
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactive fragments targeting carboxylate residues employing direct to biology, high-throughput chemistry†
Ross P.
Thomas
ab,
Emma K.
Grant
a,
Eleanor R.
Dickinson
 a,
Francesca
Zappacosta
c,
Lee J.
Edwards
a,
Michael M.
Hann
a,
David
House
a,
Nicholas C. O.
Tomkinson
*b and
Jacob T.
Bush
*a
a,
Francesca
Zappacosta
c,
Lee J.
Edwards
a,
Michael M.
Hann
a,
David
House
a,
Nicholas C. O.
Tomkinson
*b and
Jacob T.
Bush
*a
aGlaxoSmithKline, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK. E-mail: Jacob.x.Bush@GSK.com
bDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: Nicholas.Tomkinson@strath.ac.uk
cGlaxoSmithKline, South Collegeville Road, Collegeville, PA 19426, USA
First published on 22nd February 2023
Abstract
The screening of covalent or ‘reactive’ fragment libraries against proteins is becoming an integral approach in hit identification, enabling the development of targeted covalent inhibitors and tools. To date, reactive fragment screening has been limited to targeting cysteine residues, thus restricting applicability across the proteome. Carboxylate residues present a unique opportunity to expand the accessible residues due to high proteome occurrence (∼12%). Herein, we present the development of a carboxylate-targeting reactive fragment screening platform utilising 2-aryl-5-carboxytetrazole (ACT) as the photoreactive functionality. The utility of ACT photoreactive fragments (ACT-PhABits) was evaluated by screening a 546-membered library with a small panel of purified proteins. Hits identified for BCL6 and KRASG12D were characterised by LC-MS/MS studies, revealing the selectivity of the ACT group. Finally, a photosensitised approach to ACT activation was developed, obviating the need for high energy UV-B light.
Introduction
In recent years, small molecules with a covalent mechanism of action have emerged as powerful modalities in medicinal chemistry and chemical biology.1–6 A key benefit of covalent inhibition is the ability to target shallow binding pockets and protein–protein interaction (PPI) interfaces through enhanced potency and prolonged target occupancy.7,8 Recently, there has been a growing interest in the application of reactive fragments for the development of targeted covalent inhibitors (TCIs).9–12 Reactive fragments combine the efficient coverage of chemical space offered by fragments with a reactive moiety to enable robust covalent capture of weak fragment-protein binding interactions.13–16 Early reactive fragment screening focussed on targeting cysteine residues (Fig. 1a).4,10,12,13,17 Notably, cysteine reactive fragments were employed for targeting the challenging oncology target KRASG12C, culminating with the development of sotorasib (AMG510).1 The low frequency of cysteine (∼2%)18,19 in the proteome has prompted research into alternative covalent modalities that can selectively target less nucleophilic amino acids including tyrosine and lysine (Fig. 1a).20–28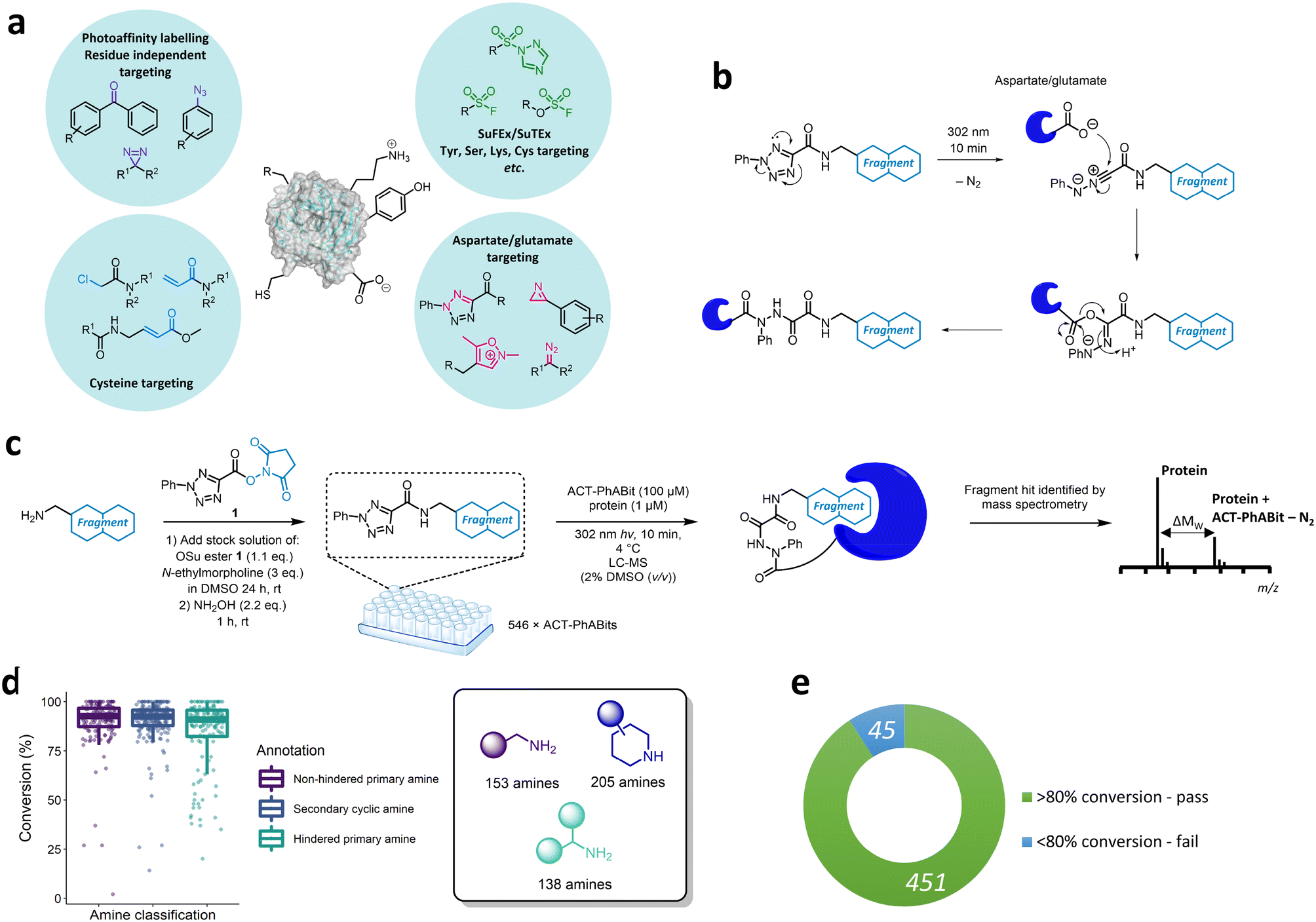 | ||
| Fig. 1 a) Methods for covalent modification of different amino acid side chains. b) Proposed mechanism for the photoactivated covalent capture of carboxylate residues using the 2-aryl-5-carboxytetrazole (ACT) functionality. c) Direct-to-biology high-throughput chemistry (D2B-HTC) synthesis of a 546-membered ACT-PhABit library in 384-well plates and direct screening of crude reaction products without purification with a protein target. d) Boxplot for the conversion of the ACT-PhABit reactions grouped by the classification of the amine structure. Amines were classified according to the following categories: hindered primary amines, unhindered primary amines and secondary cyclic amines. 50 reactions were omitted on account of impure amine starting materials or an inability to calculate conversion. e) Overall success rate of the ACT-PhABit synthetic protocol. | ||
To date, there has been limited research on selectively targeting aspartate (Asp) and glutamate (Glu) residues (Fig. 1a).29–33 The high proteome occurrence of Asp and Glu residues (>12% combined) combined with the propensity for these polar residues to be located on the surface of proteins suggests they could offer a useful handle for the development of covalent ligands.18,19,34 Recently a number of carboxylate targeting groups have been employed in the development of covalent ligands, including diazo modalities,33,35,36 isoxazolium derivatives30 and 2H-azirines (Fig. 1a).29 Tetrazole-moieties offer a photoactivable approach towards irreversible carboxylate modification.31,34,37 Diarylsubstituted tetrazoles were reported by Bach et al. to enable proteome-wide profiling of aspartate and glutamate residues in bacteria.31 Similarly, 2-aryl-5-carboxytetrazoles (ACTs) represent a photoactivatable carboxylate-targeting covalent functionality that features a carboxylic acid handle for facile derivatisation with suitable recognition elements (Fig. 1a and b).32,38 Photolysis using ultraviolet (UV) light (302 nm) affords the nitrilimine reactive species, which can react with a carboxylate residue via an O → N acyl shift to furnish the corresponding 1,2-diacylhydrazine (Fig. 1b).31,32 The ACT-derived nitrilimine intermediate exhibits a longer lifetime than common photoactivable diazirines (>500 μs cf. carbene <2 ns), which can lead to enhanced crosslinking yields.32,39–41
