
Dimeric polyphenols to pave the way for new antimalarial drugs†
Gilles
Degotte
 *ab,
Hélène
Pendeville
c,
Carla
Di Chio
d,
Roberta
Ettari
d,
Bernard
Pirotte
a,
Michel
Frédérich
b and
Pierre
Francotte
a
*ab,
Hélène
Pendeville
c,
Carla
Di Chio
d,
Roberta
Ettari
d,
Bernard
Pirotte
a,
Michel
Frédérich
b and
Pierre
Francotte
a
aLaboratory of Medicinal Chemistry, CIRM, Department of Pharmacy, University of Liège, Quartier Hôpital – B36 Tower 4, +5, Avenue Hippocrate 15, 4000 Liège, Belgium. E-mail: dgilles@hotmail.be
bLaboratory of Pharmacognosy, CIRM, Department of Pharmacy, University of Liège, Quartier Hôpital – B36 Tower 4, +5, Avenue Hippocrate 15, 4000 Liège, Belgium
cPlatform Zebrafish facility & transgenics, GIGA, University of Liège, Quartier Hôpital – B34, +2, Avenue de l'Hôpital 11, 4000 Liège, Belgium
dDipartimento di Scienze chimiche, biologiche, farmaceutiche e ambientali, Università degli Studi di Messina, Viale Annunziata, 98168 Messina, Italy
First published on 10th February 2023
Abstract
Because of the threat of resistant Plasmodium sp., new orally active antimalarials are urgently needed. Inspired by the structure of ellagic acid, exhibiting potent in vivo and in vitro antiplasmodial effects, polyphenolic structures possessing a similar activity-safety profile were synthesized. Indeed, most exhibited a marked in vitro effect (IC50 < 4 μM) on resistant P. falciparum, without any detrimental effects reported during the toxicity assays (hemolysis, cytotoxicity, in vivo). In addition, they possessed a greater hydrosolubility (from 7 μM to 2.7 mM) compared to ellagic acid. Among them, 30 is the most promising for antimalarial purposes since it displayed a significant parasitaemia reduction after oral administration in mice (50 mg kg−1) compared to the orally ineffective ellagic acid. In conclusion, our investigations led to the identification of a promising scaffold, which could bring new insights for malaria treatment.
Introduction
Among the leading causes of mortality for under five children, the World Health Organization (WHO) lists several infectious diseases, among which malaria remains one of the deadliest with around 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 annual infant deaths in 2020.1,2 This blood infection, caused by a protozoan belonging to the Plasmodium genus, is transmitted to humans by some species of Anopheles mosquitoes.3
000 annual infant deaths in 2020.1,2 This blood infection, caused by a protozoan belonging to the Plasmodium genus, is transmitted to humans by some species of Anopheles mosquitoes.3
Nowadays, five Plasmodium are recognized to parasitize humans, P. vivax, P. malariae, P. ovale, P. knowlesi, and P. falciparum. The former is considered as the main cause of concern since it is responsible for more than 50% of the cases in each WHO subregion, especially in Africa where it reached 99.7% in 2019.4,5
The main symptoms are headaches, fevers, chills, and nausea, but it could lead to other clinical signs, including anaemia, haemoglobinuria, or even worse, if it remains untreated, i.e., cerebral malaria, kidney failure, or death.6,7
Fortunately, this zoonosis benefits of high research interest and billions of investment, contrary to other tropical diseases.5,8 Consequently, efficient therapies are available and have led to a constant mortality decrease, reaching a 400000 per year death rate.8 However, the recent SARS-Cov-2 pandemic and the induced shortage in supplies and help seemed to have been responsible for a first increment in malaria mortality with 627000 deaths in 2020 & 619000 in 2021.2,9
In addition, since the discovery of artemisinin derivatives (Chart 1) and despite the recent approval of the RTS, S vaccine, there is a lack of innovations in the antimalarial therapeutic arsenal. Moreover, as for most antimalarials, a partial resistance to artemisinins, defined as an in vitro delayed clearance of the parasite, was recently reported in Africa, years after its appearance in South-East Asia.10,11
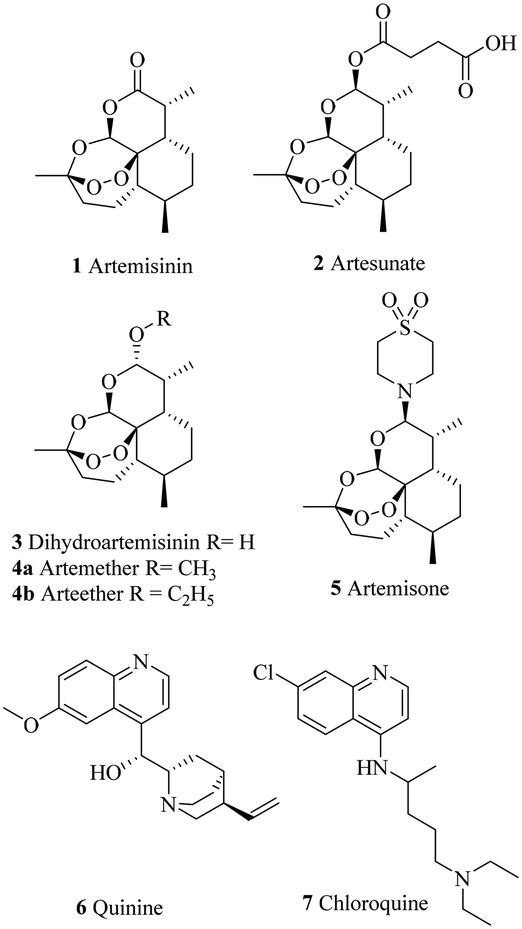 | ||
| Chart 1 Antimalarial drugs employed against Plasmodium falciparum. | ||
Consequently, it is urgent to develop new treatment alternatives. As seen with the discovery of previous antimalarials (1 and 6), and highlighted by Newman & Cragg (2020), most of the anti-infectious drugs could have been inspired by natural compounds (NPs).12 Thus, it seems logical to search for drug candidates in Nature, especially in medicinal plants recommended by traditional healers.
Indeed, dozens of highly active compounds are reported annually, as demonstrated by the numerous reviews about this particular subject.13–15 Unfortunately, most could not be directly translated to a drug because of a combination of various issues, including a complex structure, weak hydrosolubility, toxicity, or scarce availability.
During our previous investigations on small polyphenolic acids, such as gallic acid (8), we demonstrated that their limited antiplasmodial effect could be significantly improved by partial phenol substitution with lipophilic moieties. This is mainly explained by an easier penetration inside the RBC. Furthermore, the logP increment did not induce a significant cytotoxicity or a loss of hydrosolubility.16
However, despite this promising profile, the antiplasmodial activity remained limited (IC50 > 10 μM) and should be further improved since a low to submicromolar in vitro potency is recommended before in vivo experiments.17
Among the most promising identified polyphenols, ellagic acid (9, Chart 2), a widely distributed molecule, has been known for centuries (1831) and possesses high pharmacological interest. Indeed, aside from the classical antioxidant effect, it also exhibited anti-inflammatory, antidiabetic, antiviral, or antiplasmodial activities, for example.18–22 The former was extensively studied in vitro (IC50 = 110–330 nM), but also in vivo after intraperitoneal injection in a malaria mouse model, using P. vinckei petteri (ED50 < 1 mg kg−1 i.p.).23–26 Considering the biosynthesis of ellagic acid in plants, this structure could be defined as a dimer of gallic acid (8). In fact, its precursor, 6-HHDP (10, Chart 2), which originated from 2 ortho C–C bound gallates, esterified with a sugar moiety.
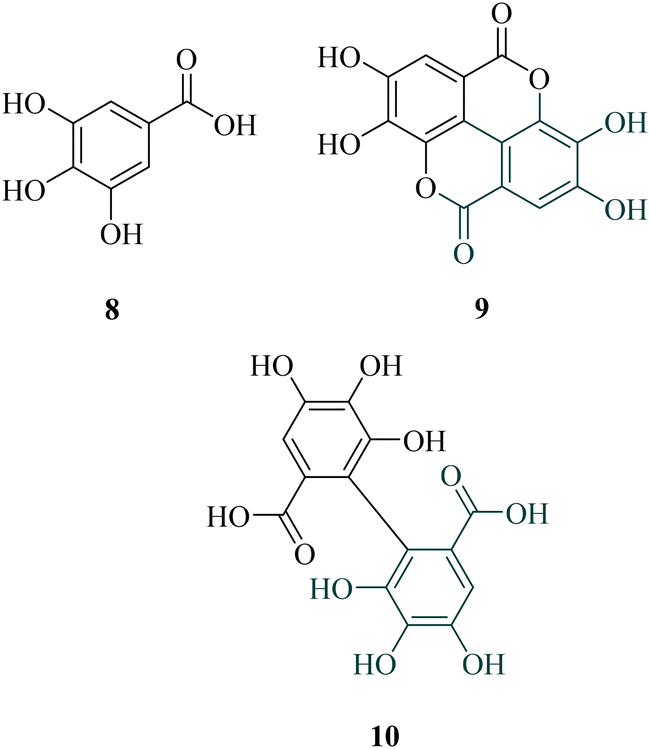 | ||
| Chart 2 Ellagic acid (9), a condensed dimer of gallic acid (8), and its precursor 6-HHDP (10). | ||
Therefore, since ellagic acid exhibits a potent antiplasmodial and antimalarial effect, the dimerization of our previously reported gallates as ellagic acid derivatives could be promising to obtain new drug candidates.
Unfortunately, the antimalarial potential of ellagic acid is limited through the parenteral route since its oral administration was inefficient (ED50 > 100 mg kg−1p.o.).23 This loss of activity could be mainly explained by a low per os bioavailability, only 1% in humans.27
This detrimental pharmacokinetics could originate from the low permeability combined to a weak hydrosolubility (Cmax = 9.7 μg mL−1), and an extensive metabolization into urolithins by the gut microflora.28–30
We suggest first that this low water solubility could result from the planar structure, allowing a strong crystal packing because of intra- & intermolecular bonds. In addition, it seems that there is a high enzyme specificity for the gut metabolization of ellagic acid (9).31–33 Therefore, we focused the synthesis of gallic acid dimers on the scaffold lacking the ortho C–C bond, which is one of the most detrimental factors for tridimensionality.
Herein, we report for the first time the synthesis and evaluation of polyhydroxybenzoic acid dimers (Chart 3) as promising scaffolds for the development of new drug candidates against malaria.
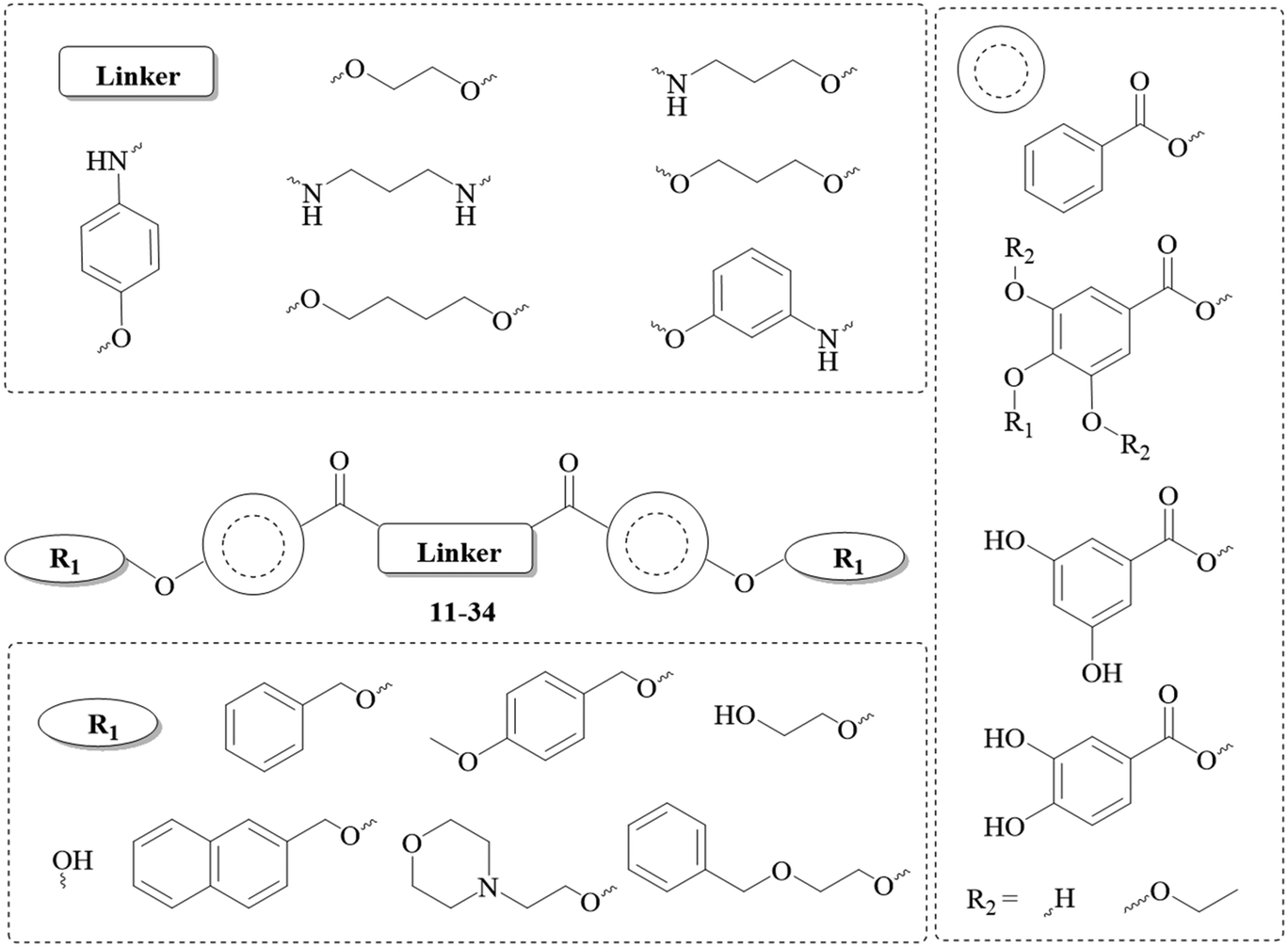 | ||
| Chart 3 General structure of the dimers and employed motifs. | ||
Results and discussion
Synthesis
