
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo potential of polymeric N-acryloyl-glycine nanoparticles with anti-inflammatory activities for wound healing†
Prem Shankar Gupta a,
Kirti Wasnika,
Gurmeet Singha,
Sukanya Patraa,
Divya Pareeka,
Desh Deepak Yadava,
Munendra Singh Tomarb,
Somedutta Maitic,
Monika Singha and
Pradip Paik*a
a,
Kirti Wasnika,
Gurmeet Singha,
Sukanya Patraa,
Divya Pareeka,
Desh Deepak Yadava,
Munendra Singh Tomarb,
Somedutta Maitic,
Monika Singha and
Pradip Paik*a
aSchool of Biomedical Engineering, Indian Institute of Technology (BHU), Varanasi, India. E-mail: paik.bme@iitbhu.ac.in
bDepartment of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado, Aurora, USA
cSchool of Engineering Science and Technology, University of Hyderabad, Hyderabad, India
First published on 24th August 2023
Abstract
An optimal wound-healing process requires highly coordinated and regulated interactions between the immune and biological systems since long-term wounds are severe clinical issues which make patients vulnerable to potentially fatal microbial infections. Therefore, it is essential to develop new treatments and therapeutic strategies. We aimed to develop a poly(N-acryloyl glycine) nanoparticle (PNAG NP) based formulation, which is effective to accelerate skin restoration. PNAG NPs ca. 35 nm in size (in dia.) have been synthesised through the mini-emulsion radical polymerisation technique and formulated with an ointment base for the establishment of wound healing application. Biocompatible, hemocompatible, proliferative and migration enhancer PNAG NPs were formulated as a PNAG nanoformulation, which showed a ∼31% higher wound healing efficiency compared to the control group of samples in the in vivo assessment. Periodic evaluation of TNF-α and IL6 for the treatment group shows the change in their levels from 4.10 ± 3.18 to 5.17 ± 3.27 pg mL−1 and 11.93 ± 2.34 pg mL−1 to 10.33 ± 3.31 pg mL−1, respectively, consistent with the reduction in inflammation. The increased level of IGF-1 from 25.41 ± 13.56 ng mL−1 to 64.22 ± 16.93 ng mL−1 in the PNAG test group could be correlated with the increase in angiogenesis followed by an increase in the wound healing efficiency. This study showed that PNAG NPs could execute rapid coordination between cell proliferation and migration, anti-inflammatory actions, and simultaneous regeneration of skin tissues without the use of any of the external synergistic factors like drugs, genes or cells. Thus, the intriguing results showed that the PNAG NPs could address the existing wound healing challenges and can serve as a potential material for regenerative purposes.
1. Introduction
The skin tissue serves as a vital organ to protect the body from the loss of fluids, electrolytes, and nutrients, as well as from external threats.1,2 Excessive damage of tissues by mechanical, chemical, physical, or biological threats can lead to cutaneous wound followed by multi-organ failure.3 Although the innate immune system gets activated immediately in response to the damage,4,5 several internal (diabetes, malnutrition) and external (infections, secondary trauma) conditions impair the wound healing mechanism6 and create a severe issue for health care worldwide.7 Annual projected costs for impaired wound healing are estimated to be 28.1 to 96.8 billion USD by 2030.8 It is also reported that more than 38 million chronic wound infections are linked to poor prognosis.9 Persistent wounds in older people can raise co-morbidity and death rates.10 For instance, the 5-year death rate from diabetic foot ulcers (DFU) alone is 43–55%, which is higher than the death rate of cancer.11 Therefore, healthy skin recovery with low-cost therapeutic is always a thrust area of research along with the increased search for the best wound-healing biomaterials.Porosity, adhesion and high mechanical strength are crucial properties of wound healing materials12 and play an essential role in selecting them to avoid toxicity issues. Conjugation of these materials, polymers, and nanomaterials could promote healing efficiency. Nanomaterials such as silver nanoparticles-composing alginate/gelatin hydrogel13 impart some advantages over conventional wound healing materials.14 Yet, due to their inability to adequately acclimatise to the wound healing process, these materials do not increase the efficiency of healing process as anticipated. Furthermore, natural polymers have been recently explored to treat wounds due to their biocompatibility, biodegradability, and resemblance to biological tissues.15 Delivery of antibiotics, growth factors or therapeutic stem cells by polymeric carriers16–18 is also an option for wound healing. However, drug-associated side effects and high costs often complicate these approaches. Similarly, conventional dressing materials, including gauze, bandage and cotton wool, cannot prevent wound beds from drying because they absorb a lot of exudates from the wound site and attract microbes, leading to infections, often damaging the regenerative tissues. Similarly, synthetic polymers have very high hydrophobic characteristics and cause cell adhesion problems during wound healing.19 For instance, most clinical wound dressings now in use cannot promote the controlled growth of wound-healing cells.20 Thus, there are still huge needs that existing wound healing materials cannot satisfy, despite the manufacture and use of several wound dressings in current clinical settings. At this stage, polymeric nanoparticles are one of the developing solutions as wound healing materials that have gained increasing interest due to their ability to combine extraordinary therapeutic qualities. Wound-healing polymeric nanoparticles can modulate many cellular and molecular mechanisms that support the wound microenvironment through anti-inflammatory and angiogenic actions, potentially transforming the environment from non-healing to healing.21
Here we have tried to eliminate a few of the drawbacks and to increase the efficiency of wound healing in terms of repair/healing time; we have synthesised the poly(N-acryloyl-glycine) (PNAG) polymer and fabricated it into nanoparticles (NPs) through free radical emulsion polymerisation. The PNAG polymer is a polymer of weak polyacid monomers synthesised through the Schotten–Baumann reaction mechanism by using acryloyl chloride and glycine amino acid. PNAG NPs were fabricated to preserve the haemostatic activity of carboxyl groups present in the PNAG polymer and have been examined for better efficiency in wound healing. We have studied the biocompatibility and hemocompatibility of PNAG NPs to ensure the non-toxic or antigenic properties of PNAG NPs, when used in association with living tissues or living systems. Herein, the proliferative and cell migratory performance of PNAG NPs have been studied by performing scratch wound assay using mouse fibroblast (L929) cell lines to evaluate efficiency. The angiogenic effect of PNAG NPs was studied using the chicken embryo membrane angiogenesis (CEMA) assay under in ovo conditions to ensure PNAG NPs’ potential in the development of blood vasculature, which is essential for effective and early wound healing.
Further, we developed the nanoformulation from PNAG NPs, and the in vivo dermal irritation study was carried out. We also assessed the in vivo wound healing efficiency of PNAG NPs in the rat model. Further, the pro-inflammatory cytokine and chemokine profiling was performed during the wound healing. Finally, the results were compared with the results of control samples to determine the effectiveness of the nanoformulation in wound healing.
2. Experimental
Materials
The following materials were used: glycine (98%) (Sigma Aldrich, Germany), acryloyl chloride (96%) stabilised with 400 ppm phenothiazines (Alfa-Aesar), methyl thiazole tetrazolium (MTT > 99.9%, Himedia), hexadecane anhydrous (99%, Sigma-Aldrich), divinylbenzene (Alfa-Aesar), chloroform, ethyl acetate, 1,4-dioxane (99%) extra pure (Merck), 2,2-azobisisobutyronitrile (AIBN, 98%, SRL), SDS (90%, Merck), DMEM, fetal bovine serum (Gibco), penicillin–streptomycin cocktail and gentamicin. Ultrapure water (18.2 M) (Pure Lab Ultra water system (ELGA, High Wycombe, United Kingdom)) was used for all sample processing. Sisco Research Laboratories (SRL) Pvt. Ltd., Mumbai, India, supplied all other analytical grade reagents. All the chemicals were used without further purification.Synthesis of the monomer
The N-acryloyl glycine monomer (NAG) was synthesised through a modified approach.22–24 In brief, a required amount of glycine was dissolved in a cold 2 M KOH solution by stirring in a round bottom flask in an ice bath (designated solution ‘A’). Then cold 1,4-dioxane was mixed with acryloyl chloride (designated solution ‘B’). Further, solution ‘B’ was slowly added to cooled solution ‘A’ at 0 °C (ice bath) for 2 h under vigorous stirring (∼600 rpm) followed by 12 h of incubation at room temperature (25 °C) with stirring. The pH of the mixture was kept above 12 by gradually adding 2 M KOH. After 2 h of continuous stirring at room temperature (25 °C), the mixture was rinsed thrice with 20 mL of diethyl ether. Then the mixture was saturated with NaCl after being acidified with 5 M HCl to achieve pH 2; the obtained aqueous solution was extracted 3–4 times with 100 mL of ethyl acetate. The ethyl acetate layer was dried by adding anhydrous magnesium sulphate and then filtered, and the monomer was obtained by evaporating ethyl acetate solvent using a rotary evaporator at reduced pressure. From a cooled combination of diethyl ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (1:1), with melting temperature 132–134 °C, 10.2 gm of crude product (NAG) (yield ∼63.75%) was crystallised out.
:ethyl acetate (1:1), with melting temperature 132–134 °C, 10.2 gm of crude product (NAG) (yield ∼63.75%) was crystallised out.
Preparation of polymeric NAG nanoparticles (PNAG NPs)
The polymeric NAG nanoparticles (PNAG NPs) were synthesised by the mini-emulsion radical polymerisation technique as described in our previous reports22,23 with modifications according to the need. Briefly, the NAG monomer and hexadecane were dispersed in toluene to form an emulsion of the monomer, where the monomer is present as a dispersed phase (droplets). To this emulsion, an organic soluble radical initiator, AIBN, was added and sonicated for 3 minutes on a bath sonicator (Elmasonics, S 30H, Germany), which allows radical polymerisation within the oil/monomer droplets. Further, cross-linking agent divinylbenzene (DVB) was added and sonicated to increase the stability of nanoparticles and to impart cross-linked net-like structure, porosity and elasticity of nanoparticles. Hexadecane (HD) is a long-chain alkane used as a co-stabiliser. In another vial, sodium dodecyl sulphate (SDS) was dissolved in H2O using an ultrasonic bath sonicator at room temperature. Then the SDS solution was added dropwise under vigorous stirring.Further, the reaction mixture was sonicated using an ultra-probe sonicator (750 W, 30% power, 45:15 cycles) at 25 °C. The obtained emulsion was kept at 80–85 °C in an oil bath for complete polymerisation for 12 h with continuous stirring; stable nanoparticles were formed, and residual toluene was evaporated by adding a sufficient amount of water. The nanoparticle suspension was separated using a cooling centrifuge at 14000 rpm followed by freeze-drying. The details of the formulation have been filed for an Indian patent (Ref. Patent Application No. 202311041625, June 19, 2023).
