
Design, synthesis and biological evaluation of pyrazolo[3,4-b]pyridine derivatives as TRK inhibitors†
Nian
Liu
,
Xin
Wang
,
Qinglin
Fu
,
Qiaohua
Qin
,
Tianxiao
Wu
,
Ruicheng
Lv
,
Dongmei
Zhao
 * and
Maosheng
Cheng
* and
Maosheng
Cheng
Key Laboratory of Structure-Based Drug Design and Discovery, Ministry of Education, School of Pharmaceutical Engineering, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenhe District, Shenyang, 110016, PR China
First published on 18th October 2022
Abstract
Tropomyosin receptor kinases (TRKs) are associated with the proliferation and differentiation of cells, and thus their continuous activation and overexpression cause cancer. Herein, based on scaffold hopping and computer-aid drug design, 38 pyrazolo[3,4-b]pyridine derivatives were synthesised. Further, we evaluated their activities to inhibit TRKA. Among them, compound C03 showed acceptable activity with an IC50 value of 56 nM and it inhibited the proliferation of the Km-12 cell line with an IC50 value of 0.304 μM together with obvious selectivity for the MCF-7 cell line and HUVEC cell line. Furthermore, compound C03 possessed good plasma stability and low inhibitory activity to a panel of cytochrome P450 isoforms except CYP2C9. Overall, C03 has potential for further exploration.
1 Introduction
TRKs, tropomyosin receptor kinases, have three subtypes, namely TRKA, TRKB and TRKC, which belong to the receptor tyrosine kinase (RTK) family.1 As a member of the transmembrane protein kinase family, TRKs are composed of an extracellular domain, which combines with endogenous ligands, an intercellular domain and a transmembrane domain.2,3 TRKA mainly combines with nerve growth factor (NGF), while TRKB is mainly activated by brain-derived neurotrophic factor (BDNF) and TRKC is activated by neurotrophin-3 (NT-3).4–9 Once activated, the intramembrane kinase domain is phosphorylated and the downstream signal transduction pathways (including Ras/Erk, PLC-γ, and PI3K/Akt) are triggered, which are associated with the proliferation, differentiation and survival of cells.10–13 TRKs are encoded by neurotrophic receptor tyrosine kinase 1 (NTRK1), NTRK2 and NTRK3 genes, respectively.1 Although NTRK fusion is the primary mechanism of TRK mutations, NTRK fusion means that the 3′ sequences of the NTRK gene fuse with the 5′ sequences of other genes that encode irrelevant proteins. Structurally, mutant TRK proteins lack an extracellular domain. Functionally, TRK proteins are manifested as continuous activation of the intracellular kinase domain, further leading to cancers,14 including colorectal cancer,15 non-small cell lung cancer (NSCLC),16 glioblastoma17 and head and neck squamous cell carcinoma.18 Thus, small-molecule TRK inhibitors are important for the treatment of these cancers.Recently, many potent TRK inhibitors have been reported (Fig. 1). In 2018, larotrectinib (1) was the first TRK inhibitor to be approved by the Food and Drug Administration (FDA) for the treatment of patients with tumours harbouring NTRK fusions. It is a highly selective TRK inhibitor with low nanomole potency (IC50 < 20 nM).19–21 In 2019, the multiple target inhibitor entrectinib (2) discovered by Roche was approved by the FDA. It exhibited strong inhibitory activities towards ALK (IC50 = 12 nM), ROS1 (IC50 = 7 nM) and all the subtypes of TRKs (IC50 values for TRKA/B/C were 1 nM, 3 nM, and 5 nM, respectively).22,23 Compound 3, which was reported by Duan et al., exhibited inhibitory activities on TRKA/B/C with IC50 values of 1.6 nM, 2.9 nM, and 2.0 nM, respectively. Moreover, 3 exhibited strong activity for the suppression of the growth of BaF3 cells harboring xDFG motif mutations.24 Compounds 4 and 5 were reported by our research group recently, which are both pan-TRK inhibitors with low nanomole enzyme activities (4, IC50 values for TRKA/B/C were 17 nM, 28 nM and 11 nM and 5, IC50 values for TRKA/B/C were 12 nM, 22 nM and 15 nM, respectively). Meanwhile, 5 showed a potent anticancer effect in vitro and in vivo.25,26 To suppress on the resistant TRK mutants, LOXO-195 (6) was developed as a next-generation TRK inhibitor, which has advanced to a phase II clinical trial for patients with TRK fusion-positive cancers that acquired resistant mutations to larotrectinib.27
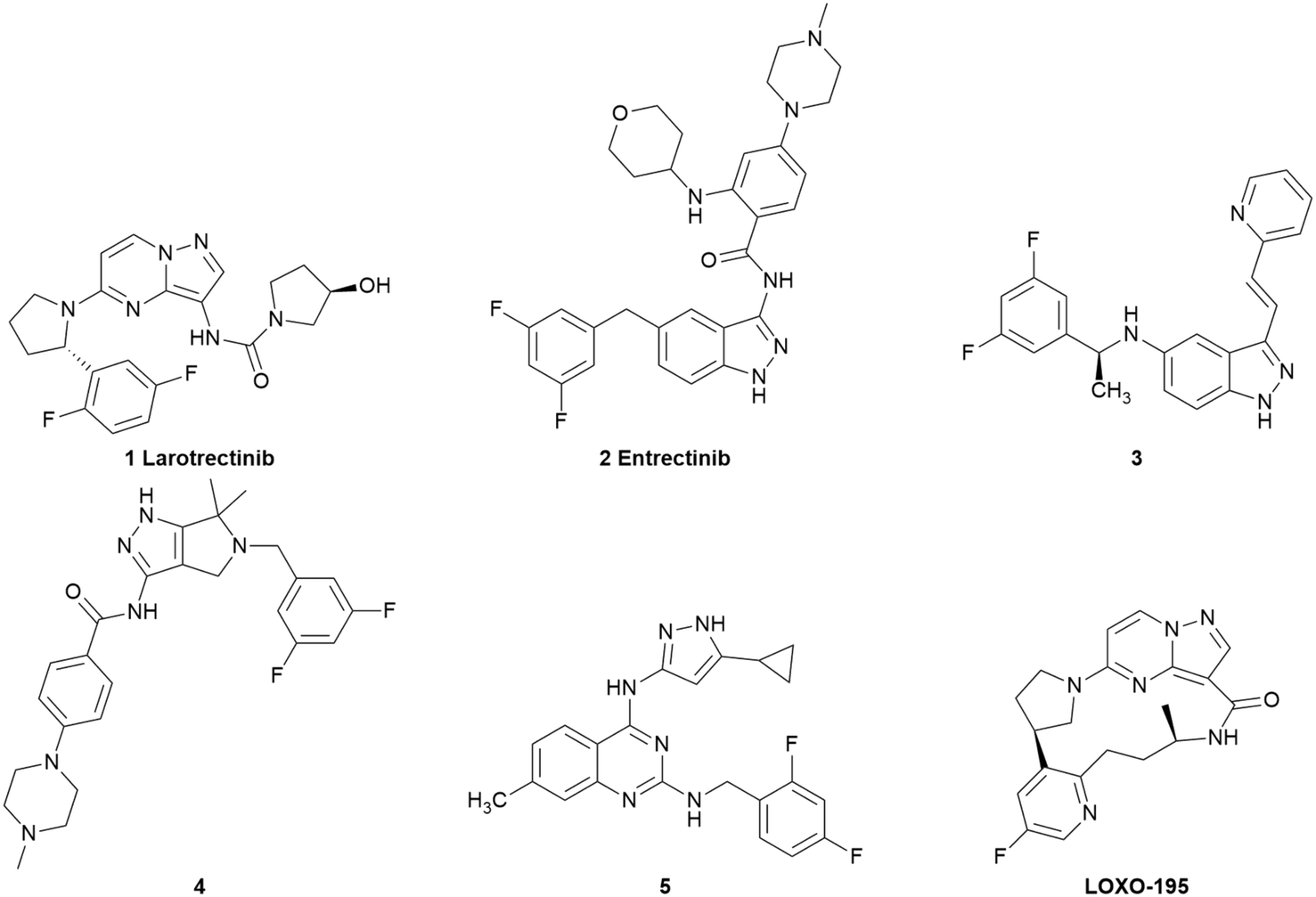 | ||
| Fig. 1 Chemical structure of potent TRK inhibitors. | ||
Herein, based on scaffold hopping, rational drug design and computer-aid drug design (CADD), 38 pyrazolo[3,4-b]pyridine derivatives were synthesised. In addition, most of them exhibited nanomole inhibitory activities against TRKA.
2 Chemistry
As shown in Scheme 1, the commercially available 5-bromo-1H-pyrazolo [3,4-b]pyridine (7) was iodized by NIS to obtain intermediate 8, and then the NH of intermediate 8 was protected by PMB-Cl to produce the key intermediate 9. Simultaneously, meta-aminobenzoic acid was reacted with morpholine to produce intermediate 12. Intermediate 12 and intermediate 9 were coupled through Buchwald–Hartwig reaction to get intermediate 13. The Miyaura borylation reaction and Suzuki reaction were performed in one pot in sequence with intermediate 13 to produce intermediates 15a–15o, and the protecting group was removed by trifluoroacetic acid to obtain the target compounds A01–A15.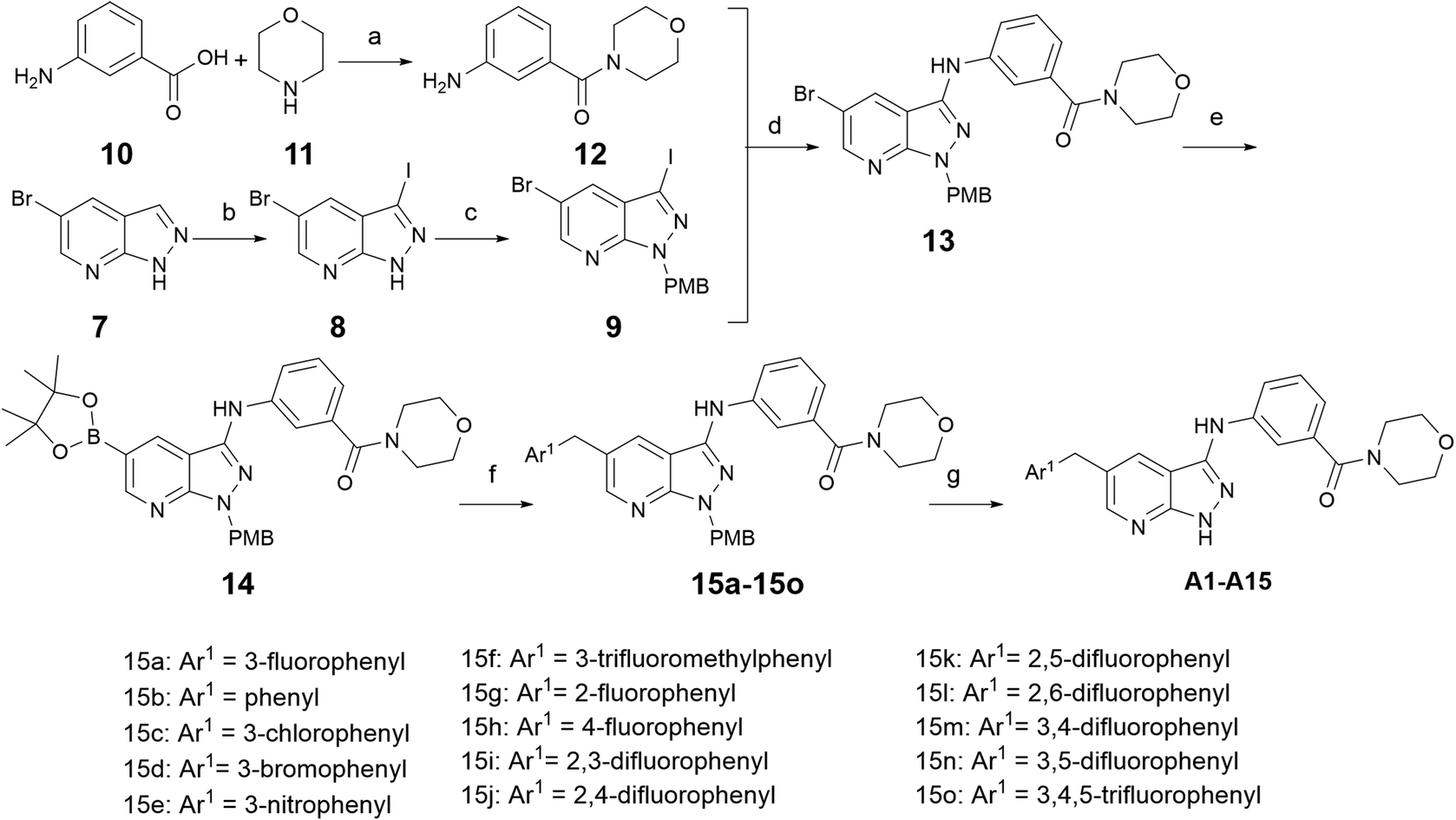 | ||
Scheme 1 Synthesis of compounds A01–A15. Reagents and conditions: (a) HOBt, EDCI, DIPEA, DMF, r.t., 82.3%; (b) NIS, DMF, 60 °C, 12 h, 82.4%; (c) NaH, DMF, PMBCl, 0–25 °C, 50.2%; (d) Pd2(dba)3, xantphos, Cs2CO3, dry dioxane, Ar, 90 °C, 8 h, 48.5%; (e) bis(pinacolato)diboron, Pd(dppf)Cl2, KOAc, dry dioxane, Ar, 100 °C, 4 h; (f) substituted benzyl bromide, Pd(PPh3)4, Cs2CO3, dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 5:1, Ar, 80 °C, 2 h, 30.3–34.7%; and (g) CF3COOH, 60 °C, 8 h, 48.5–52.3%. :H2O = 5:1, Ar, 80 °C, 2 h, 30.3–34.7%; and (g) CF3COOH, 60 °C, 8 h, 48.5–52.3%. | ||
Compounds B01–B13 were synthesized, as shown in Scheme 2. Intermediate 9 was coupled with ethyl 3-aminobenzoate or ethyl 4-aminobenzoate through Buchwald–Hartwig reaction to produce intermediate 16 or 22, respectively. The palladium-catalyzed Miyaura borylation reaction and Suzuki reaction were performed in one pot by sequence to produce intermediate 18 or 24. Trifluoroacetic acid was used to deprotect the para-methoxy benzyl and NaOH was used to deprotect the ethyl, and then crucial intermediate 20 or 26 was obtained, respectively. Condensation reactions occurred between 20(26) and different amino segments, together with deprotection reactions, if necessary, to obtain the target compounds B01–B13.
