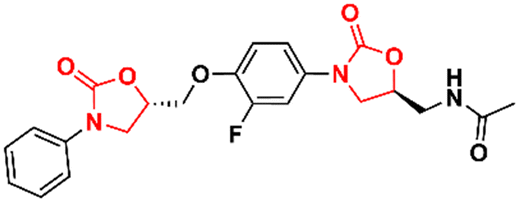
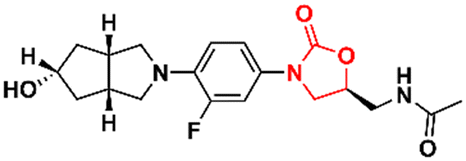
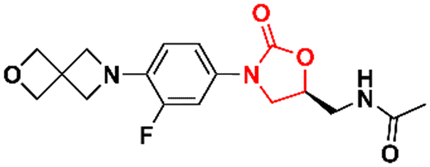
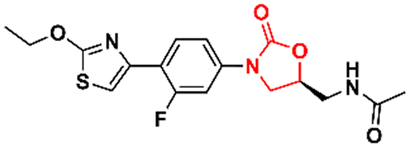
DOI:
10.1039/D2MD00415A
(Review Article)
RSC Med. Chem., 2023,
14, 823-847
Oxazolidinones as versatile scaffolds in medicinal chemistry
Received
23rd November 2022
, Accepted 6th February 2023
First published on 8th February 2023
Abstract
Oxazolidinone is a five-member heterocyclic ring with several biological applications in medicinal chemistry. Among the three possible isomers, 2-oxazolidinone is the most investigated in drug discovery. Linezolid was pioneered as the first approved drug containing an oxazolidinone ring as the pharmacophore group. Numerous analogues have been developed since its arrival on the market in 2000. Some have succeeded in reaching the advanced stages of clinical studies. However, most oxazolidinone derivatives reported in recent decades have not reached the initial stages of drug development, despite their promising pharmacological applications in a variety of therapeutic areas, including antibacterial, antituberculosis, anticancer, anti-inflammatory, neurologic, and metabolic diseases, among other areas. Therefore, this review article aims to compile the efforts of medicinal chemists who have explored this scaffold over the past decades and highlight the potential of the class for medicinal chemistry.
Introduction
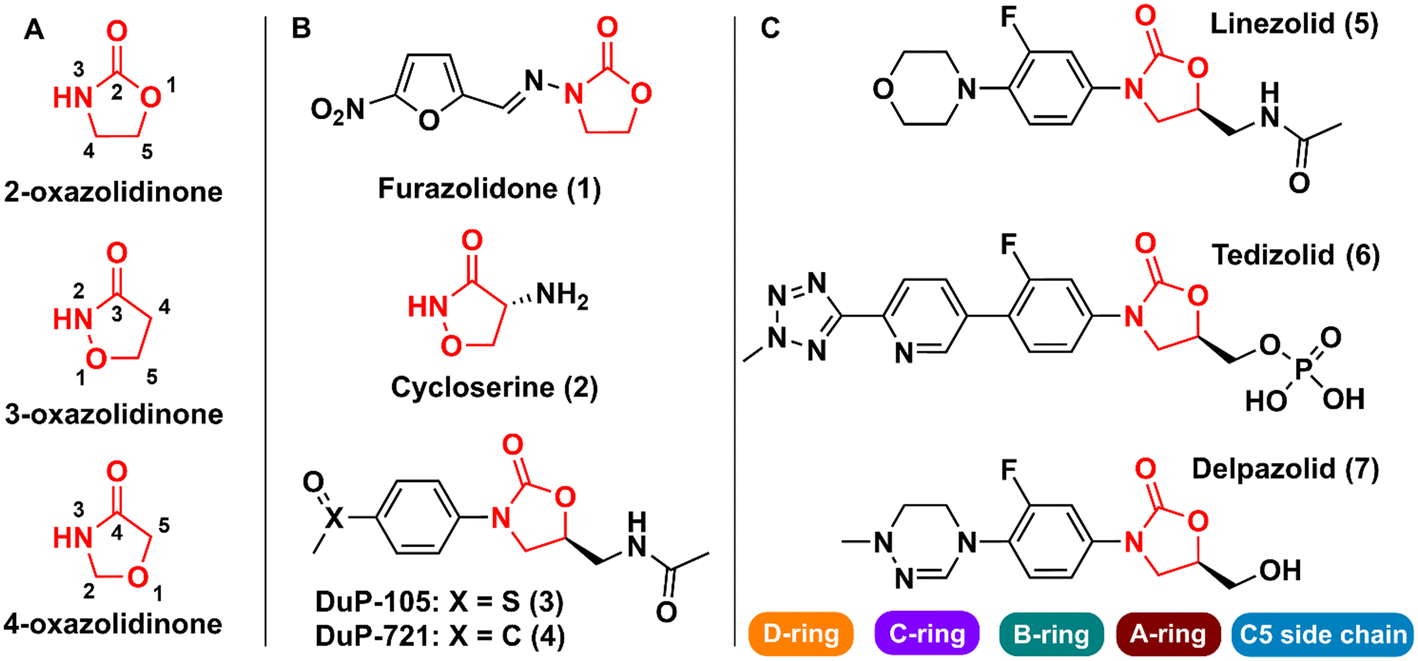
Oxazolidinones are a class of five-membered heterocyclic compounds containing both nitrogen and an oxygen atom in their structure and find broad application in organic and medicinal chemistry. Oxazolidinones can exist as different structural isomers depending on the reciprocal position of oxygen and nitrogen atoms in the five-membered ring. Consequently, different nomenclatures are used to represent the different isomers, namely 2-, 3-, and 4-oxazolidinone. Oxazolidinones constitute an important class in medicinal chemistry, since they can act as bioisosteres of different chemical groups, e.g., carbamates, thiocarbamates, ureas, and amides. This structural similarity between oxazolidinones (especially 2-oxazolidinone) and the aforementioned groups confers on the former certain critical druglike characteristics such as the formation of hydrogen bonds with amino acid residues and a higher metabolic and chemical stability because carbamate is cyclized in a five-membered ring, unlike noncyclic carbamates, for example, that are more prone to hydrolysis.1
The first drug that contained the oxazolidinone scaffold was furazolidone (2-oxazolidinone) (1) (Fig. 1). Furazolidone, a synthetic nitrofuran-oxazolidinone, was discovered in the 1940s as an antimicrobial agent targeting bacterial DNA.2 Furazolidone was likely the initial candidate that led to the genesis of further development of the class of oxazolidinone as antibacterials.3 Later, the oxazolidinone cycloserine (3-oxazolidinone) (2) (Fig. 1), was developed and used as an antitubercular drug since 1956.4 Nevertheless, it was only after the discovery in the late 1980s of N-aryl-oxazolidinones, a new class of synthetic antibacterial agents, that oxazolidinones gained notoriety in medicinal chemistry.5 The first representatives of this new class of antibacterial agents were compounds (3) (Dup-105) and (4) (DuP-721) (Fig. 1), formally reported by E.I. du Pont de Nemours & Company (DuPont) at the 1987 Interscience Conference on Antimicrobial Agents and Chemotherapy.6 Subsequently, researchers from the Pharmacia & Upjohn company started an iterative medicinal chemistry campaign aimed at further developing novel oxazolidinones as commercially available antibiotics.7 Linezolid (LZD) (5) (Fig. 1), the first commercially available member of this new class of compounds, was discovered in 1996 by Pharmacia & Upjohn and approved in 2000 for clinical use by the US Food and Drug Administration (FDA) for the treatment of infectious diseases caused by Gram-positive bacteria resistant to other antibiotics.8,9 LNZ displays potent activity against a wide range of Gram-positive bacteria, including streptococci, vancomycin-resistant Enterococci (VRE), and methicillin-resistant Staphylococcus aureus (MRSA).7 Recently, LZD was included in the list of antitubercular drugs recommended by the World Health Organization (WHO) for use in long multidrug-resistant tuberculosis (MDR-TB) treatment regimens.10 The mechanism of action of LZD involves binding to the 50S subunit of the bacterial ribosome, more specifically, at the A site of the peptidyl transferase active centre, thus preventing the interface with the 30S subunit, and consequently the formation of the 70S initiation complex, ultimately leading to the inhibition of bacterial protein synthesis.11–14 In 2014, the FDA approved the second-generation representative of the oxazolidinone class, namely tedizolid (6) (Fig. 1), for the treatment of acute bacterial skin and skin structure infections caused by certain susceptible bacteria, including Staphylococcus aureus, several Streptococcus species, and Enterococcus faecalis.15
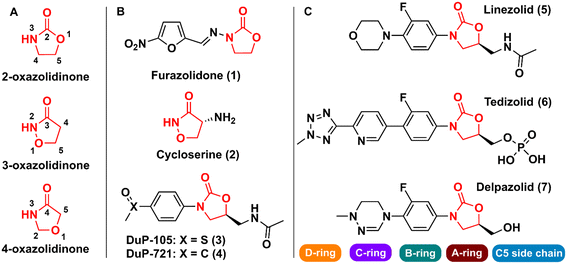 |
| | Fig. 1 A) Oxazolidinone isomers; B) first reported bioactive oxazolidinone derivatives reported; C) drugs containing the oxazolidinone scaffold. | |
Shortly after LZD approval, drug-resistant strains began to appear. This phenomenon led to the research of a new generation of oxazolidinones.16 Various analogues were developed, and some were able to reach clinical studies, including posizolid, ranbezolid, radezolid, and cadazolid, just to name a few. Currently, three new derivatives of oxazolidinones are undergoing clinical trials for the treatment of drug-resistant tuberculosis (TB), namely delpazolid (7) (Fig. 1), sutezolid, and TBI-223.17 The structural difference between these drug candidates and LZD is mainly in the C- and D-rings (Fig. 1).
During last decades, multiple studies have highlighted the potential of oxazolidinones to exert multifaceted biological activities. The majority of the studies reported in the literature concern oxazolidinones designed as antibacterial and/or antimycobacterial agents. Several reviews have already been published addressing these derivatives.18–20 Nevertheless, there is a lack of reviews covering the oxazolidinones developed for application in other therapeutic areas and, to the best of our knowledge, no review article to date has fully covered these compounds. Therefore, this review will address the oxazolidinone-based derivatives reported since 2011 and discovered for the most diverse therapeutic applications, including antibacterial, antitubercular, anticancer, anti-inflammatory, antiviral, and neurological and metabolic disorders. It should be noted that this review will cover only fully synthetic (so-called small molecules) 2-oxazolidinone derivatives. Compounds obtained from natural sources21,22 are not part of the scope of this review.
Oxazolidinones as antibacterial agents
Bacteria naturally develop resistance over time as they are exposed to antibiotics, which has been observed since antibiotics were first introduced. However, the emergence and spread of antibiotic resistance have been accelerated by the overuse and misuse of antibiotics while a lack of new antibiotics is being developed.23 The ever-increasing number of resistant bacteria is described as a crisis by many public health organizations. In 2019, it was reported that drug-resistant bacterial infections directly caused 1.27 million deaths worldwide.24 The urgency of this matter was emphasized in the review on antimicrobial resistance, as it predicts 10 million deaths per year by 2050 if there is no intervention.25 There is a growing concern about multidrug-resistant pathogens, Enterococcus spp, S. aureus, K. pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp, which were termed ESKAPE, as they have been shown to cause most nosocomial infection.26 Carbapenem-resistant Gram-negative bacteria, classified as critical in the pathogen prioritization report by WHO, drastically increase the cause of severe infection, which represents a global emergency worldwide.27 The rise of the third-generation cephalosporin in E. coli and K. pneumonia leaves us with limited antibiotic therapy for Gram-negative bacterial infections.28 Bacterial infections, which have been susceptible to standard antibiotic regimes for decades, are now becoming resistant to all major antimicrobial treatments, undermining the efficacy of available antibiotics. This highlights the urgent need for novel therapeutic agents to tackle drug-resistant bacteria. Due to the success of LZD, oxazolidinones have been extensively investigated as antibacterials.18,20 In this section, we will highlight oxazolidinone-based derivatives that have demonstrated potent and promising activity against several Gram-positive and Gram-negative bacteria.
Oxazolidinone derivatives active against Gram-positive bacteria
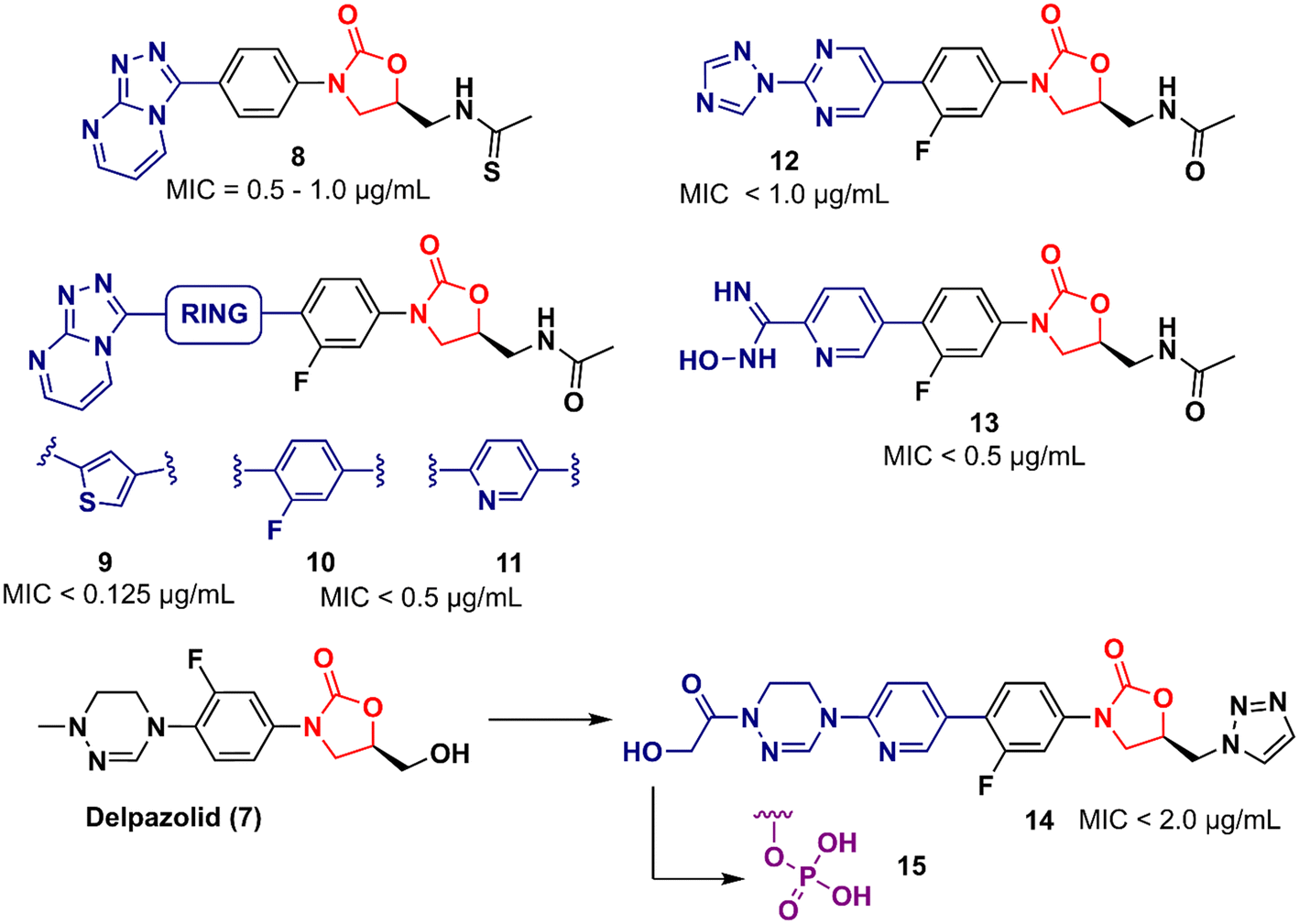
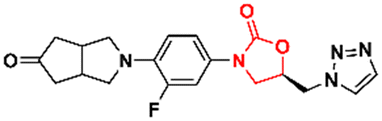
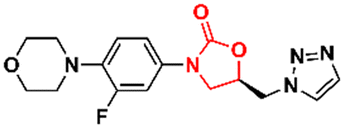
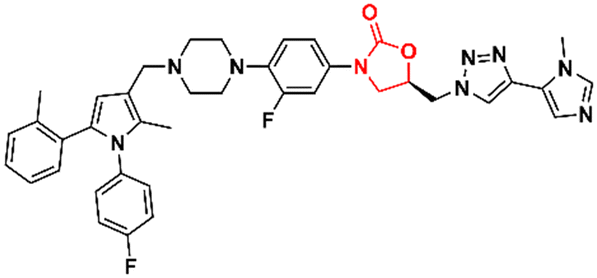
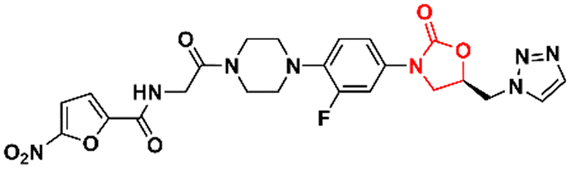
In 2011, Khera et al. described a series of 1,2,4-triazolo[4,3-α]pyrimidine oxazolidinone with potent inhibitory activity against Gram-positive bacteria.29 Initially, they disclosed a series of 22 derivatives and found compound 8 (Fig. 2) to be the most promising with minimum inhibitory concentration (MIC) values of 1.0 μg mL−1 against E. faecalis ATCC 29212 and S. aureus ATCC 25923 and 0.5 μg mL−1 against S. aureus MRSA 43300 and S. epidermedis ATCC 12228. Compound 8 also showed metabolic stability in the human liver microsome. Subsequently, the same authors reported a second series of related analogues in which an additional aromatic ring (C-ring) was inserted into the 1,2,4-triazolo[4,3-α]pyrimidine and the 3-fluorophenyl oxazolidinone subunit.30 Several aromatic rings were assessed, including 2-fluorophenyl, pyridine, phenyl, methoxy phenyl, and thiophene. Compound 9 (Fig. 2) containing the 2,4-disubstituted thiophene system was shown to be highly potent against Gram-positive strains with MIC values below 0.125 μg mL−1. Furthermore, analogues containing 2-fluorophenyl (10), and the pyridyl group (11) (Fig. 2) also displayed remarkable activity with MIC values below 0.5 μg mL−1.30 Compound 12 (RBx 11760) (Fig. 2) is another biaryl oxazolidinone developed against Clostridium difficile with potent in vivo activity.31In vitro evaluation demonstrated MIC90 below 1.0 mg L−1 (range 0.25–1.0 mg L−1) against C. difficile ATCC 43255, C. difficile ATCC 43596, C. difficile NCTC 11223, C. difficile NAP1/027/BI type strain 2009155 and clinical isolates from 2004118, and C. difficile (45 strains). Further studies revealed that compound 12 exhibited concentration-dependent bactericidal activity and reduced de novo toxin production, as well as sporulation in several isolates of C. difficile. In vivo studies using a hamster model of C. difficile gastrointestinal infection demonstrated that compound 12 (at a dose of 50 mg kg−1) led to the survival of 100% of treated animals, while the positive control group treated with metronidazole and vancomycin resulted in 0 and 50%, respectively. Similarly to LZD, compound 12 was also found to be a potent protein synthesis inhibitor.31
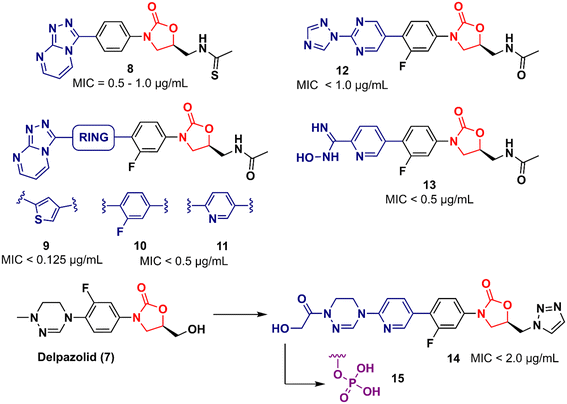 |
| | Fig. 2 Oxazolidinones as antibacterial agents. | |
LegoChem Biosciences, Inc. (Daejeon, South Korea) has developed several oxazolidinone derivatives as antibacterial agents in the last decade.32–35 For example, compound 13 (LCB01-0062) (Fig. 2) showed in vitro activity against clinical isolates of Gram-positive bacteria compared to several antibacterial drugs, including linezolid, erythromycin, ciprofloxacin, and vancomycin. In a massive effort, compound 13 was evaluated against 400 clinical isolates of Gram-positive bacteria obtained from general hospitals in South Korea. Compound 13 displayed MIC90 values below 0.5 mg L−1 against all strains tested with bacteriostatic activity similar to that of linezolid. Furthermore, in vivo studies in a mouse model of infection caused by S. aureus Giorgio (methicillin-susceptible) showed that compound 13 was more active than linezolid.35 Compound 14 (LCB 01-0648) (Fig. 2) is another representative that was developed after a lead optimization campaign on delpazolid (7) (LCB01-0371).33 The medicinal chemistry effort aimed to increase the activity against linezolid-resistant strains by modifying the C-5 side chain and the C-ring by adding a pyridinyl ring attached to a cyclic amidrazone subunit on the D-ring. Compound 14 displayed MIC values below 2.0 μg mL−1 against S. aureus, S. aureus MR, E. faecalis linezolid resistant and E. faecium linezolid resistant. Although this compound was not the most potent in the series, it was selected for further evaluation because it had the best safety profile in the inhibition and myelotoxicity of monoamine oxidase (MAO). Further in vitro studies exhibited MIC90 values below 0.5 μg mL−1 against 184 clinical isolates of Enterococci, highlighting its potential against multidrug-resistant (MDR) Gram-positive cocci.33 Further development of compound 14 led to its prodrug, compound 15 (Fig. 2).32 Pharmacokinetic (PK) studies in a rat model demonstrated that most of compound 15 was converted to the parent compound 14. The AUClast (area under the concentration–time curve from time zero to the last sampling time) of compound 15 was found to be 5.08 mg h L−1 whilst the AUClast of compound 14 was 85.0 mg h L−1. Compound 15 was more potent than linezolid in both a mouse model with systemic infection and a soft tissue infection model against S. aureus Giorgio and S. aureus P125 (methicillin-resistant), respectively. Furthermore, it did not show any change in reticulocyte count, suggesting that it was not associated with myelosuppression.32