Characterisation of the NAG monomer and PNAG NPs
The chemical functionality of the NAG monomer and PNAG NPs was characterised through FTIR spectroscopy (Nicolet iS5, 4 Thermo Fisher Scientific Inc., USA) using the KBr pellet and an 1H-NMR spectrometer (500 MHz) (One Bay NMR Spectrometer, Bruker Bio Spin International AG) using DMSO-D6 solvent and tetramethylsilane as an internal standard in NMR. HRTEM (Nova Nano SEM 450, FEI, USA) was used to examine the size and shape of the nanoparticles. The crystallinity of the NAG monomer and PNAG NPs was obtained through powder XRD (XRD; Rigaku SmartLab, RIGAKU Corporation, Japan). The average particle size and zeta potential (ζ) of PNAG NPs were measured using dynamic light scattering (DLS; Nano-ZS ZEN3600, Malvern, UK) at 25 °C (n = 3).In vitro cytotoxicity and hemocompatibility study
A material's biosafety is crucial for its therapeutic uses. Therefore, the NAG monomer and PNAG NPs were assessed for proliferation efficiency and cell viability; assays were conducted on L929 and HUVEC cell lines. Cells were grown and maintained in rich cell culture DMEM medium supplemented with 100U of penicillin–streptomycin cocktail and 10% FBS at 37 °C in a humidified environment of 95% air and 5% CO2. In the experimental procedure, cells (5 × 103) were seeded into each well of a 96-well plate and cultured for 24 h, followed by the addition of different concentrations of the NAG monomer and PNAG NPs from 2 to 250 μg mL−1 (dispersed in complete DMEM medium) and incubated for another 24 h. After incubation, the complete medium was removed from each well, and the MTT reagent (concentration = 5 μg mL−1) containing medium was added and incubated for 4 h. After incubation, the MTT reagent was removed, and formazan crystals were dissolved by adding 200 μl of DMSO. Similarly, the experiment was performed using HUVEC cell lines with different concentrations of PNAG (5–200 μg mL−1). After 20 minutes of incubation at 37 °C, absorbance was recorded at 570 nm using a microplate spectrofluorometer. Untreated cells were employed as a control. The cell viability was estimated using eqn (1):
 | (1) |
Hemocompatibility
Venous blood was collected to test the hemocompatibility of PNAG NPs. Sodium citrate was used as an anticoagulant in tubes holding whole venous blood.Blood coagulation study
The impact of PNAG NPs on the blood coagulation cascade was assessed by evaluating 2 blood coagulation pathways. After incubating plasma with PNAG NPs (1 to 200 μg mL−1), the activated partial thromboplastin time (APTT) and prothrombin time (PT) were measured independently. The plasma fraction was separated from whole blood by centrifugation (2500 rpm, 10 minutes, 25 °C). PNAG NPs were incubated with isolated plasma for 30 minutes at 37 °C with constant stirring. The incubated samples were placed in a coagulometer (Hemostar XF 2.0, Tulip Diagnostics Pvt. Ltd India), followed by the addition of phospholipid calcium thromboplastin (200 μL) to trigger clot formation and then the prothrombin time (PT) for the sample was calculated. To determine the APTT, 100 μL of cephalin was added to the previously placed sample in the coagulometer. After incubating, it was mixed for 2 min, and then 100 μL of calcium chloride was added under stirring to stimulate the clot formation, followed by monitoring the clotting time (APTT). The process was repeated three times.Haemolysis study
As previously indicated, RBCs were isolated from whole blood and the homogeneous suspension was prepared by dispersing in PBS (5 × 105 cells mL−1). A spectroscopic test was utilised to analyse the haemolytic impact of PNAG NPs in the 1 to 1000 μg mL−1 concentration range, and the haemolysis caused by 0.1% dispersion of PNAG NPs was compared to the positive control (0.1% Triton X-100 in PBS) and negative control (PBS). In a 1.5 mL microcentrifuge tube, 500 μL of each PNAG nanosuspension was combined with 500 μL of RBC suspension. The mixture was incubated for 24 h at 37 °C and 100 rpm. Following the incubation, the samples were centrifuged (2500 rpm for 20 min at 25 °C), and the percentage of haemolysis was determined spectrophotometrically by considering the absorption maxima for the sample at λ = 540 nm, and the results were compared with the positive control (Triton X-100) and negative control (PBS).In vitro wound healing assay
The scratch wound healing assay is a standard in vitro method for probing collective cell migration and proliferation in 2 dimensions. This assay was used to identify the cellular healing ability of PNAG NPs in vitro in a controlled environment. The procedure was performed following the reported method.25 Briefly, 2 × 105 L929 cells per well were seeded in a 12-well plate and cultured for 24 h to get 80% confluency; a linear scratch wound on the surface of each well was made using a sterile 20 μL pipette tip and cells were cultured in supplemented media with (test) and without (untreated control) PNAG NPs (50 to 200 μg mL−1). Further, images were acquired at different time intervals, 0 h, 24 h, and 48 h, and the closing wound area (%) and the closure rate (%) were calculated using ImageJ software.Chicken embryo membrane angiogenesis (CEMA) assay
The in vivo angiogenesis tests were performed to examine the vascular sprouting potential of PNAG NPs using a chicken embryo membrane angiogenesis (CEMA) assay. The fertilised eggs from a certified and trusted vendor (Ramana Hatchery, Varanasi, Uttar Pradesh, India) were purchased. The eggs were incubated in an egg incubator for 96 h in a controlled environment (38 ± 0.5 °C and 50–55% humidity). On the day of the experiment, the eggs were examined for successful embryogenesis using the light and shadow approach. A little examination window was made on the top of the eggshell. Further, suspensions of various concentrations of PNAG NPs (100 and 200 μg mL−1) in PBS were added, compared to the reference (PBS), and examined periodically for 8 h. Using a stereo zoom microscope-mounted Magnus camera (Magcam DC Plus 10, Magnus Opto Systems India Pvt. Ltd, New Delhi, India) with a resolution of 10 megapixels, pictures were taken at various time intervals (0, 2, 4, and 8 h) of incubation. Then, with the help of a vasculature analysis plug-in, images were examined for angiogenesis in terms of vascular density (VDM), vascular length density measurement (VLDM), and developmental area (DA) using Fiji J software.Preparation of the PNAG nanoformulation and qualitative evaluation
PNAG NPs were used to prepare the nanoformulation for the smooth application on the wound. PNAG NPs were dispersed into an oleaginous ointment base prepared as mentioned in ointment bases26 (ESI,† Table S1). Briefly, the quantity of PNAG NPs was taken on an ointment slab and triturated with a small amount of base ointment (already prepared through the melt fusion method) with the help of a spatula having a long and broad blade. After proper trituration with base ointment, glycerine was incorporated. Both PNAG nanoformulation and ointment base (formulation base) were evaluated for observational parameters such as appearance, odour, colour, and homogeneity by visual inspections and measurable parameters such as spreadability by the “slip and drag” approach using Mutimer's recommended apparatus (ESI,† Fig. S1).27 Spreadability was computed using eqn (2):
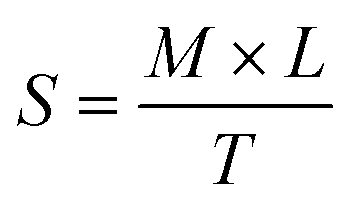 | (2) |
Animal studies
All the animal studies were examined and approved by the Institutional Animal Ethical Committee (IAEC; Approval No. IIT(BHU)/IAEC/2022/078, dated 03/05/2022, registration no. 2123/GO/Re/S/21/CPCSEA) and the use of animals in this work was carried out in compliance with IAEC's recommendations. The detailed animal protocol is described in the ESI.†Dermal irritation study
PNAG nanoformulation samples were applied to rat skin for a predetermined period to check for any irritation or erythematous reaction. Each rat had about 4 cm2 of intact shaved skin, treated with the PNAG nanoformulation and base formulation of about 1 gm, coupled with the positive control (1% formalin solution as a common irritant). The treated area was wrapped in an occlusive bandage and confined separately. The wrappings were removed after 24 h and any leftover testing materials were carefully removed and washed with water. At the end of the study, the skin of rats was examined visually for any indications of skin irritation and sensitisation and simultaneously imaged digitally. Further, rats under the experiments were observed for 7 more days to check for indications of oedema and erythema. The Draize scoring system (ESI,† Table S2) was used to calculate the skin irritation score using the primary dermal irritation index (PDII), calculated using eqn (3):
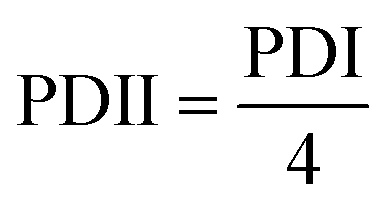 | (3) |
In vivo wound healing study
The rat excisional wound splinting model28 was used to evaluate the wound healing ability of formulations containing PNAG NPs. Splinting rings firmly cling to the skin around the wound, preventing wound closure brought on by skin contraction and enabling wound healing by granulation and re-epithelialization. A 10–12 mm diameter full-thickness skin wound on the dorsal area was created using a sterile 10 mm diameter biopsy punch. Then, the rats were put on sterile sheets. To reduce contraction throughout the healing process, sterile silicone rings with an internal diameter of 12–15 mm were firmly clung to the skin around the wound. The wounds were dressed with self-adhesive waterproof tape gauze. After each procedure, the rats were put on a heating pad until they had fully recovered from anaesthesia. The base formulation and PNAG nanoformulation were applied as soon as the wound was created, preventing the wound region from drying up and ensuring it was protected from the outside environment. On day 0, 1st, 3rd, 5th, 7th, 9th, 11th, and 13th after wounding, 100 mg of base formulation and PNAG nanoformulation was applied to the wound lesions. The wounds were imaged digitally on day 0, 3rd, 7th, 9th, and 14th with proper scaling and at an identical height. Self-adhesive sterile gauze tape was used to cover each wound and new dressings were applied after each treatment on alternate days. Rats receiving just the base formulation were employed as the control group. The therapy regimen was evaluated using ImageJ software to quantify the wound area using eqn (4), and the percentage of wound closure was calculated as
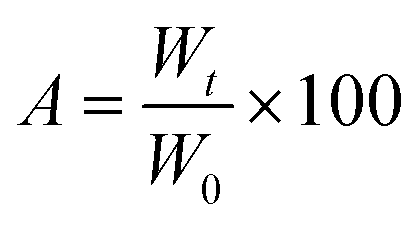 | (4) |
Histological analysis