While a photoreactive mechanism does not offer the boost in potency observed with electrophilic covalent mechanisms, it can be advantageous in enabling the discovery of ligands with good non-covalent affinity. Previously we have reported the screening of diazirine containing fragments for the discovery of ligands for proteins of interest.14,42 A challenge with this approach can be the low crosslinking yields achieved with carbenes, and it was anticipated that the nitrilimine furnished by the photoactivation of the ACT reactive group may offer improved crosslinking yields.
Herein, we describe the development of an ACT reactive fragment platform that allows the capture of fragment–protein interactions via selective carboxylate side chain modification. A library of ACT-derived fragments was synthesised using a direct-to-biology, high-throughput chemistry approach and screened against a panel of therapeutically relevant proteins including BRD4-BD1, BCL6, carbonic anhydrase I & II (CAI/CAII) and KRASG12D. Intact-protein mass spectrometry enabled identification of hits which included established chemotypes as well as novel binding motifs for these therapeutically relevant proteins. The hits were further characterised to determine the site of modification, revealing a potentially novel binding site on KRAS. Finally, an energy transfer (EnT) approach towards activation of the ACT moiety was also developed, allowing the use of longer wavelength light that is less damaging to the protein.
Results and discussion
Synthesis of an ACT-PhABit library
We anticipated that a direct-to-biology high-throughput chemistry (D2B-HTC) platform would enable access to a library of ACT-derived reactive fragments for expedient evaluation as a carboxylate-targeting screening strategy (Fig. 1c).42 This methodology employs 384-well plate-based amide couplings of amine fragments with an activated succinimide ester bearing the reactive functionality (1, Fig. 1c). The crude reaction products were screened without purification with the protein target in a ‘direct-to-biology’ approach.To generate the ACT-PhABit library, we selected 546 amine fragments that afforded high conversion to the corresponding amides in a HTC-PhABit library.42 These included primary unhindered-, primary hindered- and secondary cyclic-alkylamines (ESI† section 3.1) (for library properties see Fig. S1†).42 Amines were plated as singletons in 384-well plates (10 mM DMSO) followed by addition of a DMSO solution of OSu ester 1 and N-ethylmorpholine (NEM). The plates were sealed and incubated at room temperature for 24 h to afford the crude ACT-PhABits. The reactions were quenched with NH2OH(aq) (2.2 eq., 1 h, rt) to remove unreacted OSu ester 1, preventing potential non-specific labelling of nucleophilic amino acid residues on the protein surface.42 LC-MS analysis of the plates indicated a high success rate of 91% for the 496 reactions quantified (Fig. 1d and e, for full breakdown of reaction qualification see Fig. S2†). Of the remaining 50, conversion could not successfully be calculated or the amine starting material was impure.
Screening the ACT-PhABit library
Following the generation of the D2B-ready ACT-PhABits, a panel of proteins, BCL6, BRD4-BD1, CAI & CAII, KRASG12D and myoglobin, were selected for screening. BRD4, CAI and CAII were selected since they were known to bind to chemotypes present in the library which could serve as positive controls. The KRASG12D mutant is a common mutation in cancers and was therefore selected to investigate whether Asp12 could be specifically modified by the ACT-derived nitrilimine reactive species.43 BCL6 is a transcription factor that is often dysregulated in B-cell lymphomas and autoimmune diseases.22,44 A number of inhibitors have been reported that bind in the shallow groove formed at the BCL6 homodimeric interface, disrupting co-repressor PPIs and restoring BCL6 target genes.22,44–47 BCL6 therefore served as a target to investigate whether the ACT-fragment could ligate a shallow PPI pocket.The proteins were incubated with the library (100 μM ACT-PhABit, 1 μM protein) and irradiated (10 min, 302 nm). Intact-protein LC-MS was used to identify any light-induced covalent crosslinking events by detection of a mass shift corresponding to [protein + ACT-PhABit − N2]. Hits were characterised as ACT-PhABits that modified the protein with a crosslinking yield >mean + 2 × standard deviation (mean + 2SD). The mean crosslinking that was observed across the panel of proteins ranged from 1.9% (BRD4-BD1) to 7.1% (BCL6) (Fig. 2a and b and S3†), indicating a low extent of background, non-specific labelling. A heatmap of crosslinking in all screens highlighted selective crosslinking between specific fragments for each protein, supporting that crosslinking is driven by recognition mediated by the fragment moiety (Fig. 2a). Validation that the platform could identify true hits was provided by the identification of known binders, such as acetyl lysine mimetics (e.g. dimethylisoxazole) for BRD4-BD1 (Fig. 2a and c, 2a–d)48–50 and a number of primary sulfonamide hits (Fig. 2a and c, 3a–e) for CAII, which are known to bind in the Zn2+ binding site.51 The screen identified 14 ACT-PhABit hits for BCL6, 9 of which were not hits for any other protein, whilst 26 were identified for KRASG12D, 17 of which were selective (Fig. 2b and S4 and S5†).