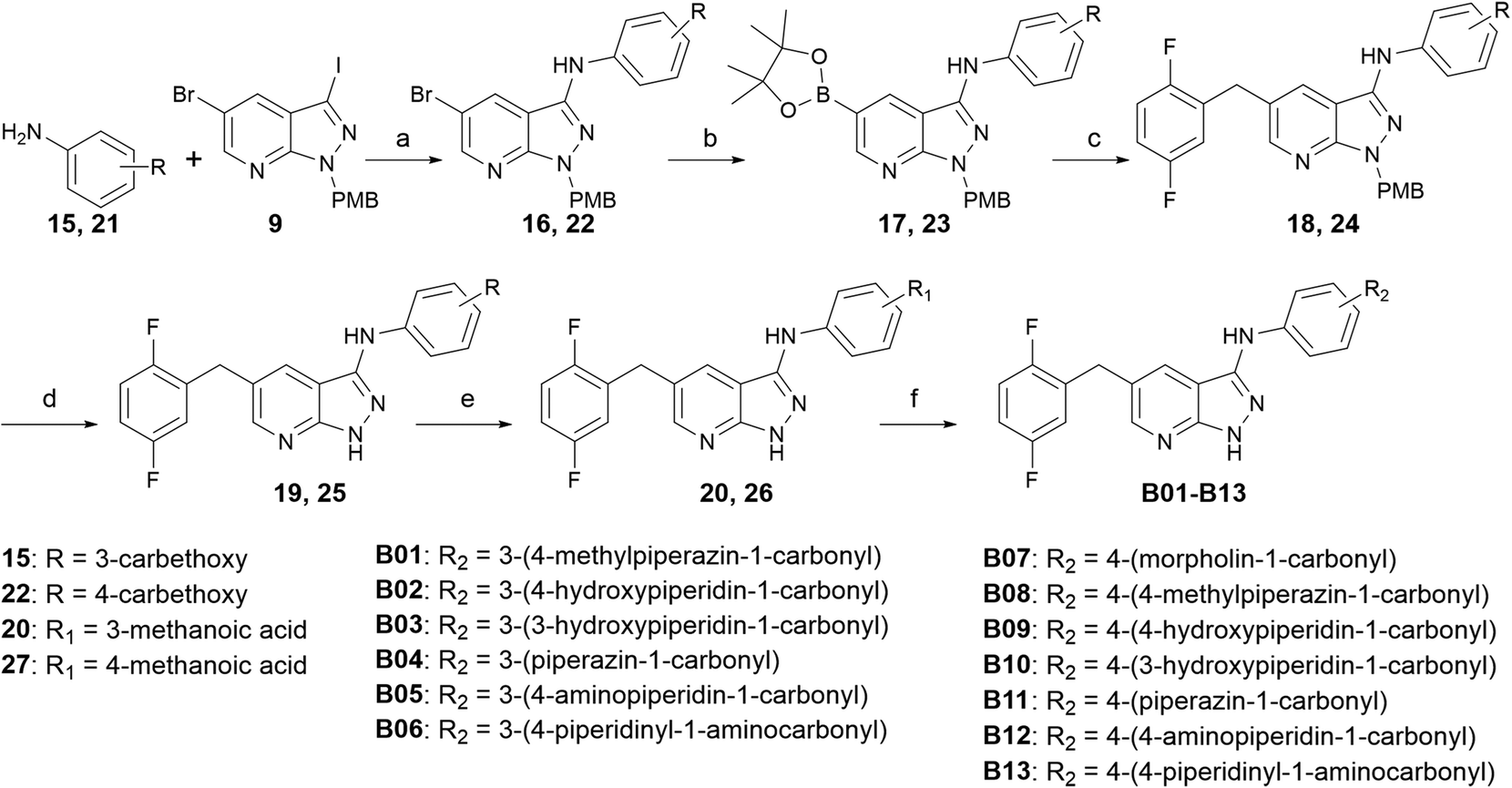 | ||
| Scheme 2 Synthesis of compounds B01–B13. Reagents and conditions: (a) Pd2(dba)3, xantphos, Cs2CO3, dry DMF, Ar, 120 °C, 8 h, 65.9%; (b) bis(pinacolato)diboron, Pd(dppf)Cl2, Cs2CO3, dry dioxane, Ar, 100 °C, 4 h; (c) 2,5-difluorobenzyl bromide, Pd(PPh3)4, Cs2CO3, dioxane:H2O = 5:1, Ar, 80 °C, 4 h, 48.6%; (d) CF3COOH, 60 °C, 5 h; (e) NaOH, methanol:H2O = 3:1, 60 °C, 4 h, 65.7%; and (f) I) appropriate amine, PyBop, DIPEA, THF, 0 °C, 2 h and II) 4 N HCl/ethyl acetate, r.t., 4 h, 28.8–55.5%. | ||
Scheme 3 shows the synthetic route for compounds C01–C10. Starting with intermediate 9, it was coupled with 3-ethoxycarbonylphenyl-boronic acid or 4-ethoxycarbonylphenyl-boronic acid through Suzuki reaction to produce intermediate 28 or 34, respectively. Intermediate 28 or 34 was coupled with 2,5-difluorobenzyl through Miyaura borylation reaction and Suzuki reaction in sequence in one pot to generate intermediate 30 or 36, then deprotection was performed to generate the key intermediate 32 or 38, respectively. Condensation reactions or condensation reactions followed by deprotection reactions occurred to yield the target compounds C01–C10.
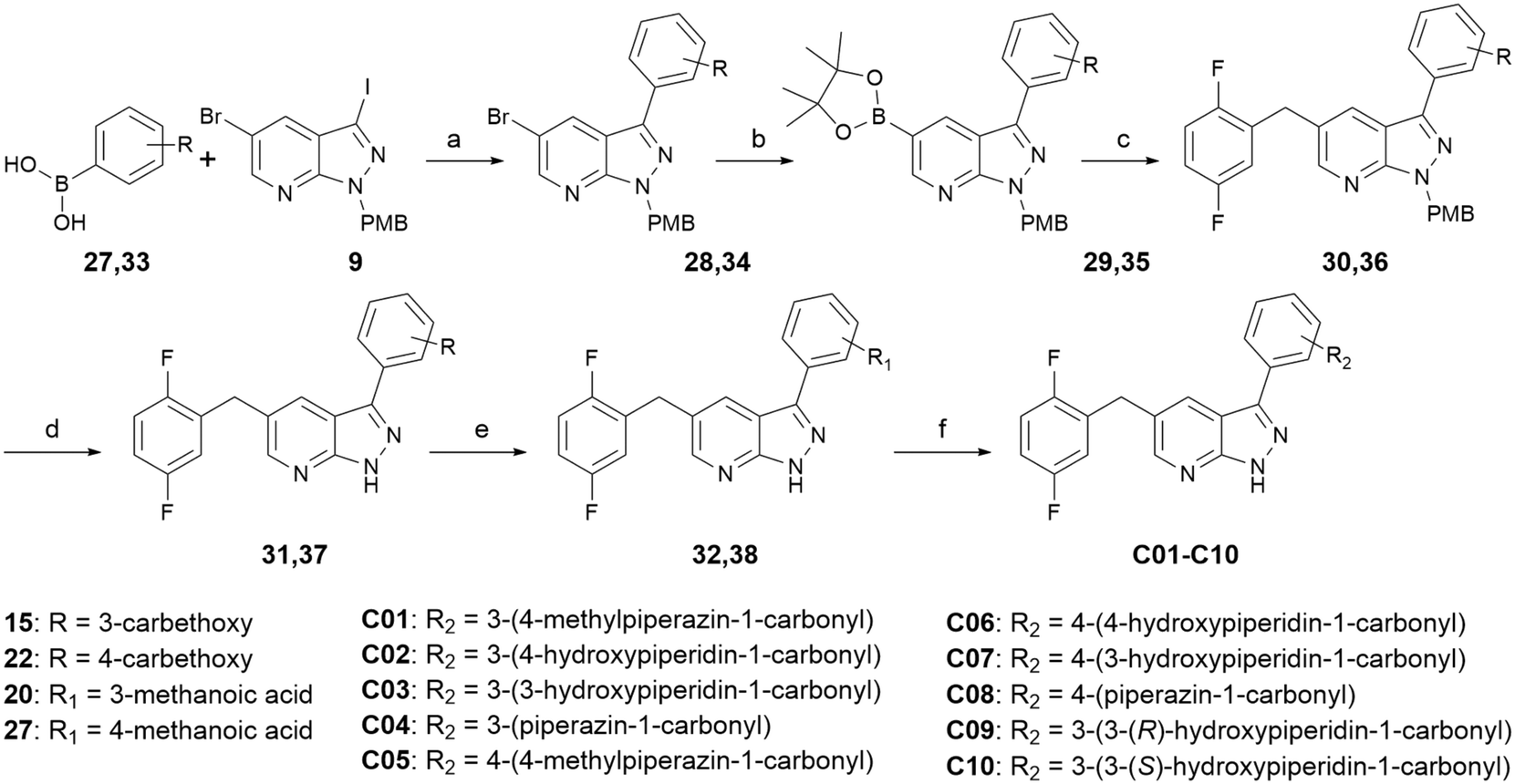 | ||
| Scheme 3 Synthesis of compounds C01–C10. Reagents and conditions: (a) Pd(pph3)4, S-Phos, KF, dioxane:H2O = 5:1, Ar, 100 °C, 24 h, 53.7%; (b) bis(pinacolato)diboron, Pd(OAc)2, X-Phos, KOAc, dry dioxane, Ar, 100 °C, 14 h; (c) 2,5-difluorobenzyl bromide, Pd(PPh3)4, Cs2CO3, dioxane:H2O = 5:1, Ar, 100 °C, 7 h, 52.3%; (d) CF3COOH, 60 °C, 5 h; (e) NaOH, methanol:H2O = 3:1, 60 °C, 4 h, 54.2%; and (f) I) appropriate amine, PyBop, DIPEA, THF, 0 °C, 2 h and II) 4 N HCl/ethyl acetate, r.t., 4 h, 31.6–59.0%. | ||
3 Results and discussion
3.1 Design and in vitro activities of target compounds against TRKA
All the novel synthesized compounds were evaluated for their in vitro inhibitory activities against TRKs using homogeneous time-resolved fluorescence (HTRF) assays and larotrectinib (1) was employed as a positive comparison. Under the experimental conditions, larotrectinib exhibited suppressive activity on TRKA, TRKB and TRKC with IC50 values of 3.0, 13 and 0.2 nM, respectively, which were similar to previously reported data.The analysis of the co-crystal structure between entrectinib and TRKA, as shown in Fig. 2A, indicated that the aminopyrazole fragment is anchored to the hinge region through three hydrogen bonds with Glu590 and Met592 residues; the fluorine of the 3,5-difluorophenyl moiety creates favorable fluorine bonds with Asn655, Cys656 and Gly667 which comes from the DFG motif; the benzene ring of the indazole scaffold has a π–π stacking interaction with gatekeeper Phe589; and N-methylpiperazine is oriented toward the solvent zone. Guided molecular docking showed that larotrectinib has a similar binding mode, while the hydroxy orients toward Asp596 and formed a hydrogen bond (Fig. 2B). Observing the structure of entrectinib and larotrectinib, it can be found that they both have a hydrophilic head extending into the solvent zone, a fluorine substituted aromatic ring as hydrophobic tail, which extends to the DFG motif, and a linker that binds to the hinge area as a hydrogen bond center (Fig. 3).28,29
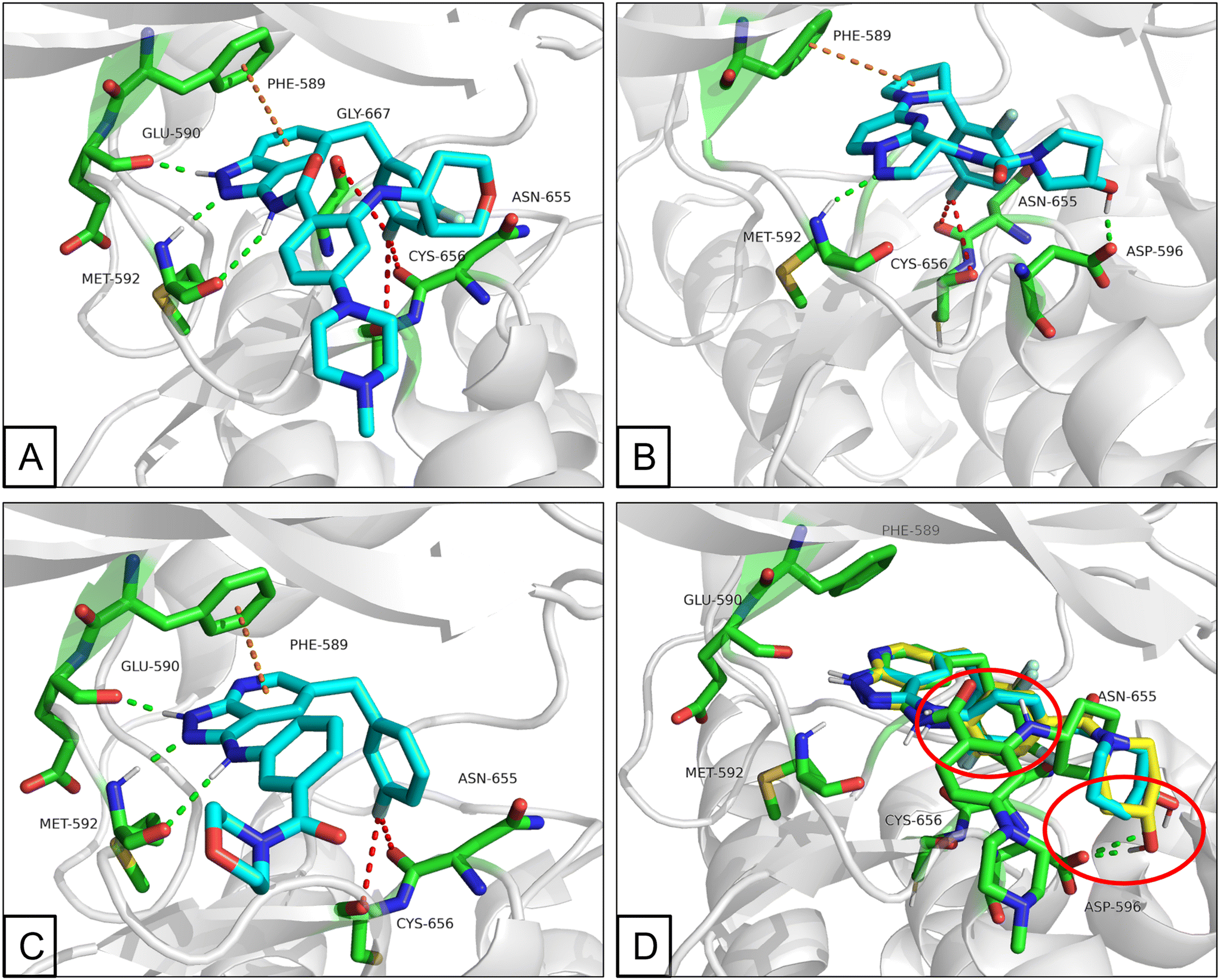 | ||
| Fig. 2 (A) Binding mode of entrectinib (blue) and TRKA. (B) Predicted binding mode of larotrectinib (blue) and TRKA. (C) Predicted binding mode of A01 (blue) and TRKA. (D) Overlap of B09 (yellow) and B10 (blue) with entrectinib (green). (PDB code: 5KVT. Residues are depicted in green; the interactions are depicted in dash lines with different colours, hydrogen bond is depicted in green, fluorine bond is depicted in red and π–π stacking interaction or δ–π interaction is depicted in orange). | ||
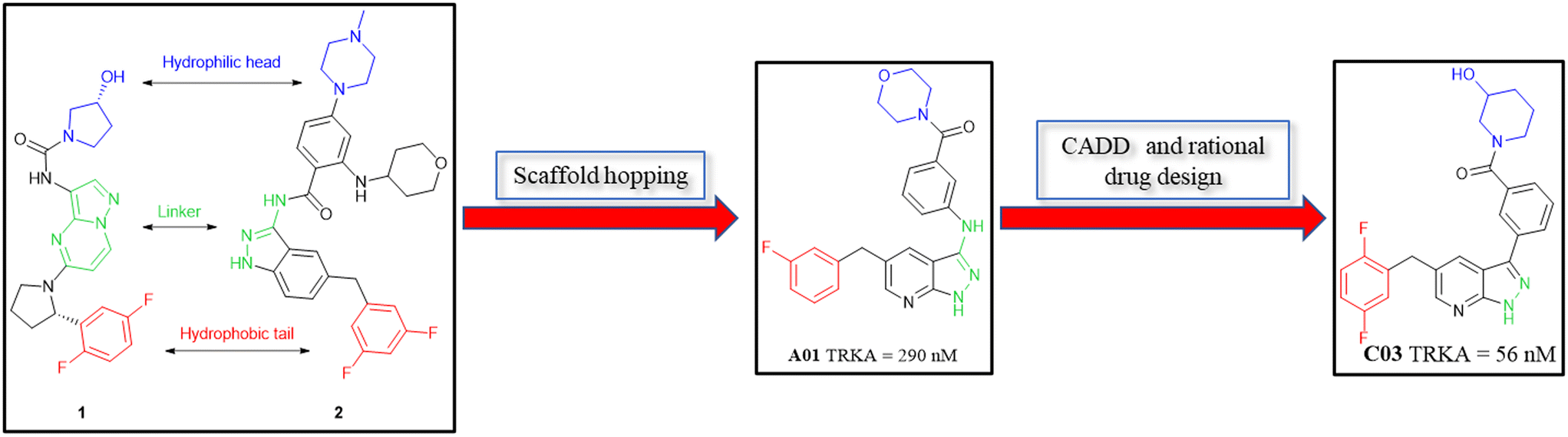 | ||
| Fig. 3 Structural analysis of larotrectinib and entrectinib, and the design strategy of target compounds. | ||
Pyrazolo[3,4-b]pyridine is a common fragment used in the synthesis of kinase inhibitors, whose pyrazolo portion is suitable as a hydrogen bond center. Simultaneously, the pyridine is thought to have a π–π stacking interaction with Phe589. Thus, the scaffold hopping strategy was used to investigate a structurally novel TRK inhibitor. Firstly, we designed and synthesized compound A01 (Table 1), which had weak inhibitory activity on TRKA (IC50 = 0.293 μM). Also, the molecular docking study preliminarily revealed the possible binding mode (Fig. 2C), where the aminopyrazole fragment is bound to Glu590 and Met592 with three hydrogen bonds; the pyridine forms a π–π stacking interaction with Phe589; the 3-fluorophenyl is oriented toward the hydrophobic pocket near to the DFG motif; and simultaneously the morpholine extends to the solvent zone. Next, we investigated the hydrophobic tail and the structures are shown in Table 1. When we removed the fluorine atom, compound A02 showed weaker potency (IC50 = 0.479 μM), which indicates that a suitable substituent group is necessary to achieve the desired activity. Subsequently, chlorine, bromine, and stronger electrophilic substituents such as nitro and trifluoromethyl were introduced in the meta-position of the benzene ring to generate compounds A03–A06, respectively; unfortunately they all showed reduced potency. This indicated that the fluorine is important for the hydrophobic tail. Then, we investigated the effect of the number and position of the fluorine on the benzene ring, obtaining compounds A07–A15. All the compounds had various degrees of activity decline except compound A11 with 2,5-difluorophenyl, which showed a moderate IC50 value of 0.178 μM. This can be ascribed to its suitable orientation and electrical distribution of the meta-fluorine on the benzene ring.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R 1 | TRKA | Entry | R 1 | TRKA |
