
Chemical genetic approaches for the discovery of bacterial cell wall inhibitors
Rinki
Gupta
 ,
Mangal
Singh†
and
Ranjana
Pathania
*
,
Mangal
Singh†
and
Ranjana
Pathania
*
Department of Biosciences and Bioengineering, Indian Institute of Technology Roorkee, Roorkee – 247 667, Uttarakhand, India. E-mail: ranjana.pathania@bt.iitr.ac.in
First published on 30th August 2023
Abstract
Antimicrobial resistance (AMR) in bacterial pathogens is a worldwide health issue. The innovation gap in discovering new antibiotics has remained a significant hurdle in combating the AMR problem. Currently, antibiotics target various vital components of the bacterial cell envelope, nucleic acid and protein biosynthesis machinery and metabolic pathways essential for bacterial survival. The critical role of the bacterial cell envelope in cell morphogenesis and integrity makes it an attractive drug target. While a significant number of in-clinic antibiotics target peptidoglycan biosynthesis, several components of the bacterial cell envelope have been overlooked. This review focuses on various antibacterial targets in the bacterial cell wall and the strategies employed to find their novel inhibitors. This review will further elaborate on combining forward and reverse chemical genetic approaches to discover antibacterials that target the bacterial cell envelope.
 Rinki Gupta | Rinki Gupta received her Master of Engineering degree in Biotechnology from Birla Institute of Technology and Science, Pilani, India (2018). During her master's dissertation, she worked on the antimicrobial activity and mode of action studies of biologically synthesized silver nanoparticles. Presently, she is a Ph.D. candidate under the supervision of Prof. Ranjana Pathania at the Department of Biosciences and Bioengineering, Indian Institute of Technology (IIT) Roorkee. Her project is focussed on the characterization and target identification of novel antibacterial small molecules for the treatment of resistant bacterial infections. Her research interests are antimicrobial resistance, drug discovery and bacterial physiology. |
 Mangal Singh | Mangal Singh received his Master's degree in Biochemistry from the Department of Biochemistry, Kurukshetra University, Kurukshetra in 2011. He worked in the area of lantibiotics for his Ph.D. at the Department of Biochemistry, Panjab University Chandigarh in 2018. For his post-doctoral studies, he joined Prof. Pathania's group at IIT Roorkee. His project was focused on the structure–activity relationship studies of novel carbapenemase inhibitors. Recently in July 2023, he joined the Regional Research Institute of Unani Medicine, Patna, India as an Assistant Research Officer of Biochemistry. |
 Ranjana Pathania | Dr. Ranjana Pathania is a professor and DBT-Wellcome Trust India Alliance Senior Fellow at the Department of Biosciences and Bioengineering at IIT Roorkee, India. She leads the Molecular Bacteriology and Chemical Genetics (MBCG) group. Her research focus is to understand molecular mechanisms of antibiotic resistance development in bacterial pathogens and discovery of novel antibacterial compounds. Her group uses several tools of chemical biology to understand and overcome the AMR problem, such as chemical genetic strategies, deciphering the role of small RNA in the success of Acinetobacter baumannii as a MDR pathogen, and novel antibiotic combinations to treat bacterial resistant populations. |
Introduction
Human society has consistently utilized antibacterial medications since the introduction of penicillin in the 1940s to treat infections caused by pathogenic microorganisms.1 Although these antibiotics have saved countless lives and are a vital component of modern medicine, they have also exacerbated the evolutionary stress on microbes leading to the rise of multidrug-resistant (MDR) pathogens.2 This rapid emergence of drug-resistant bacteria threatens the effectiveness of antibiotics, thereby reducing their widespread use.3,4 The World Health Organization (WHO) has enlisted six such MDR bacteria designated as priority pathogens under the acronym “ESKAPE” (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.). These pathogens are associated with the highest mortality rate and are of great concern in clinics.5 The multidrug-resistant pathogens evade the antibiotic action by constantly mutating the target sites, overexpressing efflux pumps, or acquiring antibiotic-modifying or -degrading enzymes, among other complex resistance strategies.6,7 With these factors in play, we are gradually transitioning into the “post-antibiotic era”, a looming threat to the society where even mild injuries would be life-harming as a result of the lack of therapeutic alternatives to tackle the current challenge of antibiotic resistance.8,9 While preventing overuse/misuse of antibiotics is necessary for combating antibiotic resistance, the drive to develop new classes of drugs is also needed.10,11 Thus, there is an urgent need to find new chemical classes or alter the present ones for greater efficacy, given the widespread rise of resistance to existing antibiotics.12The search for novel small-molecule drugs and probes has shifted in recent years to a paradigm of screening a substantial number of compounds to identify those that elicit the required biological response.13–15 Two approaches are employed in drug discovery and development to study the biological activity of small molecule probes (Fig. 1). The phenotype-based strategy, also referred to as forward chemical genetics by analogy to forward genetics, begins with the desired phenotype, followed by target identification and validation.16 Since the approach determines the phenotypic biological response without prejudice for a particular target protein, it can lead to the discovery of novel therapeutic agents. However, target identification in a phenotype-based approach requires substantial efforts and thus becomes challenging.17
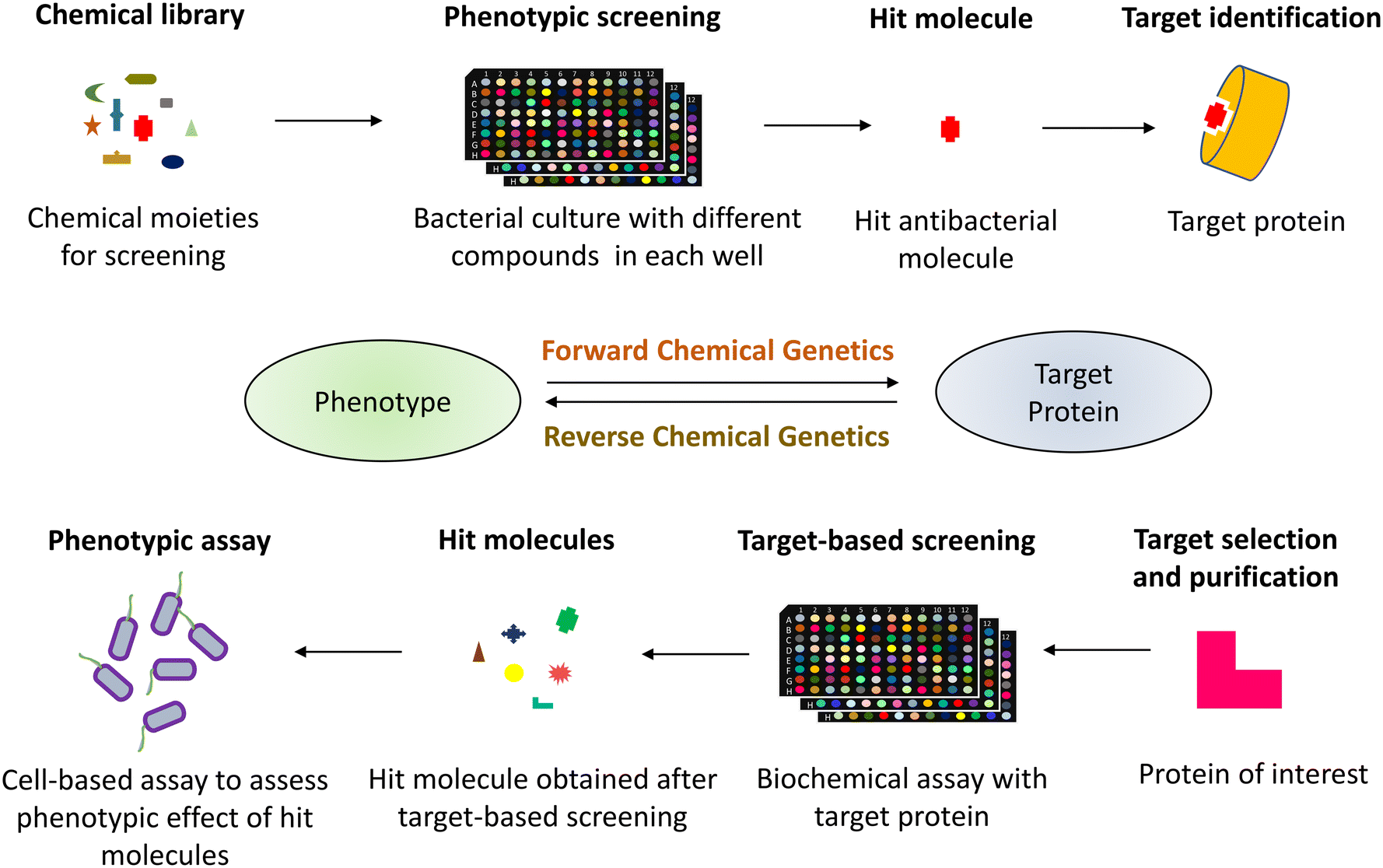 | ||
| Fig. 1 The process of chemical genetic screening in drug discovery comprises forward and reverse chemical genetics. The forward chemical genetics involves phenotypic screening of the chemical library followed by target identification of the hit molecule with desired phenotypic implications. The reverse chemical genetics is a target-based approach involving screening the compound library against the purified protein of interest, followed by evaluation of their phenotypic outcomes in cell-based assays. | ||
In contrast, target-based strategies undertake biochemical methods to screen small molecules against a particular target protein, followed by studying their physiological response.18,19 Accordingly, it is termed reverse chemical genetics by analogy to reverse genetics. This approach solves the problem of target validation as faced in the forward chemical approach. Still, it may only sometimes result in the desired phenotypic effect when used in a cell-based assay, owing to the physiological and biochemical barriers.20 As phenotypic and target-based drug discovery approaches have their own pros and cons, combining both strategies to identify new drugs that can act upon the desired molecular target is imperative. The pathway-directed approach has recently been used to screen a library of small molecules and discern inhibitors of a target pathway using a cell-based screening.21,22 Another strategy may involve a set of phenotypic and biochemical assays in series for high-throughput screens, for example, cell growth inhibition followed by a multi-well plate-based penicillin-binding protein (PBP) binding biochemical assay for specific PBP inhibitors in the initial phase of screens. With technological and biological developments, cell-based assays are found to be more favourable to discover new physiologically active small molecules.23,24 Additionally, pathway-directed cell-based assays are preferable since they allow for an early achievement of the desired phenotype and identifying the hit molecules. These hit molecules can be optimized for lead generation, resulting in a suitable drug candidate.25–27
Antibacterial drug targets
The major classes of in-clinic antibiotics target pathways essential for bacterial survival, like cell wall synthesis, DNA/RNA, and protein synthesis.28 The currently used antibiotics have been found to be prone to resistance mechanisms developed intrinsically or acquired by bacterial pathogens.29 Based on the mechanism of action, antibiotics have been classified into five categories, i.e., cell wall synthesis inhibitors, membrane depolarising molecules, inhibitors of protein synthesis, antibiotics targeting DNA/RNA synthesis, and metabolic pathway inhibitors (Fig. 2). Antibiotics, in addition to their target, are also distinguished based on their ability to induce cell death or inhibit the bacterial cell growth commonly termed as bactericidal and bacteriostatic, respectively.30 Bactericidal antibiotics follow a common cellular oxidative stress-inducing mechanism by disrupting the electron transport chain and tricarboxylic acid cycle.31,32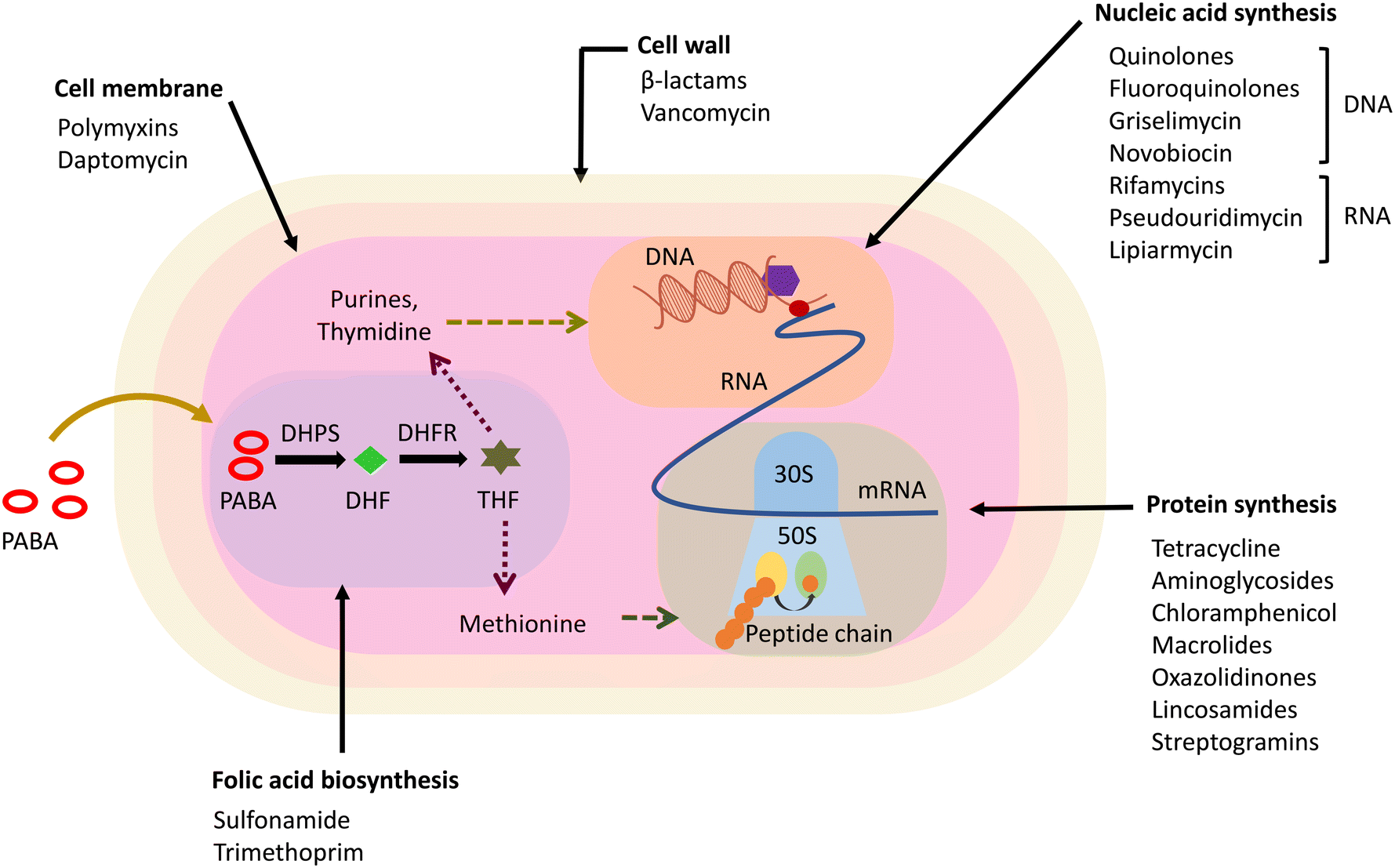 | ||
| Fig. 2 Antibacterial drug targets and the classes of antibiotics that target them. PABA – p-aminobenzoic acid, DHF – dihydrofolic acid, THF – tetrahydrofolate, DHPS – dihydropteroate synthase, DHFR – dihydrofolate reductase. | ||
Antibacterials hindering cell wall synthesis majorly belong to the β-lactam class of antibiotics and glycopeptides, which target the components involved in the cell wall synthesis machinery.33 The β-lactam class of antibiotics are inhibitors of PBPs, enzymes involved in the late stages of peptidoglycan (PG) synthesis, and inhibit their function of transpeptidation, thereby disrupting the PG synthesis.34,35 Glycopeptides like vancomycin and teicoplanin target and sequester lipid II, an essential precursor molecule in PG biosynthesis, and bind to the D-Ala-D-Ala terminal of the pentapeptide in Gram-positive bacteria.36,37 Some constituting members of lantibiotics, a class of bacteriocins active against Gram-positive bacteria, are also known to target lipid II.38 These are ribosomally synthesized polycyclic peptides that are post-translationally modified to a biologically active form containing thioether amino acids lanthionine and methyllanthionine and hence termed lantibiotics.39 Nisin is the first and most studied lantibiotic, which shows a dual mode of action by targeting cell wall synthesis via lipid II sequestration and alteration of the cell membrane permeability.40,41 The lipid II–lantibiotic complex formed by the action of lantibiotics leads to pore formation in the bacterial membrane by incorporating more peptide entities.42,43 The pore-forming ability of lantibiotics depends on their size and the lipid composition of the bacterial membrane.44,45 Other lipid II targeting classes of antibiotics are cyclic depsipeptides, such as ramoplanin and teixobactin, and non-lantibiotic bacteriocins like colicin M and defensins.46–49 Additionally, several compounds restrict the synthesis of lipid II by interacting with lipid II cycle intermediates, including fosfomycin, D-cycloserine, bacitracin and tunicamycin.50,51
Lipopeptide antibiotics, including polymyxins and daptomycin, cause alteration in the cell membrane.52,53 These are cationic or anionic cyclic/linear peptides covalently attached with a fatty acid moiety at the N-terminal. The cell membrane integrity is affected by the binding of these lipopeptide antibiotics to the cell surface. Despite having similar structures, polymyxins and daptomycin show different antibacterial spectra as polymyxins are active against Gram-negative pathogens, while lipopeptide daptomycin is known to target only Gram-positive bacteria.54 This is due to the different target sites of these antibiotics within the cell membrane. Polymyxin B and colistin (polymyxin E) target lipid A in the lipopolysaccharide (LPS), explaining their activity against Gram-negative strains, whereas daptomycin interacts with phosphatidylglycerol in the cell membrane leading to membrane damage and further inhibits cell wall synthesis by binding to the division septum.
The discriminative presence of cell wall components in bacteria and not in humans implies their prominence as valuable drug targets.55 However, the increased prevalence of resistance determinants of cell wall targeting antibiotics has raised the concern of AMR. The most common cause of AMR against such antibiotics is the growing pervasiveness of antibiotic degrading enzymes such as β-lactamases. β-Lactamases can hydrolyze β-lactam antibiotics by covalently binding to the carbonyl group thereby inhibiting their antibacterial action. Resistance to almost all the generations of β-lactams including the last line of defence drugs, carbapenems, has been observed in the clinic due to evolution of carbapenemases, β-lactamases capable of hydrolysing carbapenems.56 The catalytic division of carbapenemases into serine and metallo β-lactamases based on the presence of an active serine or zinc at the active site further complicates the available treatment options. β-lactamase inhibitors (BLIs) have been introduced in clinics to resist the action of β-lactamases and resensitize the bacteria to β-lactam antibiotics. However, none of the BLIs under clinical use target metallo-β-lactamases. Vaborbactam and avibactam, the clinically approved BLIs, show narrow-spectrum carbapenemase inhibitory activity.57
Recently, a new boronic acid-based BLI, xeruborbactam (QPX7728), has been discovered that shows broad-spectrum inhibitory activity and is under clinical trials for carbapenem-resistant infections.58 The discovery of antibiotic adjuvants, though important, can still lead to further resistance development as has been previously observed with the BLIs. Another important mechanism of resistance to cell wall targeting antibiotics in clinics is target modification which is seen in inhibiting the activity of vancomycin. Vancomycin has been found to be effective against Gram-positive pathogens including methicillin-resistant S. aureus (MRSA) infections. However, the presence of a van gene cluster in resistant bacteria causes a modification in the pentapeptide chain in PG and is the major cause of vancomycin resistance.59 The terminal D-alanine is replaced by D-lactate, reducing its affinity to vancomycin and thus, giving rise to resistant population like vancomycin-resistant S. aureus (VRSA) and vancomycin-resistant Enterococci (VRE). Furthermore, the emergence of vancomycin-intermediate S. aureus (VISA) and heterogeneous VISA (hVISA) pathogens with a thickened cell wall and the presence of free murein monomers have proved to be problematic in limiting the efficacy of vancomycin. Such instances of growing resistance towards most widely used cell wall inhibiting drugs in clinics pose a threat to their use as therapeutics. While resistance development is crucial to bacteria for survival, it comes with a fitness cost as seen in β-lactam and vancomycin-resistant strains.60,61 This further lays down the importance of cell wall components for bacterial survival along with their exclusive nature. Hence, it is vital to look for antibiotics that target novel entities in the bacterial cell wall or novel antibacterials which evade resistance development to curb AMR.
