
Semisynthetic blasticidin S ester derivatives show enhanced antibiotic activity†
Cole
Gannett
 ac,
Paige
Banks
a,
Christina
Chuong
bc,
James
Weger-Lucarelli
bc,
Emily
Mevers
acd and
Andrew N.
Lowell
*acd
ac,
Paige
Banks
a,
Christina
Chuong
bc,
James
Weger-Lucarelli
bc,
Emily
Mevers
acd and
Andrew N.
Lowell
*acd
aDepartment of Chemistry, Virginia Polytechnic Institute and State University (Virginia Tech), Blacksburg, VA 24061, USA. E-mail: alowell@vt.edu; Tel: (540) 231 5842
bDepartment of Biomedical Sciences and Pathobiology, VA-MD Regional College of Veterinary Medicine, Virginia Polytechnic Institute and State University (Virginia Tech), Blacksburg, VA 24061, USA
cCenter for Emerging, Zoonotic, and Arthropod-borne Pathogens, Virginia Polytechnic Institute and State University (Virginia Tech), Blacksburg, VA 24061, USA
dFaculty of Health Sciences, Virginia Polytechnic Institute and State University (Virginia Tech), Blacksburg, VA 24061, USA
First published on 8th March 2023
Abstract
A rich potential source of new antibiotics are undeveloped natural product cytotoxins, provided they can be derivatized to restrict their activity to bacteria. In this work, we describe modification of one such candidate, the broad-spectrum, translation termination inhibitor, blasticidin S. By semisynthetically modifying blasticidin S, we produced a series of ester derivatives of this highly polar, zwitterionic compound in a single step. These derivatives showed a marked increase in activity against Gram-positive bacteria and an increase in selectivity index for pathogenic bacteria over human cells. The results of this study suggest that semisynthetic derivatization of blasticidin S and other neglected natural product antimicrobials has the potential to increase their activity against and selectivity for bacteria, an approach that can be leveraged for the development of leads against antimicrobial resistant pathogens.
Introduction
The discovery of new antibiotics has sharply declined in the last three decades.1 With antimicrobial resistance (AMR) making our current treatments ineffective against many resistant bacteria,2 this discovery void is a serious public health concern.3 Natural products have provided the majority of antibiotic scaffolds,4 but with the current pace of discovery and development,5 additional strategies are needed. A rich source of potential antibiotics are the plethora of previously identified, but undeveloped, antimicrobial natural products.6 Many of these antimicrobial agents were neglected because of a lack of specificity for bacteria (cytotoxicity) or suboptimal physical properties, such as high polarity or poor solubility, that seemingly precluded facile semisynthetic derivatization.7 Semisynthesis, however, remains an integral pillar of generational antibiotic advancement8 and recent technological improvements, such as reverse-phase HPLC and automated flash chromatography, have dramatically improved medicinal chemists' abilities when it comes to working with challenging natural-product substrates. Thus, development of neglected natural products with semisynthesis would provide leads to help fill the discovery void for the treatment of resistant bacteria.Blasticidin S (1, Fig. 1), a peptidyl nucleoside, is one example of a neglected natural product with potential for development into a bacteria-specific agent. It was discovered in 1958 as part of a campaign to identify compounds that inhibit the growth of Piricularia oryzae, a phytopathogenic fungus responsible for rice blast disease.9 Blasticidin S is known to bind the peptidyl transferase center of the ribosome, but lacks selectivity, acting against both eukaryotes and prokaryotes.10 While highly conserved, topological differences in the peptidyl transfer center could be exploited to create bacteria-specific antibiotics. In bacteria, blasticidin S was shown to bind in a unique position that stabilizes a distorted P-site configuration, preventing termination.11 Further, the weakly active translation inhibitor amicetin (2) is hypothesized to occupy the same binding site as blasticidin S and shows selectivity for bacteria, supporting the potential to narrow blasticidin S's inhibitory effects to bacteria.12 Further, our analysis of the crystal structures of blasticidin S in the bacterial11 and fungal13 and rabbit10 ribosomes supports the possibility that semisynthesis will improve blasticidin S's selectivity for the bacterial peptidyl transferase.