Tricyclic oxazolidinone derivatives have also been reported to be promising scaffolds for the development of antibacterial agents. Yang's group has published several analogues containing this nucleus.36–39 For instance, benzoxazinyl-oxazolidinone 16 (Fig. 3) was discovered after an extensive medicinal chemistry campaign as a highly potent antibacterial 3 to 4 times more active than linezolid in vivo.38 Compound 16 displayed MIC values below 0.5 μg mL−1 against a large panel of Gram-positive bacteria, including Staphylococcus aureus ATCC 29213, MRSA, methicillin-resistant Staphylococcus epidermidis (MRSE), penicillin-resistant Streptococcus pneumoniae (PRSP), and Enterococcus faecalis. In addition, it also showed potent activity against a panel of linezolid-resistant bacteria with MIC values ranging from 0.125 to 2.0 μg mL−1. Compound 16 was also evaluated in a mouse systemic infection model and exhibited an efficacy of 4 times (ED50: 2.5 mg kg−1) and 3-times (ED50: <5.0 mg kg−1) superior to LZD against S. aureus and MRSA, respectively. PK studies demonstrated a favourable profile for compound 16 for oral and intravenous administration.38
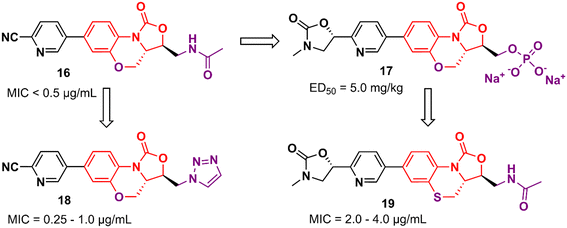 |
| | Fig. 3 Tricyclic oxazolidinone derivatives with activity against Gram-positive bacteria. | |
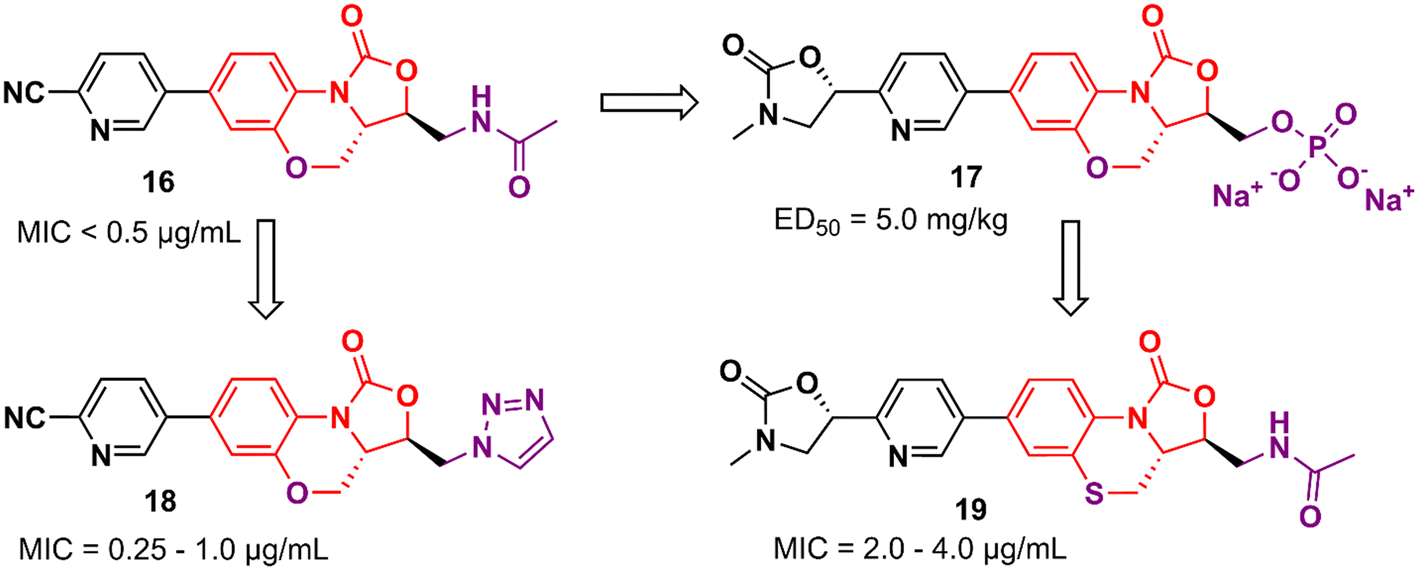
However, compound 16 showed poor solubility that limited its development in an intravenous formulation. Therefore, a lead optimization strategy was carried out to avoid the issue. Three approaches were used for this purpose: the incorporation of polar groups, the disruption of molecular planarity, and the prodrug strategy. Prodrug 17 (YG-056SP) (Fig. 3) emerged as the most promising derivative with improved water solubility (>22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold greater than compound 16), good PK profile, and superior in vivo activity than linezolid. The ED50 of compound 17 was 5.0 mg kg−1 (2-times higher than that of linezolid). Additionally, it showed a significant reduction in hERG inhibition compared to compound 16 (IC50 > 40 μM for the bioactive parent).39 Compounds 18 and 19 (Fig. 3) were later reported as a continuing effort to identify new lead derivatives.40,41 The drug design behind these two analogues relied on modification of the C-5 side chain (18) and isosteric substitution of previously benzoxazinyl-oxazolidinone with benzothiazinyl-oxazolidinone (19). Oxazolidinone 18 exhibited an increase in activity against a panel of resistant and susceptible Gram-positive bacteria compared to LZD. The MIC values against MRSA (six strains), MRSE (five strains), and PRSP (four strains) ranged between 0.25 and 1.0 μg mL−1.40 Benzothiazinyl-oxazolidinone 19, however, showed a decrease in antibacterial activity (compared to LZD) with MIC values ranging from 2.0 to 4.0 μg mL−1 against the panel. Interestingly, the substitution of oxygen on the tricyclic ring was also evaluated with different groups, including NH, N-Boc, S
000-fold greater than compound 16), good PK profile, and superior in vivo activity than linezolid. The ED50 of compound 17 was 5.0 mg kg−1 (2-times higher than that of linezolid). Additionally, it showed a significant reduction in hERG inhibition compared to compound 16 (IC50 > 40 μM for the bioactive parent).39 Compounds 18 and 19 (Fig. 3) were later reported as a continuing effort to identify new lead derivatives.40,41 The drug design behind these two analogues relied on modification of the C-5 side chain (18) and isosteric substitution of previously benzoxazinyl-oxazolidinone with benzothiazinyl-oxazolidinone (19). Oxazolidinone 18 exhibited an increase in activity against a panel of resistant and susceptible Gram-positive bacteria compared to LZD. The MIC values against MRSA (six strains), MRSE (five strains), and PRSP (four strains) ranged between 0.25 and 1.0 μg mL−1.40 Benzothiazinyl-oxazolidinone 19, however, showed a decrease in antibacterial activity (compared to LZD) with MIC values ranging from 2.0 to 4.0 μg mL−1 against the panel. Interestingly, the substitution of oxygen on the tricyclic ring was also evaluated with different groups, including NH, N-Boc, S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and SO2, and all cases led to a significant loss of antibacterial activity highlighting the importance of the oxygen atom at this position.41
O, and SO2, and all cases led to a significant loss of antibacterial activity highlighting the importance of the oxygen atom at this position.41
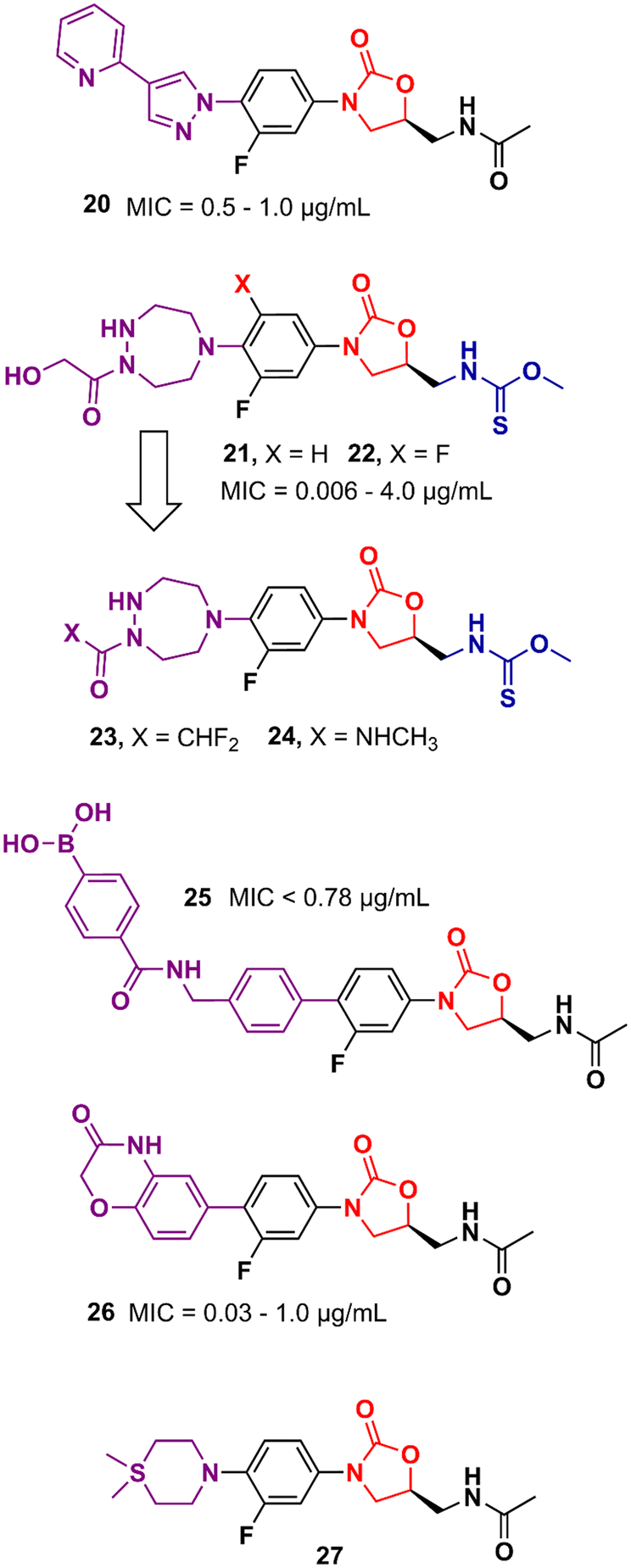
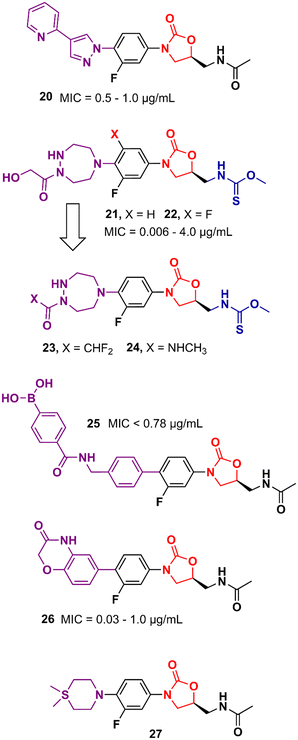
Gong et al. reported in 2013 a carrier-free nano-assemble of an oxazolidinone derivative with promising antibacterial activity.42 The authors aimed to develop an analogue and manipulate it into nano-assembles using a self-assembly method without using any carrier to improve the water solubility of the compound. Compound 20 (Fig. 4) showed MIC values between 0.5 and 1.0 μg mL−1 against a panel of MSSA and MRSA. Furthermore, in vitro, and in vivo toxicity studies showed that the compound was not toxic to HEK293 and L02 cells (IC20 > 25 and 50 μM, respectively) and did not show signs of toxicity in mice. In vivo efficacy studies of compound 20 in MSSA and MRSA, systemic infection mouse models demonstrated efficacy greater than 3-times and 2-times greater than LZD against MSSA or MRSA, respectively.42 Moreover, compound 20 displayed efficacy in inhibiting biofilm formation in vitro and in vivo. It reduced the biofilm mass of MSSA (ATCC 25923) by 50% compared to the control group. In vivo studies using catheter-associated infection in a murine model showed that compound 20 removed surface bacteria more effectively than LZD.43 More studies were conducted to investigate the metabolism of compound 20. Amide hydrolysis in the acetyl group of compound 20 was shown to be the main metabolic biotransformation in phase I metabolism.44 Subsequent studies turned compound 20 into a phosphate salt to address the solubility problem. The new phosphate salt showed MIC values ranging from 0.25 to 2.0 μg mL−1 against 120 clinically isolated strains. Furthermore, it did not show significant cytotoxicity in HEK293, L02, THP-1, and K462 cells (>70 μM) nor showed inhibition of the hERG K+ channel (>40 μM). In vivo studies highlighted its high oral bioavailability (F = 99.1%) and significantly enhanced efficacy compared to LZD.45 In 2013, Suzuki et al. reported a series of oxazolidinone derivatives with modifications in the C-ring. Several analogues were identified as potent antibacterials with in vitro and in vivo activity.46,47 For example, compounds 21 and 22 (Fig. 4) containing an N-hydroxyacetyl-substituted [1,2,5]triazepane C-ring unit and a thiocarbamate C5 side chain displayed MIC values ranging from 0.063 to 4.0 μg mL−1 against a panel of Gram-positive bacteria. In vivo evaluation in a systemic mouse infection model with S. aureus SR3637 showed an ED50 of 0.94 and 0.77 mg kg−1 (intravenous) and 1.05 and 2.07 mg kg−1 (oral) for compounds 21 and 22, respectively. These results highlighted the higher efficacy of the compounds tested compared to that of LZD. Moreover, both compounds displayed a reduction in MAO-A and B inhibition compared to LZD.46 Further optimization studies identified compounds 23 and 24 (Fig. 4) as new leads with reduced activity against MAO-A and B while maintaining a potent antibacterial effect.47 Several other studies have pursued the idea of developing LZD analogues with modifications on the C-ring. For example, compound 25 (Fig. 4) was developed by adding an arylboronic acid moiety to the C- and D-ring sections. It showed MIC values <0.78 μg mL−1 against selected Gram-positive strains and E. coli JW5503.48 Compound 26 is another example with modifications to the C-ring. This compound was designed with a heterocyclic benzoxazinone ring at that position. Compound 26 showed MICs ranging from 0.03 to 1.0 μg mL−1 against a panel of Gram-positive and negative bacteria. This in vitro activity was superior to that of LZD. Additionally, it did not show significant inhibition of cytochrome P450 (CYP450) enzymes (10 μM), and showed metabolic stability in mice liver microsomes.49 Compound 27 (Fig. 4) is another LZD analogue with modifications to the C-ring. This compound presents a silicon atom incorporated into the C-ring. According to the study, this modification led to a compound with enhanced brain exposure compared to the parental drug, which could be a therapeutic alternative for brain infections. This improvement was related to the strong bond to plasma proteins by the compound, which could be an advantage in improving the penetration of the blood–brain barrier (BBB). Furthermore, compound 27 retained potent in vitro antibacterial activity.50
 |
| | Fig. 4 Linezolid derivatives with modification in the C-ring. | |
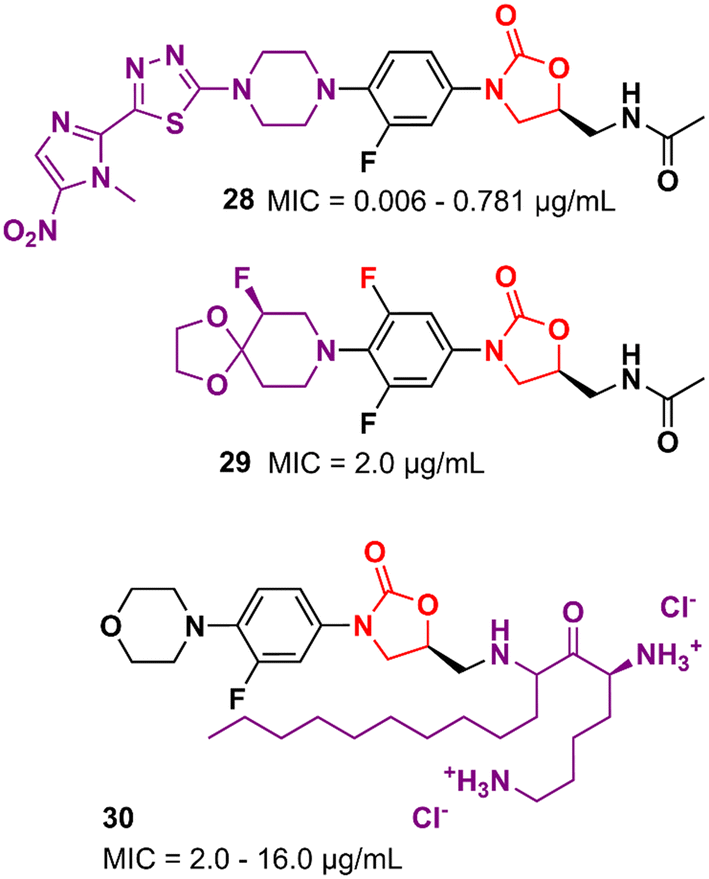
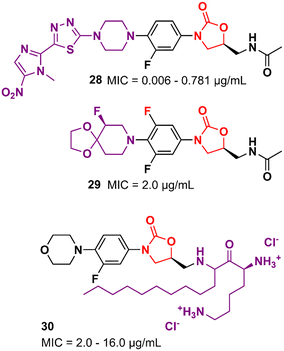
Several other oxazolidinone derivatives bearing modifications on the C-ring have been described as promising antibacterials. For example, Khalaj et al. reported a nitroimidazolyl–oxazolidinone hybrid with potent activity. The MIC values of compound 28 (Fig. 5) ranged from 0.006 to 0.781 μg mL−1 against a panel of selected strains. It did not show significant cytotoxicity activity in mouse fibroblast (NIH/3T3) cells (IC50 > 200 μg mL−1).51 Compound 29 (Fig. 5) is another example of an LZD analogue with modifications on the C-ring. It displayed a MIC of 2.0 μg mL−1 MSSA and MRSA and an ED50 of 2.5 mg kg−1 (orally, in the mouse model of septicemia). PK studies in dogs highlighted the superior profile of compound 29 over LZD.52 Analogues of LZD with modifications in the C5 side chain are less common in the literature. To date, only a few compounds have been reported. The reason behind this scarcity is mainly due to the mechanism of action of LZD-based derivatives that target the 50S subunit of the bacterial ribosome. Matsingos et al. nicely demonstrated this in an interesting study containing a series of LZD derivatives with several modifications on the C5 side chain. After an extensive medicinal chemistry campaign to synthesize and evaluate various analogues, none of the compounds obtained was identified as promising. Molecular docking studies using the 50S ribosomal subunit from S. aureus (PDB 6WRS) demonstrated that small modifications in the C5 side chain usually led to a considerable loss of antibacterial activity and in many cases to complete inactivity. This was observed when bulky groups were inserted into that position. Despite the not-so-promising results, this work brings valuable information about the structure–activity relationship (SAR) of LZD derivatives and contributes to the rational design of new generations of analogues.53 On the other hand, some compounds bearing modifications in the C5 side chain have shown remarkable activity even against resistant strains. For example, compound 30 (Fig. 5), a cationic amphiphilic derivative of LZD, showed MIC values ranging from 2.0 to 16.0 μg mL−1 against MRSA, Klebsiella pneumoniae carbapenemase (KPC) and New Delhi metallo-β-lactamase 1 (NDM-1) producing carbapenem-resistant Enterobacteriaceae (CRE), while LZD ranged from 2.0 to >64 μg mL−1. Further studies indicated that compound 30 has rapid bactericidal activity against both Gram-positive and negative bacteria and can inhibit the formation of biofilms (S. aureus and E. coli biofilms) at high concentrations (64 and 128 μg mL−1). Mechanism of action studies suggested that compound 30 acts by permeabilization and depolarization of Gram-positive and Gram-negative bacterial membranes.54
 |
| | Fig. 5 Oxazolidinone derivatives with antibacterial activity. | |
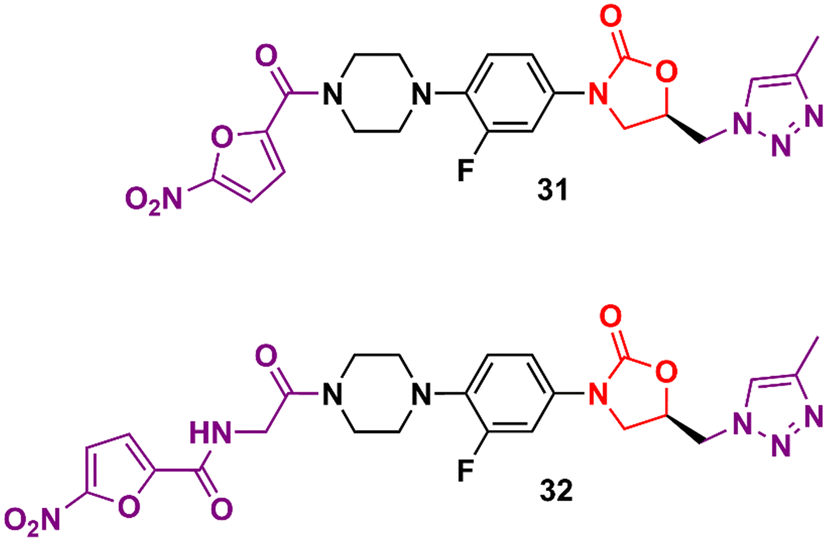
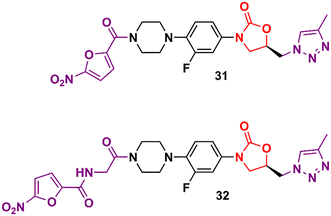
In the vast majority of cases, the oxazolidinone derivatives reported in the literature are in the early stages of drug development and frequently present only data from the initial antibacterial screening. Herein are several of these early-stage compounds that, despite their promising antibacterial activity, are still far from the most advanced stages of development due to the lack of critical studies, for example, in vitro ADME, in vivo PK, and in vitro and in vivo toxicology. Triazolylmethyl oxazolidinones 31 and 32 (Fig. 6) were identified after extensive exploration of different substituents on the D-ring. These compounds showed MIC values compared to LZD in most Gram-positive strains.55,56
 |
| | Fig. 6 Oxazolidinone as Gram-positive bacteria inhibitors. | |
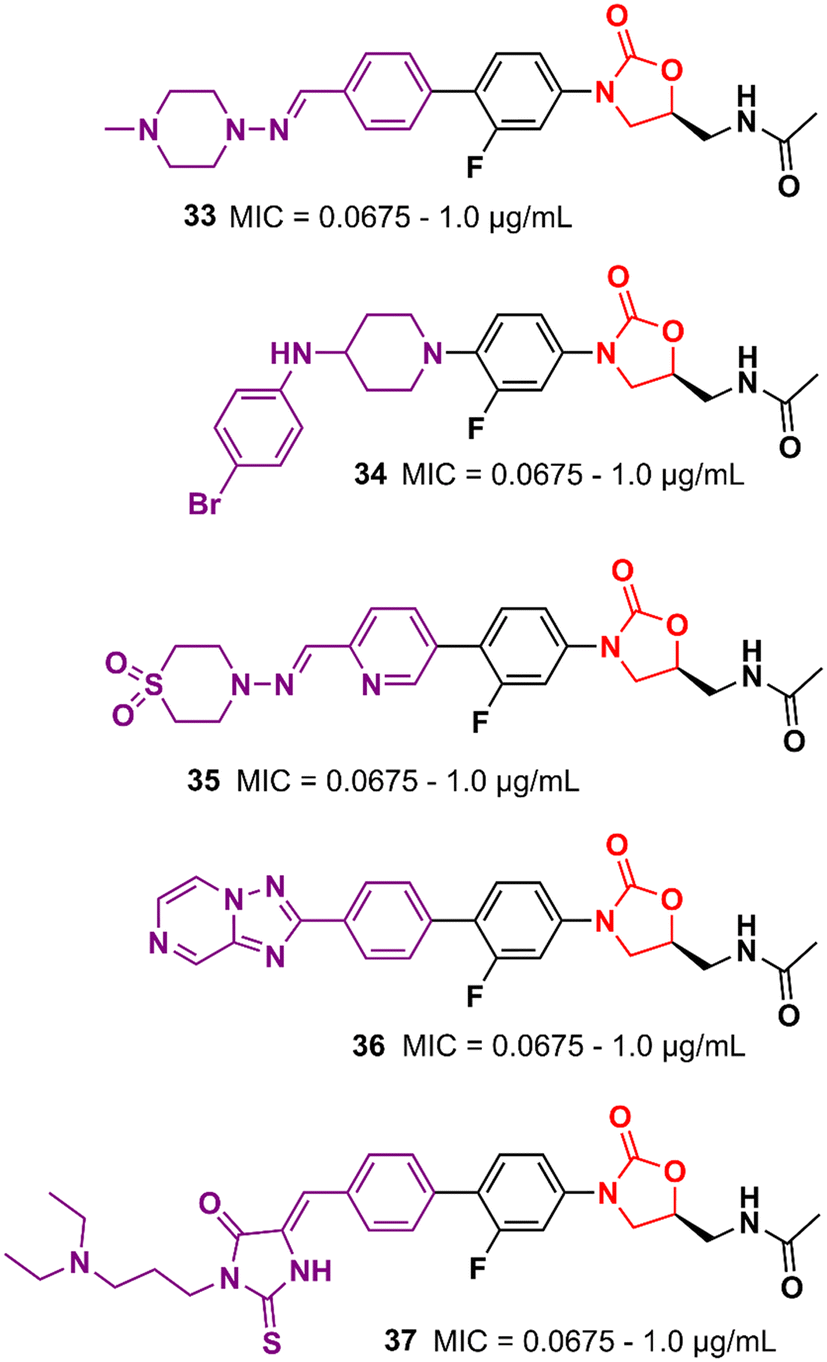
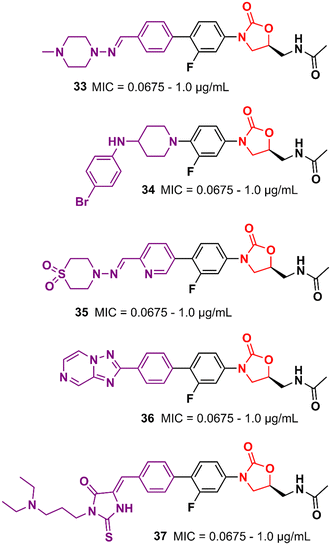
Similarly, researchers working with Shenyang Pharmaceutical University conducted an extensive medicinal chemistry project exploring variations in C- and D-rings. Several promising analogues (33–37) (Fig. 7) were identified with promising activity against Gram-positive bacteria and are many times more potent than LZD.57–61 These compounds (33–37) were tested against S. aureus, MRSA, MSSA, Linezolid-resistant Enterococcus faecalis (LREF), and VRE and MICs ranged from 0.0675 to 1.0 μg mL−1. Similarly, compounds 33–37 demonstrated similar MIC values against a panel of clinical isolates. Further in vitro cytotoxicity and ADME studies were performed on selected analogues. Compound 33 exhibited an IC50 of 0.82 μM (HepG2 cells), a low a half-life (t1/2 72.85 min), a high clearance (Clint 23.86 mL min−1 kg−1), and moderate metabolism in the human liver microsome.57 Compound 34 displayed IC50 > 64 μM (HepG2 cells), a long half-life (501.99 min), and a low human liver microsomal Clint (3.46 mL min−1 kg−1).59 Also, compound 35 exhibited IC50 > 25 μM (HepG2 cells), improved water solubility (68 μg mL−1), long half-life (409.44 min), and low Clint (4.25 mL min−1 kg−1) in human liver microsome.58
 |
| | Fig. 7 Gram-positive bacteria inhibitors. | |
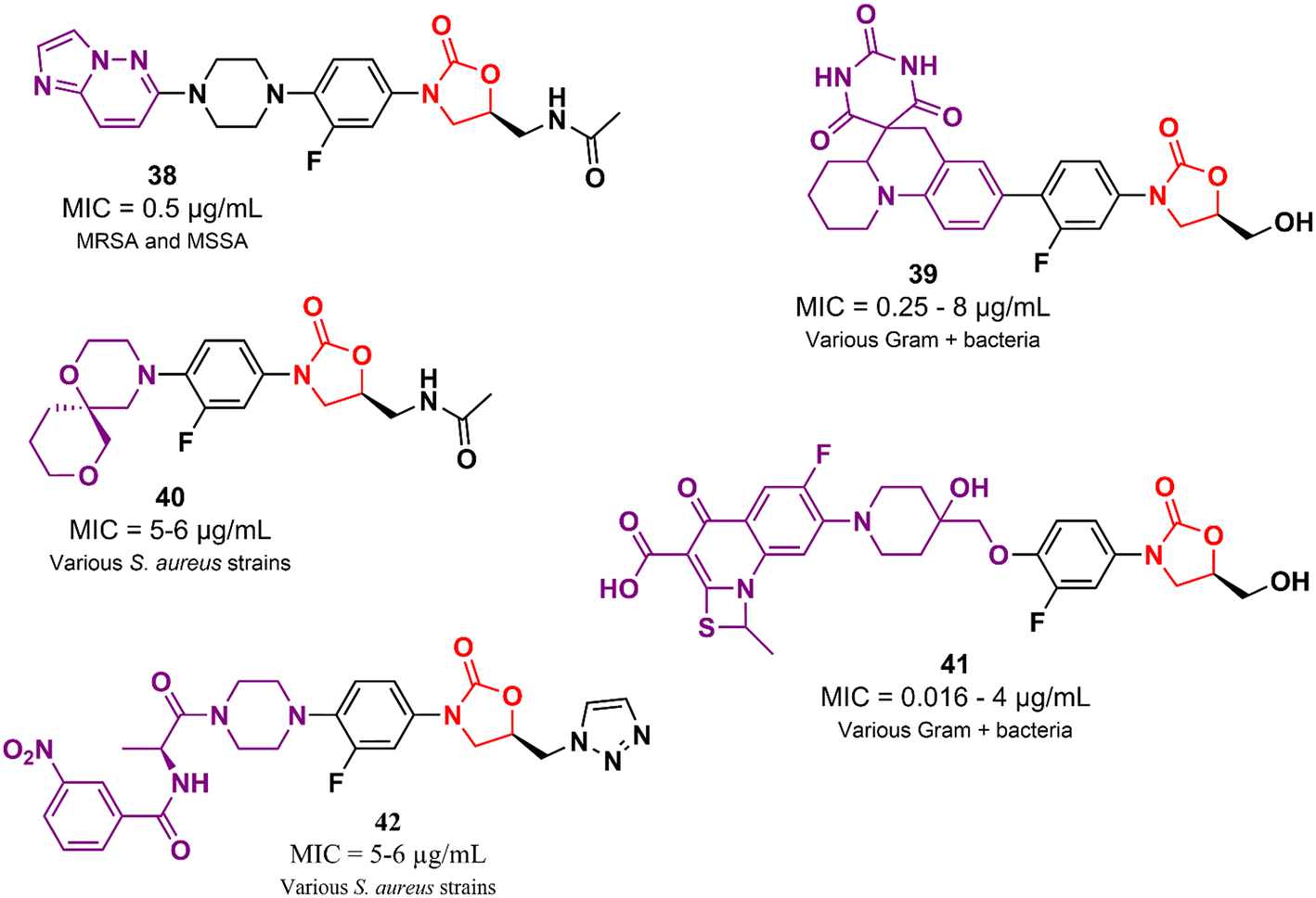
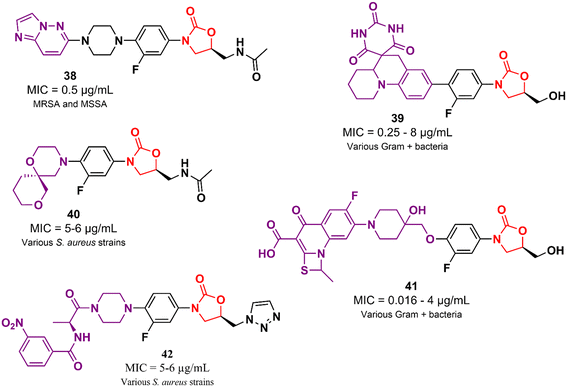
In addition to the examples highlighted above, many other oxazolidinone-based derivatives in the early stages of drug development have been reported in the literature in recent years (Fig. 8).62–66 However, despite promising initial screening, these compounds still lack several studies to advance drug development stages.