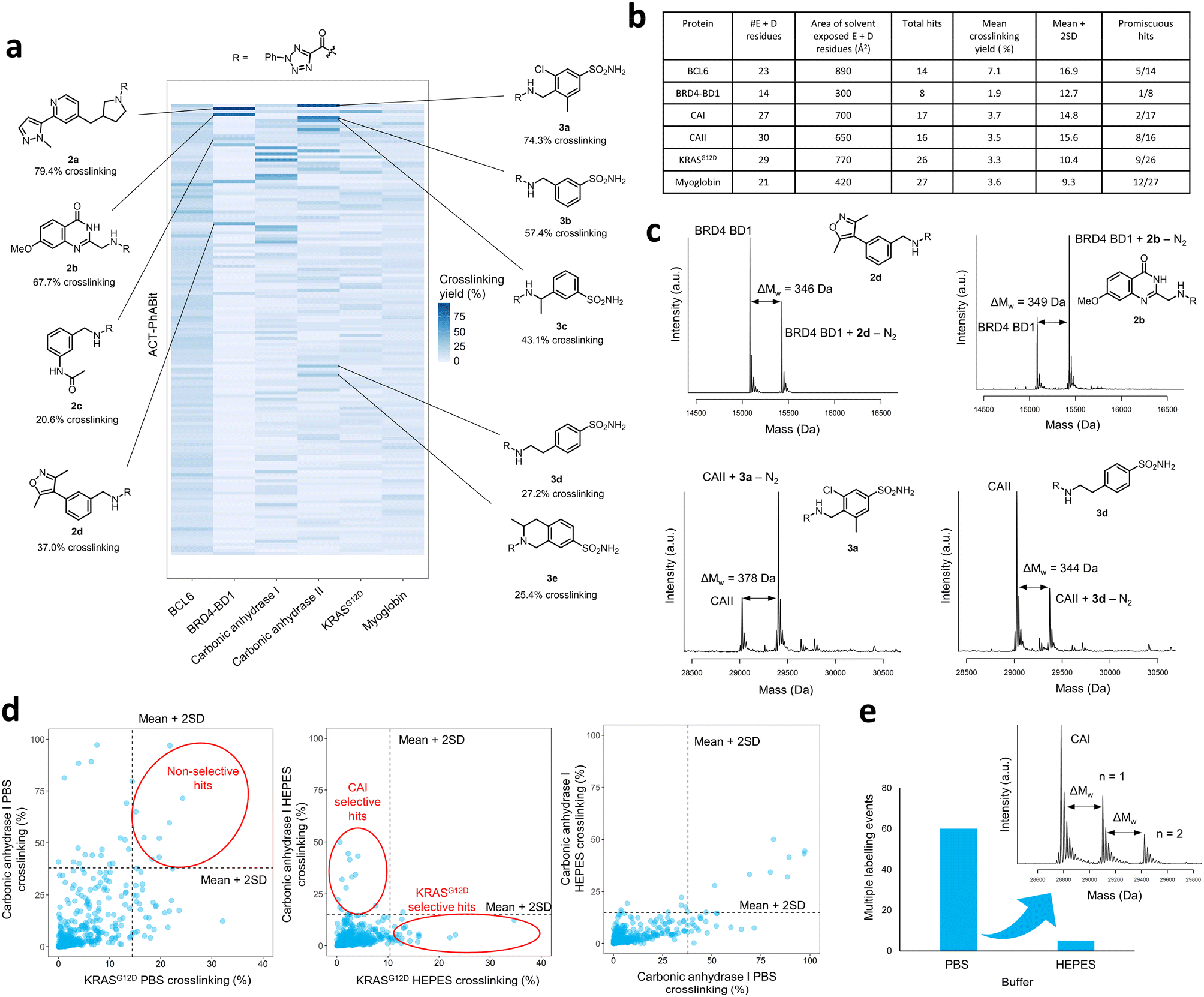 | ||
| Fig. 2 a) The ACT-PhABit library (100 μM) was irradiated (302 nm, 10 min) with protein (1 μM) in HEPES buffer, before analysis by intact-protein LC-MS. Heatmap displaying the crosslinking yields of the top 150 ACT-PhABits (ordered top to bottom by average crosslinking across the six proteins) with a panel of proteins, highlighting hit structures from the BRD4-BD1 and carbonic anhydrase II (CAII) screens. b) Screening summary data for the panel of proteins that were screened against the 546-membered ACT-PhABit library, including the total number of carboxylate residues (E = glutamate, D = aspartate) in each protein and area of solvent exposed carboxylate residues. c) Structures and corresponding mass spectra of selected BRD4-BD1 and CAII hits. d) Comparison of crosslinking yields across the 546-membered ACT-PhABit library (100 μM) with carbonic anhydrase I (CAI) and KRASG12D (each 1 μM) in two different buffers, PBS and HEPES. Hits are identified as those displaying crosslinking yields >(mean + 2 × standard deviation), indicated by the dashed lines. e) Number of multiple labelling events across the ACT-PhABit screen against CAI in both buffers, PBS and HEPES, alongside an exemplar mass spectrum displaying multiple labelling. | ||
Specificity of crosslinking and buffer selection
A key consideration in screening of reactive fragments is deconvolution of specific and non-specific binding events.52 Typically, reactive groups and screening conditions are tailored to maximise crosslinking of true binders, whilst minimising non-specific crosslinking.53–56 In the case of ACT-PhABits, the nitrilimine intermediate has a reported lifetime of >500 μs,41 which is a relatively long lifetime (cf. carbene <2 ns).39 It was therefore anticipated that the buffer could have a significant effect on the quenching of the reactive species, and thus the extent of non-specific crosslinking. The screens with CAI and KRASG12D were repeated in two buffers, phosphate buffered saline (PBS) (pH 7.2) and HEPES (50 mM, pH 7.4). The mean crosslinking yield for CAI was 8.4% in PBS and 3.7% in HEPES, and for KRASG12D was 4.0% in PBS and 3.3% in HEPES respectively. These results suggest that screening in HEPES reduces the formation of non-specific crosslinking with carboxylate residues, likely through nucleophilic quenching of the nitrilimine species by the buffer (Fig. S6†). The occurrence of non-specific crosslinking in PBS was further confirmed by the observation that several fragments crosslinked to both CAI and KRASG12D in PBS (Fig. 2d, left plot), whilst HEPES afforded selective hits for each protein (Fig. 2d, middle plot). Analysis of the CAI data in the two buffers highlighted that many additional crosslinkers were observed in PBS (Fig. 2d, right plot). Finally, the number of multiple labelling events observed was calculated to provide an indicator of non-specific crosslinking. A total of 60 multiple labelling events were identified across the CAI PBS buffer screen compared to 5 across the CAI HEPES buffer screen (Fig. 2e). The attenuation of crosslinking in HEPES was thus anticipated to limit the occurrence of false positive hits caused by non-specific crosslinking.With optimised screening conditions in place, it was observed that crosslinking yields of hits from the ACT-PhABit screen were notably improved over the previous reported screens using a diazirine library. The screen with CAII afforded 16 ACT-PhABit hits with over 10% crosslinking (cf. just 2 with a diazirine library) and with KRASG12D 26 ACT-PhABit hits yielded over 10% crosslinking (cf. just 6 with the diazirine library).14
Follow-up studies on KRASG12D and BCL6 hits