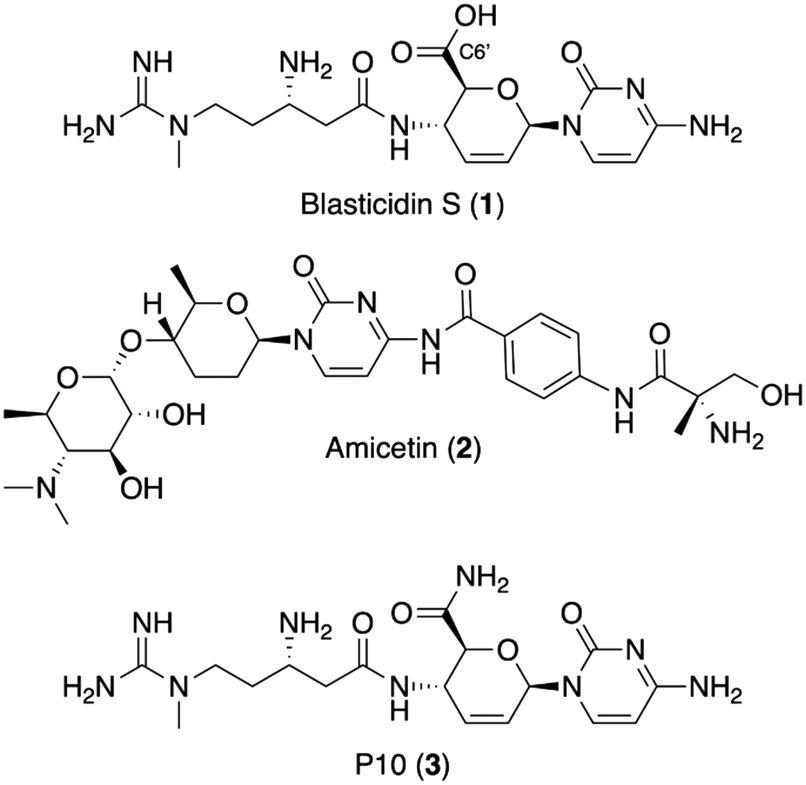 | ||
| Fig. 1 Structures of blasticidin S (1), amicetin (2) and P10 (3). | ||
The semisynthetic approach adopted for this work is based on the activity of another naturally occurring analog of blasticidin S, P10 (3). P10 was recently discovered from marine sponge extracts and has a primary amide in place of the C6′ carboxylic acid in 1.14 P10 was reported to be 8- to 16-fold more active compared to blasticidin S against strains of Staphylococcus aureus, Escherichia coli, and Acinetobacter baumannii. It was hypothesized that the change in charge state at physiological pH for P10 versus blasticidin S is responsible for the increase in activity.14 In keeping with this hypothesis, ester derivatives of commercially available blasticidin S were chosen to probe the structure activity relationship at this position as they have the same charge state as P10 at physiological pH.
Results and discussion
Ester derivatives were synthesized based on a previous report indicating that the methyl ester (4) could be prepared by treating a suspension of 1 with thionyl chloride in methanol.14 Use of this procedure yielded 97% of 4 (Fig. 2) after evaporation of the solvent. During this preparation, we observed that addition of thionyl chloride clarified the suspension. As poor solubility properties are one potential hurdle to working with neglected natural products, this result shows that this obstacle can be overcome. Based on the clarification of the solution during the process of successfully obtaining 4, we hypothesized that treating 1 with thionyl chloride in other alcohols would be a direct method to furnish additional esters. Application of this procedure using ethanol resulted in 5 precipitating from solution as the trihydrochloride salt, which could be isolated using simple filtration in an 83% yield. Use of propanol required heat to fully dissolve 1 after the addition of thionyl chloride. After cooling, 6 precipitated from solution as a white solid that was collected by filtration. The yield for propyl ester 6 dropped substantially (37%), likely because the increased temperature resulted in partial degradation of the starting material or the formation of byproducts. We attempted to synthesize the isopropyl ester derivative of 1 in the same manner, but instead isolated the acyl chloride, which was very hygroscopic and rapidly converted back to 1 upon exposure to air. From this result, we concluded that secondary alcohols are too sterically hindered to favorably interact with the acyl chloride of 1. Synthesis of the butyl ester (7) proceeded similarly to 6, requiring heat to solubilize the material after addition of thionyl chloride. Product 7 was filtered from solution