Antibacterial drug targets in the bacterial cell envelope
Bacterial cell wall synthesis and shape maintenance are multi-step processes with different essential proteins playing a crucial role, making each step of the pathway amenable to being a potential antibiotic target. Additionally, the lack of mammalian equivalents of these proteins makes them attractive drug targets. β-lactam antibiotics, the most successful and predominant cell wall-targeting antibacterials, inhibit the PG biosynthetic pathway via the enzymatic inhibition of PBPs. PG synthesis inhibitors are usually broad-spectrum antibiotics due to the presence of PG in both Gram-positive and Gram-negative bacteria. In particular, inhibitors of PBPs have been successful in clinics due to the periplasmic location and ease of access by traversing only the outer membrane (OM). The presence of the OM with LPS in Gram-negative bacteria and thick PG with wall teichoic acid (WTA) in Gram-positive bacteria limits the broad-spectrum applicability of several inhibitors that target the common proteins of bacterial membranes. In general, Gram-negative bacteria are resistant due to restriction imposed by the OM. The permeability barrier due to the LPS in the OM is species-specific depending on its architecture like thickness and surface charge distribution. It also depends on the number and size of the LPS units involved in formation of the OM. The OM obstructs the entry of non-polar molecules which are larger than general porins (>600 Da).62 The smaller hydrophobic molecules allowed through the OM are further restricted by the inner membrane. In contrast, polar solutes which are permeable through the inner membrane are blocked by lipid A. Therefore, various components that make up the bacterial cell envelope are valuable antibacterial targets. In this review, we look into the details of prominent and some of the overlooked antibacterial pathways and targets belonging to the bacterial envelope.Peptidoglycan biosynthesis
PG is an essential carbohydrate polymer in bacteria that is involved in maintaining the cell shape, resisting osmotic change, and coordinating cell division. The biosynthesis of PG is initiated in the cytoplasm with formation of uridine diphosphate N-acetyl muramic acid (UDP-MurNAc) pentapeptide also known as Park's nucleotide in a multistep enzymatic reaction catalyzed by Mur enzymes63 (Fig. 3). This is followed by Park's nucleotide ligation to the polyisoprenoid or undecaprenyl phosphate (C55-P) membrane anchor and uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) leading to lipid I and lipid II synthesis, respectively. The lipid II molecule is finally flipped to the outer leaflet, and MurNAc-pentapeptide-GlcNAc is inserted into the PG by various PBPs that catalyze glycosyltransferase and transpeptidase activity. The C55-P anchor of lipid II is recycled in the cell and is also shared between PG and WTA biosynthesis. As PG is essential for bacteria to survive, all the steps in its synthesis are recognized as important antibacterial targets.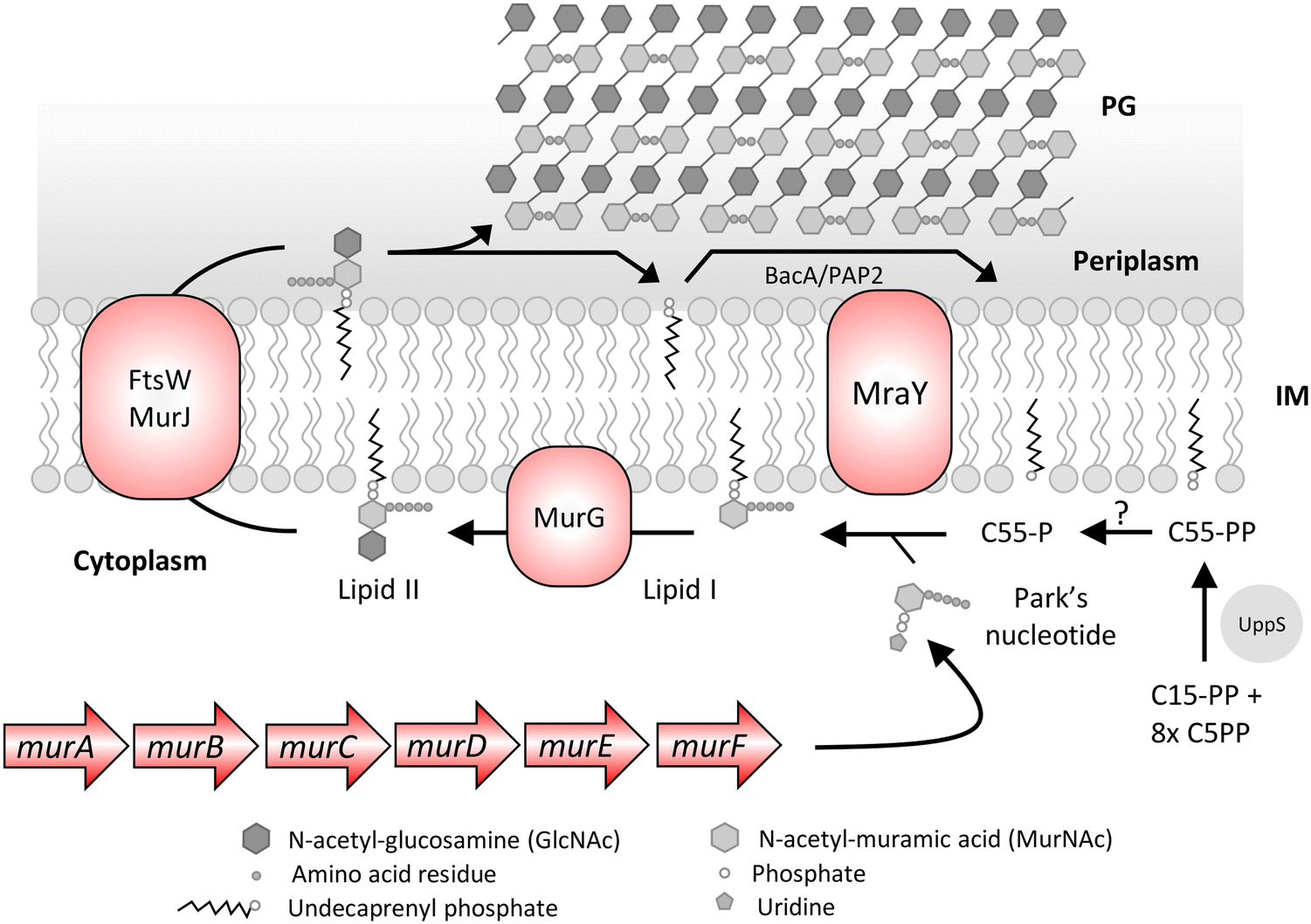 | ||
| Fig. 3 Peptidoglycan (PG) biosynthesis pathway of E. coli. The biosynthesis begins in the cytoplasm with the formation of Park's nucleotide in a multistep enzymatic pathway catalyzed by mur enzymes. The Park's nucleotide is ligated to undecaprenyl phosphate (C55-P) and uridine diphosphate N-acetylglucosamine (UDP) leading to lipid I and lipid II, respectively. The lipid II molecule is flipped across the inner membrane (IM) and the MurNAc-pentapeptide-GlcNAc component is inserted into the peptidoglycan by various penicillin binding proteins that catalyze glycosyltransferase and transpeptidase activity. The C55-PP is recycled by the BacA/PAP2 pyrophosphatases that convert it to C55-P or de novo synthesized by UppS and converted to C55-P by an unknown pyrophosphatase for another cycle of ferrying the PG precursors. | ||
| Target | Role | Inhibitor | Source | Target bacteria | Clinical stage | Ref. |
|---|---|---|---|---|---|---|
| a β-Lactam adjuvant against methicillin-resistant S. aureus (MRSA), OMP – outer membrane protein, GP – Gram-positive, GN – Gram-negative, BS – broad spectrum, NP – natural product, S – synthetic compound, *veterinary. | ||||||
| Alr | D-Alanine racemase, provides substrate for Ddl | D-Cycloserine | NP | GP | In-clinic | 64 |
| Ddl | D-Ala-D-ala ligase involved in PG synthesis | |||||
| MurA | PG precursor synthesis | Fosfomycin | NP | BS | In-clinic | 287 |
| MurC | Feglymycin | NP | GP | Preclinical | 180 | |
| MraY | Synthesis of lipid I by transfer of phospho-MurNAc-pentapeptide to the C55-P carrier | Muraymycins | NP | BS | Experimental | 67 |
| Mureidomycins | GN | |||||
| Capuramycins, liposidomycins, tunicamycins | GP | |||||
| Amphomycin | In-clinic* | 288 | ||||
| MurG | Lipid II synthesis | Ramoplanin | NP | GP | Phase III | 289 |
| Murgocil | S | GP | Experimental | 121 | ||
| Lipid II | Precursor for PG | Vancomycin | NP | GP | In-clinic | 49 |
| Nisin | In-clinic* | 49 | ||||
| Plectasin | Preclinical | 37 | ||||
| Teixobactin | 46 | |||||
| Tridecaptins | GN | 290 | ||||
| PBPs | Transpeptidases | β-Lactams | NP and S | BS | In-clinic | 81 |
| DBOs e.g. avibactam | S | |||||
| Transglycosylation | Moenomycins | NP | GP | In-clinic* | 291 | |
| C55-P | Lipid carrier for MurNAc-pentapeptide-GlcNAc | Friulimicin | NP | GP | Pre-clinical | 292 |
| C55-PP | Precursor of the lipid carrier C55-P utilized in PG and WTA biosynthesis | Bacitracin | NP | GP | In-clinic | 293 |
| Amphomycin | 288 | |||||
| UppS | Undecaprenyl phosphate synthase | Clomiphene | S | GPa | In-clinics as fertility drug | 66 |
| MAC-0547360 | S | GP | Experimental | 294 | ||
| BamA | OMP, essential component of the β-barrel assembly machinery complex | Darobactin | NP | GN | Preclinical | 295 |
| NP | ||||||
| LlpA | Experimental | 100 | ||||
| MRL-494 | S | Experimental | 102 | |||
| LpxC | Zinc dependent deacetylase, catalyzes the first committed step in lipid A biosynthesis | CHIR-090 | S | GN | Experimental | 296 |
| BM-78484, BM-78485 | 252 | |||||
| RC-01 | Phase I aborted | 107 | ||||
| ACHN-975 | Phase I | 107 | ||||
| LpxH | Fourth step of lipid A biosynthesis | Compound 1 | S | GN | Experimental | 282 |
| LptD | OMP responsible for translocation of LPS to the outer leaflet of the OM | Murepavadin (POL7080) | S | GN | Phase III – toxicity issue | 107 |
| LptB | ATPase involved in LPS extraction from the outer leaflet of the IM | Compounds 1 and 2 | S | GN | Pre-clinical | 183 |
| MsbA | ATP dependent inner membrane lipid flippase | G427, G907 | S | GN | Pre-clinical | 184 |
| TBT-1 | 105 | |||||
| MreB | Prokaryotic actin homologue involved in the maintenance of rod shape | Benzyl isothiourea derivatives of A22, MP265, CBR-4830, MAC13243 | S | GN | Experimental | 98 |
| CbtA and CptA toxin | NP | 297 | ||||
| Sceptrin | BS | 98 | ||||
| LspA | Type II signal peptidase (prolipoprotein signal peptidase) | Globomycin, myxovirescin | NP | BS | Experimental | 182 |
| Compound 1j | S | GN | ||||
| LolCDE | ABC transporter extracts mature, OM-targeted lipoproteins from the IM | G0507 | S | Inactive | 123 | |
| Compound 2 | GN | Experimental | 282 | |||
| Pyridineimidazole compounds 1 & 2 | 124 | |||||
| TarG | Involved in export of WTA | Targocil, 1835F03 | S | GP | Experimental | 134 |
| TarO | TarO catalyzes the first step of WTA biosynthesis in S. aureus | Tunicamycin | NP | GP | Experimental | 298 |
| Ticlopidine | S | In-clinics as antithrombotic drug | 109 | |||
In the rod complex, PBP2 is targeted by the β-lactam class of antibiotics, with mecillinam (or amdinocillin) as a specific inhibitor.96 Mecillinam is the active form of the prodrug pivmecillinam and is used to treat lower urinary tract infections.97 Apart from PBP2, MreB is the most studied protein, and several attempts have been made to search for drugs targeting this protein. However, despite many efforts, no clinically significant drug has been discovered to target the protein due to toxicity issues, while some are still in the initial discovery phase.98 Nevertheless, the essential role of these rod-complex proteins in lateral PG synthesis, cell morphogenesis and division makes them notable potential targets for therapeutic innovation.
OM biogenesis
An effective resistance mechanism of Gram-negative bacteria is the presence of the OM barrier, which restricts the entry of antibacterial molecules that otherwise target Gram-positive bacteria. The various components of the OM, like OMPs, LPS and lipoproteins, are synthesized and exported by dedicated multiprotein pathways. BamA and LptD are two surface-exposed proteins in Gram-negative bacteria (Fig. 4), recognized as antibacterial drug targets and antigens for vaccine development. Both proteins are involved in the maintenance of the OM in Gram-negative bacteria.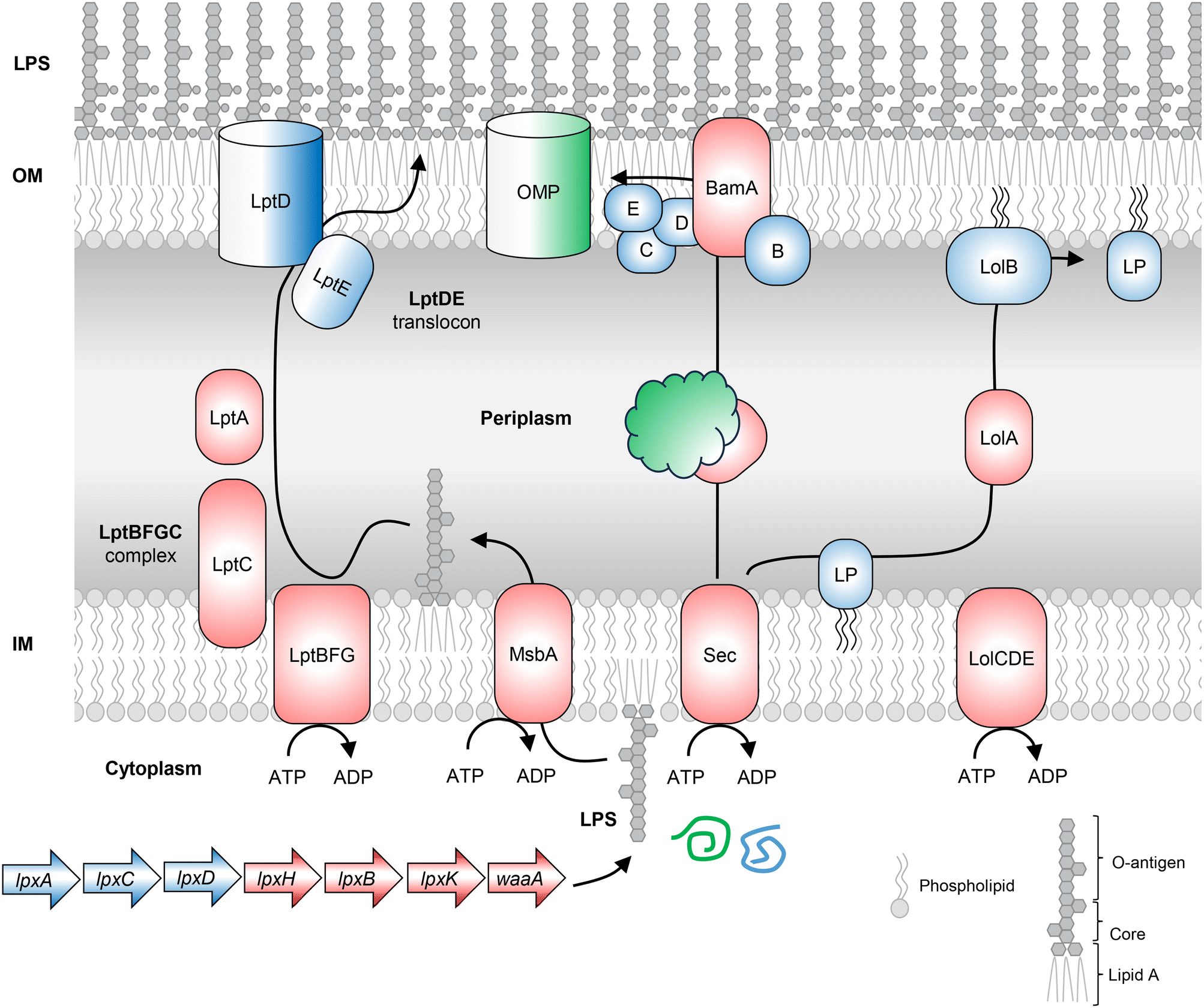 | ||
| Fig. 4 The OM components like OMPs, LPS and lipoproteins are synthesized and exported by dedicated multiprotein pathways. Lipopolysaccharide (LPS) biosynthesis in Gram-negative bacteria begins with the cytoplasmic synthesis of LPS molecules by the dispensable genes lpxA, lpxC, and lpxD and the conditionally essential genes lpxH, lpxB, lpxK and waaA. The BamABCDE β-barrel assembly machine (BAM) complex, a translocator present in the OM of Gram-negative bacteria, catalyzes the folding and insertion of beta-barrel proteins in the OM. The lipoproteins are transported from the outer leaflet of IM to the OM through the Lol system. The precursor lipoproteins carrying the signal sequence are translocated across the inner membrane via the Sec or Tat pathway. The signal sequence is cleaved and the LolCDE complex in the inner membrane extracts the lipoproteins for transfer of the OM-targeted lipoproteins to LolA, a diffusible lipoprotein-chaperone. LolB, the OM acceptor protein, integrates lipoproteins in the inner leaflet of the OM. LPS – lipopolysaccharide, OM – outer membrane, IM – inner membrane, LP – lipoprotein. | ||
Lipopolysaccharide biosynthesis
The OM present in Gram-negative bacteria is asymmetric with LPS molecules in the outer leaflet and phospholipids in the inner membrane. LPS biosynthesis begins in the cytoplasm with UDP-GlcNAc (Fig. 4). Each of the important biosynthetic genes recognized in the pathway is discussed.Wall teichoic acid biosynthesis
The cell envelope of Gram-positive bacteria comprises WTA, a cell surface phosphate-rich carbohydrate polymer and an important virulence factor that helps establish infection.66,108,109S. aureus lacking WTA exhibits compromised pathogenicity and increased sensitivity to the action of β-lactam antibiotics, and thus WTA represents an important drug target. The biosynthesis of WTA begins in the cytoplasm in a multi-enzymatic pathway (Fig. 5), where the initial enzymes (TarO and TarA) involved in polymer synthesis are found to be dispensable. At the same time, the late-stage genes (TarB, TarF, TarL, TarG and TarH) are essential unless the initial enzymes are non-functional, mutated or knocked-out, so that the polymer initiation is inhibited. As apparent, in the absence of late-stage genes, the accumulation of polymers will become lethal, and in the absence of priming enzymes, the lack of WTA leads to compromised virulence. Inhibition of TarO activity not only compromises the pathogenicity of S. aureus but is also known to resensitize methicillin-resistant S. aureus to the action of β-lactam antibiotics.109,110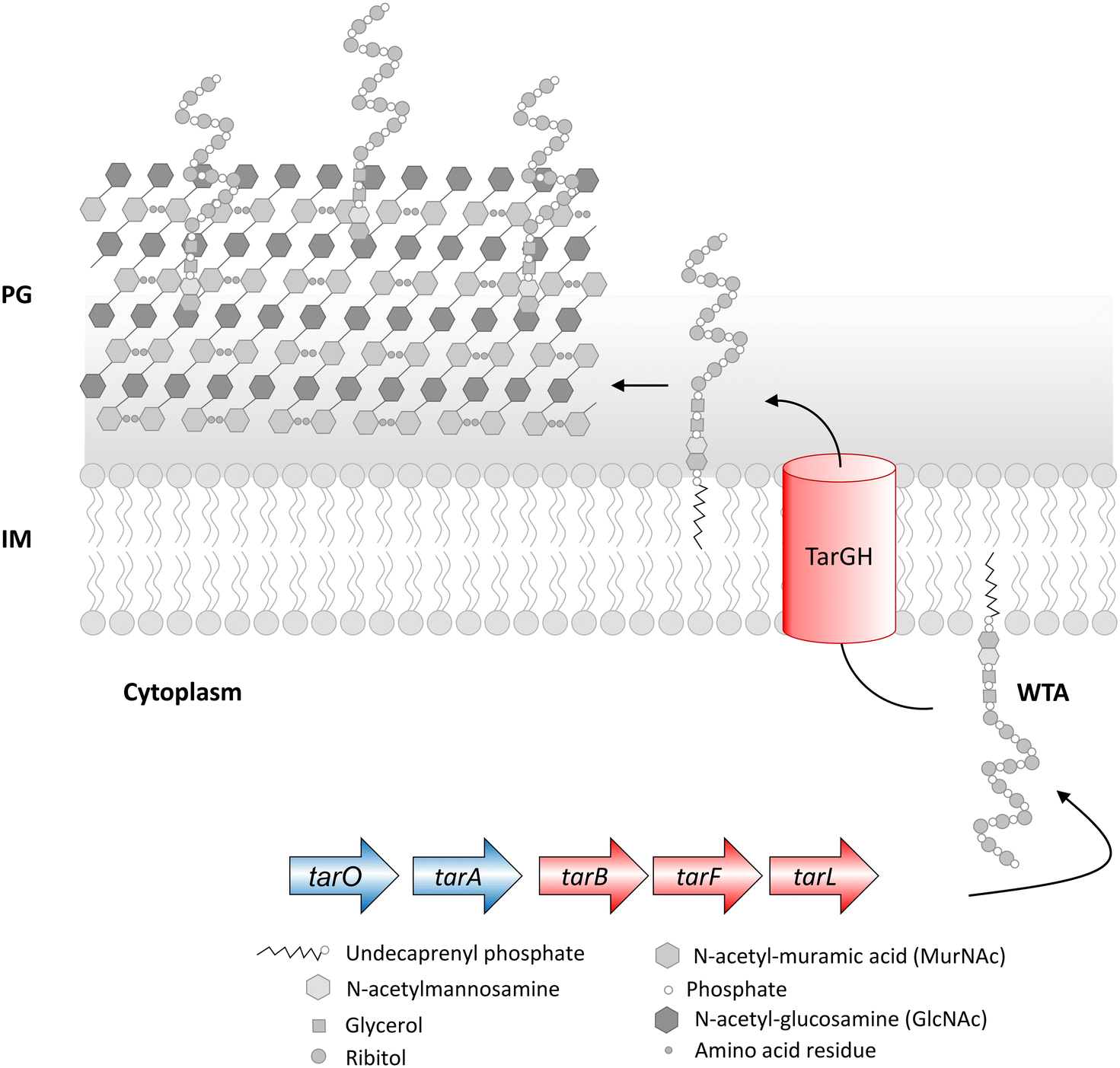 | ||
| Fig. 5 Wall teichoic acid (WTA) biosynthetic pathway in S. aureus. The biosynthesis of WTA begins in the cytoplasm in a multi-step pathway comprising TarO, TarA, TarB, TarF, TarL, TarG and TarH. Deletion of dispensable WTA biosynthetic genes (coloured in blue) leads to avirulence, and deletion of conditionally essential genes (coloured in red) in a wild-type strain leads to cell death but survival in ΔtarO and ΔtarA backgrounds. PG – peptidoglycan, IM – inner membrane. | ||
Chemical genetic approaches to discover cell wall inhibitors