Selected hits from the KRASG12D and BCL6 screens were chosen for further analysis (Fig. 3a). Compound 4a was the KRASG12D hit with the highest crosslinking yield (23.2%, Fig. 3b) and displayed good selectivity across the six proteins screened (Fig. 3a). Following resynthesis and purification, compound 4a displayed enhanced crosslinking of 42.2%, likely due to the lower concentration of the HTC compound in the crude reaction mixture. Competition studies between compound 4a (50 μM) with known switch I/II KRASG12D inhibitor BI-2852 (5 μM), a nanomolar inhibitor,57 indicated that these compounds did not compete for the same site, suggesting an alternative binding site for compound 4a (Fig. 3c). To identify the site of binding, 4a-labelled KRASG12D was digested trypsin/LysC and analysed by LC-MS/MS, which identified peptide 151QGVD*DAFYTLVR162 as carrying the modification (357.1125 Da) on Asp154 (Fig. 3d). A minor amount of modification also observed on Asp155. Residues Asp154/Asp155 are located away from the functional GDP switch I/II binding site, consistent with the competition data with BI-2852. These residues are located near the allosteric p3 KRAS site, which is formed from residues in loop 7 and the C-terminal end of the α5 helix, and therefore may present an opportunity for optimisation of a novel covalent ligand for this protein (Fig. 3d).58,59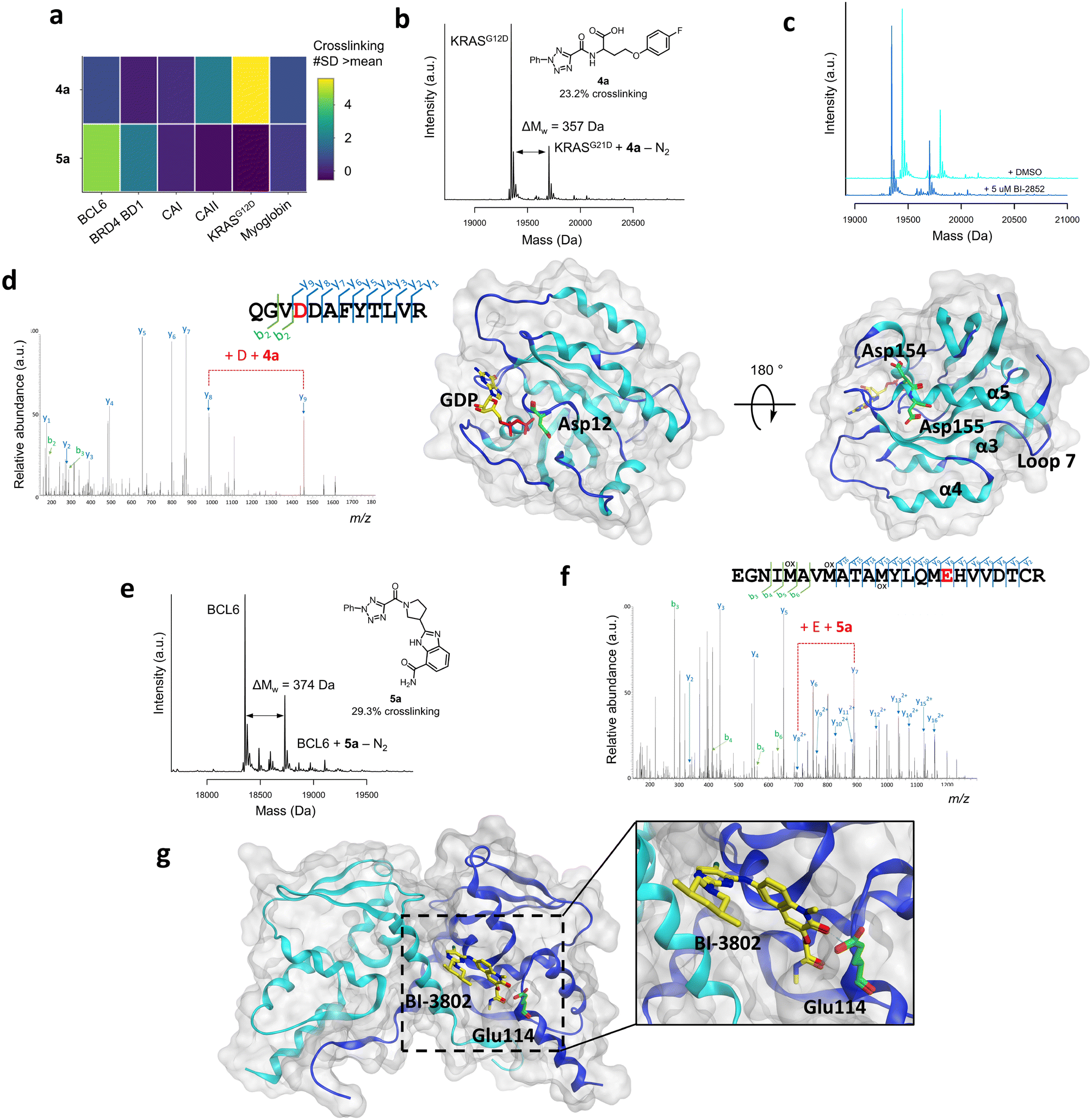 | ||
| Fig. 3 a) Crosslinking yields (#SD >mean) of compounds 4a and 5a across the panel of proteins that were screened, shown by a heatmap. b) Structure and corresponding mass spectrum of ACT-PhABit 4a (100 μM) as the crude reaction product crosslinked to KRASG12D (1 μM). c) Crosslinking competition study between purified ACT-PhABit 4a (50 μM) and known KRASG12D inhibitor, BI-2852 (5 μM). d) MS/MS spectrum of the peptide 151QGVDDAFYTLVR162 crosslinked to 4a, indicating Asp154 as the site of crosslinking and the corresponding X-ray crystal structure of GDP-bound (yellow) KRASG12D (PDB: 5US4) highlighting Asp154 (green) as the major site of crosslinking. e) Structure and corresponding mass spectrum of ACT-PhABit 5a (100 μM) as the crude reaction product crosslinked to BCL6 (1 μM). f) MS/MS spectrum of the peptide 98EGNIMAVMATAMYLQMEHVVDTCR121 crosslinked to 5a, indicating Glu114 as the site of crosslinking. g) The corresponding X-ray crystal structure of BCL6 (PDB: 5MW2) highlighting Glu114 (green) as the major site of crosslinking and known binder BI-3802 (yellow). | ||
The BCL6 hit 5a (Fig. 3a and e) was also resynthesised, purified and binding was confirmed by intact-protein LC-MS (31.7% versus 29.3% crosslinking for purified versus HTC, respectively). LC-MS/MS analysis identified peptide 98EGNIMAVMATAMYLQME*HVVDTCR121 as carrying the modification (374.1491 Da) on Glu114 (Fig. 3f). Glu114 is located at one end of the lateral grooved formed at the BCL6 homodimeric interface (Fig. 3g).22 Multiple reported inhibitors also engage with BCL6 in the lateral groove (e.g. BI-3802,47Fig. 3g), indicating that ACT-PhABit 5a is binding and crosslinking in a functional site, supporting that the covalent modification is driven by a molecular recognition event.22
These data indicate that under suitable screening conditions it is possible to capture specific fragment–protein interactions using the ACT functionality, even for challenging protein pockets, offering an opportunity for the development of specific chemical binders for therapeutically relevant proteins. The hit compounds for KRAS and BCL6 are anticipated to have, at best, weak biochemical activity and are not be expect to have sufficient potency or selectivity to show activity in cells, however, they may represent effective starting points for further development toward more potent and selective tools.