after cooling in reasonable yield (59%). Following the pattern of increasing alkyl chain length, the hexyl ester (8) was synthesized, also requiring heat to proceed. However, unlike the shorter chain esters (5–7), 8 did not precipitate from solution after cooling. Instead, the solution containing 8 was washed with ethyl acetate to remove the excess alcohol and 8 was purified using semi-preparative HPLC in a 21% yield. The 4-butynyl ester (9), benzyl ester (10), and phenethyl ester (11) were prepared in the same manner and, like the hexyl ester, they did not precipitate from solution after cooling. These derivatives also required extraction and purification via semi-preparative HPLC with correspondingly modest yields. While the larger esters proved more challenging, the relative ease of isolation/purification for the smaller derivatives (4–7) suggests that other transformations of 1 (and related polar molecules) may be achieved and isolated in a straight-forward manner.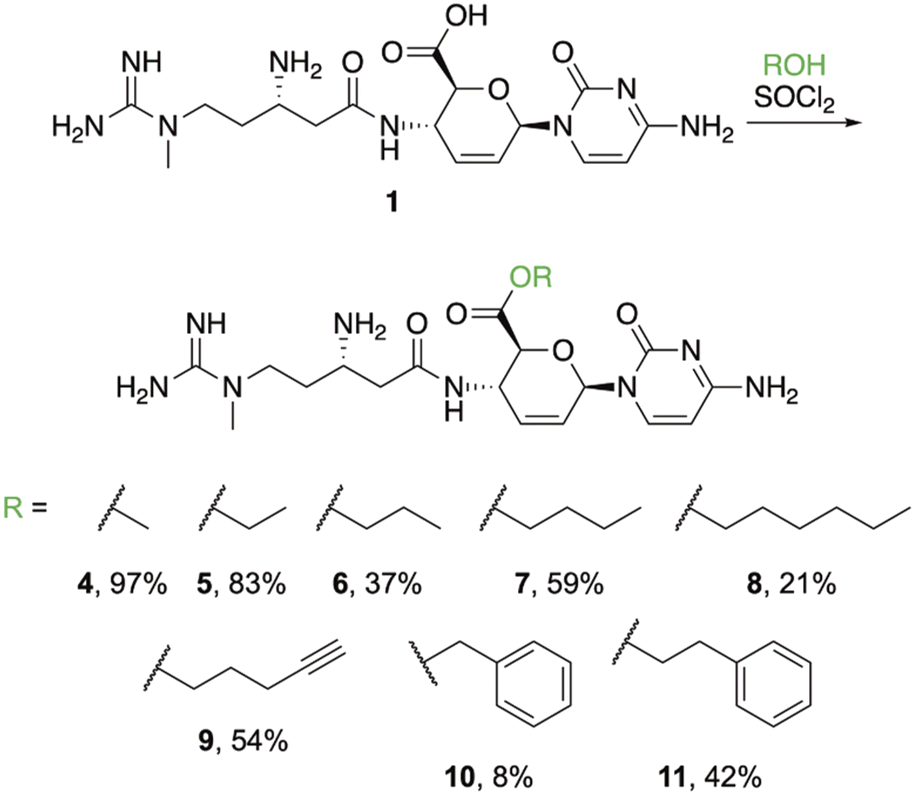 | ||
| Fig. 2 Reaction condition and ester derivative products. | ||
Blasticidin S (1) and the semisynthetic derivatives 4–7, 9, and 11 were initially assessed using a spot-on-lawn (agar spot) assay against S. aureus with the NorA efflux pump removed (ΔNorA), S. aureus, methicillin-resistant S. aureus (MRSA), Enterococcus faecalis, vancomycin-resistant Enterococcus sp. (VRE), Bacillus cereus, E. coli, Klebsiella pneumoniae, Pseudomonas aeruginosa efflux knockout (ΔMexAB-OprM), Acinetobacter baumannii, and Candida albicans. The derivatives generally showed larger zones of inhibition against the Gram-positive strains assayed and similar activity to 1 against Gram-negatives strains. Promisingly, the derivatives appeared to have no activity against the pathogenic fungus C. albicans, while blasticidin S (1) displayed inhibition.
To further assess the activity profile of our new compounds, we assayed (Table 1) blasticidin S (1), P10 (3), and all derivatives 4–11 against a subset of the pathogens above (excluding B. cereus and E. coli) as well as the non-pathogenic fungus Saccharomyces cerevisiae in broth microdilution assays. Previous semi-synthetic derivatives of blasticidin S are sparse and most were not assayed against a variety of pathogens to determine the activity profile.15–17 P10 (3),14,16 methyl ester 4 (ref. 18) and ethyl ester 5 (ref. 16) have been reported, but most of these compounds were tested in cell-free assays and displayed a decrease in activity versus blasticidin S.15,16 The exceptions are methyl ester 4 and P10 (3) which were more active than 1 against certain strains in whole-cell assays.14 Thus, we sought to make a more complete comparison of bacterial and fungal strains.