| IC50 (μM) | IC50 (μM) | ||||
| a Used as a positive comparison. | |||||
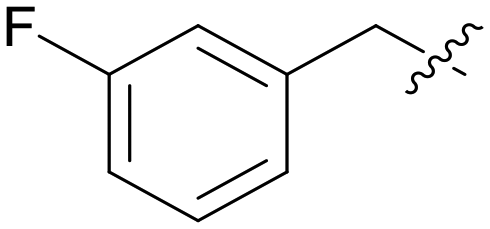
| A01 |
|
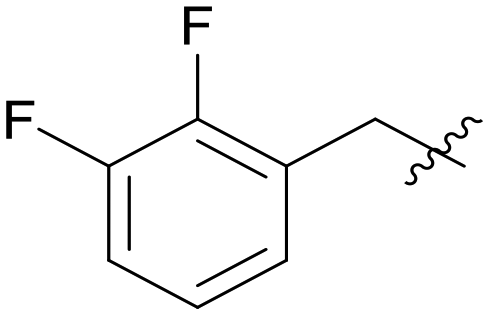
0.294 | A09 |
|
1.966 |
| A02 |
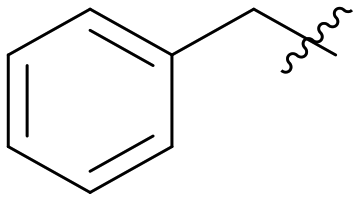
|
0.479 | A10 |
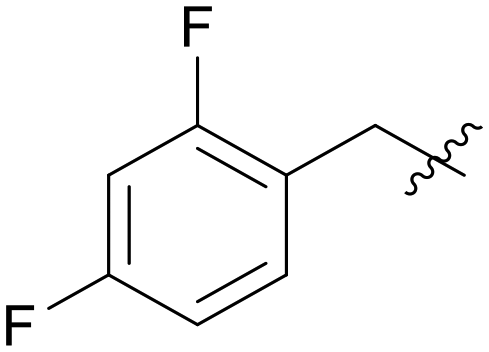
|
0.788 |
| A03 |
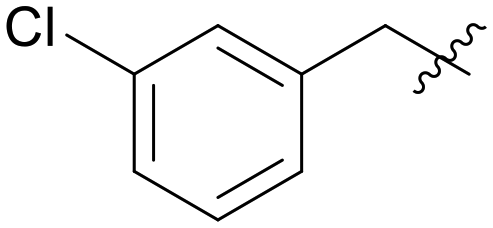
|
4.430 | A11 |
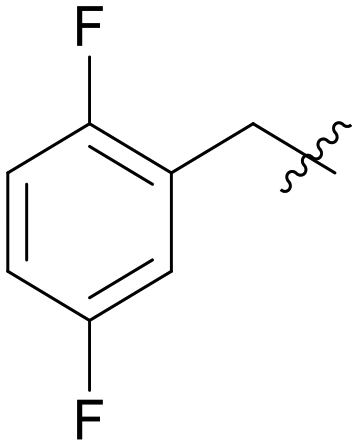
|
0.178 |
| A04 |
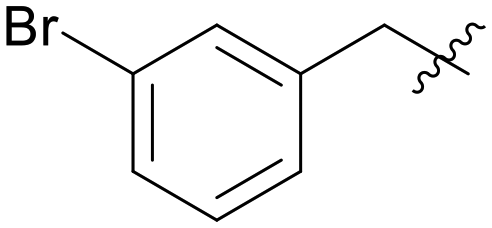
|
0.802 | A12 |
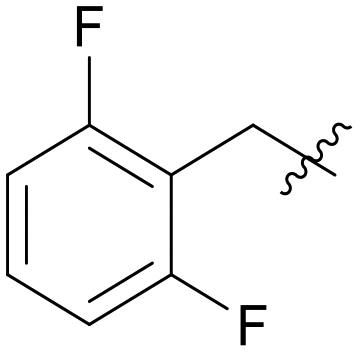
|
0.838 |
| A05 |
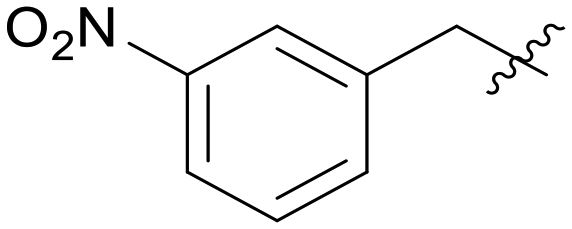
|
1.400 | A13 |
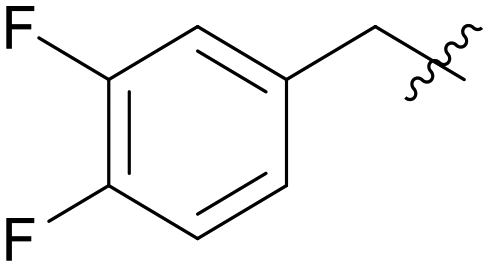
|
>100 |
| A06 |
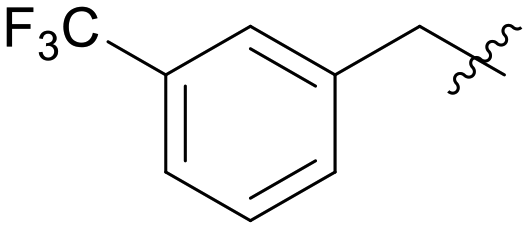
|
16.690 | A14 |
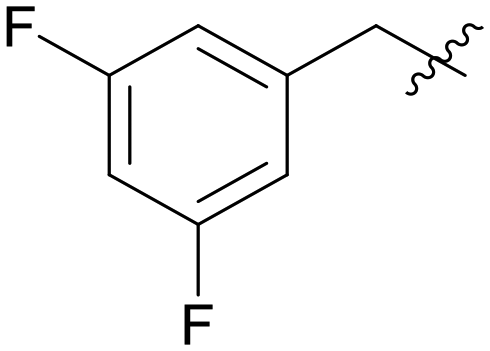
|
0.700 |
| A07 |
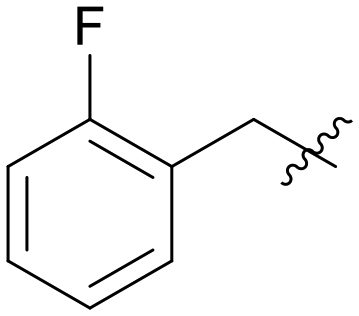
|
0.978 | A15 |
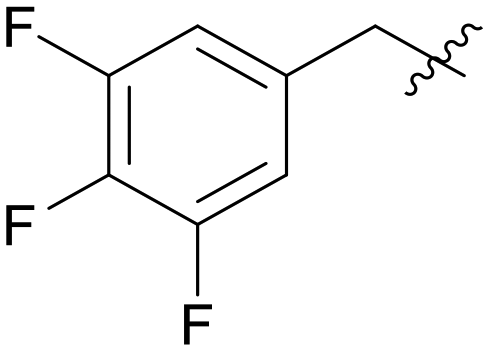
|
0.660 |
| A08 |
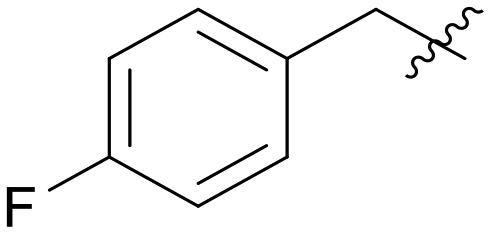
|
2.780 | 1 | 0.003 | |
Then, we fixed the hydrophobic group as 2,5-difluorophenyl and explored the types and positions of the hydrophilic fragments to get compounds B01–B13. All the structures are shown in Table 2. When the hydrophilic fragment was sited at the meta-position of the benzene ring, it was B01 with the methylpiperazinyl and B04 with piperazine, which showed a slight improvement in potency (IC50 = 0.118 and 0.117 μM, respectively). Moreover, we moved the fragments to the para-position and compounds B07–B13 were obtained, but none of them had better inhibitory activity. B09 with 4-hydroxypiperidinyl and B10 with 3-hydroxypiperidinyl showed a weak potency promotion (IC50 = 0.129 and 0.156 μM, respectively) compared with A11. Molecule docking was used to explain the possible reasons for this (Fig. 2D), where both of them showed a good binding mode at the hydrophobic tail and the hydrogen bond center; simultaneously, the hydroxy formed a hydrogen bond with Asp596, which is similar to larotrectinib. Regrettably, the benzene ring had an uncomfortable conformation, which may be due to the rotatable single bond, leading to the production of additional entropy and preventing the binding between the compound and the protein.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R 2 | TRKA | Entry | R 2 | TRKA |
| IC50 (μM) | IC50 (μM) | ||||
| a Used as a positive comparison. | |||||
| B01 |
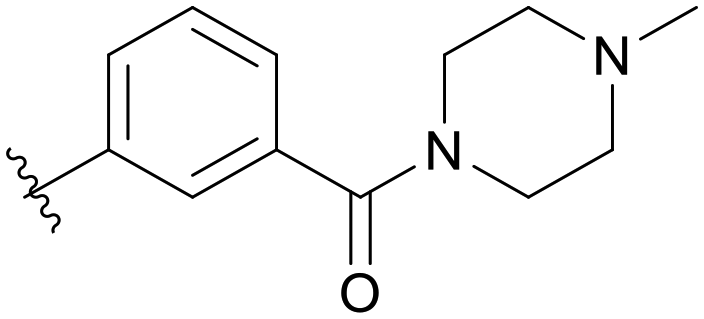
|
0.118 | B08 |
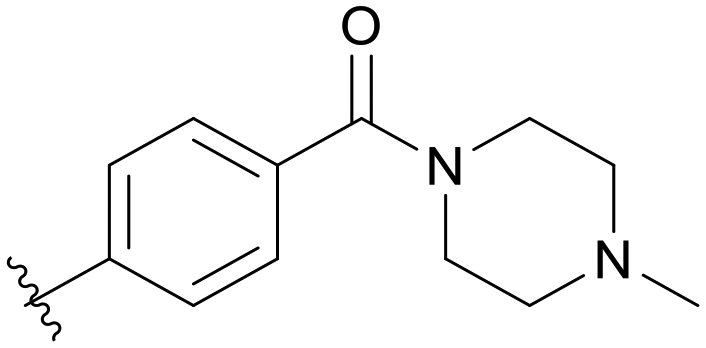
|
0.389 |
| B02 |
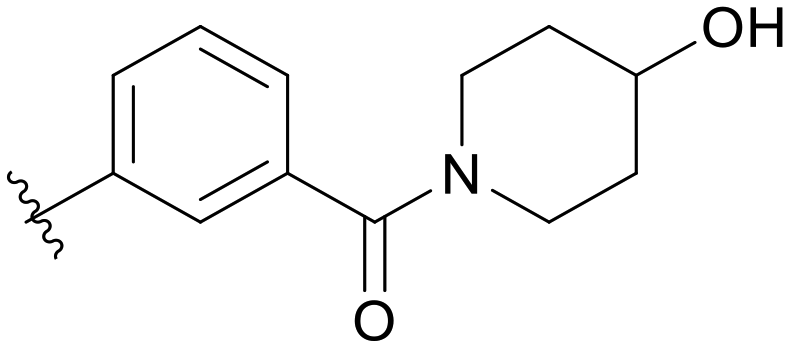
|
0.613 | B09 |
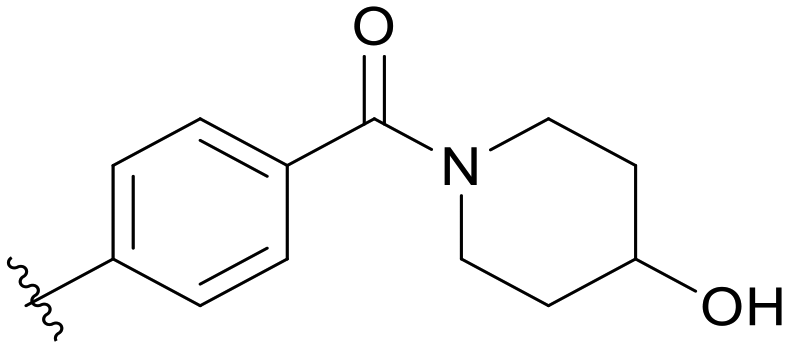
|
0.129 |
| B03 |
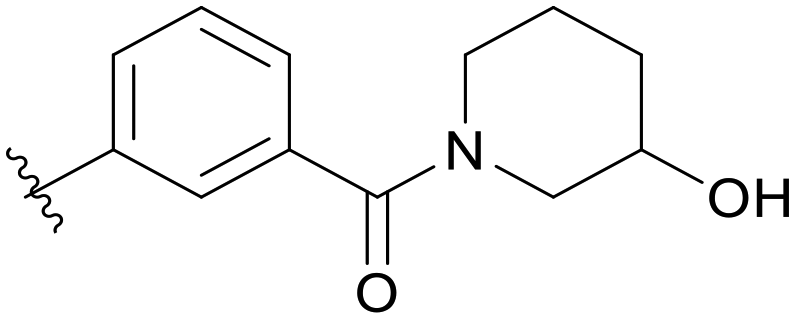
|
0.205 | B10 |
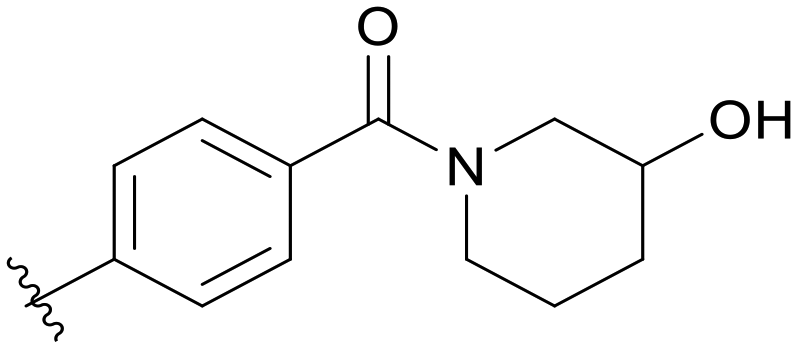
|
0.156 |
| B04 |
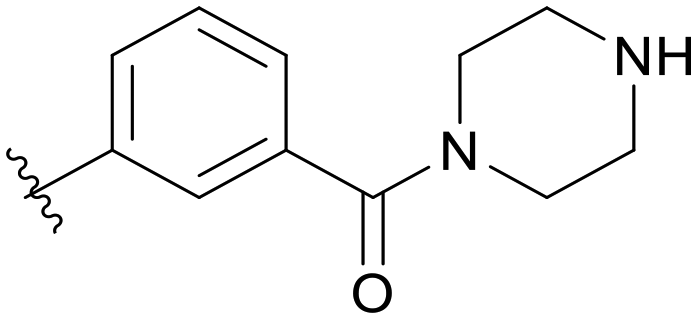
|
0.117 | B11 |
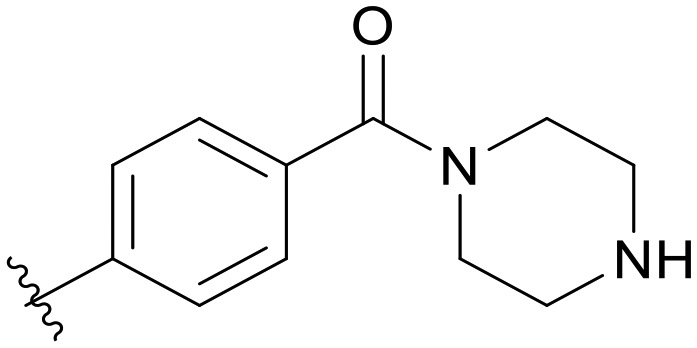
|
0.409 |
| B05 |
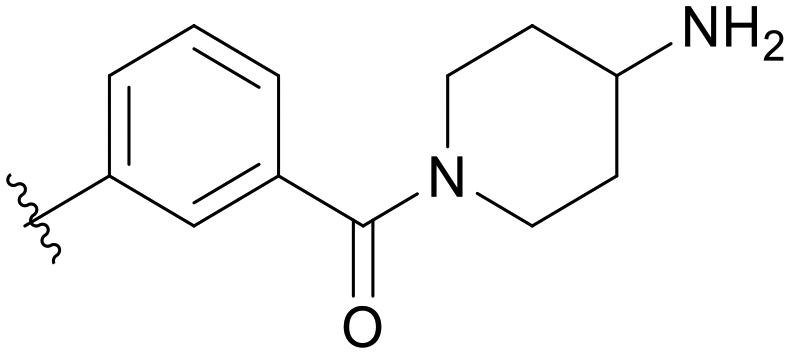
|
0.437 | B12 |
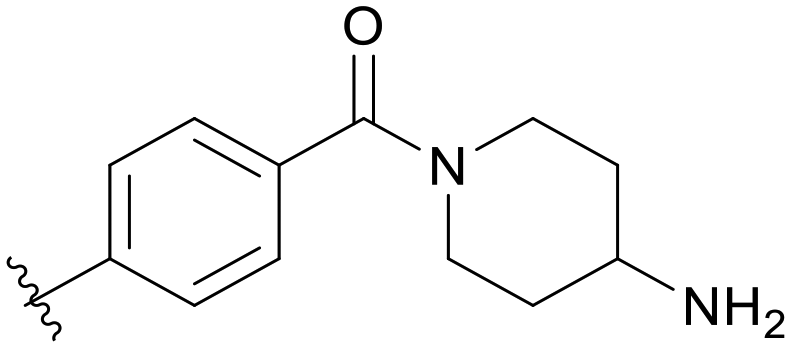
|
0.263 |
| B06 |
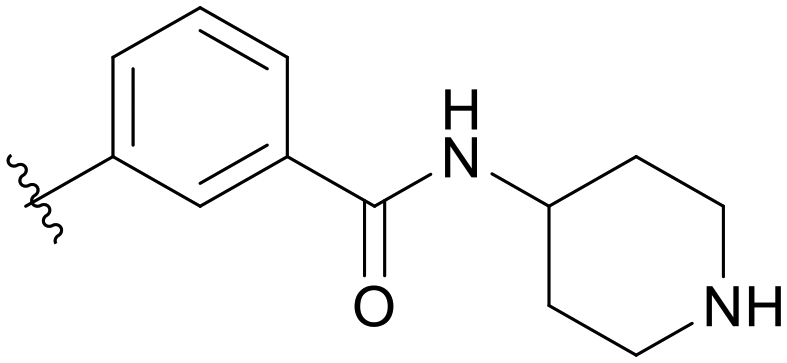
|
0.630 | B13 |
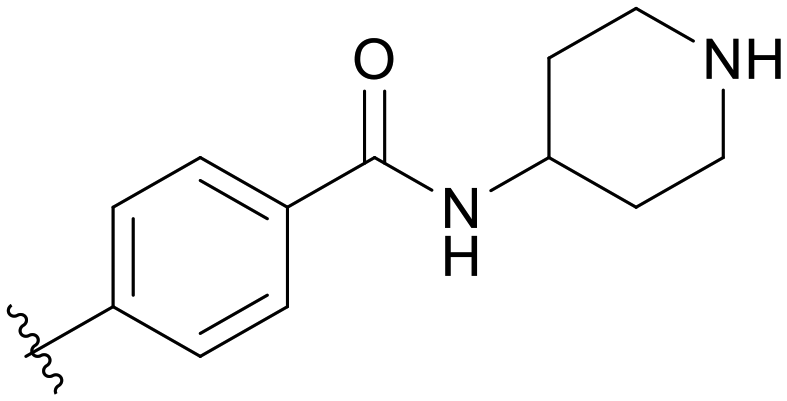
|
0.321 |
| B07 |
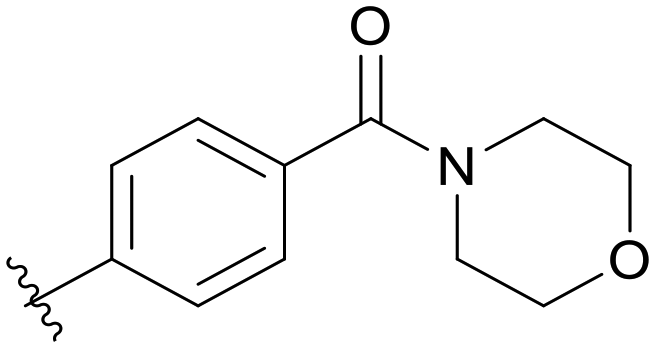
|
0.980 | 1 | 0.003 | |
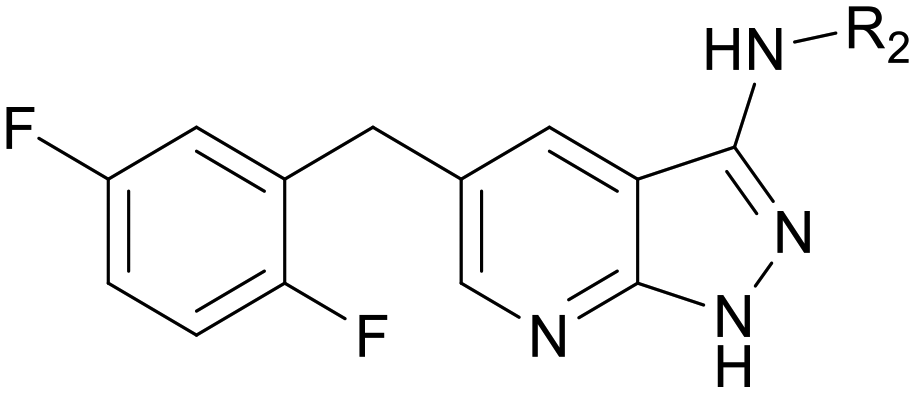
Referring to the binding mode of larotrectinib, conformational restriction was used and we removed the NH group. Compounds C01–C10 were obtained, as shown in Table 3. When the substitution was located at the para-position, compounds C05 and C08 had a slight potency improvement compared with B08 and B11, but still worse than B04, while at the meta-position, C02 and C03 had better potency. Excitedly, compound C03 with 3-hydroxypiperidinyl at the meta-position of the benzene ring showed acceptable potency with an IC50 value of 0.056 μM. Then, the chirality of C03 was considered, and C09–C10 were synthesized and evaluated for their inhibitory activities on TRKA. Alternatively, compound C10 with (S)-3-hydroxypiperidinyl showed slight superiority with an IC50 value of 0.026 μM.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R 3 | TRKA | Entry | R 3 | TRKA |
| IC50 (μM) | IC50 (μM) | ||||
| a Used as a positive comparison. | |||||
| C01 |
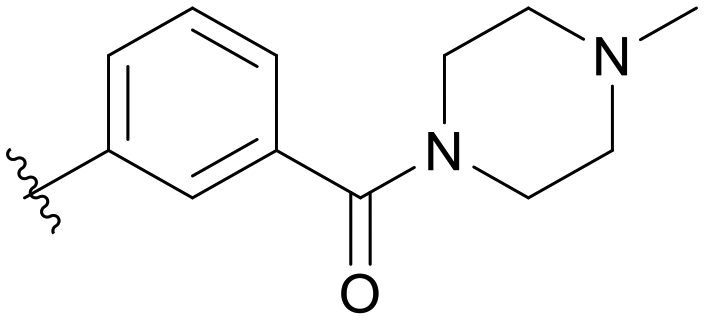
|
0.106 | C06 |
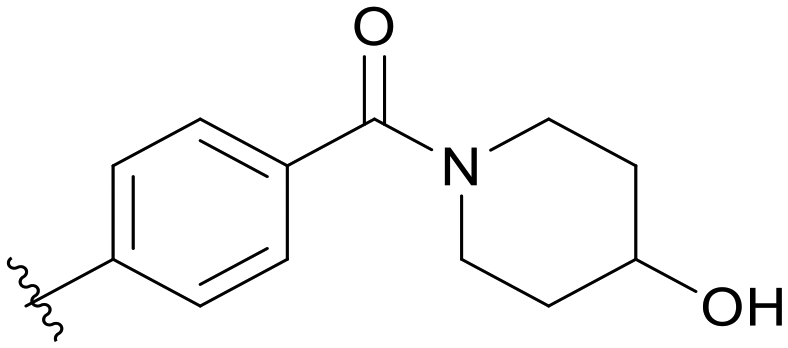
|
0.383 |
| C02 |
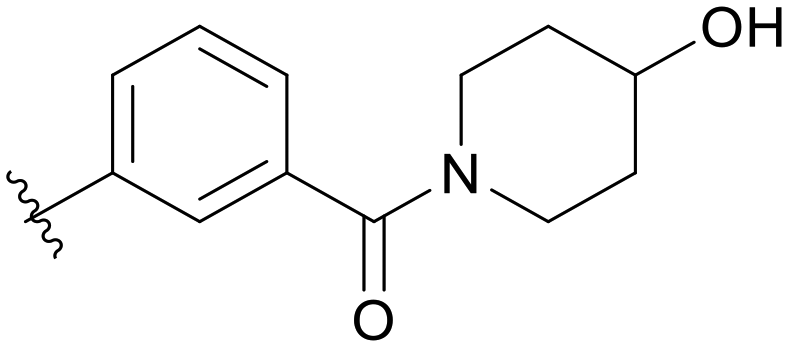
|
0.254 | C07 |
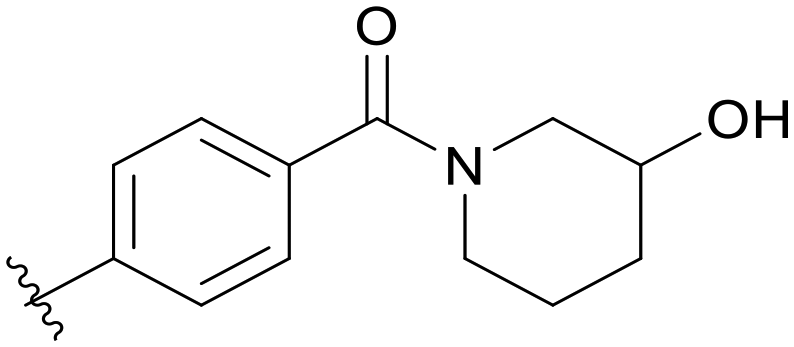
|
0.571 |
| C03 |
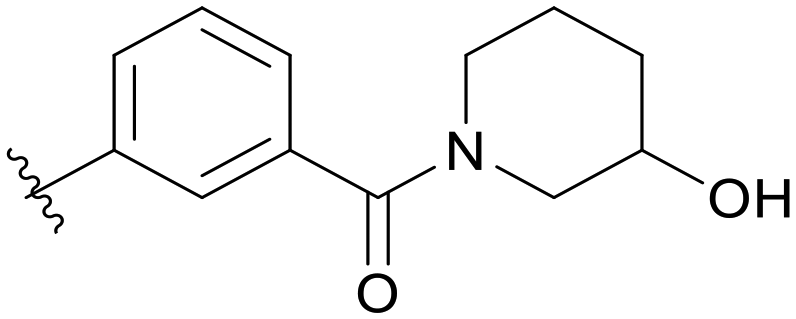
|
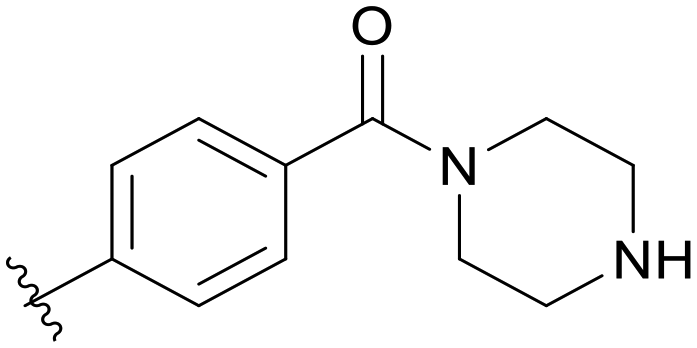
0.056 | C08 |
|
0.251 |
| C04 |
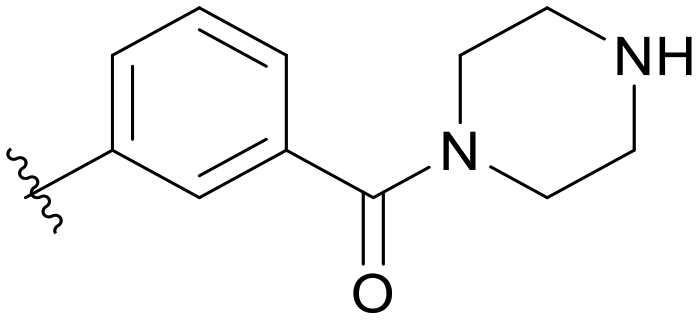
|
0.391 | C09 |
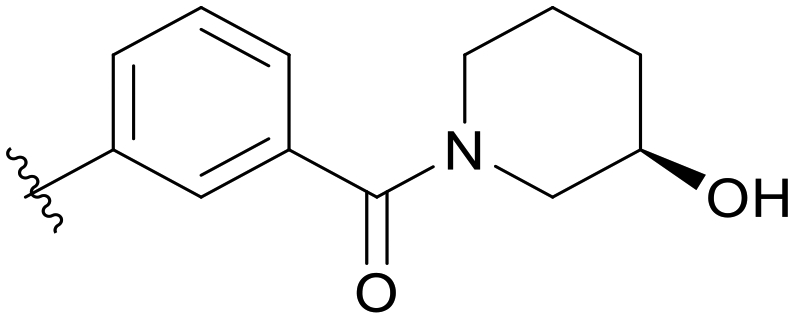
|
0.057 |
| C05 |
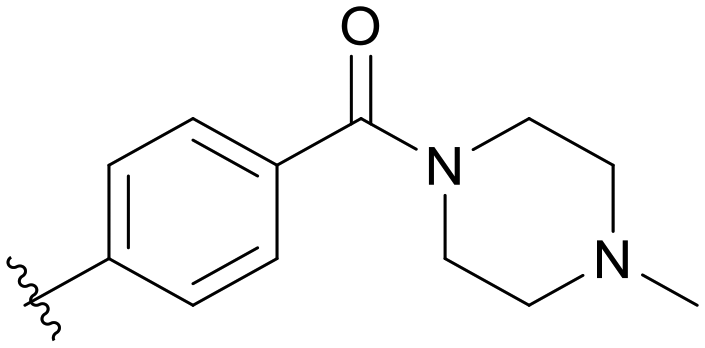
|
0.296 | C10 |
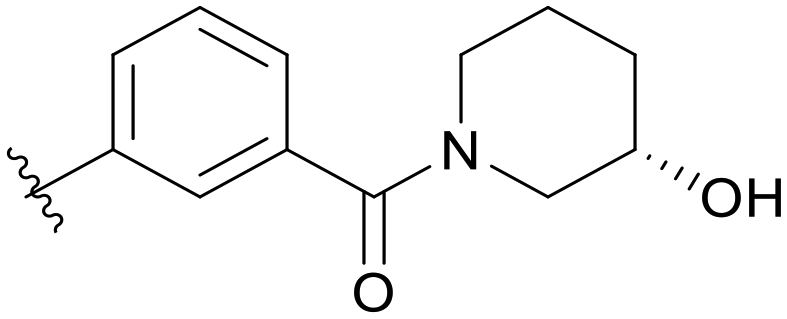
|
0.026 |
| 1 | 0.003 | ||||
3.2 Kinase selectivity
We chose compounds C03, C09 and C10 for further kinase selectivity assay. As shown in Table 4, they were all pan-TRK inhibitors with similar suppression on each subtype of TRK, while C03 had the most similar potency on each subtype of TRKs. Then, the inhibitory activities of C03, C09 and C10 on focal adhesion kinase (FAK), p21-activated kinase 4 (PAK4) and polo-like kinase 4 (PLK4) were tested. As shown in Fig. S1 (ESI†), C03, C09 and C10 exhibited low suppression on PLK4 (IC50 = 1.65, 0.69 and 1.32 μM, respectively), together with significant selectivity for FAK and PAK4 (IC50 for both was over 100 μM). Among them, considering the selectivity and synthetic cost, C03 may more suitable for further exploration.| Entry | TRKA | TRKB | TRKC |
