
The war on hTG2: warhead optimization in small molecule human tissue transglutaminase inhibitors†
Lavleen
Mader
 a,
Sarah K. I.
Watt
a,
Harish R.
Iyer
b,
Linh
Nguyen
b,
Harpreet
Kaur
b and
Jeffrey W.
Keillor
*a
a,
Sarah K. I.
Watt
a,
Harish R.
Iyer
b,
Linh
Nguyen
b,
Harpreet
Kaur
b and
Jeffrey W.
Keillor
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada. E-mail: jkeillor@uottawa.ca
bDalriada Drug Discovery, Mississauga, Ontario L5N 8G4, Canada
First published on 25th November 2022
Abstract
Human tissue transglutaminase (hTG2) is a multifunctional enzyme with protein cross-linking and G-protein activity, both of which have been implicated in the progression of diseases such as fibrosis and cancer stem cell propagation when dysregulated, prompting the development of small molecule targeted covalent inhibitors (TCIs) possessing a crucial electrophilic ‘warhead’. In recent years there have been significant advances in the library of warheads available for the design of TCIs; however, the exploration of warhead functionality in hTG2 inhibitors has remained relatively stagnant. Herein, we describe a structure–activity relationship study entailing rational design and synthesis for systematic variation of the warhead on a previously reported small molecule inhibitor scaffold, and rigorous kinetic evaluation of inhibitory efficiency, selectivity, and pharmacokinetic stability. This study reveals a strong influence on the kinetic parameters kinact and KI with even subtle variation in warhead structure, suggesting that the warhead plays a significant role in not only reactivity, but also binding affinity, which consequently extends to isozyme selectivity. Warhead structure also influences in vivo stability, which we model by measuring intrinsic reactivity with glutathione, as well as stability in hepatocytes and in whole blood, giving insight into degradation pathways and relative therapeutic potential of different functional groups. This work provides fundamental structural and reactivity information highlighting the importance of strategic warhead design for the development of potent hTG2 inhibitors.
Introduction
Human tissue transglutaminase (hTG2) is one of eight enzymes comprising the transglutaminase (TGase) family that are characterized by their ability to cross-link proteins in the extracellular matrix. Using a catalytic cysteine residue, these enzymes form Nε(γ-glutaminyl)lysine bonds via an acyl transfer (transamidation) mechanism between acyl donor and acyl acceptor substrates. This transamidase site is also capable of isopeptidic cleavage and hydrolysis.1–4 Among this family, hTG2 is uniquely multifunctional, in that it can also bind and hydrolyze guanosine triphosphate (GTP) in its function as a G-protein in cell signalling pathways.1,5,6 These two biological activities are mutually exclusive as they are dependent on the enzyme's ability to adopt remarkably distinct conformations associated with each.6,7 In the extracellular environment, high Ca2+ concentrations favour a linear ‘open’ conformation, stabilized by the binding of these ions, in which the transamidase site is formed and the GTP binding site is distorted, through which G-protein function is lost. Intracellularly however, lower Ca2+ concentrations favour a compact ‘closed’ conformation in which the enzyme folds to expose a GTP binding site, while abolishing the transamidase site and cross-linking activity.6–9Dysregulation of both enzymatic functions has been implicated in the progression of a wide range of diseases such as fibrosis,10–12 cancer,13–15 and celiac disease,16,17 all while hTG2 knockout mice appear developmentally and reproductively viable.18 Thus, hTG2 serves as an attractive target for inhibition and has been extensively studied for decades with promising results in many therapeutic avenues, such as the recent progression of a peptidomimetic inhibitor to phase II clinical trials for the treatment of celiac disease.17
Considering the mutual exclusivity of the two binding sites of hTG2, we, and many others, have developed libraries of irreversible, targeted covalent inhibitors (TCIs) that selectively bind to the transamidase site of hTG2, locking the enzyme in an open conformation and thereby abolishing both G-protein and transamidase activity.19–22 These inhibitors vary in structure from small molecule to larger peptidomimetic scaffolds; however, a critical feature they all share is an electrophilic ‘warhead’ that forms a covalent bond with the catalytic cysteine residue. This warhead must be designed to fit in the enzyme's active site in the correct conformation, and be electrophilic enough to react with the cysteine, while also possessing enough stability to survive cellular conditions and prevent off-target reactions.23 Over the years we and others have investigated many different warheads for their suitability based on these criteria, including acrylamides, dihydroisoxazoles, thiadiazoles, and epoxides, but often on different scaffolds (Fig. 1), making it difficult to assess the influence of the warhead alone.24–30 In addition, many studies outside of our lab rely predominantly on IC50 values to assess inhibitor efficiency, leaving the distinct parameters of kinact and KI unaccounted for, along with a full characterization of the impact that a warhead may have on reactivity and binding affinity, respectively.
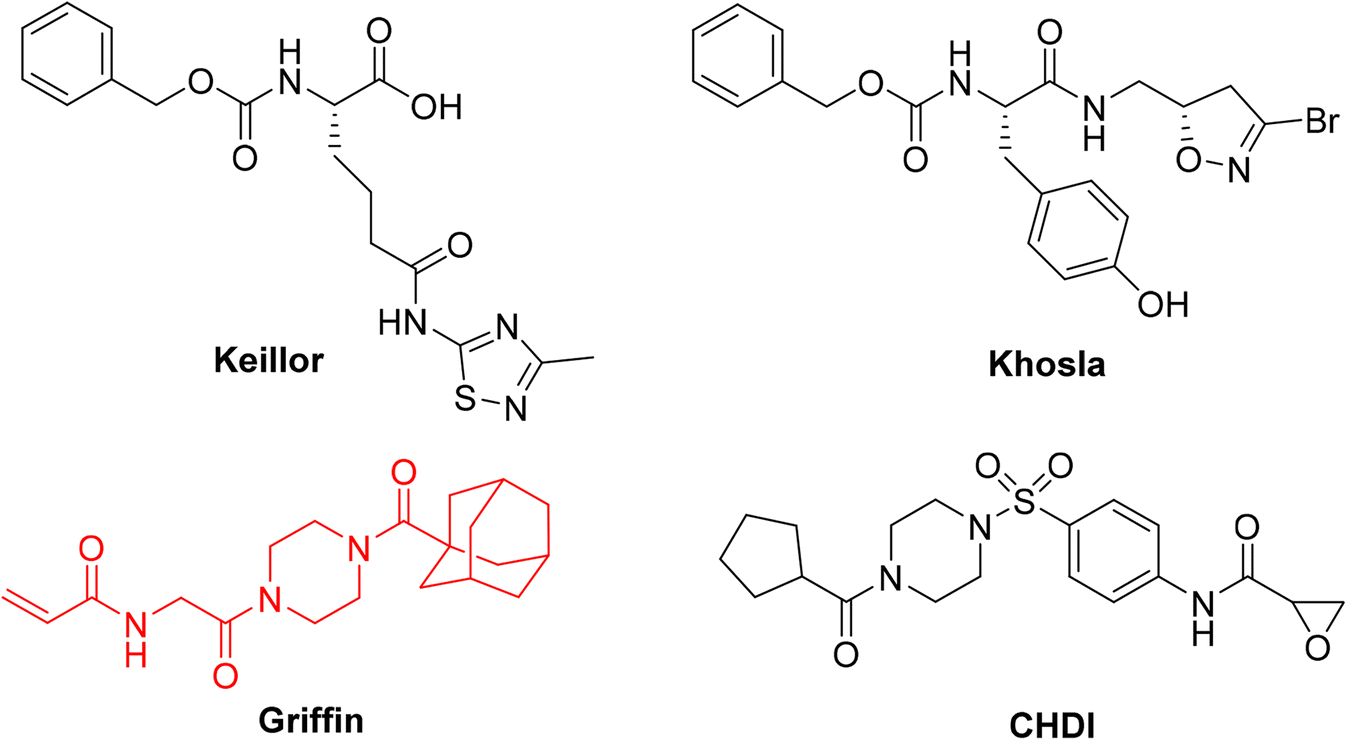 | ||
| Fig. 1 Representative small molecule hTG2 inhibitors with different warheads.25,28–30 The Ad-Pip-Gly scaffold used in this study is shown in red. | ||
So far, the acrylamide group appears to provide the best balance between reactivity and stability and is the most commonly used warhead for hTG2 inhibition.19 Since these initial studies, significant advances have been made in the development of warheads available for the design of TCIs.23,31,32 Recognizing the lack of a current and systematic structure–activity relationship study of warheads for hTG2 inhibition, in this work we used a previously reported, potent, adamantyl-piperazine-glycine (Ad-Pip-Gly) scaffold (Fig. 1),22,28 to incorporate and screen a variety of different warheads. These inhibitors were rigorously characterized with respect to their enzymatic efficiency, intrinsic reactivity, pharmacokinetic stability and isozyme selectivity, revealing interesting trends relevant to the design of inhibitors targeting hTG2, as well as other TGases.
Results and discussion
Design
For this type of systematic study in which the most reactive group of the inhibitor is varied, it was important to select a scaffold that is not only efficient for inhibition, but also synthetically accessible and enables late-stage diversification. Recently we confirmed that the Ad-Pip-Gly scaffold originally published by Badarau et al. confers excellent affinity and efficiency to inhibitors.22,28 This small molecule scaffold is also very convenient to synthesize as it allows the warhead to be incorporated in the very last step (Scheme 1), as opposed to other peptidomimetic scaffolds we have developed,19–21 making it ideal for this study.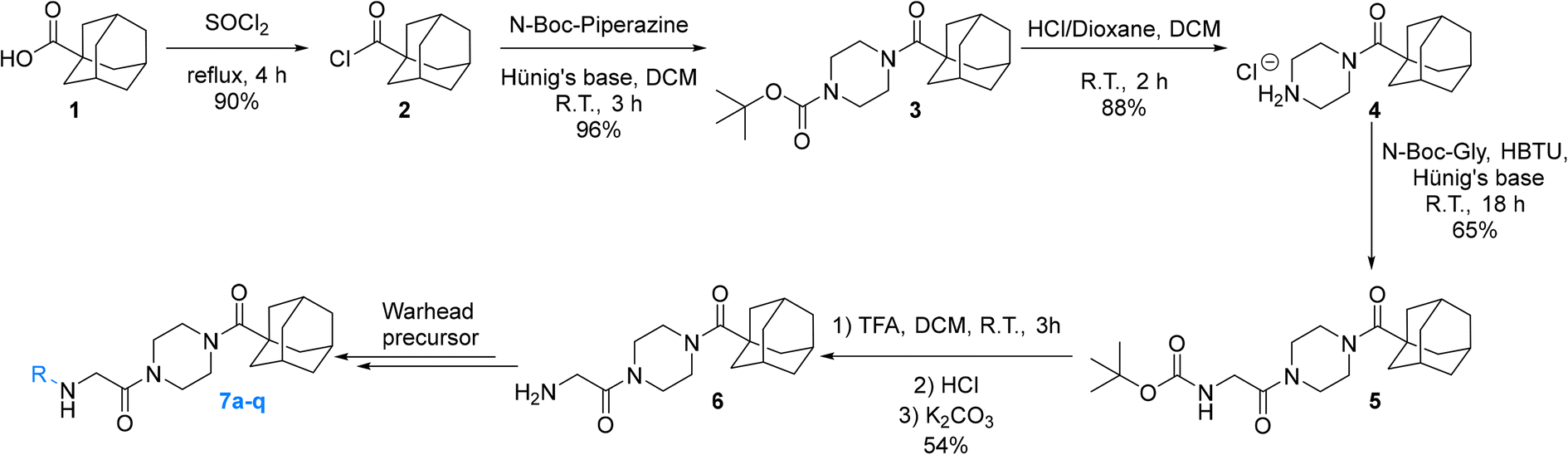 | ||
| Scheme 1 Synthesis of Ad-Pip-Gly scaffold enabling late-stage warhead diversification. | ||
Our subsequent selection of warheads (Scheme 2) was guided by a collection of works characterizing warheads for intrinsic reactivity and/or enzymatic inhibition, some of which have never been investigated for hTG2 inhibition in any scaffold that we know of. Results from previous hTG2 warhead studies,24–30 coupled with our molecular modelling of small molecule inhibitors,22 led us to believe that relatively small warheads were more suitable for inhibition as they could be better accommodated by the narrow ‘tunnel’ leading from the acyl-donor substrate binding site, down to the catalytic cysteine residue (Cys277) in the enzyme's active site (Fig. 2).7 Hence, we took size and electrophilicity into consideration to see how expected trends in sterics and electronics would correlate with inhibition parameters kinact and KI.
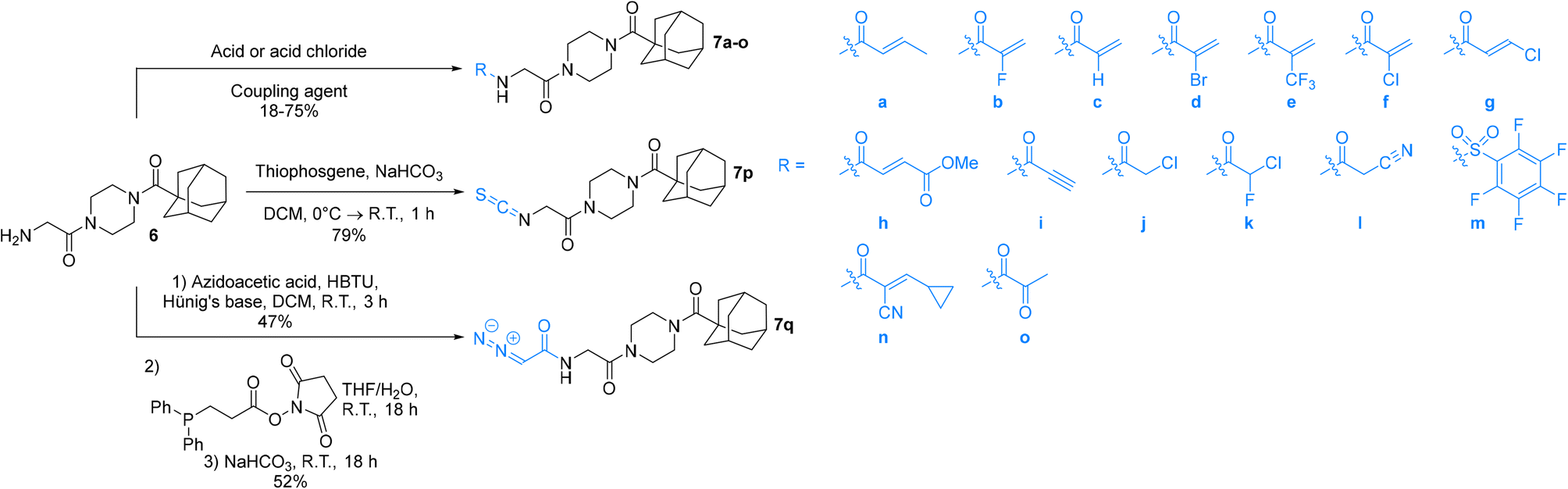 | ||
| Scheme 2 Synthesis of final inhibitors with varying warheads. | ||
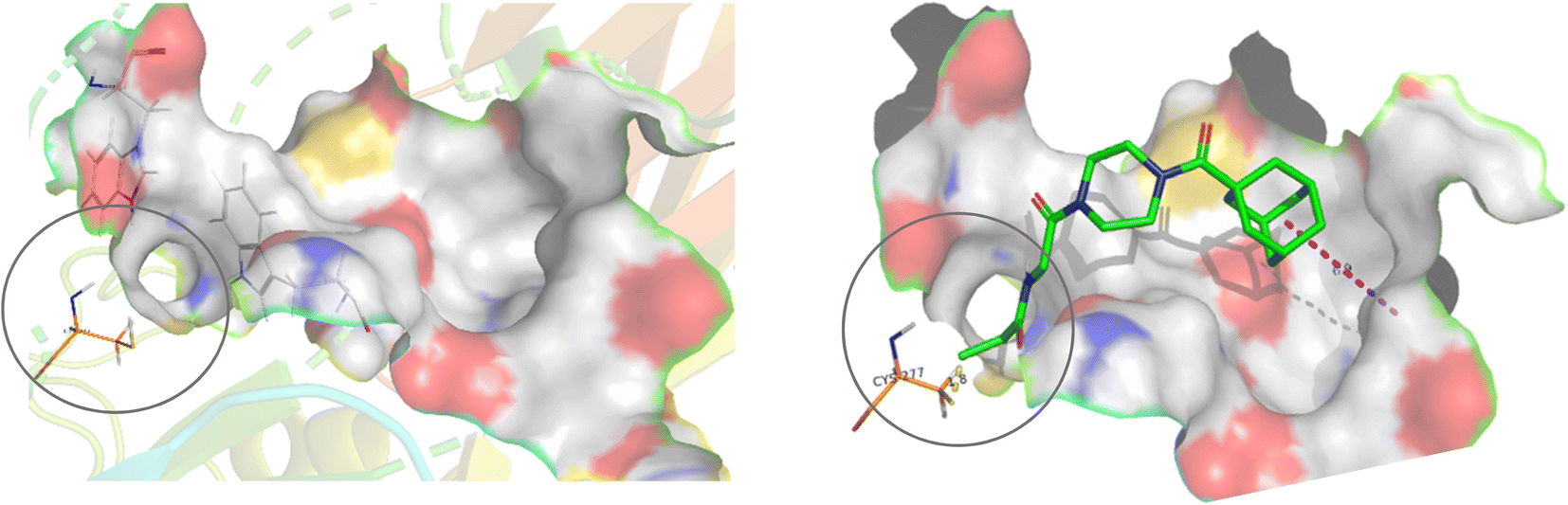 | ||
| Fig. 2 Active site of hTG2 (PDB: 2Q3Z) with narrow ‘tunnel’ leading to Cys277, shown with and without inhibitor. | ||
We have previously shown that current hTG2 crystal structures are not completely appropriate for predicting binding affinity for small molecule inhibitors.22 Our attempts at docking molecules with different warheads using our established methods did not reveal any significant trends or correlation to kinetic parameters, so we did not use molecular modelling to refine our choice of warheads, apart from considering the general structure of the active site as a guide.