To obtain the library reported in Chart 4, two major multistep synthetic pathways have been selected (Scheme 1). A, adapted from Hirokane et al. (2014), allowed the nature of the para-substituents on the 3,4,5-trihydroxybenzoate moieties to be modulated, while B was used to obtain rapidly dimeric products with modification of the number of phenolic functions and the nature of the linker (X).34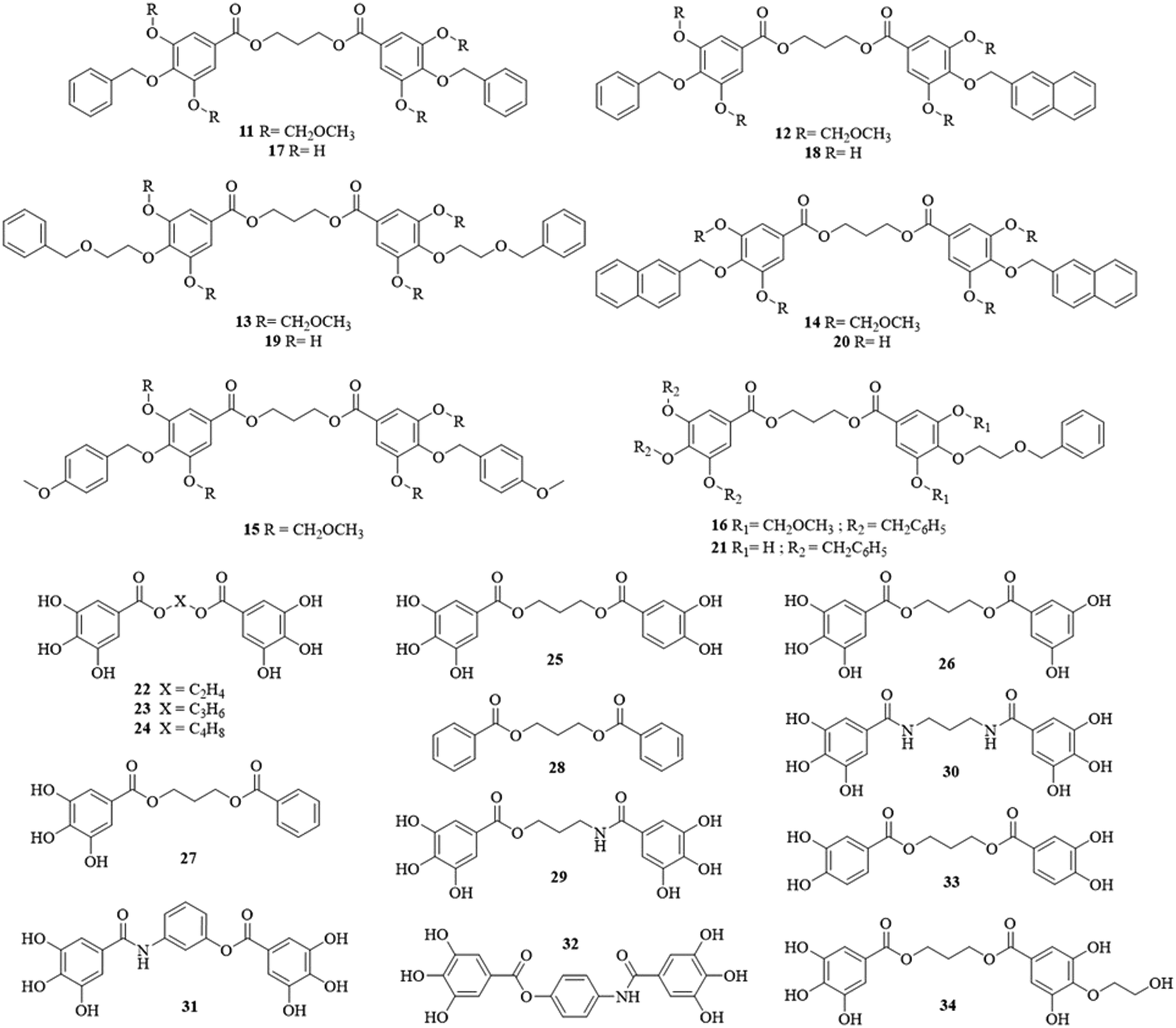 | ||
| Chart 4 Polyhydroxybenzoate dimers assessed against Plasmodium falciparum. | ||
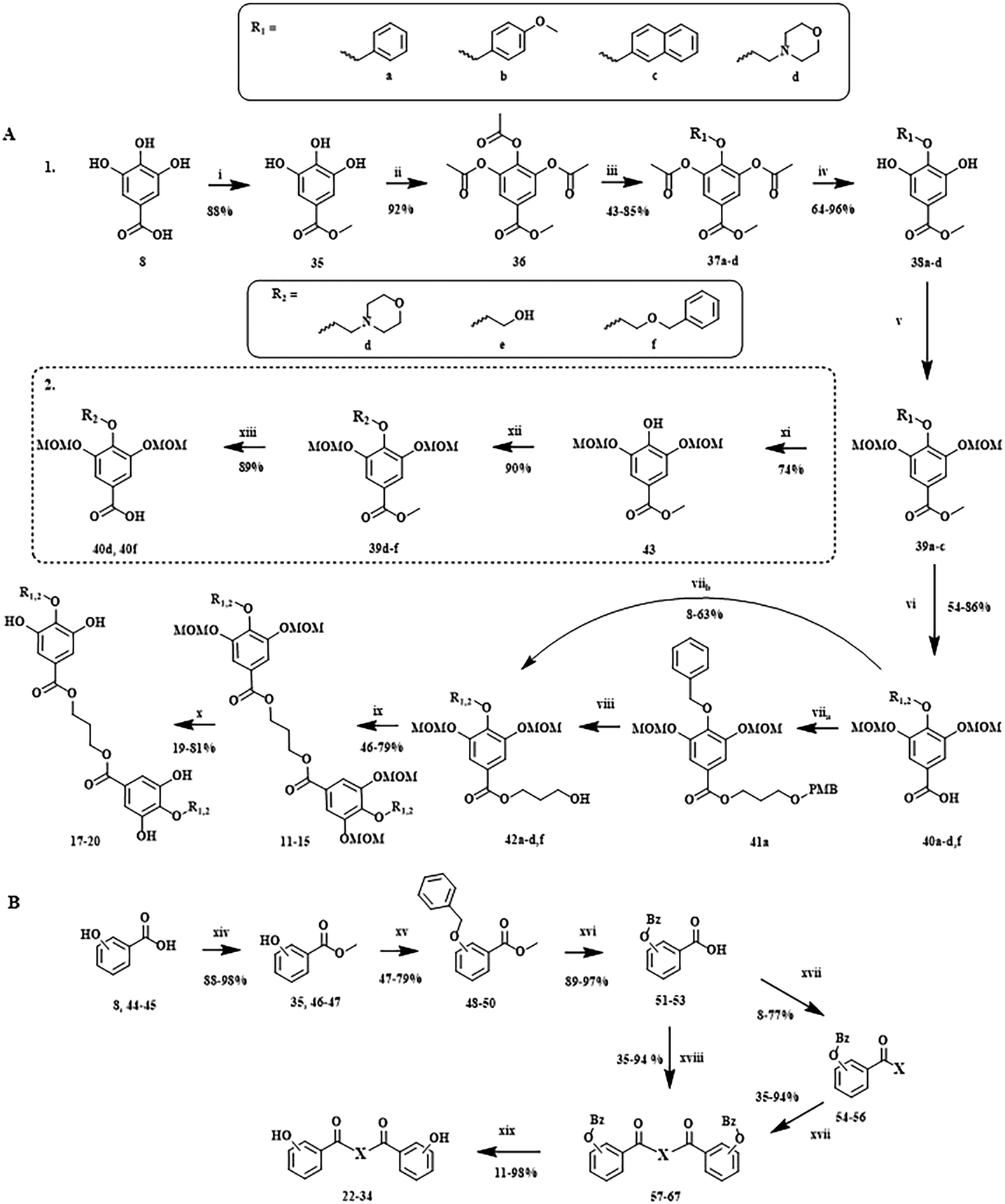 | ||
| Scheme 1 Multistep synthesis protocols explored in this work. Reagents/conditions: A1 i CH3OH, H2SO4; ii Ac2O, H2SO4; iii BnBr, KI, K2CO3, acetone; iv K2CO3, CH3OH, H2O; v NaH, MOMCl, DMF; vi LiOH, CH3OH, THF, H2O; viia 1,3-propanediol-PMB, DMAP, EDCI-HCl, CH2Cl2; viib 1,3-propanediol, DMAP, EDCI-HCl, CH2Cl2 viii DDQ, Sorenson's buffer, CH2Cl2; ix DMAP, EDCI-HCl, CH2Cl2; x THF, IPA/HCl; A2 xi H2, Pd/C, EtOH; xii KI, K2CO3, acetone; xiii LiOH, CH3OH, THF, H2O. B xiv CH3OH, H2SO4; xv BnBr, KI, K2CO3, acetone; xvi LiOH, CH3OH, THF, H2O; xvii linker (X), DMAP, EDCI-HCl, CH2Cl2; xviii linker (X), DMAP, EDCI-HCl, CHCl3; xix H2, Pd/C, THF.34–37 | ||
Interestingly, the introduction of more hydrophilic substituents, such as morpholine, on the 4-O position has required significant adaptations of the A1 protocol.
Indeed, even if the use of NaBH4 in MeOH allowed the removal of the acetyls to generate 38a, the most efficient procedure to introduce hydrophilic substituents was to remove the benzyl of 39a in a reduction protocol with H2 (xi) before the introduction of the desired alkyl chain using the previous substitution conditions (A2).
Furthermore, in contrast to the previously reported protocol by Hirokane et al. (2014),34 the use of a PMB-protected linker (viia) seems not necessary for the synthesis of asymmetric dimers. In fact, for unknown reasons, the esterification between benzoic acid and a linear diol, in the presence of DMAP and EDCI–HCl in CH2Cl2, preferentially generated the monomer (42) instead of the corresponding dimer (11–15), even under reflux (viib).
Interestingly, a similar phenomenon of selective synthesis of a monomeric vs. dimeric compound was observed in the B multistep pathway (Scheme 1). Indeed, the reaction between the benzyl protected carboxylic acid (51–53) and the linker (X) produced preferentially the monomer (54–56) in CH2Cl2 under reflux (xvii), and the symmetric dimer in CHCl3 (xviii).
This benzyl protecting group was chosen because it could be easily removed with Pd/C (10%) and hydrogen at the end of the synthetic scheme (xix). Moreover, the benzyl-protected molecules could be easily isolated by recrystallization in EtOH on ice (48–50).
These multistep synthesis protocols provided a small library of 24 dimeric structures (11–34, Chart 4), with a great structural variability. Those were further evaluated in various pharmacological assays, to establish their potential as antimalarials.
Hydrosolubility
Ellagic acid (9) is defined as a BCS IV product.38,39 Thus, this substance possesses a weak water solubility (<10 mg mL−1) combined to a low permeability. Consequently, its per os administration is greatly compromised, which impedes its medicinal use. Indeed, it has been already demonstrated that its antimalarial in vivo effect was significant only after intraperitoneal injection (100% parasitaemia reduction at 50 mg kg−1) while rather limited for the oral route (ED50 > 100 mg kg−1).23In addition, the molecules need to cross several lipophilic barriers, e.g., cellular membrane, to reach their site of action. Therefore, a good balance between lipophilicity and hydrophilicity is crucial to observe the pharmacological effect in vivo.
Consequently, the maximal concentration at room temperature (Cmax, 25 °C) of the synthesized dimers was measured, following the previously reported protocol using a UV-spectrophotometer and the shake-flask method (Table 1).30
| Compounds | 3D7a | FcB1b | W2c | HUVECd | SI | Solubilitye | Solubility increment | clogPf |
|---|---|---|---|---|---|---|---|---|
|
a IC50 value in μM against Plasmodium falciparum (chloroquine-sensitive) in triplicate with standard deviation.
b IC50 value in μM against Plasmodium falciparum (chloroquine-resistant) in triplicate with standard deviation.
c IC50 value in μM against Plasmodium falciparum (multi-resistant) in triplicate with standard deviation.
d IC50 value from the cell toxicity assay in triplicate with standard deviation.
e
C
max value in mM in triplicate with standard deviation.
f Calculated with ChemDraw 12.0.
g Experimental logP (ESI†).
h IC50 value in nM on P. falciparum in triplicate with standard deviation.
|
||||||||
| 8 | 68 ± 20 | — | — | >294 | >4 | 70 ± 8.2 | 3879 | 0.63g |
| 9 | 2.8 ± 1.9 | 7.5 ± 2.8 | 8.9 ± 5.9 | >165 | >59 | 0.018 ± 0.003 | 1 | 1.1 |
| 11 | 4.5 ± 3.1 | — | — | — | — | 0.003 ± 0.00006 | 0.2 | 6.7 |
| 12 | 21 ± 1.1 | — | — | — | — | — | — | — |
| 17 | 1.8 ± 0.9 | 1.7 ± 1.1 | 1.9 ± 1.1 | 14 ± 5.8 | 8 | 0.026 ± 0.004 | 2 | 5.2 |
| 18 | 0.9 ± 0.4 | 0.9 ± 0.2 | 0.4 ± 0.2 | 6.9 ± 0.2 | 5 | — | — | 7.2 |
| 19 | 3.2 ± 0.9 | — | — | 15 ± 3.6 | 5 | — | — | 4.9 |
| 20 | 4.0 ± 0.7 | 1.1 ± 0.6 | 1.8 ± 1.0 | 9.7 ± 1.0 | 2.4 | — | — | 6.2 |
| 22 | 3.7 ± 1.1 | 3.7 ± 1.2 | 6.7 ± 3.2 | 69 ± 12 | 19 | 0.43 ± 0.25 | 24 | 1.04g |
| 23 | 5.8 ± 1.9 | 3.2 ± 1.4 | 4.1 ± 2.9 | >132 | >23 | 2.7 ± 0.64 | 150 | 1.13g |
| 24 | 3.7 ± 1.1 | 5.7 ± 3.4 | 5.0 ± 4.2 | >127 | >35 | 0.15 ± 0.05 | 8 | 1.30g |
| 25 | 5.5 ± 1 | 4.9 ± 2.5 | 7.6 ± 4.4 | 84 ± 5.4 | 41 | 0.28 ± 0.04 | 15 | 2.4 |
| 26 | 4.6 ± 0.6 | 3.8 ± 2.0 | 3.6 ± 1.2 | — | >21 | 1.2 ± 0.14 | 64 | 0.6 |
| 27 | 27 ± 1.9 | 11 ± 1.5 | 13 ± 2.4 | 78 ± 41 | 3 | 0.6 ± 0.014 | 33 | 2 |
| 28 | > 352 | — | — | — | — | 0.007 ± 0.004 | 0.4 | 3.6 |
| 29 | 6.3 ± 4.8 | 4.7 ± 2.9 | 6.8 ± 3.2 | 59 ± 9.9 | 11 | 2.1 ± 0.27 | 114 | 1.6 |
| 30 | 9.9 ± 3.2 | 26 ± 6.7 | 12 ± 1.5 | 130 ± 6.8 | 28 | 0.61 ± 0.006 | 34 | 1.6 |
| 31 | 2.1 ± 1.0 | 3.7 ± 1.9 | 3.4 ± 0.9 | 54 ± 2.5 | 6 | 0.30 ± 0.11 | 16 | 2.0 |
| 32 | 3.9 ± 2.8 | 3.1 ± 1.1 | 2.9 ± 0.7 | >121 | >13 | 0.066 ± 0.003 | 4 | −0.1 |
| 33 | 9.0 ± 4.3 | — | — | >144 | >42 | 0.033 ± 0.008 | 2 | 2 |
| 34 | 20 ± 3.0 | 4.3 ± 1.7 | 7.6 ± 4.5 | >118 | >14 | — | — | 0.98 |
| Artemisinin (1) | 13 ± 3.5h | 17 ± 6 | 24 ± 5 | — | — | — | — | 2.9 |
| Quinine (6) | 0.6 ± 0.1 | — | — | — | — | — | 86 | 3.4 |
| Chloroquine (7) | 0.5 ± 0.2 | 1.9 ± 0.3 | 1.5 ± 0.9 | — | — | — | — | — |
As expected, 9 displayed a scarcely soluble profile in MilliQ water at 25 °C (Cmax = 18 μM or 6 μg mL−1) compared to its monomeric unit, gallic acid (8) with Cmax = 70 mM. Indeed, 8 was more than 3800-fold more soluble under these conditions. This could confirm the major role of the phenolic hydroxyl groups, as discussed before, and the intramolecular bonds in the aqueous solubility.16
As observed for the monogallate derivatives, the impact of lipophilic substituents and the availability of the hydrophilic functions (phenols and carboxylic acid) for solvent interactions seemed to be confirmed for the dimers.16 Indeed, the substitution of all phenolic functions impeded the water solubility as illustrated by the Cmax difference between 11 and 17, 3 μM and 26 μM, respectively (Table 1).
In addition, the presence of aromatic para-substituents greatly impeded the water solubility, since 17, one of the most lipophilic compounds, was 100-fold less soluble than 23, Cmax = 0.026 vs. 2.7 mM, respectively.
However, the dimerization seemed to negatively impact the dissolution as most of the dimeric structures exhibited a Cmax lower than 1 mg mL−1 compared to the 10 mg mL−1Cmax for gallic acid (Table 1). The dimeric structures (11–34) exhibited a highly variable water solubility, from 0.066 to 2.7 mM. Therefore, most of them were more soluble than ellagic acid (9, Cmax = 18 μM).
As illustrated in Fig. 1, the increase of maximal concentration was often slight (<10, Fig. 1). However, several products exhibited a significant increase, between 30 and 150-fold, especially for 23 and 29 (150 and 114-fold, respectively), despite very-close structures.