|---|---|---|---|
| IC50 (μM) | IC50 (μM) | IC50 (μM) | |
| C03 | 0.056 | 0.090 | 0.058 |
| C09 | 0.057 | 0.090 | 0.083 |
| C10 | 0.026 | 0.118 | 0.076 |
| LOXO-101 | 0.003 | 0.013 | 0.0002 |
3.3 Molecular docking study
The molecular docking study was used to evaluate the binding mode between C03, C09 and C10 and TRKA. Given that compounds C09 and C10 are isomers of C03, both of them were separately used for the molecular docking study. As shown in Fig. 4, compounds C09 and C10 had good overlap with entrectinib and suitably occupied the ATP pocket. Simultaneously, the chiral center of 3-hydroxypiperidinyl did not have an obvious influence on the binding mode. Similar to entrectinib and larotrectinib, the pyrazolyl is bound to the hinge region with Glu590 and Met592 residues through two hydrogen bonds; the 2,5-difluorophenyl is oriented toward the DFG motif and bound to Asn655 and Cys656 with fluorine bonds; pyridine forms a π–π stacking interaction with Phe589; and the 3-hydroxy on the piperidine is bound to Asp596 with a hydrogen bond. Consequently, compound C03 was chosen for further assessments.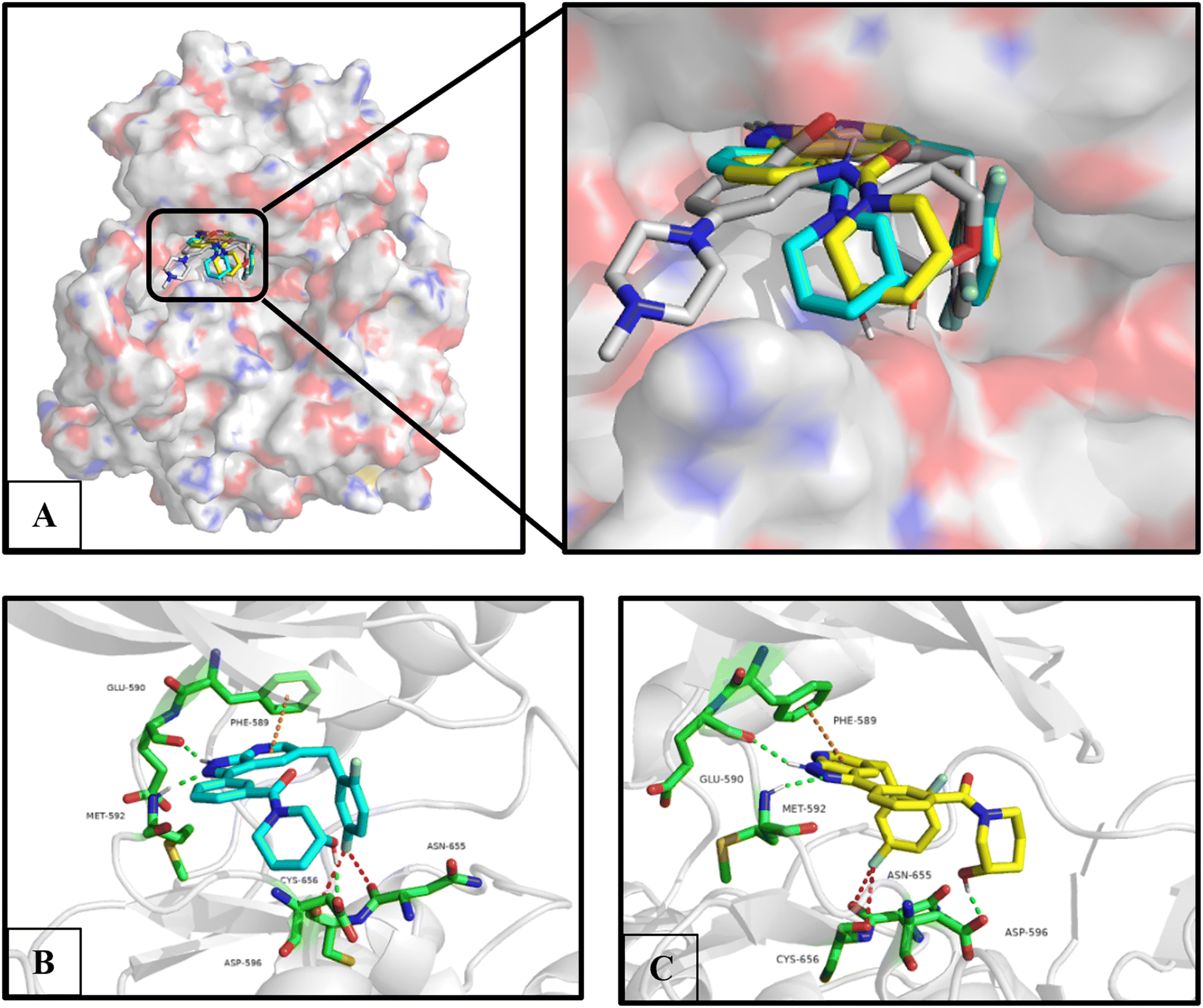 | ||
| Fig. 4 Molecular docking between C09 and C10 with TRKA (PDB code: 5KVT). (A) Overlap of C09 (blue) and C10 (yellow) with entrectinib (white). (B) Predicted binding mode between C09 and TRKA. (C) Predicted binding mode between C10 and TRKA. The residues are depicted in green; the interactions are depicted as dash lines with different colours, hydrogen bond is depicted in green, fluorine bond is depicted in red and π–π stacking interaction is depicted in orange. | ||
3.4 Antiproliferative activities
Compound C03 was further evaluated for its antiproliferative activities against three different cell lines, including Km-12, MCF-7 and HUVEC. Km-12 is a natural colorectal cancer line with TPM3-NTRK1 gene fusion. The breast cancer cell line MCF-7 does not harbour any rearrangement of TRKs. Also, HUVEC are human umbilical vein endothelial cells, a normal cell line without cancer. As shown in Table 5, compound C03 inhibited the proliferation of Km-12 cells with the IC50 value of 0.304 μM. Furthermore, the inhibitory activity against the TRKA-independent cell lines MCF-7 and HUVEC was weak (IC50 > 40 μM and IC50 > 36.69 μM, respectively). This indicated that C03 may have a low risk of off-target effects.3.5 The plasma stability and CYPs inhibition
The plasma stability and CYPs inhibition of C03 were evaluated. As shown in Table 6, compound C03 possessed excellent plasma stability with t1/2 > 289.1 min. Then, compound C03 was evaluated for its inhibition rates on a panel of cytochrome P450 isoforms (1A2, 2C9, 2C19, 2D6 and 3A4). As shown in Table 7, C03 exhibited relatively strong suppression on only CYP2C9 at the concentration of 10 μM, which means that C03 may have a low risk of drug–drug interactions (DDIs).| Time (min) | 0 | 10 | 30 | 60 | 120 |
|---|---|---|---|---|---|
| Remaining (%) | 100.0 | 103.7 | 88.0 | 93.6 | 89.1 |
| T 1/2 (min) | >289.1 |
| Isozyme | CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 |
|---|---|---|---|---|---|
| Inhibition at 10 μM (%) | 16.6 | 66.6 | 48.6 | 25.8 | 0.0 |
4 Conclusion
Herein, based on scaffold hopping, rational drug design and CADD, we designed and synthesized 38 novel TRKA inhibitors with a pyrazolo[3,4-b]pyridine core. The in vitro enzyme activity assays suggested that most of them had nanomole inhibitory activity against TRKA, while compounds C03, C09 and C10 showed acceptable potency with IC50 values of 56/57/26 nM, respectively. The kinase selectivity assays suggested that C03, C09 and C10 are pan-TRK inhibitors combined with significant selectivity for FAK, PAK4 and PLK4. The proliferation assay revealed that C03 also had acceptable antiproliferative activity in the Km-12 cell line with an IC50 value of 0.304 μM, together with selectivity for the MCF-7 cell line and HUVEC cell line. Furthermore, compound C03 exhibited good plasma stability and low inhibitory activities to CYPs except CYP2C9. Thus, this compound has potential for further exploration and related works are undergoing.5 Experiment
The starting materials, reagents and solvents were obtained from a commercial supplier and used without further purification unless otherwise indicated. Anhydrous solvents were dried and stored according to standard procedures. All reactions were monitored by thin-layer chromatography (TLC) on silica gel plates with fluorescence F-254 and visualized with UV light (254 nm or 365 nm). Column chromatography was performed on silica gel (200–300 mesh). 1H NMR and 13C NMR spectra were recorded in DMSO-d6 on Bruker ARX-600 NMR and Bruker ARX-400 NMR spectrometers with TMS as the internal standard. Coupling constants (J) are expressed in hertz (Hz). The chemical shifts (δ) of NMR are reported in parts per million (ppm). Liquid chromatography-mass spectrometry (LC-MS) (Agilent, Palo Alto, CA, USA) or high-resolution accurate mass spectrometry (HRMS) (Agilent, Q-TOF6550) was used in the determination of the structure of all the final compounds. Reverse-phase high-performance liquid chromatography (HPLC, SHIMADAZU SPD-20A) was performed to measure the purities of the target compounds (the flow rate was 1.0 mL min−1 and the eluents were 100% methanol and purified water).5.1 Chemistry
:EA = 1:1, and then filtered under reduced pressure. The crude solid was recrystallized using 200 mL DMF at 120 °C to obtain 9.
