; the motile flagellated promastigote form (present in the vector), and the intracellular non-flagellated amastigote form (present in mammalian host cells).50T. cruzi presents in three different forms throughout their life cycle: the epimastigote form (present in the vector), the trypomastigote form (present in the host), and the amastigote form (present in the host).51 Of these, the amastigotes are the clinically relevant forms for drug development against Leishmania and T. cruzi infections, due to their responsibility in the development of the infection and progression to the disease state.52 However, trypomastigotes of T. cruzi are also valuable as a drug target due to them being the infective, non-proliferative blood circulating form, and thus disease progression may be prevented through their death.53 Nonetheless, it is still beneficial to screen compounds against in vitro promastigote and epimastigote forms, along with trypomastigote and amastigote forms, to identify possible antiprotozoal hits.54 The synthesised derivatives were therefore assessed for their in vitro antileishmanial activity against the promastigotes and amastigotes of two Leishmania strains, L. donovani 9515 and L. major IR-173. These Leishmania spp. were selected to determine the specificity of the synthesised derivatives against CL- and VL-causing species, L. major and L. donovani, respectively.5 The in vitro antitrypanosomal activity of the compounds was also assessed against the epimastigotes and trypomastigotes of T. cruzi strain, the causative agent of Chagas disease. Amphotericin B (AmB) and benznidazole were used as standard drugs for antileishmanial and antitrypanosomal activity, respectively, alongside the reference cNFs, nifuroxazide, furazolidone, nitrofurazone and nitrofurantoin. The parent compounds, NFA, NTA were also screened for comparison. General cytotoxicity profiles of these derivatives were determined using Vero cells with emetine (EM) as a positive control. Specific host macrophage cytotoxicity was assessed using the human leukemia monocytic cell line, THP-1.All the compounds were initially screened at 10 μM for potential antipromastigote and anti-amastigote activities. Compounds with antipromastigote growth inhibition of >70% qualified for IC50 determination,54 whereas those with anti-amastigote growth inhibition of >60% qualified for IC50 determination.55
For the anti-amastigote IC50 determination, a modified method of Jain et al.56 on parasite rescue and transformation was used. The anti-amastigote activity was measured 72 hours post lysis to detect signs of amastigote recovery and subsequent transformation to and proliferation as promastigotes. Compounds that retained significant antileishmanial activity up to 72 hours post macrophage lysis, indicative of loss in amastigote recovery to proliferative promastigotes, were thus deemed leishmanicidal. The antileishmanial activities and cytotoxicity of the screened compounds are summarised in Tables 2 and 3.
| Compd. | General cytotoxicity Vero IC50 (μM) | Host cell cytotoxicity THP-1 IC50 (μM) | Anti-promastigote activity | Anti-amastigote activity | ||||
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SI1a | IC50 (μM) | SI2b | SI3c | SpI1d | |||
| a Selectivity indexes of L. major: SI1 = IC50 Vero/IC50 promastigote. b SI2 = IC50 Vero/IC50 amastigote. c SI3 = IC50 THP-1/IC50 amastigote. Specificity index (SpI) < 0.4 indicates more antipromastigote activity, 0.4 < SpI < 2.0 indicates activity against both forms, SpI > 2.0 indicates more anti-amastigote activity.55 d Specificity index of L. major: SpI1 = IC50 promastigote/IC50 amastigote. Selectivity Indexes of L. donovani. | ||||||||
| NFA | 17.82 ± 0.25 | 80.92 ± 6.37 | 7.94 ± 0.79 | 2 | 5.60 ± 0.44 | 3 | 14 | 1.42 |
| 1a | 16.29 ± 3.50 | >100 | 1.19 ± 0.29 | 14 | >10 | — | — | — |
| 2a | 47.42 ± 1.12 | 63.37 ± 5.49 | 0.73 ± 0.09 | 65 | 3.69 ± 0.24 | 13 | 17 | 0.20 |
| 3a | >100 | >100 | 0.42 ± 0.07 | 238 | 2.47 ± 0.43 | 40 | 40 | 0.17 |
| 4a | >100 | >100 | 0.75 ± 0.18 | 133 | 0.63 ± 0.02 | 159 | 159 | 1.19 |
| 5a | >100 | >100 | 0.42 ± 0.08 | 238 | >10 | — | — | — |
| 6a | 51.01 ± 1.26 | >100 | 0.89 ± 0.12 | 57 | >10 | — | — | — |
| 7a | >100 | >100 | 0.45 ± 0.02 | 222 | >10 | — | — | — |
| 8a | 18.66 ± 1.31 | 46.20 ± 5.02 | 0.64 ± 0.10 | 29 | >10 | — | — | — |
| 9a | 11.56 ± 1.12 | 16.75 ± 1.14 | 0.70 ± 0.10 | 17 | 7.90 ± 0.19 | 1 | 2 | 0.09 |
| NTA | 12.42 ± 0.88 | 26.69 ± 0.72 | 3.75 ± 0.39 | 3 | >10 | — | — | — |
| 1b | 42.00 ± 8.79 | >100 | 1.43 ± 0.19 | 29 | >10 | — | — | — |
| 2b | >100 | >100 | 3.79 ± 0.70 | 26 | >10 | — | — | — |
| 7b | >100 | >100 | 4.39 ± 0.24 | 23 | >10 | — | — | — |
| 8b | 33.12 ± 1.22 | 22.87 ± 0.13 | 2.22 ± 0.00 | 15 | >10 | — | — | — |
| 11 | >100 | — | >10 | >10 | ||||
| 12 | >100 | — | >10 | >10 | ||||
| NFZ | >100 | >100 | 1.85 ± 0.06 | 54 | >10 | — | — | — |
| FZD | >100 | >100 | 0.34 ± 0.03 | 294 | 5.61 ± 1.89 | 18 | 18 | 0.06 |
| Em | 0.08 ± 0.009 | — | — | — | — | — | — | |
| Bzn | >100 | >100 | — | — | — | — | — | |
| AmB | 57.80 ± 3.20 | 14.86 ± 0.09 | 0.03 ± 0.01 | 1927 | 0.47 ± 0.01 | 123 | 32 | 0.06 |
| Compd. | Anti-promastigote activity | Anti-amastigote activity | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) | SI4a | IC50 (μM) | SI5b | SI6c | SpI2d | |
| a SI4 = IC50 Vero/IC50 promastigote. b SI5 = IC50 Vero/IC50 amastigote. c SI6 = IC50 THP-1/IC50 amastigote. d Specificity index of L. donovani: SpI2 = IC50 promastigote/IC50 amastigote. Selectivity indexes of T. cruzi. | ||||||
| NFA | >10 | — | 2.68 ± 0.68 | 2 | 30 | — |
| 1a | 1.45 ± 0.19 | 11 | >10 | — | — | — |
| 2a | 1.18 ± 0.14 | 40 | >10 | — | — | — |
| 3a | 0.67 ± 0.04 | 149 | >10 | — | — | — |
| 4a | 3.56 ± 0.00 | 28 | 6.41 ± 0.00 | 16 | 16 | 0.56 |
| 5a | 0.76 ± 0.05 | 132 | >10 | — | — | — |
| 6a | 1.16 ± 0.11 | 44 | 2.28 ± 0.07 | 22 | 44 | 0.51 |
| 7a | 1.61 ± 0.22 | 62 | 0.25 ± 0.09 | 400 | 400 | 6.44 |
| 8a | 1.02 ± 0.08 | 18 | 2.70 ± 0.62 | 7 | 17 | 0.38 |
| 9a | 0.70 ± 0.06 | 17 | 0.53 ± 0.22 | 22 | 32 | 1.32 |
| NTA | 5.07 ± 0.45 | 2 | >10 | — | — | — |
| 1b | 0.93 ± 0.06 | 45 | 7.26 ± 2.56 | 6 | 14 | 0.13 |
| 2b | 2.00 ± 0.15 | 50 | >10 | — | — | |
| 8b | 1.66 ± 0.18 | 20 | 0.34 ± 0.03 | 97 | 67 | 4.88 |
| 11 | >10 | >10 | ||||
| 12 | >10 | >10 | ||||
| NFZ | 1.85 ± 0.14 | 54 | 1.69 ± 0.36 | 59 | — | 1.09 |
| FZD | 0.28 ± 0.28 | 357 | 2.70 ± 0.81 | 37 | — | 0.10 |
| AmB | 0.02 ± 0.009 | 2890 | 0.45 ± 0.05 | 128 | — | 0.04 |
All compounds were also initially screened at 10 μM for potential anti-epimastigote and antitrypomastigote activities. Currently, there are no published cut-off values for these assays. Accordingly, as the epimastigotes are cultured and assayed similarly to Leishmania promastigotes, the growth inhibition cut-off of >70% was also applied for the selection of anti-epimastigote compounds for IC50 determination. For the trypomastigote screening cut-off, >70% was also applied due to the axenic/host-free nature of the trypomastigote cultures. The antitrypanosomal activities of the screened compounds are summarised in Table 4.