The cicatricial tissue and cross-sectional full-thickness skin specimens were obtained after scarifying the experimental rats by deepening anaesthesia using diethyl ether and cervical dislocation on the 14th day of the experiment. The entire wound area was then carefully excised and fixed in 4% formalin for 24 h and then immersed in ethanol and xylol solutions with progressively increasing concentrations from 70%, 80%, 95%, and 100% before being embedded in paraffin blocks. Five-micron vertical slices were fixed to glass slides and stained with haematoxylin and eosin (H&E) to scrutinise the stages of wound healing. Bright-field microscopy assessed the slides and digitally captured images for histological inspection.29Immunochemical analysis
As a whole, the body interacts constantly with its local tissues. If the local tissue is compromised in any way, the usual molecular changes in body markers will also be affected.30 Therefore, blood plasma was collected at various intervals and kept at −80 °C until further processing to ascertain the extent of systemic change in wound healing indicators. The serum level of cytokines and chemokines was determined during various phases of wound healing. The enzyme-linked immunosorbent assay (ELISA) was used to measure the levels of wound healing indicators (IL-6, TNF-α and IGF-1) in serially collected blood samples on the 2nd, 7th, and 12th day following wounding. Commercially available ELISA test kits were used to check the antigen levels of IL-6, TNF-α and IGF-1. In short, previously collected and stored blood (−80 °C) was thawed by storing it at 4 °C for the whole night (12 h); the serum was collected by centrifugation (3500 rpm, 15 min, 4 °C). After the preparation, 100 μL of each sample and standard was taken in the wells of a 96-well plate, and then incubated for 90 min at 37 °C. After adding the antibodies and biotin in 100 μL aliquots to each well, the samples were kept for 60 min at 37 °C. Then the samples were washed five times with 0.01 M TBS, and 100 μL of avidin–biotin complex was added to each well and kept at 37 °C for 30 min. To the above mixture, tetramethyl benzidine (TMB) was added at 37 °C in the dark, allowing the reaction to complete for 30 min. Then, TMB termination solution was added. The relative protein concentrations were estimated by taking the absorbance at 450 nm using a microplate reader.Statistical analysis
Using the Origin 2021 software (OriginLab Corporations, USA), the independent Student's t-test and one-way ANOVA with the Tukey test were used to statistically analyse the in vitro and in vivo data. Statistical significance was defined at a p-value < 0.05. The data are shown as mean ± SD.3. Results and discussion
Synthesis of PNAG NPs and characterisation
The Schotten–Baumann reaction mechanism, which enables the synthesis of amides from acylchlorides and amines in an alkaline medium was adapted for the monomer synthesis. The NAG monomer was synthesised as shown in Scheme 1. First, acryloyl chloride (I) and glycine amino acid (II) reacted in an alkaline solution to form acryloylated amino acid (III). Then acryloylated amino acid underwent a charge rearrangement to produce the NAG monomer (IV) (Scheme 1). Further, the synthesis of NAG monomer are confirmed by analysing the functional group through the FTIR spectrum (Fig. 1a), the bands for functional group appeared at 3321 cm−1 for secondary amine (–NH–), 2990 cm−1 for alkane (–CH–), 2728 cm−1 for the carboxylic group (–COOH), 1723 cm−1 for the carbonyl ketone (–CO) group attached to an acid group, 1553 and 1545 cm−1 for the carbonyl ketone attached to the amide group (Fig. S2, ESI†) 1H-NMR and 13C-NMR (Fig. S3, ESI†).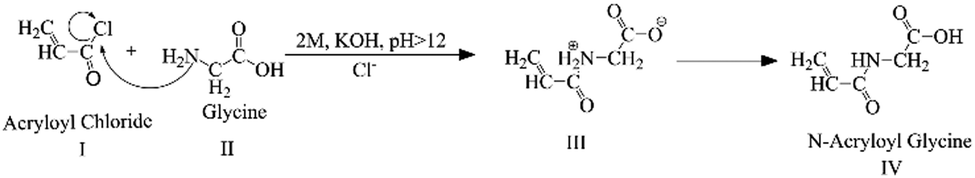 | ||
| Scheme 1 Synthesis mechanism of the N-acryloyl glycine (NAG) monomer. | ||
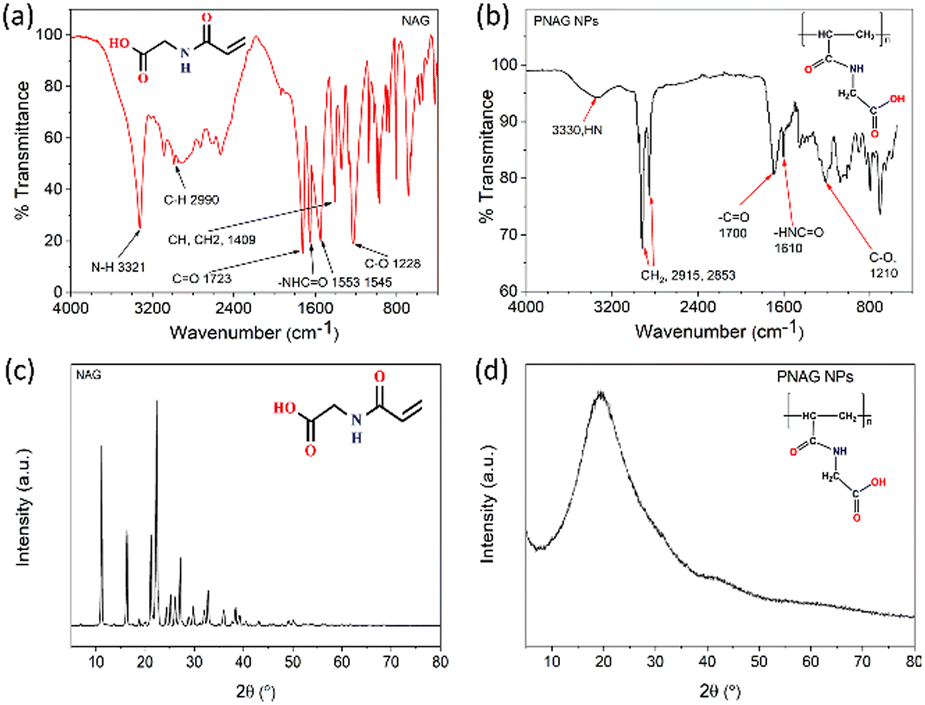 | ||
| Fig. 1 FTIR spectra and XRD pattern of the NAG monomer and PNAG NPs. FTIR spectra show the absorption bands for the (a) NAG monomer and (b) PNAG polymer; the XRD pattern (c) shows that the NAG monomer is highly crystalline, while (d) the synthesised PNAG NPs show reduced crystallinity compared to the NAG monomer. | ||
This synthesised NAG monomer undergoes a mini-emulsion radical polymerisation process. Complex polymeric particle structures are formed during the mini-emulsification process. This process was carried out by dispersing the polar molecule NAG in an oil phase (toluene) and forming tiny monomer particle droplets. AIBN is an organic soluble radical initiator, which is activated at a temperature between 60 and 80 °C allowing radical polymerisation of NAG monomer droplets at their interfaces. Radical polymerisation forms oily core-surrounded particles which have a thin polymeric linear membrane. The polymeric linear membrane around the oily core was stabilised by cross-linking with DVB, which provides particle stability and produces porosity. The long-chain hexadecane (HD) alkane is a co-stabiliser which prevents the Ostwald ripening. Polymerisation was conducted at a high temperature (70–75 °C), which also helps to increase the porosity of NPs by the evaporation of the oily core. A sufficient amount of surfactant (SDS) was used to prevent coalescence in the emulsion and to act as a primary stabiliser for particles. The preparation of nanoparticles can also be carried out without the surfactant. However, it affects the physical stability of prepared NPs as they tend to form aggregates during storage.
The structural transformations of PNAG NPs were confirmed by further characterisation by obtaining the FTIR spectrum (Fig. 1b), 1H-NMR (ESI,† Fig. S4) and 13C-NMR (ESI,† Fig. S5). PNAG NPs shows characteristics band at 3330 cm−1 for secondary amine (–NH–), 2915 and 2853 cm−1 for alkane (–CH–), 1700 cm−1 for the carbonyl ketone (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group attached to an acid group and 1610 cm−1 for carbonyl ketone attached to the amide group (Fig. 1b). The characteristic peaks are assigned as 1H-NMR (DMSO-D6): 12.57 (1H, s), 8.41 (–CONH, t) 6.26 and 6.09 (–CONH–, 1H, cis, t), 5.63 and 5.61 (–CONH–, 1H, trans, t), 3.844 and 3.832 (–CH2, CH–, 2H, vinyl hydrogen, d), 3.5 (–CH2–, 2H, d)2.56 (CH–, 1H, t) are the corresponding ppm values and for 13C-NMR: 171.66 (–CO–OH), 165.37 (–CO–NH), 131.77 and 126.12 (H2CCH–), and 41.10 (–N–CH2) (ESI,† Fig. S2 and S3).
O) group attached to an acid group and 1610 cm−1 for carbonyl ketone attached to the amide group (Fig. 1b). The characteristic peaks are assigned as 1H-NMR (DMSO-D6): 12.57 (1H, s), 8.41 (–CONH, t) 6.26 and 6.09 (–CONH–, 1H, cis, t), 5.63 and 5.61 (–CONH–, 1H, trans, t), 3.844 and 3.832 (–CH2, CH–, 2H, vinyl hydrogen, d), 3.5 (–CH2–, 2H, d)2.56 (CH–, 1H, t) are the corresponding ppm values and for 13C-NMR: 171.66 (–CO–OH), 165.37 (–CO–NH), 131.77 and 126.12 (H2CCH–), and 41.10 (–N–CH2) (ESI,† Fig. S2 and S3).
XRD patterns (Fig. 1c and d) of the NAG monomer and PNAG NPs were recorded. The amorphous form in the final formulated product enhances the material's apparent solubility and bioavailability.31 Here, it is evident from the XRD pattern of the NAG monomer (Fig. 1c) and PNAG NPs (Fig. 1d) that the NAG monomer was relatively more crystalline than the PNAG NPs. The crystallinity index (CI) for the NAG monomer was found to be 81.40%. High crystallinity ensures structural stability during transportation, packaging or storage, while amorphous solids have comparatively good oral bioavailability.32 From the XRD results, it is evident that PNAG NPs are semi-crystalline in nature. The more amorphous PNAG compared to the NAG monomer may have better bioavailability over the NAG monomer.
The morphology and particle size of the PNAG NPs were investigated through HRTEM (Fig. 2). It is clear from TEM micrographs that the particles tend to form co-connected porous network structures with huge void space. Particles are ca. ∼35 nm in diameter with pores of 2–3 nm in size (Fig. 2b). From SAED (Fig. 2d), it is confirmed that the PNAG NPs are semi-crystalline in nature which can be observed from the diffused ring pattern, as earlier confirmed through the XRD results (Fig. 1d).
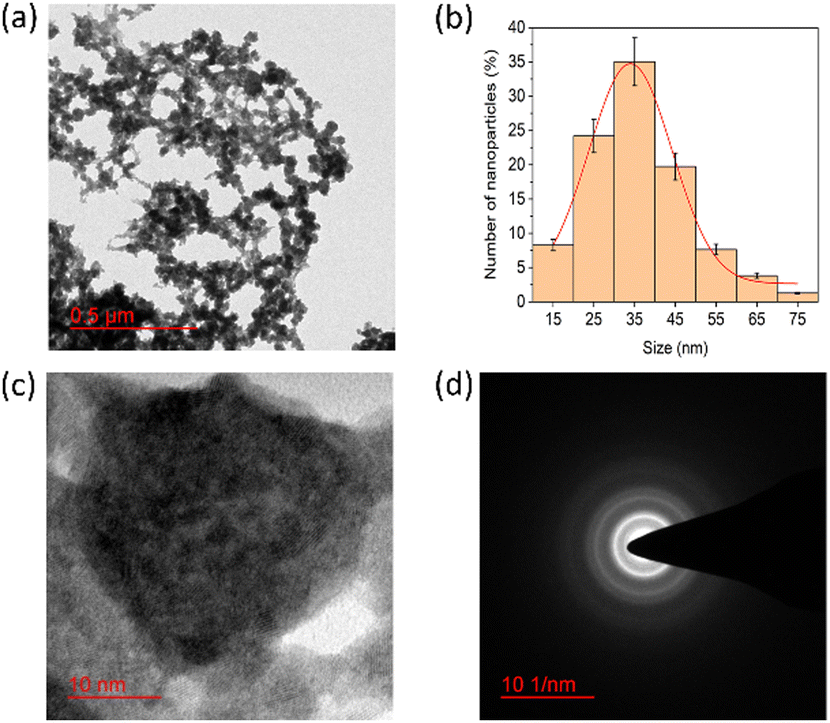 | ||
| Fig. 2 TEM micrograph and particle size distribution of PNAG NPs; (a) net-like co-connect arrangements; (b) particle size distribution of PNAG NPs calculated considering 300 particles; (c) porous particle structure and (d) SAED pattern shows diffused rings and confirms the semi-crystalline nature of PNAG NPs. (hydrodynamic size and zeta potential data: see ESI,† Fig. S2a and b) | ||
DLS was used to determine the hydrodynamic diameter of PNAG NPs (ESI,† Fig. S5), and the average size of PNAG NPs was found to be 40.2 nm in diameter, congruent with the HRTEM results (ca. ∼35 nm).
Further, PNAG NPs' negative zeta potential value (ξ) of −21.9 mV (ESI,† Fig. S6) implies colloidal stability.33
Biocompatibility and hemocompatibility of PNAG NPs
In mammals, fibroblast cells are the most abundant cells in the skin, which play a significant role in extracellular matrix development and epithelial–mesenchymal interactions,34 and hence, cytotoxicity assay of PNAG NPs was performed on L929 cells.Similarly, polymeric biomaterials must have a low cytotoxic profile on fibroblasts, whenever we focus on skin regenerative applications.35 Increased vasculature is an important phenomenon in regenerative medicine because it improves the blood, nutrients, oxygen and stem cell supply at the regenerative site. Human umbilical vein endothelial cells (HUVECs) are widely used in in vitro angiogenic studies in normal and tumour-associated angiogenesis.36 Therefore, we assessed the toxicity using MTT assay. Fig. 3a demonstrates the cytotoxicity results for the NAG monomer and PNAG NPs in L929 cells and Fig. 3b for PNAG NPs in HUVEC cells. PNAG NPs demonstrated excellent cytocompatibility compared to NAG monomers in L929 cells with a concentration of 150 μg mL−1 PNAG NPs, at which almost 100% of cells are viable, while NAG monomers decreased cell viability by nearly 85%. The viability with the NAG monomer and PNAG NPs at the highest tested concentration (250 μg mL−1) was found to be 77.01 ± 11.16 and 88.92 ± 9.43% (Fig. 3a), respectively. The viability of HUVEC cells (Fig. 3b) treated with PNAG NPs at higher concentration (200 μg mL−1) was found to be more than 80% (80.13 ± 3.75), while with the decrease in the concentration of PNAG up to 5 μg mL−1, an increase in HUVEC cell proliferation up to 115% was observed. And hence we considered that PNAG NPs could show the angiogenic potential at lower concentrations when tested in in ovo systems.