| Organism | Compound number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| a Results are based on one biological replicate. | |||||||||||
| Gram positive | S. aureus ΔNorA | >256 | 64 | 32–64 | 32 | 32–64 | 64 | 32–64 | 64–128 | 128 | 64 |
| S. aureus | 128–256 | 32 | 32 | 32 | 32 | 64–128 | 64–128 | 64 | 128 | 64 | |
| MRSA | 128 | 32 | 16–32 | 32–64 | 32–64 | 64–128 | 128 | 64–128 | 32–64 | 32–64 | |
| E. faecalis | 128 | 32 | 16 | 16 | 16 | 32 | 16 | 32 | 16–32 | 8 | |
| VRE | >256 | 128 | 64 | 64 | 128 | 256 | 256 | 128 | 128 | 32 | |
| Gram negative | K. pneumoniae | 128 | 16 | 64 | >128 | 128 | >128 | 128 | >128 | >128 | >128 |
| P. aeruginosa | 128 | 64 | 128 | >128 | >128 | >128 | >128 | >128 | >128 | >128 | |
| ΔMexAB-OprM | |||||||||||
| A. baumannii | 128 | 8 | 32 | 64 | 128 | >128 | 128 | >128 | >128 | >128 | |
| Fungi | C. albicans | 64 | 256 | >256 | >256 | >256 | >256 | >32 | >64 | >256 | >256 |
| S. cerevisiae | 16 | 64 | 32 | 32 | 32 | 64 | 16 | 64 | 128 | 64 | |
Derivatization of the carboxylic acid of 1 to alkyl esters increased activity against all Gram-positive bacteria with the biggest difference observed for S. aureus ΔNorA (up to 8-fold) and E. faecalis. Increasing the length of the alkyl chain of the ester typically resulted in a reduction in activity, suggesting longer chains are not well accommodated. However, of the two aromatic esters assayed, 11 showed modest activity, suggesting that pi interactions may aid in binding. Compound 11 was also the most active compound against E. faecalis, exhibiting up to a 16-fold increase in potency against this opportunistic pathogen.
Blasticidin S (1) exhibited minimal activity against the Gram-negative pathogens, and only the shorter chain alkyl esters (4 and 5) displayed an increase in activity against any of the Gram-negative bacteria assayed. P10 (3) was verified to be much more active (up to 16-fold) against the Gram-negative pathogens.
Compounds 1, 3, and the ester derivatives (4–11) were also tested against C. albicans and Saccharomyces cerevisiae, a representative, non-pathogenic fungi. Only 1 had an MIC below 256 μg mL−1 against C. albicans and the ester derivatives were all inactive at the concentrations used in this screen. S. cerevisiae was susceptible to all the compounds tested. Apart from 8, which was equally potent to blasticidin S, the ester derivatives were all less toxic (2- to 8-fold) to S. cerevisiae than 1.
Encouraged by the general increase in potency of the derivatives against bacteria and the abolishment of toxicity against C. albicans, we tested 1, 3, and 4–11 for cytotoxicity against MRC-5 human lung fibroblast cells (Fig. 3). Generally, our derivatives exhibited similar patterns of cell toxicity; however, 9 was found to be significantly less toxic compared to blasticidin S (50 ± 4 μg mL−1versus 16 ± 0.004 μg mL−1, ***p = 0.005). The data suggest that modification of the C6′ acid alone is not sufficient to eliminate cytotoxicity, but can begin to reduce it.