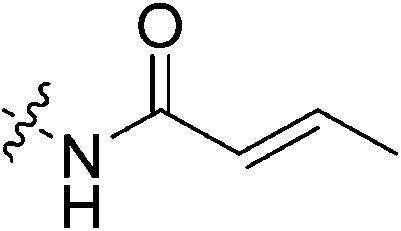
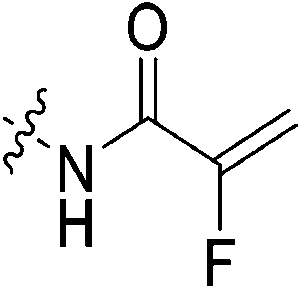
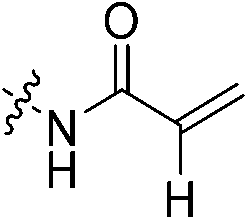
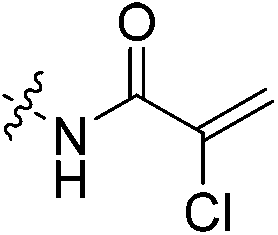
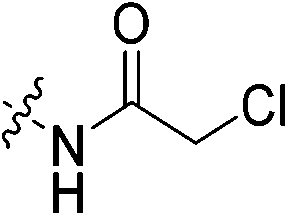
An obvious place to start was an acrylamide series with various substituents at the α and β positions (7a–h) to investigate the effect of increasing steric demand and/or electrophilicity on this commonly used warhead. Of particular interest were certain ‘mediator’ warheads that are known for their intrinsic stability,33 such as α-fluoroacrylamide (7b) or for their improved selectivity, such as sterically hindered fumarates (7h).34 We also included propiolamide (7i) as our previous studies suggest that alkyne-based inhibitors are quite potent for hTG2 inhibition.35,36 Chloroacetamide (7j) has been previously reported by Badarau et al.,28 but not characterized fully with respect to individual kinetic parameters. While chloroacetamides are excellent electrophiles,37 they are believed to be too reactive to survive cellular conditions, for this reason we investigated a less reactive counterpart, chlorofluoroacetamide (7k), which has been shown to be less electrophilic than chloroacetamides, possibly due to the strong electron-withdrawing effect of the fluorine inducing a larger partial positive charge on the α-carbon atom and thereby shortening/strengthening the C–Cl bond.38
We also studied some other warheads commonly employed in the design of TCIs, including cyanoacetamide (7l) and isothiocyanate (7p), the latter of which is present in an anticancer agent that can be used to inhibit TG2.23,39 Pentafluorobenzenesulfonamide (7m), bearing a warhead that reacts with thiolates by SNAr but has never been studied with TG2,40 and diazoacetamide (7q),41 an amide version of the 6-diazo-5-oxo-L-norleucine (DON) warhead that was used in inhibitor-hTG2 crystallization studies,7 were also studied herein.
Finally, this Ad-Pip-Gly scaffold, traditionally used for irreversible inhibitors, was also used to investigate potentially reversible covalent inhibition with α-cyanoacrylamide (7n) and α-ketoamide (7o), warheads known to react reversibly with thiolates,42,43 the latter of which has even previously been explored specifically for hTG2 inhibition.44
With this series of inhibitors, we aimed to rigorously investigate a broad range of warheads with potential to confer potency, while still exploring specific trends that would provide fundamental information, rather than screening a large number of warheads at random.
Synthesis
The synthesis of the Ad-Pip-Gly scaffold follows previously reported routes developed by our lab and Badarau et al. (Scheme 1).22,28 For this study it was necessary to stockpile pure 6 from which late-stage diversification takes place for maximum synthetic efficiency. It is noteworthy that the final deprotection step to produce amine precursor 6 works optimally with TFA followed by extraction of the free base, as reported by Badarau et al.,28 rather than with HCl/dioxane. Typically, we have observed much cleaner deprotection products with HCl/dioxane; however, in this specific case HCl/dioxane deprotection led to very poor yields due to the formation of unidentified side products. With TFA we were able to scale this reaction up to several grams, which proved to be necessary, since some of the warhead attachments were found to be difficult and gave poor yields, requiring larger amounts of starting material.Inhibitors 7a–o were synthesized by coupling amine 6 to either the carboxylic acid or the acid chloride of each warhead precursor (see Materials and methods) (Scheme 2). All of these precursors are commercially available; however, we opted to synthesize α-fluoroacrylic acid (I2) and α-cyano-β-cyclopropylacrylic acid (I3) by literature routes (see Materials and methods). Since the stereochemical configuration of I3 cannot be established by NMR, and is described rather ambiguously in previous methods,45,46 we confirmed the E-configuration of this compound by X-ray crystallography, as shown in the ESI.†
While performing these seemingly straightforward amide couplings for inhibitors 7a–o, we found that certain acrylic acids did not react well with certain amide coupling agents. More specifically, many of them were sensitive to pre-activation, gave low yields, or simply did not react at all. Hence the use of a diverse array of coupling agents, rather than one general synthetic method, was necessary for the different warheads. Ultimately, we did not focus on synthetic optimization and did not perform extensive screening on coupling conditions but did obtain acceptable yields for all reactions.
Inhibitor 7p was synthesized easily by reacting amine 6 with thiophosgene, while inhibitor 7q was obtained by first synthesizing the azidoacetamide (I5) by an amide coupling with azidoacetic acid (I4), followed by a ‘deimidogenation’ reaction using a phosphine reagent developed by Myers and Raines (Scheme 2).47
It is worth noting that originally, we wanted to synthesize less sterically hindered α-cyanoacrylamides from inhibitor 7l by a Knoevenagel condensation with either formaldehyde or acetaldehyde (Scheme 3); however, we were not able to successfully isolate these compounds under a variety of attempted conditions as they would immediately degrade upon contact with water during work up, suggesting they would not have been stable to subsequent assay conditions. These types of compounds are known to be very reactive,31 and it is possible that without a relatively bulky alkyl group at the β-position, they are too unstable to be used as viable warheads. However, the α-cyano-β-cyclopropylacrylamide warhead has been previously reported several times and was relatively easy to synthesize, so we decided to incorporate that instead.45,46,48
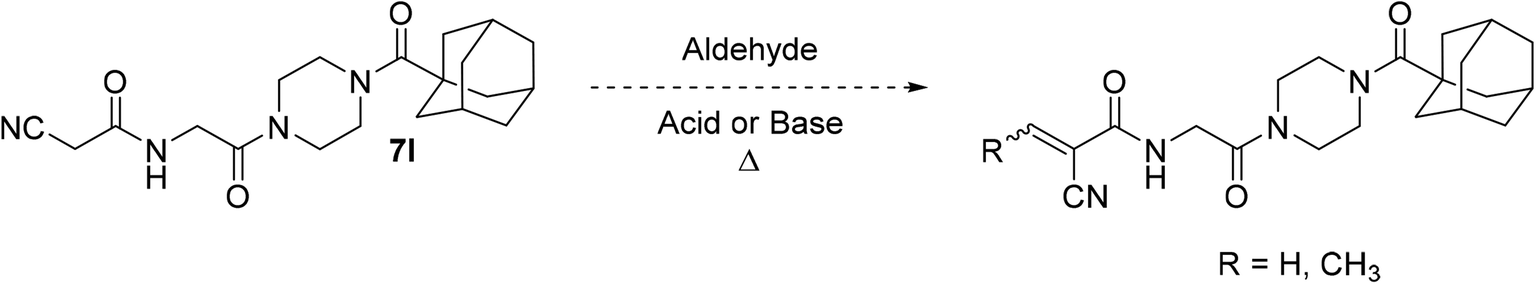 | ||
| Scheme 3 Attempted synthesis of some α-cyanoacrylamides. | ||
We also synthesized an inhibitor with an α-methylacrylamide warhead (see ESI,† compound 7r), in order to serve as a good comparison to its isostere α-chloroacrylamide (7f) and its isomer β-methylacrylamide (7a). However, we found that its solubility in polar solvents such as DMSO, DMF, ACN, and MeOH was surprisingly poor, and therefore this analogue could not be analyzed for purity or evaluated in our kinetic assay.
When necessary, inhibitors could be purified easily by flash chromatography generally up to >95% purity, typically using MeOH/EtOAc as the mobile phase for optimal separation, taking care to dry load the crude products as they had poor solubility in this solvent system. In some cases, further washes with water and/or pentane were necessary to remove residual by-products from coupling agents and contaminant grease. Overall, the synthesis of these inhibitors followed very standard conditions after some necessary troubleshooting, and the majority of issues were posed by acrylic acid precursors rather than the Ad-Pip-Gly scaffold.
Kinetic evaluation
All inhibitors were then kinetically evaluated for their inhibitory action against recombinant hTG2 using our previously developed continuous kinetic assay (Table 1).19,49 hTG2-mediated hydrolysis of substrate AL5 was analyzed by monitoring the release of chromogenic product p-nitrophenolate. Incubation with irreversible inhibitors resulted in time-dependent inactivation of the enzyme. Under Kitz and Wilson conditions,50 each inhibitor was tested at varying concentrations and the corresponding curves of absorbance versus time were fitted to a mono-exponential association equation to give observed first order rate constants of inhibition (kobs) (Fig. 3a). These rate constants were then plotted against inhibitor concentrations corrected for the presence of the competitive substrate, and then fitted to a hyperbolic equation representative of saturation kinetics, giving inhibition parameters kinact and KI (Fig. 3b).50–52 We also performed a simple linear regression of the low inhibitor concentration data points, from which the ratio of kinact/KI could be obtained directly with less relative error and compared to the ratio calculated using the individually obtained parameters.51| Compound | Warhead | k inact (min−1) | K I or Ki (μM)a | k inact/KI (×103 M−1 min−1) | k inact/KI (×103 M−1 min−1) (linear regression) |
|---|---|---|---|---|---|
| a K I for irreversible inhibitors and Ki for reversible inhibitors (7n and 7o). b No inhibition detected up to 50 μM. c No inhibition detected up to 10 μM. Note: a threshold inhibitor concentration at which no inhibition was observed was chosen depending on solubility constraints | |||||
| 7a |
|
0.51 ± 0.05 | 61.8 ± 10.2 | 8.3 ± 1.6 | 6.7 ± 0.7 |
| 7b |
|
1.17 ± 0.12 | 38.4 ± 6.2 | 30.4 ± 5.9 | 28.5 ± 1.9 |
| 7c |
|
2.64 ± 0.39 | 3.98 ± 0.79 | 662 ± 164 | 580 ± 40 |
| 7d |
|
4.69 ± 0.65 | 1.95 ± 0.32 | 2412 ± 522 | 2320 ± 81 |
| 7e |
|
5.22 ± 0.96 | 30.6 ± 6.9 | 171 ± 50 | 209 ± 13 |
| 7f |
|
7.28 ± 1.41 | 2.32 ± 0.52 | 3134 ± 921 | 3141 ± 143 |
| 7g |
|
n.d.b | n.d. | n.d. | n.d. |
| 7h |
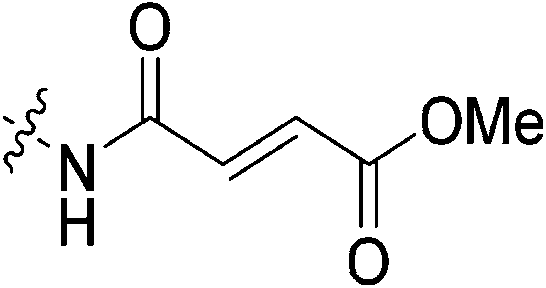
|
n.d. | n.d. | n.d. | n.d. |
| 7i |
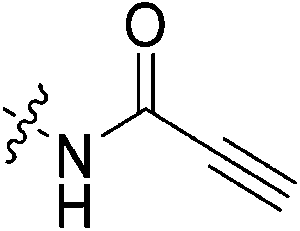
|
6.83 ± 1.75 | 70.1 ± 25.0 | 97.5 ± 42.8 | 87.7 ± 4.5 |
| 7j |
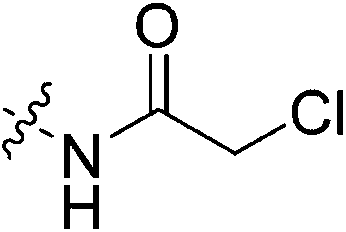
|
1.23 ± 0.30 | 0.15 ± 0.09 | 8047 ± 4887 | 4545 ± 289 |
| 7k |
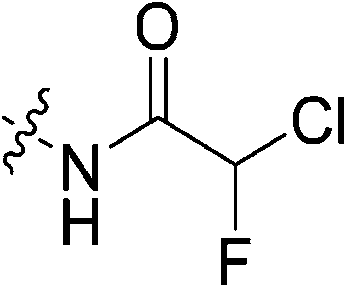
|
1.57 ± 0.29 | 88.5 ± 21.5 | 17.8 ± 5.4 | 16.0 ± 1.3 |
| 7l |
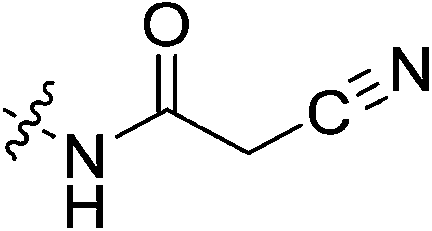
|
n.d. | n.d. | n.d. | n.d. |
| 7m |
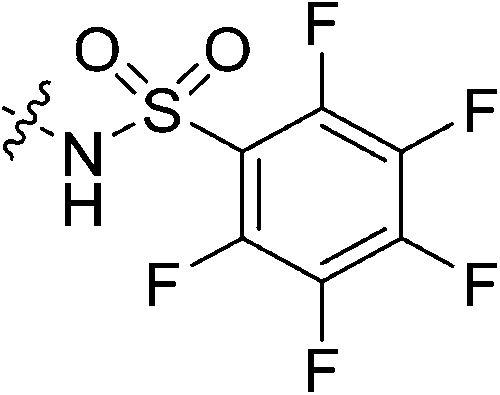
|
n.d.c | n.d. | n.d. | n.d. |
| 7n |
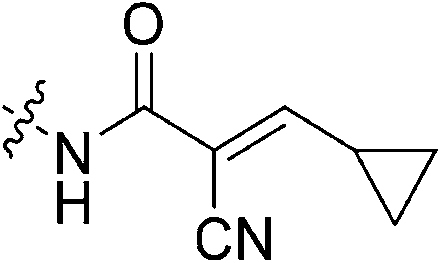
|
— | n.d. | — | — |
| 7o |
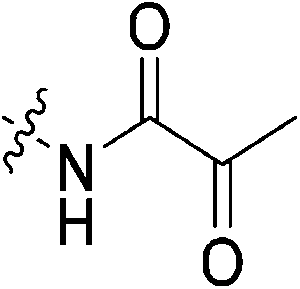
|
— | 154 ± 60 | — | — |
| 7p |
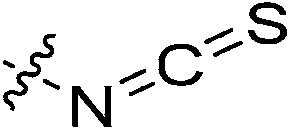
|
0.23 ± 0.03 | 8.83 ± 2.41 | 26.3 ± 8.1 | 17.7 ± 2.4 |
| 7q |
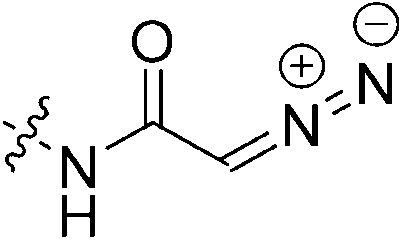
|
2.82 ± 0.49 | 26.3 ± 6.5 | 107 ± 53 | 95.0 ± 2.4 |
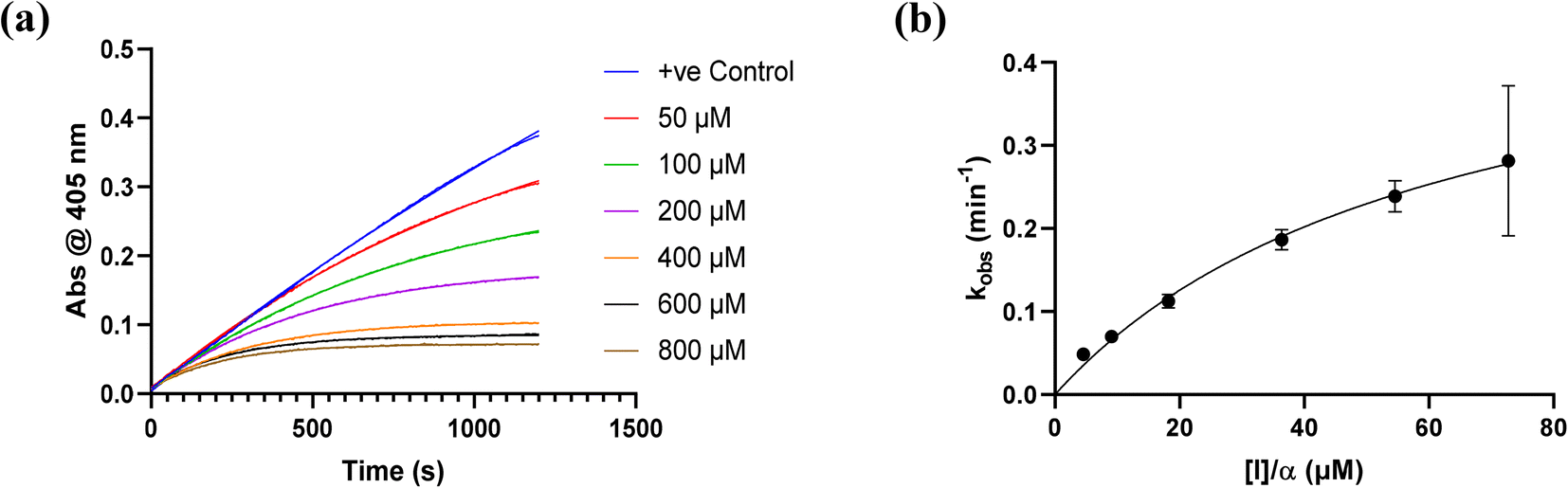 | ||
| Fig. 3 Kinetic curves obtained during hTG2 inhibition with representative inhibitor 7a. (a) kobs for the loss of activity and (b) hyperbolic fitting to saturation kinetics model (see Materials and methods and ESI†). | ||
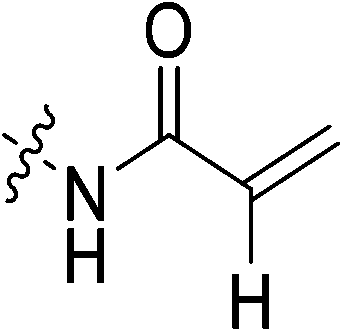
Starting with the acrylamide series, a rough correlation can be seen between expected electrophilicity of the β carbon, where the thiolate attacks via Michael addition, and kinact values ranging between 0.51–7.28 min−1 (Table 1, 7a–f). As the electronegativity of the substituent at the α-position increases, the π-bond between the α and β carbon becomes more polarized, making the β carbon more electrophilic. As a result, the kinact value (the first order rate constant of covalent bond formation within the enzyme-inhibitor complex) also increases. The apparent exception to this is α-fluoroacrylamide (7b), whose intrinsic stability (i.e., lower reactivity) has been well studied and is thought to be a result of the mesomeric electron donating ability of fluorine, which compensates at least partially for its electron withdrawing inductive effects.53 This warhead gives a kinact of 1.17 min−1, compared to a value of 2.64 min−1 for acrylamide 7c. Beyond this general trend in the kinact values, the construction of Hammett and Charton plots for substituents at the alpha position of inhibitors 7b–f did not provide any correlation (not shown).