The bacterial cell wall is crucial for survival and has served as a drug target since the discovery of penicillin.111 The clinical success of cell wall inhibitors like β-lactams and glycopeptide drugs signifies the importance of cell wall proteins as predominant antibacterial drug targets.112 The marked difference in the cell wall composition from eukaryotes is one of the crucial factors which makes them an appealing drug target.113 More importantly, Gram-negative bacteria impede the entry of antibacterial molecules to reach the target.114,115 The ever-emerging antibiotic resistance in clinics and the importance of cell wall drug targets warrant the discovery of novel bacterial cell wall inhibitors.116–118 However, the target-based approach in discovering cell wall antibacterials becomes unfavourable when the inhibitors fail to act in vivo after showing promising inhibitory activity in vitro.119 Hence, it is required to incorporate both cell-based screens (phenotypic screens) and target-based approaches in discovering bacterial cell wall inhibitors to overcome the challenges independently posed by phenotypic and target-based approaches.Chemical–chemical interaction profiling
Chemical–chemical interaction profiling is an attractive strategy to elucidate the mechanism of action (MOA) of antibacterial molecules by exploiting the fact that molecules with a similar MOA show unique synergistic interactions.120 The approach utilizes combination ratio profiling to screen molecules with unknown MOA against known bioactives. The ratio of the percentage growth of bacterial cells, when treated with a combination, to the percentage growth of cells treated alone by a known bioactive, is known as the combination ratio. A growth inhibition of ≥75% corresponds to a combination ratio of ≤0.25, which indicates synergy (Fig. 6). Moreover, using a two-point dose matrix makes it easier to perform the assay in a high-throughput manner. The two-point dose matrix assay essentially tests the combination ratio of the drugs at two concentrations, i.e., ¼ X and ⅛ X MIC. It reduces the complexity of the conventional checkerboard assay to assess synergism. The study highlighted the importance of synergistic interactions in unraveling the inhibitors of DHFR and DNA gyrase.120 When a panel of 14 known antibiotics was screened against a library of 200 antibacterial molecules, two compounds, MAC-0038968 and MAC-0003199, with unique synergies were discovered. MAC-0038968 synergistically acted with sulfamethoxazole, a folate pathway inhibitor. Although, it is suggested that synergistic interaction may arise from inhibitors of the same pathways, MAC-0038968 did not synergise with trimethoprim as they both bind to DHFR. It is a disadvantage in such screens when the molecular target of two molecules is common as they face a limitation in available target sites for their action. Further cellular and biochemical experimentations validated DHFR as the target of MAC-0038968. The other inhibitory molecule, MAC-0003199, was discovered as a DNA gyrase targeting molecule. MAC-0003199, a quinone class compound, displayed synergy with norfloxacin, a known DNA gyrase inhibitor. The chemical nature of MAC-0003199 along with its interaction profiling with norfloxacin suggested that the molecule inhibits the DNA gyrase function and thus causes DNA damage. However, the MOA of both norfloxacin and MAC-0003199 must be different otherwise it may not have resulted in synergy, as observed with MAC-0038968. Besides norfloxacin, MAC-0003199 exhibited synergistic interactions with lincosamine and triclosan as well but the exact mechanism behind these synergies is not understood. Although these chemical interactions assist in uncovering the MOA of novel antibacterials, indiscriminate interactions with multiple antibiotics pose a problem in such strategies. Hence, assessing the unique interaction profiles along with analysis of the chemical structures helps in elucidating the MOA of novel hit molecules.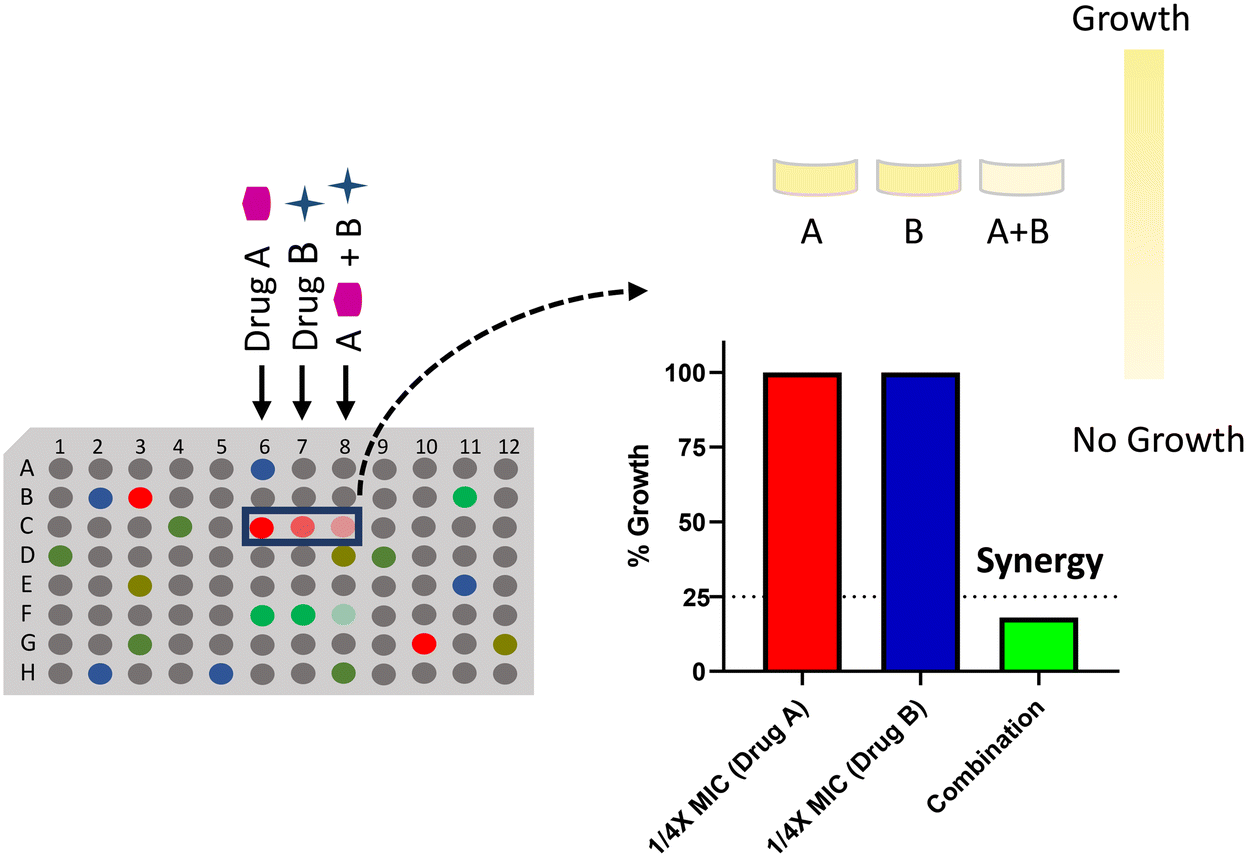 | ||
| Fig. 6 Combination ratio profiling of the antibacterial molecules to determine the mode of action of a drug using synergistic chemical–chemical interactions. | ||
In another study, Mann et al. utilized a phenotypic synergy screen to identify a PG synthesis inhibitor, murgocil.121 They screened for compounds that synergize with β-lactam antibiotics against MRSA. Firstly, a screen for molecules that potentiate the activity of imipenem was carried out, leading to 134 hit molecules. A screen for the target biosynthetic pathway involved a macromolecular labelling (MML) experiment where the relative incorporation of radiolabeled precursors in the DNA, RNA, protein, phospholipid, and cell wall is measured in the presence of the inhibitor molecules.121–124 In the MML experiment, murgocil was observed to inhibit PG synthesis specifically. Further, gene dosage perturbation studies with several PG synthesis genes were conducted with murgocil. Fourteen different xylose-inducible antisense interference plasmids targeting a specific gene were utilized, which depleted the target gene in MRSA upon induction. The observed hypersensitivity of MRSA to murgocil upon MurG depletion and reduction of the antibacterial effect upon overexpression of MurG indicated that murgocil targets MurG. Isolation of the resistant mutants and in vitro inhibition of purified MurG activity confirmed MurG as the target of murgocil.
These studies imply that the synergistic profiling strategy can be used to discover inhibitors of many essential pathways in bacteria. To identify cell wall inhibitors, a screen of a chemical library can be carried out with a panel of known cell wall inhibitors. The exact MOA of potential cell wall targeting antibacterials can be further evaluated using biochemical methods such as MML, antisense-induction, resistant mutant generation combined with NGS, etc.125–127
Pathway-directed or antagonistic whole-cell screening
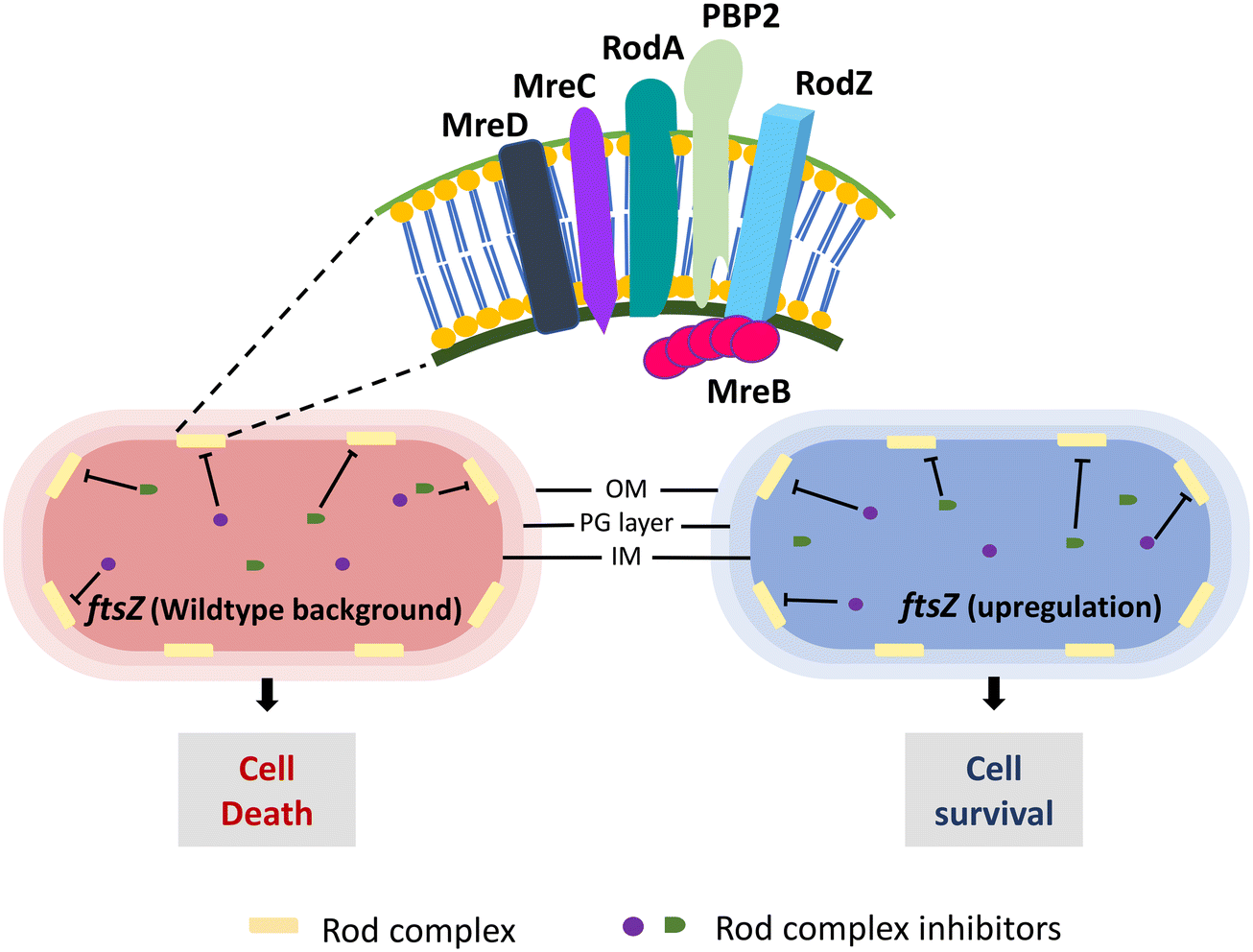 | ||
| Fig. 7 Conditional essentiality of the E. coli rod complex (RodA, PBP2, MreB, MreC, MreD, RodZ). The essential phenotype of the rod complex becomes dispensable when FtsZ expression is upregulated. | ||
Mecillinam treatment initiates an unproductive cycle of PG synthesis (due to the transglycosylase activity of RodA) and degradation (absence of PBP2 transpeptidase activity) causing rod complex malfunctioning.132 A recent study has shown that the energy demand from the metabolic processes due to this malfunctioning induces toxic shifts in the metabolic by-products causing mecillinam lethality.133 Considering these, a total of three HTS were set up, first to discover potential antibacterial molecules followed by identifying suppressors of mecillinam toxicity, and then from these molecules, rod complex inhibitors whose lethality was suppressed in ftsZ upregulated E. coli cells were identified. The screen revealed eight potent rod complex inhibitors from a total of ∼690![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules. These eight molecules were further characterized and identified as A22 analogues, active against MreB. The importance of FtsZ overexpression in rod complex non-essentiality makes it an excellent approach to identify inhibitors of rod complex proteins. However, it should be noted that such a screen may result in inhibitors of FtsZ itself. Hence, as discussed above, a counter screen to validate the target is essential, like a mecillinam toxicity suppression assay for rod complex inhibitors.
000 molecules. These eight molecules were further characterized and identified as A22 analogues, active against MreB. The importance of FtsZ overexpression in rod complex non-essentiality makes it an excellent approach to identify inhibitors of rod complex proteins. However, it should be noted that such a screen may result in inhibitors of FtsZ itself. Hence, as discussed above, a counter screen to validate the target is essential, like a mecillinam toxicity suppression assay for rod complex inhibitors.
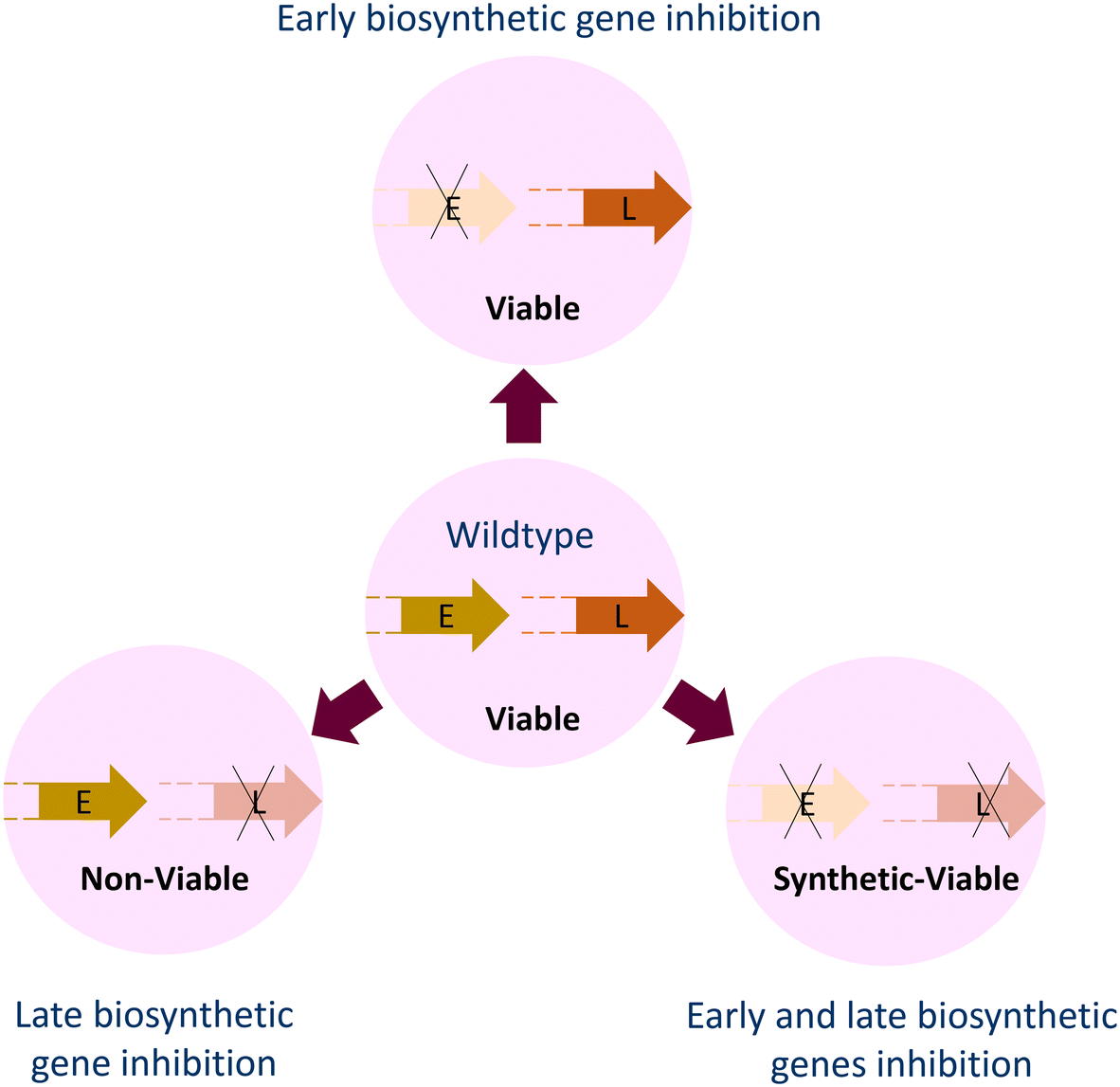 | ||
| Fig. 8 Loss of the early-stage (E) genes of lipopolysaccharide (LPS) and wall teichoic acid (WTA) biosynthesis leads to loss of virulence but the survival of bacterial cells under in vitro conditions. Loss of late-stage (L) biosynthetic genes is deleterious to cells unless polymer synthesis is halted with the deletion or mutations in early-stage genes. | ||
The C55-P carrier molecule is essential for the translocation of PG precursors and WTA molecules from the cytoplasmic to the periplasmic leaflet of the inner membrane. Clomiphene, a US FDA-approved molecule, is an inhibitor of the UppS involved in C55-P synthesis. It was discovered in a cell-based screen for molecules that antagonize the activity of targocil, a late-stage WTA biosynthesis inhibitor that targets TarG leading to accumulation of WTA molecules and consequent death of the cell.66 The screen was setup for identification of molecules that would rescue the cells from the toxicity of targocil by inhibiting the early stage enzymes that initiate WTA synthesis. Clomiphene, the obtained hit, antagonized not only targocil but also ticlopidine, an early-stage WTA biosynthesis inhibitor that inhibits TarO. Antagonism towards both, and antibacterial activity in isolation, suggested that WTA synthesis is not the actual target of clomiphene. Utilizing antisense induction for depletion of the genes involved in PG synthesis and the consequent phenotype, clomiphene was observed to synergize with UppS downregulation. This screen for WTA inhibitors turned out to identify an inhibitor of the target, common to both PG and WTA biosynthesis. Clomiphene being a US-FDA approved fertility drug has the potential for development as an antibacterial drug.
Another useful strategy to target the OM of Gram-negative pathogens is by inhibiting the surface-exposed proteins. BamA is an essential protein of the BAM complex present in the OM. The molecule MRL-494 was identified from a screen for small molecules that targeted the OM and exhibited antibacterial activity unaffected by efflux pumps.102 Antibacterial hits first identified against wild-type E. coli with an intact OM and efflux were counter-screened against efflux-compromised tolC deletion mutants. The similar antibacterial potency in both strains suggested that MRL-494 is not a substrate for efflux. Additionally, the reduced activity of MRL-494 against the BamA mutant validated it as the molecular target of the compound. Cellular thermal shift analysis of BamA with MRL-494 corroborated these results. MRL-494 further showed potentiation of rifampicin in E. coli. Besides targeting Gram-negative bacteria, MRL-494 exhibited antibacterial potential against Gram-positive bacteria. Since Gram-positive bacteria lack an OM, the antibacterial activity was accounted to the membrane permeabilizing action of the molecule. This MOA of MRL-494 in Gram-positive bacteria was similar to nisin due to the cationic and amphiphilic peptide nature of the molecule. Wade and coworkers designed a synthetic route for MRL-494 and synthesized three analogues (compounds 13, 16 and 17).142 The triazine chlorine and the ester methoxy moieties were modified with guanidine (compound 13) or ammonia (compounds 16 and 17). The methyl ester group in compound 16 was further modified with guanidine. These analogues and the parent compound MRL-494 were evaluated for antibacterial activity, OM permeabilization, rifampicin potentiation and Rcs envelope stress response generation potential. Compound 17 lost the antibacterial potential whereas compounds 13 and 16 retained the potency and their physiological effects but these effects were not better than MRL-494. Even so, the presence of the guanidine group was found to be important for the activity. However, further studies on expanding this chemical space and toxic potential of MRL-494 are warranted as it has the ability to inhibit both Gram-negative and Gram-positive bacteria with two different modes of action.