:MeOH = 150:1) to obtain 13.
:MeOH = 150:1) to give 15a–15o.
5.1.7.1 (3-((5-(3-Fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A01). Yield: 49.9%, 30 mg, faint yellow solid. m.p. 208.5–209.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.55 (s, 1H), 9.19 (s, 1H), 8.45 (d, J = 2.0 Hz, 1H), 8.18 (d, J = 1.8 Hz, 1H), 7.80(t, J = 3.2, J = 1.6, 1H), 7.61 (dd, J = 8, J = 1.6, 1H), 7.30–7.39 (m, 2H), 7.11 (dd, J = 8.8, J = 1.2, 2H), 7.06–7.01 (m, 1H), 6.83 (dt, J = 7.4, 1.3 Hz, 1H), 4.11 (s, 2H), 3.60–3.57 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.94, 163.61, 161.99, 151.25, 151.00, 144.79 (d, J = 7.6 Hz), 143.81, 143.06, 136.57, 130.98 (d, J = 8.8 Hz), 129.29 (d, J = 3.3 Hz), 127.68, 125.26 (d, J = 3.2 Hz), 117.93, 117.31, 115.87 (d, J = 20.5 Hz), 114.69, 113.44 (d, J = 21.3 Hz), 106.94, 66.68 (2C), 38.06. HRMS (ESI, m/z) calcd for C24H23FN5O2 [M + H]+, 432.1836; found 432.1836. HPLC purity (MeOH
:H2O = 80:20): 99.6%, tR = 12 min.
5.1.7.2 (3-((5-Benzyl-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A02). Yield: 51.4%, 18 mg, faint yellow solid. m.p.: 190.3–191.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.70 (s, 0.2H), 12.54 (s, 0.8H), 9.44 (s, 0.2H), 9.25 (s, 0.8H), 8.80 (s, 0.2H), 8.75 (s, 0.2H), 8.43 (s, 0.8H), 8.21 (s, 0.8H), 7.89 (s, 0.2H), 7.81 (s, 0.8H), 7.75–7.68 (m, 0.8H), 7.62 (d, J = 8.3 Hz, 0.8H), 7.54–7.51 (m, 0.4H),7.41–7.39 (m, 0.2H), 7.36 (d, J = 7.8 Hz, 0.2H), 7.33 (s, 0.4H), 7.31 (s, 0.8H), 7.30 (s, 0.8H), 7.28 (s, 0.8H), 7.26 (s, 0.8H), 7.20 (t, J = 7.3 Hz, 1H), 6.87 (d, J = 7.8 Hz, 0.2H), 6.83 (d, J = 7.5 Hz, 0.8H), 4.23–4.08 (m, 2H), 3.60 (s, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.95, 151.21, 151.01, 143.80, 143.09, 141.87, 136.54, 129.64, 129.11 (2C), 129.08 (2C), 128.26, 127.19, 126.62, 117.89, 117.30, 114.67, 106.94, 66.68 (2C), 38.54. Ms (ESI) m/z 413.2, [M + H]+ 414.2.
5.1.7.3 (3-((5-(3-Chlorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A03). Yield: 52.1%, 22 mg, faint yellow solid. m.p.: 195.9–197.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.23 (s, 1H), 8.46 (d, J = 1.8, 1H), 8.18 (s, 1H), 7.81 (s, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.36–7.31 (m, 3H), 7.28 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 7.2, 1H), 6.84 (d, J = 7.2 Hz, 1H), 4.11 (s, 2H), 3.62–3.39 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.93, 151.25, 151.01, 144.47, 143.81, 143.05, 136.57, 133.65, 130.95, 129.30, 128.95, 127.90, 127.64, 126.66, 117.93, 117.31, 114.70, 106.94, 66.68 (2C), 37.94. Ms (ESI) m/z 447.1, [M + H]+ 448.1.
5.1.7.4 (3-((5-(3-Bromobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A04). Yield: 49.1%, 25 mg, faint yellow solid. m.p.: 187.8–189.0 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.57 (s, 1H), 9.23 (s, 1H), 8.45 (d, J = 1.9 Hz, 1H), 8.17 (d, J = 1.7 Hz, 1H), 7.81 (t, J = 1.8, 1H), 7.61 (dd, J = 7.8, J = 1.2, 1H), 7.50 (s, 1H), 7.41 (dq, J = 5.6, 3.6, 2.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.30–7.23 (m, 2H), 6.83 (d, J = 7.8 Hz, 1H), 4.10 (s, 2H), 3.62 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.94, 151.25, 151.00, 144.78, 143.81, 143.05, 136.56, 131.81, 131.26, 129.55, 129.31 (2C), 128.29, 127.64, 122.36, 117.93, 117.32, 114.70, 106.94, 66.68 (2C), 65.39 (2C), 37.91. Ms (ESI) m/z 492.0, [M + H]+ 493.0.
5.1.7.5 Morpholino(3-((5-(3-nitrobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)methanone (A05). Yield: 51.2%,31 mg, faint yellow solid. m.p.: 221.3–222.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.60 (s, 1H), 9.25 (s, 1H), 8.50 (d, J = 1.8 Hz, 1H), 8.22 (d, J = 2.2 Hz, 1H), 8.16 (t, J = 2.0 Hz, 1H), 8.09 (dd, J = 8.4, J = 1.8, 1H), 7.81 (s, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.64–7.60 (m, 2H), 7.32 (t, J = 7.8 Hz, 1H), 6.83 (m, 1H), 4.26 (s, 2H), 3.62–3.38 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.93, 151.27, 151.06, 148.44, 144.23, 143.84, 143.04, 136.56, 136.08, 130.62, 129.52, 129.29, 127.23, 123.66, 121.78, 117.94, 117.32, 114.70, 106.97, 66.68 (2C), 37.74. Ms (ESI) m/z 458.2, [M + H]+ 459.1.
5.1.7.6 Morpholino(3-((5-(3-(trifluoromethyl)benzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)methanone (A06). Yield: 50.1%, 25 mg, faint yellow solid. m.p.: 218.7–219.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.59 (s, 1H), 9.25 (s, 1H), 8.48 (d, J = 2.0 Hz, 1H), 8.20 (s, 1H), 7.81 (t, J = 1.9 Hz, 1H), 7.66 (s, 1H), 7.62–7.55 (m, 4H), 7.32 (t, J = 7.8 Hz, 1H), 6.83 (d, J = 7.8 Hz, 1H), 4.21 (s, 2H), 3.66–3.40 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.93, 151.25, 151.06, 143.82, 143.40, 143.04, 136.56, 133.38, 130.16 (2C), 129.86 (d, J = 31.2 Hz), 129.40 (d, J = 16.2 Hz), 127.51, 125.56 (m), 123.60 (m), 117.93, 117.31, 114.70, 106.95, 66.68 (2C), 37.98. Ms (ESI) m/z 481.2, [M + H]+ 482.1.
5.1.7.7 (3-((5-(2-Fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A07). Yield: 49.6%, 35 mg, faint yellow solid. m.p.: 287.7–288.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.56 (s, 1H), 9.24 (s, 1H), 8.44 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 2.1 Hz, 1H), 7.81 (t, J = 1.9 Hz, 1H), 7.62 (dd, J = 8.2, 2.3 Hz, 1H), 7.37 (td, J = 7.7, 1.9 Hz, 1H), 7.33–7.28 (m, 2H), 7.20–7.16 (m, 2H), 6.83 (d, J = 7.8 Hz, 1H), 4.11 (s, 2H), 3.66–3.61 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.94, 161.64, 160.02, 151.23, 150.85, 143.77, 143.06, 136.55, 131.64 (d, J = 4.4 Hz), 129.28, 129.04 (d, J = 10.0 Hz), 128.45 (d, J = 15.6 Hz), 126.89, 125.15, 117.91, 117.32, 115.87 (d, J = 21.8 Hz), 114.71, 106.85, 66.68 (2C), 31.82. Ms (ESI) m/z 431.2, [M + H]+ 432.1.