More interesting and perplexing results are found in the corresponding KI values. For inhibitor 7a, a large decrease is observed in the binding affinity compared to parent compound 7c, with KI values of 61.8 versus 3.98 μM respectively. We believe this is due to the increased steric demand at the β position, resulting in the warhead not being able to fit properly in the active site ‘tunnel’. Surprisingly, inhibitors 7b and 7e, both containing fluorine, also display KI values an order of magnitude higher than 7c, while other halo-acrylamide inhibitors 7d and 7f have relatively unchanged binding affinity despite posing increased steric demand at the α-position. These results suggest that the α-position seems to tolerate bulkier substituents more readily than the β-position, further supported by the fact that 7g and 7h show no significant inhibition. However, the binding affinity cannot be explained by steric factors alone. It seems that fluorine-based warheads are not well tolerated in the enzyme's active site. We believe that this may be due to some fluorine mediated polar interaction or halogen–hydrogen bonding that disorients the inhibitor. Although there is currently no crystallographic evidence to support this hypothesis, we have previously discovered that fluorines on the N-terminus of our peptidomimetic inhibitors actually increase binding affinity.21 It is possible that the fluorine on the warheads studied herein directs these inhibitors to the same substrate binding site interaction as the peptidomimetic inhibitors, instead of down the active site ‘tunnel’.
Due to the increase in kinact values of 7d and 7f, while maintaining modest KI values, these inhibitors show an order of magnitude greater overall inhibitory efficiency, with kinact/KI values of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 412000 M−1 min−1 and 3134000 M−1 min−1 respectively, compared to 7c (kinact/KI = 662000 M−1 min−1), which is on par with chloroacetamide 7j and quite remarkable.
412000 M−1 min−1 and 3134000 M−1 min−1 respectively, compared to 7c (kinact/KI = 662000 M−1 min−1), which is on par with chloroacetamide 7j and quite remarkable.
Propiolamide 7i gives a kinact value (6.83 min−1) that is much higher than that of 7c. This is consistent with intrinsic reactivity studies performed by Flanagan et al. which show that this warhead is more reactive towards thiol addition than both acrylamides and chloroacetamides, presumably due to the sp hybridization state of this warhead which stabilizes negative charge more effectively.32 In fact, this is also reminiscent of our previous studies on alkyne inhibitors, which showed similarly high kinact values.36 Regardless, the low binding affinity of this inhibitor, possibly due to the inappropriate orientation of this linear warhead, makes it a relatively poor inhibitor.
Outside of the acrylamide and propiolamide warheads, kinact does not strictly correlate to the electrophilicity of a warhead, indicating that it is likely influenced by both intrinsic reactivity and orientation relative to the thiolate in the active site. Chloroacetamides are known to be highly reactive, and within this series of compounds 7j displays the best overall efficiency (kinact/KI = 8047000 M−1 min−1). However, this is largely due to a relatively high binding affinity rather than high reactivity, as shown by the relative KI and kinact values, respectively. Intrinsically it is known that chloroacetamides are more reactive than acrylamides,32 so it is interesting that the kinact values do not correlate with this intrinsic reactivity, as the kinact value (1.23 min−1) for 7j is smaller than that of 7c. Furthermore, chlorofluoroacetamide 7k displays an approximately equivalent kinact value to 7j, despite generally being less reactive. Note that the addition of a fluorine results in the increase of KI by two orders of magnitude (0.15 vs. 88.5 μM) consistent with the observation noted above, that fluorine may be detrimental to binding affinity.
Inhibitors 7l and 7m showed no significant inhibition. We believe this is due to the nitrile of the cyanoacetamide not being electrophilic enough to react efficiently. This observation is also aligned with previous hTG2 inhibition studies in which inhibitors with any type of nitrile warhead showed no detectable inhibition.30 It also comes as no surprise that the pentafluorobenzenesulfonamide warhead is inadequate likely due to its large size and perhaps abundance of fluorine atoms.
With isothiocyanate inhibitor 7p, we again observed a low kinact value (0.23 min−1) for a notoriously reactive warhead, but in this case 7p displayed a relatively high KI as well at 8.83 μM. Nonetheless, this is consistent with recently published inhibition parameters for sulforaphane, a naturally-occurring anticancer agent with an isothiocyanate warhead, with TG2.39 Sulforaphane gives values of kinact = 0.27 ± 0.07 min−1 and KI = 366 ± 157 μM; comparing these values with those of 7p, it is evident that kinact is approximately the same while the binding affinity has been improved as a result of the Ad-Pip-Gly scaffold. It is likely that, in part, the binding affinity is still low due to the inhibitor being too small in length to properly allow the warhead to reach into the active site ‘tunnel’.
The last irreversible warhead we tested was diazoacetamide 7q. We expected this warhead to show attenuated reactivity relative to its keto version, due to the additional resonance donating abilities of nitrogen which may impact the conjugation and reactivity of the diazo-carbonyl system, but we also observed a decrease in binding affinity. Evidently, any linear elongation of the warhead leads to a dramatic decrease in binding affinity. This is consistent with our previous observation that elongation of the glycine linker leads to a decrease in affinity.22 This phenomenon may also be responsible for the fact that the highly reactive reversible warhead of inhibitor 7n displayed no detectable inhibition.
We did have moderate success with reversible α-ketoamide 7o, for which we were able to extract a Ki value (referred to expressly as Ki instead of KI to distinguish between reversible and irreversible inhibition). As expected, this inhibitor displayed time-independent inhibition of hTG2 consistent with rapid binding, competitive, reversible inhibition. We performed the same kinetic experiment described for irreversible inhibition at different inhibitor concentrations and obtained initial rates at each from the absorbance versus time plot (Fig. 4a). This experiment was then repeated at a different substrate concentration and the data from the two experiments were used to construct a Dixon plot, from which the Ki was extrapolated by the point of intersection of the two resulting lines (Fig. 4b). We also performed a simple test to validate that this was in fact reversible inhibition by running the assay for an extended period of time and allowing the substrate to be completely consumed in both the uninhibited and inhibited reaction. If inhibition is reversible, both reactions should plateau at the same final absorbance value corresponding to the same initial concentration of substrate, albeit at different times. This is exactly what we observed, as shown in the ESI.† Although the binding affinity for this inhibitor is poor (Ki = 154 μM), it does represent one of the first fully characterized covalent reversible inhibitors of hTG2, demonstrating that it is a viable avenue to explore for hTG2 inhibition.
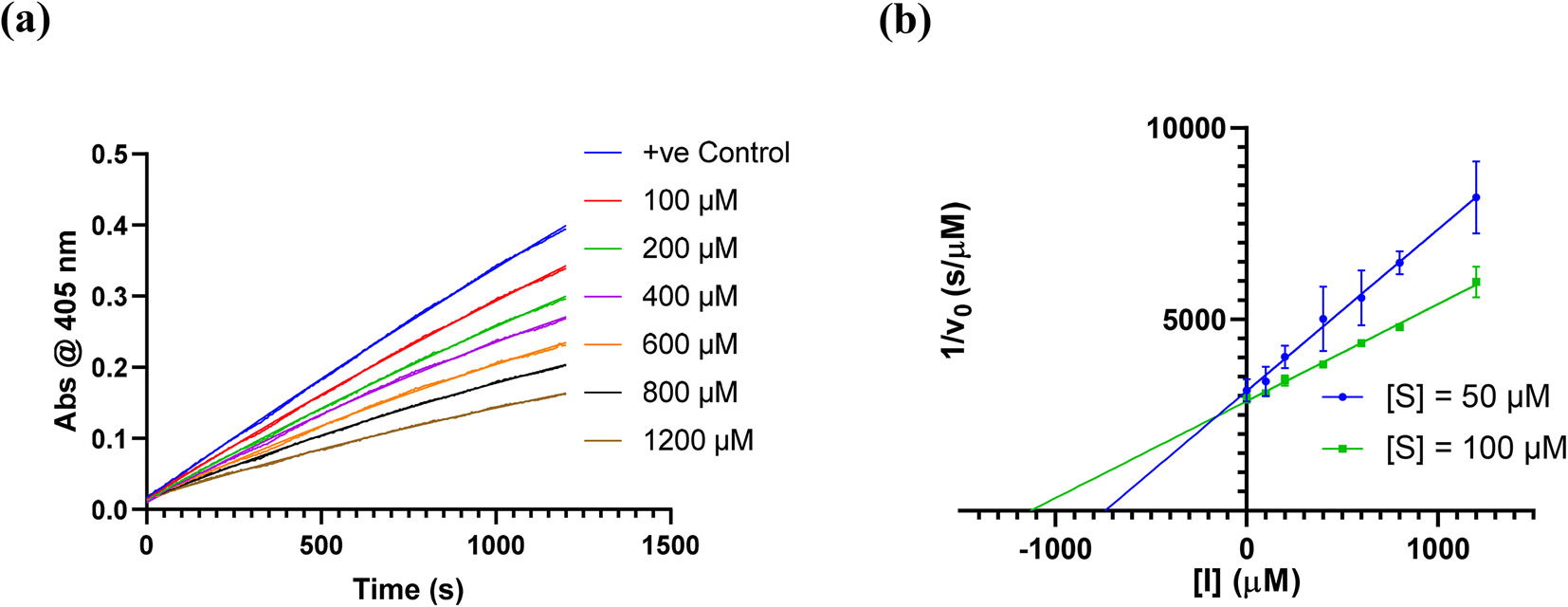 | ||
| Fig. 4 Kinetic analysis of inhibitor 7o. (a) Representative time-independent rate decrease at [S] = 100 μM. Average initial rates (v0 for the uninhibited reaction and for the inhibited reactions) were determined over the first 10% of conversion of the uninhibited reaction. (b) Dixon plot used to determine Ki. See ESI† and Materials and methods for details. | ||
Taken together, this thorough kinetic evaluation has revealed some interesting trends in the reactivity and affinity of different warheads for hTG2 and highlights how subtle changes in warhead structure can dramatically influence inhibitory efficiency. It appears acrylamides are still the best suited class of warheads for targeting hTG2, and we have identified two analogues that confer potency far greater than that of an unsubstituted acrylamide group and have never been reported for hTG2 inhibition elsewhere.
Isozyme selectivity
We also tested our top four inhibitors against other therapeutically relevant TGases, in order to assess the relative selectivity imparted by the warhead (Table 2). The assay described above for hTG2 inhibition was also used to monitor hTG1 and hTG6 inhibition,19 whereas for hTG3a and hFXIIIa, a commercially available continuous fluorescence assay that uses a peptidic FRET-quenched substrate was used.54,55 In each case, we chose a corrected inhibitor concentration ([I]/α) that gave a kobs value of approximately 0.5 min−1 with hTG2, and then adjusted inhibitor concentrations to give an identical [I]/α value for each enzyme/substrate pair, in order to permit comparison between isozymes. In this fashion, kobs values were obtained whenever time-dependent inhibition was observed. Interestingly, some isoforms displayed time-independent inhibition (i.e., instantaneous and constant decrease in rate), in which case a percent inhibition (vinhibited/vuninhibited × 100%) was measured instead. We recognize that % inhibition values cannot be directly compared to kobs values to assess selectivity for hTG2; however, they may be used to compare selectivity between different warheads (see ESI†).| Compound | Warhead | [I]/α | k obs TG2 (min−1) | k obs TG1 (min−1) | k obs TG6 (min−1) | % inhibition TG3a | hFXIIIa |
|---|---|---|---|---|---|---|---|
| 7c |
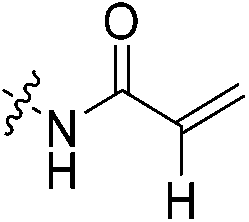
|
0.91 | 0.500 ± 0.016 | 0.023 ± 0.003 | 0.020 ± 9.00 × 10−4 | 59.1 ± 0.7 | k obs = 0.051 ± 0.001 min−1 |
| 7d |
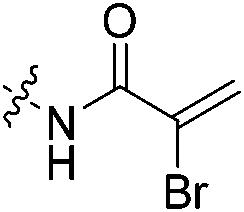
|
0.23 | 0.497 ± 0.017 | 0.018 ± 0.002 | n.d. | n.d. | n.d. |
| 7f |
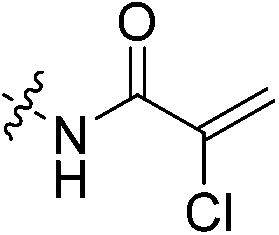
|
0.18 | 0.509 ± 0.002 | 0.022 ± 9.00 × 10−5 | n.d. | n.d. | % inhibition = 76.0 ± 3.3 |
| 7j |
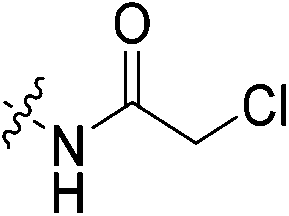
|
0.14 | 0.537 ± 0.019 | 0.018 ± 0.003 | n.d. | 41.5 ± 0.9 | k obs = 0.013 ± 0.002 min−1 |
Previously reported inhibitor 7c (Fig. 5) shows roughly 20-fold selectivity for hTG2, over hTG1 and hTG6, consistent with the findings of Badarau et al.28 However, in our assay this inhibitor is far less selective for hTG2 relative to hTG3a and hFXIIIa than reported in the original study, to the point where it appears to be a rather efficient inhibitor of these isozymes. We believe this may be due to the different assays used to measure inhibition. Indirect, coupled-enzyme assays are prone to initial lag periods that may lead to underrepresented inhibition,22 which may be further exacerbated by the intrinsically slow reactivity of hTG3a and hFXIIIa. The sensitive continuous fluorescence assay used herein may allow for more accurate measurement of inhibition of hTG3a and hFXIIIa.