 |
| | Fig. 8 Antibacterial agents containing the oxazolidinone scaffold as the pharmacophore. | |
Oxazolidinone derivatives active against Gram-negative bacteria
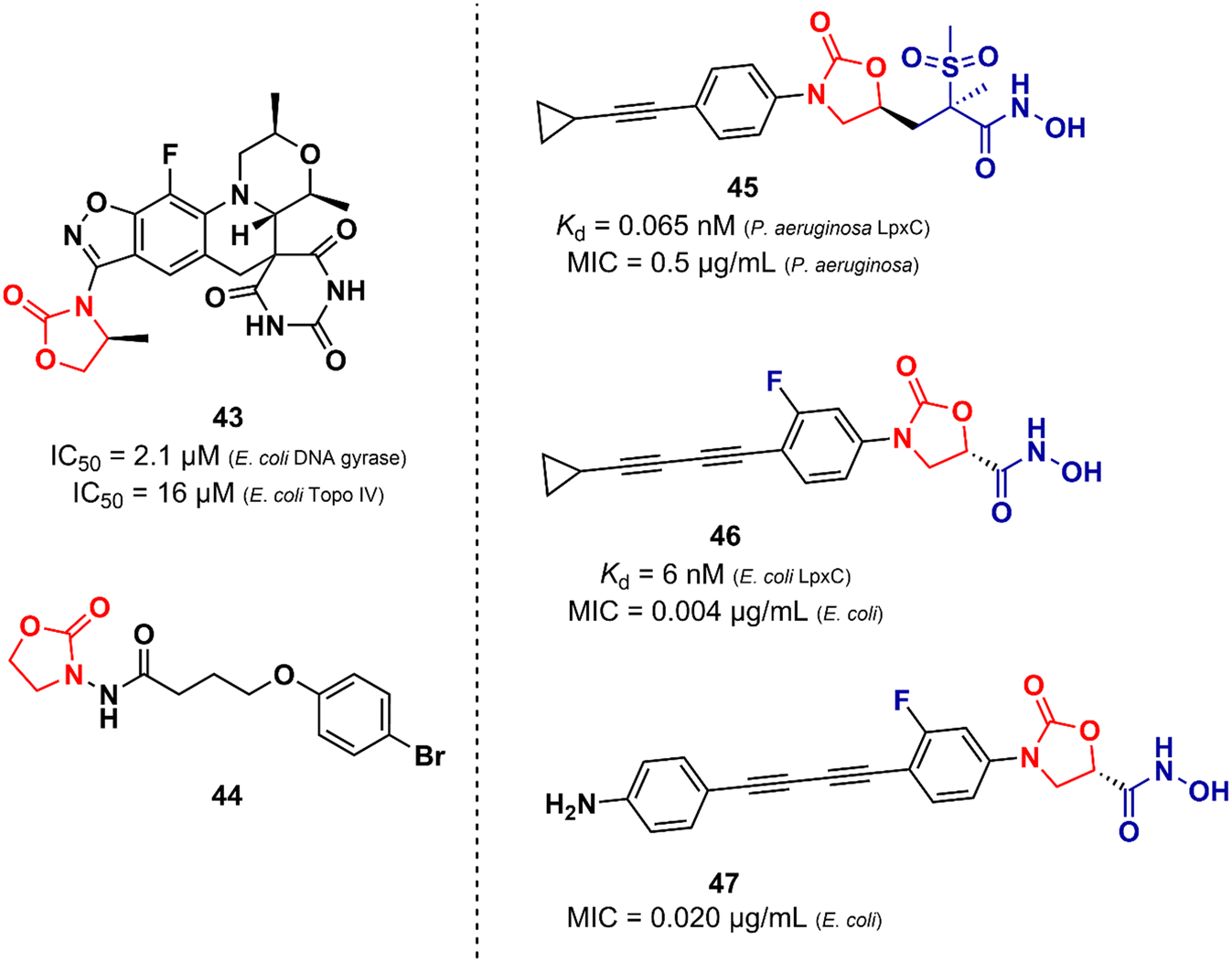
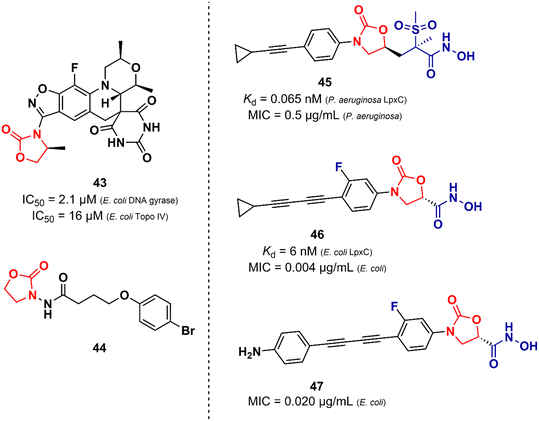
Unlike the oxazolidinones presented in the previous section that were mainly derivatives of LZD, the oxazolidinones disclosed here display a greater structural variety in relation to their peers developed against Gram-positive bacteria. Basarab et al. working at AstraZeneca identified a promising oxazolidine clinical candidate for the treatment of uncomplicated gonorrhoea with DNA Gyrase inhibitory activity.67 Compound 43 (Zoliflodacin, ETX0914) (Fig. 9) presents in its structure an oxazolidinone linked to a benzisoxazole fused with a spiropyrimidinetrione. It showed MICs of 0.39 and 6.2 μM against N. gonorrheoea and E. coli, respectively. Furthermore, it displayed potent activity against selected Gram-positive bacteria with MIC values below 0.78 μM. Structure–activity relationships demonstrated the importance of the methyl group attached to the position 4 of the oxazolidinone for the antibacterial activity. Compound 43 showed a favourable PK profile in rats, dogs, and humans. In vitro genotoxic and cytotoxic studies demonstrated its safety profile. Compound 43 was shown to act by inhibiting DNA gyrase and topoisomerase IV of Gram-positive and Gram-negative bacteria.67,68 In 2018, compound 43 completed phase II clinical trials69 and is currently being developed in phase III by Entasis Therapeutics in partnership with the Global Antibiotic Research and Development Partnership (GARDP). Compound 44 (Fig. 9) is another example of an oxazolidinone derivative with activity against Gram-negative bacteria and is not similar to the structure of LZD.70 This compound (44) was identified as a quorum-sensing inhibitor of Pseudomonas aeruginosa PAO1. Quorum-sensing is a system that controls biofilm formation and the release of virulence factors. In fact, in vitro studies showed that compound 44 inhibited the biofilm formation of P. aeruginosa PAO1 in a concentration-dependent manner. For instance, at 162.5 μM, it inhibited biofilm formation by 40.39%. In vivo studies using Caenorhabditis elegans N2 infected with P. aeruginosa demonstrated that inhibition of QS by compound 44 (165.2 μM) led to an improvement in the survival rates of C. elegans N2 infected by 13.33%.70 Oxazolidinone derivatives have also been developed as UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) inhibitors. LpxC is a zinc metalloenzyme that plays an important role in the biosynthesis of lipid A, a glucosamine disaccharide that anchors lipopolysaccharide (LPS) to the outer membrane of Gram-negative bacteria.71,72 Le et al. working at Novartis identified by virtual screening an oxazolidine derivative with potent LpxC inhibitory activity (Kd = 0.065 nM).73 Compound 45 (Fig. 9) displayed MIC values of 0.125, 0.5 and 1.0 μg mL−1 against E. coli ATCC 25922, P. aeruginosa ATCC 27853, and K. pneumoniae ATCC 43816, respectively. The cocrystal structure of compound 45 complexed with the P. aeruginosa LpxC enzyme revealed that the oxazolidinone scaffold is critical for the activity by linking the sulfone/hydroxamic acid moiety of the inhibitor with the hydrophobic pocket while retaining a low-energy conformation. In vivo studies using a mouse neutropenic thigh infection model infected with P. aeruginosa revealed that compound 45 (160 mg kg−1) reduced the bacterial burden to undetectable levels. Interestingly, compound 45 was also shown to have a potent synergic effect in vitro and in vivo with other antibiotics.73 In another article, Kurasaki et al. working at Kyorin Pharmaceutical,74 discovered a similar oxazolidinone with LpxC inhibitory activity (IC50 = 6 nM). Compound 46 (Fig. 9) has a great structural similarity to 45. However, some differences are worth noting, such as the stereochemistry of the substituent on the C5 side chain and the presence of a fluorine atom on the phenyl ring, which was shown to play an important role in the activity by making additional interactions with C207 and C63 at the binding site. Oxazolidinone 46 showed remarkable in vitro activity against E. coli TG1, E coli KAM3, and K. pneumoniae ATCC 13883 with MICs lower than 0.016 μg mL−1.74 Compound 47 (Fig. 9) is another example of an LpxC inhibitor with potent antibacterial activity against Gram-negative strains. It showed MIC values ranging from 0.020 to 16.0 μg mL−1 against a panel of wild-type and clinical isolate strains.75
 |
| | Fig. 9 Oxazolidinone with activity against Gram-negative bacteria. | |
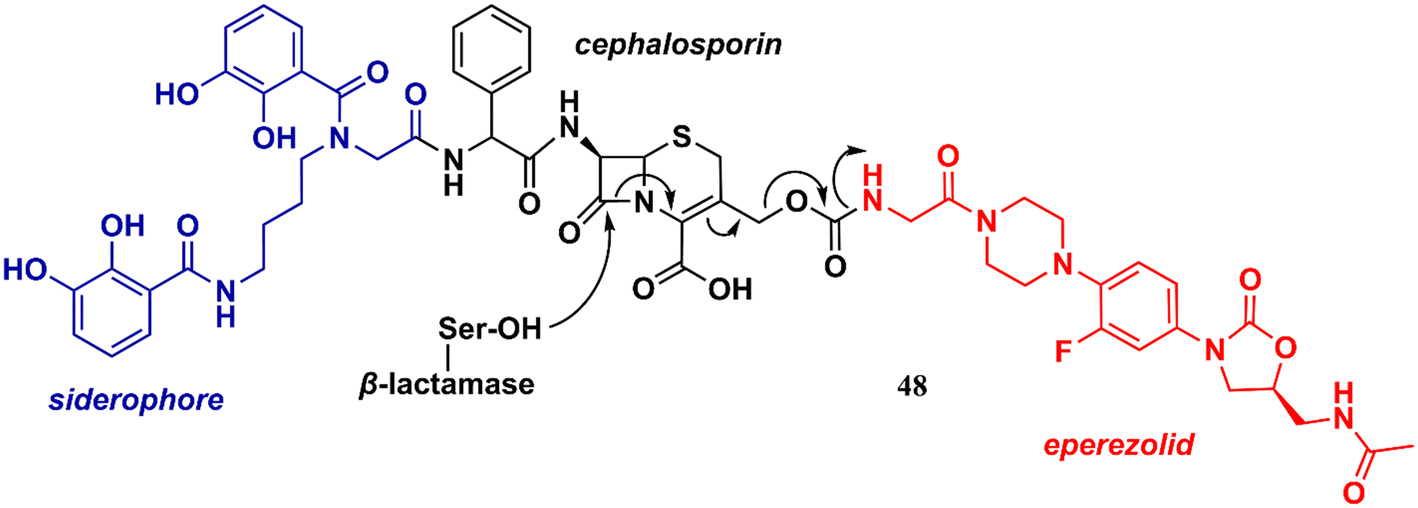
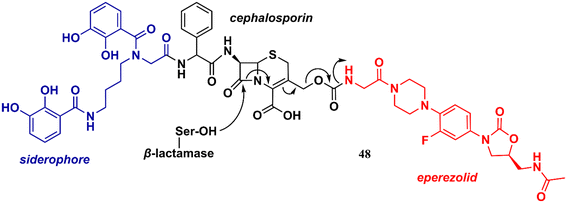
Liu et al. reported in 2018 an interesting study on an innovative concept of an oxazolidinone-based derivative linked to a cephalosporin with an attached siderophore.76 The design of the not-so-small molecule 48 (Fig. 10) can be divided into three parts. Initially, the siderophore moiety acts as a delivery system for antibiotics, a strategy known as the ‘Trojan Horse’.76,77 Subsequently, cephalosporin acts as a release system for the oxazolidinone antibiotic after hydrolysis by bacterial β-lactamases. Finally, the released oxazolidinone, a Gram-positive-only drug, can reach its ribosomal target. This approach allowed the delivery of eperezolid to its target in Gram-negative bacteria. It is worth noting that eperezolid cannot cross Gram-negative outer membranes and is quickly effluxed when it does.78 Compound 48 was active against clinical isolates of Acinetobacter baumannii and strains producing considerable amounts of ADC-1 β-lactamase. MICs against these strains ranged from 0.4 to 6.0 μM. On the other hand, oxazolidinone and cephalosporin alone, siderophore–oxazolidinone, and siderophore–cephalosporin conjugates had no activity (MIC > 50 μM).76
 |
| | Fig. 10 The trojan horse approach: siderophore–cephalosporin–eperezolid conjugate. | |
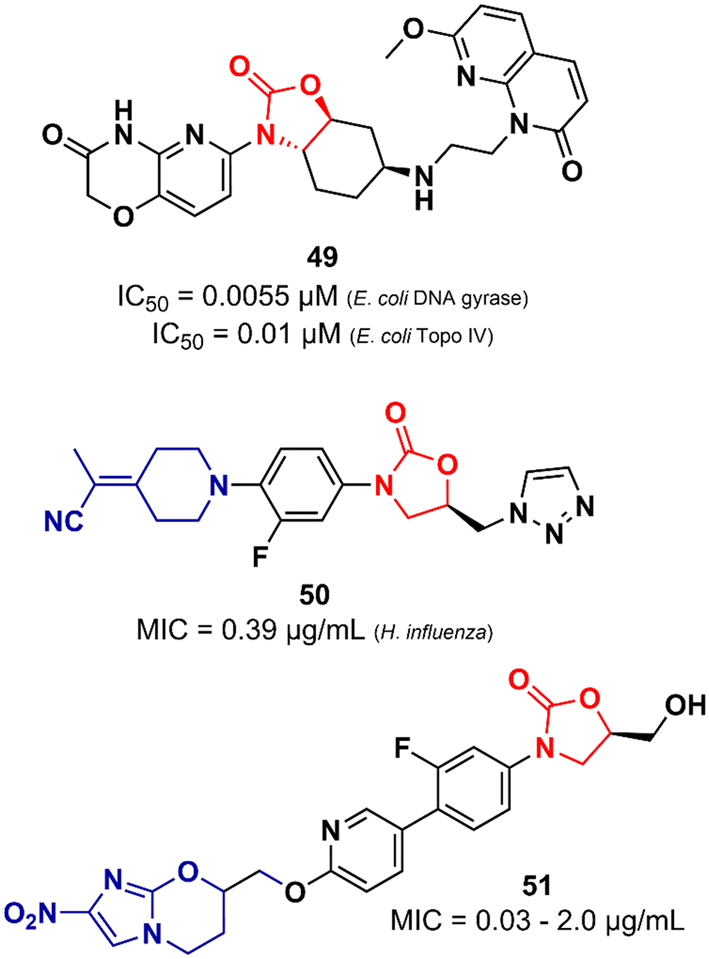
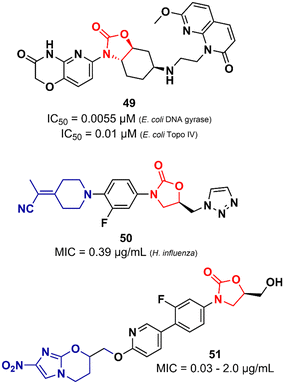
Lyons et al. also reported an oxazolidinone derivative not related to LZD as a potent Gram-negative antibacterial. Compound 49 (Fig. 11) was identified as a potent bacterial type II topoisomerase inhibitor (DNA gyrase and topoisomerase IV) with IC50 values of 0.055 and 0.01 μM against E. coli gyrase and topo IV, respectively. It showed MIC values ranging from 0.5 to 4.0 μg mL−1 against a large panel of MDR Gram-negative clinical isolates (A. baumannii, P. aeruginosa, E. cloacae, E. coli and, K. pneumoniae). Additional in vivo studies using a mouse thigh infection model of A. baumannii NCTC13420 (MDR) showed that compound 49 reduced the bacterial burden by >1log CFU g−1 at 60 mg kg−1. However, 49 did not show favourable in vitro DMPK properties, displaying high clearance in both mouse and human liver microsomes (264 and 110 μL min−1 mg−1, respectively) and demonstrated high inhibitory activity of the hERG K+ channel (IC50 = 5.0 μM).79 LZD analogues have also been reported as antibacterials with potent activity against Gram-negative pathogens. Appropriate modifications on the C-ring appear to be essential for LZD analogues to overcome the permeability barrier of the Gram-negative outer membrane whilst minimizing active efflux. Aggen et al. demonstrated that polar and/or charge-carrying groups seem to contribute greatly to the ability of LZD analogues to cross the outer barrier. Furthermore, the zwitterionic character and the presence of ≤4 rotatable bonds were also shown to be beneficial in overcoming the outer barrier.80,81 Nam et al. also identified LZD analogues bearing polar groups in their structures with activity against Gram-negative pathogens. Compound 50 (Fig. 11) showed a MIC of 0.39 μg mL−1 against Haemophilus influenzae and a low IC50 (213 μM) against the hERG K+ channel. It also showed potent activity against a panel of Gram-positive bacteria (MIC < 1.56 μg mL−1).82 Zhuang et al. demonstrated the synergistic activity of a series of nitroimidazole–oxazolidinone conjugates against Gram-negative anaerobic pathogens. Specifically, two well-known drugs were used as starting points for the design of the series, namely LZD and pretomanid. For example, compound 51 (Fig. 11), an LZD–pretomanid conjugate, showed MICs ranging from 0.03 to 2.0 μg mL−1 against selected clinically important anaerobic bacterial strains.83
 |
| | Fig. 11 Oxazolidinones as Gram-negative bacteria inhibitors. | |
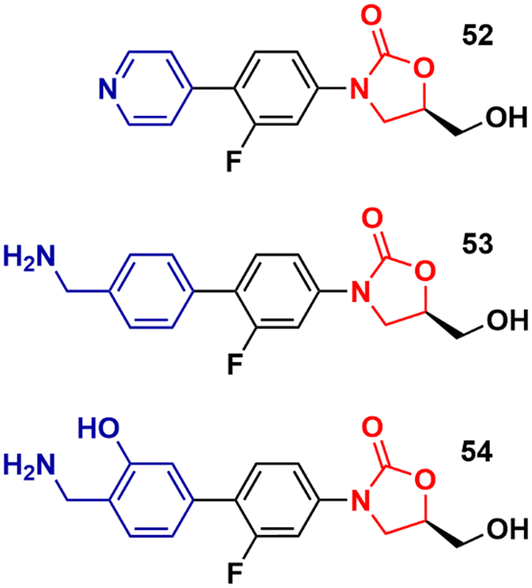
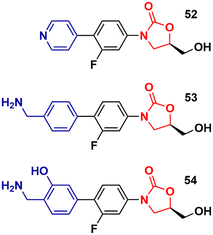
Recently, Duerfeldt et al. reported a library of oxazolidinone derivatives developed against ESKAPE bacteria.84 The drug designing direction focused on increasing the accumulation of the new analogues in Gram-negative bacteria by circumventing efflux and increasing their ability to penetrate membranes. The structural modifications focused on the C-ring and a large number of compounds were synthesized and evaluated against wild-type E. coli, A. baumannii, and P. aeruginosa, and isogenic mutants with different grades of efficiency of the outer membrane and/or efflux pump. Three oxazolidinones, 52, 53 and 54 (Fig. 12), were identified as potent analogues displaying potent activity against all three bacteria. It is worth mentioning that LZD did not show activity against these bacteria. These results highlight how small structural modifications can broaden the spectrum of action of oxazolidinones against Gram-negative bacteria.84
 |
| | Fig. 12 Oxazolidinone analogues active against ESKAPE bacteria. | |
Oxazolidinones as antitubercular agents
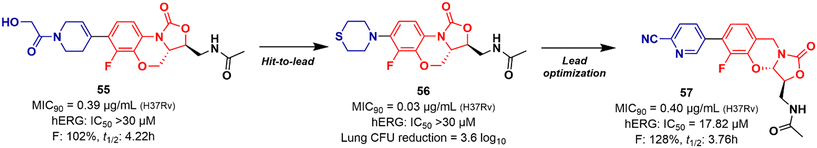
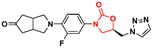
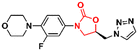
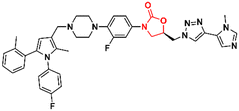
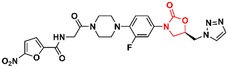
Mycobacterium tuberculosis is one of the deadliest pathogens documented in the history of human civilization. TB is currently considered the second leading cause of death from an infectious disease behind only COVID-19.17 However, this position is likely to reverse as the number of deaths caused by COVID-19 decreases due to the success of immunization programs. Oxazolidinone derivatives, especially LZD analogues, have been extensively studied as anti-TB agents. Huang et al. recently reported several of these derivatives.85–87 In 2017, benzoxazinyl–oxazolidinone 55 (Fig. 13) was discovered as a potent anti-TB compound with MIC90 of 0.391 μg mL−1 against M. tuberculosis H37Rv and less than 0.50 μg mL−1 against drug-resistant strains. It did not show toxicity against HepG2 cells (IC50 > 64 μg mL−1) nor inhibitory activity against the hERG K+ channel (IC50 > 30 μM). In vitro evaluation of ADME and in vivo PK demonstrated its favourable profile with high maximum plasma concentration (Cmax = 30.0 μg mL−1), long elimination half-life (t1/2 = 4.22 h), and outstanding oral bioavailability (F = 102%) after oral administration in Balb/c mice.85 Subsequently, compound 55 went through a hit-to-lead optimization medicinal campaign in an attempt to identify an analogue with improved anti-TB activity, reduced toxicity, and improved drug-like properties. The campaign was successful in identifying compound 56 (Fig. 13) as a new lead oxazolidinone. Analogue 56 displayed improved in vitro and in vivo efficacy compared to LZD and compound 52. For instance, it showed MIC90 of 0.03 μg mL−1 against M. tuberculosis H37Rv, 13 times lower than the parental compound 55. The drugabillity of 56 was extensively evaluated in microsomal stability, cytotoxicity, cytochrome P450 enzyme inhibition, and in vivo PK. Compound 56 displayed excellent metabolic stability in liver microsomes of different species, high IC50 values (>45 μM) against several isoforms of CYP450 enzymes, and a favourable PK profile in mice. A further in vivo evaluation of efficacy in a mouse model of acute M. tuberculosis infection demonstrated the potent activity of 56 (dose 100 mg kg−1 per day) after reducing the bacterial lung load in 3.6 log10 CFU compared to the untreated group. LZD (dose 100 mg kg−1 per day) on the other hand, achieved a reduction of 1.7 log10 CFU. Subsequently, a dose–response study also demonstrated the superior efficacy of compound 56 compared to LZD and sutezolid at different doses.86 Recently, the same group reported the discovery of a new tricyclic benzo[1,3]oxazinyl–oxazolidinone 57 (Fig. 13) as a new anti-TB lead. Compound 57 displayed MIC90 values of 0.40, 0.48, and 0.82 μg mL−1 against M. tuberculosis H37Rv, and two strains of MDR-TB, respectively. Interestingly, the benzo[1,3]oxazinyloxazolidinone derivative showed superior activity than the benzo[1,4]oxazinyloxazolidinone isomer. Similarly, to its predecessors, 57 exhibited excellent metabolic stability in human and mouse liver microsomes and no toxicity to Vero cells. However, it showed a lower IC50 value (IC50 = 17.82 μM) against the hERG K+ channel compared to analogues 55 and 56. Further evaluation of the PK profile demonstrated its excellent profile with a high maximum plasma concentration (Cmax = 10.2 μg mL−1), appropriate elimination half-life (t1/2 = 3.76 h), and good oral bioavailability (F = 128%) after oral administration in Balb/c mice.87 In summary, these results highlight the tricyclic benzo[1,3]oxazinyl–oxazolidinone as a promising moiety for the discovery of anti-TB drugs.