| Compd. | Anti-epimastigote activity | Anti-trypomastigote activity | ||||
|---|---|---|---|---|---|---|
| IC50 (μM) | SI5a | IC50 (μM) | SI6b | SI7c | SpI3d | |
| a SI5 = IC50 Vero/IC50 epimastigote. b SI6 = IC50 Vero/IC50 trypomastigote. c SI7 = IC50 THP-1/IC50 trypomastigote. Specificity index (SpI) < 0.4 indicates more anti-epimastigote activity, 0.4 < SpI < 2.0 indicates activity against both forms, SpI > 2.0 indicates more antitrypomastigote activity.55 d Specificity index of T. cruzi: SpI3 = IC50 epimastigote/IC50 trypomastigote. Vero: African green monkey kidney epithelial cells; blue = compounds qualifying as antileishmanial/antitrypanosomal hits;52 red = compounds qualifying as potential antileishmanial leads.52 All data reported in the tables were significant at p < 0.05. NFA: 5-nitro-2-furaldehyde; NTA: 5-nitro-2-thiophenecarboxaldehyde; FZD: furazolidone; NFZ: nitrofurazone; Em: emetine; Bzn: benznidazole; AmB: amphotericin B; —: not determined; >10: compound did not qualify for IC50 determination due to low growth inhibition presented during single-point screening. | ||||||
| NFA | >10 | — | >10 | — | — | — |
| 1a | 7.66 ± 0.37 | 2 | 2.31 ± 0.11 | 7 | 42 | 3.32 |
| 2a | 3.56 ± 0.46 | 13 | 1.95 ± 0.09 | 24 | 51 | 3.32 |
| 3a | 2.41 ± 0.05 | 41 | 3.05 ± 0.06 | 33 | 33 | 0.79 |
| 4a | 46.15 ± 9.12 | 2 | >10 | — | — | — |
| 5a | 3.69 ± 0.60 | 27 | 4.04 ± 0.09 | 25 | 25 | 0.91 |
| 6a | 4.92 ± 0.47 | 10 | 6.14 ± 0.01 | 8 | 16 | 0.80 |
| 7a | 95.24 ± 4.03 | 1 | >10 | — | — | — |
| 8a | 7.30 ± 2.77 | 3 | 1.74 ± 0.01 | 11 | 27 | 4.20 |
| 9a | 2.63 ± 0.01 | 4 | 0.78 ± 0.01 | 15 | 21 | 3.37 |
| NTA | >10 | — | 1.62 ± 0.05 | 8 | 16 | — |
| 1b | 9.21 ± 1.03 | 5 | 2.50 ± 0.05 | 17 | 40 | 3.68 |
| 2b | >10 | — | 6.56 ± 0.22 | 15 | 15 | — |
| 8b | >10 | — | 3.04 ± 0.06 | 11 | 8 | — |
| 11 | >10 | — | >10 | |||
| 12 | >10 | — | >10 | |||
| NFZ | 7.83 ± 1.04 | 13 | 2.23 ± 0.06 | 45 | 45 | 3.51 |
| FZD | 2.01 ± 0.41 | 50 | 3.30 ± 0.25 | 30 | 30 | 0.61 |
| Bzn | — | — | 4.79 ± 0.56 | 21 | 21 | — |
As suggested by the logP values hinting towards lipophilic properties, the compounds presented with significantly more solubility problems compared to the reference drugs when solutions were prepared in both 1% DMSO and growth medium, forming quickly separating suspensions that made uniform sampling challenging. This resulted in relatively significant standard deviations for several compounds.
The synthesised intermediates (1–9) and several of the azine derivatives exhibited no antiprotozoal activity, thus were not reported in Tables 1–3 as they did not qualify for IC50 determination. The cytotoxicity data indicated that nitrofuran derivatives 1a, 2a, 6a, 8a and 9a (series A) were moderately toxic to mammalian cells (10 μM < IC50 < 50 μM),57,58 whereas compounds 3a, 4a, 5a and 7a had low toxicity (IC50 > 100 μM).59,60 The four nitrothiophene chalcogen counterparts that qualified for antiparasitic IC50 determination (series B) showed a similar trend, with the 1b and 8b presenting with moderate toxicity, whereas 2b and 7b had low toxicity toward mammalian cells, inferring that their observed antileishmanial activities were intrinsic. The comparison of cytotoxicity is depicted in Fig. 5.
 | ||
| Fig. 5 Comparison of general cytotoxicity in Vero and electrochemistry. EWGs are indicated in green, whereas EDGs were indicated in orange and the neutral group was indicated in grey. | ||
Comparison of the cytotoxicity with the E1/2 values did not show any correlation.
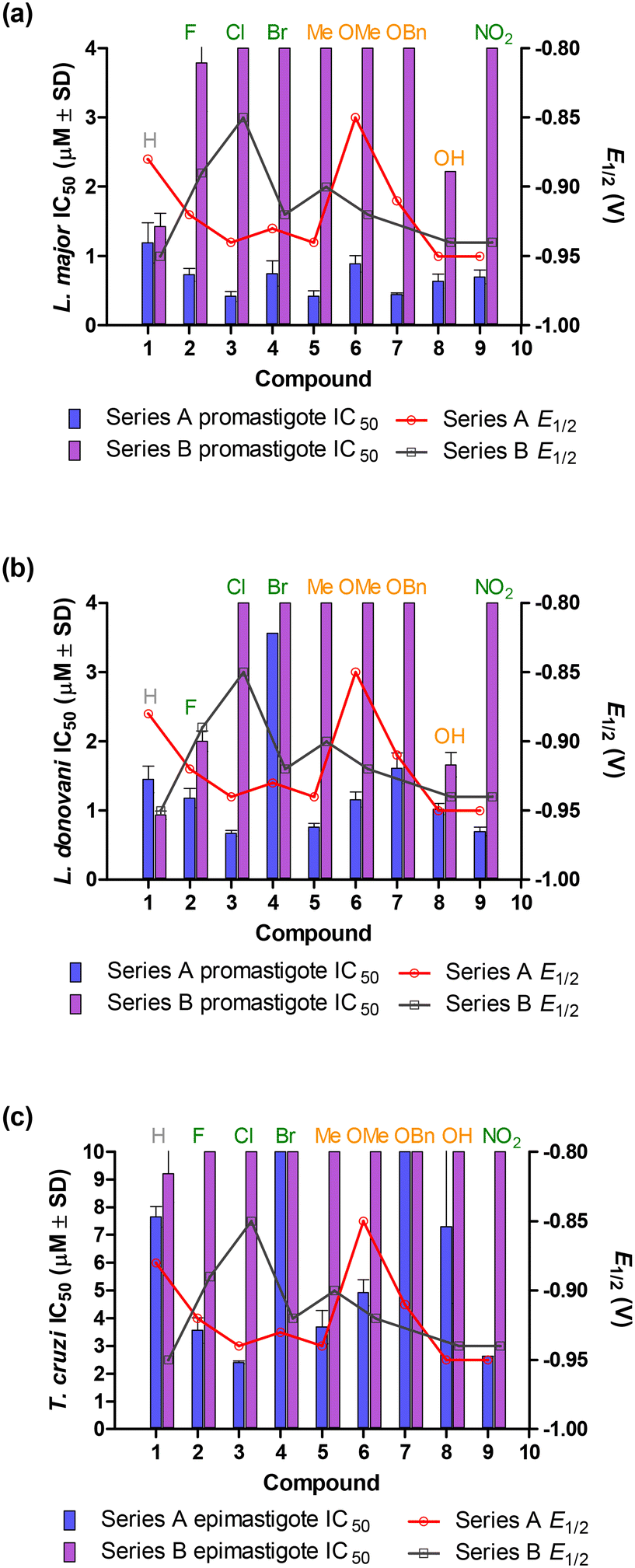 | ||
| Fig. 6 Comparison of antipromastigote and anti-epimastigote activities and electrochemistry. (a) Antipromastigote activity of L. major strain IR-173. (b) Antipromastigote activity of L. donovani strain 9515. (c) Anti-epimastigote activity of T. cruzi strain CL. EWGs are indicated in green, whereas EDGs were indicated in orange and the neutral group was indicated in grey. | ||
Nitrofuran aldazines, 2a, 3a, 4a and 9a presented with cidal anti-amastigote activity against L. major. Of these, 2a and 3a qualified as antileishmanial hits (IC50 < 10 μM; SI > 10).52 However, azine 9a presented with moderate to significant cytotoxicity against Vero and host THP1 cells, hence it does not qualify as an antileishmanial hit. The activities of these compounds were comparable to those of NFA and the nitrofuran antibiotic FZD, indicating that the activity of these azines did not improve on that of two references and the standard drug, AmB. However, the cytotoxicity of 2a and 3a were less than those of NFA and AmB. The antileishmanial activity of compounds 2a, 3a and 9a were more selectively directed towards the promastigote form (SpI < 0.4).55 Conversely, nitrofuranylazine 4a qualified as potential antileishmanial lead (IC50 < 1 μM; SI > 100)52 with specificity towards both parasite forms (0.4 < SpI < 2),55 significantly improving on the cytotoxicity and antileishmanial activity of the reference drugs against L. major.