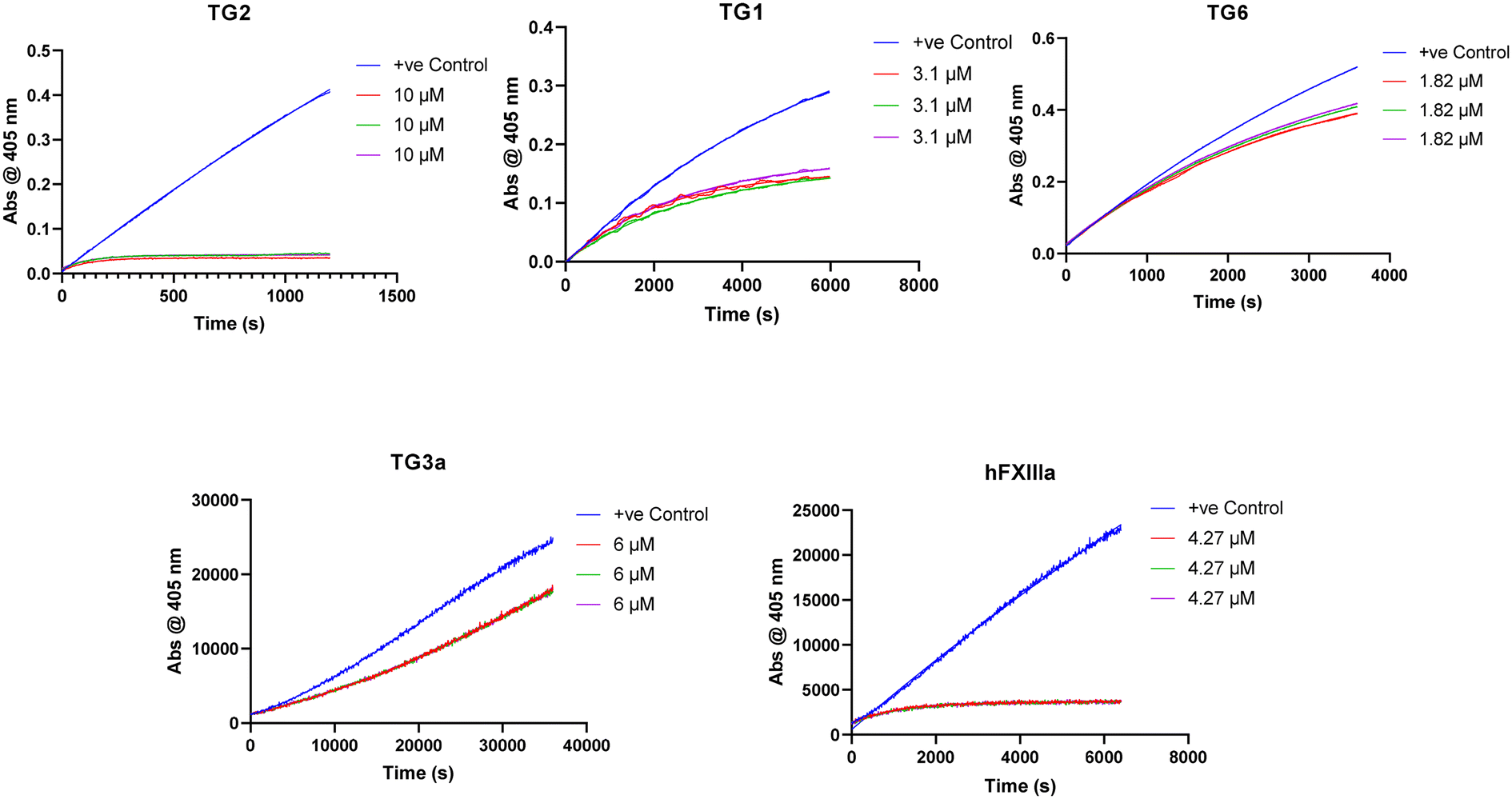 | ||
| Fig. 5 Isozyme selectivity data for inhibitor 7c. | ||
While some isozymes seem to have lower affinity for the scaffold, it is interesting to note that within the same scaffold, certain isozymes show selectivity for different warheads. All of these inhibitors target hTG1 in a similar manner and show approximately the same selectivity for hTG2 over this isozyme, whereas the selectivity is more diverse with other isozymes. No inhibitor, other than 7c, inhibits hTG6. Similarly, hTG3a is only inhibited reversibly by previously reported inhibitors 7c and 7j, but not inhibited by our new inhibitors 7d and 7f. hFXIIIa is inhibited irreversibly by 7c and 7j, reversibly by 7f, and not inhibited by 7d. It is possible that the α-chloroacrylamide exhibits reversible covalent inhibition with hFXIIIa due to increased acidity of the α-hydrogen as a result of the electron withdrawing presence of the chlorine. More explicitly, there may be an amino acid residue on hFXIIIa near this hydrogen that is basic enough to facilitate the reverse E1cB reaction. A related change in inhibition mechanism has been reported for the inhibition of rhodesain, a cysteine protease; while a vinylsulfone inhibitor was irreversible, its α-fluoro analogue was found to be slowly reversible.56 On the other hand, for inhibitors that show reversible inhibition with other TGases but not with hTG2, it is also possible that their warheads are simply not oriented in the isozyme's active site in an appropriate manner to react, and therefore they act as reversible non-covalent inhibitors. The reversibility observed in this work is clearly related to a function of the isozyme, and not to the intrinsic reactivity of the warhead.
Importantly, these results clearly demonstrate that increasing warhead reactivity does not necessarily have a negative impact on selectivity. To this point, our new halo-acrylamide inhibitors show the most selectivity overall among the top inhibitors, suggesting that it is possible to tune both reactivity and isozyme selectivity through strategic warhead design.
Intrinsic reactivity
The same top inhibitors, excluding 7d due to its poor aqueous solubility, were then evaluated further for their intrinsic reactivity, offering insight into the efficiency of the enzymatic reaction relative to reaction with a thiol in solution by simple comparison of rate constants (Table 3). We chose GSH as a representative thiol in order to also provide a rough idea of the expected reactivity of a given inhibitor with the millimolar concentration of GSH found intracellularly and therefore the potential for off-target reactions.57| Compound | Warhead | k inact/KI (×103 M−1 min−1) | k corr2 (×10−3 M−1 min−1) | (kinact/KI)/kcorr2 (×109) |
|---|---|---|---|---|
| 7c |
|
662 ± 164 | 0.079 ± 0.009 | 0.843 ± 0.231 |
| 7f |
|
3134 ± 921 | 59.387 ± 1.610 | 0.053 ± 0.016 |
| 7j |
|
8047 ± 4887 | 1.480 ± 0.191 | 5.437 ± 3.376 |
Each inhibitor was incubated with GSH under pseudo-first order conditions at room temperature and at a pH that allowed sufficient reactivity. The formation of an inhibitor-GSH adduct and the corresponding disappearance of inhibitor were both monitored simultaneously using HPLC analysis. The resulting concentration versus time data were used to measure pseudo-first order rate constants which were then converted to intrinsic second order rate constants (kcorr2) by taking into account the pH dependent concentration of excess thiolate in solution (see ESI†).
The kinetic parameter kinact/KI demonstrates overall efficiency of inhibition against hTG2. Intrinsically this is the second order rate constant of the reaction between the enzyme and the inhibitor, accounting for not only the rate of the covalent reaction (kinact) but also binding affinity (KI) and orientation/proximity effects that the enzyme's active site provides. This parameter is most relevant to compare to the second order rate constant of reaction with GSH (kcorr2), as opposed to kinact which is a first order rate constant. By calculating the ratio between the two second order rate constants we arrived at a parameter ((kinact/KI)/kcorr2) that quantifies enzymatic efficiency relative to reaction with GSH in solution. All of the inhibitors demonstrate the extreme acceleration an enzymatic reaction provides over a typical chemical reaction in solution, with enzymatic efficiency ratios of at least 10 million. In fact, reactions with GSH were generally observed to be very slow at a pH of 7.4 and an increased pH of 8.0 was required to drive the reaction. With acrylamide inhibitor (7c) we measured an enzymatic efficiency ratio of just under a billion (0.849 ± 0.232 × 109) as well as the slowest reaction with GSH, as expected based on the inherent stability of the acrylamide group compared to the other two warheads. The α-chloroacetamide (7j) displays reactivity with GSH that is two orders of magnitude faster than that of 7c, highlighting the intrinsic electrophilic superiority of α-halocarbonyls compared to the acrylamide group. This compound reacts roughly 5 billion times faster with hTG2 than with GSH.
We were particularly interested in comparing the reactivity profiles of chloroacetamide 7j and chloroacrylamide 7f, as it was not clear to us which electrophile is intrinsically more reactive. When we reacted 7f with GSH at a pH of 8.0 we observed such rapid consumption of the warhead that it was difficult to characterize, and the pH had to be adjusted back down to 7.4. Interestingly, we also noticed the formation of multiple products, instead of just one GS-inhibitor adduct. Upon careful inspection, we noted that while one product was formed at the beginning of the reaction, presumably the GS-inhibitor mono-addition product, over time this adduct reacted further to produce other products, even after the original inhibitor had been entirely consumed (Scheme 4). The initial product was also eventually consumed in this second reaction. We hypothesized that the mono-addition product (8) can react further with another molecule of GSH through nucleophilic displacement of the chloride on the α-carbon to form a di-substituted product (9). Both mono and di-substitution would produce diastereomeric products which may be separated by HPLC, leading to the formation of multiple broad peaks on the chromatogram. Another possibility is the attack of a second equivalent of GSH on the sulfur of the initial adduct, leading to formation of glutathione disulfide and displacement of chloride in an elimination reaction that would lead to the formation of acrylamide 7c. This acrylamide would itself react slowly with another equivalent of GSH at this pH to form adduct 10.
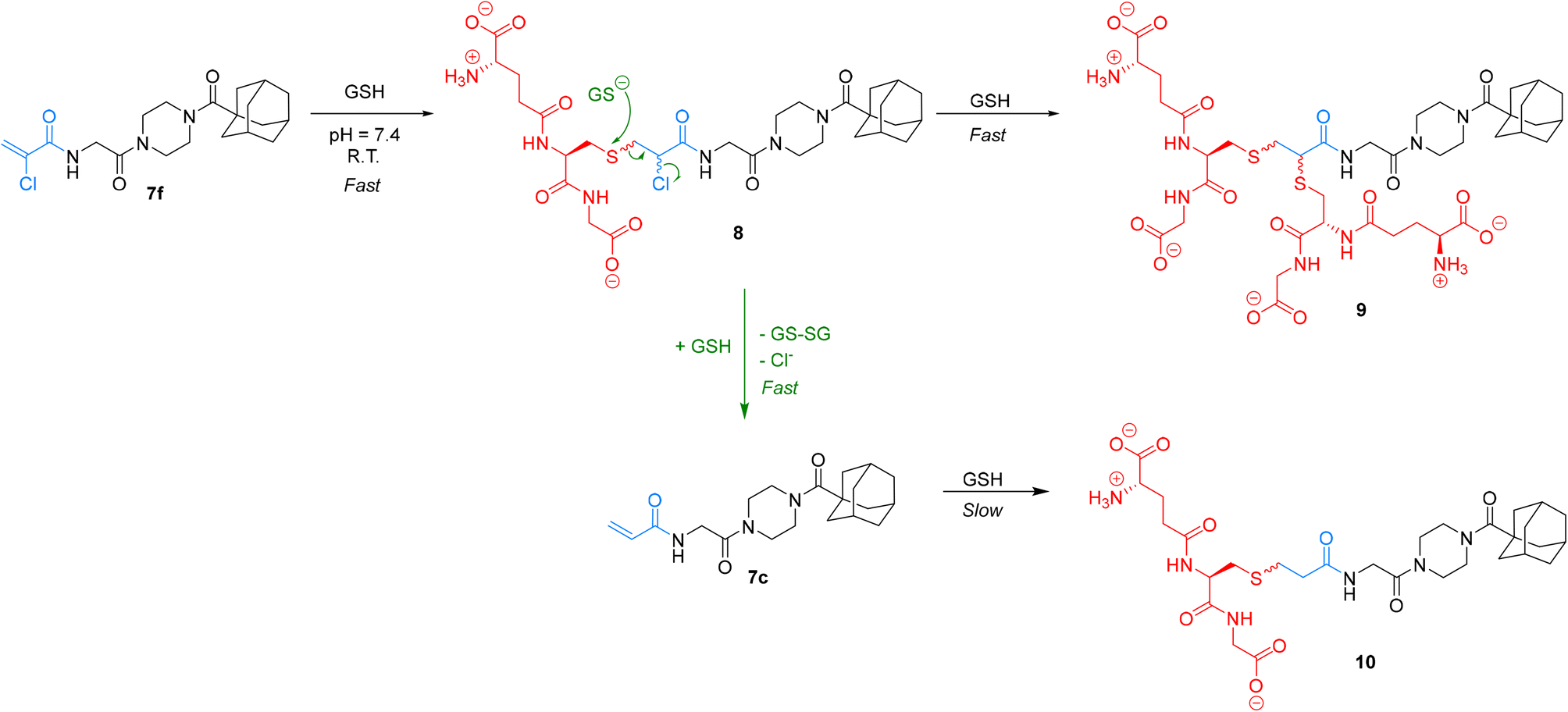 | ||
| Scheme 4 Side products of the reaction between GSH and 7f. | ||
We characterized these products using LC-MS analysis and were able to monitor and confirm the disappearance of inhibitor 7f, the formation and subsequent disappearance of mono-substituted adduct 8, the corresponding formation of double-substituted adduct 9 and formation of inhibitor 7c, confirming our hypothesis (see ESI†). However, there were also other side products that we could not identify based on the mass to charge ratio. Although we could not analyze α-bromoacrylamide inhibitor 7d in this assay, it is reasonable to assume that it could behave in a similar fashion, especially considering that bromide is a better leaving group than chloride.
Ultimately, by simply monitoring the disappearance of inhibitor 7f by our original HPLC method, we determined that this warhead reacts an order of magnitude faster than the chloroacetamide. Owing to this relatively high and multifaceted reactivity with GSH, its reaction with hTG2 is only 0.053 ± 0.016 × 109 times more efficient, which is the lowest efficiency ratio of the top warheads. However, it is particularly interesting to note that inhibitor 7f can react with GSH, in a metabolically relevant reaction, to form another potent and more stable inhibitor 7c.
Pharmacokinetic stability
The conjugation of electrophilic agents with GSH during phase 2 metabolism is most often mediated by one or more isozymes of glutathione S-transferase (GST).51 Taking this into account is necessary to assess the stability and suitability of a drug from a biochemical perspective. Therefore, we studied the stability of the same top three representative inhibitors in hepatocytes and whole blood, as in vitro models of in vivo conjugation mediated by the highly relevant α and π isozymes of human GST, respectively.58,59Inhibitors were incubated in human hepatocytes and human whole blood at 37 °C over a 120-minute period. Aliquots were taken over time and the amount of inhibitor remaining was determined by LC-MS/MS. In cases where decay was observed over this time frame, the half-life of each inhibitor was determined to model their relative stabilities (Table 4). In hepatocytes, acrylamide inhibitor 7c and chloroacetamide 7j show excellent stability with half-lives >120 min, whereas α-chloroacrylamide 7f is notably less stable, with a half-life of 92 min. In human whole blood we observed no degradation of inhibitors 7c and 7j over 120 minutes; however, 7f showed a lower half-life of about 32 min, similar to that observed in hepatocytes. Overall, these relative stabilities are consistent with our in-solution inhibitor-GSH intrinsic reactivity studies, underlining the instability of the α-chloroacrylamide warhead compared to the chloroacetamide, and the superiority of the acrylamide warhead from an in vivo viability perspective. Although the degradation of inhibitors in these media cannot be directly linked only to GST activity, as there are many other proteins and compounds that may contribute, it is important to consider that while a given scaffold may help to lower affinity for GST, the warhead can also be optimized for low reactivity within the active site of GST, contributing to the overall in vivo stability of an inhibitor.
| Compound | Warhead | HH t1/2 (min) | HWB t1/2 (min) |
|---|---|---|---|
| 7c |
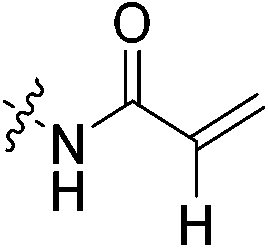
|
>120 | >120 |
| 7f |
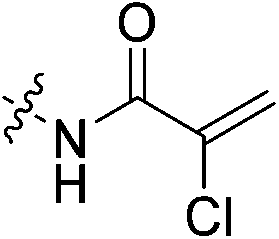
|
92 ± 16 | 32 ± 4 |
| 7j |
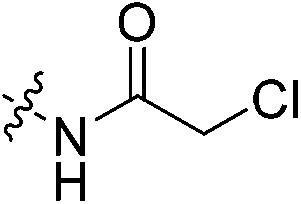
|
>120 | >120 |
Conclusions
Rigorous kinetic evaluation of small molecule hTG2 inhibitors has revealed a strong influence on inhibitory parameters with even subtle variation in warhead structure, highlighting that it is equally important to optimize the warhead as it is the inhibitor scaffold. While the latter is clearly important for initial non-covalent binding and orientation of the warhead, the warhead itself may also contribute to binding affinity and the efficiency of the subsequent reaction. We have demonstrated the opportunity for tuning (isozyme) selectivity with varying warhead structure and identified two new inhibitors with excellent overall inhibitory efficiency and modest selectivity.In addition, the pharmacokinetic stability of various functional groups must also be considered early in drug development. Warhead optimization provides an additional avenue to improve in vivo stability, particularly if reactivity with GST isoforms is taken into consideration. Here we confirm that the acrylamide warhead is still superior in terms of off-target stability and is most suitable for therapeutic inhibitor design. Other warheads we have incorporated can be applied to study hTG2 in a more fundamental manner, such as in the development of chemical probes, and provide interesting new insights for tuning reactivity, selectivity, and stability of hTG2 inhibitors.
Materials and methods
Synthesis
536 points. An automatic phase correction was applied and manually refined to provide a flatter baseline. A baseline correction, employing a Whittaker smoothing function with a 2.60 Hz filter and a smoothing factor of 16384 was employed on the spectrum between −2.00 and 15.00 ppm. Integration was measured using the manual integration tool and referenced to a relevant aliphatic proton. Multiplet analysis was conducted using the algorithm as implemented in the software, confirmed by manual analysis. High resolution mass spectrometry (HRMS) was performed using a quadrupole time-of-flight (QTOF) analyzer and electrospray ionization (ESI).