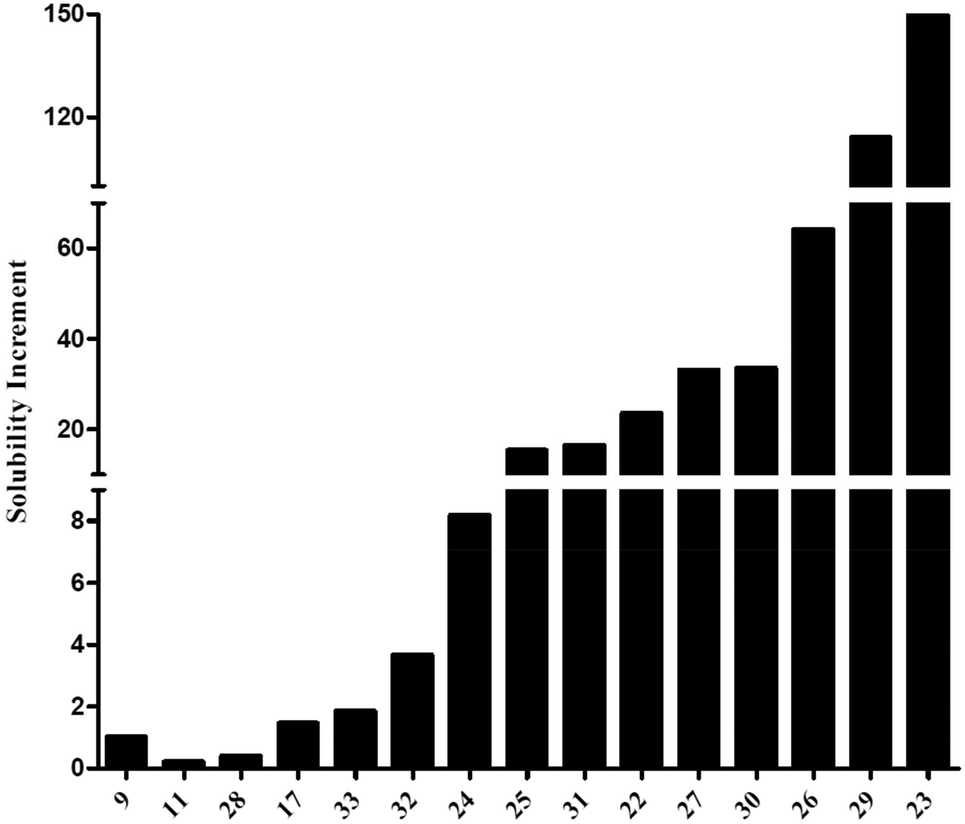 | ||
| Fig. 1 Solubility increase for the dimers compared to ellagic acid (9). | ||
This variability could certainly find its origin in the presence, the availability, and the number of phenolic functions, since 11, 17 and 28 were the less water-soluble molecules. But the position of the aromatic hydroxyls seemed to possess an even greater role. Indeed, 25 and 26, highly similar in structure, displayed quite different solubility profiles, which could only be explained by the formation of detrimental intramolecular bonds for 25, as reported before for ellagic acid.
However, the main structural parameter for the solubility seemed to be the molecular arrangement in water, illustrated by the variation of Cmax with the linker nature. Indeed, close structures exhibited highly different Cmax values, as illustrated by 22, 23, and 24.
Indeed, contrary to what was suggested by their logP values, measured as described by Liang et al. (2017), 23 was much more soluble than 24 (5-fold).40 Another example of this conformation impact was compound 32 (Cmax = 66 μM) which was 4 to 5-fold less soluble in water than 31 (Cmax = 298 μM), despite their identical theoretical lipophilicity–hydrophilicity balance (clogP = 1.99).
Therefore, the solubility, a key parameter for drug development, seemed not only driven by the lipophilicity (clogP), as for the small polyhydroxybenzoic acid analogues.16 But mainly, by the conformation adopted by the molecule in the solvents. In conclusion, a high number of available phenolic functions or a low clogP value seemed not to guarantee a great hydrosolubility for the dimers.
In vitro antiplasmodial activity
As previously reported, a great difference in antiplasmodial activity was observed between gallic (8) and ellagic acid (9), IC50 = 68 vs. 2.8 μM, respectively.16 Meanwhile nearly all the dimers (Chart 4) exhibited a significant antiplasmodial effect against the 3D7 strain, with an IC50 value around 4.5 μM for most compounds. This is despite their structural diversity in the linker, the phenolic content, and the presence of a para-substituent on the monomeric units.Therefore, most of the dimers possessed an equivalent potency to ellagic acid against Plasmodium falciparum. But the slight differences in the activity allow the determination of some crucial structural features for the activity.
First, this demonstrated the great potential of the dimerization of polyhydroxybenzoic acids. Indeed, in contrast to their corresponding monomers (8, 44–45), in which only 8 was active on Plasmodium, all the dimers exhibited an equivalent antiplasmodial efficacy.16 Therefore, the dimer is equally potent, even if the scaffold possesses a 3,4- or 3,5-dihydroxybenzoate moiety. This suggested that the dimerization of these structures created new beneficial interactions with the targets since this effect was not the result of the cumulative activity of two polyhydroxybenzoic acid molecules but instead of one.
Secondly, it confirmed the importance of the phenols. Indeed, only 27–28 had a strongly reduced effect on Plasmodium falciparum, and they possessed at least one “nude” benzoic moiety, which significantly impeded the antiplasmodial activity, especially for 28, which was totally inactive.
Third, as observed for small polyphenolic acids, the most active molecules seemed to be the most lipophilic ones (17–20), suggesting a higher cell permeation rate.41–44 However, this positive effect of the lipophilicity seemed lower in this case because of the role played by the phenolic content and the dimerization.
Interestingly, the introduction of MOM protecting groups on the phenolic functions did not impede the antiplasmodial effect. Indeed, 11 was equivalent to its corresponding unprotected structure 17 (4.5 μM and 1.8 μM, respectively). Consequently, this could suggest that the MOM protecting groups could be easily removed inside the erythrocytes, or that it could create new interactions between the molecules and the targets.
However, further studies would be necessary to confirm the incidence of this protecting group. But, because of the fragility of this moiety under strong acidic conditions, its medicinal use through oral administration is compromised without an appropriate gastro-resistant formulation.34
Finally, the antiplasmodial effect seemed not affected by previous existing resistance mechanisms. Indeed, most of the dimers exhibited a similar activity on resistant strains of Plasmodium falciparum (FcB1 and W2).
However, 30 was 2-fold less efficient on the FcB1 (chloroquine-resistant) parasite than on the other strains. This suggests that it could be recognized by the Plasmodium falciparum chloroquine resistance transporter, and could no longer accumulate in the digestive vacuole, for example. Consequently, we could suggest that the site of action of the dimers could be this particular cellular compartment.45
Moreover, because of the similar efficacy for most of the dimers, the site of action of these scaffolds could support a great structural diversity without a significant loss of efficiency. This is highly promising for future investigations since these molecules could be further modulated to optimize their potency and ADME profile.
In addition, because of the particular interest developed regarding the pan-assay interference (PAINS) potential of the polyphenolic structures, and the risk of false positives during in vitro assays,46,47 the antiplasmodial effect of the dimers was confirmed through a microscopy evaluation of the parasitaemia after 48 hours of incubation.16,48 This method allows a visual evaluation of the parasitic burden during the in vitro experiments. Fortunately, the test compounds have obtained similar IC50 values with this method to that with the enzymatic revelation.
Hemolysis
All the compounds have been tested to estimate their hemolysis induction potential with particular interest in molecules exhibiting a significant antiplasmodial effect. Indeed, a part of the developmental cycle of Plasmodium falciparum is intraerythrocytic (asexual stages).4 Consequently, in the case of erythrocytic membrane disruption, the parasites will not be able to grow and multiply. For that reason, all the products exhibiting a great toxic effect on Plasmodium falciparum could be suspected to interfere with the antiplasmodial assay because of RBC destruction.Therefore, we have submitted a 10% RBC suspension to the highest tested concentration against Plasmodium falciparum (100 μg mL−1) of each compound in duplicate for 1 hour to evaluate their hemolytic activity. The resulting absorbance of the supernatant at 550 nm was compared to a similar sample exposed to Triton X, a well-known hemolytic product (100% hemolysis).
Fortunately, none of our test samples seemed to cause a significant hemolysis at 100 μg mL−1, despite the suspension state obtained for the less soluble compounds. In fact, the resulting percentage of hemolysis was always lower than 1%. In conclusion, the antiplasmodial effect exhibited by our dimeric structures could not be assigned to a toxic effect on the erythrocytes.
Cytotoxicity
After the identification of significant inhibitors of Plasmodium falciparum and the confirmation of the absence of hemolysis, a toxicity assay on healthy human cells was realized. Indeed, it was necessary to determine the selectivity of the molecules between the host's cells and the infectious agent.Since the site of action of most antimalarials is mainly located in the blood vessels, we have selected a cell line which could be “malaria-related”, venous endothelial cells. Therefore, HUVEC (human umbilical vein endothelial cells) were exposed to different concentrations of our derivatives to observe any potential cytotoxic effects.
Moreover, since these cells are not physiologically in a constant multiplication process, we have observed the effect of some compounds on multiplying and confluent cells in parallel. This experiment was performed with ellagic acid (9) and 17 using an Incucyte® apparatus. For these two compounds, no significant differences were observed for the IC50 value under both conditions.
Indeed, 9 exhibited a non-toxic profile with an IC50 value superior to 165 μM (SI > 59) under the two conditions, whereas the dimer had an IC50 value of 14.0 μM on the growing cells and 12.1 μM on the confluent cells. Consequently, the other products have only been assessed in the classical multiplying cell assay.
As a result, most of the dimers were significantly selective for Plasmodium, with a selectivity index (SI) often superior to 10. However, some compounds possessed a medium to great toxicity against HUVEC. Among them, the most lipophilic structures (17–20), clogP between 4.9 and 7.2, were the most cytotoxic.
Consequently, if a higher partition coefficient was beneficial for the antiplasmodial activity due to a greater RBC permeation rate, it seemed also linked to a higher cytotoxicity on healthy human cells because of this easier cell membrane penetration.42–44
Interestingly, thanks to the Incucyte®, we have observed for 17 the fast formation of crystals in the culture media, in less than 1 h (Fig. 2). This crystallization could be explained by the low water solubility of the compound (Cmax = 26 μM). In fact, at some tested concentrations, the dose was superior to Cmax.
 | ||
| Fig. 2 Crystals of 17 in the cell toxicity assay on HUVEC. | ||
The presence of these solid formations could impede the cell multiplication owing to physical effects. However, a reduced water solubility did not always lead to a significant cytotoxicity since 9, 32 or 33 exhibited low Cmax values but were also selective to Plasmodium falciparum (SI > 6).
In conclusion, even if their toxicity was not completely understood, it seemed more logical to discontinue the investigations on the para-substituted dimers, especially those with a lipophilic substituent (17–20).
In vivo toxicity
Therefore, following the recommendations of the OECD, some dimers were selected: 17, 22–24, 29–30.49 Thus, they have been administered to 24 hour synchronized embryos (in duplicate, n = 30–40) at 4 different concentrations (100, 50, 25, and 10 μg mL−1 in the culture media). Afterwards, the development, hatching, and mortality rate were assessed for 3 days with a daily treatment, by renewal of the culture media, and compared to control zebrafish, treated with 0.1% DMSO.
Fortunately, none of the tested compounds exhibited significant toxic effects on Danio rerio compared to the controls (>90% mean survival at the highest concentrations). In addition, no major morphological alterations, i.e., brain size reduction, misshape, modified heart rate, were observed at any moment during the experiment.
However, the amide derivatives (29–30, Chart 4) displayed a negative effect on the hatching of the zebrafish (Fig. 3). Indeed, a concentration-dependent reduction of the hatching rate was observed for these products compared to the other products (100% hatching after 72 hpf), especially for 30 at 100 μg mL−1 where only 20% of the embryos were able to disrupt the eggshell. Consequently, the embryos seemed correctly developed but unable to hatch for unidentified reasons, even 5 days post-fertilization.
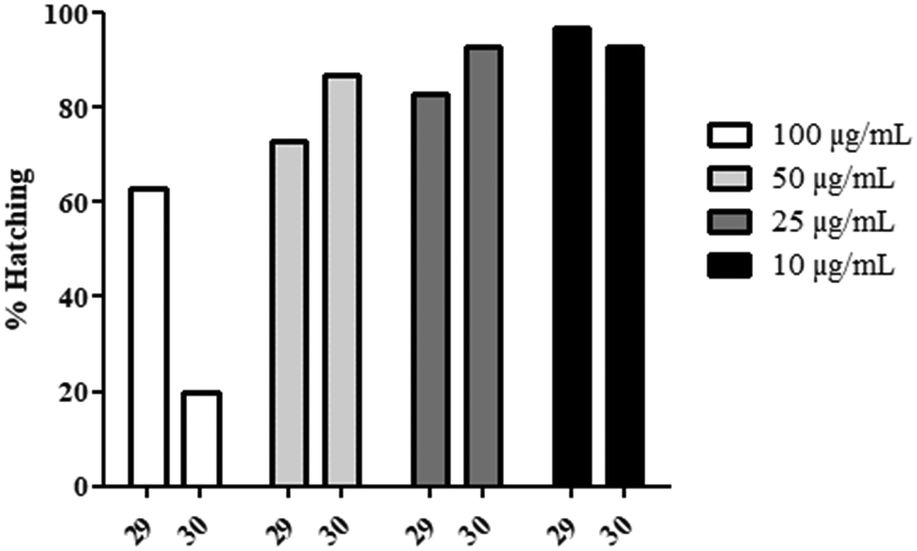 | ||
| Fig. 3 Hatching rate of Danio rerio embryos when exposed to 29–30. | ||
This phenomenon could have various explanations, from an accumulation of the product on the chorionic membrane to an impaired development.50–53 Thus, we have rerun the experiment, but the chorion was removed at 24 h post-fertilization using enzymatic digestion (pronase).54
Hopefully, despite the early removal of this protective barrier, the embryos were not subject to any signs of developmental defects or excess mortality (>95% survival).
In conclusion, the decrease of the hatching rate with these amide derivatives could be preferentially explained by the accumulation of products on the eggshell.
This phenomenon could find its origin in the precipitation of the molecules because of their reduced water solubility. However, no similar hatching reduction was observed with 17, which possessed a much lower Cmax value.
Consequently, further studies could be necessary to better understand the absence of hatching. Similarly, because of this precipitation, the real concentration, at which the embryos were exposed, was unknown. It could be beneficial to measure the dissolved fraction in the culture media.
However, since no major toxic events happened, we concluded on the global non-toxic character of our dimeric scaffolds on Danio rerio embryos.
Therefore, for each administration routes (intraperitoneal and oral) and all the test samples (23 and 30), the recommended acute toxicity assay was performed on 2 mice. Ellagic acid (9) was not evaluated here because of the extensive literature demonstrating its lack of toxicity in rodents.23,55
The assay consists in the administration of cumulative doses of the products during a short time (4 shots in 6 h) and the follow-up of the animals for 24 hours.17
Since no toxic event was observed after the last administration, the doses of 150 mg kg−1 i.p. and 200 mg kg−1per os were defined as safe for mice malaria treatment. Therefore, the recommended concentration of 50 mg kg−1 for the preliminary in vivo evaluation of the antimalarial effect could be employed without any concerns.17
In vivo antimalarial potential
Following the classical guidelines for antimalarial development, the in vivo efficacy of several promising gallic acid dimers (23 and 30) was evaluated in a malaria murine-model, the Peters' 4 day suppressive test.17 Thus, a comparison of the in vivo effects according to two routes of administration was performed, intraperitoneally (IP) to assess the in vivo efficacy, and orally (PO) since it was defined as the preferred way for drug administration.In parallel, the reference compound, ellagic acid (9), was also tested. Indeed, this molecule exhibited a potent antimalarial effect during a similar experiment after i.p. injection, leading to a 100% growth inhibition without recrudescence for 60 days with 50 and 100 mg kg−1 doses. Consequently, its ED50 was established as inferior to 1 mg kg−1 after intraperitoneal administration.23
Thus, the in vivo evaluation (Fig. 4) was made with a murine strain of Plasmodium, P. berghei NK173, in 6 female Swiss CD1 mice per compound and administration route, as recommended by the guidelines.17 As expected, 9 displayed no decrease of the parasitaemia after the oral administration of a 50 mg kg−1 dose. However, the injection exhibited only a 30% reduction only at D4 and a fast recrudescence at D7, in two independent experiments.