 |
| | Fig. 13 Tricyclic oxazolidinones as antituberculosis agents. | |
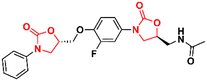
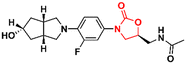
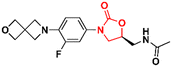
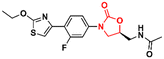
Several other oxazolidinone derivatives have been reported to act as anti-TB agents. However, most of these compounds have only been evaluated against the standard strain of M. tuberculosis H37Rv. Drug discovery of new anti-TB agents is highly unfruitful, and currently, the greatest challenge lies in the identification of new active agents against resistant strains (MDR- and XDR-TB).88,89 Often, oxazolidinone derivatives discovered as anti-TB agents show great similarity to LZD, a drug already used in the treatment of TB. Therefore, to avoid extending this section's discussion with limited information regarding only the initial screening of these compounds, we have condensed these LZD derivatives into Table 1 with their respective MIC90 values. Importantly, we converted all the MIC values from μg mL−1 to μM to facilitate the comparison between the compounds.
Table 1 Oxazolidinones with activity against M. tuberculosis
| Cmpd |
Structure |
MIC90 (μM) |
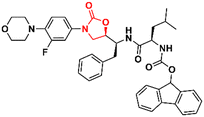
Ref. |
|
58
|
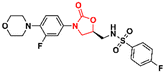
|
2.2 |
90
|
|
59
|
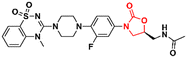
|
1.8 |
90
|
|
60
|
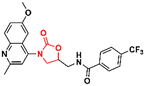
|
1.3 |
91
|
|
61
|
|
1.2 |
92
|
|
62
|
|
1.3 |
93
|
|
63
|
|
2.8 |
94
|
|
64
|
|
0.9 |
95
|
|
65
|
|
0.2 |
96
|
|
66
|
|
0.6 |
97
|
|
67
|
|
7.3 |
98
|
|
68
|
|
2.6 |
99
|
|
69
|
|
2.6 |
100
|
|
70
|
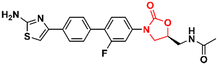
|
0.9 |
101
|
|
71
|
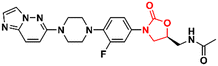
|
1.1 |
102
|
Oxazolidinones as anti-cancer agents
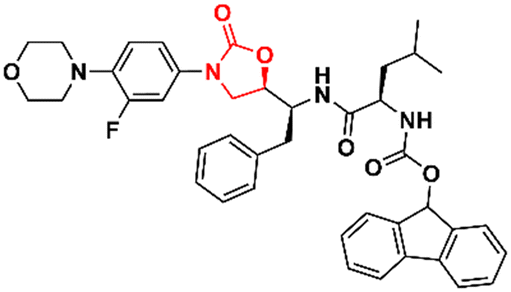
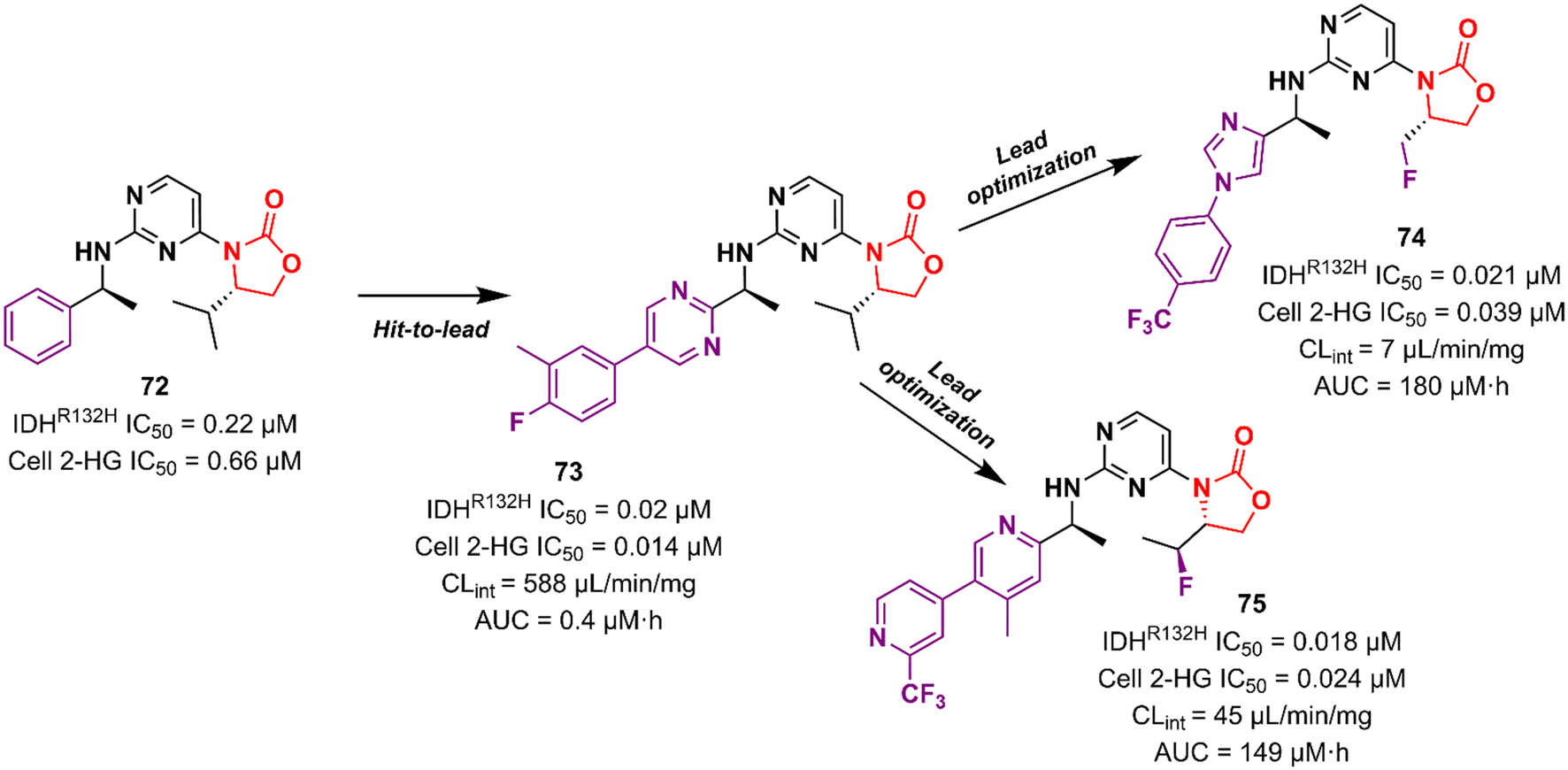
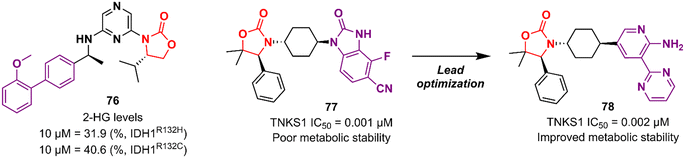
Cancer is undoubtedly one of the leading causes of death in modern times and it ranks second only to cardiovascular diseases.103 Naturally, this scenario attracts the attention of medicinal chemists from both academia and industry, who are constantly searching for new therapeutic alternatives for the treatment of the most diverse types of cancer. Oxazolidinones are one of the classes within the large chemical universe that are systematically researched for the treatment of cancer.104 Although the number of oxazolidinone derivatives reported as anticancer agents are incredibly lower than that of anti-infective compounds. Mutant isocitrate dehydrogenases 1 (mIDH1) and 2 (mIDH2) have been extensively studied as promising anticancer targets since their identification.105,106 Briefly, wild-type IDH1 catalyzes the conversion of isocitrate to α-ketoglutarate (α-KG), using NADP+ as a cofactor. Heterozygous mutations at the R132 (Arg132) position of IDH1 and R140 or R172 of IDH2 have been associated with various cancers, including low-grade glioma, glioblastoma, acute myelogenous leukaemia, and solid tumours such as chondrosarcoma, cholangiocarcinoma, colon, pancreatic, and prostate cancer.107 This mutation (gain-of-function) leads to a loss of normal enzymatic function and reduces α-KG to R-2-hydroxyglutarate (2-HG), resulting in increased levels of this metabolite, which is linked to widespread alterations in histone and DNA methylation, which can contribute to tumorigenesis.108,109 Novartis Institutes for BioMedical Research have pioneered the development of the oxazolidinone class as mutant IDH1 inhibitors. In 2017/18, Novartis reported the discovery of several oxazolidinone derivatives with promising anticancer activity. Compound 72 (IDH125) (Fig. 14) was the first hit identified by high-throughput screening (HTS).110 The X-ray cocrystal structure of 72 in the homodimer of IDH1R132H demonstrated the importance that the oxazolidinone ring has in the active binding conformation. Following an iterative hit-to-lead medicinal chemistry campaign exploring the phenyl ring, the researchers developed compound 73 (IDH889) (Fig. 14) as a new lead with improved activity and PK properties. Compound 73 exhibited equipotent inhibition of the homodimer IDH1R132H (IC50 0.02 μM) and IDH1R132C (IC50 0.072 μM) proteins and was less potent for the heterodimer IDH1wt–IDH1R132H (IC50 1.38 μM) protein. In the HCT116–IDH1R132H± xenograft model, 73 (at 200 mg kg−1) inhibited 2-HG production. PK studies highlighted its excellent permeability through brain tissue.111 Despite its potent activity, 73 demonstrated low aqueous solubility (39 μM at pH 6.8) and high in vivo clearance in rodents. A lead optimization program that focused on fluoromethyl and fluoroethyl substituted oxazolidinones to improve metabolic stability without compromising physicochemical properties led to the identification of compounds 74 and 75 (IDH305) (Fig. 14).112,113 The X-ray structure of compound 75 bound to IDH1R132H homodimer demonstrated the importance of the carbonyl group of the oxazolidinone in binding the backbone amide of Leu120. Both analogues exhibited an outstanding improvement in metabolic stability while maintaining their potent inhibitory efficacy in vitro and in vivo. Compound 75 exhibited selectivity greater than 200 times for mutant IDH1 isoforms compared to wild-type (IDH1R132H IC50, 0.027 μM; IDH1R132C IC50, 0.028 μM; IDH1WT IC50, 6.14 μM). In the HCT116–IDH1R132H/+ xenograft model, 75 dosed at 200 mg kg−1, reduced 2-HG concentration in the tumour with a maximum inhibition of 2-HG of 87.2% compared to the untreated tumours.113 The promising results of the PK and pharmacodynamic studies of oxazolidinone 75 have paved the way for clinical investigation. Currently, compound 75 has been evaluated in a phase 1 study in patients with advanced malignancies that harbour IDH1R132 mutations.114 Zha et al. reported a similar derivative during the same period, but independently of Novartis. Compound 76 (Fig. 15) reduced 2-HG production in cells transfected with IDH1R132H and IDH1R132C mutations at 10 and 50 μM. It was also shown to have a good ability to penetrate the BBB.115
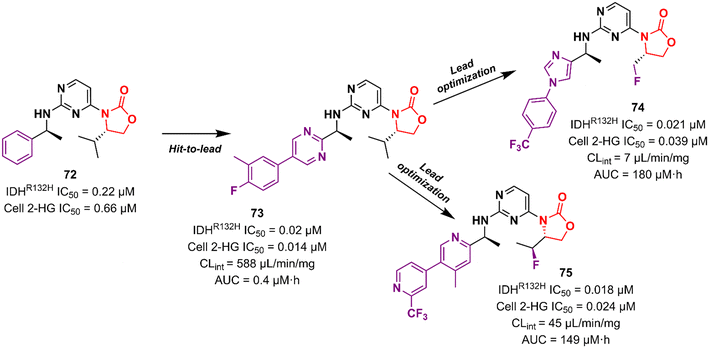 |
| | Fig. 14 Oxazolidinone as anticancer agents. | |
 |
| | Fig. 15 Anticancer oxazolidinones. | |
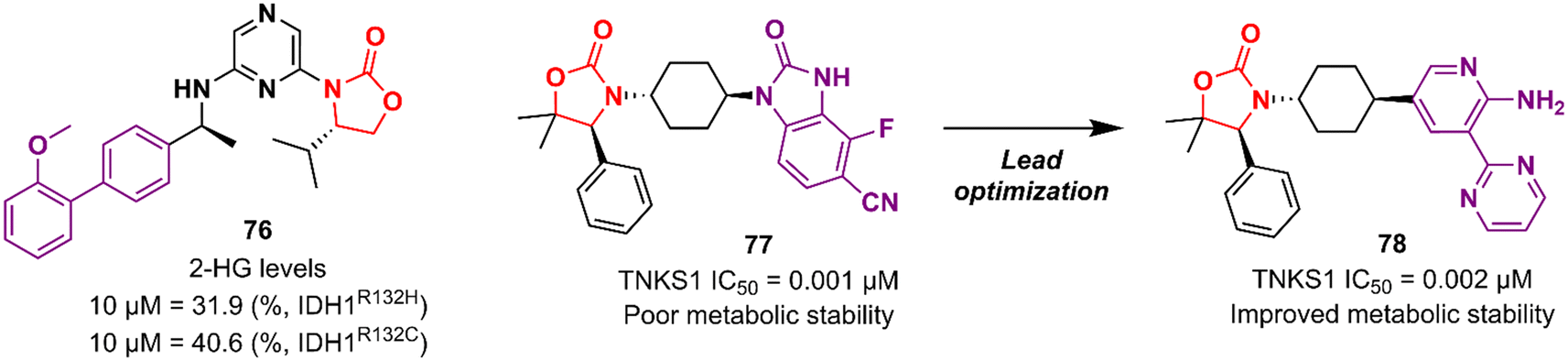
Researchers working at Amgen Inc. have also paved the way for oxazolidinone for anticancer therapy.116,117 DiMauro et al. reported in 2013 the discovery of a new oxazolidinone derivative as a potent, selective, and orally bioavailable tankyrase inhibitor.116 Tankyrases (TNKS1, PARP5a and TNKS2, PARP5b) are members of the poly-ADP-ribose polymerase (PARP) family.118 Tankyrases perform regulatory roles in controlling (in a context-dependent manner) numerous cellular pathways, including the WNT/β-catenin signalling pathway, which abnormal activation has been associated with the development and formation of several cancers.119,120 Compound 77 (Fig. 15) was identified through an iterative structure-based design effort where several scaffolds were tested before landing on the oxazolidinone moiety. It showed IC50 values of 0.001 μM against TNKS1, 0.15 μM against SW480 β-catenin, and >85 μM against PARP1 and PARP2, highlighting its selectivity PARP1/2. Nevertheless, 77 exhibited poor metabolic stability, which was linked to the hydrolysis of the aminoquinazoline amide group.116 Shortly thereafter, Amgen researchers reported the lead optimization work that aimed to improve the metabolic stability of compound 77. The drug design was based on the hypothesis of replacing the aminoquinazoline amide group with a 2-aminopyridine, thus circumventing the stability problem, while persevering two essential hydrogen bond interactions with the protein at Asp1198 and Gly1196. The approach effectively managed to improve the PK properties of the new lead compound 78 (Fig. 15). PK studies in rats and mice revealed moderate clearance and volumes of distribution (Vss). For example, after intravenous administration to rats, 78 (dose 0.5 mg kg−1) exhibited Clint 0.75 L h−1 kg−1, Vss 2.85 L kg−1, and t1/2 3.6 h. Importantly, compound 78 also maintained its selectivity and potency against TNKS1/2 with an IC50 of 0.002 μM against both isoforms. Furthermore, in vivo studies revealed that a dose of 78 at 50 mg kg−1 reduced Wnt-dependent reporter activity.117
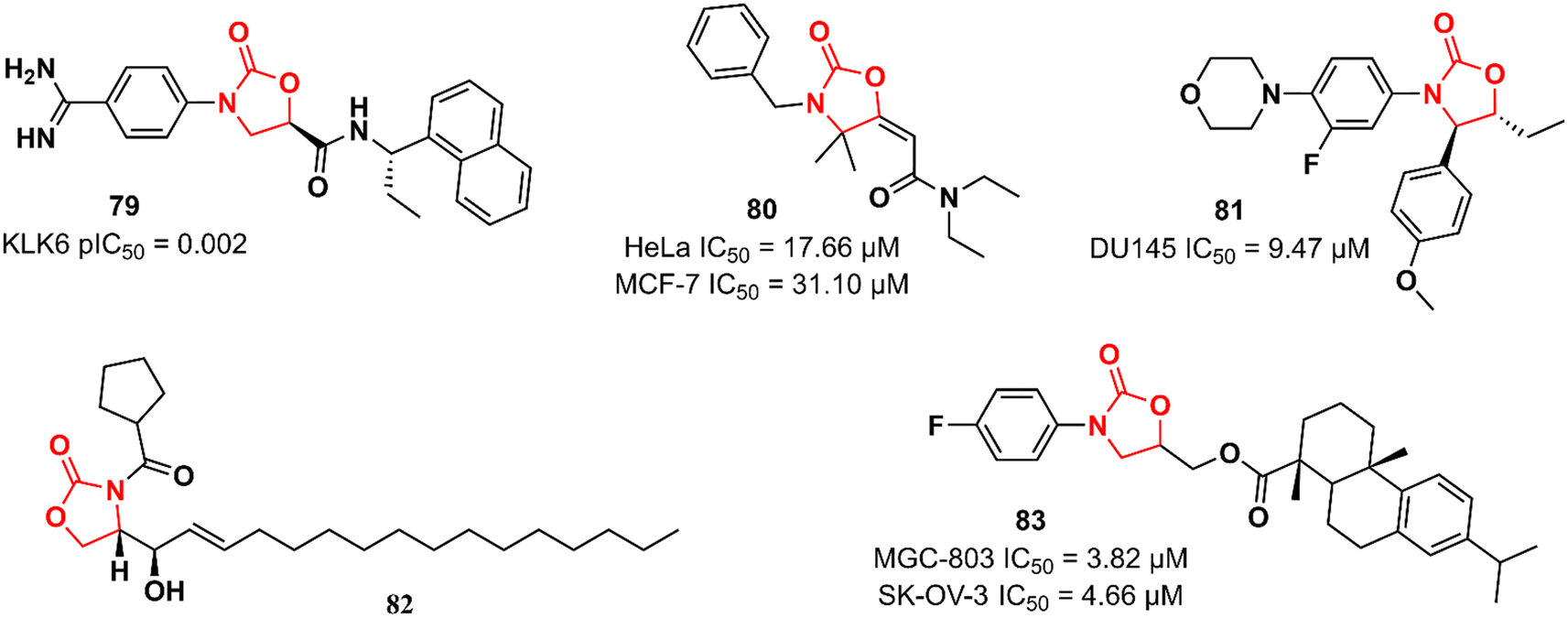
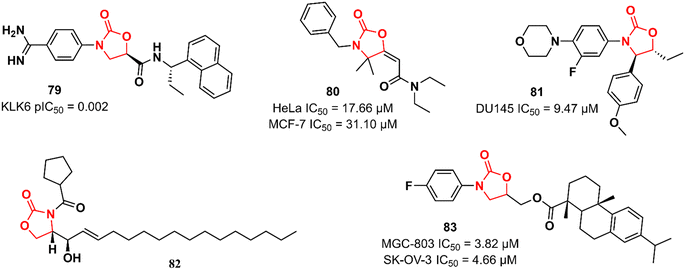
Recently, Miller et al. reported the discovery of an N-(4-benzamidino)–oxazolidinone with potent inhibitory activity against kallikrein-related peptidase 6 (KLK6).121 Kallikreins, trypsin-like serine proteases, are a group of trypsin-like serine proteases that are associated with several normal and pathological processes.122 KLK6 exerts an essential role in the degradation of extracellular matrix proteins and its differential expression has been associated with carcinogenesis in several cancers, including carcinomas, melanomas, colon, ovarian, and breast cancers.123,124 It has been hypothesized that targeting KLK6 inhibition could be a promising therapeutic strategy against some types of cancer, such as colon and squamous cell carcinomas. Compound 79 (Fig. 16) was discovered by an HTS where approximately 350000 compounds were evaluated for their ability to reduce KLK6-catalyzed hydrolysis of a fluorogenic peptide. After several rounds of funnelling, 76 was identified as a potent KLK6 inhibitor with a pIC50 of 8.6. Additional ADME profiling of 79 showed excellent solubility, high metabolic stability in human and mouse microsomes (Clint 3.1 and 2.4 mL min−1 g−1, respectively), t1/2 > 180 min, and no significant inhibition of P450 enzymes. Furthermore, compound 79 (at 50 nM and 500 nM) reduced the invasion of HCT116 tumour cells. The following oxazolidinones presented in this section have not reported their target. Nevertheless, they still highlight the application of the oxazolidinone class in cancer therapy. Compound 80 (Fig. 16) was shown to induce apoptosis in estrogen receptor-positive (ER+) breast cancer (MCF-7) and uterin cervix adenocarcinoma (HeLa) cells by increasing reactive oxygen species (ROS) production and affecting mitochondrial processes. It showed IC50 values of 17.66 and 31.10 μM against HeLa and MCF-7 cells, respectively. Interestingly, the IC50 of 80 against non-tumorigenic MCF-10A breast epithelial cells was 351.3 μM, highlighting its potential application as an anticancer agent. Further studies have demonstrated that 80 interrupts the cell cycle in the G1-S phase by inhibiting cyclin D1 expression and phosphorylation of Rb, thereby triggering apoptotic cell death. It was suggested that high levels of ROS, together with mitochondrial dysfunction and cell death, are responsible for 80 antiproliferative activity.125 Naresh et al. reported a hybrid cytoxazone–LZD that inhibited cell proliferation and induced apoptosis by activating caspase-3 and -9 in DU145 prostate cancer cells. Compound 81 (Fig. 16) showed an IC50 of 9.47 μM DU145 cell lines. Flow cytometry studies indicated that 81 arrests DU145 cells in the G0/G1 phase of the cell cycle, thus inducing cell death.126 In another study, oxazolidinone 82 (Fig. 16) (at 20 μM) was identified as an inhibitor of human leukaemia HL-60 cell proliferation by triggering apoptotic cell death.127 Oxazolidinone 83 (Fig. 16) also showed activity against four human cancer cell lines (MGC-803 human gastric cancer cells, CNE-2 human nasopharyngeal carcinoma cells, SK-OV-3 human ovarian carcinoma cells, and NCI-H460 human lung cancer cells). The IC50 values against these cells ranged from 3.82 to 17.76 μM. In particular, it did not show cytotoxicity in the human normal liver cell line LO2 (IC50 > 100 μM). It was shown to induce cell cycle arrest (G1 phase) and apoptosis in MGC-803 cells.128