Against L. donovani, azines 4a, 6a, 9a, 1b and 8b displayed with cidal anti-amastigote activity against antimonial-resistant L. donovani.61,62 Of these, 4a, 6a, 9a and 8b qualified as antileishmanial hits (IC50 < 10 μM; SI > 10).52 However, 8a and 1b presented with moderate to significant cytotoxicity against Vero and host THP-1 cells and therefore do not stand as antileishmanial hits. Compound 9a had submicromolar anti-amastigote activity and a favourable selectivity index, which qualify it as an antileishmanial hit despite its toxicity. Overall, azines, 4a, 6a, 8a and 1b did not improve on the activity of the reference compounds and AmB. However, they exhibit specificity towards the parasite. Azines 4a and 6a presented with affinity for both parasite developmental forms while 8a and 1b acted more selectively against the promastigotes. On the other hand, the activity of 9a and 8b improved significantly compared to those of the reference drugs with both compounds being selective towards the amastigote form (SpI > 2).55 Their toxicity, however, was higher than those of the reference drugs. Conversely, azine 7a qualified as potential antileishmanial lead (IC50 < 1 μM; SI > 100)52 with specificity towards the amastigote form, significantly improving on the cytotoxicity and antileishmanial activity of the reference drugs, including AmB (though marginally) against antimonial-resistant L. donovani.
To confirm that the nitrofuran moiety serves as pharmacophore, analogues of a small selection of the most active antileishmanial compounds were synthesised and screened for antiparasitic activity and cytotoxicity. Omission of the nitro-substituent in the nitrofuran moiety of the antileishmanial hits 7a and 9a, resulted in azines 11 and 12, respectively, that displayed a complete loss of both anti-amastigote activity (IC50 > 10 μM) and the cytotoxicity (IC50 > 100 μM) (Tables 2 and 3). The latter confirmed the nitro group acting as toxicophore.
 | ||
| Fig. 7 Comparison of anti-amastigote and antitrypomastigote activities and electrochemistry. (a) Anti-amastigote activity of L. major strain IR-173. (b) Anti-amastigote activity of L. donovani strain 9515. (c) Antitrypomastigote activity of T. cruzi strain CL. EWGs are indicated in green, whereas EDGs were indicated in orange and the neutral group was indicated in grey. | ||
Similarly, to the loss of antileishmanial activity, the analogue 12 of the antitrypanosomal cytotoxic hit 9a, also resulted in a complete loss of both anti-trypomastigote activity and cytotoxicity (Table 3), confirming the nitrofuran moiety as both pharmacophore and toxicophore for this nitrofuranylazine.
Due to the limited biological activity of the nitrothiophene azines, no correlations were detected between antiparasitic activity and electrochemical data. However, except for azines 7a, 8a and 9a, there was a visible correlation between the antiparasitic (Leishmania antipromastigote and T. cruzi anti-epimastigote) IC50 of the nitrofuranylazines and their E1/2 values (Fig. 6). This was in line with the following electronic effect observations, confirming a correlation between the electrochemical characteristics of the compounds and the resulting antipromastigote and anti-epimastigote activities.
Furthermore, due to a lack of significant differences between the IC50 ranges of the EDG and EWG-containing compounds, it was not possible to sufficiently determine whether the compounds' tendency to be reduced affected their resulting antipromastigote and anti-epimastigote activities.
When considering the electronic effect and strength, the R groups on the Ph ring can be divided, and in order of increasing strength, into EWGs (Br < Cl < F < NO2), neutral groups (H), and EDGs (CH3(Me) < OH < OCH3(OMe) < OCH2C6H5(OBn).
The general pattern observed for EWG-containing azines against L. major IR-173 (Fig. 6a) consists of an increase in antipromastigote activity (though marginally) as the strength of the EWG increases (except 3a). This is evident by the activity of 9a, bearing the strongest of these EWGs (i.e., NO2), that exhibits IC50 0.70 μM, compared to 0.75 μM of 4a that has the weakest EWG (i.e., Br). The EDG-harbouring azines, on the other hand, display a marginal decrease in activity as the strength of the EDG increases (Fig. 6a). This can be seen by the strongest of these EDGs (the OBn group), azine 7a, exhibiting an IC50 0.45 μM, compared to 0.42 μM of the weakest EDG Me-bearing azine 5a. The neutral group, H-containing 1a exhibited the weakest antipromastigote activity, with an IC50 value of 1.19 μM.
Scrutiny of the activity against L. donovani 9515 promastigotes shows a broadly evident trend similar to that of L. major (Fig. 4b), with an increase in activity of EWG azines as the electronic effect strengthens. The increasing activity order was: 4a (IC50 3.56 μM) < 2a (IC50 1.18 μM) < 9a (IC50 0.70 μM) with 3a (IC50 0.67 μM) being an outlier. The same trend is evident with the EDG azines, where a decrease in activity is observed with an increase in electronic strength in the order: 5a (IC50 0.76 μM), 8a (IC50 1.02 μM), 6a (IC50 1.16 μM) and 7a (IC50 1.61 μM). Thus, the strongest EDG containing azine 7a had the weakest activity while the weakest EDG bearing 5a possessed the strongest antipromastigote activity. The neutral group bearing 1a exhibited the second weakest activity (IC50 1.45 μM) of the subseries against L. donovani.
Analysis of the activity against T. cruzi reveals that the neutral 1a again exhibits weaker activity (IC50 7.66 μM). With the EWG azines, an increase in activity followed an increase in electronic strength, 3a being an outlier, as seen with the antipromastigote activities (Fig. 6c). The EDG-harbouring nitrofuranylazines also displayed the same trend of activity against T. cruzi as with L. major, where a decrease in activity is observed with an increase in electronic strength, azine 8a being the exception (Fig. 6c). Fig. 8 depicts the summary of SAR in this study.
 | ||
| Fig. 8 Summary of uncovered SAR of the study. | ||
With regards to the more clinically relevant parasite forms, there were no visible correlations between the antiparasitic IC50 values and E1/2 values (Fig. 7). However, there were several seemingly species and/or parasite form-dependent correlations between the antiparasitic activities and compound electronic effects.
In Leishmania amastigotes, azines 1 (i.e., 1a & 1b) with neutral group generally presented with IC50 > 7 μM. For L. major, an increase in EWG strength in nitrofuran derivatives resulted in significantly decrease anti-amastigote activity compared to 1a and non-significant nitrothiophene derivative activity when compared to 1b (Fig. 7a), whereas in L. donovani this activity and pattern were lost in both series (Fig. 7b). The addition of EDGs, on the other hand, did not improve activity against L. major, whereas an increase in nitrofuran derivative EDG strength resulted in an increase in activity against L. donovani amastigotes. These observations contrast with the observations of the antipromastigote activities.
In T. cruzi trypomastigotes, azines 1 (1a & 1b) were significantly more active (IC50 < 2.5 μM) than in Leishmania. An increase in EWG strength resulted in increased nitrofuran derivative anti-trypomastigote activity, whereas an increase in EDG strength resulted in activity, with the exception of 5a, but no clear patterns for the nitrothiophene series (Fig. 7c). These observations are in line with the observations of the anti-epimastigote activities.
Fig. 8 depicts a summary of discovered SAR of the study.
When comparing ring equivalents 1a and 1b, there was comparable lipophilicity with logPo/w values of 2.01 and 2.66, respectively. However, differences in toxicity and activity against all parasite strains were observed, with 1a being more potent against L. major and T. cruzi, while 1b was more potent against L. donovani.
Comparison of bioisosteres 3a and 4a, revealed that both azines had comparable lipophilicity with logPo/w values of 2.52 and 2.64, respectively, and were found to be non-toxic to mammalian cells, but exhibited different activity against parasites, with 3a being the more potent azine. Therefore, bioisosterism proved beneficial in this study.
Conclusions
Post COVID-19, there has been significant interest in the development of multifunctional, broad-spectrum antipathogenic drugs to improve global responsiveness to non-traditional diseases and future health emergencies.66 Furthermore, broad-spectrum antipathogenic drugs can also address the logistical and economic challenges associated with infectious diseases with overlapping geographical distributions.67 Accordingly, this study utilised the multifunctional clinical nitrofuran scaffold as pharmacophore to synthesise new nitrofuran derivatives with the aim of improving its biological antiparasitic activity. A series of nitrofuran-based azines were synthesised in poor to excellent yields following a two-step process that included hydrazone formation and a Schiff base reaction. Most of the compounds were found to have low cytotoxicity, apart from a handful that exhibited moderate toxicity. It was observed that the nitrofuran sub-series was more potent than the nitrothiophene sub-series, having due favourable electrochemical properties like nifurtimox. Additionally, the superior activity of nitrofuranylazines might be related to the oxygen atom of the furan ring's ability to form hydrogen bonds. The electronic effect analysis showed species and parasite form-specific effects. Nitrofuranylazines, 4a and 7a were uncovered as safe antileishmanial early leads while 9a stood out as a trypanocidal hit. All three azines will undergo future investigation to dissect their exact mechanism of action and to identify their molecular targets within the parasites. It will also be worth establishing whether the promising in vitro parasiticidal activity can be translated in vivo. Furthermore, nitrofuran was confirmed as the pharmacophore as analogues (11, 12) of the most active derivatives (7a, 9a), wherein the nitro-group of the nitrofuran was removed, presented with a complete loss in biological activity. Moreover, the antiparasitic activities of the synthesised azines were compared and hits, 2a, 3a, 9a and 8b were identified as potential dual-active compounds due to their activity against both Leishmania and T. cruzi. These parasite species are, however, taxonomically related, and accordingly share similar biochemical and structural characteristics. Further assessment against non-related pathogens will therefore be required to determine their broad-spectrum antiparasitic activity. In theory, there is potential for broad-spectrum activity as the nitrofuran pharmacophore is known to promote oxidative stress, a species-independent drug target.Further, this study focused exclusively on unsymmetrical aromatic azines that proved to be cytotoxic in general due to their lipophilicity. Hence, it will be of interest in future endeavour to explore nitrofuran azines containing less lipophilic building blocks, alkyl, heterocycles etc.