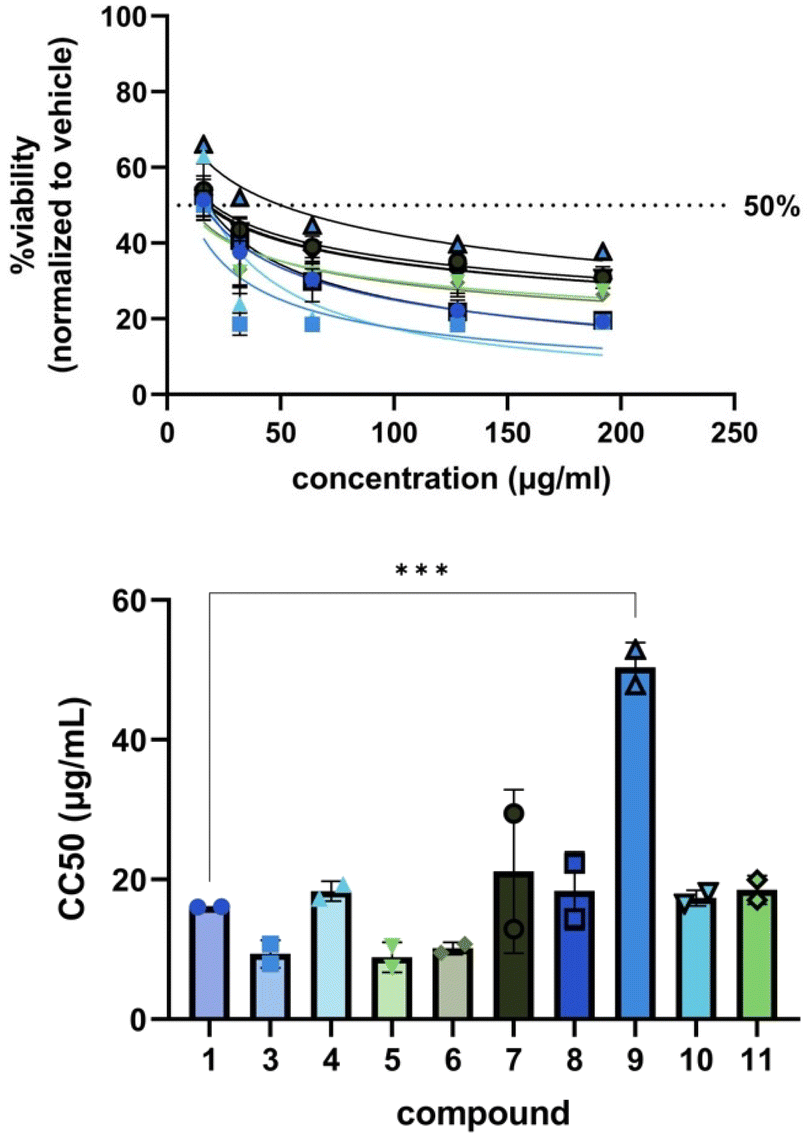 | ||
| Fig. 3 Cytotoxicity curves (top) and CC50 values (bottom) for BLS (1), P10 (3), and compounds 4–11. | ||
To compare the cytotoxicity data to the bacterial MIC data, an average IC50 value for each compound against each pathogenic bacteria tested (Table 2) was computed from the normalized inhibition curves (one for each biological replicate, Fig. S17–166.†) generated from the OD600 readings collected during the antibacterial assays. As blasticidin S (1) did not inhibit S. aureus ΔNorA nor VRE at the highest concentration in our broth microdilution assays, its IC50 was assumed to be >256 μg mL−1 for these pathogens. The selectivity index (SI) was generated for each compound-pathogen pair (Table 3) by computing the CC50/IC50 ratio assessing the selectivity for whole cell pathogenic bacteria versus human lung fibroblast cells.
| Compound number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Human cells CC50 (μg mL−1) | ||||||||||
| a Results are based on one biological replicate and standard deviation was not determined. | ||||||||||
| MRC-5 (lung fibroblast) | 16.1 ± 0.04 | 9.4 ± 2.0 | 18.3 ± 1.4 | 8.9 ± 2.2 | 10.2 ± 0.9 | 21.2 ± 12 | 18.4 ± 5.6 | 50.4 ± 3.5 | 17.4 ± 1.2 | 18.5 ± 2.0 |
| Organism | Compound number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| Gram positive | S. aureus ΔNorA | <0.1 | 0.2 | 0.8 | 0.4 | 0.5 | 0.6 | 1.0 | 1.4 | 0.4 | 0.7 |
| S. aureus | 0.2 | 0.6 | 1.6 | 0.8 | 0.7 | 0.9 | 0.7 | 2.6 | 0.5 | 1.0 | |
| MRSA | 0.2 | 0.6 | 1.7 | 0.5 | 0.6 | 0.7 | 0.4 | 1.7 | 0.7 | 1.2 | |
| E. faecalis | 0.3 | 0.7 | 2.0 | 0.9 | 1.0 | 1.3 | 1.6 | 4.1 | 1.2 | 2.7 | |
| VRE | <0.1 | 0.1 | 0.6 | 0.3 | 0.4 | 0.2 | 0.1 | 0.7 | <0.3 | 1.4 | |
| Gram negative | K. pneumoniae | 0.2 | 1.0 | 0.5 | 0.1 | 0.2 | 0.3 | 0.3 | 0.5 | 0.3 | 0.3 |
| P. aeruginosa | 0.2 | 0.5 | 0.5 | 0.1 | 0.2 | 0.3 | 0.2 | 0.7 | 0.2 | 0.2 | |
| ΔMexAB-OprM | |||||||||||
| A. baumannii | 0.2 | 3.0 | 1.8 | 0.3 | 0.2 | 0.3 | 0.3 | 0.8 | 0.2 | 0.2 | |
Relative to 1, the SIs for the derivatives increased for the Gram-positive bacteria and were similar or slightly improved for Gram-negative strains. As 1 was shown to enter S. aureus via the NorA efflux pump,14 the enhanced selectivity of 3 and the esters for this strain shows that entry is facilitated by a different mechanism. For the S. aureus strains with an intact NorA efflux pump, the SI improved 2.5- to 13-fold. The derivative with the largest selectivity index (SI = 4.1) was the 4-pentynyl ester (9) against E. faecalis followed by the phenethyl ester (11) with an SI of 2.7, improvements up to 13-fold over 1. The enhancement in the SI of 11 against VRE was also significantly improved relative to 1, a result that suggests additional modification will continue to enhance the selectivity of blasticidin S derivatives against this pathogen. Selectivity indices for Gram-negative bacteria were about the same as for 1 or marginally improved. P10 (3) showed the largest improvement followed by the methyl ester (4), both against A. baumannii. While more work is needed to reduce the cytotoxicity of our derivatives, the results of our study clearly indicate that the selectivity of our compounds for Gram-positive bacteria over human cells has increased compared to 1, verifying the hypothesis that these types of compounds can potentially be developed into antibiotics.