Developing a photosensitiser-based approach for ACT-based photoaffinity labelling
The high energy UV-B light required for the activation of ACT-PhABits (302 nm) is highly damaging to proteins, which absorb light <320 nm.60,61 Amongst the most chromophoric amino acids are tryptophan, tyrosine, histidine, cysteine and phenylalanine, which absorb UV-B light (280–320 nm), leading to radical-based side reactions and scission pathways.60–62 MacMillan and co-workers recently reported that blue light (450 nm) in combination with a suitable photosensitiser could be used for the activation of diazirines via Dexter energy transfer (EnT).63 It was therefore postulated that the ACT functionality could be activated in a similar fashion via a Dexter energy transfer (EnT) mechanism using a combination of longer wavelength, lower energy light (hv) and a suitable photosensitiser (PS).32,63–65 Energy transfer is a photophysical process whereby a photosensitiser (donor), upon excitation and intersystem crossing (ISC) to a long lived (>100 ns) triplet excited state (T1), is able to sensitise a substrate (acceptor) to its corresponding excited state (T1) via a double electron exchange (Fig. 4a).64,65 Such an EnT approach, which would utilise longer wavelength light, was anticipated to offer more tolerant and less damaging irradiation conditions towards larger, more chromophoric, proteins, whilst also being more tolerant of photo-sensitive functional groups in the ACT-PhABits.66,67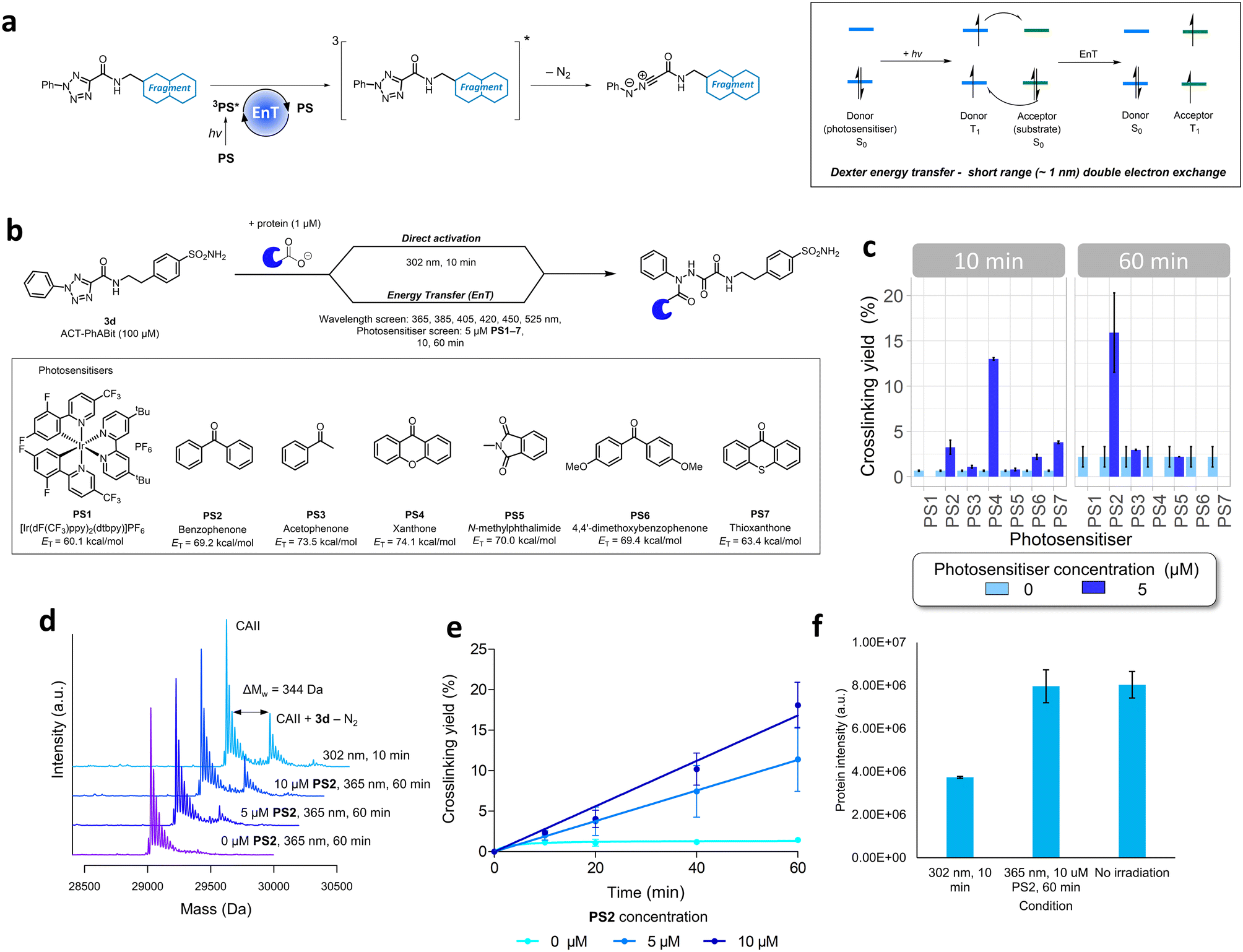 | ||
| Fig. 4 a) Alternative pathway of ACT activation via Dexter energy transfer (EnT) mechanism. An EnT step from a suitably excited photosensitiser was anticipated to indirectly photolyse the ACT functionality and form the respective nitrilimine reactive species. b) Proposed EnT screening strategy to identify suitable photosensitisers and associated wavelengths of activation compared to traditional high energy UV light (302 nm) direct activation. Wavelengths and photosensitisers that were screened are shown. c) Crosslinking yields observed when ACT-PhABit 3d (100 μM) was irradiated at 365 nm (10 & 60 min) with CAII (1 μM) in the presence of each of the seven photosensitisers (PS1–7, 5 μM). Some data has been omitted on account of protein damage, confounding analysis. d) Comparison of crosslinking yields between direct activation (302 nm, 10 min) and energy transfer pathways (0, 5, 10 μM PS2, 365 nm, 60 min) of ACT-PhABit 3d (100 μM) with CAII (1 μM). e) Time and photosensitiser concentration dependent crosslinking of ACT-PhABit 3d (100 μM) with CAII (1 μM) upon activation with 365 nm light. f) Comparison of protein intensity by mass spectrometry after irradiation with 302 nm (10 min), 365 nm (60 min) + PS2 (10 μM) and no irradiation. | ||
A selection of seven photosensitisers were chosen for investigation based on their high triplet energies (ET) (PS1–7, Table S1†).64,65,68 CAII (1 μM) was incubated with ACT-PhABit 3d (100 μM) before irradiation at six wavelengths (365–525 nm, 10–60 min) in the presence of each of the seven photosensitisers (5 μM) (Fig. 4b). Crosslinking events were observed in the cases of benzophenone (PS2) and xanthone (PS4) using 365 nm light (Fig. 4c and S8 and S9†), affording 15.9% and 13.0% crosslinking (n = 2), respectively. No crosslinking was observed using 365 nm light in the absence of the photosensitisers (Fig. 4c and S8 and S9†). For comparison, ACT-PhABit 3d was also irradiated at 302 nm for 10 min as a control, affording a crosslinking yield of 23.9% (Fig. 4d). Benzophenone PS2 was selected for further investigation due to the cleaner mass spectra observed when compared to xanthone PS4 (Fig. S8 and S9†). Additional studies utilising PS2 as an ACT photosensitiser revealed the time- and concentration-dependent behaviour of the crosslinking (Fig. 4d and e and S10†). A maximum crosslinking yield of 18.1% was obtained by irradiating ACT-PhABit 3d in the presence of 10 μM PS2 for 60 min.