Experimental section
Materials
All reagents used were purchased from Sigma-Aldrich (Johannesburg, South Africa), and solvents from Associated Chemical Enterprises, ACE (South Africa). All chemicals and reagents were of analytical grade, and as such, no further purification was needed.General procedures
The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance™ III 600 spectrometer at a frequency of 600 and 151 MHz, respectively, in deuterated dimethyl sulfoxide (DMSO-d6). The chemical shifts were reported in parts per million (ppm) with the residual protons of the solvent as a reference. The abbreviations of the splitting patterns were as follows: singlet (s), doublet (d), doublet of doublet (dd), doublet of triplets (dt), triplet (t), triplet of doublets (td), triplet of triplets (tt), quartet of doublets (qd), and multiplet (m). High resolution mass spectrometry (HRMS) spectra were recorded on a Bruker MicroTOF Q II mass spectrometer. It was equipped with an atmospheric pressure chemical ionisation (APCI) source set at 200 °C or 180 °C and analysed using Bruker Compass Data Analysis 4.0 software. A full scan ranged from 50–1500 m/z at a capillary voltage of 4500 V, an end plate offset voltage of −500 V, with the nebuliser set at 1.6 and 0.4 bar, respectively, and a collision cell RF voltage of 100 Vpp. Infrared (FTIR) spectra were recorded on a Bruker Alpha-P FTIR instrument. A BÜCHI melting point B-545 instrument was used to determine melting points (mp) and were uncorrected. Thin layer chromatography (TLC) was performed on silica gel plates (60F254) that were acquired from Merck (South Africa).Syntheses
:ethyl acetate (EtOAc) (7:3, v/v or 1:1 v/v). Upon completion, the reaction mixture was extracted with DCM (2 × 30 mL) and water (2 × 40 mL). The organic phase was dried over anhydrous magnesium sulphate (MgSO4), filtered, and concentrated in vacuo to yield a crude product, which was then recrystallised in EtOAc to form the phenylhydrazone. Characterisation data for all the intermediates is reported as ESI.†
:EtOAc (7:3, v/v or 1:1 v/v). Upon completion, the reaction was quenched with water (50 mL) and the precipitate that formed was filtered and recrystallised from EtOAc to afford the desired azines (1a–9a, 1b–9b). Compounds 6a and 7b were further purified by column chromatography on silica gel eluting with Hex:EtOAc (4:1, v/v).
In the case of azines, 5a, 9a, 5b, 6b and 9b, the acid method was found unsuitable as the product could not be formed, hence the sulphuric acid was substituted for anhydrous potassium carbonate (6 mmol, 2 eq.).
Characterisation spectra of all compounds are provided as ESI.†
(1E,2E)-1-Benzylidene-2-[(5-nitrofuran-2-yl)methylene]hydrazine 1a. Brown powder. Yield: 715 mg (98%); mp 169–171 °C; Rf 0.7 (Hex
:EtOAc 1:1, v/v). IR νmax (cm−1): 1629 (CN), 1565 (CC), 1471 (N–O), 1246 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.78 (s, 1H, H-1′), 8.64 (s, 1H, H-6), 7.90 (d, J = 6.9 Hz, 2H, H-3′), 7.81 (d, J = 3.8 Hz, 1H, H-4), 7.56 (t, J = 7.1 Hz, 1H, H-5′), 7.53 (d, J = 6.9 Hz, 2H, H-4′), 7.40 (d, J = 3.8 Hz, 1H, H-3); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.8, 152.5, 150.7, 149.6, 133.3, 132.0, 129.0, 128.8, 118.5, 114.1. HRMS-APCI (pos) m/z 244.0717 [M + H]+ (Calcd. for C12H9N3O3+, 244.0722).
(1E,2E)-1-(4-Fluorobenzylidene)-2-[(5-nitrofuran-2-yl)methylene]hydrazine 2a. Yellow powder. Yield: 517 mg (66%); mp 191–193 °C; Rf 0.39 (Hex
:EtOAc 7:3, v/v). IR νmax (cm1): 3113 (C–H), 1631 (CN), 1597 (CC), 1516 (N–O), 1342 (C–F), 1247 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.81 (s, 1H, H-1′), 8.65 (s, 1H, H-6), 7.97 (dd, JH–F = 8.8, 5.7 Hz, 2H, H-3′), 7.82 (d, J = 3.9 Hz, 1H, H-4), 7.41 (d, J = 3.9 Hz, 1H, H-3), 7.38 (t, JH–F = 8.8 Hz, 2H, H-4′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 164.2 (d, 1JC–F = 250.3 Hz), 162.6, 151.4, 150.6, 149.6, 131.2 (d, 2JC–F = 9.1 Hz), 130.0 (d, 3JC–F = 2.9 Hz), 118.5, 116.2 (d, 4JC–F = 22.0 Hz), 114.1. HRMS-APCI (pos) m/z 262.0635 [M + H]+ (Calcd. for C12H8FN3O3+, 262.0628).
(1E,2E)-1-(4-Chlorobenzylidene)-2-[(5-nitrofuran-2-yl)methylene]hydrazine 3a. Yellow powder. Yield: 500 mg (60%); mp 175–177 °C; Rf 0.53 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3092 (C–H), 1630 (CN), 1562 (CC), 1352 (N–O), 1246 (C–O), 820 (C–Cl); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.81 (s, 1H, H-1′), 8.65 (s, 1H, H-6), 7.92 (d, J = 8.5 Hz, 2H, H-3′), 7.82 (d, J = 3.9 Hz, 1H, H-4), 7.61 (d, J = 8.5 Hz, 2H, H-4′), 7.42 (d, J = 3.9 Hz, 1H, H-3); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 162.6, 152.5, 150.6, 149.8, 136.6, 132.2, 130.3, 129.2, 118.7, 114.1. HRMS-APCI (pos) m/z 278.0334 [M + H]+ (Calcd. for C12H8ClN3O3+, 278.0332).
(1E,2E)-1-(4-Bromobenzylidene)-2-[(5-nitrofuran-2-yl)methylene]hydrazine 4a. Yellow powder. Yield: 754 mg (78%); mp 196–198 °C; Rf 0.55 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3092 (C–H), 1629 (CN), 1563 (CC), 1475 (N–O), 1243 (C–O), 513 (C–Br); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.79 (s, 1H, H-1′), 8.65 (s, 1H, H-6), 7.84 (d, J = 8.5 Hz, 2H, H-3′), 7.82 (d, J = 3.9 Hz, 1H, H-4), 7.75 (d, J = 8.5 Hz, 2H, H-4′), 7.42 (d, J = 3.9 Hz, 1H, H-3); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 162.7, 152.6, 150.6, 149.8, 132.5, 132.1, 130.5, 125.6, 118.7, 114.1. HRMS-APCI (pos) m/z 321.9824 [M + H]+ (Calcd. for C12H8BrN3O3+, 321.9827).
(1E,2E)-1-(4-Methylbenzylidene)-2-[(5-nitrofuran-2-yl)methylene]hydrazine 5a. Yellow powder. Yield: 100 mg (13%); mp 167–169 °C; Rf 0.68 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3118 (C–H), 1626 (CN), 1571 (CC), 1336 (N–O), 1241 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.76 (s, 1H, H-1′), 8.64 (s, 1H, H-6), 7.82 (d, J = 3.9 Hz, 1H, H-4), 7.79 (d, J = 8.1 Hz, 2H, H-3′), 7.39 (d, J = 3.9 Hz, 1H, H-3), 7.34 (d, J = 8.1 Hz, 2H, H-4′), 2.38 (s, 3H, H-6′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.9, 152.5, 150.8, 149.3, 142.3, 130.6, 129.6, 128.8, 118.3, 114.1, 21.2. HRMS-APCI (pos) m/z 258.0880 [M + H]+ (Calcd. for C13H11N3O3+, 258.0879).