5.1.7.8 (3-((5-(4-Fluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A08). Yield: 50.5%, 37 mg, faint yellow solid. m.p.: 194.5–195.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.56 (s, 1H), 9.21 (s, 1H), 8.44 (d, J = 1.8, 1H), 8.15 (s, 1H), 7.81 (s, 1H), 7.61 (dd, J = 8.1, 2.3 Hz, 1H), 7.33–7.29 (m, 3H), 7.16–7.13 (m, 2H), 6.83 (d, J = 7.2 Hz, 1H), 4.08 (s, 2H), 3.61 (s, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.93, 162.08, 160.48, 151.22, 150.99, 143.78, 143.07, 138.00 (d, J = 2.8 Hz), 136.56, 130.95 (d, J = 7.9 Hz, 2C), 129.30, 129.16, 128.19, 117.91, 117.30, 115.84 (d, J = 21.3 Hz, 2C), 114.67, 66.68 (2C), 37.56. Ms (ESI) m/z 431.2, [M + H]+ 432.2.
5.1.7.9 (3-((5-(2,3-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A09). Yield: 51.3%, 40 mg, faint yellow solid. m.p.: 151.3–152.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.59 (s, 1H), 9.24 (s, 1H), 8.47 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 2.1 Hz, 1H), 7.82 (t, J = 1.9 Hz, 1H), 7.62 (ddd, J = 8.3, 2.4, 1.0 Hz, 1H), 7.34–7.30 (m, 2H), 7.20–7.18 (m, 2H), 6.84 (dt, J = 7.8, 1.8 Hz 1H), 4.17 (s, 2H), 3.51 (m, 8H). 13C NMR (151 MHz, DMSO) δ 169.93, 151.23, 151.16 (dd, J = 245.3, 12.4 Hz) 150.83, 149.36, (dd, J = 245.3, 12.4 Hz), 143.81, 143.00, 136.56, 131.15 (d, J = 12.4 Hz), 129.29, 129.14, 126.76 (t, J = 3.3 Hz), 126.32, 125.48 (dd, J = 6.7, 4.6 Hz), 117.96, 117.34, 116.19 (d, J = 16.8 Hz), 114.73, 106.85, 66.68 (2C), 31.53. Ms (ESI) m/z 449.2, [M + H]+ 450.1.
5.1.7.10 (3-((5-(2,4-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A10). Yield: 52.3%, 30 mg, faint yellow solid. m.p.: 212.4–213.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.57 (s, 1H), 9.23 (s, 1H), 8.44 (s, 1H), 8.15 (s, 1H), 7.81 (s, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.43 (q, J = 7.8 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.26–7.22 (m, 1H), 7.09–7.06 (m, 1H), 6.84 (m, 1H), 4.09 (s, 2H), 3.66–3.62 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.94, 162.45 (dd, J = 128.9, 12.4 Hz), 160.83 (dd, J = 131.0, 12.3 Hz), 151.22, 150.83, 143.79, 143.03, 136.56, 132.63 (dd, J = 9.8, 6.1 Hz), 129.29, 128.99, 126.73, 124.81 (dd, J = 15.7, 3.9 Hz), 117.94, 117.33, 114.73, 112.17 (dd, J = 21.0, 3.7 Hz), 106.83, 104.60 (t, J = 26.0 Hz), 66.68 (2C), 31.26. Ms (ESI) m/z 449.2, [M + H]+ 450.1.
5.1.7.11 (3-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A11). Yield: 50.1%, 27 mg, faint yellow solid. m.p.: 207.0–208.0 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.24 (s, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 2.1 Hz, 1H), 7.82 (t, J = 1.9 Hz, 1H), 7.62 (dd, J = 8.3, 2.4 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.29–7.23 (m, 2H), 7.16–7.12 (m, 1H), 6.83 (d, J = 7.2 Hz, 1H), 4.10 (s, 2H), 3.66–3.39 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.93, 159.43, 157.84 (d, J = 5.2 Hz), 156.22, 151.23, 150.84, 143.81, 143.01, 136.56, 130.59 (dd, J = 18.3, 7.9 Hz), 129.29, 129.10, 126.32, 118.01 (m), 117.85 (dd, J = 58.9, 6.8 Hz), 117.48 (m), 115.41 (dd, J = 23.8, 8.9 Hz), 114.73, 106.84, 66.68 (2C), 31.82. HRMS (ESI, m/z) calcd for C24H22F2N5O2 [M + H]+, 450.1742; found 450.1737. HPLC purity (MeOH
:H2O = 80:20): 99.5%, tR = 11 min.
5.1.7.12 (3-((5-(2,6-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A12). Yield: 49.9%, 35 mg, yellow solid. m.p.: 206.2–207.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.87 (m, 0.23H), 12.59 (s, 0.77H), 9.43 (m, 0.15H), 9.29 (s, 0.77H), 9.26 (m, 0.08H), 8.82 (m, 0.08H), 8.74 (m, 0.15H), 8.54 (d, J = 2.4 Hz, 0.08H), 8.44 (d, J = 2.1 Hz, 0.77H), 8.30 (d, J = 2.7 Hz, 0.08H), 8.19 (d, J = 2.1 Hz, 1H), 8.14 (m, 0.08H), 7.90 (m, 0.08H), 7.85(m, 1H), 7.76 (m, 0.17H), 7.70 (m, 0.08H), 7.65 (m, 1H), 7.56 (m, 0.23H), 7.38 (tt, J = 8.1, 6.5 Hz, 1H), 7.32 (t, J = 7.8 Hz, 0.77H), 7.22 (m, 0.23H), 7.15 (t, J = 7.8 Hz, 1.54H), 6.88 (d, J = 6.96 Hz, 0.23H), 6.84 (dt, J = 7.5, 1.3 Hz, 0.77H), 5.33 (m, 0.08H), 5.19 (s, 0.15H), 4.12 (s, 1.54H), 3.51 (m, 8H). Ms (ESI) m/z 449.2, [M + H]+ 450.1.
5.1.7.13 (3-((5-(3,4-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A13). Yield: 49.5%, 32 mg, faint yellow solid. m.p.: 216.9–217.7 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.20 (s, 1H), 8.45 (d, J = 2.1 Hz, 1H), 8.15 (d, J = 2.1 Hz, 1H), 7.81 (t, J = 1.9 Hz, 1H), 7.61 (dd, J = 8.2, 2.4 Hz, 1H), 7.40–7.35 (m, 2H), 7.32 (t, J = 7.9 Hz, 1H), 7.12–7.10 (m, 1H), 6.83 (d, J = 7.8 Hz, 1H), 4.09 (s, 2H), 3.74–3.61 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.94, 151.26, 150.97, 150.67 (d, J = 12.6 Hz), 149.39 (dd, J = 53.0, 12.4 Hz), 147.78 (d, J = 12.4 Hz), 143.81, 143.06, 139.64 (t, J = 4.7 Hz), 136.57, 129.30, 129.23, 127.60, 125.86, 118.12 (d, J = 10.5 Hz), 118.01(d, J = 11.7 Hz), 117.32, 114.70, 106.94, 66.68 (2C), 37.41. Ms (ESI) m/z 449.2, [M + H]+ 450.1.
5.1.7.14 (3-((5-(3,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (A14). Yield: 48.5%, 22 mg, faint yellow solid. m.p.: 218.1–219.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.57 (s, 1H), 9.21 (s, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 7.81 (t, J = 1.9 Hz, 1H), 7.62 (dd, J = 8.2, 2.3 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.10–7.05 (m, 1H), 7.05–7.00 (m, 2H), 6.83 (dt, J = 7.5, 1.2 Hz, 1H), 4.12 (s, 2H), 3.60–3.44 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.94, 163.82 (d, J = 13.5 Hz), 162.18 (d, J = 12.7 Hz), 151.24, 150.94, 146.55, 146.49 (t, J = 9.0 Hz), 143.03, 136.56, 129.42 (d, J = 17.8 Hz), 127.08, 117.95, 117.33, 114.70, 112.39 (dd, J = 19.7, 4.9 Hz), 106.97, 102.37 (t, J = 25.9 Hz), 66.68 (2C), 55.38 (2C), 37.89. Ms (ESI) m/z 449.2, [M + H]+ 450.1. HPLC purity (MeOH
:H2O = 80:20): 99.0%, tR = 11 min.
5.1.7.15 Morpholino(3-((5-(3,4,5-trifluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)methanone (A15). Yield: 49.7%, 20 mg, faint yellow solid. m.p.: 229.2–231.0 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.60 (s, 1H), 9.20 (s, 1H), 8.46 (s, 1H), 8.15 (s, 1H), 7.81 (s, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 7.27 (t, J = 7.8 Hz, 2H), 6.84 (d, J = 7.8 Hz, 1H), 4.10 (s, 2H), 3.66–3.61 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δ 169.93, 151.53 (dd, J = 9.9, 4.0 Hz), 151.28, 150.95, 149.89 (dd, J = 10.1, 3.2 Hz), 143.83, 143.04, 139.26 (m), 138.74 (dt, J = 247.0, 15.6 Hz), 138.64, 138.54, 137.11, 137.00, 136.90, 136.58, 129.31 (2C), 127.00, 117.95, 117.32, 114.70, 113.80 (d, J = 4.0 Hz), 113.69 (d, J = 4.0 Hz), 106.94, 66.68 (2C), 55.77 (2C), 37.43. Ms (ESI) m/z 467.2, [M + H]+ 468.1. HPLC purity (MeOH
:H2O = 80:20): 95.4%, tR = 10 min.
:EA = 5:1).
:MeOH = 50:1). If the compound was protected by Boc group, then the solid was treated with 4 N HCl/EA at room temperature for 1 h. The mixture was concentrated in vacuo, and then the oil was dissolved in water and saturated sodium bicarbonate solution was added to adjust the pH = 8. Subsequently, it was extracted with EA (10 mL × 3), dried with anhydrous sodium sulfate, and finally the solvent was removed to get the product.
5.1.14.1 (3-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(4-methylpiperazin-1-yl)methanone (B01). Yield: 51.4%, 25 mg, white solid. m.p.: 239.1–241.4 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.22 (s, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.18 (d, J = 2.1 Hz, 1H), 7.77 (t, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.2, 2.3 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.29–7.23 (m, 2H), 7.13 (ddd, J = 8.7, 6.4, 3.6 Hz, 1H), 6.80 (dt, J = 7.4, 1.3 Hz, 1H), 4.10 (s, 2H), 3.61 (s, 2H), 3.33 (s, 2H), 2.35–2.28 (m, 4H), 2.19 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.81, 159.43 (d, J = 240.6 Hz), 157.80 (d, J = 240.0 Hz), 151.24, 150.83, 143.82, 142.99, 136.99, 130.59 (dd, J = 18.3, 7.9 Hz), 129.28, 129.11, 126.32, 118.00 (dd, J = 24.5, 5.1 Hz) 117.81, 117.46 (dd, J = 25.2, 8.8 Hz), 117.18, 115.39, 115.28 (dd, J = 24.2, 8.5 Hz), 114.58, 106.86, 55.38–54.83 (m, 2C), 49.08 (2C), 46.07, 31.83. HRMS (ESI, m/z) calcd for C25H25F2N6O [M + H]+, 463.2058; found 463.2069. HPLC purity (MeOH
:H2O = 80:20): 98.1%, tR = 11 min.
5.1.14.2 (3-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(4-hydroxypiperidin-1-yl)methanone (B02). Yield: 55.4%, 27 mg, white solid. m.p.: 169.9–171.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.57 (s, 1H), 9.23 (s, 1H), 8.46 (d, J = 1.8, 1H), 8.19 (s, 1H), 7.76 (s, 1H), 7.63 (d, J = 7.7 Hz, 1H), 7.32–7.23 (m, 3H), 7.14 (tt, J = 8.3, 3.6 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 4.80 (d, J = 3.7 Hz, 1H), 4.10 (s, 2H), 4.02 (s, 1H), 3.75–3.73 (m, 1H), 3.55 (s, 1H), 3.20–3.16 (m, 2H), 2.02–1.70 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 169.77, 159.42(d, J = 240.2 Hz), 157.80 (d, J = 240.7 Hz), 151.22, 150.82, 143.84, 142.96, 137.44, 130.60 (dd, J = 18.3, 7.9 Hz), 129.26, 129.14, 126.29, 118.01 (dd, J = 24.2, 4.8 Hz), 117.54, 117.47 (dd, J = 24.9, 8.9 Hz), 117.01, 115.40 (dd, J = 23.8, 8.9 Hz), 114.31, 106.85, 66.04, 45.12 (2C), 31.82. Ms (ESI) m/z 463.2, [M + H]+ 464.1.
5.1.14.3 (3-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(3-hydroxypiperidin-1-yl)methanone (B03). Yield: 47.2%, 23 mg, white solid. m.p.: 197.5–199.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.57 (s, 1H), 9.25 (s, 1H), 8.45 (s, 1H), 8.21 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.99 (s, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.27–7.24 (m, 2H), 7.15–7.12 (m, 1H), 4.10 (s, 2H), 3.82–3.78 (m, 1H), 3.00 (m, 4H), 2.96–2.94 (m, 1H), 1.73–1.71 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 170.16, 159.43 (d, J = 240.0 Hz), 157.82 (d, J = 243.6 Hz), 151.23, 150.81, 143.85, 142.96, 137.52, 130.60 (dd, J = 18.4, 7.7 Hz), 129.15 (2C), 126.30, 118.00 (dd, J = 24.3, 4.9 Hz), 117.59, 117.46 (dd, J = 24.9, 9.0 Hz), 116.97, 115.39 (dd, J = 24.1, 8.6 Hz), 114.51 (d, J = 45.9 Hz), 106.87, 65.74–65.58 (m), 49.13, 47.64, 31.82, 27.29, 23.94. Ms (ESI) m/z 463.2, [M + H]+ 464.1.
5.1.14.4 (3-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(piperazin-1-yl)methanone (B04). Yield: 48.7%, 23 mg, faint yellow solid. m.p.: 234.9–236.0 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.24 (s, 1H), 8.46 (d, J = 2.1 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 7.77 (t, J = 1.9 Hz, 1H), 7.62 (dd, J = 8.2, 2.3 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.28–7.23 (m 2H), 7.13 (tt, J = 8.2, 3.6 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 4.10 (s, 2H), 3.54 (s, 2H), 3.28–3.26 (m, 2H), 2.72–2.64 (m, 4H), 2.43 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 169.79, 159.42 (d, J = 240.7 Hz), 157.80 (d, J = 240.8 Hz), 151.23, 150.82, 143.84, 142.97, 137.25, 130.60 (dd, J = 18.9, 7.9 Hz), 129.25, 129.15, 126.30, 118.00 (dd, J = 24.3, 5.1 Hz), 117.77, 117.46 (dd, J = 24.9, 9.0 Hz), 117.05, 115.39 (dd, J = 23.9, 8.2 Hz), 114.54, 106.86, 46.51, 46.34, 46.31, 46.03, 31.82. HRMS (ESI, m/z) calcd for C24H23F2N6O [M + H]+, 449.1901; found 449.1900. HPLC purity (MeOH
:H2O = 85:15): 94.5%, tR = 15 min.
5.1.14.5 (4-Aminopiperidin-1-yl)(3-((5-(2,5-difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)methanone (B05). Yield: 41.1%, 20 mg, faint yellow solid. m.p.: 163.8–164.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.58 (s, 1H), 9.22 (s, 1H), 8.46 (s, 1H), 8.18 (s, 1H), 7.76 (s, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.31–7.25 (m, 3H), 7.14 (s, 1H), 6.78 (d, J = 7.5 Hz, 1H), 4.28 (s, 1H), 4.10 (s, 2H), 3.61–3.33 (m, 4H), 1.99–1.66 (m, 4H). Ms (ESI) m/z 462.2, [M + H]+ 463.2.
5.1.14.6 3-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)-N-(piperidin-4-yl)benzamide (B06). Yield: 51.4%, 25 mg, faint yellow solid. m.p.: 173.4–174.5 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.57 (s, 1H), 9.25 (s, 1H), 8.45 (s, 1H), 8.21 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.99 (s, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.27–7.23 (m, 3H), 7.15–7.12 (m, 1H), 4.10 (s, 2H), 3.81 (s, 1H), 3.01 (s, 4H), 1.72 (s, 4H). 13C NMR (151 MHz, DMSO-d6) δ 166.47, 159.43 (d, J = 239.4 Hz), 157.81 (d, J = 240.5 Hz), 151.27, 150.74, 143.95, 142.96, 136.26, 130.60 (dd, J = 18.8, 7.7 Hz), 129.20, 128.96, 126.24, 118.41, 118.11, 118.00 (dd, J = 24.4, 5.0 Hz), 117.46 (dd, J = 24.9, 9.0 Hz), 115.91, 115.39 (dd, J = 23.9, 8.3 Hz), 106.91, 46.34, 46.31, 45.82, 31.8, 26.41, 26.36. Ms (ESI) m/z 462.2, [M + H]+ 463.1.
