
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Removal of methyl red from wastewater using a NiO@Hyphaene thebaica seed-derived porous carbon adsorbent: kinetics and isotherm studies†
Abdelaal S. A.
Ahmed
 *a,
Moustafa M. S.
Sanad
b,
Ahmed
Kotb
a,
Ahmed N. R. M.
Negm
a and
Mohamed H.
Abdallah
a
*a,
Moustafa M. S.
Sanad
b,
Ahmed
Kotb
a,
Ahmed N. R. M.
Negm
a and
Mohamed H.
Abdallah
a
aChemistry Department, Faculty of Science, Al-Azhar University, Assiut 71524, Egypt. E-mail: abdelaalsaiyd@gmail.com; abdelaalsaiyd@azhar.edu.eg
bCentral Metallurgical Research and Development Institute, P.O. Box 87 Helwan, 11421, Cairo, Egypt
First published on 13th June 2023
Abstract
In this study, the adsorption of methyl red (MR) dye from wastewater on nickel oxide (NiO) doped in porous carbon derived from Hyphaene thebaica (HT) seeds (NiO@HT derived C) as an adsorbent material was investigated. The prepared NiO@HT derived C was characterised by Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), and X-ray photoelectron spectroscopy (XPS). Moreover, the effects of solution pH, contact time, amount of the adsorbent, and the initial concentration of MR on the adsorption efficiency were consistently examined. The adsorption capacity of NiO@HT derived C at room temperature (25 °C ± 2) could reach 129.87 mg g−1 in an acidic medium after shaking for 60 min. Furthermore, the adsorption kinetics and isotherms of adsorption were investigated. The results indicated that the adsorption isotherm of reactive MR on the NiO@HT derived C surface follows the Langmuir isotherm model, which revealed that the adsorption of MR is a chemisorption process. On the other hand, the kinetic data fit well with the pseudo-second-order kinetic model, indicating that both NiO@HT derived C and MR influence the adsorption process.
1. Introduction
Dyes are chemical molecules that are unpleasant and hazardous to the environment.1 Usually, dyes are extensively used in many industrial applications, such as cosmetics, plastics, paper, printing, pharmaceuticals, and textiles.2 Textile industries are the greatest generators of dye wastewater that is discharged into the environment.3,4 Removing or minimizing dyes from wastewater has attracted attention worldwide due to the emission hazards, which are harmful to humans and the aquatic environment.2,5,6 Even if a small amount of coloured dye is discharged into the water, it is visually unappealing.7 The dyes used in industries are generally classified as cationic, anionic, or natural dyes.8 MR is an anionic azo dye that is important for various industrial and laboratory purposes; however, the presence of MR in water can cause health issues related to stomach, eye and skin irritation.9,10 Furthermore, the presence of benzene rings renders MR mutagenic under aerobic conditions and results in low biodegradability.11 Moreover, the presence of MR in water may cause digestive tract, eye and skin discomfort.12 Thus, there is an urgent need to remove MR from the polluted water before it is discharged into the environment. It is possible to remove colours from effluents using chemical, physical, or biological methods.13–15 Various techniques such as coagulation,16 photocatalytic oxidation,17 adsorption,12 ozone treatment, and others have been employed for recovering dyes from wastewater. Among all, the adsorption process is currently considered the most utilized process for the efficient removal of dyes from wastewater due to its various advantages such as ease of use, low cost, and relatively high efficiency.2 Choosing an appropriate adsorbent plays a key role in determining the total performance of the adsorption process. To date, various adsorbent materials, such as waste biomass9,18–20 and activated carbon,18 have been used for MR adsorption. Due to their large surface area and potent physical and chemical influences on the adsorption of dye effluents, metal oxide nanoparticles have been widely exploited as adsorbents toward synthetic dyes from wastewater in the last few decades.21,22 Nickel oxide (NiO) is an important transition-metal oxide widely used in various applications such as solar cells,23 energy storage systems,24 and catalysis25 due to its high chemical and thermal stability, and eco-friendly nature. For wastewater treatment, NiO is also widely used as an adsorbent for synthetic dyes.26 For example, Riaz et al.26 reported