(1E,2E)-1-(4-Methoxybenzylidene)-2-[(5-nitrofuran-2-yl)methylene]hydrazine 6a. Orange powder. Yield: 221 mg (27%); mp 169–171 °C; Rf 0.35 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 2921 (C–H), 1706 (CN), 1600 (CC), 1347 (N–O), 1239 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.75 (s, 1H, H-1′), 8.62 (s, 1H, H-6), 7.86 (d, J = 8.8 Hz, 2H, H-3′), 7.82 (d, J = 3.9 Hz, 1H, H-4), 7.38 (d, J = 3.9 Hz, 1H, H-3), 7.09 (d, J = 8.8 Hz, 2H, H-4′), 3.84 (s, 3H, H-6′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.8, 162.4, 151.0, 148.9, 130.7, 125.9, 118.0, 114.6, 114.6, 114.2, 55.5. HRMS-APCI (pos) m/z 274.0840 [M + H]+ (Calcd. for C13H11N3O4+, 274.0828).
(1E,2E)-1-[4-(Benzyloxy)benzylidene]-2-[(5-nitrofuran-2-yl)methylene]hydrazine 7a. Yellow powder. Yield: 786 mg (75%); mp 175–177 °C; Rf 0.59 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3102 (C–H), 1618 (CN), 1573 (CC), 1352 (N–O), 1246 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.74 (s, 1H, H-1′), 8.61 (s, 1H, H-6), 7.86 (d, J = 8.8 Hz, 2H, H-3′), 7.80 (d, J = 3.9 Hz, 1H, H-4), 7.47 (d, J = 7.5 Hz, 2H, H-8′), 7.41 (t, J = 7.5 Hz, 2H, H-9′), 7.37 (d, J = 3.9 Hz, 1H, H-3), 7.35 (t, J = 7.5 Hz, 1H, H-10′), 7.16 (d, J = 8.8 Hz, 2H, H-4′), 5.21 (s, 2H, H-6′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.5, 161.4, 152.3, 150.9, 148.7, 136.5, 130.6, 128.4, 127.9, 127.7, 126.0, 117.9, 115.3, 114.1, 69.5. HRMS-APCI (pos) m/z 350.1151 [M + H]+ (Calcd. for C19H15N3O4+, 350.1141).
4-{(E)-{(E)-[(5-Nitrofuran-2-yl)methylene]hydrazono}methyl}phenol 8a. Red powder. Yield: 109 mg (14%); mp 216–218 °C; Rf 0.39 (Hex
:EtOAc 1:1, v/v). IR νmax (cm−1): 3426 (O–H), 3112 (C–H), 1598 (CN), 1565 (CC), 1347 (N–O), 1211 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 10.27 (s, 1H, H-6′), 8.69 (s, 1H, H-1′), 8.59 (s, 1H, H-6), 7.81 (d, J = 3.9 Hz, 1H, H-4), 7.75 (d, J = 8.6 Hz, 2H, H-3′), 7.35 (d, J = 3.9 Hz, 1H, H-3), 6.89 (d, J = 8.6 Hz, 2H, H-4′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 164.2, 161.3, 152.4, 151.1, 148.4, 131.0, 124.3, 117.8, 115.9, 114.2. HRMS-APCI (pos) m/z 260.0686 [M + H]+ (Calcd. for C12H9N3O4+, 260.0671).
(1E,2E)-1-(4-Nitrobenzylidene)-2-[(5-nitrofuran-2-yl)methylene]hydrazine 9a. Light brown powder. Yield: 536 mg (62%); mp 170–172 °C; Rf 0.42 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3111 (C–H), 1633 (CN), 1572 (CC), 1526 (N–O), 1342 (N–O), 1251 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.92 (s, 1H, H-1′), 8.69 (s, 1H, H-6), 8.37 (d, J = 8.7 Hz, 2H, H-4′), 8.15 (d, J = 8.7 Hz, 2H, H-3′), 7.83 (*d, J = 3.9 Hz, 1H, H-4), 7.46 (*d, J = 3.9 Hz, 1H, H-3); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 161.4, 150.5, 150.3, 149.1, 139.2, 137.5, 129.7, 124.1, 119.3, 114.0. HRMS-APCI (pos) m/z 289.0577 [M + H]+ (Calcd. for C12H8N4O5+, 289.0573). *d is a coalesced doublet.
(1E,2E)-1-Benzylidene-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 1b. Yellow powder. Yield: 436 mg (56%); mp 166–168 °C; Rf 0.63 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3096 (C–H), 1610 (CN), 1531 (CC), 1495 (N–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.94 (s, 1H, H-1′), 8.75 (s, 1H, H-6), 8.19 (d, J = 4.3 Hz, 1H, H-4), 7.90 (d, J = 7.5 Hz, 2H, H-3′), 7.72 (d, J = 4.3 Hz, 1H, H-3), 7.56 (d, J = 7.5 Hz, 1H, H-5′), 7.52 (d, J = 7.5 Hz, 2H, H-4′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.6, 155.3, 152.3, 145.0, 133.3, 132.4, 132.0, 130.4, 129.0, 128.8. HRMS-APCI (pos) m/z 260.0512 [M + H]+ (Calcd. for C12H9N3O2S+, 260.0494).
(1E,2E)-1-(4-Fluorobenzylidene)-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 2b. Yellow powder. Yield: 458 mg (55%); mp 187–189 °C; Rf 0.53 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3082 (C–H), 1613 (CN), 1505 (CC), 1335 (N–O), 1221 (C–F); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.93 (s, 1H, H-1′), 8.76 (s, 1H, H-6), 8.19 (d, J = 4.3 Hz, 1H, H-4), 7.97 (dd, JH–F = 8.8, 5.7 Hz, 2H, H-3′), 7.72 (d, J = 4.3 Hz, 1H, H-3), 7.37 (t, JH–F = 8.8 Hz, 2H, H-4′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 164.2 (d, 1JC–F = 250.4 Hz), 162.5, 157.3, 155.4, 152.3, 144.9, 132.4, 131.2 (d, 2JC–F = 9.0 Hz), 130.4, 116.2 (d, 3JC–F = 22.0 Hz). HRMS-APCI (pos) m/z 278.0404 [M + H]+ (Calcd. for C12H8FN3O2S+, 278.0400).
(1E,2E)-1-(4-Chlorobenzylidene)-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 3b. Yellow powder. Yield: 238 mg (27%); mp 179–181 °C; Rf 0.62 (Hex
:EtOAc 1:1, v/v). IR νmax (cm−1): 3094 (C–H), 1604 (CN), 1522 (CC), 1321 (N–O), 815 (C–Cl); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.77 (s, 1H, H-1′), 8.57 (s, 1H, H-6), 8.17 (d, J = 4.4 Hz, 1H, H-4), 8.02 (d, J = 8.5 Hz, 2H, H-3′), 7.74 (d, J = 4.4 Hz, 1H, H-3), 7.70 (d, J = 8.5 Hz, 2H, H-4′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 161.8, 156.0, 150.9, 137.1, 136.4, 133.7, 132.0, 130.9, 129.5, 129.1. HRMS-APCI (pos) m/z 294.0097 [M + H]+ (Calcd. for C12H8ClN3O2S+, 294.0104).
(1E,2E)-1-(4-Bromobenzylidene)-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 4b. Yellow powder. Yield: 416 mg (41%); mp 193–195 °C; Rf 0.21 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3083 (C–H), 1610 (CN), 1550 (CC), 1332 (N–O), 536 (C–Br); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.77 (s, 1H, H-1′), 8.59 (s, 1H, H-6), 8.18 (d, J = 4.4 Hz, 1H, H-4), 7.95 (d, J = 8.4 Hz, 2H, H-3′), 7.85 (d, J = 8.4 Hz, 2H, H-4′), 7.75 (d, J = 4.4 Hz, 1H, H-3); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 161.9, 156.0, 150.8, 136.3, 133.7, 132.4, 132.3, 131.0, 129.1, 126.2. HRMS-APCI (pos) m/z 337.9592 [M + H]+ (Calcd. for C12H8BrN3O2S+, 337.9599).
(1E,2E)-1-(4-Methylbenzylidene)-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 5b. Yellow powder. Yield: 197 mg (24%); mp 166–168 °C; Rf 0.66 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 2920 (C–H), 1609 (CN), 1532 (CC), 1327 (N–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.92 (s, 1H, H-1′), 8.71 (s, 1H, H-6), 8.18 (d, J = 4.1 Hz, 1H, H-4), 7.80 (d, J = 8.0 Hz, 2H, H-3′), 7.70 (d, J = 4.1 Hz, 1H, H-3), 7.34 (d, J = 8.0 Hz, 2H, H-4′), 2.38 (s, 3H, H-6′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.7, 155.0, 152.2, 145.1, 142.3, 132.2, 130.6, 130.4, 129.6, 128.8, 21.2. HRMS-APCI (pos) m/z 274.0644 [M + H]+ (Calcd. for C13H11N3O2S+, 274.0650).
(1E,2E)-1-(4-Methoxybenzylidene)-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 6b. Orange powder. Yield: 399 mg (46%); mp 171–173 °C; Rf 0.44 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 2924 (C–H), 1608 (CN), 1529 (CC), 1329 (N–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.90 (s, 1H, H-1′), 8.69 (s, 1H, H-6), 8.17 (d, J = 4.3 Hz, 1H, H-4), 7.86 (d, J = 8.7 Hz, 2H, H-3′), 7.68 (d, J = 4.3 Hz, 1H, H-3), 7.08 (d, J = 8.7 Hz, 2H, H-4′), 3.84 (s, 3H, H-6′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.5, 162.4, 154.5, 152.0, 145.4, 132.0, 130.7, 130.4, 125.9, 114.6, 55.5. HRMS-APCI (pos) m/z 290.0595 [M + H]+ (Calcd. for C13H11N3O3S+, 290.0599).