5.1.14.7 (4-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(morpholino)methanone (B07). Yield: 46.6%, 22 mg, faint yellow solid. m.p.: 227.5–229.4 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.65 (s, 1H), 9.36 (s, 0.87 H),8.99 (s, 0.13 H), 8.70 (s, 0.13 H), 8.47 (s, 0.87 H), 8.20 (s, 0.87 H), 8.12 (s, 0.13 H), 7.83 (d, J = 8.4 Hz, 0.26H), 7.69 (d, J = 8.3 Hz, 1.74H), 7.44 (d, J = 7.8 Hz, 0.26 H), 7.37 (d, J = 8.3 Hz, 1.74 H), 7.26 (m, 2H), 7.13 (tt, J = 8.2, 3.5 Hz, 1H), 4.11 (s, 2H), 3.60–3.51 (m, 8H). Ms (ESI) m/z 449.2, [M + H]+ 450.1.
5.1.14.8 (4-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(4-methylpiperazin-1-yl)methanone (B08). Yield: 55.5%, 27 mg, faint yellow solid. m.p.: 168.7–170.4 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.64 (s, 1H), 9.33 (s, 1H), 8.47 (d, J = 2.1 Hz, 1H), 8.19 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.29–7.23 (m, 2H), 7.16–7.12 (m, 1H), 4.11 (s, 2H), 3.51 (s, 4H), 2.33–2.31 (m, 4H), 2.20 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.84, 159.43 (d, J = 240.0 Hz), 157.80 (d, J = 240.1 Hz), 151.23, 150.86, 144.30, 143.56, 130.59 (dd, J = 18.8, 7.9 Hz), 129.15, 129.04 (2C), 126.41, 126.29, 118.01 (dd, J = 24.3, 4.9 Hz), 117.47 (dd, J = 24.8, 9.0 Hz), 115.48 (2C), 115.40 (dd, J = 23.9, 8.9 Hz), 115.34, 115.24, 115.18, 106.92, 55.07 (2C), 49.07 (2C), 46.08, 31.82. Ms (ESI) m/z 462.2, [M + H]+ 463.2.
5.1.14.9 (4-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(4-hydroxypiperidin-1-yl)methanone (B09). Yield: 43.1%, 21 mg, faint yellow. m.p.: 171.4–172.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.63 (s, 1H), 9.31 (s, 1H), 8.47 (d, J = 2.2 Hz, 1H), 8.19 (d, J = 2.0 Hz, 1H), 7.67 (m, 2H), 7.33 (m, 2H), 7.26 (m, 2H), 7.14 (m, 1H), 4.77 (d, J = 4.0 Hz, 1H), 4.11 (s, 2H), 3.82–3.70 (m, 2H), 3.19–3.15 (m, 2H), 1.74 (s, 2H), 1.38–1.23 (m, 3H). HRMS (ESI, m/z) calcd for C25H24F2N5O2 [M + H]+, 464.1898; found 464.1894. HPLC purity (MeOH
:H2O = 80:20): 98.6%, tR = 12 min.
5.1.14.10 (4-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(3-hydroxypiperidin-1-yl)methanone (B10). Yield: 36.9%, 18 mg, faint yellow solid. m.p.: 178.7–179.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.75 (s, 0.25H), 12.62 (s, 0.75H), 9.41 (s, 0.25H), 9.30 (s, 0.75H), 8.81 (s, 0.25H), 8.71 (s, 0.25H), 8.47 (s, 0.75H), 8.19 (s, 0.75H), 7,74 (m, 0.5H), 7.67 (d, J = 8.4 Hz, 1.5H), 7.53 (t, J = 7.5 Hz, 0.5H), 7.42 (d, J = 7.2 Hz, 0.25H), 7.38 (d, J = 8.2 Hz, 0.5H), 7.33 (d, J = 8.1 Hz, 1.5H), 7.29–7.24 (m, 1.5H), 7.13 (dq, J = 8.2, 3.9, 3.5 Hz, 0.75H), 4.90 (s, 1H), 4.11 (s, 2H), 3.81–3.48 (m, 3H), 3.09–2.88 (m, 2H), 1.87 (s, 1 H), 1.70 (S, 1H), 1.41–1.38 (m, 2H). HRMS (ESI, m/z) calcd for C25H24F2N5O2 [M + H]+, 464.1898; found 464.1893. HPLC purity (MeOH
:H2O = 90:10): 96.4%, tR = 10 min.
5.1.14.11 (4-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)(piperazin-1-yl)methanone (B11). Yield: 48.8%, 23 mg, faint yellow solid. m.p.: 218.8–220.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.63 (s, 1H), 9.31 (s, 1H), 8.47 (s, 1H), 8.19 (s, 1H), 7.67 (d, J = 8.2 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.26 (m, 2H), 7.14 (m, 1H), 4.11 (s, 2H), 3.43 (s, 4H), 2.68 (s, 4H), 1.23 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 169.82, 159.42 (d, J = 240.7 Hz), 157.81 (d, J = 242.1 Hz), 151.23, 150.84, 144.16, 143.59, 130.59 (dd, J = 18.6, 7.9 Hz), 129.15, 128.98, 126.59, 126.39, 118.00 (dd, J = 23.9, 3.6 Hz), 117.46 (dd, J = 24.9, 9.1 Hz), 115.48, 115.40 (dd, J = 24.5, 9.4 Hz), 106.92, 46.27 (2C), 31.82. Ms (ESI) m/z 448.2, [M + H]+ 449.4.
5.1.14.12 (4-Aminopiperidin-1-yl)(4-((5-(2,5-difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)phenyl)methanone (B12). Yield: 32.9%, 16 mg, faint yellow solid. m.p.: 234.4–235.7 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.63 (s, 1H), 9.33 (s, 0.8H), 8.99 (s, 0.2H), 8.69 (s, 0.2H), 8.46 (s, 0.8H), 8.20 (s, 0.8H), 8.12 (s, 0.2H), 7.80 (d, J = 8.4 Hz, 0.4H), 7.67 (d, J = 8.2 Hz, 1.6H), 7.37 (d, J = 8.4 Hz, 0.4H), 7.31 (d, J = 8.3 Hz, 1.6H), 7.25 (m, 2H), 7.13 (tt, J = 8.2, 3.5 Hz, 1H), 4.11 (s, 2H), 3.33 (s, 2H), 2.97 (s, 2H), 2.82–2.77 (m, 1H), 1.71 (s, 2H), 1.19–1.14 (m, 2H). Ms (ESI) m/z 462.2, [M + H]+ 463.2. HPLC purity (MeOH
:H2O = 85:15): 98.9%, tR = 28 min.
5.1.14.13 4-((5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)amino)-N-(piperidin-4-yl)benzamide (B13). Yield: 28.8%, 14 mg, faint yellow solid. m.p.: 245.6–247.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.78 (s, 0.2H), 12.66 (s, 0.8H), 9.51 (s, 0.2H), 9.40 (s, 0.8H), 8.82 (s, 0.2H), 8.73 (s, 0.2H), 8.47 (s, 0.8H), 8.21 (s, 0.8H), 7.97 (d, J = 7.8 Hz, 0.4H), 7.85 (d, J = 8.4 Hz, 0.4H), 7.80 (d, J = 8.3 Hz, 2H), 7.75–7.72 (m, 1H), 7.67 (d, J = 8.3 Hz, 1.6H), 7.54–7.52 (m, 0.4H), 7.42–7.39 (m, 0.2H), 7.29–7.24 (m, 1.6H), 7.15–7.12 (m, 0.8H), 4.11 (s, 2H), 3.81 (s, 1H), 3.33 (s, 2H), 2.95 (d, J = 11.9 Hz, 2H), 1.99 (m, 0.5H), 1.72–1.70 (m, 2H), 1.43–1.38 (m, 2H). Ms (ESI) m/z 462.2, [M + H]+ 463.1. HPLC purity (MeOH
:H2O = 90:10): 97.5%, tR = 25 min.
:EA = 5:1).
5.1.21.1 (3-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(4-methylpiperazin-1-yl)methanone (C01). Yield: 51.0%, 25 mg, white solid. m.p.: 122.3–123.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.89 (s, 1H), 8.48 (m, 2H), 8.08 (d, J = 7.8 Hz, 1H), 7.93 (s, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.27–7.21 (m, 2H), 7.12–7.09 (m, 1H), 4.18 (s, 2H), 3.66 (s, 4H), 2.42–2.32 (m, 4H), 2.22 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.11, 159.43 (d, J = 240.0 Hz), 157.67 (d, J = 239.9 Hz), 152.48, 150.56, 142.02, 137.19, 133.79, 130.59 (dd, J = 18.2, 7.9 Hz), 130.01, 129.75, 128.89, 127.93, 127.03, 125.00, 117.85 (dd, J = 23.9, 4.6 Hz), 117.38 (dd, J = 24.9, 9.0 Hz), 115.36 (dd, J = 23.9, 8.9 Hz), 112.35, 46.17 (2C), 45.97 (3C), 31.79. Ms (ESI) m/z 447.2, [M + H]+ 448.2.
5.1.21.2 (3-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(4-hydroxypiperidin-1-yl)methanone (C02). Yield: 59.0%, 29 mg, white solid. m.p.: 120.6–121.0 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.88 (s, 1H), 8.51 (d, J = 2.0 Hz, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.07 (dt, J = 7.8, 1.4 Hz, 1H), 7.92 (t, J = 1.7 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.42 (dt, J = 7.6, 1.4 Hz, 1H), 7.28 (ddd, J = 9.1, 5.9, 3.2 Hz, 1H), 7.23 (td, J = 9.2, 4.6 Hz, 1H), 7.13–7.09 (m, 1H), 4.80 (d, J = 4.0 Hz, 1H), 4.18 (s, 2H), 4.05 (s, 1H), 3.78–3.74 (m, 1H), 3.56 (s, 1H), 3.26–3.16 (m, 2H), 1.83–1.71 (m, 2H), 1.44–1.35 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 169.05, 159.43 (d, J = 240.6 Hz), 157.69 (d, J = 240.2 Hz), 152.46, 150.54, 142.08, 137.63, 133.77, 130.58 (dd, J = 18.2, 7.9 Hz), 129.99, 129.73, 128.91, 127.78, 126.78, 124.75, 117.89 (dd, J = 24.4, 4.9 Hz), 117.40 (dd, J = 25.2, 8.7 Hz), 115.37 (dd, J = 24.2, 8.5 Hz), 112.35, 65.9, 46.22 (2C), 31.80. Ms (ESI) m/z 448.2, [M + H]+ 449.1.
5.1.21.3 (3-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(3-hydroxypiperidin-1-yl)methanone (C03). Yield: 57.0%, 28 mg, white solid. m.p.: 185.0–186.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.87 (s, 1H), 8.51 (s, 1H), 8.45 (s, 1H), 8.06 (d, J = 7.7 Hz, 1H), 7.93 (d, J = 13.5 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.42 (s, 1H), 7.28 (ddd, J = 9.0, 5.8, 3.3 Hz, 1H), 7.23 (td, J = 9.2, 4.5 Hz, 1H), 7.13–7.09 (m, 1H), 5.01–4.85 (m, 1H), 4.18 (s, 2H), 3.82 (s, 1H), 3.57–3.42 (m, 2H), 3.31–2.87 (m, 2H), 1.90–1.40 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 169.52 (m), 159.43 (d, J = 240.1 Hz), 157.68 (d, J = 240.2 Hz), 152.46, 150.52, 142.12, 137.71, 133.70, 130.59 (dd, J = 18.3, 7.9 Hz), 130.00, 129.65, 128.91, 127.70, 127.13 (m), 125.18 (m), 117.89 (dd, J = 24.3, 4.9 Hz), 117.38 (dd, J = 24.9, 9.0 Hz), 115.36 (dd, J = 24.2, 8.5 Hz), 112.36, 65.50, 54.41 (0.5C), 49.20 (0.5C), 47.73 (0.5C), 46.20 (0.5C), 33.43 (0.5C), 32.96 (0.5C), 31.81, 23.87 (0.5C), 22.29 (0.5C). HRMS (ESI, m/z) calcd for C25H23F2N4O2 [M + H]+, 449.1789; found 449.1785. HPLC purity (MeOH
:H2O = 80:20): 98.7%, tR = 10 min.
5.1.21.4 (3-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(piperazin-1-yl)methanone (C04). Yield: 31.6%, 15 mg, white solid. m.p.: 81.0–82.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.89 (s, 1H), 8.52 (d, J = 2.0 Hz, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.92 (d, J = 1.9 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.28 (ddd, J = 9.1, 5.9, 3.2 Hz, 1H), 7.24 (dt, J = 9.2, 4.6 Hz, 1H), 7.12 (m, 1H), 4.19 (s, 2H), 3.59 (s, 4H), 2.71 (m, 4H), 2.00 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 169.08, 159.43 (d, J = 239.5 Hz), 157.68 (d, J = 240.0 Hz), 152.47, 150.54, 142.03, 137.45, 133.78, 130.58 (dd, J = 18.8, 8.1 Hz),129.97, 129.71, 128.92, 127.78, 126.99, 124.97, 117.89 (dd, J = 24.7, 4.6 Hz), 117.39 (dd, J = 24.9, 9.0 Hz), 115.38 (dd, J = 23.9, 8.8 Hz), 112.34, 65.50 (2C), 46.34 (2C), 31.80. Ms (ESI) m/z 433.2, [M + H]+ 434.1. HPLC purity (MeOH
:H2O = 90:10): 93.7%, tR = 12 min.
5.1.21.5 (4-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(4-methylpiperazin-1-yl)methanone (C05). Yield: 55.1%, 27 mg, white solid. m.p.: 209.7–210.5 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.90 (s, 1H), 8.54 (d, J = 2.0 Hz, 1H), 8.51 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.28 (ddd, J = 9.2, 5.9, 3.2 Hz, 1H), 7.24 (td, J = 9.3, 4.6 Hz, 1H), 7.13–7.09 (m, 1H), 4.18 (s, 2H), 3.64–3.42 (m, 4H), 2.37 (m, 4H), 2.23 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.14, 159.43 (d, J = 240.1 Hz), 157.66 (d, J = 240.3 Hz), 152.49, 150.54, 142.00, 135.82, 134.73, 130.62 (dd, J = 18.2, 7.8 Hz), 130.20, 128.93, 128.21 (2C), 126.80 (2C), 117.85 (dd, J = 24.3, 5.0 Hz), 117.38 (dd, J = 24.9, 9.0 Hz), 115.35 (dd, J = 24.1, 8.6 Hz), 112.44, 49.20 (2C), 46.00 (2C), 31.82, 27.30 (3C). Ms (ESI) m/z 447.2, [M + H]+ 448.1.
5.1.21.6 (4-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(4-hydroxypiperidin-1-yl)methanone (C06). Yield: 40.8%, 20 mg, white solid. m.p.: 155.1–156.8 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.89 (s, 1H), 8.53 (d, J = 2.0 Hz, 1H), 8.50 (d, J = 1.9 Hz, 1H), 8.07 (d, J = 7.9 Hz, 2H), 7.53 (d, J = 7.9 Hz, 2H), 7.27 (ddd, J = 9.4, 5.9, 3.4 Hz, 1H), 7.22 (dd, J = 9.2, 4.5 Hz, 1H), 7.12–7.08 (m, 1H), 4.81 (d, J = 3.6 Hz, 1H), 4.17 (s, 2H), 4.06–4.04 (m, 1H), 3.78–3.73 (m, 2H), 3.22–3.16 (s, 2H), 1.81–1.73 (m, 2H), 1.39–1.34 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 169.11, 159.43 (d, J = 240.0 Hz), 157.66 (d, J = 239.9 Hz), 152.47, 150.51, 142.08, 136.29, 134.58, 130.63 (dd, J = 18.9, 7.9 Hz), 130.23, 128.91, 127.95 (2C), 126.79 (2C), 117.85 (dd, J = 24.3, 4.9 Hz), 117.37 (dd, J = 24.9, 9.0 Hz), 115.34 (dd, J = 24.2, 8.6 Hz), 112.45, 65.98 (2C), 31.82. Ms (ESI) m/z 448.2, [M + H]+ 449.1. HPLC purity (MeOH
:H2O = 80:20): 97.8%, tR = 11 min.
5.1.21.7 (4-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(3-hydroxypiperidin-1-yl)methanone (C07). Yield: 44.8%, 22 mg, white solid. m.p.: 230.9–232.7 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.88 (s, 1H), 8.52 (m, 2H), 8.07 (d, J = 7.9 Hz, 2H), 7.54 (s, 2H), 7.28 (m, 1H), 7.22 (dd, J = 9.2, 4.5 Hz, 1H), 7.10 (m, 1H), 5.01–4.86 (m, 1H), 4.18 (s, 2H), 3.83 (s, 1H), 3.54 (s, 2H), 3.16–2.86 (m, 2H), 1.86–1.78 (m, 2H), 1.45 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 169.50, 159.43 (d, J = 240.3 Hz), 157.66 (d, J = 240.0 Hz), 152.47, 150.51, 142.09, 136.38, 134.52, 130.64 (dd, J = 18.2, 7.9 Hz), 130.23, 128.92, 128.28, 127.96, 126.73, 117.86 (dd, J = 24.3, 4.9 Hz), 117.38 (dd, J = 24.9, 9.0 Hz), 115.34 (dd, J = 23.9, 8.8 Hz), 112.45, 65.52 (2C), 31.82. Ms (ESI) m/z 448.2, [M + H]+ 449.1.