Conclusion
In this study, we describe the functionalization of the neglected natural product, blasticidin S, to form a small library of alkyl esters (4–11). Investigation of the SAR of these compounds showed a marked improvement in MIC against Gram-positive bacteria. Blasticidin S was minimally active against Gram-negatives and, apart from methyl ester 4, most ester derivatives were not active. Blasticidin S was active against the fungal pathogen C. albicans while none of the derivatives were. Cytotoxicity in mammalian cells generally showed similar inhibition for the derivatives when compared to the parent, except for one derivative, which was less toxic. While the mammalian cytotoxicity was not substantially reduced in most of these compounds, they did display an increased specificity for certain bacterial pathogens, as highlighted by the MIC's and selectivity indices for each. This study shows that simple chemical derivatization of broadly cytotoxic compounds can narrow the activity profile toward prokaryotes. Forgotten natural product antimicrobials represent a vast chemical space in which to search for lead compounds for antibiotic development if mammalian cytotoxicity can be decreased. Subsequent investigations will seek to pinpoint the structural features of prokaryotic/eukaryotic differences in activity in blasticidin S and other neglected antimicrobials, harnessing these differences to create leads for viable, bacteria-selective drugs.Experimental
General experimental
Blasticidin S hydrochloride was purchased from GoldBio. Unless otherwise specified, all reagents and solvents were purchased commercially and used as received from Sigma Aldrich, Fisher Scientific, or Oakwood Chemical. Deionized water was obtained from in-house. Unless otherwise stated, all media components were purchased from Sigma Aldrich.Specific rotations were obtained on a Jasco P-2000 polarimeter. 1H NMR spectra were recorded on a Bruker Avance II 500 MHz spectrometer or Agilent U4-DD2 400 MHz spectrometer. Chemical shifts are reported in parts per million (ppm) using the solvent resonance as an internal standard (CD3OD 3.31 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constants (Hz), and number of protons. Proton decoupled 13C NMR were recorded on a Bruker Avance II 500 MHz (13C 125 MHz) spectrometer. Chemical shifts are reported in ppm using the solvent resonance as an internal standard (CD3OD 49.0 ppm). High resolution mass spectra were obtained on an Agilent Technologies 6220 TOF LC/MS or a Waters Synapt Q-TOF G2 in the Department of Chemistry and the VT-mass spectrometry incubator at the Virginia Polytechnic Institute and State University. Semi-preparative HPLC was performed using a Shimadzu system equipped with a manual injector, CBM-20A communication bus module, DGU-20A degassing unit, LC-20AR liquid chromatography binary pump, SPD-20A UV/Vis detector, and FRC-10A fraction collector.