Furthermore, for potential molecules that target LPS, Zhang et al. devised a cell-based screen based on the conditional essentiality of intermediate genes of LPS biogenesis. The intermediate genes are non-essential when the LPS flux through the inner membrane is prohibited whereas the early genes (lpx A/C/D) are non-essential in A. baumannii. They identified LPS inhibitors by comparing the growth of an engineered hyper-permeable strain (termed as Δ5) vs. an isogenic LPS-null strain (Δ5 ΔlpxA) of A. baumannii in the presence of small molecules.105 The increased permeability improved the screen sensitivity compared to a wild-type strain. The hyper-permeability was achieved (Δ5) by deleting three central efflux pumps involved in multidrug resistance (adeABC, adeFGH and adeIJK) and two acetyltransferases involved in lipid A modification.143–145 The screen of 150000 small molecules against Δ5 resulted in 1100 growth inhibitory molecules. A counter screen against Δ5 ΔlpxA identified 11 compounds with reduced inhibitory activity. The screen resulted in TBT scaffold molecules as active LPS biogenesis inhibitors. Further, the pathway specificity of these compounds was confirmed by a dose-dependent loss of inhibitory activity against the Δ5 strain in the presence of an LpxC inhibitor PF-5081090.146 Further, resistant mutants of the TBT molecule identified mutations in the early-stage biosynthetic genes of lipid A – lpxA, lpxC or lpxD. More studies with resistant mutants, in an effort to identify the actual target, revealed missense mutations in msbA. MsbA is an OM transporter protein responsible for LPS flux through the inner membrane. Moreover, the presence of the MsbA L150V mutated gene in trans diminished the activity of the inhibitor. Further experimentation with the purified protein identified MsbA, an essential OM protein, as the target of these inhibitors.
These strategies emphasise the importance of pathway-directed screen approaches in the discovery of cell wall inhibitors. Since these strategies are based on the conditional essentiality of cell wall biogenesis-related proteins, these cannot be used to identify inhibitors of other pathways.
Purified target protein-molecule binding
This strategy is a target-based screening with a purified cell wall protein of interest. Most target-based screens are performed virtually using docking software on an extensive library of molecules.147–149 The resulting hit molecules from virtual screening are then assayed against bacterial cells for their inhibitory potential. Such screens are helpful as they are less time-consuming and reduce the burden of resource wastage as compared to in vitro assays. However, these may not always result in a bacterial cell-active lead molecule. Hence, it is always preferable to conduct target-based studies in vitro. Cell wall metabolism is a multi-step complex phenomenon, and most of the proteins involved in the process do not offer a simple high throughput assay to perform.150 However, high throughput assays for antibacterial screening against targets like PBPs and Mur proteins involved in cell wall biogenesis have been developed.151,152 Such an approach is suitable when it is incorporated with a cell-based phenotypic screening at an early stage of drug discovery to prevent the failures associated with each approach individually. A PBP-Bocillin FL competitive assay has been employed for evaluating PBP inhibitors, which involves using Bocillin FL, a non-radioactive synthetic fluorescent penicillin molecular probe.153 Bocillin FL binds to isolated PBPs (membrane fractions or purified proteins) and forms a complex with all the classes of PBPs which become fluorescent.154,155 Bocillin FL, when incubated in combination with β-lactam antibiotics to target PBPs, results in a reduced fluorescence signal of either single or multiple PBPs based on its binding specificity.156,157 The change in fluorescence can be detected using various techniques like flow cytometry, SDS-PAGE analysis, fluorescence anisotropy/polarisation, microscopy and/or UV-vis spectrophotometry.158–160 SDS-PAGE-based fluorescence detection is a widely used, simple and traditional method for PBP-Bocillin FL binding studies depicted in Fig. 9. However, due to the low throughput nature of the technique, it cannot be implemented in the screening of large libraries of molecules.72 Hence, high throughput-compatible fluorescence detection methods are essential for screening purposes. A microtitre plate-based method of β-lactam-biotin conjugate (BIO-AMP) labelled PBP coupled with fluorescence detection has been developed.161 Other HTS methods are based on fluorescence anisotropy.162 Fedarovich et al. performed a Bocillin FL-based fluorescence polarization assay and screened 50000 molecules against PBP2 of N. gonorrhoeae, resulting in 24 molecules active against purified PBP2.163 Out of these, seven molecules exhibited in vivo antibacterial potential against penicillin- or cephalosporin-resistant strains of N. gonorrhoeae. The Thresher group utilized a similar strategy with a purified periplasmic domain of PBP3 from A. baumannii and P. aeruginosa as well as PBP2 from P. aeruginosa and showed that the assay is favourable for measuring acylation and deacylation rate constants of the β-lactams with respect to competitive Bocillin FL binding to the PBPs.164,165 Furthermore, fluorescence microscopy has also been utilized to evaluate PBP-β-lactam fluorescent probe binding in cell-based assays.166 Meropenem (MEM) pretreated B. subtilis cells have been shown to confer low MEM-BODIPY FL fluorescence owing to their low binding compared to the untreated cells.167 López-Pérez et al. demonstrated the use of yet another fluorescence-based S2d assay and screened a library of 2455 compounds against PBP3 of P. aeruginosa.168,169 The screen identified pyrrolidine-2,3-dione as a potent non-β-lactam PBP3 inhibitor. The class A HMM PBPs exhibit both transglycosylase and transpeptidase activities and the assays discussed above only indicate the binding of molecules to PBPs, not the loss of the specific catalytic activity. Thus, these assays cannot distinguish the inhibitory action of the molecules against bifunctional PBPs. Huang et al. designed and validated a fluorescence resonance energy transfer (FRET) based assay with lipid II, the substrate for the transglycosylase activity of PBPs, and used it to screen a library of 120000 compounds.170 The assay resulted in confirmed six hit transglycosylase inhibitors which were active against Mycobacterium smegmatis and S. aureus. This strategy is useful while working with bifunctional PBPs so as to assess the mode of inhibition of such PBPs.
 | ||
| Fig. 9 Schematic representation for identification of penicillin-binding protein (PBP) inhibitors in the presence of a competitor – Bocillin FL. Binding of Bocillin FL to PBP (e.g. PBP3 in the figure) is reduced in the presence of a PBP inhibitor which can be analyzed by fluorescence scanning of the Bocillin FL bound protein profile on a SDS-PAGE gel. | ||
Mur enzymes are also involved in the PG synthesis pathway as discussed in the previous section against which HTS has been carried out. The Mur pathway enzymes are widely studied for their physiological function in bacteria. Besides PG synthesis, MurA and MurC have also been implicated in spore formation in Clostridioides difficile.171 These essential enzymatic proteins MurA–F are associated with cytoplasmic steps of lipid II synthesis, while MurG is a membrane-bound protein.172 Previous reviews have covered the inhibitors screened for each enzyme in detail.173–175 However, some of the important screens are discussed here. Mur ligases (MurC–F) are ATP-dependent enzymes that sequentially ligate amino acid residues to the stem peptide.176 This ATP hydrolyzing activity of the Mur ligases has been exploited to assay hit compounds obtained from a virtually screened National Cancer Institute Diversity set against MurD and MurF of Mycobacterium tuberculosis (Mtb).177 The most promising MurD and MurF inhibitors from the in vitro phosphate release assay were further probed for their inhibitory potential by using radiolabelled nucleotide substrates. This secondary assay resulted in confirmed four MurD novel inhibitors and one MurF novel inhibitor. Structural analysis of these inhibitors revealed that the compounds containing a three-cyclic xanthene ring with a carboxylic group separated by two carbons are active MurD inhibitors. NSC 209931 displayed promising MurF inhibitory activity and the docked structure of the molecule with MurF showed similar binding patterns of the molecule compared to the known Abbott inhibitor 1 even though the scaffolds are different. Similarly, Kristan and coworkers utilized the ATP hydrolysis activity of the MurD ligase.178 However, instead of quantifying Pi release, they measured the ADP production by coupling it with another enzymatic method using an ADP Quest™ assay kit to produce a fluorometric readout. This fluorometric assay revealed 27 MurD inhibitors from a focused library of 1000 compounds. However, only two compounds – 1 and 2 – were tested further. Compound 1 was a glutamic acid derivative that is known to inhibit MurD. In contrast, compound 2 had a novel chemical structure but low enzymatic activity. The ease of experimentation and availability of commercial kits for the quantification of ATP hydrolysis make HTS for Mur ligases a viable strategy. Although these assays can be performed in a high throughput manner, they rely on the ATP hydrolysis activity. Hence, they are only amenable to MurC, MurD, MurE and MurF inhibitors. The inhibitors of the other enzymes MurA (transferase) and MurB (reductase) of the Mur pathway cannot be discovered using this strategy. Hu et al. developed an HTS for MurG inhibitors based on the displacement of the fluorescently labelled UDP-GlcNAc substrate from the active site of MurG upon inhibitor binding.179 A library of 64000 small molecules was screened using fluorescence polarization. The displacement of bound fluorescent UDP-GlcNAc in the presence of an inhibitor causes a drop in polarization. After subtracting the molecules with inherent fluorescence, a total of 220 hits were obtained, out of which 55 exhibited an IC50 of 1–10 micromoles in an in vitro assay that monitored the incorporation of radiolabelled UDP-GlcNAc into lipid II. Scaffold analysis observations revealed the presence of 1,3-disubstituted heterocyclic cores in most of the compounds. Further probing of some of the hit molecules represented their selective binding nature to MurG due to rigid structures. Another important HTS assay has been developed by reconstituting the whole pathway in vitro in a one-pot assay.151 The one-pot assay only requires the initial sugar substrate of MurA while the product from the previous reaction acts as the substrate for subsequent enzymes of the pathway. The inhibition of a particular step is confirmed with mass spectrometry to identify the reaction intermediate accumulated. This approach makes it easier to carry out the whole Mur pathway in a single experiment and allows the identification of the inhibitors of the Mur pathway. It also eliminates the labour-intensive process usually required to screen molecules for each enzyme which makes it cost-effective. Feglymycin, a natural peptide, was found to target the MurA and MurC enzymes of E. coli using a one-pot assay.180 Similarly, the Bajpai group reconstructed the in vitro Mtb Mur pathway and evaluated 50 potential MurB and MurE inhibitors obtained from an in silico screening of 684 antitubercular compounds.151,181 Out of these 50 hit molecules, seven compounds showed promising results from the in vitro one-pot assay and were further tested with MD simulation studies. The inhibitory molecules obtained from the one-pot assay have not been validated for their cell-based activity. Therefore, the efficacy of these molecules cannot be commented upon. Taking into consideration the disadvantages associated with the in vitro biochemical assays, it is important to multi-screen the compounds by executing these biochemical assays with virtual screening or a secondary confirmatory test and cell-based assays to rule out false positives. Nonetheless, the significant standing of the widely conserved Mur pathway with its role in sporulation in anaerobic bacteria C. difficile makes the proteins of this pathway propitious drug targets.
Some of the HTS have been performed with purified OM biogenesis proteins like LspA, LptB and MsbA using biochemical assays. Kitamura et al. developed a FRET based assay for HTS of LspA inhibitors.182 A synthetic acylated hexapeptide with an N-terminal quencher and a C-terminal fluorophore was designed to fluoresce upon cleavage by LspA. An HTS for LspA inhibitors was carried out which identified 17 molecules with potent LspA inhibitory activity. With additional medicinal chemistry efforts, the most active molecule against E. coli was discovered which synergized with OM weakening antibiotic derivative polymyxin B nonapeptide (PMBN). Similarly, Gronenberg and Kahne screened a collection of 244 small molecule kinase inhibitors against purified LptB and identified two compounds belonging to two different structural classes.183 The molecules exhibited activity in micromolar range IC50 with purified LptB but lacked cell-based activity against E. coli. Ho et al. adopted a reverse chemical genetic approach to screen compounds that inhibit the ATPase activity of purified E. coli MsbA.184 Being a membrane-bound ATPase, the activity of purified MsbA was reconstituted in amphipol, detergent or nanodiscs. A quinolone compound G592 was obtained from the screen. Further, cell-based on-target inhibitory activity was confirmed by loss of potency (EC50) against a permeability-improved E. coli strain that overexpressed MsbA and whole genome sequencing of resistant mutants of G592. The hyper-permeable E. coli carried a deletion in LptD to improve outer-membrane permeability. G592 was further optimized to improve potency in a medicinal chemistry effort to obtain G247 and G907, which were active against wild-type Enterobacteriaceae. Although these quinolones are active against whole cells, the hydrophobic nature of these compounds led to high serum protein binding and loss of inhibitory activity when tested for MIC in the presence of 50% serum in media.185
These studies emphasize that diverse biochemical/biophysical techniques can be implemented individually or in combination to uncover potent inhibitors of bacterial cell wall proteins. However, these studies may not always result in molecules that show cell-based activity. Hence, it should be noted that such research requires counter cell-based phenotypic screens before moving forward with potent chemical moieties so as to reduce the chances of ending up with inactive inhibitors. Nonetheless, this approach is good in lead optimization investigations to enhance the activity of existing pharmacophores.
Microscopic indications
Rapid acquisition of multiple images has recently been exploited to visualize cellular characteristics (Fig. 10).186 Phenotypic responses of bacteria upon treatment with antibacterials have been studied with high content imaging.187–189 High content imaging has further been found to be helpful in screening of a humanized monoclonal antibody (mAb) against a panel of E. coli clinical isolates.190 The O25b-O antigen in E. coli ST131 is a target of humanized mAb, 3E9-11. A variant of this humanized mAb-KM467 was synthesized and screened against 86 clinical isolates of E. coli ST131 O25b:H4, for a clinical perspective, using a high content imaging screen. The images after antibody binding to the O-antigen were captured on an Opera Phenix High-content screening system analyzed by the Perkin Elmer Harmony software. After image analysis, the phenotypes were divided into four groups based on the binding affinity of KM467. These phenotypes were further assayed to assess these characteristic results, which is out of the scope of the present discussion. Nevertheless, in this context, it should be noted that high content imaging is a viable option to screen large libraries of molecules as well as genomic libraries to expand the activity spectrum of antibacterials using phenotypic signatures.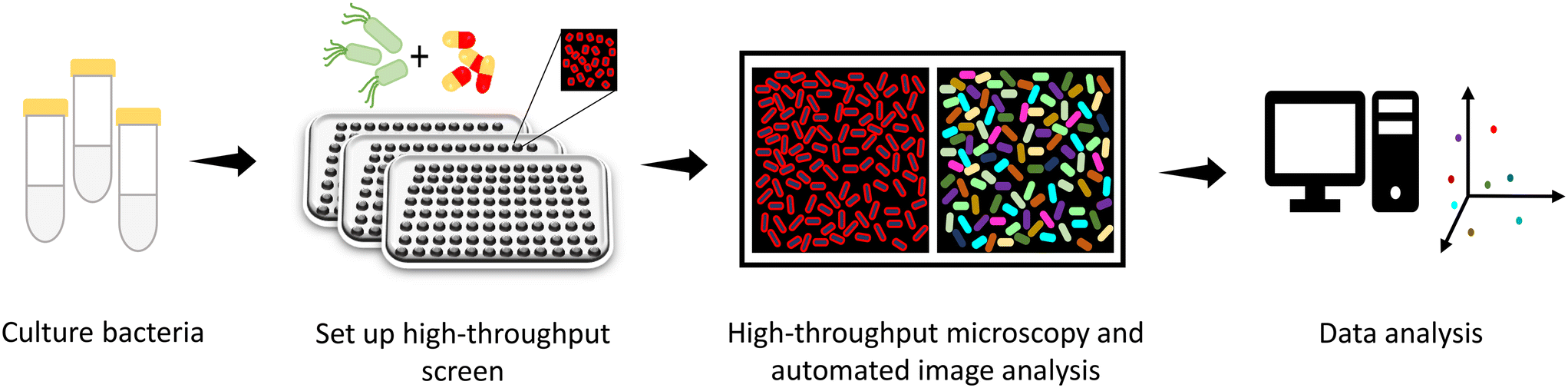 | ||
| Fig. 10 Strategy for screening of antibacterials using high throughput imaging. | ||
Bacterial cytological profiling (BCP) is one of the methods which has empowered the assessment of mechanistic action of different classes of antibiotics by studying their reference cytological profiles.191–193 Each class of antibiotics generates a clustered reference bacterial cytological profile based on the cell shape, area, amount of DNA per cell, etc. obtained by principal component analysis (PCA). The utilization of BCP to identify the signature cytological profile of antibiotics has been developed for several bacterial species. The ability of BCP to successfully differentiate between the MOA of antibiotics belonging to different classes has been further exploited to decipher the MOA of compound NSC145612.193 NSC145612 treated A. baumannii and E. coli cells revealed the presence of decondensed DNA, a hallmark of transcription inhibition. This result was consistent with the BCP fingerprint obtained when cells were treated with rifampicin, a known DNA transcription inhibitor. The molecular target of NSC145612 was further confirmed by whole genome sequencing of the isolated resistant mutant which showed mutations in the rpoB gene (DNA-dependent RNA polymerase subunit B).
In another study, the membrane permeabilizing antibacterial activity of cannabidiol was assessed using BCP based on its ability to uptake SYTOX™ green dye.194 As discussed, BCP quantitatively identifies compounds with a similar MOA, thus revealing the inhibitors of novel targets or multiple targets is incomprehensible using this method.195 An attempt to discover the molecular target of a broad-spectrum SCH-79797 compound using BCP was not successful due to the dual targeting action of the compound on DHFR and membrane integrity. Even though similarities in the phenotypic signatures of SCH-79797 and trimethoprim were not found, a somewhat close BCP of SCH-79797 to membrane-targeting antibiotics was obtained based on the SYTOX dye accumulation. Further investigations with thermal proteome profiling and CRISPRi genetics exemplified the DHFR inhibiting potential of SCH-79797. Subsequently, qualitative image analysis of SCH-79797 treated bacterial cells with nisin–trimethoprim combination treated cells demonstrated their similar BCP profiles. The dual MOA of a single scaffold prevented the resistance development in S. aureus MRSA as opposed to the individual treatments of nisin and trimethoprim which led to rapid resistance development. Dissecting the chemical structure of SCH-79797, two derivatives, IRS-10 and IRS-16, were designed to understand the dual MOA of this scaffold. IRS-10 (or Irresistin-10) is chemically the pyrroloquinazolinediamine core of SCH-79797 lacking isopropylphenyl and cyclopropyl side groups whereas IRS-16 (or Irresistin-16) is essentially a pyrroloquinazolinediamine core with a hydrophobic biphenyl group. The examination of these derivative led to the observation that the pyrroloquinazolinediamine core is responsible for the activity against DHFR while the hydrophobic group altered the membrane permeability and polarity. On the activity front, however, IRS-16 exhibited better antibacterial potential and efficacy in a mouse vaginal infection model.
Another phenotypic profiling platform called MorphEUS (Morphological Evaluation and Understanding of Stress) has been developed by Smith II and colleagues.196 This cytological profiling method also classifies antibacterials by their MOA. Further, they have shown for bedaquiline and moxifloxacin that the platform when complemented with other profiling methods like transcriptomics can unveil off-target effects. Altogether, these studies reveal that morphology profiling using high throughput microscopy can be used as a tool to screen chemical libraries for novel cell wall inhibitors. β-Lactam antibiotics are widely known to modulate the morphology dynamics in bacteria.197–200 Thus, cytological signatures of antibacterials of an unknown pathway of action when compared with cell wall inhibitors can pave the way to screen and discover novel cell wall targeting molecules. HTS for cell wall inhibitor discovery using microscopy has not been performed to date. However, considering the success of high content imaging and cytological profiling methods in MOA studies, HTS strategies can be developed for discovery of novel cell wall inhibitors.