Finally, the effect of irradiation on protein integrity was investigated. The EnT irradiation process at 365 nm (60 min) + PS2 (10 μM) resulted in negligible reduction of CAII protein MS signal whereas irradiation at 302 nm (10 min) showed a significant decrease in the protein MS signal (Fig. 4f). Furthermore, the EnT conditions were comparable to a control sample of CAII that was subjected to no irradiation. Thus, this EnT approach offers a milder method for the activation of ACT groups for photolabeling compared to the original direct activation conditions, whilst attaining comparable crosslinking yields.
Conclusion
Reactive fragment screening has become integral in hit identification in recent years. While the focus of these efforts has centred on targeting cysteine residues, modalities that enable covalent modification ‘beyond cysteine’ are highly sought after in order to expand the ligandable proteome. The high proteome occurrence of carboxylate-containing residues, aspartate and glutamate (>12%), makes them good targets for reactive fragment screening. This work describes the development of a direct-to-biology photoreactive carboxylate-targeting reactive fragment screening platform, which enables the light-induced covalent capture and identification of fragment–protein binding interactions.The implementation of direct-to-biology high-throughput chemistry (D2B-HTC) allowed the rapid synthesis of an ACT photoreactive fragment (ACT-PhABit) library for direct screening against a panel of therapeutically relevant proteins, which enabled the identification of multiple fragment hits.42 The identification of numerous chemotypes known to bind to both BRD4-BD1 and carbonic anhydrase II indicated that specific reversible recognition events were being captured. Further characterisation of BCL6 and KRASG12D hits by tandem LC-MS/MS revealed the selectivity of the ACT functionality for carboxylate residues and identified both orthosteric and allosteric sites of binding.
Hits afforded higher crosslinking yields than observed previously with diazirine-based fragment screening (PhABits), which facilitates both hit calling and follow-up studies.14,42 This is consistent with the formation of a longer-lived nitrilimine intermediate upon photoactivation of the ACT, which reacts preferentially with carboxylate protein side chains. By contrast, the diazirine forms the shorter-lived carbene upon photoactivation, which can be rapidly quenched by water, often leading to low crosslinking yields.
Finally, a novel photosensitised approach towards ACT activation was developed utilising 365 nm light and benzophenone, which obviated the need for damaging UV-B light (302 nm). This modification to the screening platform will be especially valuable with larger/low-stability proteins, which are typically more sensitive to exposure to UV light, often prohibiting the use of photoreactive screening platforms.
The ACT-PhABit screening platform offers an approach towards the development of carboxylate-targeting chemical probes, which presents a novel and complementary addition to the reactive fragment toolbox. Future opportunities will likely lie in the use of electrophilic carboxylate-targeting reactive fragments, such as reactive fragments bearing 2H-azirines or isoxazolium salts. The electrophilic nature of these groups could offer improved crosslinking yields by driving towards complete target occupancy over time and could also prove more versatile in biomedical research by obviating the requirement for high energy light.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the GlaxoSmithKline/University of Strathclyde Collaborative PhD programme for funding and chemical resources and the EPSRC for funding via Prosperity Partnership EP/S035990/1.References
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H.-L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed.
- E. V. Vinogradova, X. Zhang, D. Remillard, D. C. Lazar, R. M. Suciu, Y. Wang, G. Bianco, Y. Yamashita, V. M. Crowley, M. A. Schafroth, M. Yokoyama, D. B. Konrad, K. M. Lum, G. M. Simon, E. K. Kemper, M. R. Lazear, S. Yin, M. M. Blewett, M. M. Dix, N. Nguyen, M. N. Shokhirev, E. N. Chin, L. L. Lairson, B. Melillo, S. L. Schreiber, S. Forli, J. R. Teijaro and B. F. Cravatt, Cell, 2020, 182, 1009–1026 CrossRef CAS PubMed.
- M. D. Olp, D. J. Sprague, C. J. Goetz, S. G. Kathman, S. L. Wynia-Smith, S. Shishodia, S. B. Summers, Z. Xu, A. V. Statsyuk and B. C. Smith, ACS Chem. Biol., 2020, 15, 1036–1049 CrossRef CAS PubMed.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence, S. R. Johnson, I. Narvaiza, E. Saez and B. F. Cravatt, Cell, 2017, 168, 527–541 CrossRef CAS PubMed.
- Y. Wang, M. M. Dix, G. Bianco, J. R. Remsberg, H.-Y. Lee, M. Kalocsay, S. P. Gygi, S. Forli, G. Vite, R. M. Lawrence, C. G. Parker and B. F. Cravatt, Nat. Chem., 2019, 11, 1113–1123 CrossRef CAS PubMed.
- D. S. Johnson, E. Weerapana and B. F. Cravatt, Future Med. Chem., 2010, 2, 949–964 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- W. Lu, M. Kostic, T. Zhang, J. Che, M. P. Patricelli, L. H. Jones, E. T. Chouchani and N. S. Gray, RSC Chem. Biol., 2021, 2, 354–367 RSC.