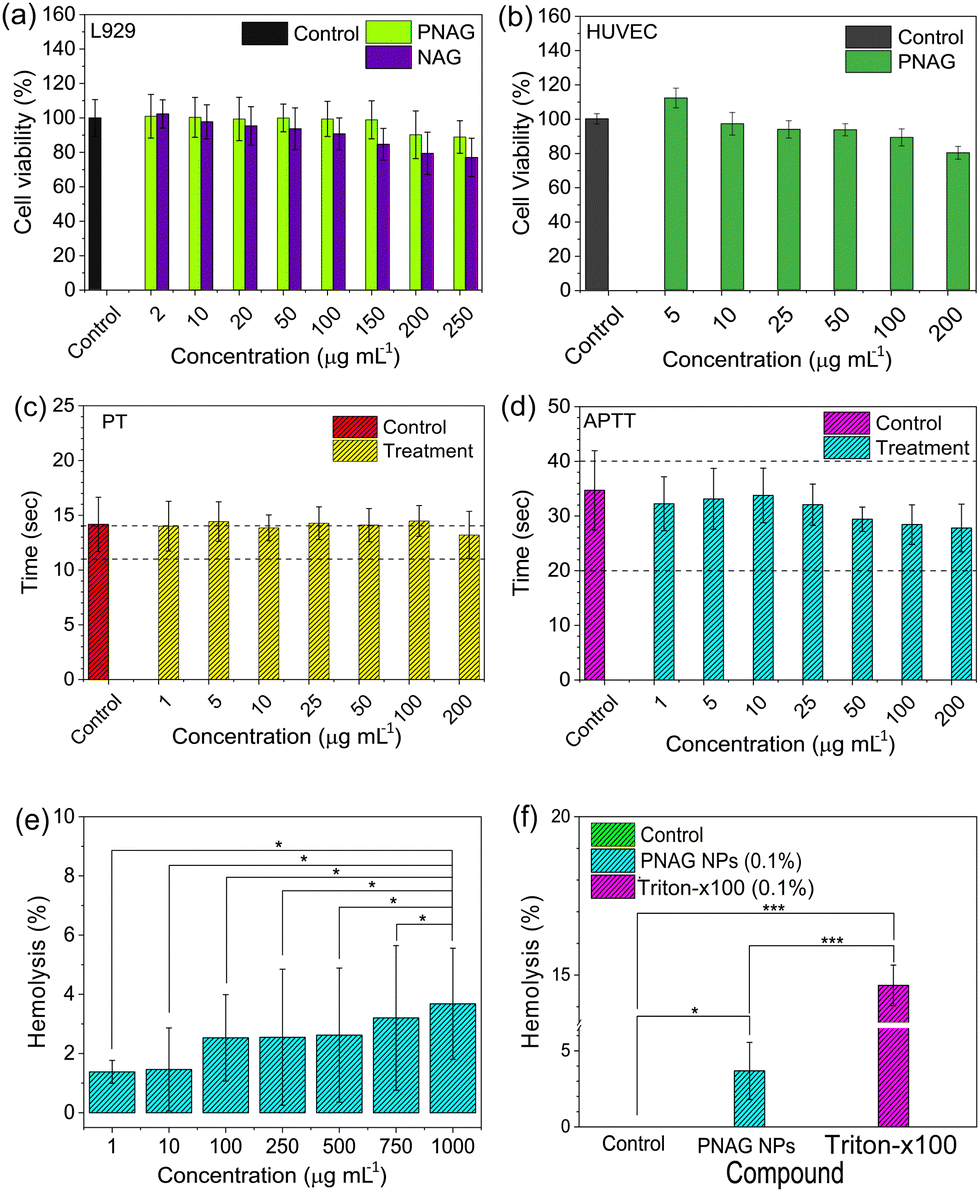 | ||
| Fig. 3 Cytotoxicity and hemocompatibility of PNAG NPs. (a) Cytotoxicity of the NAG monomer and PNAG NPs in the L929 cell line and (b) PNAG NPs in HUVEC cells. (c) and (d) Effect of different concentrations of PNAG NPs on the blood coagulation cascade in terms of PT (c) and APTT (d). The zones between the black dashed lines represent reference values for PT and APTT. Both the coagulation pathways (PT and APTT) were tested separately. (e) Dose–response curve for the haemolytic activity of PNAG NPs in rat erythrocytes, showing no significant difference (p > 0.05); (f) haemolysis (%) compared to the control caused by 0.1% dispersion of PNAG NPs and 0.1% Triton X-100 solution in PBS (p < 0.05); the outcomes represent the mean ± SD of three separate studies. | ||
Further, hemocompatibility evaluation plays a significant role in enhancing confidence to use the biomaterial for therapeutic purposes. As NPs enter the systemic circulation and cause inimical dominance, they can limit the therapeutic benefits.37,38 Therefore, examination of hemocompatibility of PNAG NPs was established by blood coagulation and hemolysis assay. Blood clotting is caused by a sequence of protein interactions known as the entire plasma coagulation cascade.39 The collected blood (Wistar rat) and the separated plasma fraction were treated with PNAG NPs at concentrations between 1 and 200 μg mL−1 and were assessed for the activated partial thromboplastin time (APTT) and the prothrombin time (PT) (Fig. 3c and d).
The PT values for PNAG NPs and control (in sec) were calculated after incubation for 30 min. The results obtained are found to be 14.17 ± 2.48 for control (PBS) and 14.01 ± 2.28, 14.43 ± 1.81, 13.86 ± 1.19, 14.29 ± 1.49, 14.11 ± 1.51, 14.48 ± 1.41 and 13.21 ± 2.16 sec for PNAG NPs at a concentration of 1, 5, 10, 25, 50, 100 and 200 μg mL−1, respectively. Similarly, the coagulation time of the intrinsic pathway of coagulation (APTT) was assessed, and APTT values were found to be 34.71 ± 8.10 for control and 32.25 ± 5.50, 33.13 ± 6.25, 33.77 ± 5.58, 32.10 ± 4.23, 29.42 ± 2.48, 28.44 ± 4.10 and 27.81 ± 4.87 sec for PNAG NPs at the above mentioned respective concentrations (Fig. 3d). Negligible impact was seen on plasma coagulation against lower concentration of PNAG NPs (suspended in PBS), as seen in Fig. 3c. However, only at higher concentrations, such as 200 μg mL−1, PNAG NPs show a decrease in PT values (13.21 ± 2.16 s) compared to control (14.17 ± 2.48 s).
Further, the hemolytic activity of PNAG NPs was evaluated in a dose-dependent manner from 1 to 1000 μg mL−1 and varied degrees of haemolysis were observed which are 1.38 ± 0.38, 1.46 ± 1.404, 2.53 ± 1.46, 2.55 ± 2.30, 2.62 ± 2.27, 3.20 ± 2.44 and 3.68 ± 1.88%, for different doses within the 24 h of treatment of control (Fig. 3e). The extent of haemolysis tends to increase with the increasing dose of PNAG NPs. However, it does not exceed more than 5% for the highest tested dose of PNAG NPs compared to control. When the hemolytic activity of the PNAG NPs treatment group was compared with the negative control (PBS), a statistically non-significant difference was observed (p > 0.05). On the other hand, the hemolytic activity of the PNAG NPs treatment group compared to the positive control (Triton X-100, 0.1% v/v) showed a statistically significant difference. Similarly, when we compared the difference between the negative control (PBS) and the positive control (Triton X-100, 0.1% v/v) statistically significant hemolysis was observed in the positive control (p < 0.05) (Fig. 3f).
In conclusion, PNAG NPs can be considered as safe regarding hemocompatibility and have no adverse effects on blood coagulation at high concentrations up to 1000 μg mL−1 and are suitable for therapeutic purposes.
PNAG NPs improve the wound closure (in vitro)
Skin cell migration is an essential facet of wound recovery.25 Herein, we have examined the pro-migratory impact of the PNAG NPs using an in vitro wound healing test in a specified period (0 to 48 h) and at varying concentration from 50 to 200 μg mL−1 (Fig. 4). For the analysis of PNAG NP induced cell migration and proliferation of fibroblasts, photographic images are acquired, as shown in Fig. 4; it is observed that PNAG NPs can efficiently induce wound closure in mouse fibroblast (L929) cells. After 24 h of wound generation, only 11.38 ± 0.42% of scratched space has been covered in control (untreated) cells, while in treated cells, the coverage of scratched space is significantly higher and is found to be 27.43 ± 1.73, 47.27 ± 4.51 and 39.08 ± 14.90%, respectively, for PNAG-50, PNAG-100 and PNAG-200 (Fig. 4). It has been noted that during the first 24 h, the PNAG-200 group of cells show lower coverage of scratched space in comparison to PNAG-100. But during the next 24 h, the rate of wound healing is higher and the healing rate at this stage is found to be dependent on the concentration of PNAG NPs (Fig. 4b and c). In control cells, even after 48 h, only 26.35 ± 2.23% of scratched space was covered. On the other hand, in treated cells, the scratched space was significantly covered due to cell migration and proliferation induction activities of PNAG NPs. After 48 h, the PNAG NP treated cells (PNAG-50, PNAG-100 and PNAG-200) show 49.37 ± 2.05, 71.46 ± 4.72 and 94.56 ± 2.12% coverage of scratched space, respectively (Fig. 4b). In the case of PNAG-200 treated cells the scratched space appears to be almost covered (94.56 ± 2.12%) and also seems to be healed completely (Fig. 4a).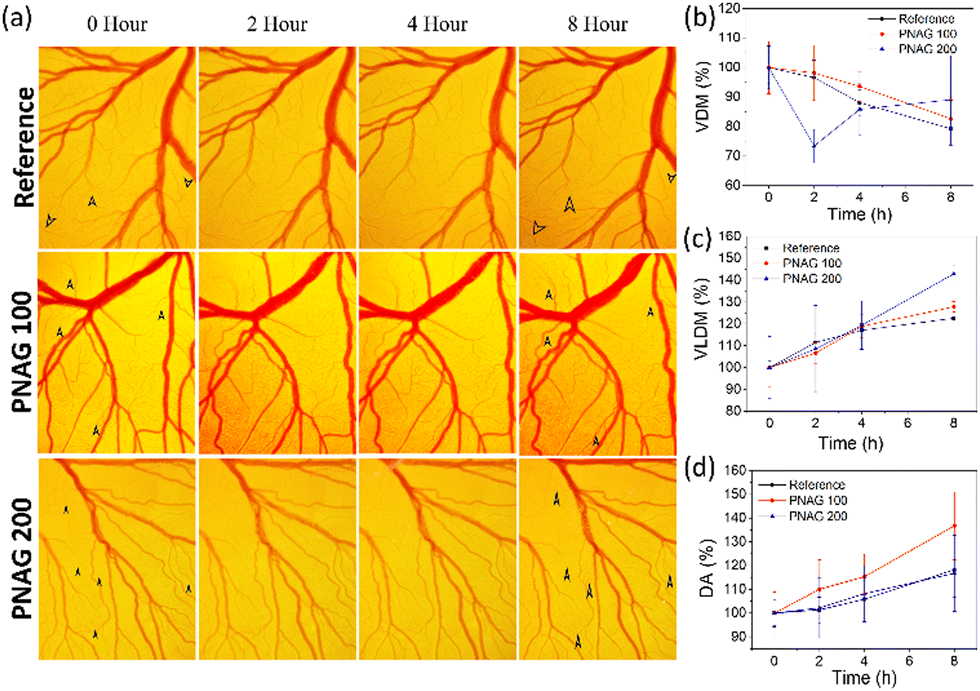 | ||
| Fig. 4 In vitro scratch wound healing assay; (a) microscopic assay images obtained at 0, 24 and 48 h. Images were captured at 10× magnification, and the scale bars indicate 100 μm. The assay was conducted using PNAG NPs in a concentration- and time-dependent manner. Treatment: control (complete cell culture media) and PNAG50, 100 and 200 (PNAG NPs in complete cell culture media at a concentration of 50, 100 and 200 μg mL−1); (b) L929 cells showing in vitro wound closure (%) and (c) rate of wound closure (%). Data are given as mean ± standard deviation (n = 3) (*P ∼ 0.05, ***P < 0.05). | ||
The results showed that administering PNAG NPs improved wound closure by 2–4 times more efficiently in treated cells than untreated cells, along with a rise in the total wound closure rate (0–48 h) (Fig. 4c) compared to the untreated cells. During the first 24 h, the rate increase corresponds to low concentrations of PNAG NPs, whereas at high concentrations, the rate was lower. After 24 h of treatment, the rates at lower concentrations were low compared to the higher concentrations (Fig. 4c). This reveals that the wound closure occurred in a concentration-dependent manner and low concentration improves the wound healing efficiency.