(1E,2E)-1-[4-(Benzyloxy)benzylidene]-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 7b. Yellow powder. Yield: 143 mg (13%); mp 136–138 °C; Rf 0.82 (Hex
:EtOAc 1:1, v/v). IR νmax (cm−1): 2921 (C–H), 1599 (CN), 1547 (CC), 1328 (N–O), 1158 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.72 (s, 1H, H-1′), 8.52 (s, 1H, H-6), 8.16 (d, J = 4.4 Hz, 1H, H-4), 7.98 (d, J = 8.8 Hz, 2H, H-3′), 7.71 (d, J = 4.4 Hz, 1H, H-3), 7.49 (d, J = 7.5 Hz, 2H, H-8′), 7.42 (t, J = 7.5 Hz, 2H, H-9′), 7.36 (t, J = 7.5 Hz, 1H, H-10′), 7.26 (d, J = 8.8 Hz, 2H, H-4′), 5.24 (s, 2H, H-6′); 13C NMR (DSMO-d6, 151 MHz) δ (ppm): 162.9, 161.9, 155.5, 150.1, 136.6, 136.5, 133.0, 131.4, 129.1, 128.5, 128.0, 127.9, 125.8, 115.7, 69.6. HRMS-APCI (pos) m/z 366.0916 [M + H]+ (Calcd. for C19H15N3O3S+, 366.0912).
4-(E)-(E)-[(5-Nitrothiophen-2-yl)methylene]hydrazonomethylphenol 8b. Orange powder. Yield: 347 mg (42%); mp 243–245 °C; Rf 0.77 (hexane
:EtOAc 1:1, v/v). IR νmax (cm−1): 3453 (O–H), 3089 (C–H), 1602 (CN), 1492 (CC), 1332 (N–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 10.22 (s, 1H, H-6′), 8.88 (s, 1H, H-1′), 8.63 (s, 1H, H-6), 8.17 (d, J = 4.4 Hz, 1H, H-4), 7.75 (d, J = 8.6 Hz, 2H, H-3′), 7.67 (d, J = 4.4 Hz, 1H, H-3), 6.88 (d, J = 8.6 Hz, 2H, H-4′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 163.8, 161.3, 154.0, 151.9, 145.6, 131.8, 131.0, 130.4, 124.3, 116.0. HRMS-APCI (pos) m/z 276.0434 [M + H]+ (Calcd. for C12H9N3O3S+, 276.0443).
(1E,2E)-1-(4-Nitrobenzylidene)-2-[(5-nitrothiophen-2-yl)methylene]hydrazine 9b. Yellow powder. Yield: 303 mg (66%); mp 239–241 °C; Rf 0.53 (Hex
:EtOAc 7:3, v/v). IR νmax (cm−1): 3108 (C–H), 1624 (CN), 1595 (CC), 1505 (N–O), 1336 (N–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.98 (s, 1H, H-1′), 8.87 (s, 1H, H-6), 8.35 (d, J = 8.7 Hz, 2H, H-4′), 8.19 (d, J = 4.3 Hz, 1H, H-4), 8.14 (d, J = 8.7 Hz, 2H, H-3′), 7.76 (d, J = 4.3 Hz, 1H, H-3); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 161.5, 156.5, 152.7, 149.1, 144.4, 139.2, 133.1, 130.3, 129.7, 124.1. HRMS-APCI (pos) m/z 305.0335 [M + H]+ (Calcd. for C12H8N4O4S+, 305.0345).
:EtOAc (3:2, v/v). Upon completion, the reaction was quenched with water (50 mL), extracted with DCM (3 × 50 mL). The organic phase was dried over MgSO4 and recrystallised from EtOAc to afford the desired azines.
(E)-1-[(E)-4-(Benzyloxy)benzylidene]-2-(furan-2-ylmethylene)hydrazine 11. Brown powder. Yield: 29%; mp 95–97 °C; Rf 0.72 (Hex
:EtOAc 3:2, v/v). IR νmax (cm−1): 2924 (C–H), 1606 (CN), 1509 (CC), 1239 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.64 (s, 1H, H-1′), 8.51 (s, 1H, H-6), 7.93 (s, 1H, H-5), 7.82 (d, J = 7.82 Hz, 2H, H-3′), 7.48 (d, J = 7.48 Hz, 2H, H-8′), 7.44 (t, J = 7.45 Hz, 1H, H-10′), 7.40 (d, J = 7.41 Hz, 2H, H-9′), 7.23 (d, J = 7.23 Hz, 1H, H-3), 7.15 (d, J = 7.14 Hz, 2H, H-4′), 6.97 (d, J = 6.96 Hz, 1H, H-4), 5.20 (s, 2H, H-6′); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 161.7, 161.4, 157.7, 150.2, 149.8, 146.8, 128.9, 128.4, 128.2, 128.0, 115.7, 114.9, 113.0, 69.9.
(E)-1-(Furan-2-ylmethylene)-2-[(E)-4-nitrobenzylidene]hydrazine 12. Yellow powder. Yield: 61%; mp 165–167 °C; Rf 0.66 (Hex
:EtOAc 3:2, v/v). IR νmax (cm−1): 3122 (C–H), 1633 (CN), 1593 (CC), 1514 (N–O), 1278 (C–O); 1H NMR (DMSO-d6, 600 MHz) δ (ppm): 8.82 (s, 1H, H-1′), 8.59 (s, 1H, H-6), 8.34 (d, J = 8.34 Hz, 2H, H-4′), 8.12 (d, J = 8.12 Hz, 2H, H-3′), 7.99 (s, 1H, H-5), 7.21 (s, 1H, H-3), 6.75 (s, 1H, H-4); 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 160.1, 152.0, 149.4, 147.6, 140.2, 129.8, 124.5, 118.8, 113.3.
The characterisation spectra of all derivatives are presented as ESI.†
Biological evaluation
For the resazurin assay, 96 well plates (Nunc, Thermofisher Scientific) were seeded with 1.25 × 106 cells per mL (50 μL per well) logarithmic phase promastigotes with 50 μL of (i) 10 μM of compound for the screening of activity, or (ii) seven two-fold dilution concentrations of compounds for the determination of IC50 values. AmB (10 μM) served as the clinically available antileishmanial (standard) drug, and the blank consisted of growth medium without parasites. After incubation at 26 °C in humidified atmosphere for 48 hours, resazurin solution (50 μL of 0.01% in phosphate-buffered saline (PBS)) was added to and the plates were further incubated in the dark for 24 hours. A Thermofisher Scientific GO Multiscan plate reader was used to measure absorbance at both 570 nm and 600 nm. SkanIt 4.0 Research Edition software was used to perform data analysis for each biological replicate. Background absorbance of resazurin (600 nm) was subtracted from the absorbance values of resorufin (570 nm).
For the single-point activity screening, the following equation was used to determine growth inhibition percentage:
| Growth inhibition% = 100 − [(ΔAbs sample − ΔAbs blank)/(ΔAbs neg control − ΔAbs blank) × 100] |
| Cell viability% = (ΔAbs sample − ΔAbs blank)/(ΔAbs neg control − ΔAbs blank) × 100 |
A cell suspension of 500000 cells per mL was prepared and 25 ng mL−1 phorbol 12-myristate 13-acetate (PMA) added before the addition of 200 μL per well to 96 well plates. The medium blank wells received only growth medium. The cells were incubated for 48 hours in a humidified atmosphere at 37 °C and 5% CO2 to differentiate the suspension cell line to adherent macrophages. The plates were subsequently carefully washed with warmed PBS to remove non-differentiated cells. Stationary phase promastigotes were then added (200 μL per well) at a multiplicity of infection (MOI) of 30:1 in RPMI 1640 growth medium with 10% FBS. Parasite blank wells consisted of THP1 cells that did not receive parasites. The plates were incubated for 24 hours at 32 °C, for IR173 strain, or 37 °C, for strain 9515, to promote infection. They were then carefully washed four times with warmed PBS to remove any remaining extracellular parasites with the wells then received 200 μL of growth medium with 10% FBS and compound: 10 μM for single-point activity screenings, seven three-fold dilution concentrations of 10 μM for IC50 determination, and 10 μM AmB for the standard drug control.