Synthetic procedures
Spot-on-lawn assays
Spot on lawn assays were carried out on disposable plastic petri dishes. Plates were prepared with sterile 1.5% agar in LB broth and allowed to cool. Individual pathogens were prepared from overnight cultures in LB broth and diluted to 0.01 OD600 using 0.7% agar in LB broth and overlaid onto each plate. Blasticidin S and derivatives 4–7, 9, and 11 as 5 mg mL−1 aqueous solutions were applied to each plate at doses of 35, 25, 10 and 5 μg using a micropipette along with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/methanol vehicle control spot. Plates were incubated at 37 °C for 18 h and diameters for the inhibition zones were measured with a ruler.
:1 water/methanol vehicle control spot. Plates were incubated at 37 °C for 18 h and diameters for the inhibition zones were measured with a ruler.
Broth microdilution assays
Media and solutions were autoclaved or sterile filtered prior to use and manipulations were carried out in a laminar flow hood. Mueller-Hinton broth was purchased from Oxoid and cation-adjusted according to CLSI specifications. Antibiotic and antifungal testing was performed in polypropylene 96-well flat bottom plates. The minimal inhibitory concentration of blasticidin S (1) hydrochloride, P10 (3) tri-hydrochloride, and derivatives 4–11 were assessed against Staphylococcus aureus ΔNorA, wild-type S. aureus 8810, methicillin-resistant S. aureus (MRSA) ATCC 43300, Enterococcus faecalis ATCC 29212, vancomycin-resistant Enterococcus, Klebsiella pneumoniae ATCC 29665, Pseudomonas aeruginosa ΔMexAB-OprM, Acinetobacter baumannii ATCC 17978, Saccharomyces cerevisiae, and Candida albicans using serial dilutions with doxycycline (Gold Bio) as a positive control for the bacterial assays, amphotericin B for the fungal assays, and the vehicle as a negative control for both. Stock solutions of each compound were prepared at 5.12 mg mL−1 and 2.56 mg mL−1 in 1:1 methanol/water and serially diluted to create master plates. From the master plates, 10 μL of each dilution was applied to well test plates. Bacteria and fungi previously grown overnight in Mueller-Hinton broth (bacteria) or yeast potato dextrose broth (fungi) prepared from individual colonies were diluted to an OD600 of 0.004 and applied to the plates (190 μL per test well) resulting in final concentrations of the test compounds of 256–2 μg mL−1 for S. aureus ΔNorA, S. aureus 8810, MRSA, and VRE and 128–1 μg mL−1 for the other microbes. The plates were incubated at 37 °C for 16 h, with the exception of VRE, E. faecalis, and S. cerevisiae; the Enterococci were incubated at 37 °C for 20 h and the S. cerevisiae was incubated at 28 °C for 16 h. For the fungal strains, MIC's were determined visually. For the bacteria, inhibition was determined by measuring the optical density at 600 nm (OD600) with a plate reader. The optical density measurements were normalized to the positive and negative controls. The MIC was the lowest concentration that inhibited >90% of growth. Two biological replicates were performed in duplicate. MIC values are reported as a range from all replicates. Inhibition curves were generated using Prism (GraphPad, San Diego). The IC50 values reported in Table 2 are the average of the two biological replicates ± standard deviation.
Cytotoxicity assays
Human lung fibroblast cells, MRC-5 (ATCC CCL-171) were plated at 5000 cells per well in a 96-well plate. Growth medium was removed and replaced with medium containing compounds 1, 3, and 4–11 ranging from 16–192 μg mL−1 or equivalent concentrations of vehicle (1:1 methanol:water). Cells were incubated for 24 h before MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) was added to each well as recommended by the manufacturer's protocol (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega, Madison, WI, USA) and further incubated for 3–4 h. After incubation, absorbance was measured at 490 nm on an Infinite M plex multimode microplate reader with i-control Tecan software (Tecan Trading AG, Switzerland). Viability was calculated by comparing absorbance values to untreated cells and normalizing to the vehicle control. These data are averaged from two independent assays performed in duplicate with standard deviation indicated.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge funding from the Virginia Tech CeZAP Interdisciplinary Team-building Pilot Grant (AL), the Lay Nam Chang Dean's Discovery Fund at Virginia Tech (AL), and VT Startup Funds (EM and AL). We acknowledge the VT NMR service center and the VT mass spectrometry incubator (VT-MSI). In addition, the authors acknowledge the Mekalanos Lab at Harvard Medical School for generation of the P. aeruginosa efflux mutant used in this work (obtained by way of the Clardy Lab at Harvard Medical School).References
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, The global preclinical antibacterial pipeline, Nat. Rev. Microbiol., 2020, 18(5), 275–285 CrossRef PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, et al., Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis, Lancet, 2022, 399(10325), 629–655 CrossRef CAS PubMed.