 |
| | Fig. 16 Oxazolidinones with anticancer activity acting against different targets. | |
Oxazolidinones as antiviral agents
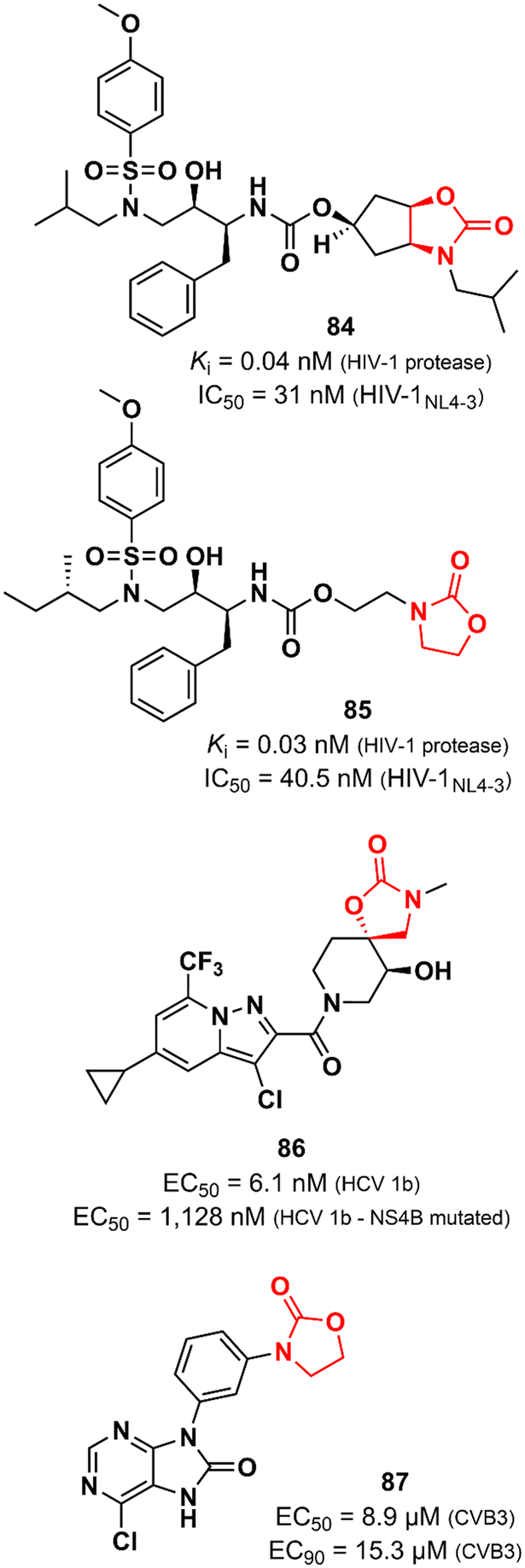
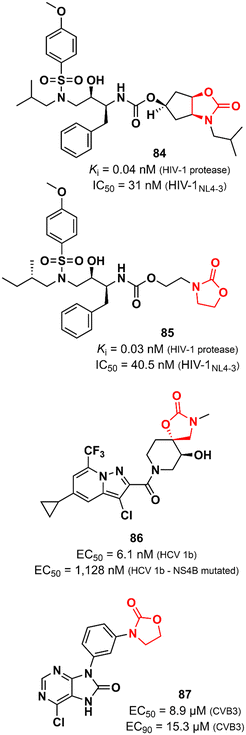
Oxazolidinones have also been explored as antiviral agents, although to a much lesser extent than antibacterials. Ghosh et al. discovered a potent HIV-1 protease inhibitor.129 In particular, the oxazolidinone scaffold was incorporated into the structure to act as a P2 ligand that would form strong hydrogen bonds with polar groups at subsite S2 of HIV-1 protease. X-ray structures of wild-type HIV-1 protease cocrystallized with some of the compounds show that the oxazolidinone group is embedded between the amide and the carbonyl oxygen of Asp29 and Gly48, respectively. Compound 84 (Fig. 17) exhibited potent enzyme inhibitory and antiviral activity (in MT-4 human T lymphocytes exposed to HIV-1NL4-3) with Ki 0.04 nM and IC50 31 nM, respectively. Furthermore, 84 maintained its potent activity against highly protease inhibitors-resistant HIV-1 variants and did not show significant cytotoxicity in MT-4 cells.129 In an independent study, compound 85 (Fig. 17) was also reported as a potent HIV-1 protease inhibitor.130 Both compounds, 84 and 85, share a great structural resemblance. The main distinction lies in the oxazolidinone group, which merges into a bicyclic scaffold in compound 84. Notwithstanding this difference, both analogues were capable to achieve the crucial hydrogen bonds with the S2 subsite of HIV-1 protease as demonstrated by X-ray crystallographic data. Oxazolidinone 85 showed Ki of 0.003 nM against wild-type HIV-1 protease and Ki values in the range of 0.003 to 1.056 nM against a panel of MDR variants of HIV-1 protease. Additionally, it exhibited EC50 values ranging from 27.8 to 57.3 nM against wild-type HIV-1 and 56.1 to 74.4 nM against MDR HIV-1 strains.130 Tai et al. working at GlaxoSmithKline reported the discovery of a spirocyclic-oxazolidinone as a hepatitis C virus (HCV) replication inhibitor. Compound 86 (Fig. 17) exhibited EC50 values of 10.9 and 6.1 nM against subgenomic replicons of genotypes 1a (HCV 1a) and 1b (HCV 1b), respectively. Further in vitro studies suggested that 86 targets the viral NS4B protein, one of the six non-structural proteins of HCV. This was supported by in vitro evaluation against two replicons that carry mutations in NS4B (H94N and V105M) with known resistance to similar analogues. There was a considerable loss of potency against these strains (27.3 times and 185 times less potent). In vitro ADME profile demonstrated excellent solubility (310 μg mL−1) and metabolic stability (t1/2 123 min). PK in vivo evaluation in rats displayed good clearance (25.7 mL min−1 kg−1) and oral exposure (Cmax 344 ng mL−1).131 Oxazolidinones have also been identified as enterovirus inhibitors. The purine–oxazolidinone hybrid 87 (Fig. 17) exhibited potent activity against the Coxsackievirus B3, Nancy strain (CVB3), a frequent cause of acute and chronic viral myocarditis. Compound 87 showed EC50 and EC90 values of 8.9 and 15.3 μM against CVB3 in Vero cells. Notably, it did not show significant antiviral activity against other enteroviruses.132
 |
| | Fig. 17 Oxazolidinones with antiviral activity. | |
Oxazolidinones as anti-inflammatory agents
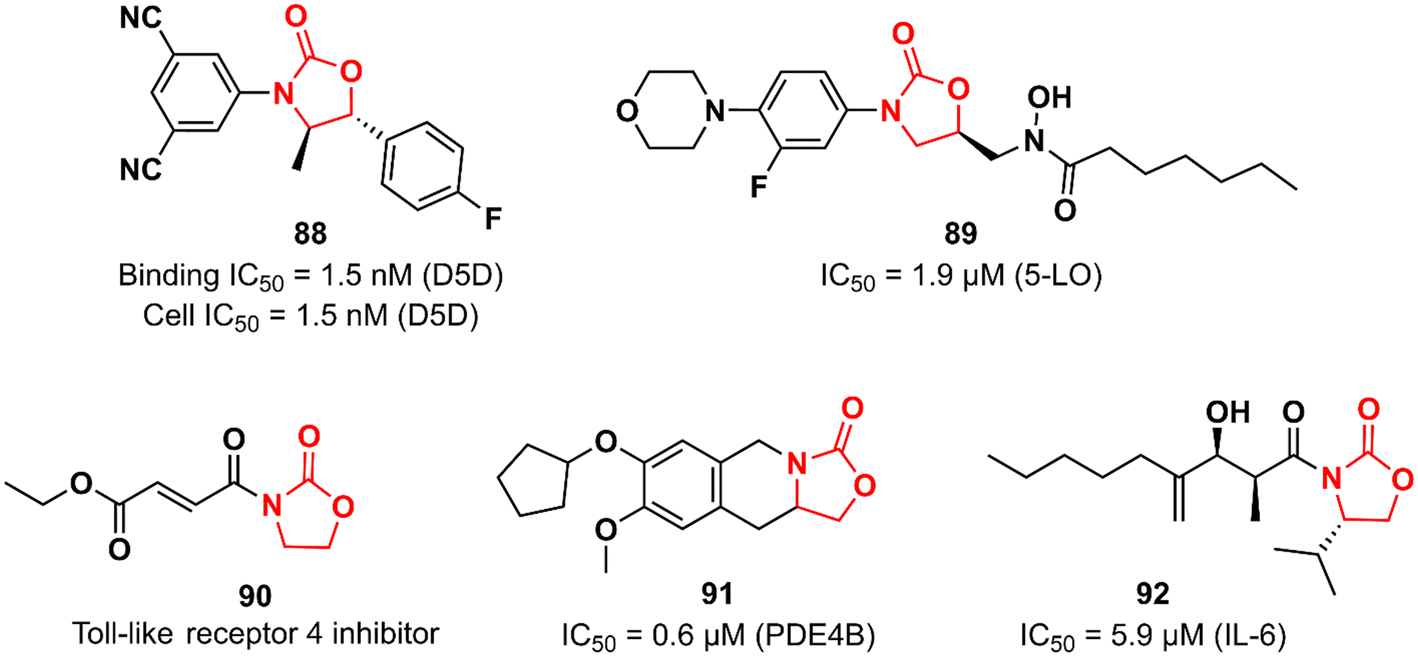
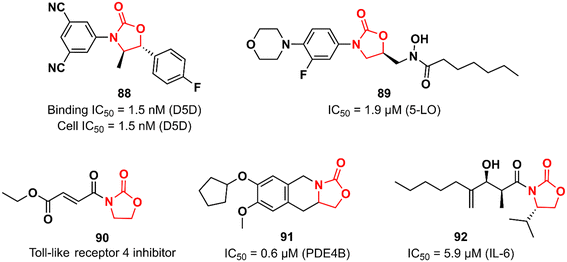
Many biological pathways are involved in an inflammatory process. Consequently, there is a wide window of opportunity for potential therapeutic targets (enzymes, G-protein-coupled receptors, nuclear hormone receptors, cytokines and cytokine receptors, cell adhesion molecules, and co-stimulatory molecules) to be explored.133 The chemical space of the oxazolidinone class has also been investigated by targeting different targets in the complex inflammatory pathways. Fujimoto et al. working at Takeda discovered an oxazolidinone with potent activity against Δ-5 desaturase (D5D).134 D5D is a transmembrane protein associated with polyunsaturated fatty acid biosynthesis, such as dihomo-γ-linolenic acid, which in turn participates in the biosynthesis of arachidonic acid that is ultimately used as a precursor for the synthesis of various unsaturated inflammatory fatty acids, including prostaglandins, leukotrienes, and thromboxanes.135 Compound 88 (Fig. 18) was developed in a medicinal chemistry campaign using HTS coupled with pharmacophore modelling. The first hits identified in the HTS were characterized by aromatic rings at each end of the molecule linked by a carbonyl-containing group. Pharmacophore modelling led the researchers to design new analogues containing oxazolidinone as a spacer between the aromatic rings. The rational drug design was successful in identifying compound 88 with high potency against D5D. It exhibited IC50 of 1.5 nM in both D5D binding activity (in rat liver microsomes) and D5D inhibitory activity (in HepG2 cells) assays. Furthermore, 88 displayed a high in vivo inhibition index for D5D in normal C57BL/6J mice (74%, 3 mg kg−1, po) and an atherosclerosis mouse model (66%, 3 mg kg−1, po). Compound 88 did not show inhibitory activity against CYP isoforms (3A4, 2C8, 2C9, and 2D6). Interestingly, 88 exhibited moderate hERG inhibitory activity (30.7% inhibition at 10 μM), which naturally raised a red flag; however, it was shown to be safe when its hERG inhibitory effect was evaluated in vivo in an electrocardiogram study with dogs.134 The LZD derivative 89 (Fig. 18) was discovered as an inhibitor of 5-lipoxygenase (5-LO).136 5-LO plays an essential role in the biosynthesis of leukotrienes (LT) from arachidonic acid.137 Compound 89 bears a hydroxamic acid group attached to the oxazolidinone at position 5, which is suggested to act as an iron-chelating moiety of an iron at the active site of the enzyme. A series of in vitro assays were carried out to assess the activity of 89. For example, in cell-based test systems (human whole blood and human monocytes), 89 inhibited the synthesis of 5-LO products (LTB4 and LTC4) with IC50 values of 0.7 and 0.9 μM, respectively. In the direct inhibition assay against 5-LO, the IC50 was 1.9 μM. Furthermore, compound 89 was active in a mouse model of peritoneal inflammation (a known model in which LT is known to play an essential role), significantly reducing the LTC4 content of the lavage fluid.136 Oxazolidinones have also been investigated as inhibitors of the toll-like receptor 4 (TLR4),138 a critical receptor involved in many inflammatory responses, such as LPS recognition.139 Oxazolidinone 90 (Fig. 18) was first described as a suppressor of LPS-induced NF-κB activation.140 Subsequently studies revealed that compound 90 acts by suppressing LPS-induced dimerization of TLR4, a key step to trigger the activation of TLR4 signalling. Interestingly, it was hypothesized that the α,β-unsaturated carbonyl group presented in 90 could play an essential role in inhibiting TLR4 dimerization by acting as a Michael acceptor.138 The oxazolidinone-fused tetrahydroisoquinoline derivative 91 (Fig. 18) was reported as a phosphodiesterase 4 (PDE4) inhibitor. It exhibited IC50 values of 0.6 and 5.13 μM against PDE4B and PDE4D, respectively. In vivo profiling demonstrated that 91 has potent suppressing activity against TNF-α release (48%) and LPS-induced neutrophilia inhibition (42%). Importantly, 91 exhibited high selectivity for PDE4, as it was inactive against other human PDE isoforms. Molecular docking analysis suggested that the catechol group plays an important role in the interaction with the enzyme.141 Choi et al. reported the first synthetic small-molecule inhibitor of interleukin 6 (IL-6), a glycoprotein that plays a critical role in inflammatory processes.142,143 Oxazolidinone 92 (Fig. 18) emerged as a hit in the screening of a large chemical library tested against IL-6-induced luciferase expression in human hepatocarcinoma HepG2 cells. Compound 92 (50 μM) decreased IL-6-induced luciferase activity by approximately 90% and showed an IC50 of 5.9 μM. It also inhibited the proliferation of the IL-6-induced human erythroleukemic cell line TF-1 with an IC50 of 7.5 μM. Furthermore, an in vivo (mice model) investigation showed that 92 inhibited IL-6-induced TNF-α production, decreased collagen-induced arthritis, and demonstrated a therapeutic effect against pancreatitis by reducing the expression of pro-inflammatory cytokines, including IL1β, TNF- α, and IL-6. Further studies revealed that compound 92 targets the β subunit of the IL-6 receptor (gp130), responsible for initiating downstream signal transduction.142
 |
| | Fig. 18 Oxazolidinones with anti-inflammatory activity. | |
Oxazolidinones in neurological disorders
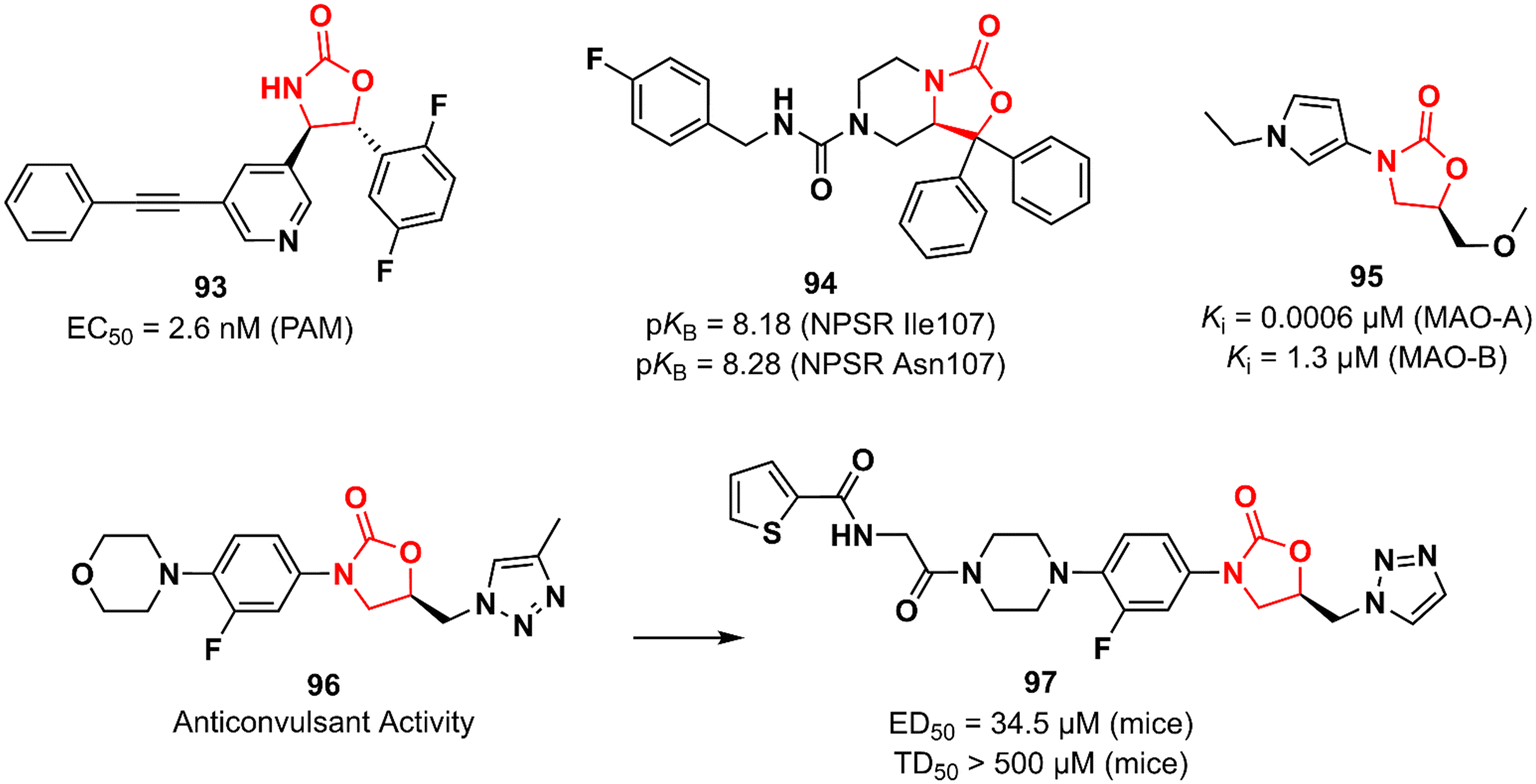
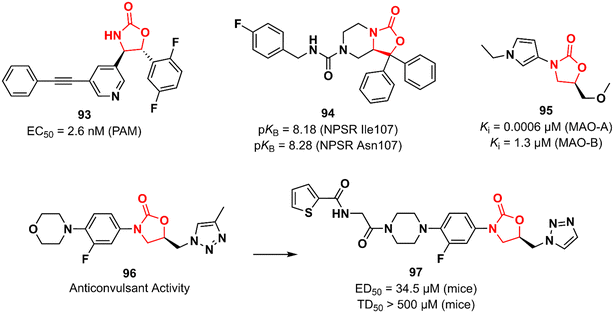
Huang et al. working at Bristol-Myers Squibb Research & Development reported an oxazolidinone acting as a positive allosteric modulator (PAM) of the metabotropic glutamate receptor subtype 5 (mGluR5) for the treatment of schizophrenia.144,145 mGluR5 is associated with N-methyl-D-aspartate receptors (NMDA) in GABAergic interneurons and its activation has been associated with increased NMDA function, which in turn can restore the imbalance between excitation and inhibition in glutamatergic synapses.146 Oxazolidinone 93 (BMS-955829) (Fig. 19) exhibited an EC50 value of 2.6 nM against PAM, excellent mGluR5 binding affinity (Ki 1.6 nM), and high selectivity for the mGluR5 subtype. In vivo investigation demonstrated that 93 (30 mg kg−1, ip) prevented convulsions in mice, which was associated with its lack of agonist activity (EC50 > 30 μM). Furthermore, it showed in vivo efficacy in mice models of schizophrenia. Further drugabillity profiling highlighted its safety profile and favourable brain penetration.145 Neuropeptide S receptor antagonist (NPSR) 94 (SHA 68) (Fig. 19) was first described by Takeda researchers as having anxiolytic-like effects.147,148 Subsequently, Guerrini et al. reported the synthesis and separation of the active enantiomer (R). Oxazolidinone 94 showed a high potency (pKB) in HEK293 cells expressing murine NPSR and human NPSR isoforms (Ile107 and Asn107) with pKB values of 8.29, 8.18, and 8.28, respectively. Conversely, the S enantiomer was devoid of biological activity.149 Another oxazolidinone 95 (Fig. 19) was described as a potent and reversible monoamine oxidase A (MAO-A) inhibitor.150 MAO inhibition is a well-known effect of certain oxazolidinone derivatives and is frequently applied as a toxicity measurement in the early stages of development.151,152 Compound 95 exhibited high selectivity towards MAO-A with a Ki value of 0.0006 versus 1.3 μM against MAO-B.150 MAO-A catalyzes the oxidation of serotonin and norepinephrine and its inhibition is associated with antidepressant effects.152 Anticonvulsant activity has also been described for oxazolidinones. The LZD derivative 96 (Fig. 19) suppressed chemically induced seizures (in vitro seizure model in the rat hippocampus) by reducing action potential firing and excitatory postsynaptic transmission highlighting the potential of the class as antiepileptic agents.153,154 In 2019, the same research group reported an analogue of 96 with in vivo anticonvulsant activity in rats and mice. Compound 97 (Fig. 19) showed ED50 values of 34.5 and 90 mg kg−1 in mice and rats, respectively, and TD50 > 500 mg kg−1 in mice. The efficacy produced was comparable to that of levetiracetam (ED50 19.4 mg kg−1).155