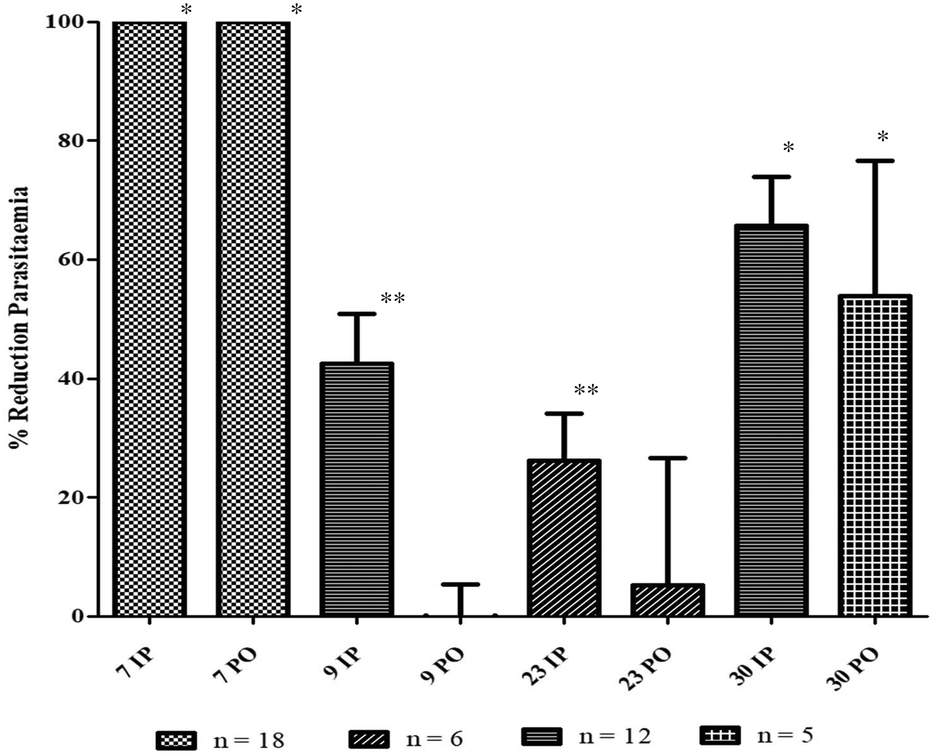 | ||
| Fig. 4 Reduction of parasitaemia obtained during in vivo studies in a murine malaria model. Chloroquine (7, 4 mg kg−1 per day i.p. or 25 mg kg−1 per day p.o.); 9, 23, 30 (50 mg kg−1 per day i.p. & p.o.); IP = intraperitoneal (i.p.); PO = per os (p.o.); * p-value < 0.005, compared to untreated mice; ** p-value < 0.01, compared to untreated mice. These have been obtained owing to Student's t-test on parasitaemia percentage at D4. | ||
This great disparity with the literature could find its origin in the slight protocol differences, leading to variations in the bioavailability. Indeed, the previous results were obtained with another solvent mixture (50% DMSO in physiological serum) which could better dissolve 9 and increase its blood level after intraperitoneal injection. In our case, since DMSO is often recognized as toxic for living organisms, the recommended mixture (saline physiological, EtOH, Tween 80%, 90/7/3) has been preferred.17
Furthermore, the previous study tested another species of Plasmodium, P. vinckei petteri, while ours was a murine P. berghei NK173.23 Consequently, the lack of activity of 9 after i.p. administration could also be partially explained by the differences between these two species of Plasmodium.56,57
Considering our original compounds, similar to 9, 23 displayed a non-active profile per os (only 5% inhibition) with a low parasitaemia reduction after intraperitoneal administration (± 26%).
The failure of this compound to cure the animals, despite its higher hydrosolubility and in vitro efficacy, demonstrated that the improvement of the oral potency did not only stand in the increase of the water solubility, but also in other pharmacokinetic parameters, notably influenced by the structure, such as the metabolization after absorption. Indeed, 23 is a diester which could be easily cleaved in its inactive monomers by the physiological esterases. Consequently, its corresponding amide dimer (30) could be metabolically more stable, and thus, more promising.
At 50 mg kg−1 per day for 4 days, 30 induced a 66% mean reduction of parasitaemia after i.p. treatment, in two independent experiments (n = 12). In addition, the oral administration led to a 54% mean reduction at D4. It is worth noting that the standard deviation for the oral administration was quite important (± 43%). This phenomenon could be explained by the incomplete solubilisation of the product in the physiological saline because of its medium solubility (0.6 mM). In addition, force-feeding could be easily impaired by regurgitation events. Therefore, an increased solubility using galenic formulation could be interesting to reduce this lack of reproducibility and confirmed the oral activity.
Unfortunately, as for 9, a fast recrudescence of the parasites was observed at D7 with no more differences between treated and untreated animals. However, this phenomenon was also observed in mice treated with the antimalarial reference drug, chloroquine (7). This quick increase of parasitaemia, even for standard antimalarials, has been regularly reported after the end of the treatment during in vivo experiments, and could originate from some residual parasites in a quiescent state.54,58
In conclusion, despite its limited in vivo efficacy, 30 seemed a promising scaffold for the development of new antimalarials since it displayed a higher antimalarial effect than ellagic acid in both administration routes at 50 mg kg−1 per day (at least 50% reduction).
However, it would be necessary to further explore this scaffold. Indeed, the information about its pharmacokinetic parameters is missing, as well as its ED50. In addition, further pharmacomodulation is necessary to enhance the in vivo efficacy and to obtain the cure of the animals.
Materials and methods
General procedures
All the chemicals employed during this research have been purchased from Fluorochem®, Sigma-Aldrich® or Abcr®. All the solvents (VWR®, Acros Organics®) have been used without further purification. MilliQ water has been obtained thanks to a Milli-Q Reference A+ system. The melting point of pure compounds was determined on the Melting point Büchi M565® apparatus and is uncorrected. The compounds were purified thanks to a Buchi Reveleris® prep on irregular silica cartridge 4–80 g. All reactions were routinely checked by TLC on silica gel Merck® 60 F254.The 1H- and 13C-NMR spectra were recorded on a Bruker Advance (500 MHz for 1H; 125 MHz for 13C) instrument using deuterated dimethyl sulfoxide (DMSO-d6), deuterated methanol (MeOD) or deuterated chloroform (CDCl3) as the solvent with tetramethylsilane (TMS) as an internal standard; chemical shifts are reported in δ values (ppm) relative to that of internal TMS.
Elemental analyses (C, H, N, S) were realized on a Thermo Scientific Flash EA1112® elemental analyzer and were within ±0.4% of the theoretical values for carbon, hydrogen, and nitrogen. This analytical method certified a purity of ≥95% for each tested compound. The UV spectra were recorded with a Hitachi® U-3010 UV/vis spectrophotometer model.
Synthesis
Methyl gallate (35), white solid, yield 88%, MP. 257 °C (decomp.)/lit. 240–242 °C,59 UV: 271 nm, 1H-NMR (in DMSO) δ 9.26 (2H, s), 8.93 (1H, s), 6.94 (2H, s), 3.75 (3H, s); 13C-NMR (in DMSO) δ 166.78, 146.04, 138.87, 119.75, 108.95, 52.05; EA Th: C, 52.18%; H, 4.38%; found: C, 52.30%; H, 4.38%.
Methyl 3,4-dihydroxybenzoate (46), white solid, yield 93%, MP. 137 °C/lit. 137–139 °C,601H-NMR (in DMSO) δ 9.77 (1H, s), 9.36 (1H, s), 7.35 (1H, s), 7.30 (1H, s), 6.79 (1H, s), 3.76 (3H, s); 13C-NMR (in DMSO) δ 166.62, 150.88, 145.54, 122.22, 120.95, 116.72, 115.78, 52.06; EA Th: C, 57.14%; H, 4.80%; found: C, 57.05%; H, 4.87%.
Methyl 3,5-dihydroxybenzoate (47), white solid, yield 98%, MP. 166 °C/lit. 169–170 °C,611H-NMR (in DMSO) δ 9.62 (2H, s), 6.81 (2H, s), 6.44 (1H, s), 3.79 (3H, s); 13C-NMR (in DMSO) δ 166.73, 159.01, 131.75, 107.63, 52.47; EA Th: C, 57.14%; H, 4.80%; found: C, 57.21%; H, 4.91%.
Methyl 3,4,5-triacetoxybenzoate (36), white solid, yield 92%, MP. 126 °C/lit. 126–128 °C,36 UV: 232 nm, 1H-NMR (in DMSO) δ 7.80 (2H, s), 3.88 (3H, s), 2.34 (3H, s), 2.30 (6H, s); 13C-NMR (in DMSO) δ 168.48, 167.39, 164.86, 143.83, 139.15, 128.05, 122.40, 53.17, 20.86, 20.32; EA Th: C, 54.20%; H, 4.55%; found: C, 54.16%; H, 4.69%.
Methyl 4-benzoxy-3,5-diacetoxybenzoate (37a), white product, yield 85%, MP. 106 °C/lit. 94–96 °C,36 UV; 254 nm, 1H-NMR (in DMSO) δ 7.69 (2H, s), 7.38 (5H, m), 5.03 (2H, s), 3.85 (3H, s), 2.25 (6H, s); 13C-NMR (in DMSO) δ 169.04, 165.05, 147.50, 144.34, 136.85, 128.92, 128.82, 128.49, 125.20, 122.78, 75.71, 52.96, 20.97; EA Th: C, 63.68%; H, 5.06%; found: C, 63.75%; H, 5.19%.
Methyl 4-(4-methoxybenzoxy)-3,5-diacetoxybenzoate (37b), white solid, yield 79%, MP. 126–132 °C, 1H-NMR (in CDCl3) δ 7.68 (2H, s), 7.28 (2H, d), 6.89 (2H, d), 4.98 (2H, s), 3.89 (3H, s), 3.82 (3H, s), 2.23 (6H, s); 13C-NMR (in CDCl3) δ 168.50, 165.24, 159.77, 147.30, 144.18, 129.66, 128.66, 125.48, 122.61, 113.89, 75.49, 55.32, 52.39, 20.68; EA Th: C, 61.85%; H, 5.19%; Found: C, 61.37%; H, 5.15%.
Methyl 4-napht-2-ylmethoxy-3,5-diacetoxybenzoate (37c), slightly brown solid, yield 61%, MP. 80 °C, 1H-NMR (in CDCl3) δ 7.85 (4H, m), 7.70 (2H, s),7.51 (2H, m), 7.47 (2H, dd), 5.22 (2H, s), 3.89 (3H, s), 2.17 (6H, s); 13C-NMR (in CDCl3) δ 168.55, 162.22, 147.48, 144.13, 134.12, 133.13, 128.33, 127.98, 127.76, 126.55, 126.42, 126.33, 125.64, 125.25122.70, 75.79, 52.42, 20.64; EA Th: C, 67.64%; H, 4.94%; found: C, 67.68%; H, 5.13%.
Methyl 4-(2-morpholinoethoxy)-3,5-diacetoxybenzoate (37d), white solid, yield 43% (after flash chromatography n-hexane/THF 1/0 to 0/1), MP. 92 °C, 1H-NMR (in DMSO) δ 7.67 (2H, s), 4.09 (2H, t), 3.84 (3H, s), 3.53 (4H, t), 2.59 (2H, t), 2.40 (4H, t), 2.34 (6H, s); 13C-NMR (in DMSO) δ 169.10, 165.08, 147.76, 144.08, 124.78, 122.76, 71.45, 66.60, 58.28, 53.92, 52.93, 21.08; EA Th: C, 56.69%; H, 6.08%; N, 3.67%; found: C, 56.96%; H, 6.19%; N, 3.88%.
Methyl 4-benzoxy-3,5-dihydroxybenzoate (38a), white solid, yield 96%, MP. 129 °C/lit. 133–134 °C,36 UV: 263 nm, 1H-NMR (in DMSO) δ 9.56 (2H, s), 7.50 (2H, d), 7.35 (3H, m), 6.97 (2H, s), 5.05 (2H, s), 3.77 (3H, s); 13C-NMR (in DMSO) δ 166.49, 151.35, 138.91, 138.28, 128.54, 128.50, 128.18, 124.84, 109.02, 73.54, 52.37; EA Th: C, 65.69%; H, 5.15%; found: C, 65.94%; H, 5.31%.
Methyl 4-(4-methoxybenzoxy)-3,5-dihydroxybenzoate (38b), white solid, yield 64%, MP. 140–146 °C, 1H-NMR (in DMSO) δ 9.50 (2H, s), 7.40 (2H, d), 6.94 (2H, s), 6.89 (2H, d), 4.98 (2H, s), 3.77 (3H, s), 3.74 (3H, s); 13C-NMR (in DMSO) δ 166.50, 159.41, 151.39, 138.73, 130.40, 130.13, 124.74, 113.85, 108.95, 73.16, 55.50, 52.36; EA Th: C, 63.15%; H, 5.30%; found: C, 62.7%; H, 5.30%.
Methyl 4-(2-morpholinoethoxy)-3,5-dihydroxybenzoate (38d), colourless oil, 1H-NMR (in MeOD) δ 6.92 (2H, s), 4.07 (2H, t), 3.74 (3H, s), 3.69 (4H, t), 2.56 (6H, m); 13C-NMR (in DMSO) δ 167.29, 151.31, 138.42, 126.00, 108.95, 68.63, 65.95, 57.06, 52.80, 51.27.
Methyl 4-napht-2-ylmethoxy-3,5-dihydroxybenzoate (38c), white solid, yield 13%, MP. 199 °C/lit. 203–205 °C,34 UV: 225 nm, 1H-NMR (in DMSO) δ 7.97 (1H, s), 7.90 (3H, d), 7.70 (1H, dd), 7.51 (2H, m), 6.97 (2H, s), 5.22 (2H, s), 3.76 (3H, s); 13C-NMR (in DMSO) δ 166.48, 151.42, 138.90, 135.95, 133.12, 133.04, 128.27, 128.03, 127.05, 126.79, 126.60, 126.46, 124.90, 109.01, 73.64, 52.38; EA Th: C, 70.36%; H, 4.97%; found: C, 69.96%; H, 5.06%.
Methyl 4-(benzyloxy)-3,5-bis(methoxymethoxy)benzoate (39a), colourless oil, 1H-NMR (in CDCl3) δ 7.52 (2H, s), 7.46–7.45 (2H, d), 7.36–7.28 (3H, m), 5.19 (4H, s), 5.13 (2H, s), 3.88 (3H, s), 3.49 (6H, s); 13C-NMR (in CDCl3) δ 166.43, 150.92, 142.94, 137.29, 128.40, 128.29, 128.13, 125.66, 112.02, 95.45, 75.21, 56.46, 52.22.
Methyl 4-((4-methoxybenzyl)oxy)-3,5-bis(methoxymethoxy)benzoate (39b), colourless oil, 1H-NMR (in CDCl3) δ 7.51 (2H, s), 7.36 (2H, d), 6.86 (2H, d), 5.19 (4H, s), 5.07 (2H, s), 3.88 (3H, s), 3.80 (3H, s), 3.49 (6H, s); 13C-NMR (in CDCl3) δ 166.45, 159.50, 150.94, 130.22, 129.24, 125.51, 113.54, 111.74, 95.29, 74.82, 56.40, 55.25, 52.27.
Methyl 3,5-bis(methoxymethoxy)-4-(naphth-2-ylmethoxy)benzoate (39c), colourless oil, 1H-NMR (in CDCl3) δ 7.89 (1H, s), 7.80–7.83 (3H, m), 7.36 (2H, d), 7.63–7.61 (1H, dd), 7.53 (2H, s), 7.48–7.46 (2H, dd), 5.30 (2H, s), 5.20 (4H, s), 3.87 (3H, s), 3.47 (6H, s); 13C-NMR (in CDCl3) δ 161.68, 146.19, 138.24, 130.09, 128.42, 123.25, 122.93, 122.47, 121.49, 120.95, 107.25, 90.71, 70.63, 51.67, 47.48.