After 72 hours of incubation, the wells were again washed three times with warmed PBS to remove the FBS and any remaining extracellular promastigotes. The host macrophages were then lysed with 20 μL of 0.05% sodium dodecyl sulphate (SDS; Sigma Aldrich) in growth medium without FBS for 30 seconds, followed by the addition of 180 μL promastigote growth medium containing 10% FBS. For the resazurin assay, 10 μL of resazurin solution (0.25% in PBS) was added to each well and the plates were further incubated at 26 °C for 72 hours in the dark to accommodate parasite recovery to promastigote forms. Absorbance measurements and data analysis were performed exactly as described for the antipromastigote assay. With regards to the activity screening, compounds with a growth inhibition of >60% qualified for further IC50 determination.55
T. cruzi strain CL (BEI Resources, USA) was cultured in LIT medium, consisting of 5 mg mL−1 Bacto™ tryptose (BD Biosciences), 9 mg mL−1 Difco™ liver infusion broth (BD Biosciences), 8 mg mL−1 Na2HPO4, 1 mg mL−1 NaCl, 0.4 mg mL−1 KCl, 1 mg mL−1 glucose, 10% FBS and 0.01 mg mL−1 hemin (Sigma Aldrich), pH 7.2. The epimastigotes were maintained at 25 °C. For the resazurin assay, epimastigotes (5 × 105 cells per well, final volume 100 μL per well) were seeded in 96 well plates (Nunc, Thermofisher Scientific) in the presence of compounds and blanks as described for the antipromastigote assay. The plates were then incubated at 25 °C in humidified atmosphere for 48 hours, followed by the addition of 50 μL of resazurin solution and 24-hour incubation. Absorbance and data analysis were performed for each biological replicate as indicated for the Leishmania antipromastigote assay.
With regards to the activity screening, there are currently no reports of cut-off growth inhibition values for anti-epimastigote activity. As the epimastigotes are cultured and assayed similarly to Leishmania promastigotes, the growth inhibition cut-off of >70% was also applied for qualifying compounds for IC50 determination.
Cultured epimastigotes were centrifuged and resuspended in minimal essential medium (MEM) supplemented with 3% FBS for addition to a monolayer of cultured African green monkey kidney (Vero) cells (Cellonex, South Africa), which were maintained as indicated in the following cytotoxicity section. By changing the Vero culture media to MEM, the growth rate of the Vero cells is reduced, which in turn prevents reduced infection due to host cell proliferation. The co-culture was then maintained for 5–15 days until sufficient trypomastigotes formed.
The growth medium of the flask was collected and centrifuged at 3000 × g for 10 min, the supernatant discarded and replaced with fresh growth medium with 10% FBS. The parasites were counted using a hemocytometer and the cell density adjusted to 1 × 107 cells per mL and >80% trypomastigote forms. The trypomastigotes were then seeded in 96 well plates (200 μL per well) in the presence of compounds and blanks as described for the antipromastigote assay. Benznidazole (10 μM) was used as standard drug. The plates were incubated for 48 hours in humidified atmosphere at 37 °C and 5% CO2, followed by the addition of 50 μL of resazurin solution and 24-hour incubation. Absorbance and data analysis were performed for each biological replicate as indicated for the Leishmania antipromastigote assay.
With regards to the activity screening, there are currently no reports of cut-off growth inhibition values for T. cruzi anti-trypomastigote activity. Due to the axenic/host-free nature of the trypomastigote cultures, a cut-off of >70% was selected.
Cytotoxicity assays
Vero cells were maintained in Hyclone Dulbecco's modified Eagle's medium, with high glucose (separations), 10% FBS and 1% L-glutamine, Pen/Strep, and non-essential amino acids, in a humidified atmosphere at 37 °C and 5% CO2. For the resazurin assay, 96 well plates with 100 μL of cell suspension (60000 cells per mL) were prepared and incubated for 24 hours. The cells were then treated with seven two-fold dilutions of 100 μM compound in growth medium. Emetine dihydrochloride (1 μM) served as positive control and the blanks contained growth medium without cells. The treated plates were then incubated for 48 hours and the resazurin assay initiated via the addition of 50 μL of resazurin solution and further incubation for 2 hours. Absorbance and data analysis were performed for each biological replicate as described for the Leishmania antipromastigote assay.
Statistical analysis
In vitro antileishmanial and antitrypanosomal activities and cytotoxicity (indicated as IC50 values), were derived from non-linear regression analysis. Results were represented as the mean ± the standard deviation (SD) from the triplicate biological experiments. Statistical analysis was performed using SkanIt 4.0 Research Edition software (Thermofisher Scientific) and Prism V5 software (GraphPad). All reported data is significant at p < 0.05.Disclaimer
Any opinions, findings, conclusions, and/or recommendations expressed in this material are those of the authors and therefore the NRF does not accept any liability in this regard.Ethics
Ethics approval for this study was obtained from the Human Research Ethics Committee of the North-West University (NWU-00385-20-A1).Abbreviations
| NTDs | Neglected tropical diseases |
| CL | Cutaneous leishmaniasis |
| MCL | Mucocutaneous leishmaniasis |
| VL | Visceral leishmaniasis |
| cNFs | Clinical nitrofurans |
| ROS | Reactive oxygen species |
| NFA | 5-Nitro-2-furaldehyde |
| NTA | 5-Nitrothiophene-2-carboxaldehyde |
| NFX | Nifuroxazide |
| NTX | Nifurtimox |
| FZD | Furazolidone |
| NFZ | Nitrofurazone |
| NFT | Nitrofurantoin |
| AmB | Amphotericin B |
| Em | Emetine |
| WHO | World Health Organization |
Author contributions
Conceptualisation: [David D. N'Da]; methodology: [Maryna Saayman, Christina Kannigadu, Janine Aucamp, Cassiem Joseph, Andrew J. Swarts, David D. N'Da]; formal analysis and investigation: [Maryna Saayman, Janine Aucamp, Helena D. Janse van Rensburg, Christina Kannigadu, David D. N'Da]; writing – original draft preparation: [Maryna Saayman, Christina Kannigadu, Janine Aucamp]; writing – review and editing: [Christina Kannigadu, Janine Aucamp, Cassiem Joseph, Andrew J. Swarts, David D. N'Da]; funding acquisition: [David D. N'Da]; resources: [David D. N'Da]; supervision: [David D. N'Da].Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was financially supported by the South African National Research Foundation (NRF) (UID 129324 and 148781) and the North-West University. The following reagents were obtained through BEI Resources, NIAID, NIH: Leishmania donovani, Strain 9515 (MHOM/IN/95/9515), NR-48822, Leishmania major, Strain IR173 (MHOM/IR/-173), NR-48816, Trypanosoma cruzi, Strain CL, NR-49381.References
- WHO, Neglected Tropical Diseases: Overview, 2023, https://www.who.int/health-topics/neglected-tropical-diseases#tab=tab_1, [Date of access: March 2023].
- WHO, Neglected tropical diseases: Impact, 2023, https://www.who.int/health-topics/neglected-tropical-diseases#tab=tab_2, [Date of access: March 2023].
- M. P. Barrett and S. L. Croft, Br. Med. Bull., 2012, 104, 175–196 CrossRef CAS PubMed.
- WHO, Leishmaniasis: Overview, 2023, https://www.who.int/health-topics/leishmaniasis#tab=tab_1, [Date of access: March 2023].
- N. Salam, W. M. Al-Shaqha and A. Azzi, PLoS Neglected Trop. Dis., 2014, 8, e3208 CrossRef PubMed.
- E. Torres-Guerrero, M. R. Quintanilla-Cedillo, J. Ruiz-Esmenjaud and R. Arenas, F1000Research, 2017, 6, 750 Search PubMed.
- WHO, Leishmaniasis: Key facts, 2023, https://www.who.int/news-room/fact-sheets/detail/leishmaniasis, [Date of access: March 2023].
- WHO, Number of cases of cutaneous leishmaniasis reported, 2023, https://www.who.int/data/gho/data/indicators/indicator-details/GHO/number-of-cases-of-cutaneous-leishmaniasis-reported, [Date of access: March 2023].
- WHO, Number of cases of cutaneous leishmaniasis reported, 2023, https://www.who.int/data/gho/data/indicators/indicator-details/GHO/number-of-cases-of-cutaneous-leishmaniasis-reported, [Date of access: March 2023].
- WHO, Report of WHO Expert Committee on the Control of Leishmaniases, Geneva: World Health Organization, 2010, https://apps.who.int/iris/handle/10665/44412, [Date of access: March 2023].
- WHO, Chagas disease: Key facts, 2022, https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis), [Date of access: March 2023].
- WHO, Chagas disease (American trypanosomiasis): Overview, 2023, https://www.who.int/health-topics/chagas-disease#tab=tab_1, [Date of access: March 2023].
- E. Chatelain, Comput. Struct. Biotechnol. J., 2017, 15, 98–103 CrossRef PubMed.
- D. A. Álvarez-Hernández, G. A. Franyuti-Kelly, R. Díaz-López-Silva, A. M. González-Chávez, D. González-Hermosillo-Cornejo and R. Vázquez-López, Rev. Med. Hosp. Gen., 2018, 81, 154–164 Search PubMed.
- PAHO, Chagas disease, 2023, https://www.paho.org/en/topics/chagas-disease, [Date of access: March 2023].
- S. P. Georgiadou, K. P. Makaritsis and G. N. Dalekos, J. Transl. Int. Med., 2015, 3, 43–50 CrossRef.
- M. Ghorbani and R. Farhoudi, Drug Des., Dev. Ther., 2018, 12, 25–40 CrossRef CAS.
- S. S. Braga, Eur. J. Med. Chem., 2019, 183, 111660 CrossRef CAS.
- L. H. Freitas-Junior, E. Chatelain, H. A. Kim and J. L. Siqueira-Neto, Int. J. Parasitol.: Drugs Drug Resist., 2012, 2, 11–19 CAS.