- C. Dubiella, B. J. Pinch, K. Koikawa, D. Zaidman, E. Poon, T. D. Manz, B. Nabet, S. He, E. Resnick, A. Rogel, E. M. Langer, C. J. Daniel, H.-S. Seo, Y. Chen, G. Adelmant, S. Sharifzadeh, S. B. Ficarro, Y. Jamin, B. Martins da Costa, M. W. Zimmerman, X. Lian, S. Kibe, S. Kozono, Z. M. Doctor, C. M. Browne, A. Yang, L. Stoler-Barak, R. B. Shah, N. E. Vangos, E. A. Geffken, R. Oren, E. Koide, S. Sidi, Z. Shulman, C. Wang, J. A. Marto, S. Dhe-Paganon, T. Look, X. Z. Zhou, K. P. Lu, R. C. Sears, L. Chesler, N. S. Gray and N. London, Nat. Chem. Biol., 2021, 17, 954–963 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
- Y. Shin, J. W. Jeong, R. P. Wurz, P. Achanta, T. Arvedson, M. D. Bartberger, I. D. G. Campuzano, R. Fucini, S. K. Hansen, J. Ingersoll, J. S. Iwig, J. R. Lipford, V. Ma, D. J. Kopecky, J. McCarter, T. San Miguel, C. Mohr, S. Sabet, A. Y. Saiki, A. Sawayama, S. Sethofer, C. M. Tegley, L. P. Volak, K. Yang, B. A. Lanman, D. A. Erlanson and V. J. Cee, ACS Med. Chem. Lett., 2019, 10, 1302–1308 CrossRef CAS PubMed.
- H. Johansson, Y.-C. Isabella Tsai, K. Fantom, C.-W. Chung, S. Kümper, L. Martino, D. A. Thomas, H. C. Eberl, M. Muelbaier, D. House and K. Rittinger, J. Am. Chem. Soc., 2019, 141, 2703–2712 CrossRef CAS PubMed.
- E. K. Grant, D. J. Fallon, M. M. Hann, K. G. M. Fantom, C. Quinn, F. Zappacosta, R. S. Annan, C.-w. Chung, P. Bamborough, D. P. Dixon, P. Stacey, D. House, V. K. Patel, N. C. O. Tomkinson and J. Bush, Angew. Chem., Int. Ed., 2020, 59, 21096–21105 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Nat. Chem., 2009, 1, 187–192 CrossRef CAS PubMed.
- D. E. Scott, A. G. Coyne, S. A. Hudson and C. Abell, Biochemistry, 2012, 51, 4990–5003 CrossRef CAS PubMed.
- S. G. Kathman, Z. Xu and A. V. Statsyuk, J. Med. Chem., 2014, 57, 4969–4974 CrossRef CAS PubMed.
- L. Switzar, M. Giera and W. M. A. Niessen, J. Proteome Res., 2013, 12, 1067–1077 CrossRef CAS PubMed.
- A. Jones, X. Zhang and X. Lei, Cell Chem. Biol., 2017, 24, 537–539 CrossRef CAS PubMed.
- S. Ray and A. S. Murkin, Biochemistry, 2019, 58, 5234–5244 CrossRef CAS PubMed.
- H. Mukherjee and N. P. Grimster, Curr. Opin. Chem. Biol., 2018, 44, 30–38 CrossRef CAS PubMed.
- M. Teng, S. B. Ficarro, H. Yoon, J. Che, J. Zhou, E. S. Fischer, J. A. Marto, T. Zhang and N. S. Gray, ACS Med. Chem. Lett., 2020, 11, 1269–1273 CrossRef CAS PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- J. W. Brulet, A. L. Borne, K. Yuan, A. H. Libby and K.-L. Hsu, J. Am. Chem. Soc., 2020, 142, 8270–8280 CrossRef CAS PubMed.
- X. Wan, T. Yang, A. Cuesta, X. Pang, T. E. Balius, J. J. Irwin, B. K. Shoichet and J. Taunton, J. Am. Chem. Soc., 2020, 142, 4960–4964 CrossRef CAS PubMed.
- H. S. Hahm, E. K. Toroitich, A. L. Borne, J. W. Brulet, A. H. Libby, K. Yuan, T. B. Ware, R. L. McCloud, A. M. Ciancone and K.-L. Hsu, Nat. Chem. Biol., 2020, 16, 150–159 CrossRef CAS PubMed.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- K. Gilbert, A. Vuorinen, A. Aatkar, P. Pogany, J. Pettinger, E. Grant, J. Kirkpatrick, K. Rittinger, D. House, G. Burley and J. Bush, ACS Chem. Biol. DOI:10.1021/acschembio.2c00633 , in press.
- N. Ma, J. Hu, Z.-M. Zhang, W. Liu, M. Huang, Y. Fan, X. Yin, J. Wang, K. Ding, W. Ye and Z. Li, J. Am. Chem. Soc., 2020, 142, 6051–6059 CrossRef CAS PubMed.
- P. Martín-Gago, E. K. Fansa, M. Winzker, S. Murarka, P. Janning, C. Schultz-Fademrecht, M. Baumann, A. Wittinghofer and H. Waldmann, Cell Chem. Biol., 2017, 24, 589–597 CrossRef PubMed.
- K. Bach, B. L. H. Beerkens, P. R. A. Zanon and S. M. Hacker, ACS Cent. Sci., 2020, 6, 546–554 CrossRef CAS PubMed.
- A. Herner, J. Marjanovic, T. M. Lewandowski, V. Marin, M. Patterson, L. Miesbauer, D. Ready, J. Williams, A. Vasudevan and Q. Lin, J. Am. Chem. Soc., 2016, 138, 14609–14615 CrossRef CAS PubMed.
- K. A. Mix and R. T. Raines, Org. Lett., 2015, 17, 2358–2361 CrossRef CAS PubMed.
- K. Cheng, J.-S. Lee, P. Hao, S. Q. Yao, K. Ding and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 15044–15048 CrossRef CAS PubMed.
- N. A. McGrath, K. A. Andersen, A. K. F. Davis, J. E. Lomax and R. T. Raines, Chem. Sci., 2015, 6, 752–755 RSC.
- K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233–3244 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J.-S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002–2006 CrossRef CAS PubMed.
- R. Miyajima, K. Sakai, Y. Otani, T. Wadatsu, Y. Sakata, Y. Nishikawa, M. Tanaka, Y. Yamashita, M. Hayashi, K. Kondo and T. Hayashi, ACS Chem. Biol., 2020, 15, 2364–2373 CrossRef CAS PubMed.
- J. P. Toscano, M. S. Platz and V. Nikolaev, J. Am. Chem. Soc., 1995, 117, 4712–4713 CrossRef CAS.
- J. Wang, J. Kubicki, H. Peng and M. S. Platz, J. Am. Chem. Soc., 2008, 130, 6604–6609 CrossRef CAS PubMed.