PNAG NPs assist concentration-dependent angiogenesis
As the above observation (Fig. 3b) demonstrates, PNAG promotes the proliferation of HUVEC cells at lower concentration and 100% viability at 100 μg mL−1 and slight inhibition of cells at 200 μg mL−1. Therefore, to establish the role of PNAG NPs, a standard in vivo angiogenesis assay, CEMA, was performed, and PNAG NPs demonstrated their role in vascular sprouting or angiogenesis. Herein, after 8 h of incubation of chick embryos with different concentrations (100 and 200 μg mL−1) of PNAG NPs, the presence of more mature and developed blood vasculature was observed, as denoted by black arrows (Fig. 5).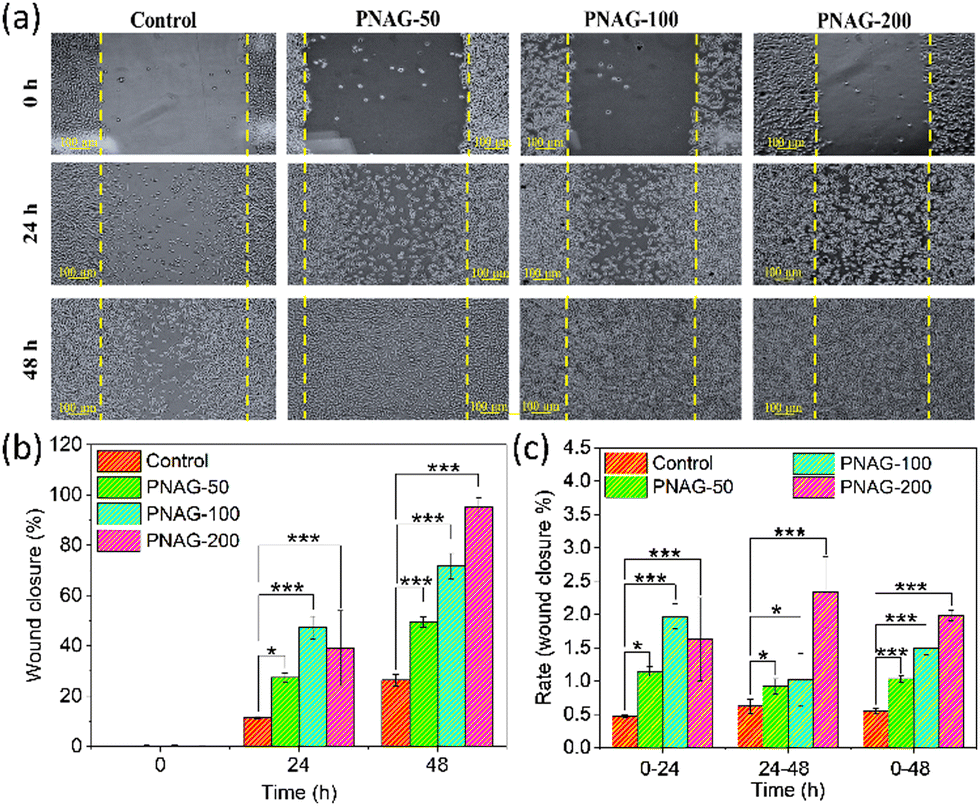 | ||
| Fig. 5 Angiogenic assay: Angiogenic effect of PNAG NPs in chicken embryo membrane assay (CEMA). (a) PNAG NPs stimulate vessel sprouting in the CAM model compared to the reference. The histogram shows the increase in vascular density (b), vascular length density (c) and mean development area (d) as a function of time for a specified vessel structure compared with the reference. The values presented are the mean ± SD of three independent experiments. *p < 0.05 is considered significant when compared to normal (n = 3). | ||
Further, for the confirmation of angiogenesis, VDM (Fig. 5b), VLDM (Fig. 5c), and DA (Fig. 5d) were measured using FijiJ software. VDM shows an increase with time from '0' h to 8 h, and a decreased vascular density was observed in the reference control as well as at lower (100 μg mL−1) and higher (200 μg mL−1) concentrations of PNAG NPs. At the higher concentration, during the initial period of 2 h, a complex behaviour was observed, but at the 8th h, the vascular density was higher in both the test groups (Fig. 5b).
The total VLDM (Fig. 5c) was found to be increased, 142.91 ± 4.05% at higher concentrations (200 μg mL−1) and 126.61 ± 0.72% at lower concentrations (100 μg mL−1) of PNAG in comparison to the reference VLDM value (122.61 ± 0.72%) with a similar pattern in all the reference and treated groups. The total length of the vessels was increased with time and concentration of PNAG NPs used, although the number density of measured vessels was decreased as a function of time. In comparison to the reference group, the VLDM enhancement in the treatment group was more and was found to be in the range of 6 ± 4.64% to 27.82 ± 2.40% and 8.56 ± 19.88% to 42.91 ± 4.05% in the case of PNAG NPs at a concentration of 100 μg mL−1 and 200 μg mL−1, respectively. Further, statistically, a significantly increased developmental area is depicted in Fig. 5d. Compared to the reference control, the increased developmental area is 18.27% within 8 h, while for the treatment group, the results are concentration-dependent. The relative increase in the developmental area in the treatment group compared to the reference group was 18.48% and −1.47% at lower (100 μg mL−1) and higher (200 μg mL−1) concentrations of PNAG NPs, respectively. This observation shows the correlation between the above in vitro wound closure results and the in vitro cytotoxicity assay. These results suggested that PNAG NPs exhibit angiogenesis properties in vivo and are helpful in tissue regeneration at lower concentrations.
Developed PNAG nanoformulation and quality evaluation
An ointment is one of the most commonly used semi-solid dosage forms for medicaments' topical (dermal) delivery. Generally, hydrocarbon-oleaginous bases deliver drugs where drugs are present inside the ointment as a dispersed phase and widely utilised as a key ingredient for the commercialised ointment. Therefore, we explored a hydrocarbon-based oleaginous base (ESI,† Table S1) for the PNAG NP formulation. Here, 5% glycerin, as a humectant, is added to the PNAG nanoformulation and in the formulation base. A qualitative formulation evaluation assessed the physical characteristics of the formulated PNAG nanoformulation and formulation base, such as colour, appearance, consistency (homogeneity), phase separation, and odour (ESI,† Table S3). The formulation base and PNAG nanoformulation were devoid of agglomeration or grittiness and appeared smooth, homogeneous, white or off-white. At 37 ± 2 °C, produced formulations' pH and spreadability were investigated. With minor deviations (±SD < 0.5), the pH of both formulations was within the desired target range of pH 6.8–7.2. For topical usage, formulations with neutral or nearly neutral pH are preferred since they do not impact the integrity of the skin and do not result in any adverse side effects such as oedema, redness, or a change in transepithelial water loss and prevent skin sensitivity and irritation.40 Spreadability, which refers to a topical formulation's capacity to distribute evenly over the skin and provide a standard dose of medication to the skin, is crucial to a topical therapy's effectiveness.41 The spreadability test was performed, and it was observed that the formulation base and PNAG nanoformulation spread at 17.65 ± 3.8 and 17.09 ± 2.6 gm cm s−1, respectively, which indicates that the spreadability of the prepared PNAG nanoformulation is close to that of the formulation base. A higher spreadability coefficient value indicates better spreadability of the formulation on the skin.PNAG nanoformulation assists in dermal safety
Predicting the adverse effects during the preclinical phase produced by any biomaterial is a powerful tool to combat the issues that arise in the drug's preclinical and clinical phases. Dermal irritation and skin sensitisation studies are crucial components and indicate the potential hazards' fundamental characteristics if the information on their relative toxicity according to the regulatory guidelines is unavailable.This investigation aimed to ascertain if the formulations may irritate rat skin following a single topical treatment. Formulations were applied to the skin of rats. Changes in skin colour (erythema) and swelling of the skin (oedema) were investigated, and Draize scoring was used to assess the primary dermal irritancy index (PDII), as shown in Table S2 (ESI†). The skin irritation results, before application (at 0 h) and after application (at 24 h), are presented in Fig. 6(A). The treatment and the control groups showed no signs and symptoms of erythema and oedema.
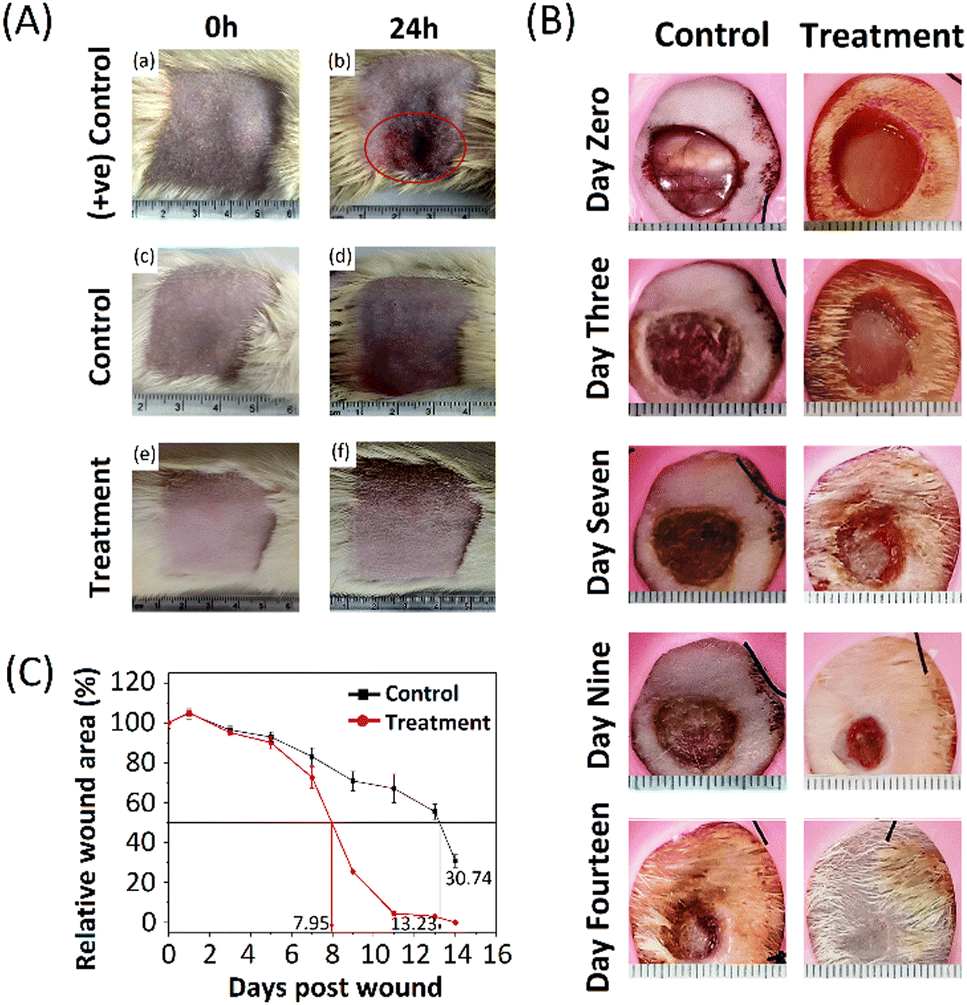 | ||
| Fig. 6 In vivo skin irritation and wound healing; (A) results show the dermal irritation study in Wistar rats after dermal application. Images were taken before (0 h) and after (24 h) the application of samples; (a) and (b) positive control (1% formalin soln), (c) and (d) control (formulation base), (e) and (f) treatment (PNAG-nanoformulation), respectively. After 24 h of application, the control group (d) and the treatment group (f) do not show any sign and symptoms of dermal toxicity, i.e., dermal irritancy, while the positive control group (b) showed significant erythema and oedema at the site of application (red circle). (B) Photo images of wound healing after 3, 7, 9 and 14 days in Wistar rats treated using the formulation base (control) and PNAG nanoformulation (treatment group). (C) Wound closure profiles of animals treated with the formulation base (control group) and PNAG nanoformulation (treatment group) (p < 0.05). Data are presented as the mean ± standard deviation (n = 3). | ||
In contrast, the positive control group showed significant erythema and oedema. Thus, these results show that the formulation base and PNAG nanoformulation were non-irritant and non-sensitive to the skin and did not produce any significant undesired effect after 24 h of application. A PDII value is found to be less than 0.25 for both the control and treatment groups, but it was more than 2 in the positive control group. After testing, the animals were kept under surveillance for another 7 days. All animals (positive control, control and treatment group) appeared active and healthy, and neither displayed any other signs and symptoms of unpleasant toxicity or abnormal behaviour nor was any treatment-related scuffle noted in the PNAG nanoformulation treated and control groups. It was also observed that the overall incidence of irritation and sensitisation severity in positive treatment group animals has decreased over time.