- H. C. Maltezou, J. Biomed. Biotechnol., 2010, 2010, 617521 Search PubMed.
- S. J. Martinez, P. S. Romano and D. M. Engman, Front. Cell. Infect. Microbiol., 2020, 10, 1–11 CrossRef PubMed.
- WHO, Chagas disease (American trypanosomiasis): Treatment, 2023, https://www.who.int/health-topics/chagas-disease#tab=tab_3, [Date of access: March 2023].
- R. Patel, L. Rinker, J. Peng and W. M. Chilian, in Reactive Oxygen Species (ROS) in Living Cells, ed. C. Filip and E. Albu, IntechOpen, London, 2018, pp. 7–20 Search PubMed.
- N. H. Zuma, J. Aucamp and D. D. N'Da, Eur. J. Pharm. Sci., 2019, 140, 105092 CrossRef CAS PubMed.
- C. Pal and U. Bandyopadhyay, Antioxid. Redox Signaling, 2012, 17, 555–582 CrossRef CAS PubMed.
- R. Sreenivasulu, K. T. Reddy, P. Sujitha, C. G. Kumar and R. R. Raju, Bioorg. Med. Chem., 2019, 27, 1043–1055 CrossRef CAS PubMed.
- V. Manikandan, S. Balaji, R. Senbagam, R. Vijayakumar, M. Rajarajan, G. Vanangamudi, R. Arulkumaran, R. Sundararajan and G. Thirunarayanan, Int. J. Adv. Chem., 2017, 5, 17–24 CrossRef.
- B. Glinma, F. A. Gbaguidi, U. C. Kasséhin, S. D. S. Kpoviessi, A. Houngbèmè, H. D. Houngue, G. C. Accrombessi and J. H. Poupaert, J. Appl. Pharm. Sci., 2015, 5, 001–007 CAS.
- V. T. Angelova, V. Valcheva, N. G. Vassilev, R. Buyukliev, G. Momekov, I. Dimitrov, L. Saso, M. Djukic and B. Shivachev, Bioorg. Med. Chem. Lett., 2017, 27, 223–227 CrossRef CAS PubMed.
- L. Dehestani, N. Ahangar, S. M. Hashemi, H. Irannejad, P. Honarchian Masihi, A. Shakiba and S. Emami, Bioorg. Chem., 2018, 78, 119–129 CrossRef CAS PubMed.
- A. Kajal, S. Bala, N. Sharma, S. Kamboj and V. Saini, Int. J. Med. Chem., 2014, 2014, 761030 Search PubMed.
- L. Navidpour, H. Shafaroodi, G. Saeedi-Motahar and A. Shafiee, Med. Chem. Res., 2014, 23, 2793–2802 CrossRef CAS.
- S. S. Chourasiya, D. Kathuria, A. A. Wani and P. V. Bharatam, Org. Biomol. Chem., 2019, 17, 8486–8521 RSC.
- B. Singh and A. Pandey, Liq. Cryst., 2009, 37, 57–67 CrossRef.
- J. Gómez, A. H. Klahn, M. Fuentealba, D. Sierra, C. Olea-Azar, J. D. Maya and M. E. Medina, J. Organomet. Chem., 2017, 839, 108–115 CrossRef.
- S. E. Ward and P. Beswick, Expert Opin. Drug Discovery, 2014, 9, 995–1003 CrossRef CAS PubMed.
- C. J. Abelt and J. M. Pleier, J. Am. Chem. Soc., 1989, 111, 1795–1799 CrossRef CAS.
- I. Alkorta, F. Blanco and J. Elguero, ARKIVOC, 2007, 2008, 48–56 Search PubMed.
- N. P. Neureiter, J. Am. Chem. Soc., 1959, 81, 2910 CrossRef CAS.
- R. Karmakar, C. R. Choudhury, S. R. Batten and S. Mitra, J. Mol. Struct., 2007, 826, 75–81 CrossRef CAS.
- J. Gómez, A. Hugo Klahn, M. Fuentealba, D. Sierra, C. Olea-Azar and M. E. Medina, Inorg. Chem. Commun., 2015, 61, 204–206 CrossRef.
- C. W. Lindsley, in Encyclopedia of Psychopharmacology, ed. I. Stolerman and L. Price, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 1–6, DOI:10.1007/978-3-642-27772-6_7015-1.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Phys. Chem. A, 1998, 102, 3762–3772 CrossRef CAS.
- C. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863–875 CrossRef CAS PubMed.
- G. Caron and G. Ermondi, Future Med. Chem., 2016, 8, 2013–2016 CrossRef CAS.
- T. Reinhardt, K. M. Lee, L. Niederegger, C. R. Hess and S. A. Sieber, ACS Chem. Biol., 2022, 17, 3077–3085 CrossRef CAS PubMed.
- J. H. Tocher, Gen. Pharmacol., 1997, 28, 485–487 CrossRef CAS PubMed.
- S. J. Teague, A. M. Davis, P. D. Leeson and T. Oprea, Angew. Chem., Int. Ed., 1999, 38, 3743–3748 CrossRef CAS PubMed.
- B. S. McGwire and A. R. Satoskar, QJM, 2014, 107, 7–14 CrossRef CAS.
- CDC, American Trypanosomiasis, 2021, https://www.cdc.gov/dpdx/trypanosomiasisamerican/index.html, [Date of access: March 2023].
- K. Katsuno, J. N. Burrows, K. Duncan, R. Hooft van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS PubMed.
- F. Villalta and G. Rachakonda, Expert Opin. Drug Discovery, 2019, 14, 1161–1174 CrossRef CAS.
- J. L. Siqueira-Neto, O.-R. Song, H. Oh, J.-H. Sohn, G. Yang, J. Nam, J. Jang, J. Cechetto, C. B. Lee, S. Moon, A. Genovesio, E. Chatelain, T. Christophe and L. H. Freitas-Junior, PLoS Neglected Trop. Dis., 2010, 4, e675 CrossRef.
- G. De Muylder, K. K. H. Ang, S. Chen, M. R. Arkin, J. C. Engel and J. H. McKerrow, PLoS Neglected Trop. Dis., 2011, 5, e1253 CrossRef PubMed.
- S. K. Jain, R. Sahu, L. A. Walker and B. L. Tekwani, J. Visualized Exp., 2012, 70, 4054 Search PubMed.
- N. S. Finiuk, V. P. Hreniuh, Y. V. Ostapiuk, V. S. Matiychuk, D. A. Frolov, M. D. Obushak, R. S. Stoika and A. M. Babsky, Biopolym. Cell, 2017, 33, 135–146 Search PubMed.
- S. Liu, M. Su, S. J. Song and J. H. Jung, Mar. Drugs, 2017, 15, 329 CrossRef PubMed.
- J. Fu, H. Chen, D. N. Soroka, R. F. Warin and S. Sang, J. Agric. Food Chem., 2014, 62, 4632–4642 CrossRef CAS.
- E. A. Adewusi, P. Steenkamp, G. Fouche and V. Steenkamp, Nat. Prod. Commun., 2013, 8, 1213–1216 CrossRef CAS PubMed.
- R. Lira, S. Sundar, A. Makharia, R. Kenney, A. Gam, E. Saraiva and D. Sacks, J. Infect. Dis., 1999, 180, 564–567 CrossRef CAS PubMed.
- J. E. Potvin, P. Leprohon, M. Queffeulou, S. Sundar and M. Ouellette, Clin. Infect. Dis., 2021, 72, e526–e532 CrossRef CAS PubMed.
- C. A. Bauer, G. Schneider and A. H. Göller, J. Cheminform., 2019, 11, 59 CrossRef PubMed.
- G. Papadatos and N. Brown, WIREs Comput. Mol. Sci., 2013, 3, 339–354 CrossRef CAS.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- A. Firth and P. Prathapan, Curr. Res. Pharmacol. Drug Discov., 2021, 2, 100011 CrossRef PubMed.
- J. Aucamp and D. D. N'Da, Exp. Parasitol., 2022, 236–237, 108249 CrossRef CAS PubMed.
- D. K. Mangwegape, N. H. Zuma, J. Aucamp and D. D. N'Da, Arch. Pharm., 2021, 354, e2000280 CrossRef PubMed.
- J. W. de Freitas Oliveira, T. M. Torres, C. J. G. Moreno, B. Amorim-Carmo, I. Z. Damasceno, A. K. M. C. Soares, J. da Silva Barbosa, H. A. O. Rocha and M. S. Silva, Sci. Rep., 2021, 11, 11200 CrossRef CAS PubMed.
- C. A. N. Njanpa, S. C. N. Wouamba, L. R. T. Yamthe, D. Dize, B. M. T. Tchatat, P. V. F. Tsouh, M. N. Pouofo, J. B. Jouda, B. L. Ndjakou, N. Sewald, S. F. Kouam and F. F. Boyom, BMC Complementary Med. Ther., 2021, 21, 106 CrossRef CAS PubMed.
- E. M. Czekanska, Methods Mol. Biol., 2011, 740, 27–32 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00220a |
| This journal is © The Royal Society of Chemistry 2023 |