Gene dosage perturbations
Alteration in gene copy numbers by modulating gene dosage has been implicated in identifying chemical–genetic interactions inside the cell.201,202 Changes in the level of target gene expression can reveal the molecular target of the antibacterials as highlighted in Fig. 11. Such changes can be achieved by gain-of-function (GOF) and loss-of-function (LOF) strategies which have been utilized as a tool to elucidate the cellular target of therapeutic agents.203–205 The construction of genome-wide mutant libraries in bacteria has made the task of performing high throughput target screening significantly easier.206–209 The utility of these libraries has also been found to be beneficial in assessing bacterial antibiotic susceptibility signatures and drug interactions.210–212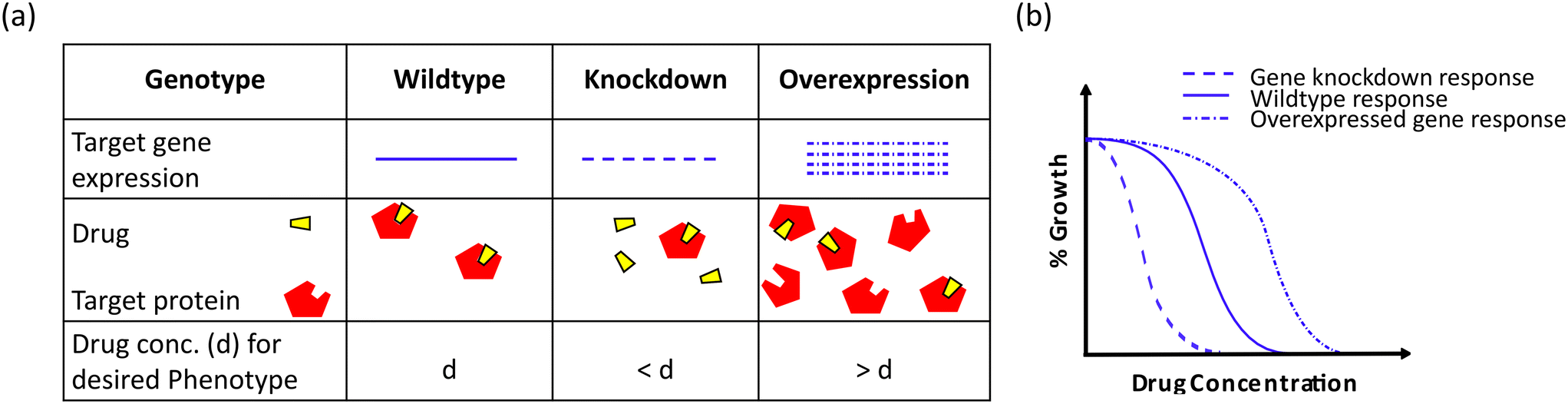 | ||
| Fig. 11 Schematic representation of the effect of gene dosage perturbations on the antibacterial activity of a drug. (a) Changes in the gene dosage of the target protein limit or increase the available binding sites of the respective target. (b) Perturbations in the gene dosage are reflected in the antibacterial efficacy of a drug. Overexpression of a target protein requires a higher number of drug molecules, whereas a lower target protein concentration requires a lower number of drug molecules as compared to the wild-type background. | ||
A multi-copy or high copy number of a gene abrogates the antibacterial potential of a drug and this phenomenon was strategized for the chemical library screening in E. coli which revealed the targets of 2,4-diaminopyrimidine, 2,4-diaminoquinazoline (DHFR) and MAC13243 (LolA).201,213 Similarly, in M. tuberculosis, GSK-710 was found to target decaprenylphosphoryl-β-D-ribose oxidase (Mt-DprE1) by applying the aforementioned multicopy suppression approach.214 Additionally, another functional genomic platform of cosmid- or plasmid-sequencing (Cos-Seq or Plas-Seq) has been beneficial in investigating resistance mechanisms, new targets and the action of novel inhibitors.215 This gene enrichment method couples functional cloning through genomic library transformation in pathogens with next-generation sequencing (NGS). The Cos-Seq is well developed as an HTS strategy for the protozoan parasite Leishmania.216 On the other hand, Plas-Seq has paved the way for elucidating antibacterial drug resistance patterns like ftsI overexpression in ceftriaxone resistance, sidA overexpression in tetracycline and ceftriaxone resistance.217
LOF mutations, as opposed to multicopy suppression, hypersensitize the bacteria to drug toxicity due to depletion in the copies of cognate drug targets and thus enhance the drug efficacy.218 The construction of single gene deletion libraries in various bacterial species has empowered the functional genomics for hypersensitive screens.207,219–222 Target-based whole-cell screening in M. tuberculosis revealed the hypersensitivity of the mutants of pantothenate synthetase (panC) and isocitrate lyase (icl1) to their specific inhibitors.223 However, hypersensitivity screens for essential genes, although successful for yeast models, are rather challenging in bacteria due to their haploid nature.224,225 Consequently, techniques, like CRISPRi, protein degradation tags, antisense RNA and transposon mutagenesis via promoter replacement, have emerged for tunable target gene expression.226 CRISPR interference (CRISPRi) has been a well-established genetic tool for the generation of LOF genome-wide mutants.227,228 This strategy has successfully led to the creation of knockdown libraries in several bacterial species, including E. coli, Streptococcus pneumoniae, and B. subtilis.229–231 The application of CRISPRi on a genome-wide scale enables the study of gene essentiality, bacterial fitness under antibiotic stress and the identification of potential drug targets.232–235 Similar to CRISPRi, the utility of transposon mutagenesis integrated with sequencing techniques (TnSeq) has been harnessed in the generation of LOF mutants.208,236–238 Transposon mutagenesis can be accomplished by random transposon insertions or defined locations in the whole genome to generate mutant libraries.237 Transposon mutagenesis for defined insertions can be carried out through promoter replacement or gene/promoter disruption.239 In the case of essential genes, gene disruption by transposon mutagenesis suffers from the same predicament as knockout single gene deletions for their inability to recover essential gene mutants.240–242 However, variable strength or inducible promoter replacement of essential genes allows the growth of the mutants.243 Therefore, the application of Tn-Seq and CRISPRi on titratable gene expression and probing essential genes have been found to be favourable in screening for therapeutics against essential genes.244 For example, FtsZ was revealed as the target of molecule C109 based on the hypersensitivity profile when screened against a transposon mutant library.245 Transposon mutagenesis has also been applied for gene overexpression by using a transposon cassette with an outward-facing promoter in S. aureus.246 A transposon library of both inactivation and overexpression mutants was created which contained 690000 unique insertions and it was integrated with a complementary machine-learning approach and lipid II was identified as the target of lysocins. Similarly, Gingras et al. employed two whole-genome screening strategies in S. pneumoniae coupled with NGS known as Int-Seq and Mut-Seq and validated both of the approaches in trimethoprim resistance with dhfr mutations.247 The Hung group executed a conditional proteolysis approach for controlled target protein expression levels and created an essential gene-depleted mutant library of M. tuberculosis.248 They fused protein degradation tags to the C-terminal of essential genes and by implementing the PROSPECT (primary screening of strains to prioritize expanded chemistry and targets) approach, they identified inhibitors targeting essential bacterial pathways. Collectively, these genome-wide perturbation screens permit the recognition of potential drug targets, gene functions, conditional essentiality and synergistic drug interactions.249–251
A good example of gene dosage perturbations used in the screening of cell wall inhibitors is the screen against LpxC protein present in the OM. LpxC is an another LPS biosynthesis related protein which has been assayed for novel inhibitors. Considering the metalloenzymatic nature of the protein, Clements et al. screened a library of metal-chelators against a hypersensitive E. coli strain carrying lpxC101 mutation with compromised LpxC activity and increased OM permeability.252 To further confirm LpxC as the target, the antibacterial hits were tested against a purified LpxC enzyme, leading to the identification of sulfonamide derivatives of α-(R)-amino hydroxamic acids – BM-78484 and BM78485. Resistance to BM-78484 was mapped to mutations in the fabZ gene and, to a lesser extent, in the lpxC itself. FabZ is a fatty acid biosynthesis enzyme which shares a common precursor (R-3-hydroxy myristoyl) with LpxA, the first enzyme in LPS biosynthesis. So, the loss in FabZ activity compensates for the loss in LpxC activity by feeding more substrate for LpxC in the LPS biosynthesis.252,253 These derivatives were potent against a wide range of Gram-negative pathogens; however, antibacterial activity against Gram-positive bacterial cells was not observed as expected.
Several inhibitors of LpxC have been discovered with or without HTS to date; however, none have made it to clinics yet. This is attributed to the fact that they are metal chelators (hydroxamate compound) and possess associated side-effects. ACHN-975 is the only candidate drug that entered clinical trials but the trials were halted due to cardiovascular toxicity.254 Recently, a non-hydroxamate molecule TP0586532 was discovered as an LpxC inhibitor with broad-spectrum activity.255 TP0586532 showed efficacy against carbapenem-resistant bacterial pathogens in murine infection models. The absence of hydroxamate in TP0586532 makes it a good LpxC inhibitor drug with minimum toxic effects; however, it has yet to be studied further for potential clinical implications.
Stress response
The importance of cell-based assays in the discovery of cell wall targeting antibacterials has already been implicated in this review. One of the rarely used approaches in screening large chemical libraries for cell wall inhibitors is stress response-related reporter genes. Bacterial biosensors or cell-based reporter assays harness the induction of the gene response under stressful situations.256,257 Various kinds of stresses like the presence of pollutants, toxins, antibiotics, nutrient availability, and change in temperature can be sensed by bacteria, which elicits a response inside the cell by modulating specific gene expressions.258,259 Reporter genes like green fluorescent protein (gfp), beta-galactosidase (lacZ), and luciferase operon (lux) genetically fused downstream to the stress-responsive gene have been used as powerful tools to detect such stress faced by bacterial cells.260–262 This strategy has been developed for DNA-damaging agents by utilizing SOS response-related genes like recA and sulA.263 This detection method using biosensors has also been employed for other antibiotics like tetracyclines, azithromycin, macrolides and other translation inhibitors.264–268 While most of the biosensors have been focused on single-reporter genes, a dual reporter system has also been developed.269 This dual fluorescent protein reporter system, pDualrep2, aims at identifying potential antibacterials that induce the SOS response via DNA damage (red) and those that can cause ribosome stalling (far-red). The red and far-red fluorescent protein genes (rfp and katushka2S) are translationally fused under the control of a sulA promoter and tryptophan attenuator, respectively. Further, screening platforms using reporter genes have been implemented to discover antibacterials. The pDualRep2 system leads to recognize and characterize the mode of action of tetracenomycin and klebsazolicin as protein synthesis inhibitors.270–272 Another HTS for DNA metabolism inhibitors from an AstraZeneca compound library was undertaken using recA-GFP fusion.273 Four phenylcyclohexylacetic acid analogs active against E. coli DNA gyrase were identified from the screen. The effectiveness of the cell reporter assay in HTS for cell wall inhibitors has also been described.274The action of β-lactams using whole-cell biosensors was reported in the study by exploiting the β-lactamase resistance mechanism of the bacterial cell. Valtonen et al. utilized the β-lactam antibiotic responsive element ampC/ampR from Citrobacter freundii.274 The AmpR protein acts as a transcriptional repressor of ampC in the presence of UDP-pentapeptides under normal conditions.275 The accumulation of cell wall breakdown products (muropeptides) due to the antimicrobial action of β-lactam antibiotics and their binding to ampR results in transcriptional activation of ampC by AmpR.276,277 This transcriptional regulation of ampR/ampC was utilized in the construction of the luminescent cell biosensor to assess the β-lactam action. The luciferase operon from Photorhabdus luminescens was used as the reporter under the control of the AmpR inducible ampC promoter. The bioluminescence produced as a result of the ampR/ampC regulation was measured using a luminometer which helped in the analysis of the antibacterial mode of action of cell wall biogenesis inhibitors. It has also been reported that ampC expression is induced in the presence of not only β-lactams but other cell wall active compounds as well due to the presence of several other regulatory proteins.278–281 Therefore, the ampR/ampC cell reporter assay in a high throughput manner resulted in the discovery of sulfonyl piperazine and pyrazole compounds which target LpxH and LolC or LolE, respectively, in the cell wall synthesis machinery in E. coli.282
The σE and Rcs envelope stress responses are also known to be induced upon inhibition of different pathways involved in the biosynthesis of the OM. A screen for molecules which were antibacterial to the efflux deficient hyperpermeable strain and induced a σE stress response identified molecule G0507 (pyrrolopyrimidinedione compound). Resistant mutations to G0507 were mapped to amino acid changes in lolC, lolD and lolE genes. Additionally, mutations were also observed in the lpp gene that encodes Braun’s lipoprotein, the most abundant OM protein in E. coli. Inhibition of the LolCDE complex leads to the accumulation and defective linkage of PG to Braun's lipoprotein trapped in the inner membrane and hence deletion of lpp alleviates the toxic effect of LolCDE inhibition.123 G0507 analogues with modifications in the thiophene ring were assessed and the derivative G0793 showed better antibacterial activity than G0507. The antibacterial potential of these molecules was still weak (MIC = 16 μg ml−1, E. coli MG1655). Furthermore, high resistance development against Lol pathway inhibitors discourages the lead optimization of such molecules. However, novel entities discovered with screening efforts may solve this problem. Another screen based on both σE and Rcs envelope stress systems was conducted to screen for OM permeabilizers using fluorescent reporter assays.283 The mNeonGreen (mNG) reporter protein was translationally fused downstream of the promoters of rpoE and rprA, key regulators of σE and Rcs envelope stress systems, respectively. The plasmid constructs PrpoE-mNG and PrprA-mNG were used in the reporter strains for HTS along with PgroES-mNG (heat shock response) for negative response. From the library of 316953 compounds, 5 molecules showed positive σE and Rcs stress responses. Further experimental validation led to BamA inhibitory activity of two structurally diverse compounds – 2 and 14. However, both of these compounds were toxic against the HEK293 cell line with IC50 values lower than the MIC values against E. coli.
These stress response-based reporter systems offer the advantage of screening large libraries which provide results in a smaller time-frame and simultaneous assessment of cell-based activity. For envelope stress in bacteria, five stress response systems, σE, Bae, Cpx, Rcs and Psp, are present. All these response systems are stimulated when the cell counters any kind of stress which can affect the cell envelope integrity or biogenesis. However, this strategy may not be useful for general stress responsive factors like σs which gets activated on the action of multiple antibiotics. Hence, specific stress response genes should be included in such studies.
Conclusions
Antimicrobial resistance is a well-recognized worldwide health problem. Various antibacterial drug targets like metabolic, nucleic acid or protein biosynthesis inhibitors have been recognized in bacteria. Screening for novel inhibitors that target a cytosolic process is limited by a complex membrane structure that restricts the entry of molecules that otherwise prove active against Gram-positive and Gram-negative bacteria. It is daunting to target Gram-negative than Gram-positive bacteria due to the additional LPS layer in the membrane architecture of the former. Identifying surface-exposed proteins such as LptD and BamA on the bacterial surface as antibacterial targets has opened new possibilities for small molecule inhibitors. These inhibitors do not need to cross the membrane, hence making them unaffected by the permeability and efflux barriers. The proven role of bacterial polymers, LPS and WTA, in bacterial virulence implies their relevance as antibacterial drug targets. The conditional essentiality of some biosynthetic proteins, like those belonging to the rod complex and WTA synthesis pathway, has allowed cell-based screening for pathway-specific inhibitors. As these targets are underexplored and largely unutilized, the developed antibiotics have the potential to evade existing resistance mechanisms in play. The importance of these drug targets combined with the unparalleled demand for novel antibiotics in clinics denotes the need to screen cell wall synthesis inhibitors. Various HTS strategies have been designed and validated to screen large compound libraries against OM and PG proteins. These strategies have utilized both cell-based phenotypic and reverse genetic approaches. However, not all of the aforementioned HTS assays have been applied for screening cell wall inhibitors. One such example is the high throughput microscopic imaging technique. High content imaging platforms have been developed and the success of cytological profiling using microscopy can be implemented to screen novel scaffolds. Similarly, multiple low and high-throughput assays have been developed to probe the activity of MraY.284–286 Still, none have been adopted so far in HTS for novel inhibitors. Several cell-based and biochemical assays have been discussed in this review which can be executed individually or in combination to reveal bacterial cell wall inhibitors, especially for assays based on purified proteins. The purified protein–inhibitor binding is the most utilized screening method for antibacterial molecules which is the hallmark of reverse chemical genetics. Although useful in primary screening, the major hurdle associated with this technique is the lack or uncertainty of cell-based activity of the hit molecules. A key example is the screens with purified Mur enzymes which have identified several inhibitors that were found to be active in vitro but exhibited weak to no antibacterial activity in cell-based assays. The cell-based screens are the most valuable in these scenarios as the inhibitory phenotype is observed in the initial phase of drug discovery. The use of these strategies in combination is beneficial for identification of the molecular target and cell-based active molecules which are the major hurdles faced with forward or reverse chemical genetic approaches, respectively. These approaches are also useful in screens for antibacterials other than cell envelope inhibitors except for pathway-directed screens. Chemical–chemical interaction profiling, gene dosage perturbations, reporter assays and purified protein assays have led to antibacterial molecules active against DNA replication, protein translation, and folate metabolism. The genetic approaches discussed in the review are advantageous in solving the MOA but some disadvantages are also associated with them (Table 2). Further, these screens have not resulted in a clinically approved drug. This may be attributed to the fact that most screens have been recently developed. Though they have led to some novel chemical scaffolds, not all displayed good antibacterial potential and toxicity profiles. Hence, medicinal chemistry efforts are required for development of potent and non-toxic chemical scaffolds. The chemical genetic strategies highlighted in this review offer great potential in the discovery of novel antibacterials. While these help in screening large libraries, validating the drug targets with multiple techniques is always recommended. The discovery of novel cell wall synthesis targeting molecules can help alleviate the existing concern of AMR. In addition to being used as stand-alone drugs, the cell wall targeting drugs can potentiate the antibiotics that alter cytosolic pathways. This allows for the repurposing of drugs that the OM barrier has restrained.| Screening strategy | Inhibitors [target] | Advantages | Disadvantages | Ref. | |
|---|---|---|---|---|---|
| Chemical–chemical interaction profiling | MAC-0038968 [DHFR], MAC-0003199 [DNA gyrase] | Cell-based assay, ease of MOA identification, can be used for multiple essential proteins | Assessment of unique synergies is required as some synergistic interactions may not arise from similar MOA | 120 | |
| Pathway-directed or antagonistic whole cell screen | FtsZ suppression screen for anti-rod system compounds | Compounds 1–8 [MreB] | Cell-based assay, combines forward and reverse genetics, specific for rod complex inhibitors | Counter-screens required as it may result in FtsZ inhibitors, cannot be used for other protein inhibitors | 131 |
| Polymer biosynthesis inhibitor screen | Clomiphene [UppS], MAC-0547630 [UppS] | Cell-based assay, combines forward and reverse genetics, specific for WTA synthesis inhibitors | Counter-screens required | 66 and 294 | |
| Outer membrane disruptor screen | Liproxstatin-1 and MAC-0568743 [LPS], TBT-1 [MsbA] | Cell-based assay, combines forward and reverse genetics, enhances the efficacy of large scaffold antibiotics | Gram-negative specific | 105 and 140 | |
| Purified target protein-molecule binding | Feglimycin [MurA and MurC], TBT-1, G592, G247 and G907 [MsbA] | Broad-spectrum application, easy elucidation of the MOA of novel antibacterials | Hit antibacterials may not have cell-based activity, can only be used for proteins with measurable activity | 180, 184, 185 and 299 | |
| Microscopic indications | KM467 [O25b O-antigen], NSC145612 [RpoB], SCH-79797 [DHFR and membrane integrity] | Narrows down MOA studies | Can only be used with inhibitors that cause morphological alterations, costly, not for molecules with multiple MOAs | 190, 193 and 195 | |
| Gene dosage perturbations | 2,4-Diaminopyrimidine and 2,4-diaminoquinazoline [DHFR], GSK-710 [Mt-DprE1], [PanC], [Icl-1], C109 [FtsZ], lysocins [lipid II] | Cell-based assay, combines forward and reverse genetics, time consuming process | Costly as it requires genome libraries, some genes cannot be used for LOF or GOF studies as they may result in lethal effects | 201, 214, 223, 245 and 246 | |
| Stress response | G0507 [Lol CDE complex], sulfonyl piperazine [LpxH], pyrazole [LolC or LolE], macrolides [MphR promoter activation], ketolides [erm© induction], amicoumacin A [protein synthesis inhibition], 2-guanidino-quinazolines [translation inhibition], tetracenomycin X and klebsazolicin [peptide exit tunnel], phenylcyclohexylacetic acids [DNA gyrase] | Broad-spectrum application, can be applied to multiple stress responsive genes, easy to perform, cost effective | Multiple biological effects may result in a similar stress response | 123, 265–267, 270–273 and 282 | |
Author contributions
R. G., M. S., and R. P. designed the content and flow of the paper. R. G. and M. S. collected the necessary information/data. R. G., M. S., and R. P. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. P. acknowledges the DBT/Wellcome Trust India Alliance Senior Fellowship (Ref. IA/S/21/1/505588). R. G. acknowledges the DBT-JRF fellowship (DBT/2018/IIT-R/1144) received from DBT, Government of India and M. S. acknowledges the Postdoctoral Fellowship received from IIT Roorkee.References
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- E. J. Klemm, V. K. Wong and G. Dougan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12872–12877 CrossRef CAS PubMed.
- R. Cantón and M. I. Morosini, FEMS Microbiol. Rev., 2011, 35, 977–991 CrossRef PubMed.
- V. Dubey, R. Gupta and R. Pathania, Antimicrob. Agents Chemother., 2021, 65 DOI:10.1128/aac.02180-20.
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Front. Microbiol., 2019, 10, 539 CrossRef PubMed.
- E. Peterson and P. Kaur, Front. Microbiol., 2018, 9, 2928 CrossRef PubMed.
- J. M. Munita and C. A. Arias, Microbiol. Spectrum, 2016, 4, 481–511 Search PubMed.
- M. Hutchings, A. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed.
- J. H. Kwon and W. G. Powderly, Science, 2021, 373, 471 CrossRef CAS PubMed.
- C. Llor and L. Bjerrum, Ther. Adv. Drug Saf., 2014, 5, 229–241 CrossRef PubMed.
- M. Miethke, M. Pieroni, T. Weber, M. Brönstrup, P. Hammann, L. Halby, P. B. Arimondo, P. Glaser, B. Aigle, H. B. Bode, R. Moreira, Y. Li, A. Luzhetskyy, M. H. Medema, J. L. Pernodet, M. Stadler, J. R. Tormo, O. Genilloud, A. W. Truman, K. J. Weissman, E. Takano, S. Sabatini, E. Stegmann, H. Brötz-Oesterhelt, W. Wohlleben, M. Seemann, M. Empting, A. K. H. Hirsch, B. Loretz, C. M. Lehr, A. Titz, J. Herrmann, T. Jaeger, S. Alt, T. Hesterkamp, M. Winterhalter, A. Schiefer, K. Pfarr, A. Hoerauf, H. Graz, M. Graz, M. Lindvall, S. Ramurthy, A. Karlén, M. van Dongen, H. Petkovic, A. Keller, F. Peyrane, S. Donadio, L. Fraisse, L. J. V. Piddock, I. H. Gilbert, H. E. Moser and R. Müller, Nat. Rev. Chem., 2021, 5, 726–749 CrossRef CAS PubMed.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackerman, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay and J. J. Collins, Cell, 2020, 180, 688–702 CrossRef CAS PubMed , e13.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- D. Schnappinger, Cold Spring Harbor Perspect. Med., 2015, 5, 1–16 CAS.
- L. Miesel, J. Greene and T. A. Black, Nat. Rev. Genet., 2003, 4, 442–456 CrossRef CAS PubMed.
- B. R. Stockwell, Nat. Rev. Genet., 2000, 1, 116–125 CrossRef CAS PubMed.
- D. E. Williams and R. J. Andersen, Nat. Prod. Rep., 2020, 37, 617–633 RSC.
- M. Kawasumi and P. Nghiem, J. Invest. Dermatol., 2007, 127, 1577–1584 CrossRef CAS PubMed.