Crystallographic measurements on I3 were performed using a Bruker AXS SMART single-crystal diffractometer equipped with an APEX II CCD detector, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) from a sealed tube source. The crystal chosen was mounted on the tip of a 200 μM MicroLoop using Parabar 10312 oil, and the data collection was performed at room temperature. Raw data collection and processing were performed with the Bruker APEX II software package. A multi-scan absorption correction was applied (SADABS). The crystal structure was solved via direct methods and refined using a full-matrix least-squares method on F2 using the SHELXTL program suite and the Olex2 user interface. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms bonded to carbon were included at geometrically idealized positions [riding model] and were not refined. The isotropic thermal parameters of these hydrogen atoms were fixed at 1.2 Ueq of the parent carbon atom. The hydrogen atom bonded to oxygen in the carboxylic acid moiety was located in the Fourier difference map, allowing isotopic refinement with the O–H distance restrained to 0.86 (0.01) Å. A relatively large crystal (with dimensions 0.268 × 0.447 × 0.461 mm3) was purposely chosen to compensate for the somewhat weak diffraction, caused by the lack of atoms heavier than oxygen in the compound. This compound crystallized in the monoclinic space group P21/c. The extended structure of this crystal comprises hydrogen-bonded dimers of the compound via the carboxylic acid moiety. Short contacts (2.5351(22) Å) are also present between H4 (alkene proton) and N1 (nitrile N).
N-(tert-Butoxycarbonyl)glycine (I1)
To a stirring solution of NaOH (1.60 g, 39.96 mmol, 1.5 eq) in 1:1 H2O/dioxane (50 mL) was added glycine (2.00 g, 26.64 mmol, 1.0 eq). The solution was cooled to 0 °C and stirred for 15 min. Di-tert-butyl dicarbonate (8.72 g, 39.96 mmol, 1.5 eq) was dissolved in dioxane (10 mL) and added dropwise to the solution. The reaction mixture was allowed to warm to R.T. and stir for 16 h until the reaction was confirmed to be complete by TLC analysis (10% MeOH in DCM, Rf = 0.46). The reaction mixture was concentrated in vacuo and the residue was redissolved in H2O (50 mL). The aqueous phase was washed with Et2O (3 × 50 mL) and then acidified with 12 M HCl to pH 2. The aqueous phase was then extracted with EtOAc (3 × 50 mL). The organic phase was washed with brine (25 mL), dried with MgSO4, and concentrated in vacuo to give a clear oil which dried into a crystalline white solid overnight (4.26 g, 91%). 1H NMR (400 MHz, CDCl3) δ 3.97–3.89 (m, 2H, rotamers), 1.44 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 175.0/174.1 (1C, rotamers), 157.4/156.1 (1C, rotamers), 81.9/80.6 (1C, rotamers), 43.5/42.3 (1C, rotamers), 28.4. Characterization data are consistent with previously reported values.22
Adamantanecarbonyl chloride (2)
1-Adamantanecarboxylic acid (1.8 g, 10 mmol) and thionyl chloride (5 mL) were stirred at reflux under an inert atmosphere with a bubbler attached to monitor gas evolution. After 4 h the reaction was complete as confirmed by 13C NMR analysis (shift in carbonyl carbon resonance). The reaction was evaporated to dryness under reduced pressure to give the product as a white crystalline solid (1.77 g, 90%). 1H NMR (300 MHz, CDCl3) δ 2.14–2.11 (m, 3H), 2.03–2.02 (m, 6H), 1.82–1.69 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 180.0, 51.2, 39.1 (3C), 36.2 (3C), 28.0 (3C). Characterization data are consistent with previously reported values.22N-(Adamantanecarbonyl)-N-(tert-butoxycarbonyl)-piperazine (3)
To a stirring solution of N-Boc-piperazine (0.446 g, 2.396 mmol, 1.0 eq) and Hünig's base (0.835 mL, 4.792 mmol, 2.0 eq) in dry DCM (10 mL) at 0 °C was added 2 (0.500 g, 2.516 mmol, 1.05 eq) portion wise. The ice bath was removed, and the reaction was stirred at R.T. for 3 h when it was confirmed to be complete by TLC analysis (1:1 hexanes/EtOAc, Rf = 0.67). The reaction mixture was diluted with DCM and washed with 5% AcOH (3 × 20 mL) and brine (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give the product as a pale-yellow solid (0.797 g, 96%). 1H NMR (400 MHz, CDCl3) δ 3.66–3.63 (t, J = 5.6 Hz, 4H), 3.42–3.38 (t, J = 5.6 Hz, 4H), 2.05–2.00 (m, 3H), 1.98–1.97 (m, 6H), 1.76–1.68 (m, 6H), 1.46 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 176.1, 154.8, 80.3, 45.2, 41.8, 39.2, 36.7, 28.6, 28.5. Characterization data are consistent with previously reported values.22
N-(Adamantanecarbonyl)piperazine hydrochloride (4)
To a stirring solution of 3 (0.797 g, 2.287 mmol, 1.0 eq) in DCM (5 mL) was added 5 mL of HCl (4 M in dioxane). A precipitate formed as the reaction progressed and the disappearance of starting material was monitored by TLC (1:1 hexanes/EtOAc), completely disappearing in 2 h. The reaction mixture was concentrated in vacuo and the resulting solid was resuspended in DCM (7 mL) and Et2O (7 mL), filtered, and washed with Et2O (3 × 5 mL) to give the product as a white solid (0.575 g, 88%). 1H NMR (300 MHz, DMSO-d6) δ 9.33 (br s, 2H), 3.82–3.78 (t, J = 5.1 Hz, 4H), 3.05–3.01 (t, J = 5.1 Hz, 4H), 2.00–1.93 (m, 3H), 1.88–1.80 (m, 6H), 1.74–1.58 (m, 6H). 13C NMR (75 MHz, DMSO-d6) δ 174.8, 43.0, 41.7, 41.0, 38.3, 36.0, 27.9. Characterization data are consistent with previously reported values.22
N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] carbamic acid tert-butyl ester (5)
Compound 1 (0.244 g, 1.389 mmol, 1.1 eq), HBTU (0.765 g, 2.022 mmol, 1.6 eq), and Hünig's base (0.9 mL, 5.052 mmol, 4.0 eq) were stirred in DCM (20 mL) at R.T. After 1 h, 4 (0.360 g, 1.263 mmol, 1.0 eq) was added. The reaction was left to stir overnight (∼18 h). After confirmed to be complete by TLC (10% MeOH/DCM, Rf = 0.71) the reaction was concentrated in vacuo and the residue was redissolved in EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 25 mL), brine (25 mL), NaHCO3 (50 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a pale-yellow solid, which was washed with Et2O to give a white solid (0.330 g, 65%). 1H NMR (400 MHz, CDCl3) δ 5.47 (br s, 1H), 3.97–3.96 (d, J = 4.0 Hz, 1H), 3.71–3.68 (m, 4H), 3.63–3.59 (m, 2H), 3.39–3.39 (t, J = 5.2 Hz, 2H), 2.06–2.04 (m, 3H), 1.98–1.97 (m, 6H), 1.77–1.68 (m, 6H), 1.44 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 176.2, 167.4, 155.9, 79.9, 45.4, 44.8, 44.6, 42.4, 42.3, 41.9, 39.1, 36.7, 28.5, 28.4. Characterization data are consistent with previously reported values.221-[4-(Adamantane-1-carbonyl)piperazin-1-yl]-2-amino-ethanone (6)
Compound 5 (0.203 g, 0.5 mmol. 1.0 eq) was dissolved in DCM (7.0 mL) and treated with TFA (0.31 mL, 4.0 mmol, 8.0 eq) until the starting material was completely consumed by TLC (10% MeOH/DCM) in 3 h. The reaction mixture was diluted with DCM (15 mL) and washed with 0.5 M HCl (3 × 25 mL). The pH of the combined aqueous layer was adjusted to 9–10 using sat. K2CO3 and then extracted using DCM (3 × 35 mL). The combined organic layer was dried over MgSO4 and concentrated in vacuo to give a white solid (0.0827 g, 54%) (10% MeOH/DCM, Rf = 0.27). 1H NMR (300 MHz, CDCl3) δ 3.75–3.59 (m, 6H), 3.49 (apparent s, 2H), 3.39–3.34 (m, 2H), 2.05 (apparent s, 3H), 1.99–1.98 (m, 6H), 1.79–1.67 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 171.6, 45.7, 44.8, 44.5, 43.7, 42.3, 41.9, 39.2, 36.7, 28.5. Characterization data are consistent with previously reported values.28N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-3-methyl-propenamide (7a)
Crotonic acid (0.032 g, 0.363, 1.0 eq), Hünig's base (0.15 mL, 0.871 mmol, 2.4 eq), EDCI–HCl (0.084 g, 0.436 mmol, 1.2 eq), and HOBt·H2O (0.067 g, 0.436 mmol, 1.2 eq) were stirred in anhydrous DCM (4 mL) under an atmosphere of nitrogen at R.T. 6 (0.100 g, 0.327 mmol, 0.9 eq) was then added as a solution in DCM (1 mL). The reaction was stirred for 18 h at R.T. when TLC analysis confirmed the reaction as complete (7.5% MeOH/EtOAc, Rf = 0.51). The reaction mixture was diluted with DCM (7 mL) and washed with 5% AcOH (3 × 7 mL), brine (7 mL), sat. NaHCO3 (2 × 10 mL), and brine again (10 mL). The organic phase was then dried over MgSO4 and concentrated in vacuo to give a white solid which was purified by silica gel column chromatography (7.5% MeOH/EtOAc, Rf = 0.51, dry loading using silica) to yield the product as a white chalk-like solid (0.0632 g, 52%). 1H NMR (300 MHz, CDCl3) δ 6.93–6.81 (dq, J = 15.3, 6.9 Hz, 1H), 6.52 (br s, 1H), 5.92–5.86 (dq, J = 15.3, 1.8 Hz, 1H), 4.14–4.12 (d, J = 4.2 Hz, 2H), 3.73–3.70 (m, 4H), 3.65–3.62 (m, 2H), 3.44–3.41 (m, 2H), 2.06 (apparent s, 3H), 1.99–1.98 (m, 6H), 1.88–1.85 (dd, J = 6.9, 1.8 Hz, 3H), 1.78–1.68 (m, 6H, overlapping with H2O signal). 13C NMR (75 MHz, CDCl3) δ 176.2, 167.1, 166.0, 140.7, 124.7, 45.3, 44.9, 44.7, 42.4, 41.9, 41.4, 39.2, 36.7, 28.5, 17.9. HRMS (ESI) calc'd for C21H31N3O3Na [MNa]+: 396.2263, found: 393.2260.2-Fluoroacrylic acid (I2)
2-Fluoroacrylic acid methyl ester (0.500 g, 4.80 mmol) was dissolved in EtOH/H2O (8.7:1.3 mL, 10 mL). NaOH (2 M) was then added dropwise until pH 11 was reached, and the resulting mixture was stirred for 30 min, after which the solution was concentrated in vacuo to yield the sodium salt as a white solid. The salt was suspended in Et2O (20 mL) and HCl (6 M) was added dropwise until the solid dissolved. The layers were separated, and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic layer was dried over MgSO4 and evaporated in vacuo to yield the desired acid as a white solid (0.306 g, 71%). 1H NMR (CDCl3, 300 MHz) δ 11.01 (br s, 1H), 5.93–5.77 (dd, JHF = 42.9, JHH = 3.3 Hz, 1H), 5.52–5.47 (dd, JHF = 12.6, JHH = 3.3 Hz, 1H). 13C NMR (CDCl3, 75 MHz) δ 165.6 (d, JCF = 36.9 Hz), 152.6 (d, JCF = 259.2 Hz), 105.4 (d, JCF = 14.6 Hz). 19F NMR (CDCl3, 283 MHz) δ −118.3. Characterization data are consistent with previously reported values.60
N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-2-fluoro-propenamide (7b)
2-Fluoroacrylic acid (0.062 g, 0.684 mmol, 1.1 eq), HBTU (0.378 g, 0.995 mmol, 1.6 eq), and Hünig's base (0.433 mL, 2.49 mmol, 4.0 eq) were stirred in anhydrous DCM (5 mL) at 0 °C for 30 min. 6 (0.190 g, 0.622 mmol, 1.0 eq) was then added dropwise as a solution in DCM (2 mL) at R.T. and the reaction was stirred for 5 h at R.T. at which point it was confirmed complete by TLC analysis (2% MeOH/EtOAc, Rf = 0.60). The reaction was concentrated in vacuo and the residue was redissolved in EtOAc (25 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (15 mL), NaHCO3 (15 mL), and brine again (15 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give an orange solid, which was washed with Et2O/hexanes (10 mL:10 mL) and then further purified by silica gel column chromatography (2% MeOH/EtOAc, dry loading using celite) to give the product as a pale-yellow solid (0.055 g, 24%). 1H NMR (300 MHz, CDCl3) δ 7.35 (br s, 1H), 5.76–5.60 (dd, JHF = 46.8, JHH = 3.3 Hz, 1H), 5.52–5.47 (dd, JHF = 14.7, JHH = 3.3 Hz, 1H), 4.15–4.14 (d, J = 4.2 Hz, 2H), 3.74–3.71 (m, 4H), 3.66–3.63 (m, 2H), 3.44–3.41 (m, 2H), 2.06 (apparent s, 3H), 1.99 (apparent s, 6H), 1.78–1.68 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 166.1, 159.7 (d, JCF = 31.2 Hz), 156.1 (d, JCF = 268.9 Hz), 99.3 (d, JCF = 14.6 Hz), 45.4, 44.8, 44.7, 42.4, 41.9, 41.2, 39.2, 36.7, 28.5. 19F NMR (CDCl3, 283 MHz) δ −120.5. HRMS (ESI) calc'd for C20H28FN3O3Na [MNa]+: 400.2012, found: 400.2010.