 |
| | Fig. 19 Oxazolidinones discovered for neurological disorders. | |
Oxazolidinones in metabolic disorders
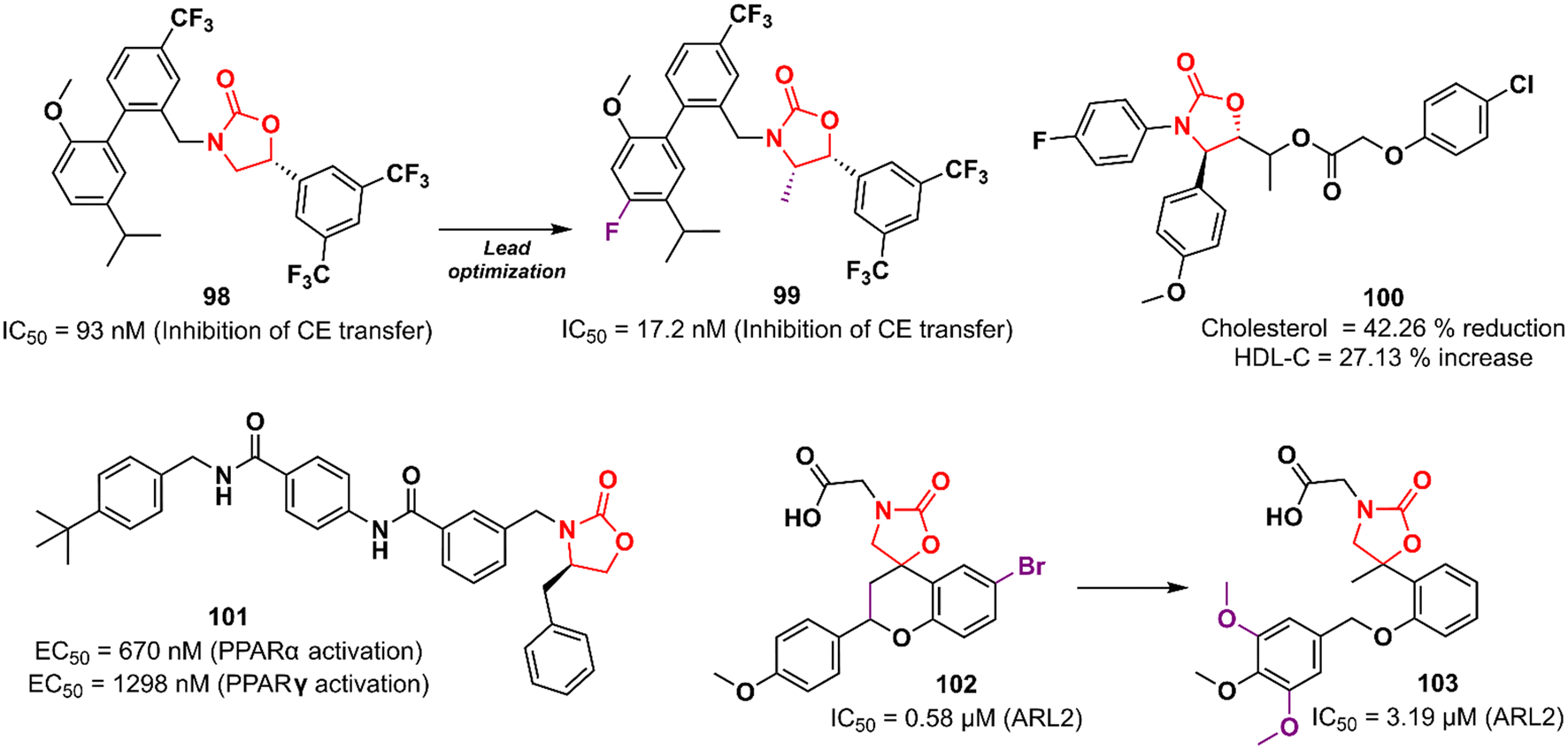
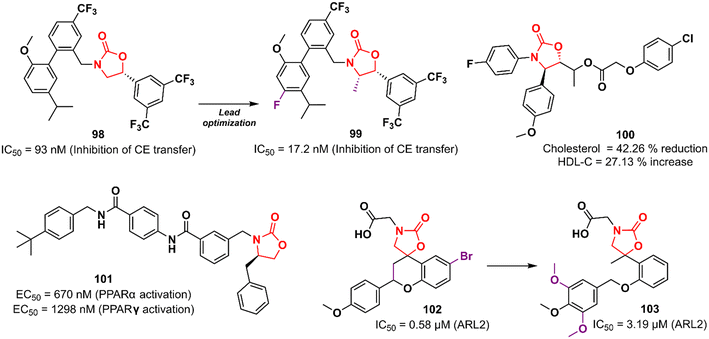
Cardiovascular diseases (CVDs) are the leading cause of death worldwide. According to the WHO, approximately 17.9 million people die each year from CVDs, an estimated 32% of all deaths globally.156,157 Elevated levels of low-density lipoprotein-cholesterol (LDL-C) and low levels of high-density lipoprotein-cholesterol (HDL-C; the so-called good cholesterol) are considered the main risk factors for CVD. Therefore, drugs that aim to increase HDL-C levels could have a potential therapeutic application. Oxazolidinones have been studied for this purpose. In 2011, researchers working at Merck Research Laboratories discovered a substituted biphenyl oxazolidinone 98 (Fig. 20) as a cholesteryl ester transfer protein (CETP) inhibitor.158 CETP acts by decreasing HDL-C levels and increasing LDL-C levels.159 Compound 98 was designed based on a previously identified inhibitor of cholesteryl ester (CE) transfer. Although oxazolidinone 98 had shown good potency in vitro in CE transfer inhibition (IC50 93 nM), it failed to increase HDL-C levels in vivo, possibly due to its poor PK profile.158 Shortly after, Merck disclosed the clinical candidate anacetrapib 99 (Fig. 20) as an optimized lead from 98.160 This compound exhibited potent CETP inhibitory activity (IC50 17.2 nM) and good in vivo efficacy in the B6-Tg(CETP) mouse model. PK studies in mice, rats, and rhesus monkeys demonstrated moderate profiles.160 However, Merck abandoned its development in 2017 due to a lack of efficacy or safety issues in clinical trials, just as several CETP inhibitors have been abandoned since 2006, including torcetrapib (Pfizer), evacetrapib (Lilly), and dalcetrapib (Roche). Oxazolidinone 100 (Fig. 20) was discovered as a cholesterol absorption inhibitor through a pharmacophore modelling study using ezetimibe as a model. In vivo studies with cholesterol-fed Sprague–Dawley rats demonstrated that the reduction in total cholesterol and triglyceride levels and increased HDL-C levels after treatment with compound 100 was compared to that of ezetimibe.161 In another study, oxazolidinone 101 (Fig. 20) was identified by molecular docking studies using the peroxisome proliferator-activated receptor (PPARα) receptor.162 PPARs are nuclear receptors involved in numerous cellular processes, including adipogenesis, lipid and glucose metabolism, energy levels, and inflammation.163 Compound 101 emerged as the most potent PPARα/γ agonist with EC50 values of 670 and 1298 nM in PPARα and PPARγ activation, respectively. In vivo investigation revealed that 101 (3.0 mg kg−1) induced a 50% reduction in food intake in food-deprived obese rats.162 Rapposelli et al. reported the spiro-oxazolidinone derivative 102 (Fig. 20) as an aldose reductase (ARL2) inhibitor.164 ARL2 is a key enzyme of the aldo–keto reductase superfamily, and its activity in pathological conditions is associated with diabetic complications.165 Compound 102 exhibited an IC50 value of 0.58 μM against ARL2, which was comparable to sorbinil (IC50 0.65 μM), a well-known ARL2 inhibitor.164 Subsequently, an analogue of 102 lacking the spiro scaffold was reported. However, the new compound 103 (Fig. 20) was shown to be less active against the enzyme (IC50 3.19 μM), highlighting the importance of the spiro-oxazolidinone moiety.166 Together, these results highlight the potential of oxazolidinones in the discovery of drugs for the treatment of metabolic syndromes.
 |
| | Fig. 20 Oxazolidinones developed for the treatment of metabolic syndromes. | |
Miscellaneous oxazolidinones
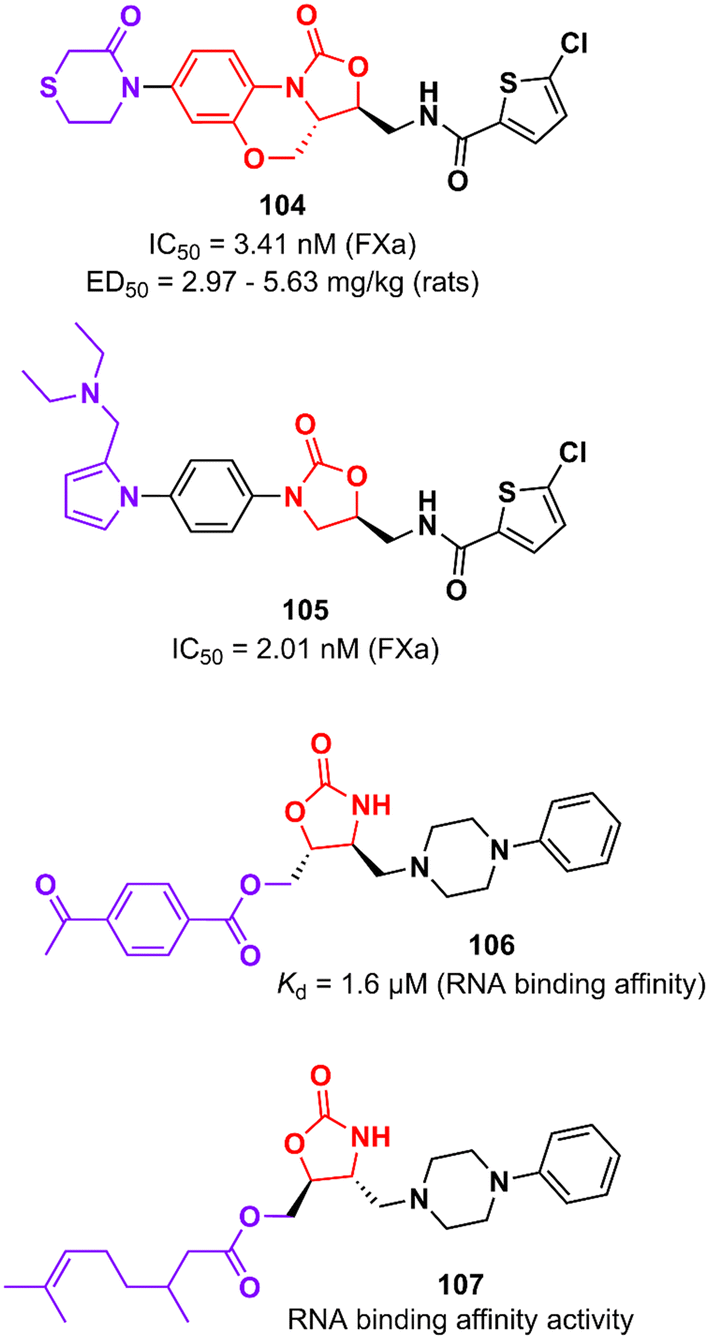
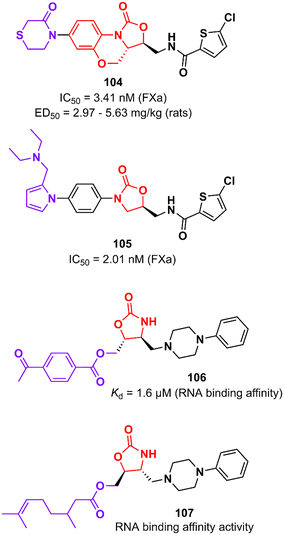
Rivaroxaban (Xarelto®, Janssen Pharmaceuticals) is another drug representative of the oxazolidinone class that is used as an anticoagulant agent to treat deep vein thrombosis and pulmonary emboli by inhibiting protein factor Xa (FXa). FXa is a serine endopeptidase that plays a critical role in the coagulation pathway by cleaving prothrombin to generate thrombin.167 Yang et al. reported in 2015 an analogue of rivaroxaban in which oxazolidinone was merged into a tricyclic scaffold. Compound 104 (Fig. 21) exhibited an IC50 value of 3.41 nM against FXa (selectivity >20000) and high anticoagulant activity in vitro. PK studies in dogs demonstrated an excellent profile for compound 104 with high bioavailability (F = 92%) and excellent oral exposure (Cmax = 530 ng mL−1). It is worth mentioning that 104 was superior to rivaroxaban in almost all PK properties. Moreover, compound 104 had excellent antithrombotic activity in three different models of thrombosis in rats: FeCl3-induced venous thrombosis (ED50 5.63 mg kg−1), arteriovenous shunt (ED50 5.28 mg kg−1), and electrically induced rat carotid artery thrombosis (ED50 2.97 mg kg−1). It is noteworthy that its efficacy in vivo was compared with that of rivaroxaban. Additionally, compound 104 did not show inhibition of the hERG K+ channel (IC50 79.75 μM) and CYP450 isozymes (IC50 > 40 μM), was not mutagenic in the Ames test and it was well-tolerated in rats upon oral administration of high doses (100 mg kg−1).168 The oxazolidinone moiety present in rivaroxaban and 104 plays a critical role in the binding affinity to the enzyme by forming two hydrogen bonds with Gly219.168 Subsequently, Gong et al. designed new analogues by replacing the morpholin-3-one moiety with different water-soluble groups. Oxazolidinone 105 (Fig. 21) was identified as the most promising analogue with an IC50 of 2.01 nM against FXa and anticoagulant activities in human and rabbit plasma. This compound has a pyrrole-alkylamino group at the S4 ligand position, which was suggested to be essential for anticoagulant activity.169
 |
| | Fig. 21 Oxazolidinones targeting FXa and T-box riboswitch RNA. | |
Oxazolidinones have also been developed as ligands that target the T-box riboswitch of non-coding RNA,170–173 which in turn could disrupt RNA function. RNA riboswitches control RNA transcription by forming a complex with metabolic effector molecules that ultimately lead to transcription.171The T-box transcription antitermination riboswitch presented in the 5′-untranslated mRNA region (5′-UTR) presented in several essential genes in Gram-positive bacteria is an example of noncoding RNA with potential application in drug discovery. This non-coding RNA is involved in controlling the transcription of the protein-encoding region of the mRNA.174 Hines et al. described an oxazolidinone derivative 106 (Fig. 21) that binds to the T-box riboswitch RNA (AM1A model), thus altering the function of the riboswitch. RNA binding affinity studies demonstrated that compound 106 has a Kd value of 1.6 μM, which was correlated with the most energetically favourable docking score (−117.4 kcal mol−1).173 Subsequently, the same group reported an iterative medicinal chemistry campaign of a series of oxazolidinones with RNA-binding affinity. Specifically, a SAR study was carried out at the C5 position of the oxazolidinone ring where several substituents were evaluated, eventually leading to compound 107 (Fig. 21) with the strongest binding affinity to AM1A RNA. As a result of the SAR analysis, the importance of hydrogen bonding and hydrophobic properties in oxazolidine binding was demonstrated.172 These results showing that oxazolidinones bind to AM1A RNA via surface binding highlight their application for the discovery of RNA-binding drugs focusing on other medicinally relevant RNAs.
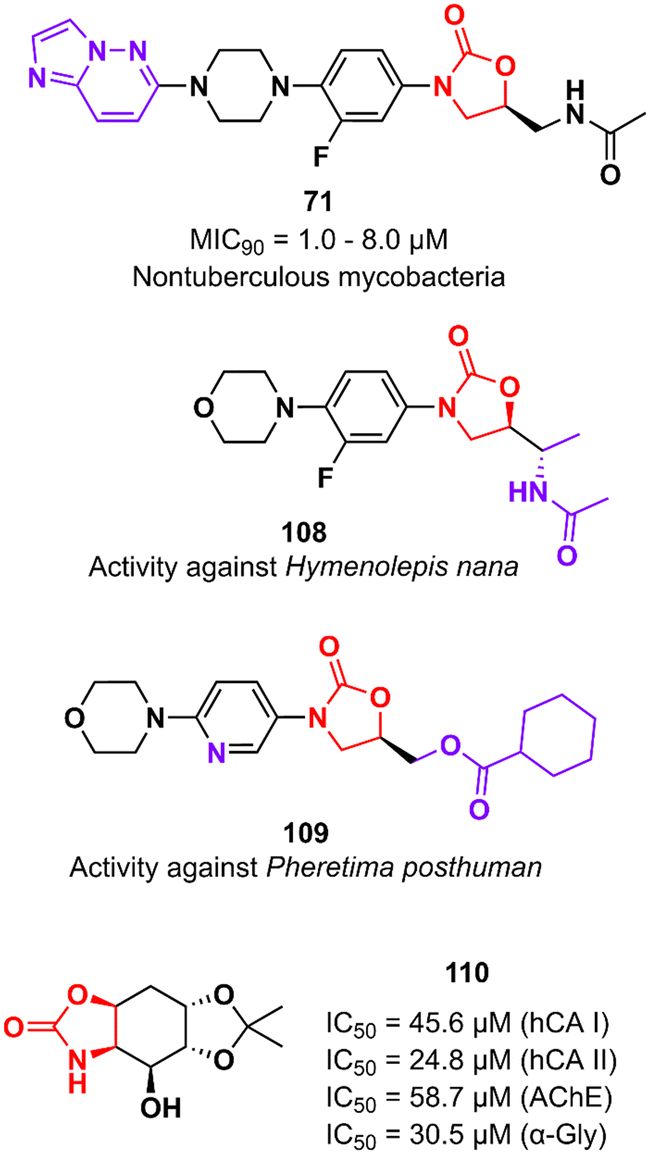
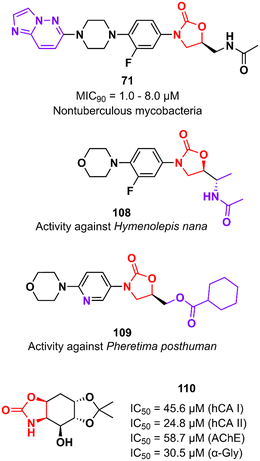
In addition to classical Gram-positive and Gram-negative bacteria and M. tuberculosis, LZD analogues have also been investigated against other microorganisms. For example, compound 71 (Fig. 22) was tested against a panel of non-tuberculous mycobacteria strains, including M. avium, M. kansasii, M. intracellulare, M. chelonae, M. abscessus, and M. fortuitum. The treatment of infections caused by these mycobacteria is often difficult due to the lack of activity of traditional antitubercular drugs. Oxazolidinone 71 was particularly active against M. kansasii, M. fortuitum and M. chelonae with MIC90 values of 1.0, 8.0 and 8.0 μM, respectively.175 Compound 108 (Fig. 22) was identified with antiparasitic activity against Hymenolepis nana, a common parasite responsible for hymenolepiasis, an intestinal parasitosis. In vitro analysis showed that after exposure to 108 (20 mg mL−1), the Hymenolepis nana worms were paralyzed in 6 min and died after 18 min.176 Similarly, compound 109 (Fig. 22) was reported with anthelmintic activity against the Indian earthworm Pheretima posthuman. This compound, an LZD analogue, induced paralysis in worms in 3.6 min and subsequent death after 8.1 min.177 It is worth noting that none of the compounds presented significant activity against Gram-positive bacteria, highlighting their high selectivity against parasites. Finally, oxazolidinone 110 (Fig. 22) was found to be a potent inhibitor against several enzymes, including human carbonic anhydrase I and II isoenzymes (hCA I and hCA II), acetylcholinesterase (AChE), and α-glycosidase (α-Gly). These enzymes are involved in a variety of cellular processes, and their inhibition by small molecules has been investigated for the treatment of several diseases, such as glaucoma, leukaemia, epilepsy, Alzheimer's disease, and type-2 diabetes mellitus. Compound 110 exhibited IC50 values of 45.6 μM (hCA I), 24.8 μM (hCA II), 58.7 μM (AChE), and 30.5 μM (α-Gly).178
 |
| | Fig. 22 Oxazolidinone derivatives with various activities. | |
Future perspectives and conclusions
The discovery of LZD and its approval by the FDA in the 2000 revealed oxazolidinones as important and promising scaffolds in the toolbox of medicinal chemistry. Ever since then, oxazolidinones have been extensively investigated in medicinal chemistry, and numerous LZD analogues have been developed, mainly as antibacterials. LZD was indeed a pioneering drug, being the first fully synthetic small molecule able to inhibit the bacterial protein synthesis. Notwithstanding the great success of oxazolidinones as antibacterial agents, there is still wide potential for this scaffold to be exploited in other therapeutic areas. Indeed, the design and synthesis of LZD analogues, as well as analogues of other oxazolidinone drugs like tedizolid and sutezolid, is an attractive field since such derivatives may find application in the treatment of different diseases. As an example, the tricyclic benzo[1,3]oxazinyl-oxazolidinone 57 shows promise as a potential preclinical anti-TB agent, while Novartis and Amgen have demonstrated the potential of oxazolidinones 74, 75 and 78 in anticancer therapy. The drug candidate 75 has completed a phase I clinical trial for the treatment of mutant acute myeloid leukemia and myelodysplastic syndrome, and inavolisib (GDC-0077), an oxazolidinone derivative discovered by Genentech, has found application in the treatment of breast cancer and it is now being evaluated in a phase III clinical trial.179 Finally, oxazolidinones 93 and 96 have found application in the treatment of schizophrenia and epilepsy, highlighting the versatility of this class of compounds.