- W. Wohlleben, Y. Mast, E. Stegmann and N. Ziemert, Microb. Biotechnol., 2016, 9, 541–548 CrossRef PubMed.
- M. Schenone, V. Dančík, B. K. Wagner and P. A. Clemons, Nat. Chem. Biol., 2013, 9, 232–240 CrossRef CAS PubMed.
- C. Landeta and A. Mejia-Santana, J. Bacteriol., 2022, 204, e00477-21 CrossRef PubMed.
- L. M. Matano, H. G. Morris, B. M. K. Wood, T. C. Meredith and S. Walker, Bioorg. Med. Chem., 2016, 24, 6307–6314 CrossRef CAS PubMed.
- E. Michelini, L. Cevenini, L. Mezzanotte, A. Coppa and A. Roda, Anal. Bioanal. Chem., 2010, 398, 227–238 CrossRef CAS PubMed.
- G. Nierode, P. S. Kwon, J. S. Dordick and S.-J. Kwon, J. Microbiol. Biotechnol., 2016, 26, 213–225 CrossRef CAS PubMed.
- L. R. de Souza Neto, J. T. Moreira-Filho, B. J. Neves, R. L. B. R. Maidana, A. C. R. Guimarães, N. Furnham, C. H. Andrade and F. P. Silva, Front. Chem., 2020, 8, 93 CrossRef PubMed.
- E. M. E. Dokla, N. S. Abutaleb, S. N. Milik, E. A. E. A. Kandil, O. M. Qassem, Y. Elgammal, M. Nasr, M. J. McPhillie, K. A. M. Abouzid, M. N. Seleem, P. Imming and M. Adel, Eur. J. Med. Chem., 2023, 247, 115040 CrossRef CAS PubMed.
- M. Xu, P. Wu, F. Shen, J. Ji and K. P. Rakesh, Bioorg. Chem., 2019, 91, 103133 CrossRef CAS PubMed.
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482–501 CAS.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer, B. Hayete, C. A. Lawrence and J. J. Collins, Cell, 2007, 130, 797–810 CrossRef CAS PubMed.
- S. Kalghatgi, C. S. Spina, J. C. Costello, M. Liesa, J. R. Morones-Ramirez, S. Slomovic, A. Molina, O. S. Shirihai and J. J. Collins, Sci. Transl. Med., 2013, 5, 192ra85 Search PubMed.
- W. Wang and B. Sun, J. Antimicrob. Chemother., 2021, 76, 1712–1723 CrossRef CAS PubMed.
- J. Flores-Kim, G. S. Dobihal, A. Fenton, D. Z. Rudner and T. G. Bernhardt, eLife, 2019, 8, 1–23 CrossRef PubMed.
- J. F. Fisher and S. Mobashery, Protein Sci., 2020, 29, 629–646 CrossRef CAS PubMed.
- J. J. Malin and E. De Leeuw, Infect. Drug Resist., 2019, 12, 2613–2625 CrossRef CAS PubMed.
- T. Schneider and H. G. Sahl, Int. J. Med. Microbiol., 2010, 300, 161–169 CrossRef CAS PubMed.
- R. Bauer and L. M. T. Dicks, Int. J. Food Microbiol., 2005, 101, 201–216 CrossRef CAS PubMed.
- O. McAuliffe, R. P. Ross and C. Hill, FEMS Microbiol. Rev., 2001, 25, 285–308 CrossRef CAS PubMed.
- D. Field, P. D. Cotter, R. P. Ross and C. Hill, Bioengineered, 2015, 6, 187–192 CrossRef CAS PubMed.
- A. Prince, P. Sandhu, P. Kumar, E. Dash, S. Sharma, M. Arakha, S. Jha, Y. Akhter and M. Saleem, Sci. Rep., 2016, 6, 1–15 CrossRef PubMed.
- R. Dickman, S. A. Mitchell, A. M. Figueiredo, D. F. Hansen and A. B. Tabor, J. Org. Chem., 2019, 84, 11493–11512 CrossRef CAS PubMed.
- G. Bierbaum and H.-G. Sahl, Curr. Pharm. Biotechnol., 2009, 10, 2–18 CAS.
- D. N. Chernyshova, A. A. Tyulin, O. S. Ostroumova and S. S. Efimova, Membranes, 2022, 12, 1166 CrossRef CAS PubMed.
- R. Pokhrel, N. Bhattarai, P. Baral, B. S. Gerstman, J. H. Park, M. Handfield and P. P. Chapagain, Phys. Chem. Chem. Phys., 2019, 21, 12530–12539 RSC.
- R. Shukla, F. Lavore, S. Maity, M. G. N. Derks, C. R. Jones, B. J. A. Vermeulen, A. Melcrová, M. A. Morris, L. M. Becker, X. Wang, R. Kumar, J. Medeiros-Silva, R. A. M. van Beekveld, A. M. J. J. Bonvin, J. H. Lorent, M. Lelli, J. S. Nowick, H. D. MacGillavry, A. J. Peoples, A. L. Spoering, L. L. Ling, D. E. Hughes, W. H. Roos, E. Breukink, K. Lewis and M. Weingarth, Nature, 2022, 608, 390–396 CrossRef CAS PubMed.
- K. M. Varney, A. M. J. J. Bonvin, M. Pazgier, J. Malin, W. Yu, E. Ateh, T. Oashi, W. Lu, J. Huang, M. Diepeveen-de Buin, J. Bryant, E. Breukink, A. D. MacKerell and E. P. H. de Leeuw, PLoS Pathog., 2013, 9, e1003732 CrossRef PubMed.
- T. Touzé, H. Barreteau, M. El Ghachi, A. Bouhss, A. Barnéoud-Arnoulet, D. Patin, E. Sacco, D. Blanot, M. Arthur, D. Duché, R. Lloubès and D. Mengin-Lecreulx, Biochem. Soc. Trans., 2012, 40, 1522–1527 CrossRef PubMed.
- E. Breukink and B. de Kruijff, Nat. Rev. Drug Discovery, 2006, 5, 321–323 CrossRef CAS PubMed.
- H. Piepenbreier, A. Diehl and G. Fritz, Nat. Commun., 2019, 10, 2733 CrossRef PubMed.
- D. J. Scheffers and M. B. Tol, PLoS Pathog., 2015, 11, 1–12 Search PubMed.
- M. Saini, A. Gaurav, A. Kothari, B. J. Omar, V. Gupta, A. Bhattacharjee and R. Pathania, ACS Infect. Dis., 2023, 9, 692–705 CrossRef CAS PubMed.
- W. R. Miller, A. S. Bayer and C. A. Arias, Cold Spring Harbor Perspect. Med., 2016, 6, a026997 CrossRef PubMed.
- E. V. K. Ledger, A. Sabnis and A. M. Edwards, Microbiology, 2022, 168, 001136 CrossRef CAS PubMed.
- J. Zhou, Y. Cai, Y. Liu, H. An, K. Deng, M. A. Ashraf, L. Zou and J. Wang, Front. Microbiol., 2022, 13, 952633 CrossRef PubMed.
- P. Nordmann, L. Dortet and L. Poirel, Trends Mol. Med., 2012, 18, 263–272 CrossRef CAS PubMed.
- G. W. Langley, R. Cain, J. M. Tyrrell, P. Hinchliffe, K. Calvopiña, C. L. Tooke, E. Widlake, C. G. Dowson, J. Spencer, T. R. Walsh, C. J. Schofield and J. Brem, Bioorg. Med. Chem. Lett., 2019, 29, 1981–1984 CrossRef CAS PubMed.
- S. J. Hecker, K. R. Reddy, O. Lomovskaya, D. C. Griffith, D. Rubio-Aparicio, K. Nelson, R. Tsivkovski, D. Sun, M. Sabet, Z. Tarazi, J. Parkinson, M. Totrov, S. H. Boyer, T. W. Glinka, O. A. Pemberton, Y. Chen and M. N. Dudley, J. Med. Chem., 2020, 63, 7491–7507 CrossRef CAS PubMed.
- G. Dhanda, P. Sarkar, S. Samaddar and J. Haldar, J. Med. Chem., 2019, 62, 3184–3205 CrossRef CAS PubMed.
- M. L. Foucault, P. Courvalin and C. Grillot-Courvalin, Antimicrob. Agents Chemother., 2009, 53, 2354–2359 CrossRef CAS PubMed.
- D. C. Marciano, O. Y. Karkouti and T. Palzkill, Genetics, 2007, 176, 2381–2392 CrossRef CAS PubMed.
- Key reference: S. Zhao, J. W. Adamiak, V. Bonifay, J. Mehla, H. I. Zgurskaya and D. S. Tan, Nat. Chem. Biol., 2020, 16, 1293–1302 CrossRef CAS PubMed.
- A. D. Radkov, Y. P. Hsu, G. Booher and M. S. Vannieuwenhze, Annu. Rev. Biochem., 2018, 87, 991–1014 CrossRef CAS PubMed.
- S. Batson, C. De Chiara, V. Majce, A. J. Lloyd, S. Gobec, D. Rea, V. Fülöp, C. W. Thoroughgood, K. J. Simmons, C. G. Dowson, C. W. G. Fishwick, L. P. S. De Carvalho and D. I. Roper, Nat. Commun., 2017, 8, 1–7 CrossRef CAS PubMed.
- M. El Ghachi, N. Howe, C. Y. Huang, V. Olieric, R. Warshamanage, T. Touzé, D. Weichert, P. J. Stansfeld, M. Wang, F. Kerff and M. Caffrey, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- Key reference: M. A. Farha, T. L. Czarny, C. L. Myers, L. J. Worrall, S. French, D. G. Conrady, Y. Wang, E. Oldfield, N. C. J. Strynadka and E. D. Brown, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11048–11053 CrossRef CAS PubMed.
- E. H. Mashalidis, B. Kaeser, Y. Terasawa, A. Katsuyama, D. Y. Kwon, K. Lee, J. Hong, S. Ichikawa and S. Y. Lee, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- K. J. Hetrick and W. A. van der Donk, Curr. Opin. Chem. Biol., 2017, 38, 36–44 CrossRef CAS PubMed.
- I. Wiedemann, T. Böttiger, R. R. Bonelli, T. Schneider, H. G. Sahl and B. Martínez, Appl. Environ. Microbiol., 2006, 72, 2809–2814 CrossRef CAS PubMed.
- V. M. Hernández-Rocamora, N. Baranova, K. Peters, E. Breukink, M. Loose and W. Vollmer, eLife, 2021, 10, 1–79 CrossRef PubMed.
- O. Kocaoglu, H. C. T. Tsui, M. E. Winkler and E. E. Carlson, Antimicrob. Agents Chemother., 2015, 59, 3548–3555 CrossRef CAS PubMed.
- D. S. Sutaria, B. Moya, K. B. Green, T. H. Kim, X. Tao, Y. Jiao, A. Louie, G. L. Drusano and J. B. Bulitta, Antimicrob. Agents Chemother., 2018, 62 DOI:10.1128/aac.00282-18.
- A. Zapun, C. Contreras-Martel and T. Vernet, FEMS Microbiol. Rev., 2008, 32, 361–385 CrossRef CAS PubMed.
- C. Urban, E. Go, N. Mariano and J. J. Rahal, FEMS Microbiol. Lett., 1995, 125, 193–197 CrossRef CAS.
- D. Bellini, L. Koekemoer, H. Newman and C. G. Dowson, J. Mol. Biol., 2019, 431, 3501–3519 CrossRef CAS PubMed.
- S. Bobba and W. G. Gutheil, Int. J. Biochem. Cell Biol., 2011, 43, 1490–1499 CrossRef CAS PubMed.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS PubMed.
- M. Haenni and P. Moreillon, Antimicrob. Agents Chemother., 2006, 50, 4053–4061 CrossRef CAS PubMed.
- R. Maya-Martinez, J. A. N. Alexander, C. F. Otten, I. Ayala, D. Vollmer, J. Gray, C. M. Bougault, A. Burt, C. Laguri, M. Fonvielle, M. Arthur, N. C. J. Strynadka, W. Vollmer and J. P. Simorre, Front. Microbiol., 2019, 10, 3223 CrossRef PubMed.
- D. A. Dik, J. F. Fisher and S. Mobashery, Chem. Rev., 2018, 118, 5952–5984 CrossRef CAS PubMed.
- M. Mora-Ochomogo and C. T. Lohans, RSC Med. Chem., 2021, 12, 1623–1639 RSC.
- G. A. Jacoby, Clin. Microbiol. Rev., 2009, 22, 161–182 CrossRef CAS PubMed.
- R. Ago and D. Shiomi, AIMS Microbiol., 2019, 5, 358–367 CAS.
- X. Liu, J. Biboy, E. Consoli, W. Vollmer and T. den Blaauwen, PLoS Genet., 2020, 16, e1009276 CrossRef CAS PubMed.
- D. Shiomi, M. Sakai and H. Niki, EMBO J., 2008, 27, 3081–3091 CrossRef CAS PubMed.
- H. Li and T. Gao, Microbiol. Res., 2021, 243, 126655 CrossRef CAS PubMed.
- A. Vigouroux, B. Cordier, A. Aristov, L. Alvarez, G. Özbaykal, T. Chaze, E. R. Oldewurtel, M. Matondo, F. Cava, D. Bikard and S. van Teeffelen, eLife, 2020, 9, e51998 CrossRef CAS PubMed.
- S. Tamaki, H. Matsuzawa and M. Matsuhashi, J. Bacteriol., 1980, 141, 52–57 CrossRef CAS PubMed.
- G. Özbaykal, E. Wollrab, F. Simon, A. Vigouroux, B. Cordier, A. Aristov, T. Chaze, M. Matondo and S. van Teeffelen, eLife, 2020, 9, e50629 CrossRef PubMed.
- N. Levy, J. M. Bruneau, E. Le Rouzic, D. Bonnard, F. Le Strat, A. Caravano, F. Chevreuil, J. Barbion, S. Chasset, B. Ledoussal, F. Moreau and M. Ruff, J. Med. Chem., 2019, 62, 4742–4754 CrossRef CAS PubMed.
- A. J. Meeske, E. P. Riley, W. P. Robins, T. Uehara, J. J. Mekalanos, D. Kahne, S. Walker, A. C. Kruse, T. G. Bernhardt and D. Z. Rudner, Nature, 2016, 537, 634–638 CrossRef CAS PubMed.
- F. O. Bendezú, C. A. Hale, T. G. Bernhardt and P. A. J. De Boer, EMBO J., 2009, 28, 193–204 CrossRef PubMed.
- P. Nurse and K. J. Marians, J. Biol. Chem., 2013, 288, 3469–3475 CrossRef CAS PubMed.
- S. van Teeffelen, S. Wang, L. Furchtgott, K. C. Huang, N. S. Wingreen, J. W. Shaevitz and Z. Gitai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15822–15827 CrossRef CAS PubMed.
- A. Martins, C. Contreras-Martel, M. Janet-Maitre, M. M. Miyachiro, L. F. Estrozi, D. M. Trindade, C. C. Malospirito, F. Rodrigues-Costa, L. Imbert, V. Job, G. Schoehn, I. Attrée and A. Dessen, Nat. Commun., 2021, 12, 2987 CrossRef CAS PubMed.
- J. V. Cox, Y. M. Abdelrahman and S. P. Ouellette, Sci. Rep., 2020, 10, 12588 CrossRef PubMed.
- B. Å. Hansen, N. Grude, M. Lindbæk and T. Stenstad, BMC Infect. Dis., 2022, 22, 478 CrossRef PubMed.
- E. Awuni, Front. Chem., 2020, 7 Search PubMed.
- A. Konovalova, D. E. Kahne and T. J. Silhavy, Annu. Rev. Microbiol., 2017, 71, 539–556 CrossRef CAS PubMed.
- M. G. Ghequire, T. Swings, J. Michiels, S. K. Buchanan and R. De Mot, mBio, 2018, 9 DOI:10.1128/mbio.02138-17.
- K. M. Storek, M. R. Auerbach, H. Shi, N. K. Garcia, D. Sun, N. N. Nickerson, R. Vij, Z. Lin, N. Chiang, K. Schneider, A. T. Wecksler, E. Skippington, G. Nakamura, D. Seshasayee, J. T. Koerber, J. Payandeh, P. A. Smith and S. T. Rutherford, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3692–3697 CrossRef CAS PubMed.
- Key reference: E. M. Hart, A. M. Mitchell, A. Konovalova, M. Grabowicz, J. Sheng, X. Han, F. P. Rodriguez-Rivera, A. G. Schwaid, J. C. Malinverni, C. J. Balibar, S. Bodea, Q. Si, H. Wang, M. F. Homsher, R. E. Painter, A. K. Ogawa, H. Sutterlin, T. Roemer, T. A. Black, D. M. Rothman, S. S. Walker and T. J. Silhavy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21748–21757 CrossRef CAS PubMed.
- K. Lehman, H. Smith and M. Grabowicz, mBio, 2022, 13, 1–16 CrossRef CAS PubMed.
- M. T. Nguyen and F. Götz, Microbiol. Mol. Biol. Rev., 2016, 80, 891–903 CrossRef CAS PubMed.
- Key reference: G. Zhang, V. Baidin, K. S. Pahil, E. Moison, D. Tomasek, N. S. Ramadoss, A. K. Chatterjee, C. W. McNamara, T. S. Young, P. G. Schultz, T. C. Meredith and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6834–6839 CrossRef CAS PubMed.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van Der Meijden, F. Bernardini, A. Lederer, R. L. A. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Käch, L. Eberl, K. Riedel, S. J. Demarco and J. A. Robinson, Science, 2010, 327, 1010–1013 CrossRef CAS PubMed.
- N. K. Prasad, I. B. Seiple, R. T. Cirz and O. S. Rosenberg, Antimicrob. Agents Chemother., 2022, 66, e00054-22 CrossRef PubMed.
- J. G. Swoboda, J. Campbell, T. C. Meredith and S. Walker, ChemBioChem, 2010, 11, 35–45 CrossRef CAS PubMed.
- M. A. Farha, A. Leung, E. W. Sewell, M. A. D'Elia, S. E. Allison, L. Ejim, P. M. Pereira, M. G. Pinho, G. D. Wright and E. D. Brown, ACS Chem. Biol., 2013, 8, 226–233 CrossRef CAS PubMed.
- J. Campbell, A. K. Singh, J. P. Santa Maria, Y. Kim, S. Brown, J. G. Swoboda, E. Mylonakis, B. J. Wilkinson and S. Walker, ACS Chem. Biol., 2011, 6, 106–116 CrossRef CAS PubMed.
- T. Dörr, P. J. Moynihan and C. Mayer, Front. Microbiol., 2019, 10, 2051 CrossRef PubMed.
- P. Sarkar, V. Yarlagadda, C. Ghosh and J. Haldar, MedChemComm, 2017, 8, 516–533 RSC.
- A. Maitra, T. Munshi, J. Healy, L. T. Martin, W. Vollmer, N. H. Keep and S. Bhakta, FEMS Microbiol. Rev., 2019, 43, 548–575 CrossRef CAS PubMed.
- F. Ruggiu, S. Yang, R. L. Simmons, A. Casarez, A. K. Jones, C. Li, J. M. Jansen, H. E. Moser, C. R. Dean, F. Reck and M. Lindvall, ACS Infect. Dis., 2019, 5, 1688–1692 CrossRef CAS PubMed.
- D. A. C. Heesterbeek, N. I. Martin, A. Velthuizen, M. Duijst, M. Ruyken, R. Wubbolts, S. H. M. Rooijakkers and B. W. Bardoel, Sci. Rep., 2019, 9, 3074 CrossRef CAS PubMed.
- A. Kaushik, N. C. Ammerman, N. M. Parrish and E. L. Nuermberger, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.00733-19.
- V. Dubey, K. Devnath, V. K. Gupta, G. Kalyan, M. Singh, A. Kothari, B. J. Omar and R. Pathania, J. Antimicrob. Chemother., 2022, 77, 1313–1323 CrossRef CAS PubMed.
- M. Shaku, C. Ealand and B. D. Kana, Front. Cell. Infect. Microbiol., 2020, 10, 603382 CrossRef CAS PubMed.
- N. Thorne, D. S. Auld and J. Inglese, Curr. Opin. Chem. Biol., 2010, 14, 315–324 CrossRef CAS PubMed.
- Key reference: M. A. Farha and E. D. Brown, Chem. Biol., 2010, 17, 852–862 CrossRef CAS PubMed.
- Key reference: P. A. Mann, A. Müller, L. Xiao, P. M. Pereira, C. Yang, S. Ho Lee, H. Wang, J. Trzeciak, J. Schneeweis, M. M. Dos Santos, N. Murgolo, X. She, C. Gill, C. J. Balibar, M. Labroli, J. Su, A. Flattery, B. Sherborne, R. Maier, C. M. Tan, T. Black, K. Önder, S. Kargman, F. J. Monsma, M. G. Pinho, T. Schneider and T. Roemer, ACS Chem. Biol., 2013, 8, 2442–2451 CrossRef CAS PubMed.
- J. Huber, R. G. K. Donald, S. H. Lee, L. W. Jarantow, M. J. Salvatore, X. Meng, R. Painter, R. H. Onishi, J. Occi, K. Dorso, K. Young, Y. W. Park, S. Skwish, M. J. Szymonifka, T. S. Waddell, L. Miesel, J. W. Phillips and T. Roemer, Chem. Biol., 2009, 16, 837–848 CrossRef CAS PubMed.
- Key reference: N. N. Nickerson, C. C. Jao, Y. Xu, J. Quinn, E. Skippington, K. Alexander, A. Miu, N. Skelton, J. V. Hankins, M. S. Lopez, C. M. Koth and S. Rutherford, Antimicrob. Agents Chemother., 2018, 62, 10–1128 CrossRef PubMed.
- S. M. McLeod, P. R. Fleming, K. MacCormack, R. E. McLaughlin, J. D. Whiteaker, S. ichiro Narita, M. Mori, H. Tokuda and A. A. Miller, J. Bacteriol., 2015, 197, 1075–1082 CrossRef CAS PubMed.