Then, the oil (39a–c) was dissolved in MeOH/THF solvent mixture (2/1, v/v) and treated with LiOH (6.93 g, 165 mmol) in H2O (30 mL) under reflux for 2 h with stirring. After cooling to RT, HCl 1 N was carefully added to acidify the medium until pH 3. The organic solvents were partially evaporated in vacuo and the mixture was extracted with EtOAc. EtOAc was successively washed with water and brine, and then dried over MgSO4. The drying agent was removed by filtration, and the filtrate was evaporated as a white powder.
4-(Benzyloxy)-3,5-bis(methoxymethoxy)benzoic acid (40a), white solid, yield 74%, MP. 108 °C, 1H-NMR (in DMSO) δ 7.48 (2H, dd), 7.39 (2H, s), 7.37 (2H, dd), 7.34–7.31 (1H, m), 5.23 (4H, s), 5.07 (2H, s), 3.40 (6H, s); 13C-NMR (in DMSO) δ 167.15, 150.93, 142.52, 137.81, 128.57, 126.46, 111.53, 95.30, 74.78, 56.34; EA Th: C, 62.06%; H, 5.79%; found: C, 61.67%; H, 5.97%.
4-((4-Methoxybenzyl)oxy)-3,5-bis(methoxymethoxy)benzoic acid (40b), white solid, yield 54%, MP. 99–104 °C, 1H-NMR (in MeOD) δ 7.42 (2H, s), 7.28 (2H, d), 6.81 (2H, d), 5.11 (4H, s), 4.96 (2H, s), 3.70 (3H, s), 3.39 (6H, s); 13C-NMR (in DMSO) δ 167.18, 159.58, 150.99, 142.46, 130.39, 129.73, 114.03, 111.56, 95.31, 74.46, 56.32, 55.54; EA Th: C, 60.31%; H, 5.86%; found: C, 60.38%; H, 5.84%.
3,5-bis(methoxymethoxy)-4-(naphth-2-ylmethoxy)benzoic acid (40c), white solid, yield 86%, MP. 111–119 °C/102–105 °C,341H-NMR (in DMSO) δ 7.98 (1H, s), 7.94–7.91 (3H, m), 7.66 (1H, dd), 7.52 (2H, m), 7.40 (2H, s), 5.25 (4H, s), 5.25 (2H, s), 3.40 (6H, s); 13C-NMR (in DMSO) δ 167.14, 150.94, 142.53, 135.49, 133.14, 133.10, 128.32, 128.25, 128.03, 127.12, 126.72, 126.65, 126.63, 111.50, 95.32, 74.94, 56.35; EA Th: C, 66.32%; H, 5.57%; found: C, 66.87%; H, 5.57%.
Methyl 4-hydroxy-3,5-bis(methoxymethoxy)benzoate (43), white solid, yield 74%, MP. 85 °C, 1H-NMR (in DMSO) δ 9.57 (1H, bd s), 7.38 (2H, s), 5.19 (4H, s), 3.79 (3H, s), 3.42 (6H, s); 13C-NMR (in DMSO) δ 166.32, 145.63, 143.42, 119.61, 112.41, 95.56, 56.26, 52.37; EA Th: C, 52.94%; H, 5.92%; found: C, 52.76%; H, 6.07%.
Methyl 3,5-bis(methoxymethoxy)-4-(2-morpholinoethoxy)benzoate (39d), white solid, yield 90% (after DCVC petroleum ether/THF, 1/0 to 1/1), MP. 35–43 °C, 1H-NMR (in CDCl3) δ 7.51 (2H, s), 5.23 (4H, s), 4.19 (2H, t), 3.88 (3H, s), 3.74 (4H, t), 3.51 (6H, s), 2.80 (2H, t), 2.59 (4H, t); 13C-NMR (in CDCl3) δ 166.41, 150.78, 143.31, 125.59, 111.98, 95.42, 70.52, 66.98, 58.46, 56.47, 53.89, 52.24; EA Th: C, 56.10%; H, 7.06%, N, 3.63%; found: C, 55.88%; H, 7.00%, N, 4.01%.
Methyl 4-(2-hydroxyethoxy)-3,5-bis(methoxymethoxy)benzoate (39e), colourless oil, 1H-NMR (in DMSO) δ 7.40 (2H, s), 7.30 (5H, m), 5.22 (4H, s), 4.54 (2H, s), 4.20 (2H, t), 3.83 (3H, s), 3.74 (2H, t), 3.39 (6H, s); 13C-NMR (in DMSO) δ 166.07, 150.84, 143.49, 138.89, 128.65, 127.88, 124.96, 111.64, 95.36, 72.69, 72.44, 69.61, 56.34, 52.72.
Methyl 4-(2-(benzyloxy)ethoxy)-3,5-bis(methoxymethoxy)benzoate (39f), colourless oil, 1H-NMR (in DMSO) δ 7.40 (2H, s), 5.25 (4H, s), 4.03 (2H, t), 3.82 (3H, s), 3.67 (2H, m), 3.41 (6H, s); 13C-NMR (in DMSO) δ 166.07, 150.84, 124.89, 111.71, 95.37, 75.11, 65.46, 60.73, 56.36, 52.71.
3,5-Bis(methoxymethoxy)-4-(2-morpholinoethoxy)benzoic acid (40d), colourless oil, 1H-NMR (in DMSO) δ 7.38 (2H, d), 5.24 (4H, d), 4.10 (2H, dt), 3.55 (2H, t), 3.42 (4H, t), 2.67 (2H, t), 2.47 (4H, bd t); 13C-NMR (in DMSO) δ 166.08, 150.91, 150.61, 143.34, 124.98, 111.71, 111.46, 95.32, 70.93, 66.66, 58.21, 56.46, 53.92, 52.72.
4-(2-(benzyloxy)ethoxy)-3,5-bis(methoxymethoxy)benzoic acid (40f), white solid, yield 89%, MP. 95 °C, 1H-NMR (in DMSO) δ 7.38 (2H, s), 7.32 (5H, m), 5.21 (4H, s), 4.54 (2H, s), 4.19 (2H, t), 3.74 (2H, t), 3.39 (6H, s); 13C-NMR (in DMSO) δ 167.16, 150.69, 143.09, 138.91, 128.65, 127.89, 111.89, 95.36, 72.66, 72.44, 69.6, 56.30, 31.17; EA Th: C, 61.22%; H, 6.17%, found: C, 61.16%; H, 6.20%.
Methyl 3,4,5-tribenzyloxybenzoate (48), white solid, yield 79%, MP. 99 °C, 1H-NMR (in CDCl3) δ 7.45–7.25 (17H, m), 5.14 (4H, s), 5.11 (2H, s), 3.89 (3H, s); 13C-NMR (in CDCl3) δ 166.65, 152.57, 142.42, 137.45, 136.66, 128.55, 128.53, 128.19, 128.03, 127.95, 127.55, 125.23, 109.08, 75.13, 71.24, 52.24; EA Th: C, 76.63%; H, 5.77%; found: C, 76.50%; H, 5.82%.
Methyl 3,4-dibenzyloxybenzoate (49), white solid, yield 47%, MP. 62 °C, 1H-NMR (in CDCl3) δ 7.64–7.30 (12H, m), 5.21 (4H, d), 3.87 (3H, s); 13C-NMR (in CDCl3) δ 166.77, 152.90, 148.34, 136.85, 136.57, 128.60, 128.53, 128.00, 127.94, 127.41, 127.13, 124.00, 123.09, 115.47, 113.24, 71.20, 70.83, 51.99; EA Th: C, 75.84%; H, 5.79%; found: C, 75.76%; H, 5.90%.
Methyl 3,5-dibenzyloxybenzoate (50), white solid, yield 76%, MP. 79 °C, 1H-NMR (in CDCl3) δ 7.43–7.32 (10H, m), 7.30 (2H, d), 6.80 (1H, t), 5.07 (4H, s), 3.90 (3H, s); 13C-NMR (in CDCl3) δ 166.79, 159.81, 136.48, 132.07, 128.64, 128.14, 127.59, 107.28, 70.31, 52.29; EA Th: C, 75.84%; H, 5.79%; found: C, 75.88%; H, 6.02%.
3,4,5-Tribenzyloxybenzoic acid (51), white solid, yield 89%, MP. 190 °C, 1H-NMR (in CDCl3) δ 7.44–7.26 (17H, m), 5.15 (4H, s), 5.14 (2H, s); 13C-NMR (in CDCl3) δ 170.57, 152.60, 143.16, 137.37, 136.55, 128.58, 128.53, 128.22, 128.08, 128.00, 127.56, 124.06, 109.63, 75.17, 71.24; EA Th: C, 76.35%; H, 5.49%; found: C, 76.04%; H, 5.48%.
3,4-Dibenzyloxybenzoic acid (52), white solid, yield 97%, MP. 186–187.8 °C/lit. 187.9–188.4 °C,62δ1H-NMR (in DMSO) δ 7.55 (1H, s), 7.54 (1H, d), 7.47–7.31 (10H, m), 7.16 (1H, d), 5.22 (2H, s), 5.18 (2H, s); 13C-NMR (in DMSO) δ 167.43, 152.55, 148.09, 137.51, 137.21, 128.93, 128.88, 128.39, 128.28, 128.02, 127.92, 123.95, 123.78, 115.05, 113.59, 70.47; EA Th: C, 75.43%; H, 5.43%; found: C, 75.55%; H, 5.39%.
3,5-Dibenzyloxybenzoic acid (53), white solid, yield 90%, MP. 207–215 °C/lit. 208–209 °C, 1H-NMR (in DMSO) δ 7.46–7.32 (10H, m), 7.15 (2H, s), 6.92 (1H, s), 5.14 (4H, s); 13C-NMR (in DMSO) δ 167.36, 159.88, 137.22, 133.41, 128.93, 128.37, 128.15, 108.46, 107.02, 69.97; EA Th: C, 75.43%; H, 5.43%; found: C, 75.12%; H, 5.47%.
Method viia. To a solution of the protected alcohol (1.5 eq.) in CH2Cl2 (167 mL) were added the corresponding carboxylic acid (8.73 g, 25.1 mmol), DMAP (2.04 g, 16.7 mmol), and EDCI·HCl (6.41 g, 33.4 mmol). The mixture was stirred at RT until the term of the reaction (controlled by TLC). Then, the mixture was neutralized with 1 N aq. H3PO4, and diluted with H2O (25 mL). After, the solution was extracted with EtOAc. The organic layer was washed with H2O and brine. After the general drying procedure and evaporation in vacuo, the resulting product was purified by flash chromatography to obtain a colourless oil.34
3-((4-Methoxybenzyl)oxy)propyl 4-(benzyloxy)-3,5-bis(methoxymethoxy)benzoate (41a), colourless oil, 1H-NMR (in CDCl3) 7.50 (2H, s), 7.47 (2H, d), 7.36–7.31 (3H, m), 7.26 (2H, d), 6.86 (2H, d), 5.18 (4H, s), 5.13 (2H, s), 4.45 (2H, s), 4.40 (2H, t), 3.78 (3H, s), 3.59 (2H, t) 3.48 (6H, s), 2.05 (2H, m).
Method viib. Similar to method viia, the carboxylic acid was dissolved in CH2Cl2 with DMAP, EDCI–HCl and the linker. The reaction was performed under reflux until the reaction was completed. Then, water was added to the reaction medium and extracted with EtOAc. The organic layer was washed with 0.2 N NaOH, NH4Cl aq. sat., 1 N HCl and brine. After the general drying procedure, the solvents were removed in vacuo and the resulting oil was purified with flash chromatography (n-hexane/EtOAc, 1/0–1/1) to obtain a colourless oil.
3-Hydroxypropyl 4-(benzyloxy)-3,5-bis(methoxymethoxy)benzoate (42a), colourless oil, 1H-NMR (in CDCl3) δ 7.52 (2H, s), 7.46 (2H, d), 7.36–7.29 (3H, m), 5.19 (4H, s), 5.13 (2H, s), 4.46 (2H, t), 3.74 (2H, t), 3.49 (6H, s), 1.99 (2H, m); 13C-NMR (in CDCl3) δ 166.43, 150.96, 143.15, 137.26, 133.08, 128.41, 128.32, 128.16, 125.54, 112.16, 95.51, 75.25, 61.92, 59.12, 56.44, 31.97.
3-Hydroxypropyl 4-((4-methoxybenzyl)oxy)-3,5-bis(methoxymethoxy)benzoate (42b), colourless oil, 1H-NMR (in CDCl3) δ 7.52 (2H, s), 7.46 (2H, m), 7.36–7.29 (3H, m), 5.19 (4H, s), 5.13 (2H, s), 4.47 (2H, t), 3.74 (2H, t), 3.49 (6H, s), 1.99 (2H, s).
3-Hydroxypropyl 3,5-bis(methoxymethoxy)-4-(naphth-2-ylmethoxy)benzoate (42c), white solid, yield 63%, MP. 67 °C, 1H-NMR (in DMSO) δ 7.88 (1H, d), 7.81 (2H, m), 7.61 (1H, d), 7.51 (2H, d) 7.47 (2H, dd), 7.25 (2H, s), 5.29 (2H, s), 5.19 (4H, s), 4.46 (2H, t), 3.73 (2H, q), 3.46 (6H, s), 1.97 (2H, m); 13C-NMR (DMSO) δ 161.68, 146.22, 138.44, 130.05, 128.42, 123.27, 123.24, 122.94, 122.48, 121.48, 121.39, 121.36, 120.82, 107.38, 70.65, 57.16, 54.38, 51.69, 27.22; EA Th: C, 65.78%; H, 6.18%, found: C, 66.15%; H, 6.16%.
3-Hydroxypropyl 3,5-bis(methoxymethoxy)-4-(2-morpholinoethoxy)benzoate (42d), white solid, yield 8%, MP. 59.9–67.8 °C, 1H-NMR (in CDCl3) δ 7.51 (2H, s), 5.23 (4H, s), 4.47 (2H, t), 4.19 (2H, t), 3.74 (6H, t), 3.51 (6H, s), 2.80 (2H, t), 2.59 (4H, s), 1.99 (2H, m); 13C-NMR (DMSO) δ 166.38, 150.81, 143.51, 125.47, 112.10, 95.47, 70.53, 66.97, 61.92, 59.13, 107.38, 58.44, 53.88, 31.97; EA Th: C, 55.93%; H, 7.28%, N, 3.26% Found: C, 56.04%; H, 7.24%, N, 3.82%.
3-Hydroxypropyl 4-(2-(benzyloxy)ethoxy)-3,5-bis(methoxymethoxy)benzoate (42f), colourless oil, 1H-NMR (in CDCl3) δ 7.52 (2H, s), 7.34 (4H, d), 7.26 (1H, s), 5.21 (4H, s), 4.62 (2H, t), 4.47 (2H, t), 4.27 (2H, t), 3.82 (2H, t), 3.75 (2H, q), 3.49 (6H, s), 1.99 (2H, m); 13C-NMR (CDCl3) δ 165.79, 150.71, 143.68, 138.27, 128.34, 127.64, 125.42, 112.40, 95.55, 73.19, 72.69, 69.40, 61.68, 56.44, 28.28.
2-Hydroxyethyl 3,4,5-tris(benzyloxy)benzoate (54), white solid (after flash chromatography DCM/EtOAc, 1/0 to 3/1), MP. 105 °C, 1H-NMR (in DMSO) δ 7.48–7.27 (17H, m), 5.18 (4H, s), 5.05 (2H, s), 4.26 (2H, t), 3.70 (2H, q); 13C-NMR (DMSO) δ 165.84, 152.57, 141.76, 137.74, 137.20, 128.92, 128.70, 128.56, 128.44, 128.19, 125.50, 108.77, 74.69, 70.84, 67.18, 59.52.
3-Hydroxypropyl 3,4,5-tris(benzyloxy)benzoate (55), white solid, yield 77%, MP. 115 °C, 1H-NMR (in DMSO) δ 7.47–7.27 (17H, m), 5.19 (4H, s), 5.05 (2H, s), 4.30 (2H, t), 3.54 (2H, q), 1.84 (2H, m); 13C-NMR (DMSO) δ 165.75, 152.55, 141.73, 137.76, 137.23, 128.92, 128.70, 128.57, 128.42, 128.13, 125.49, 108.58, 74.69, 70.77, 62.65, 57.75, 32.05; EA Th: C, 74.68%; H, 6.07%, found: C, 74.55%; H, 6.15%.