- 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis, World Health Organization, Geneva, 2021, licence: CC BY-NC-SA 3.0 IGO Search PubMed.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83(3), 770–803 CrossRef CAS PubMed.
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Antibiotics: Past, present and future, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS.
- M. S. Butler, M. A. T. Blaskovich, J. G. Owen and M. A. Cooper, Old dogs and new tricks in antimicrobial discovery, Curr. Opin. Microbiol., 2016, 33, 25–34 CrossRef CAS.
- S. Arenz and D. N. Wilson, Blast from the Past: Reassessing forgotten translation inhibitors, antibiotic selectivity, and resistance mechanisms to aid drug development, Mol. Cell, 2016, 61(1), 3–14 CrossRef CAS PubMed.
- P. M. Wright, I. B. Seiple and A. G. Myers, The evolving role of chemical synthesis in antibacterial drug discovery, Angew. Chem., Int. Ed., 2014, 53(34), 8840–8869 CrossRef CAS PubMed.
- S. Takeuchi, K. Hirayama, K. Ueda, H. Sakai and H. Yonehara, Blasticidin S, a new antibiotic, J. Antibiot., Ser. A, 1958, 11, 1–5 CAS.
- K. T. Powers, F. Stevenson-Jones, S. K. N. Yadav, B. Amthor, J. C. Bufton, U. Borucu, D. Shen, J. P. Becker, D. Lavysh and M. W. Hentze, et al., Blasticidin S inhibits mammalian translation and enhances production of protein encoded by nonsense mRNA, Nucleic Acids Res., 2021, 49(13), 7665–7679 CrossRef CAS PubMed.
- E. Svidritskiy and A. A. Korostelev, Mechanism of inhibition of translation termination by blasticidin S, J. Mol. Biol., 2018, 430(5), 591–593 CrossRef CAS PubMed.
- C. M. Serrano, H. R. Kanna-Reddy, D. Eiler, M. Koch, B. I. C. Tresco, L. R. Barrows, R. T. VanderLinden, C. A. Testa, P. R. Sebahar and R. E. Looper, Unifying the aminohexopyranose- and peptidyl-nucleoside antibiotics: Implications for antibiotic design, Angew. Chem., Int. Ed., 2020, 59(28), 11330–11333 CrossRef CAS.
- N. Garreau de Loubresse, I. Prokhorova, W. Holtkamp, M. V. Rodnina, G. Yusupova and M. Yusupov, Structural basis for the inhibition of the eukaryotic ribosome, Nature, 2014, 513(7519), 517–522 CrossRef CAS PubMed.
- J. R. Davison, K. M. Lohith, X. Wang, K. Bobyk, S. R. Mandadapu, S.-L. Lee, R. Cencic, J. Nelson, S. Simpkins and K. M. Frank, et al., A new natural product analog of blasticidin S reveals cellular uptake facilitated by the NorA multidrug transporter, Antimicrob. Agents Chemother., 2017, 61(6), e02635/02631–e02635/02617 CrossRef PubMed.
- M. Kawana, D. G. Streeter, R. J. Rousseau and R. K. Robins, Nucleoside peptides. 4. Synthesis of certain 1-(N-4-aminoacyl-4-amino-2,3–4-trideoxy-β-D-erythro-hex-2-enopyranuronic acid) cytosine derivatives related to blasticidin S, J. Med. Chem., 1972, 15(5), 561–564 CrossRef CAS PubMed.
- H. M. Menzel and F. W. Lichtenthaler, Structure–activity relations in the aminoacyl-4-aminohexosyl-cytosine group of peptidyl transferase inhibitors, Nucleic Acids Res., Spec. Publ., 1975, 1, S155–S158 CAS.
- T. Yoshinari, Y. Sugita-Konishi, T. Ohnishi and J. Terajima, Inhibitory activities of blasticidin S derivatives on aflatoxin production by Aspergillus flavus, Toxins, 2017, 9(6), 176 CrossRef PubMed.
- N. Otake, S. Takeuchi, T. Endo and H. Yonehara, Chemical studies of blasticidin S. III. The structure of blasticidin S, Agric. Biol. Chem., 1966, 30(2), 132–141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00412g |
| This journal is © The Royal Society of Chemistry 2023 |