5.1.21.8 (4-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(piperazin-1-yl)methanone (C08). Yield: 31.6%, 15 mg, white solid. m.p.: 107.8–109.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.89 (s, 1H), 8.52 (m, 2H), 8.07 (d, J = 7.9 Hz, 2H), 7.53 (d, J = 7.9 Hz, 2H), 7.28–7.25 (m, 1H), 7.23 (td, J = 9.3, 4.5 Hz, 1H), 7.12–7.08 (m, 1H), 4.23–4.20 (m, 1H), 4.17 (s, 2H), 3.57 (s, 4H), 2.74–2.68 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 169.11, 159.42 (d, J = 240.2 Hz), 157.66 (d, J = 239.9 Hz), 152.49, 150.52, 142.03, 136.10, 134.58, 132.00, 130.63 (dd, J = 18.8, 8.0 Hz), 130.21, 129.14, 128.91, 128.17, 126.78, 117.84 (dd, J = 24.1, 4.7 Hz), 117.37 (dd, J = 24.9, 9.0 Hz), 115.34 (dd, J = 24.4, 8.7 Hz), 112.44, 65.50 (2C), 31.82. HRMS (ESI, m/z) calcd for C24H22F2N5O [M + H]+, 434.1792; found 434.1788.
5.1.21.9 (R)-(3-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(3-hydroxypiperidin-1-yl)methanone (C09). Yield: 46.9%, 23 mg, white solid. m.p.: 86.5–87.5 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.88 (s, 1H), 8.51 (d, J = 1.9 Hz, 1H), 8.45 (s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.96–7.34 (m, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.43 (t, J = 7.3 Hz, 1H), 7.27 (ddd, J = 9.1, 5.9, 3.2 Hz, 1H), 7.22 (td, J = 9.2, 4.5 Hz, 1H), 7.10 (tt, J = 8.2, 3.5 Hz, 1H), 5.03–4.874.95 (m, 1H), 4.18 (s, 2H), 3.83–3.82 (m, 1H), 3.58–3.50 (m, 2H), 3.28–2.85 (m, 2H), 1.93–1.42 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 169.55–169.31 (m), 158.63 (d, J = 240.2 Hz), 156.89 (d, J = 239.6 Hz), 152.47, 150.52, 142.13, 137.69, 133.70, 130.48 (dd, J = 18.2, 7.9 Hz), 129.99, 129.66, 128.92 (2C), 127.72, 117.78 (dd, J = 24.4, 4.9 Hz), 117.25 (dd, J = 24.9, 9.0 Hz), 115.23 (dd, J = 24.2, 8.5 Hz), 112.37 (2C), 65.50, 51.80 (d, J = 784.9 Hz), 44.97 (d, J = 831.7 Hz), 33.18 (d, J = 71.0 Hz), 31.82, 23.05 (d, J = 237.4 Hz). HRMS (ESI, m/z) calcd for C25H23F2N4O2 [M + H]+, 449.1789; found 449.1785. HPLC purity (MeOH
:H2O = 80:20): 94.0%, tR = 10 min.
5.1.21.10 (S)-(3-(5-(2,5-Difluorobenzyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)(3-hydroxypiperidin-1-yl)methanone (C10). Yield: 57.0%, 28 mg, white solid. m.p.: 110.1–111.0 °C. 1H NMR (600 MHz, DMSO-d6) δ 13.88 (s, 1H), 8.51 (d, J = 2.0 Hz, 1H), 8.45 (s, 1H), 8.07 (dt, J = 7.8, 1.5 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 7.44–7.41 (m, 1H), 7.27 (ddd, J = 9.1, 5.9, 3.2 Hz, 1H), 7.22 (td, J = 9.2, 4.6 Hz, 1H), 0.10 (ddd, J = 12.1, 8.3, 3.5 Hz, 1H), 5.03–4.87 (m, 1H), 4.18 (s, 2H), 3.82 (s, 1H), 3.59–3.50 (m, 2H), 3.20–2.85 (m, 2H), 1.92–1.41 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 169.54–169.31 (m), 158.63 (d, J = 240.5 Hz), 156.89 (d, J = 240.0 Hz), 152.46, 150.51, 137.70, 133.71, 130.49 (dd, J = 18.9, 7.9 Hz), 129.99, 129.64, 128.91, 128.29, 127.72, 119.58, 117.78 (dd, J = 23.9, 4.6 Hz), 117.25 (dd, J = 24.9, 9.0 Hz), 115.23 (dd, J = 24.2, 8.5 Hz), 112.37, 110.11, 65.50, 51.81 (d, J = 785.0 Hz), 44.97 (d, J = 831.3 Hz), 33.18 (d, J = 71.1 Hz), 31.81, 23.06 (d, J = 238.6 Hz). HRMS (ESI, m/z) calcd for C25H23F2N4O2 [M + H]+, 449.1789; found 449.1786. HPLC purity (MeOH
:H2O = 80:20): 97.6%, tR = 15 min.
5.2 Pharmacological assay
Firstly, the compounds were diluted from a concentrated stock of 8 mM in 100% DMSO with serial kinase reaction buffer dilutions. Secondly, a 5 μM TK solution, 0.5 μM XL-665 solution, kinase solution and ATP solution whose concentrations were displayed previously were prepared. For each assay, 4 μL of dispensed compound, 2 μL of ATP, 2 μL of substrate TK and 2 μL of kinase were added to the assay wells. The assay wells were incubated at 25 °C for 30–40 min. The enzymatic reaction time was related to the type of kinase (the reaction time of TRKA was 30 min, while that for TRKB and TRKC was 40 min). Finally, a mixture of 5 μL of XL-665 and 5 μL of TK-antibody-cryptate was added to terminate the reactions. The assay wells were incubated at 25 °C for another 60 min, and then HTRF signals were obtained by reading plates using an Infinite® F500 microplate reader (Tecan, Switzerland). The ratio of fluorescence at 665 nm to fluorescence at 620 nm was calculated for each well. The data were analyzed using GraphPad Prism 8. Every experiment was repeated at least two times.
5.3 Molecular docking study
The crystal structure of TRKA (PDB code: 5KVT) downloaded from the Protein Data Bank was processed with the Protein Preparation Wizard in Schrodinger suite. The protein structure was adjusted and modified, followed by the addition of hydrogen atoms, deleting solvent water molecules, and defining right bonds orders using Prime. The protonation and tautomeric states of Asp, Lys, and His were assigned at pH 7.4 state. Afterward, all the hydrogen atoms of the TRKA complexes were optimized with the OPLS_2005 force field, which minimized and converged heavy atoms to an RMSD of 0.3. The selected inhibitors were prepared by using LigPrep from the Schrodinger suite with the OPLS_2005 force field. The structure of the inhibitors was also adjusted and modified, followed by the addition of all hydrogen atoms and checking the bond order and atom types. Receptor grids were generated before docking with the allosteric site determined according to the literature. The prepared protein ligand complex was imported into Glide 9.7, which defined it as the receptor structure with a size box of (15 Å × 15 Å × 15 Å). Based on the OPLS_2005 force field, the grid of the TRKA crystal structure was generated. The extra precision (XP) mode was set for docking studies with the two crucial residues Glu-590 and Met-592 in constrained binding to get accurate results. The ligand entrectinib in co-crystallization was extracted for re-docking to evaluate the accuracy of the docking method. The results showed that the conformation obtained by docking was consistent with that in co-crystallization.Abbreviations used
| DCM | Dichloromethane |
| DIPEA | N,N-Diisopropylethylamine |
| DMF | N,N-Dimethylformamide |
| EA | Ethyl acetate |
| EDCI | N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride |
| HOBt | 1-Hydroxybenzotriazole |
| MeOH | Methanol |
| NaH | Sodium hydride |
| PE | Petroleum ether |
| THF | Tetrahydrofuran |
| PMB-Cl | 4-Methoxybenzene-1-sulfonyl chloride |
| PyBop | ((1H-Benzo[d][1,2,3]triazol-1-yl)oxy)tri(pyrrolidin-1-yl)phosphonium hexafluorophosphate |
| NIS | N-Iodosuccinimide |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Program for Innovative Research Team of the Ministry of Education and the Program for Liaoning Innovative Research Team in University.Notes and references
- E. J. Huang and L. F. Reichardt, Neurotrophins: roles in neuronal development and function, Annu. Rev. Neurosci., 2001, 24, 677–736 CrossRef CAS
.
- M. H. Ultsch, C. Wiesmann, L. C. Simmons, J. Henrich, M. Yang and D. Reilly,
et al., Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC 1 1Edited by I. A. Wilson, J. Mol. Biol., 1999, 290(1), 149–159 CrossRef CAS PubMed
- C. Wiesmann, M. H. Ultsch, S. H. Bass and A. M. de Vos, Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor, Nature, 1999, 401(6749), 184–188 CrossRef CAS
- D. R. Kaplan, B. L. Hempstead, D. Martin-Zanca, M. V. Chao and L. F. Parada, The trk Proto-Oncogene Product: a Signal Transducing Receptor for Nerve Growth Factor, Science, 1991, 252(5005), 554–558 CrossRef CAS PubMed
- D. R. Kaplan, D. Martin-Zanca and L. F. Parada, Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF, Nature, 1991, 350(6314), 158–160 CrossRef CAS PubMed
- R. Klein, S. Jing, V. Nanduri, E. O'Rourke and M. Barbacid, The trk proto-oncogene encodes a receptor for nerve growth factor, Cell, 1991, 65(1), 189–197 CrossRef CAS PubMed
- F. Lamballe, R. Klein and M. Barbacid, trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3, Cell, 1991, 66(5), 967–979 CrossRef CAS
- S. P. Squinto, T. N. Stitt, T. H. Aldrich, S. Davis, S. M. Blanco and C. RadzieJewski,
et al., trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor, Cell, 1991, 65(5), 885–893 CrossRef CAS
- E. C. Yuen and W. C. Mobley, Early BDNF, NT-3, and NT-4 Signaling Events, Exp. Neurol., 1999, 159(1), 297–308 CrossRef CAS
- C. Miranda, A. Greco, C. Miele, M. A. Pierotti and E. Van Obberghen, IRS-1 and IRS-2 are recruited by TrkA receptor and oncogenic TRK-T1, J. Cell. Physiol., 2000, 186(1), 35–46 CrossRef
- V. Ranzi, S. O. Meakin, C. Miranda, P. Mondellini, M. A. Pierotti and A. Greco, The Signaling Adapters Fibroblast Growth Factor Receptor Substrate 2 and 3 Are Activated by the Thyroid TRK Oncoproteins, Endocrinology, 2003, 144(3), 922–928 CrossRef CAS
- E. Roccato, C. Miranda, V. Ranzi, M. Gishizki, M. A. Pierotti and A. Greco, Biological activity of the thyroid TRK-T3 oncogene requires signalling through Shc, Br. J. Cancer, 2002, 87(6), 645–653 CrossRef CAS PubMed
- K. Bartkowska, A. Paquin, G. AeS, D. R. Kaplan and F. D. Miller, Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development, Development, 2007, 134(24), 4369–4380 CrossRef CAS
- W. Yan, N. R. Lakkaniga, F. Carlomagno, M. Santoro, N. Q. McDonald and F. Lv,
et al., Insights into Current Tropomyosin Receptor Kinase (TRK) Inhibitors: Development and Clinical Application, J. Med. Chem., 2019, 62(4), 1731–1760 CrossRef CAS PubMed
- E. Ardini, R. Bosotti, A. L. Borgia, C. De Ponti, A. Somaschini and R. Cammarota,
et al., The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition, Mol. Oncol., 2014, 8(8), 1495–1507 CrossRef CAS PubMed
- R. Wang, H. Hu, Y. Pan, Y. Li, T. Ye and C. Li,
et al., RET Fusions Define a Unique Molecular and Clinicopathologic Subtype of Non–Small-Cell Lung Cancer, J. Clin. Oncol., 2012, 30(35), 4352–4359 CrossRef CAS PubMed
- J. Kim, Y. Lee, H. J. Cho, Y.-E. Lee, J. An and G.-H. Cho,
et al., NTRK1 fusion in glioblastoma multiforme, PLoS One, 2014, 9(3), e91940 CrossRef PubMed
- M. Mourad, T. Jetmore, A. A. Jategaonkar, S. Moubayed, E. Moshier and M. L. Urken, Epidemiological Trends of Head and Neck Cancer in the United States: A SEER Population Study, J. Oral Maxillofac. Surg., 2017, 75(12), 2562–2572 CrossRef PubMed
- R. C. Doebele, L. E. Davis, A. Vaishnavi, A. T. Le, A. Estrada-Bernal and S. Keysar,
et al., An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101, Cancer Discovery, 2015, 5(10), 1049–1057 CrossRef CAS PubMed
- A. Drilon, T. W. Laetsch, S. Kummar, S. G. DuBois, U. N. Lassen and G. D. Demetri,
et al., Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children, N. Engl. J. Med., 2018, 378(8), 731–739 CrossRef CAS PubMed
- T. W. Laetsch, S. G. DuBois, L. Mascarenhas, B. Turpin, N. Federman and C. M. Albert,
et al., Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study, Lancet Oncol., 2018, 19(5), 705–714 CrossRef CAS PubMed
- E. Ardini, M. Menichincheri, P. Banfi, R. Bosotti, C. De Ponti and R. Pulci,
et al., Entrectinib, a Pan–TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications, Mol. Cancer Ther., 2016, 15(4), 628–639 CrossRef CAS PubMed
- M. Menichincheri, E. Ardini, P. Magnaghi, N. Avanzi, P. Banfi and R. Bossi,
et al., Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor, J. Med. Chem., 2016, 59(7), 3392–3408 CrossRef CAS
- Y. Duan, J. Wang, S. Zhu, Z.-C. Tu, Z. Zhang and S. Chan,
et al., Design, synthesis, and Structure–Activity Relationships (SAR) of 3-vinylindazole derivatives as new selective tropomyosin receptor kinases (Trk) inhibitors, Eur. J. Med. Chem., 2020, 203, 112552 CrossRef CAS
- T. Wu, C. Zhang, R. Lv, Q. Qin, N. Liu and W. Yin,
et al., Design, synthesis, biological evaluation and pharmacophore model analysis of novel tetrahydropyrrolo[3,4-c]pyrazol derivatives as potential TRKs inhibitors, Eur. J. Med. Chem., 2021, 223, 113627 CrossRef CAS
- T. Wu, Q. Qin, R. Lv, N. Liu, W. Yin and C. Hao,
et al., Discovery of quinazoline derivatives CZw-124 as a pan-TRK inhibitor with potent anticancer effects in vitro and in vivo, Eur. J. Med. Chem., 2022, 238, 114451 CrossRef CAS
- A. Drilon, R. Nagasubramanian, J. F. Blake, N. Ku, B. B. Tuch and K. Ebata,
et al., A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors, Cancer Discovery, 2017, 7(9), 963–972 CrossRef CAS PubMed
- L. S. Zhuo, M. S. Wang, F. X. Wu, H. C. Xu, Y. Gong and Z. C. Yu,
et al., Discovery of Next-Generation Tropomyosin Receptor Kinase Inhibitors for Combating Multiple Resistance Associated with Protein Mutation, J. Med. Chem., 2021, 64(20), 15503–15514 CrossRef CAS PubMed
- T. Wu, Q. Qin, N. Liu, C. Zhang, R. Lv and W. Yin,
et al., Rational drug design to explore the structure-activity relationship (SAR) of TRK inhibitors with 2,4-diaminopyrimidine scaffold, Eur. J. Med. Chem., 2022, 230, 114096 CrossRef CAS
- C. Hao, F. Zhao, H. Song, J. Guo, X. Li and X. Jiang,
et al., Structure-Based Design of 6-Chloro-4-aminoquinazoline-2-carboxamide Derivatives as Potent and Selective p21-Activated Kinase 4 (PAK4) Inhibitors, J. Med. Chem., 2018, 61(1), 265–285 CrossRef CAS PubMed
- Y. Sun, L. Wang, Y. Sun, J. Wang, Y. Xue and T. Wu,
et al., Structure-based discovery of 1-(3-fluoro-5-(5-(3-(methylsulfonyl)phenyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)phenyl)-3-(pyrimidin-5-yl)urea as a potent and selective nanomolar type-II PLK4 inhibitor, Eur. J. Med. Chem., 2022, 243, 114714 CrossRef CAS
- R. Wang, S. Yu, X. Zhao, Y. Chen, B. Yang and T. Wu,
et al., Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors, Eur. J. Med. Chem., 2020, 188, 112024 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00334a |
| This journal is © The Royal Society of Chemistry 2023 |