PNAG nanoformulation improves wound closure (in vivo)
A full-thickness in vivo skin wound model on Wistar rats was used to assess further the potential of the PNAG nanoformulation in the light of the findings mentioned above and the knowledge that PNAG NPs could encourage cell proliferation and migration, and support angiogenesis, all of which are advantageous for skin wound repair. The findings are shown in Table S4 (ESI†) and depicted in Fig. 6B and C.As PNAG NPs enhance the viability, proliferative and migratory effect of cells in vitro, here, PNAG NPs released from the nanoformulation also showed faster wound healing (Fig. 6B) and reduced wound size after treatment initiation than control groups (Fig. 6C). Fig. 6B displays the macroscopic view of the wound after treatment (0, 3rd, 7th, 9th, and 14th day) with PNAG nanoformulation and formulation base. Further the relative wound area (%) was measured by measuring the wound area (Fig. 6C). It can be concluded that the PNAG nanoformulation facilitated considerable wound size reduction in the treatment group than the control group and showed clean wound surfaces. Our findings showed that the PNAG nanoformulation was more effective in promoting wound healing because it showed a superior healing impact than the formulation base (ESI,† Table S4). On the 0, 1st, 3rd, 5th, 7th, 9th, 11th, 13th and 14th day post-surgery, the wound areas (%) in the control group (formulation base) are found to be 100.00 ± 2.80, 104.79 ± 2.74, 96.45 ± 2.38, 92.96 ± 2.47, 83.15 ± 4.28, 70.91 ± 4.81, 67.17 ± 7.17, 55.47 ± 3.83 and 30.74 ± 3.11, whereas in the treatment group (PNAG nanoformulation) the wound areas (%) are 100.00 ± 0.97, 105.08 ± 1.62, 95.12 ± 0.71, 90.19 ± 3.25, 72.63 ± 5.44, 25.33 ± 0.34, 4.49 ± 0.88, 2.93 ± 0.43 and 0.00 ± 0.1, respectively. This efficient healing effect could be due to the PNAG nanoformulation providing an appropriate suitable environment at the wound site and encouraging cell growth. Hence, from the above data, we can conclude that the PNAG nanoformulation was efficiently helpful in cutaneous wound treatment.
Histological examination
Further, histological examination was performed, and for this, the skin sections were stained with hematoxylin and eosin (H&E) to confirm skin regeneration, re-epithelization, granulation tissue development and ultimately, wound healing.42 During the healing process, histological examination confirmed the general morphology of skin layers (Fig. 7). The results show the histological evaluations and comparison of wound healing on the 14th day post-wounding with the healthy tissue. The injured tissue was embedded into paraffin blocks, and histological slides were generated by staining with H&E. Three types of tissues, namely, healthy skin tissue (a), tissue obtained from control group animals (formulation base (b)) and treatment group animals (PNAG nanoformulation (c)), were examined and compared (Fig. 7).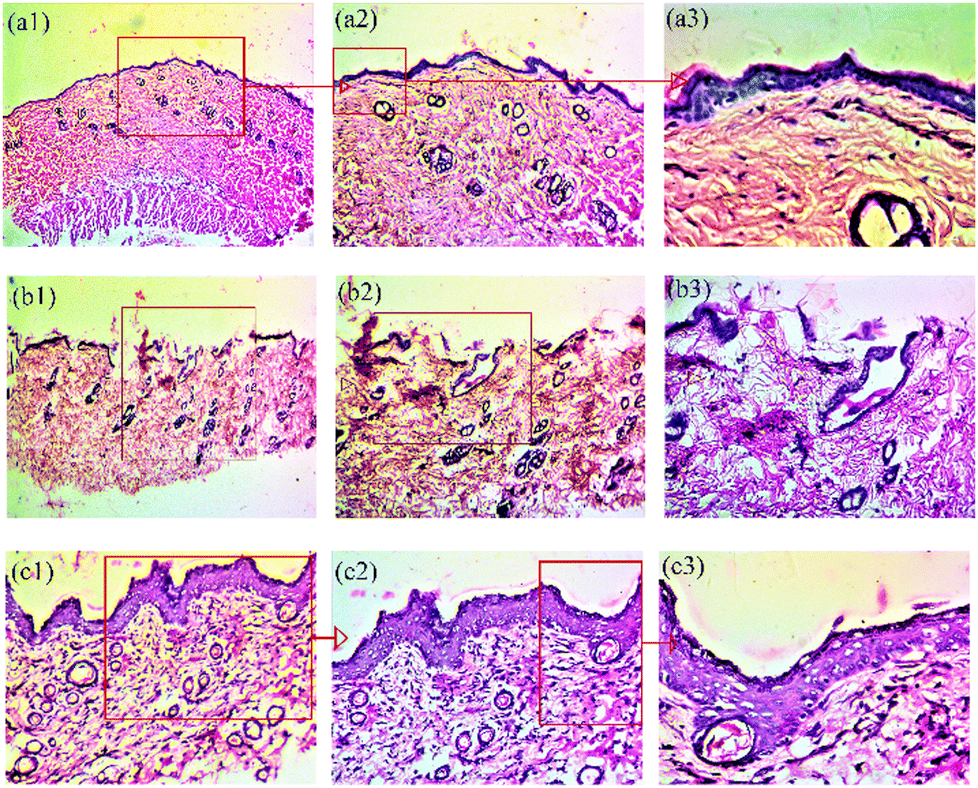 | ||
| Fig. 7 Histological evaluation and comparison of wound healing on day 14 post-wounding with healthy tissue. Healthy skin tissue (a), control tissue (base nanoformulation) (b), and PNAG nanoformulation (c). Numbers 1, 2 and 3 denote images captured at 4×, 10× and 40× magnification. Healthy skin images show the following well-defined skin structures: dead skin, epithelium, dermis (collagen fibre), hypodermis, smooth muscle fibre, sweat gland, sebaceous gland and hair follicle, larger blood vessels, nerve endings, papillary dermis, reticular dermis, rete ridges of the epidermis, the basal layer of the epidermis, thick fat collagen fibre, and blood vessels. Control skin images show broken or ruptured epithelium, underdeveloped dermis fibres, absence of hypodermis, absence of rete ridges of the epidermis, and undifferentiated reticular dermis and papillary dermis. PNAG nanoformulation treated skin images show a clear and well-differentiated epidermis, underdeveloped basal layer, fully differentiated and grown large and dilated blood vessels, a thick layer of the epidermis at some places, fully differentiated epidermis ridges, fully grown granuloma, some phases of remodelling, meshy papillary dermis and reticular dermis, and large fibres. | ||
Healthy skin tissue images [Fig. 7(a1), (a2) and (a3)] show the following structures: (a1): dead skin, epithelium, dermis (collagen fibre), hypodermis, smooth muscle fibre, (a2): sweat gland, sebaceous gland and hair follicle, larger blood vessels, nerve endings, papillary dermis, reticular dermis, rete ridges of the epidermis; (a3): the basal layer of the epidermis, thick fat collagen fibre and blood vessels.
Control group tissue images [Fig. 7(b1), (b2) and (b3)] show the following structures: (b1): broken or ruptured epithelium, (b2): underdeveloped dermis fibres, absence of hypodermis, absence of rete ridges of the epidermis, undifferentiated reticular dermis and papillary dermis, (b3): underdeveloped superficial vascular plexus, numerous blood vessels and capillaries, spongy epithelium/dermis showing oedema and inflammation.
Treatment group tissue images (Fig. 7(c1), (c2) and (c3)) show the following structures: (c1): clear and differentiated epidermis, underdeveloped basal layer, (c2): fully differentiated and grown large and dilated blood vessels, a thick layer of the epidermis at some places, fully differentiated epidermis ridges, (c3): fully grown granuloma, some phases of remodelling, meshy papillary dermis and reticular dermis, large fibres.
Thus, the results suggest that the PNAG nanoformulation treated wound demonstrated an increased number of fibroblast-like cells and decreased mononuclear inflammatory cells with healed skin structures close to the normal healthy dermis, whereas the control group demonstrated ulceration (damaged epidermis), oedema, and abundance of mononuclear inflammatory cells with deep inflammatory infiltration getting through the dermal layer.43
Further, the H&E images of the PNAG nanoformulation-treated group exhibited morphology more similar to healthy skin than the control group. In the treatment group, we observed more neo-vascularisation, complete dermal and epidermal tissue, sebaceous glands, hair follicles and a high degree of re-epithelialization (Fig. 7c).
Impact of the PNAG nanoformulation on immune response, in vivo analysis
The wound healing process was completed in four phases: homeostasis, inflammatory, proliferative and remodelling. The second inflammatory phase is essential for activating the innate immune system to remove debris and toxins, prevent coagulation and bacterial infection and initiate the fibrin plug formation. With the decrease in the inflammatory phase, the proliferative phase gets initiated from day 3 till day 10, where growth factors are released.44 To better understand the mechanism of wound healing of PNAG NPs, we evaluated the expression of proteins (TNF-α, IL-6, and IGF-1) exerting extraordinary capabilities, using the rat's blood samples collected at unique periods on the 2nd, 7th and 12th day as they represent the distinguished phases of wound healing.TNF-α entails regulated chemo-elements at the wounded site and regulates the activity of keratinocytes, fibroblasts and vascular endothelial cells.45–47 In our results, TNF-α (Fig. 8a) was found to be 4.30 ± 1.21, 9.80 ± 3.19 and 8.39 ± 2.24 pg mL−1 for the control group, whereas for the treatment group, its level was found to be 4.10 ± 3.18, 6.02 ± 2.22 and 5.17 ± 3.27 pg mL−1 on the 2nd, 7th and 12th day, respectively. TNF-α, as observed from the 7th and 12th day, shows the proliferative phase promoted by PNAG; here it produces the macrophages and encourages the production of proteoglycan and fibronectin in fibroblasts, and subsequently, these fibroblasts promote the extracellular matrix (ECM) in the damaged area increasing the wound healing efficiency.46 Thus, it can be concluded that lower levels of TNF-α for the treatment group efficiently promote wound healing and closure.
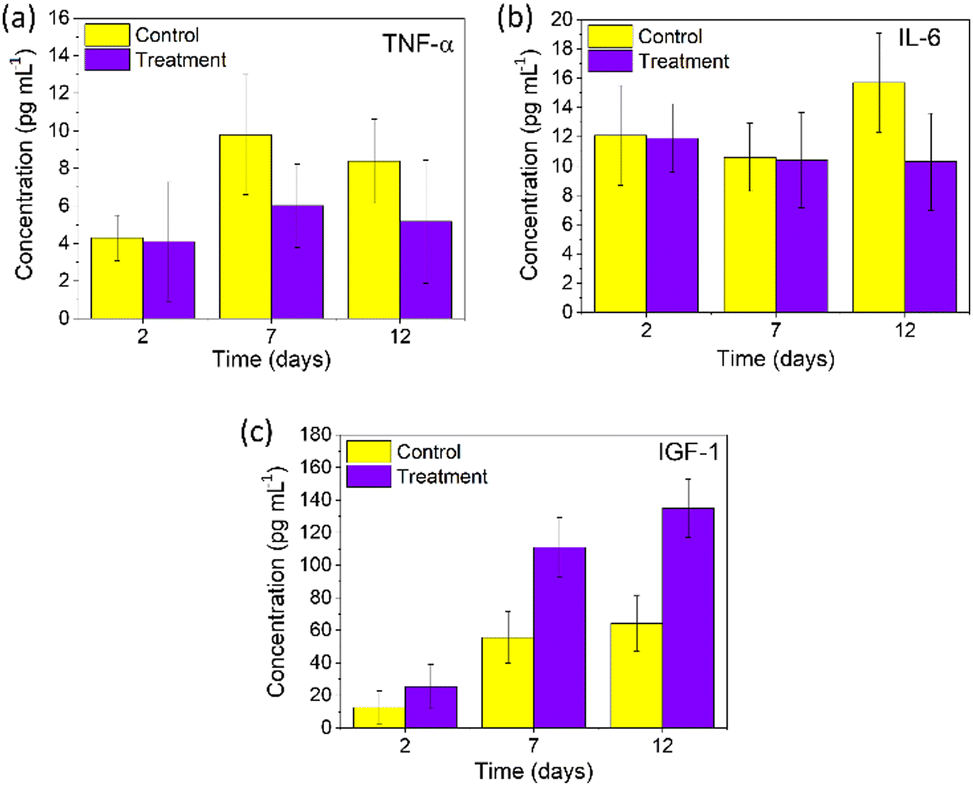 | ||
| Fig. 8 Immunochemical analysis: (a) TNF-α, (b) IL-6, and (c) IGF. PNAG NPs enhance the concentration and levels of IGF-1 (c) and regulate pro-inflammatory cytokines and chemokines TNF-α (a) and IL-6 (b). | ||
Interleukin 6 (IL-6) plays an essential role in cell signalling, lymphocyte control, and acute inflammation and is required for the rapid resolution of wound healing by regulating leukocyte infiltration, angiogenesis, and collagen build-up and release by the immune cells.48,49 Here, we observed relatively less change in the IL-6 (Fig. 8b) values on the 2nd day (11.93 ± 2.34 pg mL−1), 7th day (10.41 ± 3.26 pg mL−1), and 12th day (10.33 ± 3.31 pg mL−1) for the treatment group, whereas for the control groups, higher values were observed on the 12th day (15.7 ± 3.37 pg mL−1) compared to the 2nd (10.63 ± 2.32 pg mL−1) and 7th day (12.11 ± 3.41 pg mL−1) (Fig. 8b). It can be noted that the IL-6 expression has increased during the remodelling phase of regular wound repair. These results corroborate the anti-inflammatory response of PNAG NPs and nanoformulation along with the volume closure efficiency.