N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] propenamide (7c)
Compound 6 (0.085 g, 0.278 mmol, 1.0 eq), and NEt3 (0.2 mL, 1.390 mmol, 5.0 eq) were stirred in anhydrous DCM (5.0 mL) under an atmosphere of nitrogen at 0 °C. Acryloyl chloride (0.025 mL, 0.306 mmol, 1.1 eq) was then added dropwise. The reaction was allowed to warm to R.T. over 3 h while monitoring product formation by TLC (10% MeOH/DCM, Rf = 0.59). The reaction mixture was concentrated in vacuo and the residue was redissolved in EtOAc (100 mL) and washed with sat. NaHCO3 (3 × 50 mL) and brine (50 mL). The organic phase was then dried over MgSO4 and concentrated in vacuo. The resulting solid was purified by silica gel column chromatography (4% MeOH/EtOAc, Rf = 0.37, dry loading using celite) and then washed with pentane (3 × 5 mL) to give the product as a white solid (0.073 g, 73%). 1H NMR (300 MHz, CDCl3) δ 6.75 (br s, 1H), 6.35–6.20 (dd, J = 1.3, 17.0 Hz, 1H), 6.17–6.14 (dd, J = 10.1, 17.0 Hz, 1H), 5.70–5.66 (dd, J = 1.1, 10.1 Hz, 1H), 4.16–4.15 (d, J = 4.1 Hz, 1H), 3.72–3.70 (m, 4H), 3.65–3.62 (m, 2H), 3.45–3.42 (m, 2H), 2.05–2.04 (m, 3H), 1.98–1.97 (m, 6H), 1.77–1.67 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.2, 166.9, 165.5, 130.4, 127.1, 45.3, 44.9, 44.7, 42.4, 41.9, 41.4, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C20H29N3O3Na [MNa]+: 382.2107, found: 382.2101.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-2-bromo-propenamide (7d)
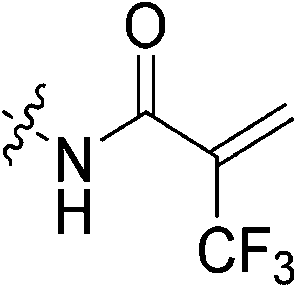
To a stirring solution of bromoacrylic acid (0.055 g, 0.360 mmol, 1.1 eq) and 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM (2 mL) under an atmosphere of nitrogen at 0 °C was added a solution of DCC (0.075 g, 0.360 mmol, 1.1 eq) and DMAP (0.001 g, 2 mol%) in anhydrous DCM (2 mL). The ice bath was then removed, and the reaction was stirred for 3 h. The mixture was then filtered through celite and the filtrate was diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid, which was purified by silica gel column chromatography (100% EtOAc, Rf = 0.55, dry loading using celite) (0.048 g, 34%). 1H NMR (300 MHz, CDCl3) δ 7.73 (br s, 1H), 7.00 (d, J = 1.5 Hz, 1H), 6.06 (d, J = 1.5 Hz, 1H), 4.12 (d, J = 4.2 Hz, 2H), 3.73–3.72 (m, 4H), 3.67–3.65 (m, 2H), 3.44–3.41 (m, 2H), 2.06 (m, 3H), 1.99–1.98 (m, 6H), 1.78–1.69 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 166.2, 161.2, 127.8, 122.5, 45.4, 44.8, 44.7, 42.4, 42.4, 41.9, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C20H28BrN3O3Na [MNa]+: 460.1212, found: 460.1210.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-2-trifluoromethyl-propenamide (7e)
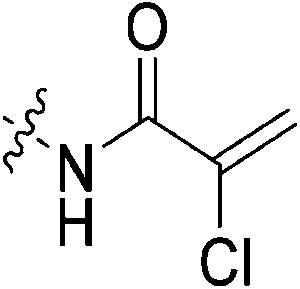
Compound 6 (0.100 g, 0.327 mmol, 1.0 eq) and Hünig's base (0.23 mL, 1.310 mmol, 4.0 eq) were dissolved in anhydrous DCM (3 mL) under an atmosphere of nitrogen. The solution was cooled to 0 °C and BOP (0.160 g, 0.360 mmol, 1.1 eq) and 2-trifluoromethylacrylic acid (0.051 g, 0.360 mmol, 1.1 eq) were added. The reaction was stirred for 1 h (note: running the reaction longer than this led to product degradation) and then poured into EtOAc (50 mL). The organic phase was washed with H2O (50 mL) and brine (50 mL) and then dried over MgSO4 and concentrated in vacuo to give a yellow oil. The crude material was purified by silica gel column chromatography (5% MeOH/EtOAc, Rf = 0.56, dry loading using celite) to give a white solid (0.025 g, 18%). 1H NMR (300 MHz, CDCl3) δ 7.21 (br s, 1H), 6.56 (quint, JHH = 1.5 Hz, JHF = 1.5 Hz, 1H), 5.52–5.47 (quint, JHH = 1.5 Hz, JHF = 1.5 Hz, 1H), 4.17 (d, J = 3.9 Hz, 2H), 3.75–3.71 (m, 4H), 3.67–3.63 (m, 2H), 3.44–3.41 (m, 2H), 2.06 (apparent s, 3H), 1.99–1.98 (m, 6H), 1.78–168 (m, 6H). 13C NMR (75 MHz, CD3OD) δ 177.2, 167.9, 163.7, 134.8 (q, JCF = 30.5 Hz), 127.5 (q, JCF = 5.3 Hz), 122.4 (q, JCF = 270.7 Hz), 54.0, 45.3, 45.0, 42.5, 42.1, 41.1, 39.2, 36.7, 28.1. 19F NMR (CDCl3, 283 MHz) δ −64.1 HRMS (ESI) calc'd for C21H8F3N3O3Na [MNa]+: 450.1980, found: 450.1972.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-2-chloro-propenamide (7f)
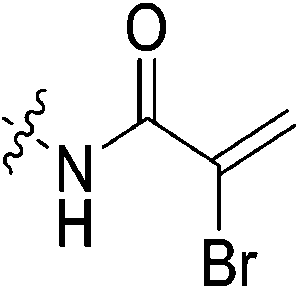
α-Chloroacrylic acid (0.073 g, 0.687 mmol, 2.1 eq) was dissolved in anhydrous DCM (2 mL) under an atmosphere of nitrogen. DCC (0.071 g, 0.343 mmol, 1.05 eq) in anhydrous DCM (1 mL) was added slowly and the reaction was stirred at R.T. for 1 h. The solution was then filtered to isolate the symmetric anhydride and the filtrate was added to a solution of 6 (0.1 g, 0.327 mmol, 1.0 eq) and NEt3 (0.091 mL, 0.654 mmol, 2.0 eq) in DCM (5 mL) under nitrogen and stirred for 6 h. The reaction was then diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid, which was purified by silica gel column chromatography (2% MeOH/EtOAc, Rf = 0.56, dry loading using celite) (0.080 g, 60%). 1H NMR (300 MHz, CDCl3) δ 7.71 (br s, 1H), 6.59 (d, J = 1.2 Hz, 1H), 5.82 (d, J = 1.2 Hz, 1H), 4.14 (d, J = 4.5 Hz, 2H), 3.73–3.64 (m, 6H), 3.45–3.41 (m, 6H), 2.06–2.04 (m, 3H), 1.99–1.98 (m, 6H), 1.78–1.69 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.2, 166.2, 161.1, 132.2, 123.0, 45.4, 44.8, 44.7, 42.5, 42.1, 41.9, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C20H28ClN3O3Na [MNa]+: 416.1717, found: 416.1706.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-3-chloro-propenamide (7g)
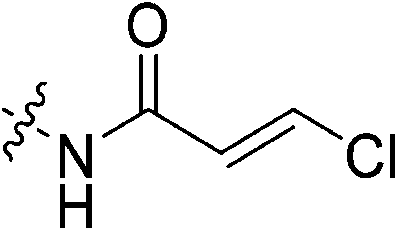
To a stirring solution of trans-3-chloroacrylic acid (0.075 g, 0.360 mmol, 1.1 eq) and 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM (2 mL) under an atmosphere of nitrogen at 0 °C was added a solution of DCC (0.075 g, 0.360 mmol, 1.1 eq) and DMAP (0.001 g, 2 mol%) in anhydrous DCM (2 mL). The ice bath was then removed, and the reaction was stirred for 3 h. The mixture was then filtered through celite and the filtrate was diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid, which was purified by silica gel column chromatography (2% MeOH/EtOAc, Rf = 0.39, dry loading using celite) (0.050 g, 39%). 1H NMR (300 MHz, CDCl3) δ 7.35 (d, J = 12.6 Hz, 1H), 6.69 (br s, 1H), 6.36 (d, J = 12.6 Hz, 1H), 4.17 (d, J = 4.2 Hz, 2H), 3.78–3.74 (m, 4H), 3.70–3.66 (m, 2H), 3.47–3.44 (m, 2H), 2.10 (m, 3H), 2.03–2.02 (m, 6H), 1.82–1.72 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.1, 166.4, 163.1, 134.6, 126.2, 45.2, 44.7, 44.6, 42.3, 41.8, 41.3, 39.1, 36.5, 28.4. HRMS (ESI) calc'd for C20H28ClN3O3Na [MNa]+: 416.1685, found: 416.1695.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-3-methylester-propenamide (7h)
To a stirring solution of methyl fumarate (0.047 g, 0.360 mmol, 1.1 eq) and 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM (2 mL) under an atmosphere of nitrogen at 0 °C was added a solution of DCC (0.075 g, 0.360 mmol, 1.1 eq) and DMAP (0.001 g, 2 mol%) in anhydrous DCM (2 mL). The ice bath was then removed, and the reaction was stirred for 3 h. The mixture was then filtered through celite, and the filtrate was diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid, which was purified by silica gel column chromatography (2% MeOH/EtOAc, Rf = 0.44, dry loading using celite) (0.067 g, 49%). 1H NMR (300 MHz, CDCl3) δ 7.02–6.98 (m, 2H, NH overlapping with olefin proton), 6.85 (d, J = 11.4 Hz, 1H), 4.17 (d, J = 3.0 Hz, 2H), 3.80 (s, 3H), 3.74–3.72 (m, 4H), 3.67–3.64 (m, 2H), 3.44–3.42 (m, 2H), 2.06–2.04 (m, 3H), 1.99–1.98 (m, 6H), 1.77–1.69 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 166.4, 166.0, 163.6, 135.9, 130.6, 52.4, 45.3, 44.8, 44.7, 42.5, 41.9, 41.7, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C22H31N3O5Na [MNa]+: 440.2161, found: 440.2150.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] propiolamide (7i)
To a stirring solution of propiolic acid (0.023 mL, 0.360 mmol, 1.1 eq) and 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM (2 mL) under an atmosphere of nitrogen at 0 °C was added a solution of DCC (0.075 g, 0.360 mmol, 1.1 eq) and DMAP (0.001 g, 2 mol%) in anhydrous DCM (2 mL). The ice bath was then removed, and the reaction was stirred for 3 h. The mixture was then filtered through celite and the filtrate was diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid, which was purified by silica gel column chromatography (2% MeOH/EtOAc, Rf = 0.59, dry loading using celite) (0.062 g, 53%). 1H NMR (300 MHz, CDCl3) δ 7.03 (br s, 1H), 4.12 (d, J = 3.9 Hz, 2H), 3.75–3.63 (m, 6H), 3.40–3.38 (m, 2H), 2.84 (s, 1H), 2.06–2.04 (m, 3H), 1.99–1.98 (m, 6H), 1.78–1.68 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 165.8, 152.0, 76.9, 74.1, 45.3, 44.8, 44.7, 42.5, 41.9, 41.6, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C20H27N3O3Na [MNa]+: 380.1950, found: 380.1953.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] chloroacetamide (7j)
Under an atmosphere of nitrogen at −78 °C, chloroacetyl chloride (0.024 mL, 0.297 mmol, 1.1 eq.) was slowly added to a solution of 6 (0.082 g, 0.270 mmol, 1.0 eq.) and NEt3 (0.190 mL, 1.350 mmol, 5.0 eq.) in anhydrous DCM (3 mL). The reaction mixture was stirred at −78 °C for 3 h, diluted with DCM (7 mL) and washed with 0.2 M HCl (3 × 10 mL). The organic layer was dried over MgSO4, and the solvent was evaporated in vacuo to give the product as an off-white solid (0.0777 g, 75%). 1H NMR (300 MHz, CDCl3) δ 7.61 (apparent s, 1H), 4.11–4.10 (d, J = 4.2 Hz, 2H), 4.08 (s, 2H), 3.75–3.70 (m, 4H), 3.66–3.63 (m, 2H), 3.43–3.40 (m, 2H), 2.06 (apparent s, 3H), 1.99–1.98 (m, 6H), 1.78–1.68 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 166.3, 166.2, 45.4, 44.8, 44.7, 42.5, 42.4, 41.9, 41.6, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C19H28ClN3O3Na [MNa]+: 404.1717, found: 404.1745.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] chlorofluoroacetamide (7k)
To a stirring solution of sodium chlorofluoroacetate (0.066 g, 0.491 mmol, 1.5 eq) and 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM:DMF (3.0:0.5 mL) under an atmosphere of nitrogen at 0 °C was added T3P (50% by weight in EtOAc) (0.3 mL, 0.491 mmol, 1.5 eq) and Hünig's base (0.12 mL, 0.654 mmol, 2.0 eq). The reaction was stirred at R.T. for 3.5 h and then diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid which was then washed with pentane (0.040 g, 31%). 1H NMR (300 MHz, CDCl3) δ 7.47 (br s, 1H), 6.33 (d, JHF = 50.4 Hz, 1H), 4.13 (d, J = 4.2 Hz, 2H), 3.76–3.71 (m, 4H), 3.67–3.64 (m, 2H), 3.43–3.40 (m, 2H), 2.06–2.04 (m, 3H), 1.99–1.98 (m, 6H), 1.78–1.68 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 165.7, 164.1 (d, JCF = 24.4 Hz), 93.9 (d, JCF = 255.2 Hz), 45.4, 44.7, 44.7, 42.5, 41.9, 41.4, 39.2, 36.7, 28.5. 19F NMR (CDCl3, 283 MHz) δ −144.3. HRMS (ESI) calc'd for C19H27ClFN3O3Na [MNa]+: 422.1623, found: 422.1623.
N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] cyanoacetamide (7l)
Cyanoacetic acid (0.031 g, 0.360 mmol, 1.1 eq), EDCI–HCl (0.075 g, 0.392 mmol, 1.2 eq), HOBt·H2O (0.060 g, 0.392 mmol, 1.2 eq), and Hünig's base (0.14 mL, 0.785 mmol, 2.4 eq) were stirred in dry DCM (1 mL) at R.T. for 30 min. Compound 6 (0.100 g, 0.327 mmol, 1.0 eq) was then added as a solution in DCM (1 mL). The reaction was left to stir for 24 h, after which point it was diluted with DCM (15 mL) and washed with H2O (2 × 15 mL), followed by brine (15 mL). The organic layer was dried over MgSO4 and concentrated in vacuo to give a yellow solid which was purified by silica gel column chromatography (7.5% MeOH/EtOAc, Rf = 0.45, dry loading using celite) to give a white solid (0.022 g, 18%). 1H NMR (300 MHz, CDCl3) δ 7.35 (br s, 1H), 4.11 (d, J = 4.1 Hz, 1H), 3.73–3.70 (m, 4H), 3.64–3.60 (m, 2H), 3.46 (s, 2H), 3.42–3.39 (m, 2H), 2.05 (apparent s, 3H), 1.97–1.96 (m, 6H), 1.77–1.67 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.2, 166.2, 161.4, 114.4, 45.2, 44.7, 44.7, 42.4, 41.9, 41.9, 39.2, 36.6, 28.5, 25.9. HRMS (ESI) calc'd for C20H23N4O3Na [MNa]+: 395.2059, found: 395.2088.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] pentafluorobenzenesulfonamide (7m)
To a stirring solution of 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM (2 mL) under an atmosphere of nitrogen at 0 °C. Was added Hünig's base (0.12 mL, 0.654 mmol, 2.0 eq) and pentafluorobenzenesulfonyl chloride (0.055 mL, 0.360 mmol, 1.1 eq). The reaction was warmed to R.T. and stirred for 2 h and then diluted with EtOAc (30 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid which was purified by silica gel column chromatography (30% EtOAc/hexanes, Rf = 0.43, dry loading using celite) (0.157 g, 90%). 1H NMR (300 MHz, CDCl3) δ 4.95 (s, 2H), 3.79–3.73 (m, 4H), 3.59–3.58 (m, 2H), 3.47–3.44 (m, 2H), 2.06–2.04 (m, 3H), 2.00–1.99 (m, 6H), 1.79–1.69 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 163.9, 147.4–147.2 (m), 143.9–143.7 (m), 139.2–139.5 (m), 136.5–136.1 (m), 115.8, 52.1, 45.4, 45.4, 44.6, 42.9, 41.9, 39.2, 36.6, 28.5. 19F NMR (CDCl3, 283 MHz) δ −132.8 (br s), −141.8–142.1 (m), −158.2–158.4 (m). HRMS (ESI) calc'd for C19H25F5N3O6S [M–H]−: 534.1486, found: 534.1457.2-Cyano-3-cyclopropylacrylic acid (I3)
To a suspension of cyanoacetic acid (0.85 g, 10 mmol, 1.0 eq) in toluene (10 mL) in a sealed tube was added cyclopropanecarboxaldehyde (0.75 mL, 10 mmol, 1.0 eq) and NH4OAc (0.039 g, 0.5 mmol, 0.05 eq). The reaction was stirred at 110 °C for 2 h and then cooled down to R.T. The resulting precipitate was filtered, washed with hexanes, and dried in vacuo to yield a yellow crystalline solid (1.04 g, 73%) which was used without further purification. 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 11.6 Hz, 1H), 1.95–1.86 (m, 1H), 1.27–1.22 (m, 2H), 1.04–1.00 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.0, 163.2, 116.0, 106.5, 25.3, 16.4, 11.5. Characterization data are consistent with previously reported values.45E configuration confirmed by X-ray crystallography (see ESI†).N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-2-cyano-3-cyclopropylpropenamide (7n)
To a stirring solution of I3 (0.054 g, 0.392 mmol, 1.2 eq) and 6 (0.1 g, 0.327 mmol, 1.0 eq) in anhydrous DCM (2 mL) under an atmosphere of nitrogen at 0 °C was added a solution of DCC (0.075 g, 0.360 mmol, 1.1 eq) and DMAP (0.001 g, 2 mol%) in anhydrous DCM (2 mL). The ice bath was then removed, and the reaction was stirred for 3 h. The mixture was then filtered through celite and the filtrate was diluted with EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (25 mL), sat. NaHCO3 (25 mL), and brine again (25 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a white solid, which was purified by preparative thin layer chromatography (0–2% MeOH/DCM, Rf = 0.43, dry loading using celite) (0.083 g, 57%). 1H NMR (300 MHz, CDCl3) δ 7.17 (br s, 1H), 7.00 (d, J = 11.4 Hz, 1H), 4.14 (d, J = 4.2 Hz, 2H), 3.75–3.63 (m, 6H), 3.43–3.40 (m, 2H), 2.07–2.02 (m, 4H, CH of cyclopropyl overlapping with adamantane protons), 1.98–1.97 (m, 6H), 1.77–1.73 (m, 6H), 1.32–1.24 (m, 2H), 0.96–0.91 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 176.2, 166.1 (2C), 160.1, 115.5, 106.6, 45.5, 44.7, 44.7, 42.5, 41.9, 41.8, 39.2, 36.7, 28.5 (4C), 16.0, 11.3. HRMS (ESI) calc'd for C24H32N4O3Na [MNa]+: 447.2362, found: 447.2353.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl]-2-oxo-propanamide (7o)