4-Hydroxybutyl 3,4,5-tris(benzyloxy)benzoate (56), white solid, yield 58% (after flash chromatography DCM/EtOAc, 1/0 to 3/1), MP. 117 °C, 1H-NMR (in DMSO) δ 7.47–7.27 (17H, m), 5.19 (4H, s), 5.05 (2H, s), 4.25 (2H, t), 3.46 (2H, q), 1.73 (2H, m), 1.52 (2H, m); 13C-NMR (DMSO) δ 165.70, 152.56, 141.75, 137.77, 137.23, 128.92, 128.70, 128.57, 128.41, 128.12, 125.48, 108.57, 74.69, 70.76, 62.65, 60.76, 29.39, 25.51; EA Th: C, 74.98%; H, 6.29%, found: C, 74.66%; H, 6.41%.
Propane-1,3-diyl bis(4-(benzyloxy)-3,5-bis(methoxymethoxy)benzoate) (11), white solid, yield 46% (after DCVC), MP. 62 °C, 1H-NMR (in DMSO) δ 7.46 (4H, d), 7.39 (4H, s), 7.37–7.30 (5H, m), 5.21 (8H, s), 5.06 (4H, s), 4.40 (4H, t), 3.39 (12H, s), 2.15 (2H, m); 13C-NMR (in DMSO) δ 165.52, 150.99, 142.97, 137.73, 128.70, 125.26, 111.4, 95.32, 74.82, 62.39, 56.37, 28.18; EA Th: C, 63.58%; H, 6.02%, found: C, 63.48%; H, 6.29%.
3-((4-(Benzyloxy)-3,5-bis(methoxymethoxy)benzoyl)oxy)propyl-3,5-bis(methoxymethoxy)-4-(naphth-2-ylmethoxy)benzoate (12), colourless oil, 1H-NMR (in DMSO) δ 7.93 (4H, m), 7.64 (1H, dd), 7.52 (2H, dd), 7.46 (2H, d), 7.40 (4H, d), 7.37–7.31 (4H, m), 5.22 (10H, d), 5.07 (2H, s), 4.40 (4H, t), 2.15 (2H, m); 13C-NMR (in DMSO) δ 165.51, 150.98, 143.02, 142.98, 137.74, 135.41, 133.14, 133.10, 128.69, 128.45, 128.32, 128.27, 128.03, 127.11, 126.72, 126.63, 126.60, 125.31, 125.27, 111.42, 74.82, 62.40, 56.37, 28.18.
Propane-1,3-diyl bis(4-(2-(benzyloxy)ethoxy)-3,5-bis(methoxymethoxy)benzoate) (13), white solid, yield 59%, 1H-NMR (in DMSO) δ 7.54 (2H, s), 7.34 (10H, d), 5.21 (8H, s), 4.62 (4H, t), 4.44 (4H, t), 4.29 (4H, t), 3.81 (4H, t), 3.48 (12H, s), 2.23 (2H, m); 13C-NMR (in CDCl3) δ 165.80, 150.72, 143.69, 138.27, 128.34, 127.60, 125.43, 112.41, 95.56, 73.20, 72.70, 69.41, 61.69, 56.44, 28.29, EA Th: C, 61.80%; H, 6.07%, found: C, 62.08%; H, 5.96%.
Propane-1,3-diyl bis(3,5-bis(methoxymethoxy)-4-(naphth-2-ylmethoxy)benzoate) (14), yellowish oil, 1H-NMR (in DMSO) δ 7.94 (2H, s), 7.92 (6H, m), 7.65 (2H, dd), 7.52 (4H, m), 7.41 (4H, s), 5.26 (12H, s), 4.3 (4H, t), 3.40 (12H, s), 1.83 (2H, m).
Propane-1,3-diyl bis(4-((4-methoxybenzyl)oxy)-3,5-bis(methoxymethoxy)benzoate) (15), colourless oil, 1H-NMR (in CDCl3) δ 7.38 (4H, s), 7.36 (4H, d), 6.91 (4H, d), 5.21 (8H, s), 4.99 (4H, t), 4.39 (4H, t), 3.74 (6H, s), 3.39 (12H, s), 2.14 (2H, m); 13C-NMR (in DMSO) δ 165.54, 159.58, 151.06, 142.96, 130.38, 129.66, 125.14, 114.03, 111.47, 95.33, 74.51, 62.36, 56.36, 55.54, 28.19.
3-((4-(2-(Benzyloxy)ethoxy)-3,5-bis(methoxymethoxy)benzoyl)oxy)propyl-3,4,5-tris(benzyloxy)benzoate (16), colourless oil, 1H-NMR (in CDCl3) δ 7.56–7.28 (24H, m), 5.19 (4H, s), 5.13 (4H, s), 5.12 (2H, s), 4.61 (2H, s), 4.44 (4H, q), 4.28 (2H, t), 3.82 (2H, t), 3.47 (6H, s), 2.21 (2H, m), 13C-NMR (in DMSO) δ 165.57, 152.52, 150.79, 143.64, 141.83, 138.87, 137.77, 137.20, 128.91, 128.66, 128.63, 128.57, 128.41, 128.10, 127.86, 127.8, 125.23, 125.04, 111.80, 108.61, 95.37, 74.70, 72.70, 70.73, 69.60, 62.30, 56.33, 28.16.
Ethane-1,2-diyl bis(3,4,5-tris(benzyloxy)benzoate) (57), white solid, yield 88%, MP. 135 °C, 1H-NMR (in DMSO) δ 7.40–7.24 (34H, m), 5.08 (8H, s), 4.97 (4H, s), 4.62 (4H, s); 13C-NMR (in DMSO) δ 165.65, 152.58, 141.98, 137.70, 137.02, 128.91, 128.85, 128.60, 128.54, 128.42, 128.37, 128.13, 128.11, 125.11, 108.71, 74.68, 70.77, 63.24; EA Th: C, 76.72%; H, 5.66%, found: C, 76.20%; H, 5.67%.
Propane-1,3-diyl bis(3,4,5-tris(benzyloxy)benzoate) (58), white solid, yield 54%, MP. 92 °C, 1H-NMR (in DMSO) δ 7.43–7.23 (34H, m), 5.09 (8H, s), 4.99 (4H, s), 4.43 (4H, t), 2.18 (2H, m); 13C-NMR (in DMSO) δ 165.67, 152.50, 141.81, 137.76, 137.14, 128.86, 128.55, 128.52, 128.38, 128.32, 128.06, 125.30, 108.58, 74.71, 70.70, 62.69, 28.10; EA Th: C, 76.94%; H, 5.69%, found: C, 76.52%; H, 5.71%.
Butane-1,4-diyl bis(3,4,5-tris(benzyloxy)benzoate) (59), white solid, yield 88%, MP. 133 °C, 1H-NMR (in DMSO) δ 7.43–7.25 (34H, m), 5.14 (8H, s), 5.02 (4H, s), 4.34 (4H, bd t), 1.85 (4H, bd t); 13C-NMR (in DMSO) δ 165.72, 152.53, 141.79, 137.75, 137.18, 128.88, 128.64, 128.55, 128.38, 128.11, 128.06, 125.38, 108.55, 74.70, 70.74, 65.01, 25.48; EA Th: C, 76.99%; H, 5.92%, found: C, 76.32%; H, 5.86%.
Propane-1,3-diyl bis(3,4-di(benzyloxy)benzoate) (60), white solid, yield 74%, MP. 123 °C, 1H-NMR (in CDCl3) δ 7.62 (2H, s), 7.60 (2H, d), 7.46–7.41 (8H, dd), 7.30–7.37 (10H, m), 6.90 (2H, d) 5.20 (4H, s), 5.17 (4H, s), 4.42 (4H, t), 2.18 (2H, m); 13C-NMR (in CDCl3) δ 166.14, 152.97, 148.32, 136.85, 136.53, 128.60, 128.54, 128.00, 127.94, 127.40, 127.13, 124.02, 122.97, 115.50, 113.20, 71.21, 70.83, 61.57; EA Th: C, 76.25%; H, 5.69%, found: C, 75.63%; H, 5.69%.
3-((3,4-Bis(benzyloxy)benzoyl)oxy)propyl 3,4,5-tris(benzyloxy)benzoate (61), white solid, yield 94%, MP. 170 °C, 1H-NMR (in CDCl3) δ 7.62 (2H, d), 7.46–7.24 (28H, m), 6.89 (2H, d), 5.17 (4H, d), 5.10 (6H, s), 4.43 (4H, t), 2.18 (2H, m); 13C-NMR (in CDCl3) δ 166.13, 166.03, 153.00, 152.54, 148.31, 142.50, 137.44, 136.82, 136.67, 136.51, 128.59, 128.55, 128.53, 128.20, 128.02, 127.99, 127.94, 127.54, 127.39, 127.13, 125.13, 124.01, 122.89, 115.49, 113.16, 109.13, 75.15, 71.23, 70.81, 61.91, 61.56, 28.27, 26.93; EA Th: C, 76.64%; H, 5.69%, found: C, 76.63%; H, 5.70%.
3-(Benzoyloxy)propyl 3,4,5-tris(benzyloxy)benzoate (62), white solid, yield 89%, MP. 117–119.5 °C, 1H-NMR (in DMSO) δ 7.95 (2H, dd), 7.64 (1H, t), 7.50–7.28 (19H, m), 5.15 (4H, s), 5.04 (2H, s), 4.42 (4H, q), 2.18 (2H, m); 13C-NMR (in DMSO) δ 166.18, 165.67, 152.52, 141.8, 137.76, 137.21, 133.80, 130.12, 129.60, 129.17, 128.91, 128.69, 128.58, 128.42, 128.12, 125.25, 108.64, 74.69, 70.74, 62.32, 28.16; EA Th: C, 75.73%; H, 5.69%; found: C, 75.62%; H, 5.70%.
3-((3,5-Bis(benzyloxy)benzoyl)oxy)propyl 3,4,5-tris(benzyloxy)benzoate (63), white solid, yield 91% (after DCVC CH2Cl2/ether, 1/0 to 9/1), MP. 128–132 °C, 1H-NMR (in DMSO) δ 7.45–7.26 (28H, m), 7.15 (2H, d), 5.12 (4H, s), 5.08 (4H, s), 5.01 (2H, s), 4.41 (4H, q), 2.17 (2H, m); 13C-NMR (in DMSO) δ 165.77, 165.67, 159.95, 152.51, 141.79, 137.77, 137.18, 137.05, 132.14, 128.92, 128.89, 128.65, 128.56, 128.40, 128.19, 128.09, 125.26, 108.57, 106.39, 107.27, 74.69, 70.70, 70.04, 62.46, 28.05; EA Th: C, 76.64%; H, 5.69%; found: C, 77.14%; H, 5.61%.
3-(3,4,5-Tris(benzyloxy)benzamido)propyl 3,4,5-tris(benzyloxy)benzoate (64), colourless oil, 1H-NMR (in DMSO) δ 8.53 (1H, s), 7.44–7.25 (34H, m), 5.13 (4H, s), 5.08 (4H, s), 5.00 (2H, s), 4.94 (2H, s), 4.33 (2H, t), 3.46 (2H, q), 1.99 (2H, m); 13C-NMR (in DMSO) δ 165.96, 165.70, 152.52, 152.37, 141.72, 139.94, 137.95, 137.77, 137.28, 137.19, 130.25, 128.88, 128.59, 128.55, 128.51, 128.39, 128.35, 128.27, 128.10, 125.45, 108.58, 106.81, 74.69, 70.70, 63.64, 37.05, 28.82.
N,N′-(Propane-1,3-diyl)bis(3,4,5-tris(benzyloxy)benzamide) (65), white solid, yield 35%, MP. 193–202 °C, 1H-NMR (in CDCl3) δ 7.42–7.24 (28H, m), 7.22 (4H, s), 7.00 (2H, t), 5.14 (8H, s), 5.09 (4H, s), 3.53 (4H, t), 1.82 (2H, m); 13C-NMR (in CDCl3) δ 167.54, 152.82, 141.28, 137.50, 136.71, 128.56, 128.53, 128.20, 128.02, 127.93, 127.63, 106.85, 75.18, 71.35, 36.26; EA Th: C, 77.10%; H, 3.52; N, 3.05%, found: C, 77.07%; H, 5.91%; N, 3.17%.
3-(3,4,5-Tris(benzyloxy)benzamido)phenyl 3,4,5-tris(benzyloxy)benzoate (66), white solid, yield 36%, MP. 160 °C, 1H-NMR (in DMSO) δ 10.04 (1H, s), 9.17 (6H, bd s), 7.72 (1H, t), 7.62–7.60 (1H, dd), 7.36 (1H, t), 7.11 (2H, s), 6.96 (2H, s), 6.91–6.89 (1H, dd); 13C-NMR (in DMSO) δ 166.19, 165.03, 151.32, 146.26, 145.98, 141.18, 139.78, 137.42, 129.72, 125.39, 125.19, 118.68, 117.55, 116.94, 113.84, 109.54, 107.70; EA Th: C, 58.12%; H, 3.9%; N, 3.39%, found: C, 57.91%; H, 4.4%; N, 3.0%.
4-(3,4,5-Tris(benzyloxy)benzamido)phenyl 3,4,5-tris(benzyloxy)benzoate (67), white solid, yield 72%, MP. 156.6–169.6 °C, 1H-NMR (in CDCl3) δ 7.64 (2H, d), 7.53 (2H, s), 7.46–7.28 (30H, m), 7.20 (2H, d), 7.14 (2H, s), 5.18–5.14 (12H, m); 13C-NMR (in CDCl3) δ 165.67, 152.50, 141.81, 137.76, 137.14, 128.86, 128.55, 128.52, 128.38, 128.32, 128.06, 125.30, 108.58, 74.71, 70.70, 62.69, 28.10; EA Th: C, 78.05%; H, 5.39%; N, 1.47%, found: C, 77.75%; H, 5.52%; N, 1.95%.
Propane-1,3-diyl bis(4-(benzyloxy)-3,5-dihydroxybenzoate) (17), white solid, yield 81%, MP. 199 °C, 1H-NMR (in DMSO) δ 9.56 (4H, bd s), 7.51 (4H, d), 7.36–7.29 (6H, m), 6.99 (4H, s), 5.04 (4H, s), 4.32 (4H, t), 2.10 (2H, m); EA Th: C, 66.42%; H, 5.03%, found: C, 66.19%; H, 5.42%.
3-((4-(Benzyloxy)-3,5-dihydroxybenzoyl)oxy)propyl-3,5-dihydroxy-4-(naphthalen-1-ylmethoxy)benzoate (18), colourless oil, 1H-NMR (in CDCl3) δ 7.87–7.81 (4H, m), 7.53–7.49 (3H, m), 7.38 (5H, s), 7.20 (4H, d), 5.29 (2H, s), 5.12 (2H, s), 4.44 (4H, t), 2.17 (2H, m); 13C-NMR (in CDCl3) δ 166.14, 148.94, 137.58, 136.35, 133.78, 133.40, 133.23, 129.05, 128.98, 128.94, 128.62, 128.12, 127.89, 127.82, 126.63, 126.54, 126.02, 125.81, 109.80, 75.65, 62.20, 28.12.
Propane-1,3-diyl bis(4-(2-(benzyloxy)ethoxy)-3,5-dihydroxybenzoate) (19), colourless oil, 1H-NMR (in CDCl3) δ 7.39–7.38 (8H, m), 7.19 (4H, s), 4.71 (4H, t), 4.43 (4H, t), 4.20 (4H, t), 3.77 (4H, t), 2.26 (2H, m); 13C-NMR (in CDCl3) δ 166.01, 149.50, 137.86, 136.36, 128.73, 128.40, 128.17, 126.85, 109.61, 73.79, 68.50, 60.43, 28.19.