Insulin-like growth factor-1 (IGF-1) is significant in repairing human tissues and organs with anabolic effects.50 According to Barreca et al., human fibroblasts produce IGF-1 peptides which stimulate keratinocyte growth in a paracrine manner, emphasising their function in regulating keratinocyte proliferation, skin growth, and wound healing.51 IGF-1 levels on the 2nd, 7th, and 12th day were determined and were found to be considerably increasing for all groups, as shown in Fig. 8c. On the 2nd day, the IGF-1 value for the treatment group was found to be 25.41 ± 13.56 ng mL−1, which further increased to 134.83 ± 17.90 ng mL−1 on the 12th day, whereas for the control group it was found to be 12.53 ± 10.32 ng mL−1 on the 2nd day and 64.22 ± 16.93 ng mL−1 on the 12th day post-treatment. From our experimental results, the IGF-1 level increased significantly and led to enhancement in keratinocyte migration, which accelerates wound re-epithelialization and wound contraction. Thus, it can be noted that during the remodelling phase of a wound on application of the PNAG nanoformulation, IGF-1 shows a potential role in keratinocyte migration.52
Discussion
Polymeric nanoparticles (NPs) are very influential because they have an essential impact without impairing healthy body functions. Nanosized polymeric particles have a higher cellular uptake and provide more interaction sites with biological units along with accessibility to various cellular and intracellular targets because of their larger surface area compared to the micro-sized or macro-sized particles. As NPs pass through the submucosal layers whereas micro-sized particles are primarily localised in the epithelial lining, nanosized particles have a greater absorption rate than micro-sized particles.53In this work, we developed PNAG NPs with significant therapeutic capabilities, biocompatibility, low cost, and high yield. PNAG NPs were synthesised using the mini emulsion radical polymerisation method22 with controlled size and shape. Organic soluble radical initiator AIBN sparked free radical production at high temperatures (over 75 °C), enabling polymerisation by generating intermediate free radicals inside the emulsion droplets. Hexadecane (HD), a long-chain alkane utilised as a co-stabiliser, boosts osmotic pressure and decreases the coalescence of oil droplets induced by Laplace pressure (Ostwald ripening).54 Thus, the concentration of the co-stabiliser is vital, and the co-stabiliser to monomer ratio must be greater than 1:250.55 PNAG NPs’ mechanical stability and porosity were produced using DVB, which imparts a sizable amount of cross-linking between the linear polymer chains. Increasing the concentration of DVB may result in nano aggregates and nanoclusters of particles. Emulsifiers (surfactants) are the ingredients which promote colloidal stability. In our synthesis, we have used SDS as an emulsifier that aids in the uniform size of PNAG NPs. In the absence of SDS, PNAG NPs show poor physical stability as they aggregate and form bigger particles during storage. A dense nano-porous web-like network arrangement of PNAG NPs (Fig. 2a) along with ca. ∼35 nm in diameter (Fig. 2b) makes them a future synergistic delivery system. The zeta potential of PNAG NPs (ESI,† Fig. S4) in an aqueous suspension was −21.9 mV, which is significantly closer to the deflocculation zone and offers colloidal stability to the PNAG NP aqueous suspension.56
In addition to the safety of the bulk material, NPs' physicochemical characteristics, such as their monodispersed size distribution, spherical shape, and surface charge, impact their biocompatibility. Here, both NAG monomer and PNAG NPs show good biocompatibility and apparent no cytotoxicity in mouse fibroblasts (L929) (Fig. 3a), while cytotoxicity evaluation on HUVEC cells was limited for PNAG NPs as they are used for nanoformulation. The same results are also depicted in the case of HUVEC cells which are highly sensitive to biomaterials and these cells tolerate PNAG NPs very well in the concentration range between 5 and 200 μg mL−1 along with the increase in proliferation at lower concentrations of 5 μg mL−1 PNAG NPs (Fig. 3b).
The NPs entering the systemic circulation carry adverse effects and can limit the therapeutic benefits.37 The hemocompatibility study confirmed that PNAG NPs have no significant hemolytic effect on rat RBCs compared to control (Fig. 3f), indicating that our PNAG NPs do not interfere with the mechanism of aquaporins and Na+/K+ ATPase, which affect water permeability, diffusion, and erythrocyte osmotic fragility.57 We also investigated the dose dependence of haemolysis by PNAG NPs and identified a progressive increase in haemolytic activity (Fig. 3e). In terms of blood coagulation, we observed that all tested concentrations of PNAG NPs (up to 200 μg mL−1) do not affect the levels of PT and APTT (Fig. 3c and d). It was also observed that the PNAG NPs did not affect the blood coagulation system, cellular system, or haemolysis at clinically relevant concentrations. PNAG NPs are biocompatible with rat RBCs, and cause no possible risk of erythrocyte toxicity.
Fibroblast migration and proliferation in the presence of biomaterials are critical steps in cutaneous wound healing. The proliferation- and migration-inducing effect of PNAG NPs (Fig. 4) supports the hypothesis of re-epithelialization and restoration of the skin functions. Activation of fibroblast proliferation and migration results in the enhancement of rate of granulation tissue formation, which ultimately leads to accelerated wound healing.58
During injury, blood vasculature is damaged, leading to nutrient and oxygen deficiency in the injured tissue. During wound repair, it is essential that a paramount neo-capillary bed must be formed and pruned back to status-co as before the injury. In our study, we observed that PNAG NPs promote vascular sprouting and angiogenesis, which accelerates the migration of blood vessels to the wound bed (Fig. 5), especially at a lower concentration.
The initial indication of non-toxicity of PNAG NPs is high vitality in L929 and HUVEC cells (Fig. 3a and b) and no substantial hemolysis (Fig. 3e and f) in mouse RBCs. Proliferation induction in HUVEC cells, biocompatibility towards fibroblasts, hemocompatibility, and pro-angiogenic behaviour validated the safety of PNAG NPs and gave the preliminary outline for the in vivo wound healing investigation.
Medical-grade materials were used to prepare the PNAG nanoformulation. To be employed in preclinical studies, the nanoformulation generated from PNAG NPs was subjected to quality control tests to determine the formulation's fitness for topical applications. Appearance, odour, colour, homogeneity, pH, spreadability, and dermal irritancy effects are significant elements that influence patient acceptability of formulations; consequently, they must remain intact during the application. Thus, the nanoformulation was adjusted for skin pH using triethanolamine.
Other key parameters are homogeneity and spreadability of the PNAG nanoformulation. If the nanoformulation is not homogeneous and easy to spread, then the friction of gritty particles causes secondary injury and pain, extending the healing time, necessitating pain management therapy, and increasing the total cost of therapy, resulting in an economic burden. For this purpose, dermal irritancy test is highly required. After applying a formulation, it is evaluated in terms of erythema and oedema and measured using the Draize scoring scale (ESI,† Table S2), which is then transformed into PDII values using eqn (3). A PDII value of ‘0’ indicates no irritation, 0.5–1.9 indicates modest discomfort, 2–4.9 indicates mild irritation and values above 5 indicate severe irritation. The formulation was evaluated and compared on rat skin against the standard irritant. All animals except the positive control group were free from cutaneous irritation. However, the positive control animals had several flares on their skin (Fig. 6A(b)). The animals were observed for 7 days, which showed no unpleasant toxicity or odd behaviour, and seemed active and healthy. PNAG NPs have an interconnected and uniform porous structure. Their shape allows for the infiltration and proliferation of epithelial cells, the absorption of exudates, and the preservation of the oxygen supply to the wound tissue. Hence, the PNAG nanoformulation is an appropriate formulation for dermal application.
In Wistar rats, an in vivo assessment of the PNAG NP's capacity for speedy wound healing was carried out (Fig. 6B and C). On a 10–12 mm diameter excisional wound, we observed that lower levels of exudates are secreted when the wound healed. These findings are further supported by the histology of 14 day old wounds, which demonstrates dense granulation and more effective re-epithelialization tissue in the wound centre when the PNAG nanoformulation is used compared to the formulation base (Fig. 7).
On the 2nd, 7th and 12th day, we studied that the topically administered PNAG nanoformulation affected numerous chemokines, cytokines, and growth factors in the systemic blood of control and treated animals. The inflammatory, proliferative, and remodelling phases of wound healing were chosen as the time points. Lower levels of TNF-α and IL-6 were reported in the treatment group (Fig. 8), showing efficient regulation of the anti-inflammatory effects of PNAG NPs. On the 12th day, in the treatment group, IGF-1 growth factor levels intensely increased, demonstrating higher potential for keratinocyte migration at the injured area during the remodelling phase. An increase in IGF-1 is also correlated with an increase in the vasculature, as IGF-1 activates the PI3-kinase/Akt signalling pathway and simultaneously up-regulates the expression of PDGFB, MMPs and other angiogenic growth factors.59 And hence, herein, we estimated that adequate wound healing occurs through the formation of new vasculature via angiogenesis which supplies nutrients and oxygen to the wound bed.60 This clarifies the role of PNAG NPs in supporting wound healing.
4. Conclusions
Biocompatibility, hemocompatibility, anti-inflammatory response and angiogenesis are the key factors for wound healing. In this work, we have synthesised PNAG NPs by the mini emulsion polymerisation method. The prepared NPs were characterised by various techniques evidencing the successful fabrication of PNAG NPs. Moreover, a wide range of in vitro analysis of PNAG NPs, such as hemocompatibility, cytotoxicity and in vitro wound healing, confirmed the suitability for testing in the in vivo wound healing model. The moderate expression of wound healing markers suggests that PNAG NPs have some extent of anti-inflammatory potential. Overall, the PNAG NPs can enhance the healing rate in a full-thickness in vivo skin wound model. Indeed, this study demonstrated that PNAG NPs can induce angiogenesis and reduce inflammation in the microenvironment of skin cells/tissues. The main hallmarks, such as angiogenesis, cell migration and proliferation and anti-inflammatory activity properties of PNAG NPs, support wound healing and salubrity, suggesting their potential use for topical application in wound healing and regenerative nanomedicine without the use of any external synergistic factors like drugs, genes, cytokines, growth factors or cells.Author contributions
Prem Shankar Gupta (investigations, formal analysis, writing manuscript), Kirti Wasnik (experimental procedure, data analysis and editing), Gurmeet Singh (performed angiogenesis), Sukanya Patra (methodology and analysis), Divya Pareek (methodology and analysis), Desh Deepak Yadav (data analysis), Munender Singh Tomer (methodology), Somedutta Maiti (DLS and zeta), Monika Singh (methodology), and Pradip Paik (PI of the project, supervision, resources, fund acquisition, data analysis and writing manuscript).Conflicts of interest
The authors have no conflicts of interest to declare.Note added after first publication
This article replaces the version published on 13th September 2023. Fig. 4 and Fig. 5 had been swapped in error during production.Acknowledgements
The authors acknowledge the financial support awarded to Prof. Pradip Paik by DST-Nanomission, India (Ref: SR/NM/NS-1005/2015), Science and Engineering Research Board (SERB), India (Ref: EEQ/2016/000040) and IIT(BHU) Seed funds.References
- A. O. Ijaola, D. O. Akamo, F. Damiri, C. J. Akisin, E. A. Bamidele, E. G. Ajiboye, M. Berrada, V. O. Onyenokwe, S. Y. Yang and E. Asmatulu, J. Biomater. Sci., Polym. Ed., 2022, 33, 1998–2050 CrossRef CAS PubMed.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS PubMed.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CrossRef CAS PubMed.