In conclusion, 2-oxazolidinones are a widely investigated and still fascinating class of compounds in drug discovery and medicinal chemistry. Several oxazolidinones have succeeded in reaching the advanced stages of clinical trials, and others have made their way to the clinic (e.g., linezolid, tedizolid, and rivaroxaban). On the other hand, even if a large number of oxazolidinone derivatives have not been successful in advancing the drug development pipeline, they still represent interesting starting points for future optimization or drug repurposing strategies. This review highlights the versatility of oxazolidinones in medicinal chemistry, as demonstrated by the wide applicability of such compounds in many therapeutic areas, including anticancer, antiviral, anti-inflammatory, neurological disorders, antitubercular, and especially antibacterial. This reinforces the distinguished quote made by Nobel Prize winner Sir James Black that “the most fruitful basis for the discovery of a new drug is to start with an old drug”. The goal of this review was thus to provide a comprehensive overview of all the oxazolidinone analogues reported in the literature in the past decade to shed light on this important chemical motif and to stimulate the discussion on their use and future application in drug discovery.
Abbreviations
| LZD | Linezolid |
| FDA | Food and Drug Administration |
| VRE | Vancomycin-resistant Enterococci |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| WHO | World Health Organization |
| MDR-TB | Multidrug-resistant tuberculosis |
| TB | Tuberculosis |
| MIC | Minimum inhibitory concentration |
| MAO | Monoamine oxidase |
| MDR | Multidrug-resistant |
| PK | Pharmacokinetic |
| AUC | Area under the concentration |
| MRSE | Methicillin-resistant Staphylococcus epidermidis |
| PRSP | Penicillin-resistant Streptococcus pneumoniae |
|
F
| Bioavailability |
| CYP450 | Cytochrome P450 |
| BBB | Blood brain barrier |
| SAR | Structure–activity relationship |
| KPC |
Klebsiella pneumoniae carbapenemase |
| NDM-1 | New Delhi metallo-β-lactamase 1 |
| CRE | Carbapenem-resistant Enterobacteriaceae |
| LREF | Linezolid-resistant Enterococcus faecalis |
|
t
1/2
| Half-life |
| Clint | Clearance |
| LpxC | Hydroxymyristoyl)-N-acetylglucosamine deacetylase |
| LPS | Lipopolysaccharide |
| mIDH | Mutant isocitrate dehydrogenase |
| α-KG | α-Ketoglutarate |
| 2-HG | R-2-Hydroxyglutarate |
| HTS | High-throughput screening |
| PARP | Poly-ADP-ribose polymerase |
|
V
ss
| Volumes of distribution |
| KLK6 | Kallikrein-related peptidase 6 |
| ER+ | Estrogen receptor-positive |
| ROS | Reactive oxygen species |
| HCV | Hepatitis C virus |
| D5D | Δ-5 desaturase |
| LT | Leukotrienes |
| 5-LO | 5-Lipoxygenase |
| TLR4 | Toll-like receptor 4 |
| PDE4 | Phosphodiesterase 4 |
| PAM | Positive allosteric modulator |
| mGluR5 | Metabotropic glutamate receptor subtype 5 |
| NMDA |
N-Methyl-D-aspartate receptors |
| CVD | Cardiovascular disease |
| LDL-C | Low-density lipoprotein-cholesterol |
| HDL-C | High-density lipoprotein-cholesterol |
| CETP | Cholesteryl ester transfer protein |
| CE | Cholesteryl ester |
| PPARα | Peroxisome proliferator-activated receptor |
| ARL2 | Aldose reductase |
| FXa | Protein factor Xa |
| 5′-UTR | 5′-Untranslated mRNA region |
| hCA | Carbonic anhydrase |
| AChE | Acetylcholinesterase |
| α-Gly | α-Glycosidase |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101027065.
References
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- W. Raether and H. Hänel, Parasitol. Res., 2003, 90, S19–S39 CrossRef PubMed.
- N. Pandit, R. K. Singla and B. Shrivastava, Int. J. Med. Chem., 2011, 2012, 159285 Search PubMed.
- W. Hong, L. Chen and J. Xie, Expert Opin. Ther. Targets, 2014, 18, 691–701 CrossRef CAS PubMed.
- S. J. Brickner, Curr. Pharm. Des., 1996, 2, 175–194 CrossRef CAS.
- A. M. Slee, M. A. Wuonola, R. J. McRipley, I. Zajac, M. J. Zawada, P. T. Bartholomew, W. A. Gregory and M. Forbes, Antimicrob. Agents Chemother., 1987, 31, 1791–1797 CrossRef CAS PubMed.
- M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010–2023 CrossRef CAS PubMed.
- C. W. Ford, G. E. Zurenko and M. R. Barbachyn, Curr. Drug Targets: Infect. Disord., 2001, 1, 181–199 CAS.
- R. C. Moellering, Ann. Intern. Med., 2003, 138, 135–142 CrossRef CAS PubMed.
-
World Health Organization, WHO Consolidated Guidelines on Tuberculosis, Module 4: Treatment - Drug-Resistant Tuberculosis Treatment, Geneva, 2020 Search PubMed.
- S. M. Swaney, H. Aoki, M. C. Ganoza and D. L. Shinabarger, Antimicrob. Agents Chemother., 1998, 42, 3251–3255 CrossRef CAS PubMed.
- D. L. Shinabarger, K. R. Marotti, R. W. Murray, A. H. Lin, E. P. Melchior, S. M. Swaney, D. S. Dunyak, W. F. Demyan and J. M. Buysse, Antimicrob. Agents Chemother., 1997, 41, 2132–2136 CrossRef CAS PubMed.
- K. Tsai, V. Stojković, D. J. Lee, I. D. Young, T. Szal, D. Klepacki, N. Vázquez-Laslop, A. S. Mankin, J. S. Fraser and D. G. Fujimori, Nat. Struct. Mol. Biol., 2022, 29, 162–171 CrossRef CAS PubMed.
- G. F. S. Fernandes, H. R. N. Salgado and J. L. Dos Santos, Crit. Rev. Anal. Chem., 2019, 50, 196–211 CrossRef PubMed.
- Z. A. Kanafani and G. R. Corey, Expert Opin. Invest. Drugs, 2012, 21, 515–522 CrossRef CAS PubMed.
- O. A. Phillips and L. H. Sharaf, Expert Opin. Ther. Pat., 2016, 26, 591–605 CrossRef CAS PubMed.
- G. F. S. Fernandes, A. M. Thompson, D. Castagnolo, W. A. Denny and J. L. Dos Santos, J. Med. Chem., 2022, 65, 7489–7531 CrossRef CAS PubMed.
- L. Foti, A. Piperno, A. Scala and O. Giuffrè, Molecules, 2021, 26, 4280 CrossRef PubMed.
- A. R. N. Ndukwe, S. Wiedbrauk, N. R. B. Boase and K. E. Fairfull-Smith, Chem. – Asian J., 2022, 17, e202200201 CrossRef CAS PubMed.
- Q. Zhao, L. Xin, Y. Liu, C. Liang, J. Li, Y. Jian, H. Li, Z. Shi, H. Liu and W. Cao, J. Med. Chem., 2021, 64, 10557–10580 CrossRef CAS PubMed.
- V. R. Macherla, J. Liu, M. Sunga, D. J. White, J. Grodberg, S. Teisan, K. S. Lam and B. C. M. Potts, J. Nat. Prod., 2007, 70, 1454–1457 CrossRef CAS PubMed.
- M. Tadesse, M. B. Strøm, J. Svenson, M. Jaspars, B. F. Milne, V. Tørfoss, J. H. Andersen, E. Hansen, K. Stensvåg and T. Haug, Org. Lett., 2010, 12, 4752–4755 CrossRef CAS PubMed.
- C. L. Ventola, Pharm. Ther., 2015, 40, 277–283 Search PubMed.
- Antimicrobial Resistance Collaborators, Lancet, 2022, 399, 629–655 CrossRef PubMed.
-
J. O'Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, The Review on Antimicrobial Resistance, London, 2016 Search PubMed.
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Front. Microbiol., 2019, 10, 539 CrossRef PubMed.
-
World Health Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, Geneva, 2017 Search PubMed.
- S. H. Park, Korean J. Intern. Med., 2014, 29, 27–30 CrossRef CAS PubMed.
- M. K. Khera, I. A. Cliffe, T. Mathur and O. Prakash, Bioorg. Med. Chem. Lett., 2011, 21, 2887–2889 CrossRef CAS PubMed.
- M. K. Khera, I. A. Cliffe and O. Prakash, Bioorg. Med. Chem. Lett., 2011, 21, 5266–5269 CrossRef CAS PubMed.
- T. Mathur, M. Kumar, T. K. Barman, G. R. Kumar, V. Kalia, S. Singhal, V. S. Raj, D. J. Upadhyay, B. Das and P. K. Bhatnagar, J. Antimicrob. Chemother., 2011, 66, 1087–1095 CrossRef CAS PubMed.
- S.-H. Oh, H.-S. Park, J.-H. Lee, S.-Y. Baek, S.-E. Chae, K. Oh, Y. L. Cho and J.-H. Kwak, Molecules, 2017, 22, 2096 CrossRef PubMed.
- S.-H. Oh, J. Kim, S.-Y. Baek, S.-E. Chae, H.-S. Park, Y.-L. Cho and J.-H. Kwak, Molecules, 2017, 22, 394 CrossRef PubMed.
- J.-W. Jeong, S.-J. Jung, H.-H. Lee, Y.-Z. Kim, T.-K. Park, Y.-L. Cho, S.-E. Chae, S.-Y. Baek, S.-H. Woo, H.-S. Lee and J.-H. Kwak, Antimicrob. Agents Chemother., 2010, 54, 5359–5362 CrossRef CAS PubMed.
- S.-J. Jung, I.-N.-R. Yun, H. S. Park, H.-H. Lee, J.-W. Jeong, Y.-Z. Kim, Y.-L. Cho and J.-H. Kwak, Int. J. Antimicrob. Agents, 2012, 40, 539–543 CrossRef CAS PubMed.
- B. Guo, H. Fan, Q. Xin, W. Chu, H. Wang and Y. Yang, Bioorg. Med. Chem. Lett., 2013, 23, 3697–3699 CrossRef CAS PubMed.
- T. Xue, S. Ding, B. Guo, W. Chu, H. Wang and Y. Yang, Bioorg. Med. Chem. Lett., 2015, 25, 2203–2210 CrossRef CAS PubMed.
- Q. Xin, H. Fan, B. Guo, H. He, S. Gao, H. Wang, Y. Huang and Y. Yang, J. Med. Chem., 2011, 54, 7493–7502 CrossRef CAS PubMed.
- B. Guo, H. Fan, Q. Xin, W. Chu, H. Wang, Y. Huang, X. Chen and Y. Yang, J. Med. Chem., 2013, 56, 2642–2650 CrossRef CAS PubMed.
- B. Guo, H. Fan, Q. Xin, W. Chu, H. Wang and Y. Yang, Bioorg. Med. Chem. Lett., 2013, 23, 3697–3699 CrossRef CAS PubMed.
- T. Xue, S. Ding, B. Guo, W. Chu, H. Wang and Y. Yang, Bioorg. Med. Chem. Lett., 2015, 25, 2203–2210 CrossRef CAS PubMed.
- C. Gong, T. Yang, X. Yang, Y. Liu, W. Ang, J. Tang, W. Pi, L. Xiong, Y. Chang, W. Ye, Z. Wang, Y. Luo, X. Zhao and Y. Wei, Nanoscale, 2013, 5, 275 RSC.
- S. Wu, T. Yang, Y. Luo, X. Li, X. Zhang, J. Tang, X. Ma and Z. Wang, J. Antimicrob. Chemother., 2014, 69, 3011–3019 CrossRef CAS PubMed.
- Z. Sang, H. Long, T. Yang, W. Ye, X. Yang, G. Chen, Z. Wang and Y. Luo, Drug Test. Anal., 2016, 8, 976–984 CrossRef CAS PubMed.
- T. Yang, G. Chen, Z. Sang, Y. Liu, X. Yang, Y. Chang, H. Long, W. Ang, J. Tang, Z. Wang, G. Li, S. Yang, J. Zhang, Y. Wei and Y. Luo, J. Med. Chem., 2015, 58, 6389–6409 CrossRef CAS PubMed.
- H. Suzuki, I. Utsunomiya, K. Shudo, N. Fukuhara, T. Iwaki and T. Yasukata, Eur. J. Med. Chem., 2013, 63, 811–825 CrossRef CAS PubMed.
- H. Suzuki, I. Utsunomiya, K. Shudo, N. Fukuhara, T. Iwaki and T. Yasukata, Eur. J. Med. Chem., 2013, 69, 262–277 CrossRef CAS PubMed.
- C. D. Cruz, P. Wrigstedt, K. Moslova, V. Iashin, H. Makkyla, L. Ghemtio, S. Heikkinen, P. Tammela and J. E. Perea-Buceta, Eur. J. Med. Chem., 2021, 211, 113002 CrossRef CAS PubMed.
- M. S. Deshmukh and N. Jain, ACS Med. Chem. Lett., 2017, 8, 1153–1158 CrossRef CAS PubMed.
- B. Seetharamsingh, R. Ramesh, S. S. Dange, P. V. Khairnar, S. Singhal, D. Upadhyay, S. Veeraraghavan, S. Viswanadha, S. Vakkalanka and D. S. Reddy, ACS Med. Chem. Lett., 2015, 6, 1105–1110 CrossRef CAS PubMed.
- A. Khalaj, M. Nakhjiri, A. S. Negahbani, M. Samadizadeh, L. Firoozpour, S. Rajabalian, N. Samadi, M. A. Faramarzi, N. Adibpour, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2011, 46, 65–70 CrossRef CAS PubMed.
- S. Bhawsar, S. Pawar, P. Deshpande, R. Yeole, R. Chavan, M. Nandanwar, S. Bhagwat and M. Patel, Bioorg. Med. Chem. Lett., 2022, 71, 128842 CrossRef CAS PubMed.
- C. Matsingos, T. Al-Adhami, S. Jamshidi, C. Hind, M. Clifford, J. M. Sutton and K. M. Rahman, Bioorg. Med. Chem., 2021, 49, 116397 CrossRef CAS PubMed.
- P.-Y. Bai, S.-S. Qin, W.-C. Chu, Y. Yang, D.-Y. Cui, Y.-G. Hua, Q.-Q. Yang and E. Zhang, Eur. J. Med. Chem., 2018, 155, 925–945 CrossRef CAS PubMed.
- O. A. Phillips, E. E. Udo, M. E. Abdel-Hamid and R. Varghese, Arch. Pharm. Chem. Life Sci., 2012, 345, 790–803 CrossRef CAS PubMed.
- O. A. Phillips, E. E. Udo, M. E. Abdel-Hamid and R. Varghese, Eur. J. Med. Chem., 2013, 66, 246–257 CrossRef CAS PubMed.
- Y. Wu, X. Ding, L. Ding, Y. Zhang, L. Cui, L. Sun, W. Li, D. Wang and Y. Zhao, Eur. J. Med. Chem., 2018, 158, 247–258 CrossRef CAS PubMed.
- Y. Wu, X. Ding, Y. Yang, Y. Li, Y. Qi, F. Hu, M. Qin, Y. Liu, L. Sun and Y. Zhao, Eur. J. Med. Chem., 2020, 185, 111781 CrossRef CAS PubMed.
- Y. Hou, Y. Dong, T. Ye, J. Jiang, L. Ding, M. Qin, X. Ding and Y. Zhao, Bioorg. Med. Chem. Lett., 2019, 29, 126746 CrossRef CAS PubMed.
- J. Jiang, Y. Hou, M. Duan, B. Wang, Y. Wu, X. Ding and Y. Zhao, Bioorg. Med. Chem. Lett., 2021, 32, 127660 CrossRef CAS PubMed.
- Y. Wu, X. Ding, S. Xu, Y. Yang, X. Zhang, C. Wang, H. Lei and Y. Zhao, Bioorg. Med. Chem. Lett., 2019, 29, 496–502 CrossRef CAS PubMed.
- A. M. Siddiqui, J. A. Sattigeri, K. Javed, S. Shafi, M. Shamim, S. Singhal and Z. M. Malik, Bioorg. Med. Chem. Lett., 2018, 28, 1198–1206 CrossRef CAS PubMed.
- A. W. Sun, P. L. Bulterys, M. D. Bartberger, P. A. Jorth, B. M. O'Boyle, S. C. Virgil, J. F. Miller and B. M. Stoltz, Bioorg. Med. Chem. Lett., 2019, 29, 2686–2689 CrossRef CAS PubMed.
- L. Liu, L. Shao, J. Li, H. Cui, B. Li, X. Zhou, P. Lv and J. Zhang, Molecules, 2019, 24, 1641 CrossRef PubMed.
- O. A. Phillips, E. E. Udo and R. J. D'silva, Sci. Pharm., 2018, 86, 42 CrossRef PubMed.
- H. Lei, Y. Jiang, D. Wang, P. Gong, Y. Li, Y. Dong and M. Dong, Jpn. J. Infect. Dis., 2014, 67, 402–404 CrossRef CAS PubMed.
- G. S. Basarab, P. Doig, V. Galullo, G. Kern, A. Kimzey, A. Kutschke, J. P. Newman, M. Morningstar, J. Mueller, L. Otterson, K. Vishwanathan, F. Zhou and M. Gowravaram, J. Med. Chem., 2015, 58, 6264–6282 CrossRef CAS PubMed.
- G. S. Basarab, G. H. Kern, J. McNulty, J. P. Mueller, K. Lawrence, K. Vishwanathan, R. A. Alm, K. Barvian, P. Doig, V. Galullo, H. Gardner, M. Gowravaram, M. Huband, A. Kimzey, M. Morningstar, A. Kutschke, S. D. Lahiri, M. Perros, R. Singh, V. J. A. Schuck, R. Tommasi, G. Walkup and J. V. Newman, Sci. Rep., 2015, 5, 11827 CrossRef CAS PubMed.
- S. N. Taylor, J. Marrazzo, B. E. Batteiger, E. W. Hook, A. C. Seña, J. Long, M. R. Wierzbicki, H. Kwak, S. M. Johnson, K. Lawrence and J. Mueller, N. Engl. J. Med., 2018, 379, 1835–1845 CrossRef CAS PubMed.
- K. Jiang, X. Yan, J. Yu, Z. Xiao, H. Wu, M. Zhao, Y. Yue, X. Zhou, J. Xiao and F. Lin, Eur. J. Med. Chem., 2020, 194, 112252 CrossRef CAS PubMed.
- J. E. Jackman, C. R. H. Raetz and C. A. Fierke, Biochemistry, 1999, 38, 1902–1911 CrossRef CAS PubMed.
- S. Basak, Y. Li, S. Tao, F. Daryaee, J. Merino, C. Gu, S. L. Delker, J. N. Phan, T. E. Edwards, S. G. Walker and P. J. Tonge, J. Med. Chem., 2022, 65, 11854–11875 CrossRef CAS PubMed.
- P. S. Lee, G. Lapointe, A. M. Madera, R. L. Simmons, W. Xu, A. Yifru, M. Tjandra, S. Karur, A. Rico, K. Thompson, J. Bojkovic, L. Xie, K. Uehara, A. Liu, W. Shu, C. Bellamacina, D. McKenney, L. Morris, G. R. Tonn, C. Osborne, B. M. Benton, L. McDowell, J. Fu and Z. K. Sweeney, J. Med. Chem., 2018, 61, 9360–9370 CrossRef CAS PubMed.
- H. Kurasaki, K. Tsuda, M. Shinoyama, N. Takaya, Y. Yamaguchi, R. Kishii, K. Iwase, N. Ando, M. Nomura and Y. Kohno, ACS Med. Chem. Lett., 2016, 7, 623–628 CrossRef CAS PubMed.
- S. Ding, J. Ji, M. Zhang, Y. Yang, R. Wang, X. Zhu, L. Wang, Y. Zhong, L. Gao, M. Lu, J. Liu and Y. Chen, Arch. Pharm. Chem. Life Sci., 2019, 352, 1900129 CrossRef CAS PubMed.
- R. Liu, P. A. Miller, S. B. Vakulenko, N. K. Stewart, W. C. Boggess and M. J. Miller, J. Med. Chem., 2018, 61, 3845–3854 CrossRef CAS PubMed.
- U. Möllmann, L. Heinisch, A. Bauernfeind, T. Köhler and D. Ankel-Fuchs, BioMetals, 2009, 22, 615–624 CrossRef PubMed.
- K. Takrouri, H. D. Cooper, A. Spaulding, P. Zucchi, B. Koleva, D. C. Cleary, W. Tear, P. J. Beuning, E. B. Hirsch and J. B. Aggen, ACS Infect. Dis., 2016, 2, 405–426 CrossRef CAS PubMed.
- A. Lyons, J. Kirkham, K. Blades, D. Orr, E. Dauncey, O. Smith, E. Dick, R. Walker, T. Matthews, A. Bunt, J. Finlayson, I. Morrison, V. J. Savage, E. Moyo, H. S. Butler, R. Newman, N. Ooi, A. Smith, C. Charrier, A. J. Ratcliffe, N. R. Stokes, S. Best, A.-M. Salisbury, M. Craighead and I. R. Cooper, Bioorg. Med. Chem. Lett., 2022, 65, 128648 CrossRef CAS PubMed.
- K. Takrouri, H. D. Cooper, A. Spaulding, P. Zucchi, B. Koleva, D. C. Cleary, W. Tear, P. J. Beuning, E. B. Hirsch and J. B. Aggen, ACS Infect. Dis., 2016, 2, 406–426 Search PubMed.
- A. Spaulding, K. Takrouri, P. Mahalingam, D. C. Cleary, H. D. Cooper, P. Zucchi, W. Tear, B. Koleva, P. J. Beuning, E. B. Hirsch and J. B. Aggen, Bioorg. Med. Chem. Lett., 2017, 27, 5310–5321 CrossRef CAS PubMed.
- H.-N. Shin, S. H. Seo, H. Choo, G. Kuem, K. Il Choi and G. Nam, Bioorg. Med. Chem. Lett., 2013, 23, 1193–1196 CrossRef CAS PubMed.
- Z. Zhuang, D. Wan, J. Ding, S. He, Q. Zhang, X. Wang, Y. Yuan, Y. Lu, C. Z. Ding, A. S. Lynch, A. M. Upton, C. B. Cooper, W. A. Denny and Z. Ma, Molecules, 2020, 25, 2431 CrossRef CAS PubMed.
- Z. Hu, I. V. Leus, B. Chandar, B. S. Sherborne, Q. P. Avila, V. V. Rybenkov, H. I. Zgurskaya and A. S. Duerfeldt, J. Med. Chem., 2022, 65, 14144–14179 CrossRef CAS PubMed.
- H. Zhao, Y. Lu, L. Sheng, Z. Yuan, B. Wang, W. Wang, Y. Li, C. Ma, X. Wang, D. Zhang and H. Huang, ACS Med. Chem. Lett., 2017, 8, 533–537 CrossRef CAS PubMed.
- H. Zhao, B. Wang, L. Fu, G. Li, H. Lu, Y. Liu, L. Sheng, Y. Li, B. Zhang, Y. Lu, C. Ma, H. Huang, D. Zhang and Y. Lu, J. Med. Chem., 2020, 63, 9316–9339 CrossRef CAS PubMed.
- Y. Wu, B. Wang, H. Lu, H. Zhao, B. Yang, L. Li, Y. Lu, D. Zhang, N. Sun and H. Huang, J. Med. Chem., 2021, 64, 3234–3248 CrossRef CAS PubMed.
- G. F. S. Fernandes, D. H. Jornada, P. C. Souza, C. Man Chin, F. R. Pavan and J. L. Santos, Curr. Med. Chem., 2015, 22, 3133–3161 CrossRef CAS PubMed.