Propane-1,3-diyl bis(3,5-dihydroxy-4-(naphth-2-ylmethoxy)benzoate) (20), white solid, yield 19%, MP. 188.2–194.7 °C, 1H-NMR (in DMSO) δ 9.61 (4H, bd s), 7.97 (2H, s), 7.90 (6H, m), 7.70 (2H, dd), 7.51 (4H, m), 6.99 (4H, s), 5.22 (4H, s), 4.31 (4H, t), 2.09 (2H, m); 13C-NMR (in DMSO) δ 165.97, 151.47, 139.01, 135.96, 133.12, 128.28, 128.03, 127.05, 126.80, 126.60, 126.46, 124.91, 109.06, 73.62, 61.68, 28.23; EA Th: C, 70.90%; H, 4.88%, found: C, 71.09%; H, 5.02%.
Ethane-1,2-diyl bis(3,4,5-trihydroxy)benzoate) monohydrate (22), white solid, yield 61%, MP. 251 °C (decomp.), 1H-NMR (in DMSO) δ 6.97–6.95 (4H, d), 4.17 (2H, t), 3.65 (2H, t); 13C-NMR (in DMSO) δ 166.22, 146.04, 139.10, 119.98, 119.48, 109.06, 66.35, 62.89, 59.65; EA Th: C, 50.01%; H, 4.20%, found: C, 50.03%; H, 4.36%.
Propane-1,3-diyl bis(3,4,5-trihydroxybenzoate) (23), white solid, yield 75%, MP. 249 °C (decomp.), 1H-NMR (in DMSO) δ 6.96 (4H, s), 4.29 (4H, t), 2.08 (2H, m); 13C-NMR (in DMSO) δ 166.28, 146.07, 139.26, 119.48, 108.96, 61.25, 28.36; EA Th: C, 53.69%; H, 4.24%, found: C, 53.78%; H, 4.48%.
Butane-1,4-diyl bis(3,4,5-trihydroxybenzoate) (24), white solid, yield 64%, MP. 224 °C (decomp.), 1H-NMR (in DMSO) δ 6.95 (4H, s), 4.22 (4H, bd t), 1.78 (4H, bd t); 13C-NMR (in DMSO) δ 166.29, 146.04, 138.92, 119.87, 108.94, 64.08, 25.61; EA Th: C, 54.83%; H, 4.60%, found: C, 54.74%; H, 4.87%.
3-((3,4-Dihydroxybenzoyl)oxy)propyl 3,4,5-trihydroxybenzoate (25), white solid, yield 95%, MP. 236–245 °C, 1H-NMR (in DMSO) δ 9.78 (1H, s), 9.36 (1H, s), 9.26 (2H, s), 8.94 (1H, s), 7.37 (1H, d), 7.32 (1H, dd), 6.90 (2H, s), 6.80 (2H, d), 4.31 (4H, q), 2.09 (2H, m); 13C-NMR (in DMSO) δ 166.25, 166.11, 150.94, 146.04, 145.54, 138.95, 122.31, 120.96, 119.76, 116.73, 115.78, 108.99, 61.36, 28.35; EA Th: C, 45.33%; H, 5.82%; N, 6.22%, found: C, 45.50%; H, 5.84%; N, 6.55%.
3-((3,5-Dihydroxybenzoyl)oxy)propyl 3,4,5-trihydroxybenzoate (26) (1/2H2O), white solid, yield 72%, MP. 221 °C (decomp), 1H-NMR (in DMSO) δ 7.97 (2H, d), 7.65 (1H, t), 7.51 (2H, t), 6.96 (2H, s), 4.40 (2H, t), 4.33 (2H, t), 2.14 (2H, m); 13C-NMR (in DMSO) δ 166.26, 166.20, 146.04, 138.96, 133.81, 130.11, 129.63, 120.96, 129.20, 119.77, 109.00, 62.27, 61.48, 28.26; EA Th: C, 54.64%; H, 4.55%, found: C, 54.78%; H, 4.54%.
3-(Benzoyloxy)propyl-3,4,5-trihydroxybenzoate (27), white solid, yield 84% (after DCVC cyclohexane/THF, 4/1 to 3/2), MP. 127–140 °C, 1H-NMR (in DMSO) δ 9.63–9.25 (5H, m), 6.96 (2H, s), 6.84 (2H, s), 6.44 (1H, s), 4.32 (4H, dt), 2.10 (2H, m); 13C-NMR (in DMSO) δ 166.25, 166.19, 146.05, 138.97, 131.79, 119.73, 108.99, 107.58, 61.76, 61.19, 28.25; EA Th: C, 61.45%; H, 4.85%, found: C, 61.20%; H, 5.20%.
Propane-1,3-diyl dibenzoate (28), white solid, yield 11%, MP. 60 °C, 1H-NMR (in CDCl3) δ 8.04 (4H, dd), 7.56 (2H, t), 7.43 (4H, t), 4.51 (4H, t), 2.28 (2H, m); 13C-NMR (in CDCl3) δ 166.54, 133.02, 130.09, 129.61, 128.39, 61.79, 28.28; EA Th: C, 71.82%; H, 5.67%, found: C, 71.81%; H, 5.87%.
3-(3,4,5-Trihydroxybenzamido)propyl 3,4,5-trihydroxybenzoate (29), white solid, yield 90%, MP. 250 °C (decomp.), 1H-NMR (in DMSO) δ 8.14 (1H, t), 6.98 (2H, s), 6.82 (2H, s), 4.19 (2H, t), 3.60 (2H, t), 3.32 (2H, q), 1.88 (2H, m); 13C-NMR (in DMSO) δ 166.94, 166.35, 146.02, 145.85, 138.90, 136.91, 125.38, 119.96, 108.98, 107.15, 62.44, 36.50, 29.09; EA Th: C, 53.83%; H, 4.52%; N, 3.69%, found: C, 53.93%; H, 4.84%; N, 3.87%.
N,N′-(Propane-1,3-diyl)bis(3,4,5-trihydroxybenzamide) (4H2O) (30), white solid, yield 98% (after crystallisation in acidified water), MP. 110.7–113 °C, 1H-NMR (in DMSO) δ 8.09 (2H, t), 6.83 (2H, s), 3.22 (4H, q), 1.66 (2H, m); 13C-NMR (in DMSO) δ 166.89, 145.88, 136.60, 125.42, 107.08, 37.09, 29.95; EA Th: C, 45.33%; H, 5.82%; N, 6.22%, found: C, 45.50%; H, 5.84%; N, 6.55%.
3-(3,4,5-Trihydroxybenzamido)phenyl 3,4,5-trihydroxybenzoate (31), lightly brown solid, yield 44%, MP. 235 °C (decomp.), 1H-NMR (in DMSO) δ 8.53 (1H, s), 7.44–7.25 (34H, m), 5.13 (4H, s), 5.08 (4H, s), 5.00 (2H, s), 4.94 (2H, s), 4.33 (2H, t), 3.46 (2H, q), 1.99 (2H, m); 13C-NMR (in DMSO) δ 165.96, 165.70, 152.52, 152.37, 141.72, 139.94, 137.95, 137.77, 137.28, 137.19, 130.25, 128.88, 128.59, 128.55, 128.51, 128.39, 128.35, 128.27, 128.10, 125.45, 108.58, 106.81, 74.69, 70.70, 63.64, 37.05, 28.82; EA Th: C, 78.05%; H, 5.39%; N, 1.47%, found: C, 77.89%; H, 5.51%; N, 1.72%.
4-(3,4,5-Trihydroxybenzamido)phenyl 3,4,5-trihydroxybenzoate (32), white solid, yield 50%, MP. 233 °C (decomp.); 1H-NMR (in DMSO) δ 9.98 (1H, s), 9.39 (2H, s), 9.15 (2 h, s), 9.12 (1H, s), 8.80 (1H, s), 7.79 (2H, d), 7.15 (2H, d), 7.10 (2H, s), 6.96 (2H, s); 13C-NMR (in DMSO) δ 166.01, 165.26, 146.61, 146.22, 145.97, 139.66, 137.58, 125.36, 122.32, 121.47, 118.41, 109.54, 107.66; EA Th: C, 58.12%; H, 3.66%; N, 3.39%, found: C, 58.43%; H, 3.9%; N, 3.26%.
Propane-1,3-diyl bis(3,4-dihydroxybenzoate) (33), white solid, yield 59%, MP. 222 °C, 1H-NMR (in DMSO) δ 7.37 (2H, d), 7.32 (2H, dd), 6.78 (2H, d), 4.31 (4H, t), 2.10 (2H, m); 13C-NMR (in DMSO) δ 166.11, 150.97, 145.55, 122.31, 120.95, 116.72, 115.76, 61.50, 28.35; EA Th: C, 58.62%; H, 4.63%, found: C, 58.78%; H, 4.79%.
3-((3,5-Dihydroxy-4-(2-hydroxyethoxy)benzoyl)oxy)propyl 3,4,5-trihydroxybenzoate (34), white solid, yield 20%, MP. 173.3–184.2 °C, 1H-NMR (in MeOD) δ 7.05 (4H, d), 4.40 (4H, dt), 4.13 (2H, t), 3.81 (2H, t), 2.19 (2H, m); 13C-NMR (in MeOD) δ 166.99, 166.46, 150.52, 145.12, 125.48, 119.99, 108.74, 108.64, 74.31, 61.41, 61.03, 60.69, 28.01; EA Th: C, 53.78%; H, 4.78%, found: C, 53.63%; H, 4.87%.
In vitro antiplasmodial assay
The Plasmodium falciparum 3D7 strain was obtained through BEI Resources, NIAID, NIH: Plasmodium falciparum, strain 3D7, MRA-102, contributed by Daniel J. Carucci. This chloroquine-sensitive strain (initially isolated in the Netherlands) was cultured in vitro thanks to a modified method from Trager and Jensen.63,64 In addition, the resistant strains W2 (multi-resistant) and FcB1 (chloroquine-resistant) were obtained from Prof. Grellier (Museum d'Histoire Naturelle, Paris).65 Consequently, the intraerythrocytic forms of this parasite were grown in a completed RPMI medium at 37 °C. Thus, the culture medium was supplemented with glucose (Sigma-Aldrich®, Belgium), hypoxanthine (Sigma-Aldrich®, Belgium), gentamycin (Gibco, Fisher Scientific®, U.K.), and 10% human pooled serum (A+/O+), as previously described.54,66The tested compounds were first dissolved in dimethylsulfoxide to reach 5–10 mg mL−1. But the maximum concentration for this solvent in the final test solutions was 1%. The samples were diluted with supplemented RPMI.
All the products were evaluated with 8 two-fold dilutions in a 96-well plate for three consecutive assays. The parasitic growth was compared to parasitized red blood cells (100% growth) and free RBC (0%) thanks to Plasmodium lactate dehydrogenase activity.67 Moreover, two positive controls (artemisinin and quinine) were employed to confirm the validity of the assays. The UV absorbance was read on a FlexStation® 3 Benchtop Multi-Mode Microplate Reader at 650 nm.
Hemolysis assay
Hemolytic potential was evaluated based on a previously reported procedure.68 Consequently, a 10% red blood cell suspension in PBS (v/v) (A+ or O+) was incubated with the compounds in duplicate. The primary solutions at 10 mg mL−1 in DMSO were diluted in PBS to reach 100 μg mL−1 as the final concentration (DMSO < 1%), similar to the doses tested for the antiplasmodial effect. After stirring at room temperature for 1 h, the mixtures were centrifuged for 5 min at 2000 rpm, and 150 μL of supernatant was transferred to a 96-well plate.The absorbance was evaluated at 550 nm with a microplate reader (FlexStation® 3 Benchtop Multi-Mode Microplate Reader, OD550). The positive control was Triton X-100 1% (v/v) (corresponding to 100% lysis) and PBS as the negative control (corresponding to 0% lysis). The percentage of red blood cell lysis (H) was calculated with the following equation H = (OD550 sample − OD550 PBS)/(OD550 Triton − OD550 PBS) × 100.54
In vitro cytotoxicity assay
The test compounds were evaluated on human umbilical vein endothelial cells (HUVEC), furnished by Lonza®. These cells were cultured in vitro in EBM-2® medium, supplemented with FBS, hydrocortisone, hFGF-B, VEGF, R3-IGF-1, ascorbic acid, hEGF, GA-1000 and heparin, known as EGM-2 Single-Quots® from Lonza®. The cells were passed on a 3-days basis but were maintained until maximum 10 passages.Samples were prepared at 10 mg mL−1 in DMSO, with a maximal test concentration of 0.5%. The samples were first diluted in EBM-2® medium. For the assay, 2.5 × 104 cells (100 μL) were seeded in a 96-well plate and incubated for 24 h at 37 °C. After that, 50 μL of test solution were poured in triplicate (six 2-fold concentrations).
The cells were incubated with the products for 48 h. Finally, the supernatant was replaced by 10-fold diluted Presto Blue® (75 μL) and left for 2–3 h at 37 °C. The plate was read in fluorescence mode (UV SpectraMax I3® at 560–590 nm). The cellular growth was compared between treated and untreated cells. Each compound was tested in 3 consecutive tests (n = 3).
For evaluation of cell toxicity on confluent cells, the plate was seeded 3 days before the treatment with the test compounds. The revelation method was similar to the previously described method.
Maximal water solubility (Cmax)
This parameter was measured to quantify the hydrosolubility of our antiplasmodial compounds. Thus, the shake-flask method in MilliQ water was employed in combination with a UV spectrophotometer.30 Therefore, the maximal wavelength was first determined thanks to a UV-scan (400–190 nm) in a 1 cm quartz cuvette. Secondly, a calibration curve was calculated by means of 3 dilutions of a standard solution prepared by dissolving an accurately weighted quantity of the products in MilliQ water or MeOH/H2O (50/50, v/v). Linearity was evaluated by linear regression analysis (R2 > 0.95).Finally, saturated solutions were prepared by overnight stirring of an excess of the product in MilliQ water at RT (around 25 °C). The solution was filtered on a 0.45 μM filter (Chromafil® XTra PVDF-45/25, Filter Service®) to remove the non-dissolved part and diluted with MilliQ water or MeOH/H2O (50/50, v/v). The absorbance was measured at the selected wavelength. This experiment was performed for 3 consecutive days with freshly prepared saturated solutions. Then, the maximal water concentration was calculated owing to the equation retrieved from the calibration curve.
Zebrafish embryo toxicity
Adult Danio rerio were maintained in GIGA Zebrafish Platform based on the ethical criteria of the Ethical Committee for the Use of Laboratory Animals at the University of Liège. Thus, they are bred at 28 °C on a 14 h day per 10 h night period. The fertilized eggs were collected at day 1, washed with sterile water, and placed in Petri® dishes for 24 h incubation at 28 °C. At day 2, synchronized larvae, without chorion removal, were selected and distributed in 12-well plates (20 embryos per conditions, in duplicate). Compounds were first dissolved in DMSO at 100 mg mL−1 before dilution in culture medium to reach less than 0.1% of DMSO at the highest test concentration (100, 50, 25 and 10 μg mL−1), similar to the antiplasmodial assay. The medium was replaced once daily for 3 days. The general aspect, hatching and mortality rate of the embryos were observed until 96 hpf. 30 embryos were used as control in sterile water with 0.1% DMSO. Zebrafish LC50 was determined based on the cumulative mortality after 72 h of exposure.54If necessary, the chorionic membrane could be removed at D2 before the first treatment. Thus, the eggs were placed for a few minutes in a diluted solution of pronase enzyme. Then, synchronized larvae were selected.54
In vivo acute toxicity test
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Liège University and approved by the Animal Ethics Committee of the University of Liège (no. 16-1873). The maximal tolerated dose during the in vivo antiplasmodial assay was determined thanks to a previously described protocol.17 Two mice per administration pathway (i.p. or p.o.) were treated with 4 increasing doses in 6 hours (every 2 hours). Samples were prepared with accurately weighted products dissolved in Tween 80/EtOH/physiological saline (7/3/90). Thus, the maximal dose was determined as 50% of the cumulative dose at which toxicity (health or behavioural problems) was observed.In vivo antiplasmodial activity assay
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Liège University and approved by the Animal Ethics Committee of the University of Liège (no. 16-1873) and was designed according to internationally recognized guidelines.17 Female Swiss CD1 mice (10 weeks old, 25 ± 2 g) were obtained from Janvier Laboratories and randomly distributed in six mice groups. After 2 weeks of acclimation, they were infested by the murine parasite Plasmodium berghei NK173, following the protocol previously described.41,54,64 Therefore, parasitized red blood cells (5%) were intraperitoneally injected into each mouse at D1. The compounds were such evaluated thanks to Peters' 4 day suppressive test as internationally recommended.69Thus, a treatment dose (50 mg kg−1 i.p. or p.o., dissolved in 7% Tween 80 and 3% EtOH in physiological saline solution) was given 2–3 h after infection (D1) and repeated on a daily base for 3 days. Then, the parasitaemia was evaluated by microscopy counting (at least 500 erythrocytes) using thin blood smears made from mouse-tail blood and stained with Giemsa. Two blood smears were performed at D4 (2 h post treatment) and D7. The vehicle solution was used as a negative control in each administration ways. On the other hand, chloroquine was a positive control with 4 mg kg−1 i.p. and 25 mg kg−1per os with the same solvents. In addition, one group remains untreated. The percentage of inhibition of Plasmodium growth was calculated by comparison of the parasitaemia between the treated groups and the negative controls.