- A. Sugimoto, A. Maeda, K. Itto and H. Arimoto, Sci. Rep., 2017, 7, 1129 CrossRef PubMed.
- Z. Gitai, N. A. Dye, A. Reisenauer, M. Wachi and L. Shapiro, Cell, 2005, 120, 329–341 CrossRef CAS PubMed.
- J. P. Hegarty and D. B. Stewart, Appl. Microbiol. Biotechnol., 2018, 102, 1055–1065 CrossRef CAS PubMed.
- F. O. Bendezú and P. A. J. De Boer, J. Bacteriol., 2008, 190, 1792–1811 CrossRef PubMed.
- G. C. Lai, H. Cho and T. G. Bernhardt, PLoS Genet., 2017, 13, e1006934 CrossRef PubMed.
- J. Wagstaff and J. Löwe, Nat. Rev. Microbiol., 2018, 16, 187–201 CrossRef CAS PubMed.
- Key reference: J. A. Buss, V. Baidin, M. A. Welsh, J. Flores-Kim, H. Cho, B. Mckay Wood, T. Uehara, S. Walker, D. Kahne and T. G. Bernhardt, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.01530-18.
- Key reference: H. Cho, T. Uehara and T. G. Bernhardt, Cell, 2014, 159, 1300–1311 CrossRef CAS PubMed.
- Key reference: M. A. Lobritz, I. W. Andrews, D. Braff, C. B. M. Porter, A. Gutierrez, Y. Furuta, L. B. G. Cortes, T. Ferrante, S. C. Bening, F. Wong, C. Gruber, C. W. Bakerlee, G. Lambert, G. C. Walker, D. J. Dwyer and J. J. Collins, Cell Chem. Biol., 2022, 29, 276–286 CrossRef CAS PubMed , e4.
- Key reference: J. G. Swoboda, T. C. Meredith, J. Campbell, S. Brown, T. Suzuki, T. Bollenbach, A. J. Malhowski, R. Kishony, M. S. Gilmore and S. Walker, ACS Chem. Biol., 2009, 4, 875–883 CrossRef CAS PubMed.
- K. Yamamoto, N. Yamamoto, S. Ayukawa, Y. Yasutake, K. Ishiya and N. Nakashima, Sci. Rep., 2022, 12, 5609 CrossRef CAS PubMed.
- C. Muheim, H. Götzke, A. U. Eriksson, S. Lindberg, I. Lauritsen, M. H. H. Nørholm and D. O. Daley, Sci. Rep., 2017, 7, 1–11 CrossRef CAS PubMed.
- E. Lei, H. Tao, S. Jiao, A. Yang, Y. Zhou, M. Wang, K. Wen, Y. Wang, Z. Chen, X. Chen, J. Song, C. Zhou, W. Huang, L. Xu, D. Guan, C. Tan, H. Liu, Q. Cai, K. Zhou, J. Modica, S. Y. Huang, W. Huang and X. Feng, J. Am. Chem. Soc., 2022, 144, 10622–10639 CrossRef CAS PubMed.
- C. M. J. Wesseling and N. I. Martin, ACS Infect. Dis., 2022, 8, 1731–1757 CrossRef CAS PubMed.
- Q. Li, R. Cebrián, M. Montalbán-López, H. Ren, W. Wu and O. P. Kuipers, Commun. Biol., 2021, 4, 1–11 CrossRef PubMed.
- Key reference: K. Klobucar, J. P. Côté, S. French, L. Borrillo, A. B. Y. Guo, M. H. Serrano-Wu, K. K. Lee, B. Hubbard, J. W. Johnson, J. L. Gaulin, J. Magolan, D. T. Hung and E. D. Brown, ACS Chem. Biol., 2021, 16, 929–942 CrossRef CAS PubMed.
- Key reference: J. M. Stokes, S. French, O. G. Ovchinnikova, C. Bouwman, C. Whitfield and E. D. Brown, Cell Chem. Biol., 2016, 23, 267–277 CrossRef CAS PubMed.
- Key reference: N. Wade, C. M. J. Wesseling, P. Innocenti, C. J. Slingerland, G. M. Koningstein, J. Luirink and N. I. Martin, ACS Infect. Dis., 2022, 8, 2242–2252 CrossRef CAS PubMed.
- L. Damier-Piolle, S. Magnet, S. Brémont, T. Lambert and P. Courvalin, Antimicrob. Agents Chemother., 2008, 52, 557–562 CrossRef CAS PubMed.
- S. Coyne, N. Rosenfeld, T. Lambert, P. Courvalin and B. Périchon, Antimicrob. Agents Chemother., 2010, 54, 4389–4393 CrossRef CAS PubMed.
- S. Magnet, P. Courvalin and T. Lambert, Society, 2001, 45, 3375–3380 CAS.
- J. I. Montgomery, M. F. Brown, U. Reilly, L. M. Price, J. A. Abramite, J. Arcari, R. Barham, Y. Che, J. M. Chen, S. W. Chung, E. M. Collantes, C. Desbonnet, M. Doroski, J. Doty, J. J. Engtrakul, T. M. Harris, M. Huband, J. D. Knafels, K. L. Leach, S. Liu, A. Marfat, L. McAllister, E. McElroy, C. A. Menard, M. Mitton-Fry, L. Mullins, M. C. Noe, J. O'Donnell, R. Oliver, J. Penzien, M. Plummer, V. Shanmugasundaram, C. Thoma, A. P. Tomaras, D. P. Uccello, A. Vaz and D. G. Wishka, J. Med. Chem., 2012, 55, 1662–1670 CrossRef CAS PubMed.
- S. O. Oselusi, A. O. Fadaka, G. J. Wyckoff and S. A. Egieyeh, ACS Omega, 2022, 7, 37896–37906 CrossRef CAS PubMed.
- M. Masumi, F. Noormohammadi, F. Kianisaba, F. Nouri, M. Taheri and A. Taherkhani, Int. J. Microbiol., 2022, 2022, 9130700 Search PubMed.
- G. P. Volynets, F. Barthels, S. J. Hammerschmidt, O. V. Moshynets, S. S. Lukashov, S. A. Starosyla, H. V. Vyshniakova, O. S. Iungin, V. G. Bdzhola, A. O. Prykhod'ko, A. R. Syniugin, V. M. Sapelkin, S. M. Yarmoluk and T. Schirmeister, J. Antibiot., 2022, 75, 321–332 CrossRef CAS PubMed.
- M. Winn, R. J. M. Goss, K. I. Kimura and T. D. H. Bugg, Nat. Prod. Rep., 2010, 27, 279–304 RSC.
- Key reference: K. Eniyan, A. Kumar, G. V. Rayasam, A. Perdih and U. Bajpai, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- S. A. Cochrane and C. T. Lohans, Eur. J. Med. Chem., 2020, 194, 112262 CrossRef CAS PubMed.
- G. Zhao, T. I. Meier, S. D. Kahl, K. R. Gee and L. C. Blaszczak, Antimicrob. Agents Chemother., 1999, 43, 1124–1128 CrossRef CAS PubMed.
- G. Zhao, T. I. Meier, J. Hoskins and K. A. Mcallister, Antimicrob. Agents Chemother., 2000, 44, 1745–1748 CrossRef CAS PubMed.
- G. Kumar, C. Galanis, H. R. Batchelder, C. A. Townsend and G. Lamichhane, mSphere, 2022, 7, e00039-22 CrossRef PubMed.
- M. Park, J. B. Sutherland and F. Rafii, Anaerobe, 2020, 62, 102179 CrossRef CAS PubMed.
- K. R. Gee, H. C. Kang, T. I. Meier, G. Zhao and L. C. Blaszcak, Electrophoresis, 2001, 22, 960–965 CrossRef CAS PubMed.
- A. Boes, S. Olatunji, T. Mohammadi, E. Breukink and M. Terrak, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed.
- T. Jarzembowski, K. Wiśniewska, A. Józwik, E. Bryl and J. Witkowski, Curr. Microbiol., 2008, 57, 167–169 CrossRef CAS PubMed.
- E. Lacasse, E. Brouillette, A. Larose, T. R. Parr and A. Rubio, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.02181-18.
- Key reference: M. Stefanova, S. Bobba and W. G. Gutheil, Annu. Rev. Biochem., 2010, 396, 164–166 CrossRef CAS PubMed.
- Key reference: X. Huang and A. Aulabaugh, Methods Mol. Biol., 2009, 565, 127–143 CrossRef CAS PubMed.
- Key reference: A. Fedarovich, K. A. Djordjevic, S. M. Swanson, Y. K. Peterson, R. A. Nicholas and C. Davies, PLoS One, 2012, 7, e44918 CrossRef CAS PubMed.
- Key reference: A. B. Shapiro, N. Gao, R. F. Gu and J. Thresher, Anal. Biochem., 2014, 463, 15–22 CrossRef CAS PubMed.
- Key reference: A. B. Shapiro, R. F. Gu, N. Gao, S. Livchak and J. Thresher, Anal. Biochem., 2013, 439, 37–43 CrossRef CAS PubMed.
- Key reference: O. Kocaoglu and E. E. Carlson, Curr. Protoc. Chem. Biol., 2013, 5, 239–250 CrossRef PubMed.
- S. Sharifzadeh, F. Dempwolff, D. B. Kearns and E. E. Carlson, ACS Chem. Biol., 2020, 15, 1242–1251 CrossRef CAS PubMed.
- Key reference: A. López-Pérez, S. Freischem, I. Grimm, O. Weiergräber, A. J. Dingley, H. Waldmann, W. Vollmer, K. Kumar and C. Vuong, Antibiotics, 2021, 10, 529 CrossRef PubMed.
- Key reference: E. Pagliero, L. Chesnel, J. Hopkins, J. Croizé, O. Dideberg, T. Vernet and A. M. Di Guilmi, Antimicrob. Agents Chemother., 2004, 48, 1848–1855 CrossRef CAS PubMed.
- Key reference: S. H. Huang, W. S. Wu, L. Y. Huang, W. F. Huang, W. C. Fu, P. T. Chen, J. M. Fang, W. C. Cheng, T. J. R. Cheng and C. H. Wong, J. Am. Chem. Soc., 2013, 135, 17078–17089 CrossRef CAS PubMed.
- M. Sapkota, R. K. R. Marreddy, X. Wu, M. Kumar and J. G. Hurdle, Anaerobe, 2020, 61, 102129 CrossRef CAS PubMed.
- F. Laddomada, M. M. Miyachiro, M. Jessop, D. Patin, V. Job, D. Mengin-Lecreulx, A. Le Roy, C. Ebel, C. Breyton, I. Gutsche and A. Dessen, Sci. Rep., 2019, 9, 1–17 CrossRef CAS PubMed.
- M. Hrast, I. Sosič, R. Šink and S. Gobec, Bioorg. Chem., 2014, 55, 2–15 CrossRef CAS PubMed.
- M. A. Azam and S. Jupudi, Chem. Pap., 2020, 74, 1697–1708 CrossRef CAS.
- N. Saha and M. A. Azam, J. Inclusion Phenom. Macrocyclic Chem., 2020, 98, 127–136 CrossRef CAS.
- T. Munshi, A. Gupta, D. Evangelopoulos, J. D. Guzman, S. Gibbons, N. H. Keep and S. Bhakta, PLoS One, 2013, 8, 1–12 Search PubMed.
- Key reference: S. Turk, A. Kovač, A. Boniface, J. M. Bostock, I. Chopra, D. Blanot and S. Gobec, Bioorg. Med. Chem., 2009, 17, 1884–1889 CrossRef CAS PubMed.
- Key reference: K. Kristan, M. Kotnik, M. Oblak and U. Urleb, J. Biomol. Screening, 2009, 14, 412–418 CrossRef CAS PubMed.
- Key reference: Y. Hu, J. S. Helm, L. Chen, C. Ginsberg, B. Gross, B. Kraybill, K. Tiyanont, X. Fang, T. Wu and S. Walker, Chem. Biol., 2004, 11, 703–711 CrossRef CAS PubMed.
- S. Rausch, A. Hänchen, A. Denisiuk, M. Löhken, T. Schneider and R. D. Süssmuth, ChemBioChem, 2011, 12, 1171–1173 CrossRef CAS PubMed.
- Key reference: K. Eniyan, J. Rani, S. Ramachandran, R. Bhat, I. A. Khan and U. Bajpai, SLAS Discovery, 2020, 25, 70–78 CrossRef CAS PubMed.
- Key reference: S. Kitamura, A. Owensby, D. Wall and D. W. Wolan, Cell Chem. Biol., 2018, 25, 301–308 CrossRef CAS PubMed , e12.
- Key reference: L. S. Gronenberg and D. Kahne, J. Am. Chem. Soc., 2010, 132, 2518–2519 CrossRef CAS PubMed.
- Key reference: H. Ho, A. Miu, M. K. Alexander, N. K. Garcia, A. Oh, I. Zilberleyb, M. Reichelt, C. D. Austin, C. Tam, S. Shriver, H. Hu, S. S. Labadie, J. Liang, L. Wang, J. Wang, Y. Lu, H. E. Purkey, J. Quinn, Y. Franke, K. Clark, M. H. Beresini, M. W. Tan, B. D. Sellers, T. Maurer, M. F. T. Koehler, A. T. Wecksler, J. R. Kiefer, V. Verma, Y. Xu, M. Nishiyama, J. Payandeh and C. M. Koth, Nature, 2018, 557, 196–201 CrossRef CAS PubMed.
- Key reference: M. K. Alexander, A. Miu, A. Oh, M. Reichelt, H. Ho, C. Chalouni, S. Labadie, L. Wang, J. Liang, N. N. Nickerson, H. Hu, L. Yu, M. Du, D. Yan, S. Park, J. Kim, M. Xu, B. D. Sellers, H. E. Purkey, N. J. Skelton, M. F. T. Koehler, J. Payandeh, V. Verma, Y. Xu, C. M. Koth and M. Nishiyama, Antimicrob. Agents Chemother., 2018, 62, 10–1128 CrossRef PubMed.
- G. Pegoraro and T. Misteli, Trends Genet., 2017, 33, 604–615 CrossRef CAS PubMed.
- T. Zahir, R. Camacho, R. Vitale, C. Ruckebusch, J. Hofkens, M. Fauvart and J. Michiels, Commun. Biol., 2019, 2, 269 CrossRef PubMed.
- S. Zoffmann, M. Vercruysse, F. Benmansour, A. Maunz, L. Wolf, R. Blum Marti, T. Heckel, H. Ding, H. H. Truong, M. Prummer, R. Schmucki, C. S. Mason, K. Bradley, A. I. Jacob, C. Lerner, A. Araujo del Rosario, M. Burcin, K. E. Amrein and M. Prunotto, Sci. Rep., 2019, 9, 5013 CrossRef PubMed.
- S. Sridhar, S. Forrest, B. Warne, M. Maes, S. Baker, G. Dougan and J. Bartholdson Scott, mSystems, 2021, 6, e00028-21 CrossRef PubMed.
- Key reference: M. Maes, Z. A. Dyson, S. E. Smith, D. A. Goulding, C. Ludden, S. Baker, P. Kellam, S. T. Reece, G. Dougan and J. B. Scott, Sci. Rep., 2020, 10, 12414 CrossRef CAS PubMed.
- P. Nonejuie, M. Burkart, K. Pogliano and J. Pogliano, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16169–16174 CrossRef CAS PubMed.
- D. T. Quach, G. Sakoulas, V. Nizet, J. Pogliano and K. Pogliano, EBioMedicine, 2016, 4, 95–103 CrossRef CAS PubMed.
- H. H. Htoo, L. Brumage, V. Chaikeeratisak, H. Tsunemoto, J. Sugie, C. Tribuddharat, J. Pogliano and P. Nonejuie, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.02310-18.
- M. A. T. Blaskovich, A. M. Kavanagh, A. G. Elliott, B. Zhang, S. Ramu, M. Amado, G. J. Lowe, A. O. Hinton, D. M. T. Pham, J. Zuegg, N. Beare, D. Quach, M. D. Sharp, J. Pogliano, A. P. Rogers, D. Lyras, L. Tan, N. P. West, D. W. Crawford, M. L. Peterson, M. Callahan and M. Thurn, Commun. Biol., 2021, 4, 7 CrossRef CAS PubMed.
- Key reference: J. K. Martin, J. P. Sheehan, B. P. Bratton, G. M. Moore, A. Mateus, S. H. J. Li, H. Kim, J. D. Rabinowitz, A. Typas, M. M. Savitski, M. Z. Wilson and Z. Gitai, Cell, 2020, 181, 1518–1532 CrossRef CAS PubMed , e14.
- T. C. Smith II, K. M. Pullen, M. C. Olson, M. E. Mcnellis, I. Richardson, S. Hu, J. Larkins-Ford, X. Wang, J. S. Freundlich, D. M. Ando and B. B. Aldridge, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18744–18753 CrossRef PubMed.
- T. P. T. Cushnie, N. H. O'Driscoll and A. J. Lamb, Cell. Mol. Life Sci., 2016, 73, 4471–4492 CrossRef CAS PubMed.
- Z. Yao, D. Kahne and R. Kishony, Mol. Cell, 2012, 48, 705–712 CrossRef CAS PubMed.
- K. C. Huang, R. Mukhopadhyay, B. Wen, Z. Gitai and N. S. Wingreen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19282–19287 CrossRef CAS PubMed.
- W. J. Godinez, H. Chan, I. Hossain, C. Li, S. Ranjitkar, D. Rasper, R. L. Simmons, X. Zhang and B. Y. Feng, ACS Chem. Biol., 2019, 14, 1217–1226 CrossRef CAS PubMed.
- Key reference: A. B. Parsons, R. L. Brost, H. Ding, Z. Li, C. Zhang, B. Sheikh, G. W. Brown, P. M. Kane, T. R. Hughes and C. Boone, Nat. Biotechnol., 2004, 22, 62–69 CrossRef CAS PubMed.
- E. Cacace, G. Kritikos and A. Typas, Curr. Opin. Syst. Biol., 2017, 4, 35–42 CrossRef PubMed.
- A. Paul, R. Anand, S. P. Karmakar, S. Rawat, N. Bairagi and S. Chatterjee, Sci. Rep., 2021, 11, 213 CrossRef CAS PubMed.
- H. Luesch, T. Y. H. Wu, P. Ren, N. S. Gray, P. G. Schultz and F. Supek, Chem. Biol., 2005, 12, 55–63 CrossRef CAS PubMed.
- S. Hoon, A. M. Smith, I. M. Wallace, S. Suresh, M. Miranda, E. Fung, M. Proctor, K. M. Shokat, C. Zhang, R. W. Davis, G. Giaever, R. P. Stonge and C. Nislow, Nat. Chem. Biol., 2008, 4, 498–506 CrossRef CAS PubMed.
- M. Kitagawa, T. Ara, M. Arifuzzaman, T. Ioka-Nakamichi, E. Inamoto, H. Toyonaga and H. Mori, DNA Res., 2005, 12, 291–299 CrossRef CAS PubMed.
- T. Baba, T. Ara, M. Hasegawa, Y. Takai, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner and H. Mori, Mol. Syst. Biol., 2006, 2, 2006.0008 CrossRef PubMed.
- V. K. Yajjala, T. J. Widhelm, J. L. Endres, P. D. Fey and K. W. Bayles, Methods Mol. Biol., 2016, 1353, 103–110 Search PubMed.
- N. Wang, E. A. Ozer, M. J. Mandel and A. R. Hauser, mBio, 2014, 5, e01163-14 CrossRef PubMed.
- K. L. Stirrett, J. A. Ferreras, S. M. Rossi, R. L. Moy, F. V. Fonseca and L. E. N. Quadri, BMC Microbiol., 2008, 8, 122 CrossRef PubMed.
- A. Liu, L. Tran, E. Becket, K. Lee, L. Chinn, E. Park, K. Tran and J. H. Miller, Antimicrob. Agents Chemother., 2010, 54, 1393–1403 CrossRef CAS PubMed.
- R. J. Nichols, S. Sen, Y. J. Choo, P. Beltrao, M. Zietek, R. Chaba, S. Lee, K. M. Kazmierczak, K. J. Lee, A. Wong, M. Shales, S. Lovett, M. E. Winkler, N. J. Krogan, A. Typas and C. A. Gross, Cell, 2011, 144, 143–156 CrossRef CAS PubMed.
- Key reference: R. Pathania, S. Zlitni, C. Barker, R. Das, D. A. Gerritsma, J. Lebert, E. Awuah, G. Melacini, F. A. Capretta and E. D. Brown, Nat. Chem. Biol., 2009, 5, 849–856 CrossRef CAS PubMed.
- Key reference: S. M. Batt, M. Cacho Izquierdo, J. Castro Pichel, C. J. Stubbs, L. Vela-Glez Del Peral, E. Pérez-Herrán, N. Dhar, B. Mouzon, M. Rees, J. P. Hutchinson, R. J. Young, J. D. McKinney, D. Barros Aguirre, L. Ballell, G. S. Besra and A. Argyrou, ACS Infect. Dis., 2016, 1, 615–626 CrossRef PubMed.
- É. Gazanion, C. Fernández-Prada, B. Papadopoulou, P. Leprohon and M. Ouellette, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3012–E3021 CrossRef PubMed.
- Key reference: C. Fernandez-Prada, M. Sharma, M. Plourde, E. Bresson, G. Roy, P. Leprohon and M. Ouellette, Int. J. Parasitol.: Drugs Drug Resist., 2018, 8, 165–173 Search PubMed.
- H. Gingras, B. Dridi, P. Leprohon and M. Ouellette, Microb. Genomics, 2018, 4(2), e000148 Search PubMed.
- P. B. Otoupal, K. A. Eller, K. E. Erickson, J. Campos, T. R. Aunins and A. Chatterjee, Commun. Biol., 2021, 4, 1267 CrossRef CAS PubMed.