- G. F. dos S. Fernandes, C. M. Chin and J. L. Dos Santos, Pharmaceuticals, 2017, 10, 51 CrossRef PubMed.
- A. Kamal, R. V. C. R. N. C. Shetti, S. Azeeza, P. Swapna, M. N. A. Khan, I. A. Khan, S. Sharma and S. T. Abdullah, Eur. J. Med. Chem., 2011, 46, 893–900 CrossRef CAS PubMed.
- K. D. Thomas, A. V. Adhikari, I. H. Chowdhury, T. Sandeep, R. Mahmood, B. Bhattacharya and E. Sumesh, Eur. J. Med. Chem., 2011, 46, 4834–4845 CrossRef CAS PubMed.
- D. Bhattarai, S. H. Lee, S. H. Seo, G. Nam, S. B. Kang, A. N. Pae, E. E. Kim, T. Oh, S.-N. Cho and G. Keum, Chem. Biol. Drug Des., 2012, 80, 388–397 CrossRef CAS PubMed.
- O. A. Phillips, E. E. Udo and R. Varghese, Tuberc. Res. Treat., 2012, 2012, 289136 Search PubMed.
- A. Kamal, P. Swapna, R. V. C. R. N. C. Shetti, A. B. Shaik, M. P. N. Rao, F. Sultana, I. A. Khan, S. Sharma, N. P. Kalia, S. Kumar and B. Chandrakant, Eur. J. Med. Chem., 2013, 64, 239–251 CrossRef CAS PubMed.
- N. F. Al-Tannak and O. A. Phillips, Sci. Pharm., 2017, 85, 34 CrossRef PubMed.
- W. Ang, W. Ye, Z. Sang, Y. Liu, T. Yang, Y. Deng, Y. Luo and Y. Wei, Bioorg. Med. Chem. Lett., 2014, 24, 1496–1501 CrossRef PubMed.
- D. Bhattarai, J. Lee, S. H. Seo, G. Nam, H. Choo, S. B. Kang, J.-H. Kwak, T. Oh, S.-N. Cho, A. N. Pae, E. E. Kim, N. Jeong and G. Keum, Chem. Pharm. Bull., 2014, 62, 1214–1224 CrossRef PubMed.
- P. K. Gadekar, A. Roychowdhury, P. S. Kharkar, V. M. Khedkar, M. Arkile, H. Manek, D. Sarkar, R. Sharma, V. Vijayakumar and S. Sarveswari, Eur. J. Med. Chem., 2016, 122, 475–487 CrossRef CAS PubMed.
- Q. Tang, Y. Zhao, B. Xu, P. Gong and D. Wang, Jpn. J. Infect. Dis., 2017, 70, 678–681 CrossRef CAS PubMed.
- G. D. García-Olaiz, E. Alcántar-Zavala, A. Ochoa-Terán, A. Cabrera, R. Muñiz-Salazar, J. Montes-Ávila, A. J. Salazar-Medina, E. Alday, C. Velazquez, J. L. Medina-Franco and R. Laniado-Laborín, Bioorg. Chem., 2020, 95, 103483 CrossRef PubMed.
- X. Ding, Y. Wu, A. Tong, B. Sun, Y. Zhao and Y. Jiang, Jpn. J. Infect. Dis., 2021, 74, 245–248 CrossRef CAS PubMed.
- D. Wang, Y. Zhao, Z. Liu, H. Lei, M. Dong and P. Gong, J. Antimicrob. Chemother., 2014, 69, 1711–1714 CrossRef CAS PubMed.
- C. Mattiuzzi and G. Lippi, J. Epidemiol. Glob. Health, 2019, 9, 217–222 CrossRef PubMed.
- E. A. V. de F. Ramalho, M. G. da R. Pitta, H. de B. S. Neto and I. da R. Pitta, Clin. Cancer Drugs, 2020, 7, 95–106 CrossRef CAS.
- L. Dang, K. Yen and E. C. Attar, Ann. Oncol., 2016, 27, 599–608 CrossRef CAS PubMed.
- D. Golub, N. Iyengar, S. Dogra, T. Wong, D. Bready, K. Tang, A. S. Modrek and D. G. Placantonakis, Front. Oncol., 2019, 9, 417 CrossRef PubMed.
- D. J. Urban, N. J. Martinez, M. I. Davis, K. R. Brimacombe, D. M. Cheff, T. D. Lee, M. J. Henderson, S. A. Titus, R. Pragani, J. M. Rohde, L. Liu, Y. Fang, S. Karavadhi, P. Shah, O. W. Lee, A. Wang, A. McIver, H. Zheng, X. Wang, X. Xu, A. Jadhav, A. Simeonov, M. Shen, M. B. Boxer and M. D. Hall, Sci. Rep., 2017, 7, 12758 CrossRef PubMed.
- X. Liu and Z.-Q. Ling, Histol. Histopathol., 2015, 30, 1155–1160 CAS.
- L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden and S. M. Su, Nature, 2009, 462, 739–744 CrossRef CAS PubMed.
- X. Xie, D. Baird, K. Bowen, V. Capka, J. Chen, G. Chenail, Y. Cho, J. Dooley, A. Farsidjani, P. Fortin, D. Kohls, R. Kulathila, F. Lin, D. McKay, L. Rodrigues, D. Sage, B. B. Toure, S. van der Plas, K. Wright, M. Xu, H. Yin, J. Levell and R. A. Pagliarini, Structure, 2017, 25, 506–513 CrossRef CAS PubMed.
- J. R. Levell, T. Caferro, G. Chenail, I. Dix, J. Dooley, B. Firestone, P. D. Fortin, J. Giraldes, T. Gould, J. D. Growney, M. D. Jones, R. Kulathila, F. Lin, G. Liu, A. Mueller, S. van der Plas, K. Slocum, T. Smith, R. Terranova, B. B. Toure, V. Tyagi, T. Wagner, X. Xie, M. Xu, F. S. Yang, L. X. Zhou, R. Pagliarini and Y. S. Cho, ACS Med. Chem. Lett., 2017, 8, 151–156 CrossRef CAS PubMed.
- Q. Zhao, J. R. Manning, J. Sutton, A. Costales, A. Sendzik, C. M. Shafer, J. R. Levell, G. Liu, T. Caferro, Y. S. Cho, M. Palermo, G. Chenail, J. Dooley, B. Villalba, A. Farsidjani, J. Chen, S. Dodd, T. Gould, G. Liang, K. Slocum, M. Pu, B. Firestone, J. Growney, T. Heimbach and R. Pagliarini, ACS Med. Chem. Lett., 2018, 9, 746–751 CrossRef CAS PubMed.
- Y. S. Cho, J. R. Levell, G. Liu, T. Caferro, J. Sutton, C. M. Shafer, A. Costales, J. R. Manning, Q. Zhao, M. Sendzik, M. Shultz, G. Chenail, J. Dooley, B. Villalba, A. Farsidjani, J. Chen, R. Kulathila, X. Xie, S. Dodd, T. Gould, G. Liang, T. Heimbach, K. Slocum, B. Firestone, M. Pu, R. Pagliarini and J. D. Growney, ACS Med. Chem. Lett., 2017, 8, 1116–1121 CrossRef CAS PubMed.
- C. D. DiNardo, A. Hochhaus, M. G. Frattini, K. Yee, T. Zander, A. Krämer, X. Chen, Y. Ji, N. S. Parikh, J. Choi and A. H. Wei, J. Cancer Res. Clin. Oncol., 2022 DOI:10.1007/s00432-022-03983-6.
- T. Ma, F. Zou, S. Pusch, L. Yang, Q. Zhu, Y. Xu, Y. Gu, A. von Deimling and X. Zha, Bioorg. Med. Chem., 2017, 25, 6379–6387 CrossRef CAS PubMed.
- H. Bregman, N. Chakka, A. Guzman-Perez, H. Gunaydin, Y. Gu, X. Huang, V. Berry, J. Liu, Y. Teffera, L. Huang, B. Egge, E. L. Mullady, S. Schneider, P. S. Andrews, A. Mishra, J. Newcomb, R. Serafino, C. A. Strathdee, S. M. Turci, C. Wilson and E. F. DiMauro, J. Med. Chem., 2013, 56, 4320–4342 CrossRef CAS PubMed.
- H. Huang, A. Guzman-Perez, L. Acquaviva, V. Berry, H. Bregman, J. Dovey, H. Gunaydin, X. Huang, L. Huang, D. Saffran, R. Serafino, S. Schneider, C. Wilson and E. F. DiMauro, ACS Med. Chem. Lett., 2013, 4, 1218–1223 CrossRef CAS PubMed.
- L. Mariotti, K. Pollock and S. Guettler, Br. J. Pharmacol., 2017, 174, 4611–4636 CrossRef CAS PubMed.
- P. Polakis, Curr. Opin. Genet. Dev., 2007, 17, 45–51 CrossRef CAS PubMed.
- N. Barker and H. Clevers, Nat. Rev. Drug Discovery, 2006, 5, 997–1014 CrossRef CAS PubMed.
- E. De Vita, N. Smits, H. van den Hurk, E. M. Beck, J. Hewitt, G. Baillie, E. Russell, A. Pannifer, V. Hamon, A. Morrison, S. P. McElroy, P. Jones, N. A. Ignatenko, N. Gunkel and A. K. Miller, ChemMedChem, 2020, 15, 79–95 CrossRef CAS PubMed.
- G. M. Yousef and E. P. Diamandis, Endocr. Rev., 2001, 22, 184–204 CAS.
- J. J. Kim, J.-T. Kim, H. R. Yoon, M. A. Kang, J. H. Kim, Y.-H. Lee, J. W. Kim, S.-J. Lee, E. Y. Song, P. K. Myung and H. G. Lee, Tumor Biol., 2012, 33, 731–738 CrossRef CAS PubMed.
- F. Yang, Z.-D. Hu, Y. Chen and C.-J. Hu, Biomed. Rep., 2016, 4, 681–686 CrossRef CAS PubMed.
- B. Armentano, R. Curcio, M. Brindisi, R. Mancuso, V. Rago, I. Ziccarelli, L. Frattaruolo, M. Fiorillo, V. Dolce, B. Gabriele and A. R. Cappello, Biomedicines, 2020, 8, 35 CrossRef CAS PubMed.
- A. Naresh, M. V. Rao, S. S. Kotapalli, R. Ummanni and B. V. Rao, Eur. J. Med. Chem., 2014, 80, 295–307 CrossRef CAS PubMed.
- A. Singh, H.-J. Ha, J. Park, J. H. Kim and W. K. Lee, Bioorg. Med. Chem., 2011, 19, 6174–6181 CrossRef CAS PubMed.
- X. Wang, F.-H. Pang, L. Huang, X.-P. Yang, X.-L. Ma, C.-N. Jiang, F.-Y. Li and F.-H. Lei, Int. J. Mol. Sci., 2018, 19, 3116 CrossRef PubMed.
- A. K. Ghosh, J. N. Williams, R. Y. Ho, H. M. Simpson, S. Hattori, H. Hayashi, J. Agniswamy, Y.-F. Wang, I. T. Weber and H. Mitsuya, J. Med. Chem., 2018, 61, 9722–9737 CrossRef CAS PubMed.
- M. K. Parai, D. J. Huggins, H. Cao, M. N. L. Nalam, A. Ali, C. A. Schiffer, B. Tidor and T. M. Rana, J. Med. Chem., 2012, 55(14), 6328–6341 CrossRef CAS PubMed.
- V. W.-F. Tai, D. Garrido, D. J. Price, A. Maynard, J. J. Pouliot, Z. Xiong, J. W. Seal III, K. L. Creech, L. H. Kryn, T. M. Baughman and A. J. Peat, Bioorg. Med. Chem. Lett., 2014, 24, 2288–2294 CrossRef CAS PubMed.
- L. Aguado, M.-D. Canela, H. J. Thibaut, E.-M. Priego, M.-J. Camarasa, P. Leyssen, J. Neyts and M.-J. Pérez-Pérez, Eur. J. Med. Chem., 2012, 49, 279–288 CrossRef CAS PubMed.
- D. L. Simmons, Drug Discovery Today, 2006, 11, 210–219 CrossRef CAS PubMed.
- J. Fujimoto, R. Okamoto, N. Noguchi, R. Hara, S. Masada, T. Kawamoto, H. Nagase, Y. O. Tamura, M. Imanishi, S. Takagahara, K. Kubo, K. Tohyama, K. Iida, T. Andou, I. Miyahisa, J. Matsui, R. Hayashi, T. Maekawa and N. Matsunaga, J. Med. Chem., 2017, 60, 8963–8981 CrossRef CAS PubMed.
-
F. Tosi, F. Sartori, P. Guarini, O. Olivieri and N. Martinelli, in Oxidative Stress and Inflammation in Non-communicable Diseases - Molecular Mechanisms and Perspectives in Therapeutics. Advances in Experimental Medicine and Biology, ed. J. Camps, Springer Cham, Cham, 2014, pp. 66–81 Search PubMed.
- O. A. Phillips, M. A. Bosso and C. I. Ezeamuzie, J. Enzyme Inhib. Med. Chem., 2020, 35, 1471–1482 CrossRef CAS PubMed.
- O. Rådmark, O. Werz, D. Steinhilber and B. Samuelsson, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2015, 1851, 331–339 CrossRef PubMed.
- S.-J. Park, S. H. Kang, Y. K. Kang, Y.-B. Eom, K. O. Koh, D. Y. Kim and H.-S. Youn, Int. Immunopharmacol., 2011, 11, 19–22 CrossRef CAS PubMed.
- R. Medzhitov, Nat. Rev. Immunol., 2001, 1, 135–145 CrossRef CAS PubMed.
- S. Yun, S. H. Kang, A. Lee, S. Park, D. Y. Kim and H. Youn, Int. Immunopharmacol., 2010, 10, 163–168 CrossRef CAS PubMed.
- G. Song, X. Zhu, J. Li, D. Hu, D. Zhao, Y. Liao, J. Lin, L.-H. Zhang and Z.-N. Cui, Bioorg. Med. Chem., 2017, 25, 5709–5717 CrossRef CAS PubMed.
- S.-S. Hong, J. H. Choi, S. Y. Lee, Y.-H. Park, K.-Y. Park, J. Y. Lee, J. Kim, V. Gajulapati, J.-I. Goo, S. Singh, K. Lee, Y.-K. Kim, S. H. Im, S.-H. Ahn, S. Rose-John, T.-H. Heo and Y. Choi, J. Immunol., 2015, 195, 237–245 CrossRef CAS PubMed.
- S. Singh, V. Gajulapati, K. Gajulapati, J.-I. Goo, Y.-H. Park, H. Y. Jung, S. Y. Lee, J. H. Choi, Y. K. Kim, K. Lee, T.-H. Heo and Y. Choi, Bioorg. Med. Chem. Lett., 2016, 26, 1282–1286 CrossRef CAS PubMed.
- H. Huang, A. P. Degnan, A. Balakrishnan, A. Easton, M. Gulianello, Y. Huang, M. Matchett, G. Mattson, R. Miller, K. S. Santone, A. Senapati, E. E. Shields, D. V. Sivarao, L. B. Snyder, R. Westphal, V. J. Whiterock, F. Yang, J. J. Bronson and J. E. Macor, Bioorg. Med. Chem. Lett., 2016, 26, 4165–4169 CrossRef CAS PubMed.
- F. Yang, L. B. Snyder, A. Balakrishnan, J. M. Brown, D. V. Sivarao, A. Easton, A. Fernandes, M. Gulianello, U. M. Hanumegowda, H. Huang, Y. Huang, K. M. Jones, Y.-W. Li, M. Matchett, G. Mattson, R. Miller, K. S. Santone, A. Senapati, E. E. Shields, F. J. Simutis, R. Westphal, V. J. Whiterock, J. J. Bronson, J. E. Macor and A. P. Degnan, ACS Med. Chem. Lett., 2016, 7, 289–293 CrossRef CAS PubMed.
- P. Paoletti, C. Bellone and Q. Zhou, Nat. Rev. Neurosci., 2013, 14, 383–400 CrossRef CAS PubMed.
- N. Okamura, S. A. Habay, J. Zeng, A. R. Chamberlin and R. K. Reinscheid, J. Pharmacol. Exp. Ther., 2008, 325, 893–901 CrossRef CAS PubMed.
- C. Ruzza, A. Rizzi, C. Trapella, M. Pela, V. Camarda, V. Ruggieri, M. Filaferro, C. Cifani, R. K. Reinscheid, G. Vitale, R. Ciccocioppo, S. Salvadori, R. Guerrini and G. Calo, Peptides, 2010, 31, 915–925 CrossRef CAS PubMed.
- C. Trapella, M. Pela, L. Del Zoppo, G. Calo, V. Camarda, C. Ruzza, A. Cavazzini, V. Costa, V. Bertolasi, R. K. Reinscheid, S. Salvadori and R. Guerrini, J. Med. Chem., 2011, 54, 2738–2744 CrossRef CAS PubMed.
- S. Valente, S. Tomassi, G. Tempera, S. Saccoccio, E. Agostinelli and A. Mai, J. Med. Chem., 2011, 54, 8228–8232 CrossRef CAS PubMed.
- T. Z. E. Jones, P. Fleming, C. J. Eyermann, M. B. Gravestock and R. R. Ramsay, Biochem. Pharmacol., 2005, 70, 407–416 CrossRef CAS PubMed.
- R. R. Ramsay and M. B. Gravestock, Mini-Rev. Med. Chem., 2003, 3, 129–136 CrossRef CAS PubMed.
- S. B. Kombian and O. A. Phillips, Med. Princ. Pract., 2013, 22, 340–345 CrossRef PubMed.
- S. B. Kombian and O. A. Phillips, Neuroscience, 2011, 180, 53–63 CrossRef CAS PubMed.
- M. G. Qaddoumi, O. A. Phillips and S. B. Kombian, Eur. J. Pharm. Sci., 2019, 130, 21–26 CrossRef CAS PubMed.
-
World Health Organization, Cardiovascular diseases, https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds), (accessed 9 October 2022) Search PubMed.
- G. D. Flora and M. K. Nayak, Curr. Pharm. Des., 2019, 25, 4063–4084 CrossRef CAS PubMed.
- C. F. Thompson, A. Ali, N. Quraishi, Z. Lu, M. L. Hammond, P. J. Sinclair, M. S. Anderson, S. S. Eveland, Q. Guo, S. A. Hyland, D. P. Milot, C. P. Sparrow and S. D. Wright, ACS Med. Chem. Lett., 2011, 2, 424–427 CrossRef CAS PubMed.
- A. R. Tall, L. Yvan-Charvet, N. Terasaka, T. Pagler and N. Wang, Cell Metab., 2008, 7, 365–375 CrossRef CAS PubMed.
- C. J. Smith, A. Ali, M. L. Hammond, H. Li, Z. Lu, J. Napolitano, G. E. Taylor, C. F. Thompson, M. S. Anderson, Y. Chen, S. S. Eveland, Q. Guo, S. A. Hyland, D. P. Milot, C. P. Sparrow, S. D. Wright, A.-M. Cumiskey, M. Latham, L. B. Peterson, R. Rosa, J. V. Pivnichny, X. Tong, S. S. Xu and P. J. Sinclair, J. Med. Chem., 2011, 54, 4880–4895 CrossRef CAS PubMed.
- X. Zhu, J. Ji, D. Huang, Y. Zhu, C. Tang, X. Yang, H. Qian and W. Huang, Chem. Biol. Drug Des., 2012, 80, 426–433 CrossRef CAS PubMed.
- N. Fresno, M. Macías-González, A. Torres-Zaguirre, M. Romero-Cuevas, P. Sanz-Camacho, J. Elguero, F. J. Pavón, F. R. de Fonseca, P. Goya and R. Pérez-Fernández, J. Med. Chem., 2015, 58, 6639–6652 CrossRef CAS PubMed.
- M. B. Wright, M. Bortolini, M. Tadayyon and M. Bopst, Mol. Endocrinol., 2014, 28, 1756–1768 CrossRef PubMed.
- S. Rapposelli, F. Da Settimo, M. Digiacomo, C. La Motta, A. Lapucci, S. Sartini and M. Vanni, Arch. Pharm. Chem. Life Sci., 2011, 11, 372–385 CrossRef PubMed.
- J. M. Jez, M. J. Bennett, B. P. Schlegel, M. Lewis and T. M. Penning, Biochem. J., 1997, 326, 625–636 CrossRef CAS PubMed.
- M. Digiacomo, S. Sartini, G. Nesi, S. Sestito, V. Coviello, C. La Motta and S. Rapposelli, Open Med. Chem. J., 2017, 11, 9–23 CrossRef CAS PubMed.
- E. W. Davie, K. Fujikawa and W. Kisiel, Biochemistry, 1991, 30, 10363–10370 CrossRef CAS PubMed.
- T. Xue, S. Ding, B. Guo, Y. Zhou, P. Sun, H. Wang, W. Chu, G. Gong, Y. Wang, X. Chen and Y. Yang, J. Med. Chem., 2014, 57, 7770–7791 CrossRef CAS PubMed.
- Y. Zhao, M. Jiang, S. Zhou, S. Wu, X. Zhang, L. Ma, K. Zhang and P. Gong, Eur. J. Med. Chem., 2015, 96, 369–380 CrossRef CAS PubMed.
- J. Means, S. Katz, A. Nayek, R. Anupam, J. V. Hines and S. C. Bergmeier, Bioorg. Med. Chem. Lett., 2006, 16, 3600–3604 CrossRef CAS PubMed.
- R. Anupam, A. Nayek, N. J. Green, F. J. Grundy, T. M. Henkin, J. A. Means, S. C. Bergmeier and J. V. Hines, Bioorg. Med. Chem. Lett., 2008, 18, 3541–3544 CrossRef CAS PubMed.
- I. Maciagiewicz, S. Zhou, S. C. Bergmeier and J. V. Hines, Bioorg. Med. Chem. Lett., 2011, 21, 4524–4527 CrossRef CAS PubMed.
- C. M. Orac, S. Zhou, J. A. Means, D. Boehm, S. C. Bergmeier and J. V. Hines, J. Med. Chem., 2011, 54, 6786–6795 CrossRef CAS PubMed.
- A. Gutiérrez-Preciado, T. M. Henkin, F. J. Grundy, C. Yanofsky and E. Merino, Microbiol. Mol. Biol. Rev., 2009, 73, 36–61 CrossRef PubMed.
- W. Zhao, Y. Jiang, P. Bao, Y. Li, L. Tang, Y. Zhou and Y. Zhao, Jpn. J. Infect. Dis., 2015, 68, 520–522 CrossRef CAS PubMed.
- E. Alcántar-Zavala, E. Hernández-Guevara, A. Ochoa-Terán, J. Montes-Ávila, E. A. Estrada-Zavala, A. J. Salazar-Medina, E. Alday, A. Cabrera, G. Aguirre, V. Miranda-Soto, C. Velazquez, S. P. Díaz-Camacho and J. L. Medina-Franco, Bioorg. Chem., 2020, 105, 104359 CrossRef PubMed.
- B. Jin, J. Chen, Z. Sheng, M. Sun and H. Yang, Molecules, 2022, 27, 1103 CrossRef CAS PubMed.
- U. Atmaca, R. Kaya, H. S. Karaman, M. Çelik and İ. Gülçin, Bioorg. Chem., 2019, 88, 102980 CrossRef CAS PubMed.
- E. J. Hanan, M.-G. Braun, R. A. Heald, C. MacLeod, C. Chan, S. Clausen, K. A. Edgar, C. Eigenbrot, R. Elliott, N. Endres, L. S. Friedman, E. Gogol, X.-H. Gu, R. H. Thibodeau, P. S. Jackson, J. R. Kiefer, J. D. Knight, M. Nannini, R. Narukulla, A. Pace, J. Pang, H. E. Purkey, L. Salphati, D. Sampath, S. Schmidt, S. Sideris, K. Song, S. Sujatha-Bhaskar, M. Ultsch, H. Wallweber, J. Xin, S. Yeap, A. Young, Y. Zhong and S. T. Staben, J. Med. Chem., 2022, 65, 16589–16621 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Cauê Benito
Scarim†
*a,
Cauê Benito
Scarim†