Conclusions
Due to the growing number of resistance to recommended drugs, the antimalarial therapeutic arsenal urgently needs new alternatives.8,11,70 Based on our previous investigations, dimers of polyhydroxybenzoic acids were investigated as new potent antiplasmodial substances, inspired by the ellagic acid (9) scaffold.These dimeric products exhibited an antiplasmodial activity at similar concentrations to 9 (IC50 = 2.8 μM) and the main scaffold could tolerate great structural variations without loss of potency. However, the presence of phenolic functions on both sides of the dimer seemed mandatory, and the introduction of aromatic lipophilic substituents seemed to slightly increase the efficacy.
In addition, most of the compounds have never displayed any signs of toxicity in the selected models. This selectivity for Plasmodium was further confirmed in vivo on zebrafish embryos and all the tested products displayed a ED50 > 100 μg mL−1.
However, a great variation of water solubility was observed for the dimers, with a maximum concentration ranging from 7 μM to 2.7 mM. Therefore, some compounds were up to 100-fold more soluble than ellagic acid (Cmax = 18 μM) under similar conditions, and the nature of the linker seemed to be the major structural component.
As a result, ellagic acid and two dimers (23 and 30) were tested on a murine model of malaria (P. berghei). Thus, a 50 mg kg−1 d−1 dose was orally administrated or by means of intraperitoneal injection. Contrary to 9 and 23, which were inactive per os and displaying a 30% transitory efficacy by the i.p. route, 30 induced a significant parasitaemia reduction in both administration routes (>50%, p < 0.0005).
In conclusion, even if this dimeric scaffold could not be considered as a new antimalarial lead because of its limited in vivo efficacy, it gives insightful structural information to design new candidates. Moreover, further experiments will be necessary to study it more deeply. In fact, its exact mode of action is not clearly defined, and it is necessary to improve the antimalarial activity before clinical experiments.
Author contributions
DG: investigation; formal analysis; writing – original draft; funding acquisition. PH: supervision; investigation. DCC: investigation. ER: supervision; data curation; formal analysis; writing – review & editing. PB: supervision; resources; writing – review & editing. FM: resources, supervision; methodology; project administration; writing – review & editing. FP: resources, supervision; methodology; project administration; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to FRS-FNRS for the financial support through a FRIA PhD Grant (FC23283), and Foundation Léon Frédéricq. The authors would also thank all the persons who have contributed to this work, especially Pauline Desdemoustier & Dr Erik Maquoi for their expertise in cell culture, Counerotte Stéphane for the elemental analysis, and Luc Duwez and Laura Senny for their help during the mice experiments.Notes and references
- World Health Organisation, Distribution of Causes of Death Among Children aged < 5 years, https://www.who.int/data/gho/data/indicators/indicator-details/GHO/distribution-of-causes-of-death-among-children-aged-5-years-(-), (accessed 8 April 2021) Search PubMed.
- World Health Organization, World malaria report 2021, 2021 Search PubMed.
- Center for Disease Control, Anopheles Mosquitoes, https://www.cdc.gov/malaria/about/biology/index.html#tabs-1-5, (accessed 27 January 2021).
- Center for Disease Control, Biology, https://www.cdc.gov/malaria/about/biology/index.html, (accessed 27 January 2021) Search PubMed.
- World Health Organization, Malaria, https://www.who.int/news-room/fact-sheets/detail/malaria, (accessed 8 April 2021) Search PubMed.
- Center for Disease Control, Disease, https://www.cdc.gov/malaria/about/disease.html, (accessed 27 January 2021) Search PubMed.
- World Health Organisation, Malaria, https://www.who.int/news-room/fact-sheets/detail/malaria, (accessed 10 November 2020) Search PubMed.
- World Health Organization, World Malaria Report 2020, 2020 Search PubMed.
- World Health Organization, World Malaria Report 2022, https://apps.who.int/iris/rest/bitstreams/1484818/retrieve, (accessed 20 January 2023) Search PubMed.
- World Health Organization, Antibiotic resistance, https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance, (accessed 10 November 2020).
- A. Uwimana, E. Legrand, B. H. Stokes, J. L. M. Ndikumana, M. Warsame, N. Umulisa, D. Ngamije, T. Munyaneza, J. B. Mazarati, K. Munguti, P. Campagne, A. Criscuolo, F. Ariey, M. Murindahabi, P. Ringwald, D. A. Fidock, A. Mbituyumuremyi and D. Menard, Nat. Med., 2020, 26, 1602–1608 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- G. Degotte, B. Pirotte, P. Francotte and M. Frédérich, Curr. Med. Chem., 2021, 28(1) DOI:10.2174/0929867328666210329112354.
- L. Mamede, A. Ledoux, O. Jansen and M. Frédérich, Planta Med., 2020, 86, 585–618 CrossRef CAS PubMed.
- N. Tajuddeen and F. R. Van Heerden, Malar. J., 2019, 18, 1–62 CrossRef PubMed.
- G. Degotte, B. Pirotte, M. Frédérich and P. Francotte, Arch. Pharm., 2021, 354, 2100190 CrossRef CAS PubMed.
- D. A. Fidock, P. J. Rosenthal, S. L. Croft, R. Brun and S. Nwaka, Nat. Rev. Drug Discovery, 2004, 3, 509–520 CrossRef CAS PubMed.
- A. Gupta, R. Kumar, R. Ganguly, A. K. Singh, H. K. Rana and A. K. Pandey, Toxicol. Rep., 2021, 8, 44–52 CrossRef CAS PubMed.
- C. Chao, M. Mong, K. Chan and M. Yin, Mol. Nutr. Food Res., 2010, 54(3), 388–395 CrossRef CAS PubMed.
- M. Ghadimi, F. Foroughi, S. Hashemipour, M. Rashidi Nooshabadi, M. H. Ahmadi, B. Ahadi Nezhad and H. Khadem Haghighian, Phytother. Res., 2021, 35, 1023–1032 CrossRef CAS PubMed.
- S. Acquadro, A. Civra, C. Cagliero, A. Marengo, M. Rittà, R. Francese, C. Sanna, C. Bertea, B. Sgorbini, D. Lembo, M. Donalisio and P. Rubiolo, Planta Med., 2020, 86, 1363–1374 CrossRef CAS PubMed.
- P. Li, R. Du, Z. Chen, Y. Wang, P. Zhan, X. Liu, D. Kang, Z. Chen, X. Zhao, L. Wang, L. Rong and Q. Cui, J. Med. Virol., 2020, 1–8 Search PubMed.
- P. N. Soh, B. Witkowski, D. Olagnier, M. L. Nicolau, M. C. Garcia-Alvarez, A. Berry and F. Benoit-Vical, Antimicrob. Agents Chemother., 2009, 53, 1100–1106 CrossRef CAS PubMed.
- R. Muganga, L. Angenot, M. Tits and M. Frederich, Planta Med., 2014, 80, 482–489 CrossRef CAS PubMed.
- D. Ndjonka, B. Bergmann, C. Agyare, F. M. Zimbres, K. Lüersen, A. Hensel, C. Wrenger and E. Liebau, Parasitol. Res., 2012, 111, 827–834 CrossRef PubMed.
- J. T. Banzouzi, R. Prado, H. Menan, A. Valentin, C. Roumestan, M. Mallie, Y. Pelissier and Y. Blache, J. Ethnopharmacol., 2002, 81, 399–401 CrossRef PubMed.
- A. W. R. Hamad, W. M. Al-Momani, S. Janakat and S. A. Oran, Pak. J. Nutr., 2009, 8, 1661–1664 CrossRef CAS.
- J. M. Landete, Food Res. Int., 2011, 44, 1150–1160 CrossRef CAS.
- V. Murugan, K. Mukherjee, K. Maiti and P. K. Mukherjee, J. Agric. Food Chem., 2009, 57, 4559–4565 CrossRef CAS PubMed.
- I. Bala, V. Bhardwaj, S. Hariharan and M. N. V. R. Kumar, J. Pharm. Biomed. Anal., 2006, 40, 206–210 CrossRef CAS PubMed.
- C. G. Wermuth, D. Aldous, P. Raboisson and D. Rognan, The Practice of Medicinal Chemistry, Academic Press, London, 4th edn, 2015 Search PubMed.
- R. García-Villalba, D. Beltrán, M. D. Frutos, M. V. Selma, J. C. Espín and F. A. Tomás-Barberán, Food Funct., 2020, 11, 7012–7022 RSC.
- M. V. Selma, D. Beltrán, R. García-Villalba, J. C. Espín and F. A. Tomás-Barberán, Food Funct., 2014, 5, 1779–1784 RSC.
- T. Hirokane, Y. Hirata, T. Ishimoto, K. Nishii and H. Yamada, Nat. Commun., 2014, 5, 2017–2019 Search PubMed.
- T. Gokcen, I. Gulcin, T. Ozturk and A. C. Goren, J. Enzyme Inhib. Med. Chem., 2016, 31, 180–188 CrossRef CAS PubMed.
- A. J. Pearson and P. R. Bruhn, J. Org. Chem., 1991, 56, 7092–7097 CrossRef CAS.
- H. Yamada, K. Nagao, K. Dokei, Y. Kasai and N. Michihata, J. Am. Chem. Soc., 2008, 130, 7566–7567 CrossRef CAS PubMed.
- I. Nyamba, A. Lechanteur, R. Semdé and B. Evrard, Eur. J. Pharm. Biopharm., 2021, 159, 198–210 CrossRef CAS PubMed.
- G. L. Amidon, H. Lennernäs, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS PubMed.
- C. Liang, J. Q. Qiao and H. Z. Lian, J. Chromatogr. A, 2017, 1528, 25–34 CrossRef CAS PubMed.
- S. G. Alson, O. Jansen, E. Cieckiewicz, H. Rakotoarimanana, H. Rafatro, G. Degotte, P. Francotte and M. Frederich, J. Pharm. Pharmacol., 2018, 70, 1349–1356 CrossRef CAS PubMed.
- R. P. Verma and C. Hansch, ChemBioChem, 2004, 5, 1188–1195 CrossRef CAS PubMed.
- X. W. Wu, W. Wei, X. W. Yang, Y. B. Zhang, W. Xu, Y. F. Yang, G. Y. Zhong, H. N. Liu and S. L. Yang, Molecules, 2017, 22, 2–11 Search PubMed.
- N. A. Al Zahrani, R. M. El-Shishtawy and A. M. Asiri, Eur. J. Med. Chem., 2020, 204, 112609 CrossRef CAS PubMed.
- S. L. Rawe and C. McDonnell, in Antimalarial Agents, Elsevier, 2020, pp. 65–98 Search PubMed.
- A. Whitty, Future Med. Chem., 2011, 3, 797–801 CrossRef CAS PubMed.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS PubMed.
- A. Arsianti, H. Astuti, F. Fadilah, D. Martin Simadibrata, Z. Marie Adyasa, D. Amartya, A. Bahtiar, H. Tanimoto and K. Kakiuchi, Orient. J. Chem., 2018, 34, 655–662 CrossRef CAS.
- Fish Acute Toxicity Test, in OECD Guidelines for the Testing of Chemicals, ed. OECD Publishing, OECD, Paris, 2018 Search PubMed.
- J. F. De La Paz, N. Beiza, S. Paredes-Zúñiga, M. S. Hoare and M. L. Allende, Int. J. Mol. Sci., 2017, 18(4), 710 CrossRef PubMed.
- X. Zhu, S. Tian and Z. Cai, PLoS One, 2012, 7, 1–6 Search PubMed.
- J. Li, Y. Zhang, K. Liu, Q. He, C. Sun, J. Han, L. Han and Q. Tian, Front. Pharmacol., 2018, 9, 1–12 CrossRef PubMed.
- A. Jaja-Chimedza, M. Gantar, P. D. L. Gibbs, M. C. Schmale and J. P. Berry, Mar. Drugs, 2012, 10, 2322–2336 CrossRef CAS PubMed.
- A. Ledoux, A. St-Gelais, E. Cieckiewicz, O. Jansen, A. Bordignon, B. Illien, N. Di Giovanni, A. Marvilliers, F. Hoareau, H. Pendeville, J. Quetin-Leclercq and M. Frédérich, J. Nat. Prod., 2017, 80, 1750–1757 CrossRef CAS PubMed.
- M. Tasaki, T. Umemura, M. Maeda, Y. Ishii, T. Okamura, T. Inoue, Y. Kuroiwa, M. Hirose and A. Nishikawa, Food Chem. Toxicol., 2008, 46, 1119–1124 CrossRef CAS PubMed.
- R. Carter, Parasitology, 1973, 66, 297–307 CrossRef CAS PubMed.
- S. Déchamps, M. Maynadier, S. Wein, L. Gannoun-Zaki, E. Maréchal and H. J. Vial, J. Lipid Res., 2010, 51, 81–96 CrossRef PubMed.
- C. Beaufay, A. Ledoux, O. Jansen, A. Bordignon, S. Zhao, C. N. Teijaro, R. B. Andrade, J. Quetin-Leclercq and M. Frédérich, Planta Med., 2018, 84, 881–885 CrossRef CAS PubMed.
- T.-H. Lee, J.-L. Chiou, C.-K. Lee and Y.-H. Kuo, J. Chin. Chem. Soc., 2005, 52, 833–841 CrossRef CAS.
- A. Mahendran, A. Vuong, D. Aebisher, Y. Gong, R. Bittman, G. Arthur, A. Kawamura and A. Greer, J. Org. Chem., 2010, 75, 5549–5557 CrossRef CAS PubMed.
- S. E. Denmark, C. S. Regens and T. Kobayashi, J. Am. Chem. Soc., 2007, 129, 2774–2776 CrossRef CAS PubMed.
- Y. Xia, Y. Jin, N. Kaur, Y. Choi and K. Lee, Eur. J. Med. Chem., 2011, 46, 2386–2396 CrossRef CAS PubMed.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CrossRef CAS PubMed.
- M. Frédérich, M. J. Jacquier, P. Thépenier, P. De Mol, M. Tits, G. Philippe, C. Delaude, L. Angenot and M. Zèches-Hanrot, J. Nat. Prod., 2002, 65, 1381–1386 CrossRef PubMed.
- F. L. Henriquez and D. R. A. M. Williams, Knowing one's enemy: the Plasmodium parasite, Elsevier Ltd, 2020 Search PubMed.
- J. Bero, M. Frédérich and J. Quetin-Leclercq, J. Pharm. Pharmacol., 2009, 61, 1401–1433 CrossRef CAS PubMed.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 205–210 CrossRef CAS PubMed.
- O. Jansen, M. Tits, L. Angenot, J. P. Nicolas, P. De Mol, J. B. Nikiema and M. Frédérich, Malar. J., 2012, 11, 289 CrossRef PubMed.
- W. Peters, J. H. Portus and B. L. Robinson, Ann. Trop. Med. Parasitol., 1975, 69, 155–171 CrossRef CAS PubMed.
- Center for Disease Control, Drug Resistance in the Malaria-Endemic World, https://www.cdc.gov/malaria/malaria_worldwide/reduction/drug_resistance.html, (accessed 27 January 2021) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Biological activities for the synthesis intermediates. NMR and UV spectra of all the reported compounds. Results and method for the logP determination by isocratic HPLC. See DOI: https://doi.org/10.1039/d2md00392a |
| This journal is © The Royal Society of Chemistry 2023 |