- K. Kobayashi, S. D. Ehrlich, A. Albertini, G. Amati, K. K. Andersen, M. Arnaud, K. Asai, S. Ashikaga, S. Aymerich, P. Bessieres, F. Boland, S. C. Brignell, S. Bron, K. Bunai, J. Chapuis, L. C. Christiansen, A. Danchin, M. Débarbouillé, E. Dervyn, E. Deuerling, K. Devine, S. K. Devine, O. Dreesen, J. Errington, S. Fillinger, S. J. Foster, Y. Fujita, A. Galizzi, R. Gardan, C. Eschevins, T. Fukushima, K. Haga, C. R. Harwood, M. Hecker, D. Hosoya, M. F. Hullo, H. Kakeshita, D. Karamata, Y. Kasahara, F. Kawamura, K. Koga, P. Koski, R. Kuwana, D. Imamura, M. Ishimaru, S. Ishikawa, I. Ishio, D. Le Coq, A. Masson, C. Mauël, R. Meima, R. P. Mellado, A. Moir, S. Moriya, E. Nagakawa, H. Nanamiya, S. Nakai, P. Nygaard, M. Ogura, T. Ohanan, M. O'Reilly, M. O'Rourke, Z. Pragai, H. M. Pooley, G. Rapoport, J. P. Rawlins, L. A. Rivas, C. Rivolta, A. Sadaie, Y. Sadaie, M. Sarvas, T. Sato, H. H. Saxild, E. Scanlan, W. Schumann, J. F. M. L. Seegers, J. Sekiguchi, A. Sekowska, S. J. Séror, M. Simon, P. Stragier, R. Studer, H. Takamatsu, T. Tanaka, M. Takeuchi, H. B. Thomaides, V. Vagner, J. M. Van Dijl, K. Watabe, A. Wipat, H. Yamamoto, M. Yamamoto, Y. Yamamoto, K. Yamane, K. Yata, K. Yoshida, H. Yoshikawa, U. Zuber and N. Ogasawara, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4678–4683 CrossRef CAS PubMed.
- V. De Berardinis, D. Vallenet, V. Castelli, M. Besnard, A. Pinet, C. Cruaud, S. Samair, C. Lechaplais, G. Gyapay, C. Richez, M. Durot, A. Kreimeyer, F. Le Fèvre, V. Schächter, V. Pezo, V. Döring, C. Scarpelli, C. Médigue, G. N. Cohen, P. Marlière, M. Salanoubat and J. Weissenbach, Mol. Syst. Biol., 2008, 4, 174 CrossRef PubMed.
- P. Xu, X. Ge, L. Chen, X. Wang, Y. Dou, J. Z. Xu, J. R. Patel, V. Stone, M. Trinh, K. Evans, T. Kitten, D. Bonchev and G. A. Buck, Sci. Rep., 2011, 1, 125 CrossRef PubMed.
- A. Muir, I. Gurung, A. Cehovin, A. Bazin, D. Vallenet and V. Pelicic, Nat. Commun., 2020, 11, 5541 CrossRef CAS PubMed.
- Key reference: G. L. Abrahams, A. Kumar, S. Savvi, A. W. Hung, S. Wen, C. Abell, C. E. Barry, D. R. Sherman, H. I. M. Boshoff and V. Mizrahi, Chem. Biol., 2012, 19, 844–854 CrossRef CAS PubMed.
- B. Christen, E. Abeliuk, J. M. Collier, V. S. Kalogeraki, B. Passarelli, J. A. Coller, M. J. Fero, H. H. McAdams and L. Shapiro, Mol. Syst. Biol., 2011, 7, 528 CrossRef PubMed.
- P. Y. Lum, C. D. Armour, S. B. Stepaniants, G. Cavet, M. K. Wolf, S. Butler, J. C. Hinshaw, P. Garnier, G. D. Prestwich, A. Leonardson, P. Garrett-Engele, C. M. Rush, M. Bard, G. Schimmack, J. W. Philips, C. J. Roberts and D. D. Shoemaker, Cell, 2004, 116, 121–137 CrossRef CAS PubMed.
- A. M. Hogan and S. T. Cardona, FEMS Microbiol. Rev., 2022, 46, fuac005 CrossRef PubMed.
- O. Shalem, N. E. Sanjana and F. Zhang, Nat. Rev. Genet., 2015, 16, 299–311 CrossRef CAS PubMed.
- A. K. Cain, L. Barquist, A. L. Goodman, I. T. Paulsen, J. Parkhill and T. van Opijnen, Nat. Rev. Genet., 2020, 21, 526–540 CrossRef CAS PubMed.
- T. Wang, C. Guan, J. Guo, B. Liu, Y. Wu, Z. Xie, C. Zhang and X. H. Xing, Nat. Commun., 2018, 9, 2475 CrossRef PubMed.
- V. de Bakker, X. Liu, A. M. Bravo and J. W. Veening, Nat. Protoc., 2022, 17, 252–281 CrossRef CAS PubMed.
- J. M. Peters, A. Colavin, H. Shi, T. L. Czarny, M. H. Larson, S. Wong, J. S. Hawkins, C. H. S. Lu, B. M. Koo, E. Marta, A. L. Shiver, E. H. Whitehead, J. S. Weissman, E. D. Brown, L. S. Qi, K. C. Huang and C. A. Gross, Cell, 2016, 165, 1493–1506 CrossRef CAS PubMed.
- C. Bock, P. Datlinger, F. Chardon, M. A. Coelho, M. B. Dong, K. A. Lawson, T. Lu, L. Maroc, T. M. Norman, B. Song, G. Stanley, S. Chen, M. Garnett, W. Li, J. Moffat, L. S. Qi, R. S. Shapiro, J. Shendure, J. S. Weissman and X. Zhuang, Nat. Rev. Methods Primers, 2022, 2, 8 CrossRef CAS.
- S. Li, N. C. Poulton, J. S. Chang, Z. A. Azadian, M. A. DeJesus, N. Ruecker, M. D. Zimmerman, K. A. Eckartt, B. Bosch, C. A. Engelhart, D. F. Sullivan, M. Gengenbacher, V. A. Dartois, D. Schnappinger and J. M. Rock, Nat. Microbiol., 2022, 7, 766–779 CrossRef CAS PubMed.
- R. G. K. Donald, S. Skwish, R. A. Forsyth, J. W. Anderson, T. Zhong, C. Burns, S. Lee, X. Meng, L. LoCastro, L. W. Jarantow, J. Martin, S. H. Lee, I. Taylor, D. Robbins, C. Malone, L. Wang, C. S. Zamudio, P. J. Youngman and J. W. Phillips, Chem. Biol., 2009, 16, 826–836 CrossRef CAS PubMed.
- J. Bai, Y. Dai, A. Farinha, A. Y. Tang, S. Syal, G. Vargas-Cuebas, T. van Opijnen, R. R. Isberg and E. Geisinger, J. Bacteriol., 2021, 203 DOI:10.1128/jb.00565-20.
- M. Foreman, M. Gershoni and D. Barkan, mSystems, 2020, 5, e00976-20 CrossRef PubMed.
- M. A. Jacobs, A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. V. Olson and C. Manoil, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14339–14344 CrossRef CAS PubMed.
- F. A. Sargison and J. R. Fitzgerald, Trends Microbiol., 2021, 29, 282–285 CrossRef CAS PubMed.
- S. A. Lee, L. A. Gallagher, M. Thongdee, B. J. Staudinger, S. Lippman, P. K. Singhc and C. Manoila, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5189–5194 CrossRef CAS PubMed.
- N. R. Salama, B. Shepherd and S. Falkow, J. Bacteriol., 2004, 186, 7926–7935 CrossRef CAS PubMed.
- H. Wang, D. Claveau, J. P. Vaillancourt, T. Roemer and T. C. Meredith, Nat. Chem. Biol., 2011, 7, 720–729 CrossRef CAS PubMed.
- Y. Le Breton, A. T. Belew, K. M. Valdes, E. Islam, P. Curry, H. Tettelin, M. E. Shirtliff, N. M. El-Sayed and K. S. McIver, Sci. Rep., 2015, 5, 9838 CrossRef PubMed.
- R. A. M. Bloodworth, A. S. Gislason and S. T. Cardona, MicrobiologyOpen, 2013, 2, 243–258 CrossRef CAS PubMed.
- R. Zhang, W. Xu, S. Shao and Q. Wang, Front. Microbiol., 2021, 12, 635227 CrossRef PubMed.
- Key reference: A. M. Hogan, V. C. Scoffone, V. Makarov, A. S. Gislason, H. Tesfu, M. S. Stietz, A. K. C. Brassinga, M. Domaratzki, X. Li, A. Azzalin, M. Biggiogera, O. Riabova, N. Monakhova, L. R. Chiarelli, G. Riccardi, S. Buroni and S. T. Cardona, Antimicrob. Agents Chemother., 2018, 62 DOI:10.1128/aac.01231-18.
- Key reference: M. Santiago, W. Lee, A. A. Fayad, K. A. Coe, M. Rajagopal, T. Do, F. Hennessen, V. Srisuknimit, R. Müller, T. C. Meredith and S. Walker, Nat. Chem. Biol., 2018, 14, 601–608 CrossRef CAS PubMed.
- H. Gingras, K. Patron, A. Bhattacharya, P. Leprohon and M. Ouellette, Antimicrob. Agents Chemother., 2019, 63 DOI:10.1128/aac.02381-18.
- E. O. Johnson, E. LaVerriere, E. Office, M. Stanley, E. Meyer, T. Kawate, J. E. Gomez, R. E. Audette, N. Bandyopadhyay, N. Betancourt, K. Delano, I. Da Silva, J. Davis, C. Gallo, M. Gardner, A. J. Golas, K. M. Guinn, S. Kennedy, R. Korn, J. A. McConnell, C. E. Moss, K. C. Murphy, R. M. Nietupski, K. G. Papavinasasundaram, J. T. Pinkham, P. A. Pino, M. K. Proulx, N. Ruecker, N. Song, M. Thompson, C. Trujillo, S. Wakabayashi, J. B. Wallach, C. Watson, T. R. Ioerger, E. S. Lander, B. K. Hubbard, M. H. Serrano-Wu, S. Ehrt, M. Fitzgerald, E. J. Rubin, C. M. Sassetti, D. Schnappinger and D. T. Hung, Nature, 2019, 571, 72–78 CrossRef CAS PubMed.
- W. Jiang, P. Oikonomou and S. Tavazoie, Cell, 2020, 180, 1002–1017 CrossRef CAS PubMed , e31.
- J. C. Evans, C. Trujillo, Z. Wang, H. Eoh, S. Ehrt, D. Schnappinger, H. I. M. Boshoff, K. Y. Rhee, C. E. Barry and V. Mizrahi, ACS Infect. Dis., 2016, 2, 958–968 CrossRef CAS PubMed.
- E. Geisinger, N. J. Mortman, Y. Dai, M. Cokol, S. Syal, A. Farinha, D. G. Fisher, A. Y. Tang, D. W. Lazinski, S. Wood, J. Anthony, T. van Opijnen and R. R. Isberg, Nat. Commun., 2020, 11, 4522 CrossRef CAS PubMed.
- Key reference: J. M. Clements, F. Coignard, I. Johnson, S. Chandler, S. Palan, A. Waller, J. Wijkmans and M. G. Hunter, Antimicrob. Agents Chemother., 2002, 46, 1793–1799 CrossRef CAS PubMed.
- S. Mohan, T. M. Kelly, S. S. Eveland, C. R. H. Raetz and M. S. Anderson, J. Biol. Chem., 1994, 269, 32896–32903 CrossRef CAS PubMed.
- A. L. Erwin, Cold Spring Harbor Perspect. Med., 2016, 6, 1–14 Search PubMed.
- K. Fujita, I. Takata, I. Yoshida, H. Okumura, K. Otake, H. Takashima and H. Sugiyama, J. Antibiot., 2022, 75, 98–107 CrossRef CAS PubMed.
- L. R. Ptitsyn, G. Horneck, O. Komova, S. Kozubek, E. A. Krasavin, M. Bonev and A. P. Rettberg, Appl. Environ. Microbiol., 1997, 63, 4377–4384 CrossRef CAS PubMed.
- E. Shapiro and F. Baneyx, J. Biotechnol., 2007, 132, 487–493 CrossRef CAS PubMed.
- S. Daunert, G. Barrett, J. S. Feliciano, R. S. Shetty, S. Shrestha and W. Smith-Spencer, Chem. Rev., 2000, 100, 2705–2738 CrossRef CAS PubMed.
- E. Shapiro and F. Baneyx, Antimicrob. Agents Chemother., 2002, 46, 2490–2497 CrossRef CAS PubMed.
- A. A. Bianchi and F. O. Baneyx, Appl. Environ. Microbiol., 1999, 65, 5023–5027 CrossRef CAS PubMed.
- T. K. Van Dyk, W. R. Majarian, K. B. Konstantinov, R. M. Young, P. S. Dhurjati and R. A. Larossal, Appl. Environ. Microbiol., 1994, 60, 1414–1420 CrossRef CAS PubMed.
- C. S. Orser, F. C. F. Foong, S. R. Capaldi, J. Nalezny, W. Mackay, M. Benjamin and S. B. Farr, In Vitro Toxicol., 1995, 8, 71–86 CAS.
- M. Kostrzynska, K. T. Leung, H. Lee and J. T. Trevors, J. Microbiol. Methods, 2002, 48, 43–51 CrossRef CAS PubMed.
- J. Kurittu, M. Karp and M. Korpela, Luminescence, 2000, 15, 291–297 CrossRef CAS PubMed.
- V. Möhrle, M. Stadler and G. Eberz, Anal. Bioanal. Chem., 2007, 388, 1117–1125 CrossRef PubMed.
- M. Bailey, T. Chettiath and A. S. Mankin, Antimicrob. Agents Chemother., 2008, 52, 866–874 CrossRef CAS PubMed.
- Y. S. Polikanov, I. A. Osterman, T. Szal, V. N. Tashlitsky, M. V. Serebryakova, P. Kusochek, D. Bulkley, I. A. Malanicheva, T. A. Efimenko, O. V. Efremenkova, A. L. Konevega, K. J. Shaw, A. A. Bogdanov, M. V. Rodnina, O. A. Dontsova, A. S. Mankin, T. A. Steitz and P. V. Sergiev, Mol. Cell, 2014, 56, 531–540 CrossRef CAS PubMed.
- I. A. Osterman, I. V. Prokhorova, V. O. Sysoev, Y. V. Boykova, O. V. Efremenkova, M. S. Svetlov, V. A. Kolb, A. A. Bogdanov, P. V. Sergiev and O. A. Dontsova, Antimicrob. Agents Chemother., 2012, 56, 1774–1783 CrossRef CAS PubMed.
- I. A. Osterman, E. S. Komarova, D. I. Shiryaev, I. A. Korniltsev, I. M. Khven, D. A. Lukyanov, V. N. Tashlitsky, M. V. Serebryakova, O. V. Efremenkova, Y. A. Ivanenkov, A. A. Bogdanov, P. V. Sergiev and O. A. Dontsova, Antimicrob. Agents Chemother., 2016, 60, 7481–7489 CrossRef CAS PubMed.
- E. S. Komarova (Andreyanova), I. A. Osterman, P. I. Pletnev, Y. A. Ivanenkov, A. G. Majouga, A. A. Bogdanov and P. V. Sergiev, Biochimie, 2017, 133, 45–55 CrossRef CAS PubMed.
- I. A. Osterman, M. Wieland, T. P. Maviza, K. A. Lashkevich, D. A. Lukianov, E. S. Komarova, Y. V. Zakalyukina, R. Buschauer, D. I. Shiriaev, S. A. Leyn, J. E. Zlamal, M. V. Biryukov, D. A. Skvortsov, V. N. Tashlitsky, V. I. Polshakov, J. Cheng, Y. S. Polikanov, A. A. Bogdanov, A. L. Osterman, S. E. Dmitriev, R. Beckmann, O. A. Dontsova, D. N. Wilson and P. V. Sergiev, Nat. Chem. Biol., 2020, 16, 1071–1077 CrossRef CAS PubMed.
- M. Metelev, I. A. Osterman, D. Ghilarov, N. F. Khabibullina, A. Yakimov, K. Shabalin, I. Utkina, D. Y. Travin, E. S. Komarova, M. Serebryakova, T. Artamonova, M. Khodorkovskii, A. L. Konevega, P. V. Sergiev, K. Severinov and Y. S. Polikanov, Nat. Chem. Biol., 2017, 13, 1129–1136 CrossRef CAS PubMed.
- J. Fan, B. L. M. De Jonge, K. MacCormack, S. Sriram, R. E. McLaughlin, H. Plant, M. Preston, P. R. Fleming, R. Albert, M. Foulk and S. D. Mills, Antimicrob. Agents Chemother., 2014, 58, 7264–7272 CrossRef PubMed.
- Key reference: S. J. Valtonen, J. S. Kurittu and M. Karp, J. Biomol. Screening, 2002, 7, 127–134 CrossRef CAS PubMed.
- Key reference: P. D. Tamma, Y. Doi, R. A. Bonomo, J. K. Johnson and P. J. Simner, Clin. Infect. Dis., 2019, 69, 1446–1455 CrossRef CAS PubMed.
- X. Zeng and J. Lin, Front. Microbiol., 2013, 4, 128 Search PubMed.
- R. E. Mendes, N. Cotroneo, I. A. Critchley, B. Roth, S. J. R. Arends, M. Castanheira and M. Castanheira, Open Forum Infect. Dis., 2021, 8, S608–S609 CrossRef.
- D. Sun, S. Cohen, N. Mani, C. Murphy and D. M. Rothstein, J. Antibiot., 2002, 55, 279–287 CrossRef CAS PubMed.
- M. Decenzoa, M. Kurandaa, S. Cohena, J. Babiakb, Z.-D. Jianga, D. Suna, M. Hickeya, P. Sanchetia, P. A. Bradfordb, P. Youngmana, S. Projanb and D. M. Rothsteina, J. Antibiot., 2002, 55, 288–295 CrossRef PubMed.
- F. Guérin, C. Isnard, V. Cattoir and J. C. Giard, Antimicrob. Agents Chemother., 2015, 59, 7753–7761 CrossRef PubMed.
- G. Torrens, S. B. Hernández, J. A. Ayala, B. Moya, C. Juan, F. Cava and A. Oliver, mSystems, 2019, 4 DOI:10.1128/msystems.00524-19.
- Key reference: A. S. Nayar, T. J. Dougherty, K. E. Ferguson, B. A. Granger, L. McWilliams, C. Stacey, L. J. Leach, S. ichiro Narita, H. Tokuda, A. A. Miller, D. G. Brown and S. M. McLeod, J. Bacteriol., 2015, 197, 1726–1734 CrossRef CAS PubMed.
- Key reference: M. Steenhuis, F. Corona, C. M. ten Hagen-Jongman, W. Vollmer, D. Lambin, P. Selhorst, H. Klaassen, M. Versele, P. Chaltin and J. Luirink, ACS Infect. Dis., 2021, 7, 2250–2263 CrossRef CAS PubMed.
- T. Stachyra, C. Dini, P. Ferrari, A. Bouhss, J. Van Heijenoort, D. Mengin-Lecreulx, D. Blanot, J. Biton and D. Le Beller, Antimicrob. Agents Chemother., 2004, 48, 897–902 CrossRef CAS PubMed.
- A. A. Branstrom, S. Midha, C. B. Longley, K. Han, E. R. Baizman and H. R. Axelrod, Anal. Biochem., 2000, 280, 315–319 CrossRef CAS PubMed.
- A. B. Shapiro, H. Jahić, N. Gao, L. Hajec and O. Rivin, J. Biomol. Screening, 2012, 17, 662–672 CrossRef CAS PubMed.
- L. L. Silver, Cold Spring Harbor Perspect. Med., 2017, 7, 1–11 Search PubMed.
- C. Dini, Curr. Top. Med. Chem., 2005, 5, 1221–1236 CrossRef CAS PubMed.
- S. Walker, L. Chen, Y. Hu, Y. Rew, D. Shin and D. L. Boger, Chem. Rev., 2005, 105, 449–475 CrossRef CAS PubMed.
- S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo, E. M. Rodriguez-Lopez, P. Mercier and J. C. Vederas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11561–11566 CrossRef CAS PubMed.
- B. Ostash and S. Walker, Nat. Prod. Rep., 2010, 27, 1594–1617 RSC.
- T. Schneider, K. Gries, M. Josten, I. Wiedemann, S. Pelzer, H. Labischinski and H. G. Sahl, Antimicrob. Agents Chemother., 2009, 53, 1610–1618 CrossRef CAS PubMed.
- K. J. Stone and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 3223–3227 CrossRef CAS PubMed.
- Key reference: T. L. Czarny and E. D. Brown, ACS Infect. Dis., 2016, 2, 489–499 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS PubMed.
- A. L. McClerren, S. Endsley, J. L. Bowman, N. H. Andersen, Z. Guan, J. Rudolph and C. R. H. Raetz, Biochemistry, 2005, 44, 16574–16583 CrossRef CAS PubMed.
- C. F. Schuster and R. Bertram, FEMS Microbiol. Lett., 2013, 340, 73–85 CrossRef CAS PubMed.
- I. C. Hancock, G. Wiseman and J. Baddiley, FEBS Lett., 1976, 69, 75–80 CrossRef CAS PubMed.
- F. A. Thélot, W. Zhang, K. Song, C. Xu, J. Huang and M. Liao, Science, 2021, 374, 580–585 CrossRef PubMed.
Footnote |
| † Present address: Biochemistry and Pathology Laboratory, Regional Research Institute of Unani Medicine, Patna – 800 008, Bihar, India. |
| This journal is © The Royal Society of Chemistry 2023 |