Pyruvic acid (0.027 mL, 0.392 mmol, 1.2 eq) was added to a flame-dried 2-neck round bottom flask, flushed with nitrogen, and dissolved in anhydrous DCM (1 mL). Anhydrous DMF (catalytic, 2 drops) was added, and the mixture was cooled to 0 °C. A bubbler was attached to the flask and oxalyl chloride (2 M in DCM) (0.2 mL, 0.392 mmol, 1.2 eq) was added dropwise, and the reaction was stirred at R.T. for 3 h. A solution of 6 (0.1 g, 0.327 mmol, 1.0 eq) and NEt3 (0.070 mL, 0.491 mmol, 1.5 eq) in DCM (1 mL) was then added to the reaction mixture at 0 °C. White fumes immediately formed along with a precipitate. An additional 2 mL of DCM was then added to the reaction mixture to enable uniform stirring. The reaction was left to stir at R.T. for 15 h, after which it was quenched with 1 M HCl (5 mL). The layers were separated, and the organic layer was washed with brine (5 mL), sat. NaHCO3 (5 mL), and brine (5 mL) again. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude material was purified by silica gel column chromatography using 0–5% MeOH/EtOAc gradient elution (100% EtOAc, Rf = 0.36, dry loading using celite) to give a white solid (0.059 g, 48%). 1H NMR (300 MHz, CDCl3) δ 7.82 (br s, 1H), 4.07 (s, 2H), 3.73–3.63 (m, 6H), 3.42 (apparent s, 2H), 2.45 (s, 3H), 2.03–2.02 (m, 3H), 1.97–1.96 (m, 6H), 1.76–1.66 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 195.8, 176.3, 165.9, 160.3, 45.4, 44.7, 44.7, 42.4, 41.9, 41.0, 39.1, 36.6, 28.4, 24.6. HRMS (ESI) calc'd for C20H29N3O4Na [MNa]+: 398.2056, found: 398.2071.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] isothiocyanate (7p)
To a solution of 6 (0.1 g, 0.327 mmol, 1.0 eq) in DCM (2 mL) was added sat. NaHCO3 (2 mL). The biphasic mixture was stirred vigorously and cooled to 0 °C before adding thiophosgene (0.028 mL, 0.360 mmol, 1.1 eq) dropwise. The reaction was stirred at 0 °C for 10 min then at R.T. for 1 h, at which point the reaction was complete by TLC analysis (10% MeOH/DCM, Rf = 0.77). The layers were separated, and the aqueous phase was extracted with DCM (3 × 5 mL). The combined organic layer was dried over MgSO4 and concentrated in vacuo to yield yellow oil which was triturated with hexanes to give a pale-yellow solid (0.09 g, 79%). 1H NMR (300 MHz, CDCl3) δ 4.31 (s, 2H), 3.75–3.69(m, 4H), 3.65–3.61 (m, 2H), 3.35–3.32 (m, 2H), 2.05–2.02 (m, 3H), 1.97–1.96 (m, 6H), 1.76–1.67 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.1, 163.6, 139.6, 47.6, 45.4, 45.2, 44.5, 42.8, 41.8, 39.1, 36.5, 28.4. HRMS (ESI) calc'd for C18H25N3O2SNa [MNa]+: 370.1565, found: 370.1558.Azidoacetic acid (I4)
To a solution of bromoacetic acid (2 g, 14.40 mmol, 1.0 eq) in H2O (5 mL) was added a solution of NaN3 (1.84 g, 28.80 mmol, 2.0 eq) in H2O (5 mL). The reaction was stirred for 24 h at room temperature, followed by dropwise addition of 12 M HCl until pH = 1 was reached. The aqueous mixture was extracted using Et2O (3 × 20 mL) and the combined organic later was dried over MgSO4 and concentrated in vacuo to obtain azidoacetic acid as a pale-yellow oil which was used without further purification (1.42 g, 98%). 1H NMR (300 MHz, CDCl3) δ 8.29 (br s, 1H), 3.95 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 173.5, 50.1. Characterization data are consistent with previously reported values.61N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] azidoacetamide (I5)
Azidoacetic acid (I4) (0.1 g, 0.98 mmol, 1.5 eq), HBTU (0.397 g, 1.05 mmol, 1.6 eq), Hünig's base (0.35 mL, 1.97 mmol, 3.0 eq), and 6 (0.2 g, 0.655 mmol, 1.0 eq) were stirred in anhydrous DCM (5 mL) at R.T. for 3 h at which point the reaction was confirmed complete by TLC analysis. The reaction was concentrated in vacuo and the residue was redissolved in EtOAc (50 mL). The organic phase was washed with 5% AcOH (3 × 15 mL), brine (15 mL), sat. NaHCO3 (15 mL), and brine again (15 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a brown solid, which was purified by silica gel column chromatography (5% MeOH/EtOAc, Rf = 0.43, dry loading using celite) and then washing with water to give a white solid (0.12 g, 47%). 1H NMR (300 MHz, CDCl3) δ 7.29 (br s, 1H), 4.10 (d, J = 3.9 Hz, 2H), 4.02 (s, 2H), 3.73–3.70 (m, 4H), 3.65–3.63 (m, 2H), 3.43–3.39 (m, 2H), 2.06–2.04 (m, 3H), 1.99–1.98 (m, 6H), 1.78–1.68 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.3, 166.9, 166.3, 52.7, 45.4, 44.8, 44.7, 42.4, 41.9, 41.2, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C19H28N6O3Na [MNa]+: 411.2107, found: 411.2121.N-2-[4-[[1-Adamantanecarbonyl]-1-piperazinyl]-2-oxoethyl] diazoacetamide (7q)
I5 (0.05 g, 0.129 mmol, 1.0 eq) was dissolved in a solution of THF/H2O (2 mL/0.3 mL) at R.T. and sparged with nitrogen for 5 minutes. N-Succinimidyl 3-(diphenylphosphino)propionate (0.048 g, 0.135, 1.05 eq) was then added and the reaction was stirred for 18 h. A solution of sat. NaHCO3 (2 mL) was added, and the reaction was stirred for another 18 h, after which the reaction mixture was diluted with brine (15 mL) and extracted with EtOAc (3 × 15 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to give a yellow solid, which was purified by silica gel column chromatography (5% MeOH/EtOAc, Rf = 0.27, dry loading using celite) to give a yellow solid (0.025 g, 52%) 1H NMR (300 MHz, CDCl3) δ 6.31 (br s, 1H), 4.87 (s, 1H), 4.13 (d, J = 4.2 Hz, 2H), 3.73–3.69 (m, 4H), 3.63–3.60 (m, 2H), 3.47–3.40 (m, 2H), 2.05–2.04 (m, 3H), 1.98–1.97 (m, 6H), 1.77–1.67 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 176.2, 167.2, 165.6, 47.6, 45.2, 44.9, 44.7, 42.4, 41.9, 41.7, 39.2, 36.7, 28.5. HRMS (ESI) calc'd for C19H27N5O3Na [MNa]+: 396.2012, found: 396.1992.TG2 inhibition assay
Recombinant TG2 was expressed from E. coli and purified as described previously.19 TG2 activity was determined according to a previously published colorimetric activity assay using the chromogenic substrate Cbz-Glu(γ-p-nitrophenylester)Gly (AL5).19,46 In order to determine irreversible inhibition parameters for each inhibitor, enzymatic assays were run under Kitz and Wilson conditions,50 in the presence of 100 μM AL5 substrate, in triplicate. A large excess of substrate is used to ensure that any curvature in the slope is not attributed to depletion of the substrate, but rather to time-dependent inhibition. Buffered solutions of 50 mM of 3-(4-morpholino)propanesulfonic acid (MOPS) (pH 6.9), 7.5 mM CaCl2, 100 μM AL5, and various concentrations of inhibitor (from 0.25 to 1200 μM, depending on the inhibitor) were prepared in a 96-well polystyrene microplate with a final volume of 200 μL at 25 °C. AL5 and inhibitor stocks were prepared in DMSO ensuring that the final concentration of this co-solvent did not exceed 5% v/v, with the exception of inhibitor 7d, for which a stock solution was prepared in DMF (due to poor solubility in DMSO) and the final volume of DMF was less than 1% v/v. If necessary, working stocks of each inhibitor were diluted with water to maintain less than 5% v/v DMSO. To initiate the enzymatic reaction, 5 mU mL−1 (0.25 μM) TG2, or water for the blank, was added to the well and the formation of the hydrolysis product, p-nitrophenolate, was followed at 405 nm for 20 min (a period of time over which the positive control is linear and not being impacted by substrate depletion) using a BioTek Synergy 4 plate reader. Background AL5 hydrolysis was corrected for by subtracting the blank from each reaction. Observed first order rate constants of inactivation (kobs) were obtained by fitting the inhibition data sets with non-linear regression to mono-exponential association eqn (1) using GraphPad Prism software.| Abst = Absmax(1 − e−kobst) | (1) |
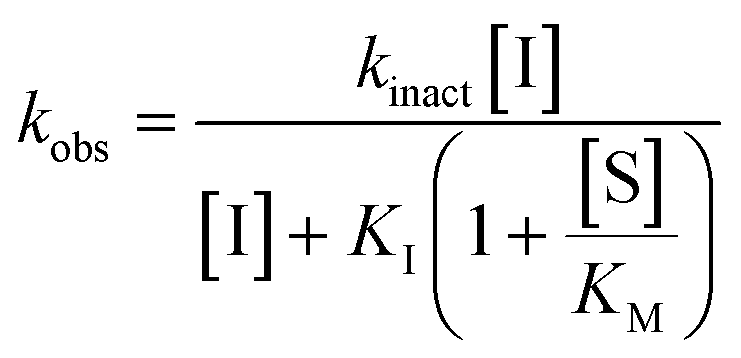 | (2) |
Simple linear regression was performed on the kobs values measured at the lowest concentrations of each inhibitor for validation of kinact/KI ratios.
For inhibitor 7o, the assay was performed in a similar manner as described above at AL5 concentrations of 100 μM and 50 μM. Initial rates of the uninhibited reaction (positive control) and the inhibited reactions (v0) were determined over the first 10% of conversion of the positive control. The reciprocals of the average initial rate (1/v0) were plotted against inhibitor concentration at each AL5 concentration to construct a Dixon plot according to eqn (3) from which the Ki was determined by the point of intersection of the two resulting lines.62
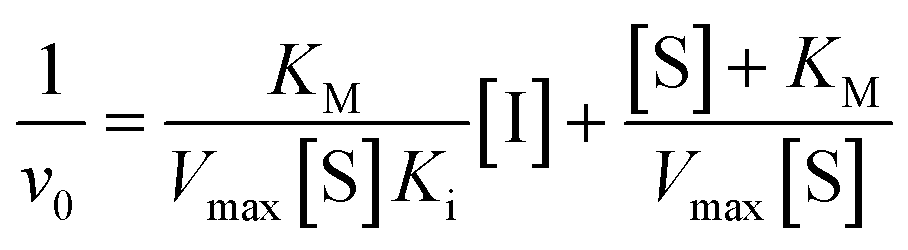 | (3) |
Isozyme selectivity
First, for each inhibitor, a single corrected inhibitor concentration ([I]/α) was chosen, which would result in the complete inactivation of TG2 in under 5 min. Then for each other isozyme, the concentration of inhibitor was adjusted according to the value of α determined by the concentration and KM value of the substrate of each isozyme assay. In this way the effective inhibitor concentration ([I]/α) remained constant for each isozyme.The activities of TG1 and TG6 were measured by a colorimetric assay using the substrate AL5. Assays were performed as previously described for TG2 under Kitz & Wilson conditions established for each transglutaminase isoform by varying the concentration of substrate to 112 μM and 436 μM of AL5 for TG1 and TG6, respectively. The reaction was initiated with the addition of enzyme, 0.10 μM TG1 or 0.32 μM TG6. Formation of the hydrolysis product, p-nitrophenolate, was followed at 405 nm for 100 min for TG1 or 60 min for TG6.19
The isopeptidase activities of activated TG3a and hFXIIIa (purchased from Zedira) were measured by a fluorescence-based assay based on the use of the peptidic FRET-quenched probe A101 (Zedira). The final assay mixture comprised 50 mM Tris (pH 7.0), 10 mM CaCl2, 100 mM NaCl, 2.8 mM TCEP, 50 μM A101 and 14 mM H-Gly-OMe. The reaction was monitored at 25 °C using a BioTek Synergy 4 plate reader (Ex/Em: 318/413 nm). Enzymatic inhibition assays were run under Kitz and Wilson conditions, which were established for TG3a and FXIIIa at a substrate (A101) concentration of 50 μM using enzyme concentrations of 0.17 μM and 0.11 μM for TG3a and FXIIIa, respectively.19,54,55 Experiments were completed at least in triplicate.
In cases where irreversible inhibition (time-dependent curvature) was clearly observed, a kobs value was obtained as described above. If the inhibition resembled a reversible mode (instantaneous decrease in rate, linear product increase), a percent inhibition (% inhibition = vinhibited/vuninhibited × 100%) was determined.
Intrinsic reactivity
HPLC traces were collected by Gilson-Mandel GXP271 high performance liquid chromatography (HPLC) with UV detection at 214 and 254 nm (Phenomenex Luna, 150 mm × 4.6 mm, 30 min, 1.5 mL min−1 flow rate, 20–95% CH3CN with 0.1% TFA in H2O with 0.1% TFA, 20 min method).Three inhibitors from the series were selected in order to study the intrinsic reaction rates with a 30-fold excess of GSH. In 2-mL HPLC vials fitted with a pre-slit screw cap, 150 μL of 0.1 M GSH stock solution in the corresponding aqueous buffer was diluted to a final concentration of 15 mM in 830 μL aq buffer. The reactions were initiated by addition of 20 μL of 25 mM warhead stock solution in DMSO to a final concentration of 0.5 mM with 2% v/v DMSO as co-solvent. Once initiated, the HPLC vials were inverted, placed on the autosampler tray and an initial aliquot was taken immediately. The vials were incubated at 22 °C with aliquots taken at pre-determined time points over the course of at least 5–6 half lives. Both the disappearance of inhibitor and appearance of adduct were monitored for inhibitors 7c and 7j. Only the disappearance of inhibitor 7f was monitored due to further reaction of the thiol adduct and formation of side products. Experiments were completed in duplicate for each inhibitor.
The increasing area under the curve (AUC) of the adduct peaks of the HPLC chromatograms were fitted to a mono-exponential association model with the constraint that Y0 = 0 (eqn (4)).
| AUC = AUCmax(1 − e−kobst) | (4) |
| AUC = AUC0(e−kobst) | (5) |
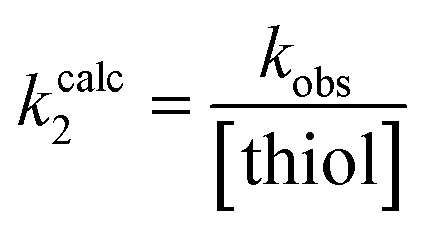 | (6) |
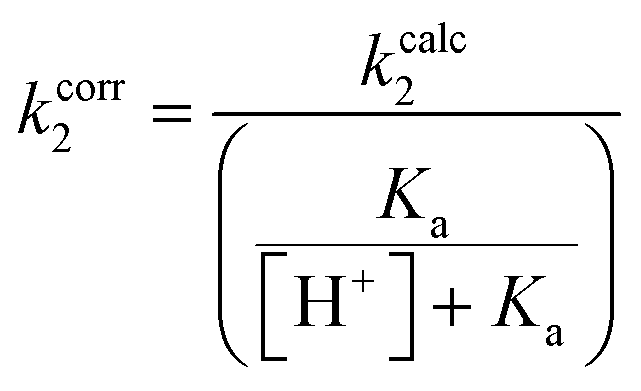 | (7) |
Pharmacokinetic stability
000 cells per well containing 1 μM test compound in a 24-well plate, were incubated in a humidified CO2 incubator for up to 2 h, in duplicate. Aliquots of 75 μL were collected at 0, 5, 15, 30, 60, 120 min to determine metabolic half-life. The samples were deproteinized with 2.5 volumes of acetonitrile containing 0.1% formic acid and 0.5 μM glyburide (as an internal standard), and the supernatant obtained by centrifugation (2500 rpm, 10 min) were analyzed by LC-MS/MS. Midazolam and 7-ethoxycoumarin were utilized as positive controls for the assay.
List of abbreviations
| DCM | Dichloromethane |
| Et2O | Diethyl ether |
| EtOAc | Ethyl acetate |
| AcOH | Acetic acid |
| HBTU | N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate |
| MeOH | Methanol |
| TFA | Trifluoroacetic acid |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| HOBt | Hydroxybenzotriazole |
| R.T. | Room temperature |
| EtOH | Ethanol |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DMAP | 4-Dimethylaminopyridine |
| BOP | (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| NEt3 | Triethylamine |
| DMF | Dimethylformamide |
| T3P | Propanephosphonic acid anhydride |
Author contributions
Conceptualization: J. W. K. and L. M.; funding acquisition: J. W. K.; supervision: J. W. K.; investigation and methodology: L. M. (synthesis, characterization, and enzymatic assays), S. K. I. W. (intrinsic reactivity), H. R. I., L. N., and H. K. (pharmacokinetic measurements); writing – original draft: L. M., writing – reviewing and editing: all authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J. W. K. is grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes of Health Research (CIHR) for funding. L. M. thanks NSERC for a CGS-M award and Roberto Diaz-Rodriguez from the Murugesu Lab at the University of Ottawa for obtaining crystallographic data.References
- S. Gundemir, G. Colak, J. Tucholski and G. V. Johnson, Transglutaminase 2: a molecular Swiss army knife, Biochim. Biophys. Acta, 2012, 1823(2), 406–419 CrossRef PubMed.
- R. L. Eckert, M. T. Kaartinen, M. Nurminskaya, A. M. Belkin, G. Colak, G. V. Johnson and K. Mehta, Transglutaminase regulation of cell function, Physiol. Rev., 2014, 94(2), 383–417 CrossRef PubMed.
- J. W. Keillor, C. M. Clouthier, K. Y. P. Apperley, A. Akbar and A. Mulani, Acyl transfer mechanisms of tissue transglutaminase, Bioorg. Chem., 2014, 57, 186–197 CrossRef PubMed.
- W. P. Katt, M. A. Antonyak and R. A. Cerione, Drug Discovery Today, 2018, 23, 575 CrossRef PubMed.
- J. S. K. Chen and K. Mehta, Tissue transglutaminase: an enzyme with a split personality, Int. J. Biochem. Cell Biol., 1999, 31(8), 817–836 CrossRef PubMed.
- S. Liu, R. A. Cerione and J. Clardy, Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(5), 2743–2747 CrossRef CAS PubMed.
- D. M. Pinkas, P. Strop, A. T. Brunger and C. Khosla, Transglutaminase 2 undergoes a large conformational change upon activation, PLoS Biol., 2007, 5(12), 2788–2796 CrossRef CAS PubMed.
- J. W. Keillor and G. V. W. Johnson, Transglutaminase 2 as a therapeutic target for neurological conditions, Expert Opin. Ther. Targets, 2021, 25, 721 CrossRef CAS PubMed.
- G. E. Begg, S. R. Holman, P. H. Stokes, J. M. Matthews, R. M. Graham and S. E. Iismaa, J. Biol. Chem., 2006, 281, 12603 CrossRef CAS PubMed.
- G. E. Begg, S. R. Holman, P. H. Stokes, J. M. Matthews, R. M. Graham and S. E. Iismaa, Mutation of a Critical Arginine in the GTP-Binding Site of Transglutaminase 2 Disinhibits Intracellular Cross-Linking Activity, J. Biol. Chem., 2006, 281(18), 12603–12609 CrossRef CAS PubMed.