- B. Alberts, A. Johnson and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 4th edn, 2002 Search PubMed.
- K. P. Krafts, Organogenesis, 2010, 6, 225–233 CrossRef PubMed.
- S. Guo and L. A. Dipietro, J. Dent. Res., 2010, 89, 219–229 CrossRef CAS PubMed.
- M. Haalboom, Curr. Med. Chem., 2018, 25, 5772–5781 CrossRef CAS PubMed.
- S. R. Nussbaum, M. J. Carter, C. E. Fife, J. DaVanzo, R. Haught, M. Nusgart and D. Cartwright, Value in health, 2018, 21, 27–32 CrossRef PubMed.
- H. Nosrati, R. Aramideh Khouy, A. Nosrati, M. Khodaei, M. Banitalebi-Dehkordi, K. Ashrafi-Dehkordi, S. Sanami and Z. Alizadeh, J. Nanobiotechnol., 2021, 19, 1 CrossRef PubMed.
- M. M. Mihai, M. Preda, I. Lungu, M. C. Gestal, M. I. Popa and A. M. Holban, Int. J. Mol. Sci., 2018, 19, 1179 CrossRef PubMed.
- Y. Ge and Q. Wang, Front. Mol. Biosci., 2022, 9, 1057766 CrossRef CAS PubMed.
- X. Deng, M. Gould and M. A. Ali, J. Biomed. Mater. Res., Part B, 2022, 110, 2542–2573 CrossRef CAS PubMed.
- F. R. Diniz, R. C. A. Maia, L. R. M. de Andrade, L. N. Andrade, M. V. Chaud, C. F. da Silva, C. B. Corrêa, R. L. C. de Albuquerque Junior, L. P. da Costa, S. R. Shin, S. Hassan, E. S. Lopez, E. B. Souto and P. Severino, Nanomaterials, 2020, 10, 4079 CrossRef PubMed.
- F. Paladini and M. Pollini, Materials, 2019, 12, 2540 CrossRef CAS PubMed.
- B. Sheokand, M. Vats, A. Kumar, C. M. Srivastava, I. Bahadur and S. R. Pathak, J. Polym. Sci., 2023, 61, 1389–1414 CrossRef CAS.
- P. W. Henderson, S. P. Singh, D. D. Krijgh, M. Yamamoto, D. C. Rafii, J. J. Sung, S. Rafii, S. Y. Rabbany and J. A. Spector, Wound repair and regene., 2011, 19, 420–425 CrossRef PubMed.
- W. Liu, W. Ou-Yang, C. Zhang, Q. Wang, X. Pan, P. Huang, C. Zhang, Y. Li, D. Kong and W. Wang, ACS Nano, 2020, 14, 12905–12917 CrossRef CAS PubMed.
- Q. Xu, S. A, Y. Gao, L. Guo, J. Creagh-Flynn, D. Zhou, U. Greiser, Y. Dong, F. Wang, H. Tai, W. Liu, W. Wang and W. Wang, Acta Biomater., 2018, 75, 63–74 CrossRef CAS PubMed.
- M. Ranjbar Mohammadi, S. Kargozar, S. H. Bahrami and S. Rabbani, Polym. Degrad. Stab., 2020, 174, 109105 CrossRef CAS.
- R. Fitridge and M. Thompson, Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists, University of Adelaide Press, Adelaide (AU), 2011 Search PubMed.
- A. Kushwaha, L. Goswami and B. S. Kim, Nanomaterials, 2022, 12, 618 CrossRef CAS PubMed.
- A. K. Yamala, V. Nadella, Y. Mastai, H. Prakash and P. Paik, Nanoscale, 2017, 9, 14006–14014 RSC.
- A. K. Yamala, V. Nadella, Y. Mastai, H. Prakash and P. Paik, J. Appl. Polym. Sci., 2020, 137, 48363 CrossRef CAS.
- A. Bentolila, I. Vlodavsky, R. Ishai-Michaeli, O. Kovalchuk, C. Haloun and A. J. Domb, J. Med. Chem., 2000, 43, 2591–2600 CrossRef CAS PubMed.
- S. Cometa, C. Licini, M. A. Bonifacio, P. Mastrorilli, M. Mattioli-Belmonte and E. De Giglio, Carbohydr. Polym., 2022, 283, 119145 CrossRef CAS PubMed.
- M. De Villiers, Pract. Guide Contemp. Pharm. Pract., 2009, 3, 277–290 Search PubMed.
- M. N. Mutimer, C. Riffkin, J. A. Hill, M. E. Glickman and G. N. Cyr, J. Am. Pharm. Assoc., 1956, 45, 212–218 CrossRef CAS PubMed.
- X. Wang, J. Ge, E. E. Tredget and Y. Wu, Nat. Protoc., 2013, 8, 302–309 CrossRef CAS PubMed.
- V. C. O. Póvoa, G. J. V. P. dos Santos, G. F. Picheth, C. P. Jara, L. C. E. da Silva, E. P. de Araújo and M. G. de Oliveira, J. Tissue Eng. Regener. Med., 2020, 14, 807–818 CrossRef PubMed.
- L. E. Lindley, O. Stojadinovic, I. Pastar and M. Tomic-Canic, Plast. Reconstr. Surg., 2016, 138, 18s–28s CrossRef CAS PubMed.
- H. S. Purohit, N. S. Trasi, D. D. Sun, E. C. Y. Chow, H. Wen, X. Zhang, Y. Gao and L. S. Taylor, J. Pharm. Sci., 2018, 107, 1330–1341 CrossRef CAS PubMed.
- A. Schittny, J. Huwyler and M. Puchkov, Drug Delivery, 2020, 27, 110–127 CrossRef CAS PubMed.
- S. Samimi, N. Maghsoudnia, R. B. Eftekhari and F. Dorkoosh, in Characterization and Biology of Nanomaterials for Drug Delivery, ed. S. S. Mohapatra, S. Ranjan, N. Dasgupta, R. K. Mishra and S. Thomas, Elsevier, 2019, ch. 3, pp. 47–76 DOI:10.1016/B978-0-12-814031-4.00003-9.
- T. Wong, J. A. McGrath and H. Navsaria, Br. J. Dermatol., 2007, 156, 1149–1155 CrossRef CAS PubMed.
- J. Lee, S. P. Hlaing, J. Cao, N. Hasan and J.-W. Yoo, J. Pharm. Invest., 2020, 50, 505–512 CrossRef CAS.
- D. J. Medina-Leyte, M. Domínguez-Pérez, I. Mercado, M. T. Villarreal-Molina and L. Jacobo-Albavera, Appl. Sci., 2020, 10, 938 CrossRef CAS.
- L. Chen, J. J. Glass, R. De Rose, C. Sperling, S. J. Kent, Z. H. Houston, N. L. Fletcher, B. E. Rolfe and K. J. Thurecht, ACS Appl. Bio Mater., 2018, 1, 756–767 CrossRef CAS PubMed.
- O. Maksimenko, J. Malinovskaya, E. Shipulo, N. Osipova, V. Razzhivina, D. Arantseva, O. Yarovaya, U. Mostovaya, A. Khalansky, V. Fedoseeva, A. Alekseeva, L. Vanchugova, M. Gorshkova, E. Kovalenko, V. Balabanyan, P. Melnikov, V. Baklaushev, V. Chekhonin, J. Kreuter and S. Gelperina, Int. J. Pharm., 2019, 572, 118733 CrossRef CAS PubMed.
- J. Laloy, V. Minet, L. Alpan, F. Mullier, S. Beken, O. Toussaint, S. Lucas and J. M. Dogné, Nanobiomedicine, 2014, 1, 4 CrossRef PubMed.
- Y. Yamamoto, T. Fukami, T. Koide, T. Suzuki, Y. Hiyama and K. Tomono, Int. J. Pharm., 2012, 426, 54–60 CrossRef CAS PubMed.
- G. E. Djiobie Tchienou, R. K. Tsatsop Tsague, T. F. Mbam Pega, V. Bama, A. Bamseck, S. Dongmo Sokeng and M. B. Ngassoum, Cosmetics, 2018, 5, 7 CrossRef.
- Y. Kang, J. Kim, Y. M. Lee, S. Im, H. Park and W. J. Kim, J. Controlled Release, 2015, 220, 624–630 CrossRef CAS PubMed.
- H. Nurhasni, J. Cao, M. Choi, I. Kim, B. L. Lee, Y. Jung and J. W. Yoo, Int. J. Nanomed., 2015, 10, 3065–3080 CAS.
- J. Davis and A. McLister, in Smart Bandage Technologies, ed. J. Davis, A. McLister, J. Cundell and D. Finlay, Academic Press, 2016, pp. 1–35 DOI:10.1016/B978-0-12-803762-1.00001-1.
- B. A. Mast and G. S. Schultz, Wound Repair Regen., 1996, 4, 411–420 CrossRef CAS PubMed.
- M. Ritsu, K. Kawakami, E. Kanno, H. Tanno, K. Ishii, Y. Imai, R. Maruyama and M. Tachi, J. Dermatol. Dermatol. Surg., 2017, 21, 14–19 CrossRef.
- I. A. Mckay and I. M. Leigh, Br. J. Dermatol., 1991, 124, 513–518 CrossRef CAS PubMed.
- D. O. Avazi, A. C. Awasum, A. Z. Hassan, J. O. Ayo, T. Aluwong, S. T. Muhammed, A. Y. Simon, M. H. Suleiman and A. C. Kudi, Cytokine, 2019, 113, 128–138 CrossRef CAS PubMed.
- Z.-Q. Lin, T. Kondo, Y. Ishida, T. Takayasu and N. Mukaida, J. Leukocyte Biol., 2003, 73, 713–721 CrossRef CAS PubMed.
- A. T. Grazul-Bilska, M. L. Johnson, J. J. Bilski, D. A. Redmer, L. P. Reynolds, A. Abdullah and K. M. J. D. T. Abdullah, Drugs Today, 2003, 39, 787–800 CrossRef CAS PubMed.
- A. Barreca, A. Voci, F. Minuto, M. de Marchis, E. Cecchelli, E. Fugassa, G. Giordano and G. Gallo, Mol. Cell. Endocrinol., 1992, 84, 119–126 CrossRef CAS PubMed.
- Y. Ando and P. J. Jensen, J. Invest. Dermatol., 1993, 100, 633–639 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard, Jr., Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- Y. Shen, C. Du, J. Zhou and F. Ma, Sci. Rep., 2018, 8, 12279 CrossRef PubMed.
- J. Jeng, C.-A. Dai, W.-Y. Chiu, C.-S. Chern, K.-F. Lin and P.-Y. Young, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4603–4610 CrossRef CAS.
- P. S. Gupta, V. Sharma and K. Pathak, Expert Opin. Drug Delivery, 2013, 10, 17–32 CrossRef CAS PubMed.
- I. Greco, N. Molchanova, E. Holmedal, H. Jenssen, B. D. Hummel, J. L. Watts, J. Håkansson, P. R. Hansen and J. Svenson, Sci. Rep., 2020, 10, 13206 CrossRef CAS PubMed.
- G. Han, L. N. Nguyen, C. Macherla, Y. Chi, J. M. Friedman, J. D. Nosanchuk and L. R. Martinez, Am. J. Pathol., 2012, 180, 1465–1473 CrossRef CAS PubMed.
- S. Lin, Q. Zhang, X. Shao, T. Zhang, C. Xue, S. Shi, D. Zhao and Y. Lin, Cell Proliferation, 2017, 50, 12478 Search PubMed.
- L. A. DiPietro, J. Leukocyte Biol., 2016, 100, 979–984 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Animal protocol and ethics, Table S1. Formulation table for PNAG nanoformulation, Table S2. Draize scoring system for skin irritation, Table S3. Qualitative characterization of nanoformulation, Table S4. In vivo wound healing result; Fig. S1: setup for spreadability measurement, Fig. S2: 1H-NMR spectra of NAG, Fig. S3: 13C-NMR spectra of the NAG monomer, Fig. S4: particle size and Fig. S5: zeta potential estimation; TOC. See DOI: https://doi.org/10.1039/d3ma00378g |
| This journal is © The Royal Society of Chemistry 2023 |