- K. Bhattacharyya, D. Ramaiah, P. K. Das and M. V. George, J. Photochem., 1987, 36, 63–84 CrossRef CAS.
- R. P. Thomas, R. E. Heap, F. Zappacosta, E. K. Grant, P. Pogány, S. Besley, D. J. Fallon, M. M. Hann, D. House, N. C. O. Tomkinson and J. T. Bush, Chem. Sci., 2021, 12, 12098–12106 RSC.
- I. A. Prior, P. D. Lewis and C. Mattos, Cancer Res., 2012, 72, 2457–2467 CrossRef CAS PubMed.
- Y. Kamada, N. Sakai, S. Sogabe, K. Ida, H. Oki, K. Sakamoto, W. Lane, G. Snell, M. Iida, Y. Imaeda, J. Sakamoto and J. Matsui, J. Med. Chem., 2017, 60, 4358–4368 CrossRef CAS PubMed.
- L. C. Cerchietti, A. F. Ghetu, X. Zhu, G. F. Da Silva, S. Zhong, M. Matthews, K. L. Bunting, J. M. Polo, C. Farès, C. H. Arrowsmith, S. N. Yang, M. Garcia, A. Coop, A. D. MacKerell, G. G. Privé and A. Melnick, Cancer Cell, 2010, 17, 400–411 CrossRef CAS PubMed.
- T. Sameshima, T. Yamamoto, O. Sano, S. Sogabe, S. Igaki, K. Sakamoto, K. Ida, M. Gotou, Y. Imaeda, J. Sakamoto and I. Miyahisa, Biochemistry, 2018, 57, 1369–1379 CrossRef CAS PubMed.
- N. Kerres, S. Steurer, S. Schlager, G. Bader, H. Berger, M. Caligiuri, C. Dank, J. R. Engen, P. Ettmayer, B. Fischerauer, G. Flotzinger, D. Gerlach, T. Gerstberger, T. Gmaschitz, P. Greb, B. Han, E. Heyes, R. E. Iacob, D. Kessler, H. Kölle, L. Lamarre, D. R. Lancia, S. Lucas, M. Mayer, K. Mayr, N. Mischerikow, K. Mück, C. Peinsipp, O. Petermann, U. Reiser, D. Rudolph, K. Rumpel, C. Salomon, D. Scharn, R. Schnitzer, A. Schrenk, N. Schweifer, D. Thompson, E. Traxler, R. Varecka, T. Voss, A. Weiss-Puxbaum, S. Winkler, X. Zheng, A. Zoephel, N. Kraut, D. McConnell, M. Pearson and M. Koegl, Cell Rep., 2017, 20, 2860–2875 CrossRef CAS PubMed.
- A. C. Belkina and G. V. Denis, Nat. Rev. Cancer, 2012, 12, 465–477 CrossRef CAS PubMed.
- J. Seal, Y. Lamotte, F. Donche, A. Bouillot, O. Mirguet, F. Gellibert, E. Nicodeme, G. Krysa, J. Kirilovsky, S. Beinke, S. McCleary, I. Rioja, P. Bamborough, C.-W. Chung, L. Gordon, T. Lewis, A. L. Walker, L. Cutler, D. Lugo, D. M. Wilson, J. Witherington, K. Lee and R. K. Prinjha, Bioorg. Med. Chem. Lett., 2012, 22, 2968–2972 CrossRef CAS PubMed.
- S. Picaud, C. Wells, I. Felletar, D. Brotherton, S. Martin, P. Savitsky, B. Diez-Dacal, M. Philpott, C. Bountra, H. Lingard, O. Fedorov, S. Müller, P. E. Brennan, S. Knapp and P. Filippakopoulos, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19754–19759 CrossRef CAS PubMed.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- J. Park, M. Koh, J. Y. Koo, S. Lee and S. B. Park, ACS Chem. Biol., 2016, 11, 44–52 CrossRef CAS PubMed.
- D. J. Fallon, S. Lehmann, C.-w. Chung, A. Phillipou, C. Eberl, K. G. M. Fantom, F. Zappacosta, V. K. Patel, M. Bantscheff, C. J. Schofield, N. C. O. Tomkinson and J. T. Bush, Chem. – Eur. J., 2021, 27, 17880–17888 CrossRef CAS PubMed.
- E. K. Grant, D. J. Fallon, H. C. Eberl, K. G. M. Fantom, F. Zappacosta, C. Messenger, N. C. O. Tomkinson and J. T. Bush, Angew. Chem., Int. Ed., 2019, 58, 17322–17327 CrossRef CAS PubMed.
- A. V. West, G. Muncipinto, H.-Y. Wu, A. C. Huang, M. T. Labenski, L. H. Jones and C. M. Woo, J. Am. Chem. Soc., 2021, 143, 6691–6700 CrossRef CAS PubMed.
- L. P. Conway, A. M. Jadhav, R. A. Homan, W. Li, J. S. Rubiano, R. Hawkins, R. M. Lawrence and C. G. Parker, Chem. Sci., 2021, 12, 7839–7847 RSC.
- D. Kessler, M. Gmachl, A. Mantoulidis, L. J. Martin, A. Zoephel, M. Mayer, A. Gollner, D. Covini, S. Fischer, T. Gerstberger, T. Gmaschitz, C. Goodwin, P. Greb, D. Häring, W. Hela, J. Hoffmann, J. Karolyi-Oezguer, P. Knesl, S. Kornigg, M. Koegl, R. Kousek, L. Lamarre, F. Moser, S. Munico-Martinez, C. Peinsipp, J. Phan, J. Rinnenthal, J. Sai, C. Salamon, Y. Scherbantin, K. Schipany, R. Schnitzer, A. Schrenk, B. Sharps, G. Siszler, Q. Sun, A. Waterson, B. Wolkerstorfer, M. Zeeb, M. Pearson, S. W. Fesik and D. B. McConnell, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15823–15829 CrossRef CAS PubMed.
- B. J. Grant, S. Lukman, H. J. Hocker, J. Sayyah, J. H. Brown, J. A. McCammon and A. A. Gorfe, PLoS One, 2011, 6, 1–10 Search PubMed.
- P. Prakash, J. F. Hancock and A. A. Gorfe, Proteins, 2015, 83, 898–909 CrossRef CAS PubMed.
- M. J. Davies and R. J. W. Truscott, J. Photochem. Photobiol., B, 2001, 63, 114–125 CrossRef CAS PubMed.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 CrossRef CAS PubMed.
- M. J. Davies, Photochem. Photobiol. Sci., 2004, 3, 17–25 CrossRef CAS PubMed.
- J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker, E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Science, 2020, 367, 1091–1097 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- S. Yakubov and J. P. Barham, Beilstein J. Org. Chem., 2020, 16, 2151–2192 CrossRef CAS PubMed.
- E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793–797 CrossRef CAS PubMed.
- Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00453d |
| This journal is © The Royal Society of Chemistry 2023 |