- S. E. Iismaa, B. M. Mearns, L. Lorand and R. M. Graham, Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders, Physiol. Rev., 2009, 89(3), 991–1023 CrossRef CAS PubMed.
- T. S. Johnson, M. Fisher, J. L. Haylor, Z. Hau, N. J. Skill, R. Jones, R. Saint, I. Coutts, M. E. Vickers, A. M. El Nahas and M. Griffin, Transglutaminase inhibition reduces fibrosis and preserves function in experimental chronic kidney disease, J. Am. Soc. Nephrol., 2007, 18(12), 3078–3088 CrossRef CAS PubMed.
- C. Kerr, H. Szmacinski, M. L. Fisher, B. Nance, J. R. Lakowicz, A. Akbar, J. W. Keillor, T. Lok Wong, R. Godoy-Ruiz, E. A. Toth, D. J. Weber and R. L. Eckert, Transamidase site-targeted agents alter the conformation of the transglutaminase cancer stem cell survival protein to reduce GTP binding activity and cancer stem cell survival, Oncogene, 2017, 36(21), 2981–2990 CrossRef CAS PubMed.
- M. L. Fisher, J. W. Keillor, W. Xu, R. L. Eckert and C. Kerr, Transglutaminase Is Required for Epidermal Squamous Cell Carcinoma Stem Cell Survival, Mol. Cancer Res., 2015, 13(7), 1083–1094 CrossRef CAS PubMed.
- Z. Szondy, I. Korponay-Szabó, R. Király, Z. Sarang and G. J. Tsay, Transglutaminase 2 in human diseases, BioMedicine, 2017, 7(3), 15 CrossRef PubMed.
- S. Caja, M. Mäki, K. Kaukinen and K. Lindfors, Antibodies in celiac disease: implications beyond diagnostics, Cell. Mol. Immunol., 2011, 8(2), 103–109 CrossRef CAS PubMed.
- D. Schuppan, M. Mäki, K. E. A. Lundin, J. Isola, T. Friesing-Sosnik, J. Taavela, A. Popp, J. Koskenpato, J. Langhorst, Ø. Hovde, M.-L. Lähdeaho, S. Fusco, M. Schumann, H. P. Török, J. Kupcinskas, Y. Zopf, A. W. Lohse, M. Scheinin, K. Kull, L. Biedermann, V. Byrnes, A. Stallmach, J. Jahnsen, J. Zeitz, R. Mohrbacher and R. Greinwald, A Randomized Trial of a Transglutaminase 2 Inhibitor for Celiac Disease, N. Engl. J. Med., 2021, 385(1), 35–45 CrossRef CAS PubMed.
- Z. Sarang, B. Tóth, Z. Balajthy, K. Köröskényi, É. Garabuczi, L. Fésüs and Z. Szondy, Some lessons from the tissue transglutaminase knockout mouse, Amino Acids, 2009, 36(4), 625–631 CrossRef CAS PubMed.
- A. Akbar, N. M. R. McNeil, M. R. Albert, V. Ta, G. Adhikary, K. Bourgeois, R. L. Eckert and J. W. Keillor, Structure–Activity Relationships of Potent, Targeted Covalent Inhibitors That Abolish Both the Transamidation and GTP Binding Activities of Human Tissue Transglutaminase, J. Med. Chem., 2017, 60(18), 7910–7927 CrossRef CAS PubMed.
- J. W. Keillor, K. Y. P. Apperley and A. Akbar, Inhibitors of tissue transglutaminase, Trends Pharmacol. Sci., 2015, 36(1), 32–40 CrossRef CAS PubMed.
- N. M. R. McNeil, E. W. J. Gates, N. Firoozi, N. J. Cundy, J. Leccese, S. Eisinga, J. D. A. Tyndall, G. Adhikary, R. L. Eckert and J. W. Keillor, Structure-activity relationships of N-terminal variants of peptidomimetic tissue transglutaminase inhibitors, Eur. J. Med. Chem., 2022, 232, 114172 CrossRef CAS PubMed.
- A. M. M. Rangaswamy, P. Navals, E. W. J. Gates, S. Shad, S. K. I. Watt and J. W. Keillor, Structure–activity relationships of hydrophobic alkyl acrylamides as tissue transglutaminase inhibitors, RSC Med. Chem., 2022, 13(4), 413–428 RSC.
- Chapter 2 - Warheads for designing covalent inhibitors and chemical probes, in Advances in Chemical Proteomics, ed. P. Ábrányi-Balogh and G. M. Keserű, Elsevier, 2022, pp. 47–73 Search PubMed.
- C. Pardin, S. M. F. G. Gillet and J. W. Keillor, Synthesis and Evaluation of Peptidic Irreversible Inhibitors of Tissue Transglutaminase, Bioorg. Med. Chem., 2006, 14(24), 8379–8385 CrossRef CAS PubMed.
- C. Marrano, P. de Macédo, P. Gagnon, D. Lapierre, C. Gravel and J. Keillor, Synthesis and Evaluation of Novel Dipeptide-Bound 1,2,4-Thiadiazoles as Irreversible Inhibitors of Guinea Pig Liver Transglutaminase, Bioorg. Med. Chem., 2002, 9(12), 3231–3241 CrossRef PubMed.
- D. Halim, K. Caron and J. W. Keillor, Synthesis and Evaluation of Peptidic Maleimides as Transglutaminase Inhibitors, Bioorg. Med. Chem. Lett., 2007, 17(2), 305–308 CrossRef CAS PubMed.
- P. de Macédo, C. Marrano and J. W. Keillor, Synthesis of Dipeptide-Bound Epoxides and α,β-Unsaturated Amides as Potential Irreversible Transglutaminase Inhibitors, Bioorg. Med. Chem., 2002, 10(2), 355–360 CrossRef PubMed.
- E. Badarau, Z. Wang, D. L. Rathbone, A. Costanzi, T. Thibault, C. E. Murdoch, S. El Alaoui, M. Bartkeviciute and M. Griffin, Development of Potent and Selective Tissue Transglutaminase Inhibitors: Their Effect on TG2 Function and Application in Pathological Conditions, Chem. Biol., 2015, 22(10), 1347–1361 CrossRef CAS PubMed.
- C. Klöck, Z. Herrera, M. Albertelli and C. Khosla, Discovery of potent and specific dihydroisoxazole inhibitors of human transglutaminase 2, J. Med. Chem., 2014, 57(21), 9042–9064 CrossRef PubMed.
- M. E. Prime, O. A. Andersen, J. J. Barker, M. A. Brooks, R. K. Y. Cheng, I. Toogood-Johnson, S. M. Courtney, F. A. Brookfield, C. J. Yarnold, R. W. Marston, P. D. Johnson, S. F. Johnsen, J. J. Palfrey, D. Vaidya, S. Erfan, O. Ichihara, B. Felicetti, S. Palan, A. Pedret-Dunn, S. Schaertl, I. Sternberger, A. Ebneth, A. Scheel, D. Winkler, L. Toledo-Sherman, M. Beconi, D. Macdonald, I. Muñoz-Sanjuan, C. Dominguez and J. Wityak, Discovery and Structure–Activity Relationship of Potent and Selective Covalent Inhibitors of Transglutaminase 2 for Huntington's Disease, J. Med. Chem., 2012, 55(3), 1021–1046 CrossRef CAS PubMed.
- N. Shindo and A. Ojida, Recent progress in covalent warheads for in vivo targeting of endogenous proteins, Bioorg. Med. Chem., 2021, 47, 116386 CrossRef PubMed.
- M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors, J. Med. Chem., 2014, 57(23), 10072–10079 CrossRef PubMed.
- G. Xia, W. Chen, J. Zhang, J. Shao, Y. Zhang, W. Huang, L. Zhang, W. Qi, X. Sun, B. Li, Z. Xiang, C. Ma, J. Xu, H. Deng, Y. Li, P. Li, H. Miao, J. Han, Y. Liu, J. Shen and Y. Yu, A Chemical Tuned Strategy to Develop Novel Irreversible EGFR-TK Inhibitors with Improved Safety and Pharmacokinetic Profiles, J. Med. Chem., 2014, 57(23), 9889–9900 CrossRef PubMed.
- J. L. Montaño, B. J. Wang, R. F. Volk, S. E. Warrington, V. G. Garda, K. L. Hofmann, L. C. Chen and B. W. Zaro, Improved Electrophile Design for Exquisite Covalent Molecule Selectivity, ACS Chem. Biol., 2022, 17(6), 1440–1449 CrossRef PubMed.
- K. Y. P. Apperley, I. Roy, V. Saucier, N. Brunet-Filion, S.-P. Piscopo, C. Pardin, É. De Francesco, C. Hao and J. W. Keillor, Development of new scaffolds as reversible tissue transglutaminase inhibitors, with improved potency or resistance to glutathione addition, MedChemComm, 2016, 8(2), 338–345 RSC.
- K. Y. P. Apperley, Reversible and Photolabile Inhibitors for Human Tissue Transglutaminase, PhD thesis, University of Ottawa, 2017 Search PubMed.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening, J. Am. Chem. Soc., 2019, 141(22), 8951–8968 CrossRef PubMed.
- N. Shindo, H. Fuchida, M. Sato, K. Watari, T. Shibata, K. Kuwata, C. Miura, K. Okamoto, Y. Hatsuyama, K. Tokunaga, S. Sakamoto, S. Morimoto, Y. Abe, M. Shiroishi, J. M. M. Caaveiro, T. Ueda, T. Tamura, N. Matsunaga, T. Nakao, S. Koyanagi, S. Ohdo, Y. Yamaguchi, I. Hamachi, M. Ono and A. Ojida, Selective and reversible modification of kinase cysteines with chlorofluoroacetamides, Nat. Chem. Biol., 2019, 15(3), 250–258 CrossRef CAS PubMed.
- E. A. Rorke, G. Adhikary, H. Szmacinski, J. R. Lakowicz, D. J. Weber, R. Godoy-Ruiz, P. Puranik, J. W. Keillor, E. W. J. Gates and R. L. Eckert, Sulforaphane covalently interacts with the transglutaminase 2 cancer maintenance protein to alter its structure and suppress its activity, Mol. Carcinog., 2022, 61(1), 19–32 CrossRef CAS PubMed.
- A. M. Ali, R. F. Gómez-Biagi, D. A. Rosa, P.-S. Lai, W. L. Heaton, J. S. Park, A. M. Eiring, N. A. Vellore, E. D. de Araujo, D. P. Ball, A. E. Shouksmith, A. B. Patel, M. W. Deininger, T. O'Hare and P. T. Gunning, Disarming an Electrophilic Warhead: Retaining Potency in Tyrosine Kinase Inhibitor (TKI)-Resistant CML Lines While Circumventing Pharmacokinetic Liabilities, ChemMedChem, 2016, 11(8), 850–861 CrossRef CAS PubMed.
- K. A. Mix, M. R. Aronoff and R. T. Raines, Diazo Compounds: Versatile Tools for Chemical Biology, ACS Chem. Biol., 2016, 11(12), 3233–3244 CrossRef CAS PubMed.
- A. Bandyopadhyay and J. Gao, Targeting biomolecules with reversible covalent chemistry, Curr. Opin. Chem. Biol., 2016, 34, 110–116 CrossRef CAS PubMed.
- M. Robello, E. Barresi, E. Baglini, S. Salerno, S. Taliani and F. D. Settimo, The Alpha Keto Amide Moiety as a Privileged Motif in Medicinal Chemistry: Current Insights and Emerging Opportunities, J. Med. Chem., 2021, 64(7), 3508–3545 CrossRef CAS PubMed.
- R. Pasternack, M. Hills and C. Büchold, INHIBITORS OF TRANSGLUTAMINASE, US Pat., WO2018122419, 2018 Search PubMed.
- F. Yang, Y. Wen, C. Wang, Y. Zhou, Y. Zhou, Z.-M. Zhang, T. Liu and X. Lu, Efficient targeted oncogenic KRASG12C degradation via first reversible-covalent PROTAC, Eur. J. Med. Chem., 2022, 230, 114088 CrossRef CAS PubMed.
- N. Arora, G. M. Bacani, G. K. Barbay, S. D. Bembenek, M. Cai, W. Chen, C. Pooley, J. P. Edwards, B. Ghosh, B. Hao, K. Kreutter, G. Li, M. S. Tichenor, J. D. Venable, J. Wei, J. J. M. Wiener, Y. Wu, Y. Zhu, F. Zhang, Z. Zhang and K. Xiao, Inhibitors of bruton's tyrosine kinase and methods of their use, U.S. Pat., WO2017100668A1, 2017 Search PubMed.
- E. L. Myers and R. T. Raines, A phosphine-mediated conversion of azides into diazo compounds, Angew. Chem., Int. Ed., 2009, 48(13), 2359–2363 CrossRef CAS PubMed.
- L. M. McGregor, M. L. Jenkins, C. Kerwin, J. E. Burke and K. M. Shokat, Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes, Biochemistry, 2017, 56(25), 3178–3183 CrossRef CAS PubMed.
- A. Leblanc, C. Gravel, J. Labelle and J. W. Keillor, Kinetic studies of guinea pig liver transglutaminase reveal a general-base-catalyzed deacylation mechanism, Biochemistry, 2001, 40(28), 8335–8342 CrossRef CAS PubMed.
- R. Kitz and I. B. Wilson, Esters of Methanesulfonic acid as Irreversible Inhibitors of Acetylcholinesterase, J. Biol. Chem., 1962, 237(10), 3245–3249 CrossRef CAS.
- S. R. Stone and J. Hofsteenge, Specificity of activated human protein C, Biochem. J., 1985, 230(2), 497–502 CrossRef CAS PubMed.
- M. Mohutsky and S. D. Hall, Irreversible Enzyme Inhibition Kinetics and Drug–Drug Interactions, in Enzyme Kinetics in Drug Metabolism: Fundamentals and Applications, ed. S. Nagar, U. A. Argikar and D. J. Tweedie, Humana Press, Totowa, NJ, 2014, pp. 57–91 Search PubMed.
- A. Birkholz, D. J. Kopecky, L. P. Volak, M. D. Bartberger, Y. Chen, C. M. Tegley, T. Arvedson, J. D. McCarter, C. Fotsch and V. J. Cee, Systematic Study of the Glutathione Reactivity of N-Phenylacrylamides: 2. Effects of Acrylamide Substitution, J. Med. Chem., 2020, 63(20), 11602–11614 CrossRef CAS PubMed.
- K. Oertel, A. Hunfeld, E. Specker, C. Reiff, R. Seitz, R. Pasternack and J. Dodt, A highly sensitive fluorometric assay for determination of human coagulation factor XIII in plasma, Anal. Biochem., 2007, 367(2), 152–158 CrossRef CAS PubMed.
- R. Király, K. Thangaraju, Z. Nagy, R. Collighan, Z. Nemes, M. Griffin and L. Fésüs, Isopeptidase activity of human transglutaminase 2: disconnection from transamidation and characterization by kinetic parameters, Amino Acids, 2016, 48(1), 31–40 CrossRef PubMed.
- S. Jung, N. Fuchs, P. Johe, A. Wagner, E. Diehl, T. Yuliani, C. Zimmer, F. Barthels, R. A. Zimmermann, P. Klein, W. Waigel, J. Meyr, T. Opatz, S. Tenzer, U. Distler, H.-J. Räder, C. Kersten, B. Engels, U. A. Hellmich, J. Klein and T. Schirmeister, Fluorovinylsulfones and -Sulfonates as Potent Covalent Reversible Inhibitors of the Trypanosomal Cysteine Protease Rhodesain: Structure–Activity Relationship, Inhibition Mechanism, Metabolism, and In Vivo Studies, J. Med. Chem., 2021, 64(16), 12322–12358 CrossRef CAS PubMed.
- D. Montero, C. Tachibana, J. Rahr Winther and C. Appenzeller-Herzog, Intracellular glutathione pools are heterogeneously concentrated, Redox Biol., 2013, 1(1), 508–513 CrossRef CAS PubMed.
- Y. C. Awasthi, R. Sharma and S. S. Singhal, Human glutathione S-transferases, Int. J. Biochem., 1994, 26(3), 295–308 CrossRef CAS PubMed.
- L. Leung, X. Yang, T. J. Strelevitz, J. Montgomery, M. F. Brown, M. A. Zientek, C. Banfield, A. M. Gilbert, A. Thorarensen and M. E. Dowty, Clearance Prediction of Targeted Covalent Inhibitors by In Vitro-In Vivo Extrapolation of Hepatic and Extrahepatic Clearance Mechanisms, Drug Metab. Dispos., 2017, 45(1), 1–7 CrossRef CAS PubMed.
- T. J. Cogswell, C. S. Donald, D.-L. Long and R. Marquez, Short and efficient synthesis of fluorinated δ-lactams, Org. Biomol. Chem., 2015, 13(3), 717–728 RSC.
- S. O. Zaitseva, S. V. Golodukhina, N. S. Baleeva, E. A. Levina, A. Y. Smirnov, M. B. Zagudaylova and M. S. Baranov, Azidoacetic Acid Amides in the Synthesis of Substituted Arylidene-1-H-imidazol-5-(4H)-ones, ChemistrySelect, 2018, 3(30), 8593–8596 CrossRef CAS.
- B. T. Burlingham and T. S. Widlanski, An Intuitive Look at the Relationship of Ki and IC50: A More General Use for the Dixon Plot, J. Chem. Educ., 2003, 80(2), 214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2213069. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2md00378c |
| This journal is © The Royal Society of Chemistry 2023 |