that NiO nanoparticles prepared by a green hydrothermal method showed an effective adsorption toward methyl orange (MO) with a maximum adsorption capacity of 97.56 mg g−1 at pH 4. Furthermore, Song et al.27 revealed that NiO nanosheets have high adsorptive affinity toward brilliant red X-3B, Congo red and fuchsine acid with maximum adsorption capabilities of 30.4 mg g−1, 35.15 mg g−1 and 22 mg g−1, respectively. However, their overall adsorption efficiency still needs to be enhanced.28 Integrating NiO nanoparticles with porous carbon materials is an effective strategy due to the synergistic effect.29 Due to their abundant availability with almost zero cost, agricultural wastes have received intensive attention as precursors for the preparation of porous carbon for various environmental applications including wastewater treatment.30 From the standpoint of waste management, converting bio-waste materials into porous carbon is a promising strategy for wastewater treatment.31,32 In Egypt, there are numerous agricultural bio-waste items that are typically burned or dumped into the environment, which causes environmental contamination and subsequent health risks. These agricultural wastes could be a resource for the synthesis of porous carbon because they come from hardwood species with lignocellulosic character. Utilizing agricultural waste for the preparation of porous carbon is a promising way to produce cheap and efficient adsorbents and a potential alternative for solving the environmental problems of Egypt. In the Nile Valley governorate of Egypt and Sub-Saharan Africa, Hyphaene thebaica (HT) is a famous fruit that belongs to the Arecaceae family. The Hyphaene thebaica fruit is a solid fruit with a solid core, and the outside layer is edible. Due to the high percentage of waste produced from this fruit (>80% of the fruit's weight), it is highly recommended to find a way to make use of such waste materials, which will be important in economic and environmental aspects.33 Herein, we used HT seeds as a source of porous carbon to prepare a NiO@HT-derived C composite to be utilized as an adsorbent for MR removal from aqueous solutions. The various parameters that affect the adsorption efficiency, such as pH, contact time, and dye concentration, were investigated. Moreover, the adsorption kinetics and isotherms were also investigated. The estimated data revealed high adsorption affinity of the NiO@HT-derived C composite toward MR, with an adsorption capacity of 129.87 mg g−1 at room temperature, which is much better than that previously reported in the literature. According to our knowledge, this is the first time that this composite is prepared and utilized for wastewater treatment.2. Experimental section
2.1. Preparation of adsorbents
Hyphaene thebaica fruits were obtained from the local market in New Valley Governorate, Egypt. The nickel oxide dispersed into HT-derived C (NiO@HT-derived C) was prepared as described in the scheme shown in Fig. 1. The whole process involves three steps: (i) a pre-treatment step, where the HT species was initially washed repeatedly with distilled water to remove soluble compounds and dust. After that, HT species was dried in an oven at 100 °C for 8 h, followed by grinding with an electric grinder and screening through a 50 mesh screen. Then, the HT powder was bathed in boiling water for 8 h to remove the soluble impurities and finally dried at 100 °C overnight. (ii) Adsorption of Ni2+ ions on the HT powder: 1.0 g of the dried HT powder was immersed in 100 mL of an aqueous solution of 0.1 M Ni(NO3)2 under a magnetic stirrer for 3 h. Subsequently, 50 mL of a 2 M NH4OH solution was stepwise dropped into the mixture and kept under a magnetic stirrer for 3 h at 80 °C. The suspension was thoroughly washed with DI water and ethanol to obtain the Ni(OH)2/HT composite; and (iii) the obtained composite (Ni(OH)2/HT) was subjected to carbonization at 500 °C for 2 h at a heating rate of 5 °C min−1. The prepared NiO@HT-derived C was thoroughly washed with dilute HCl and distilled water until the pH reached 7, followed by drying at 80 °C overnight to be ready for adsorption studies. The HT-derived C material was prepared using the same steps used for the NiO@HT-derived C composite except immersing in Ni2+ solution. The dried prepared materials were subjected to physicochemical characterization and adsorption studies.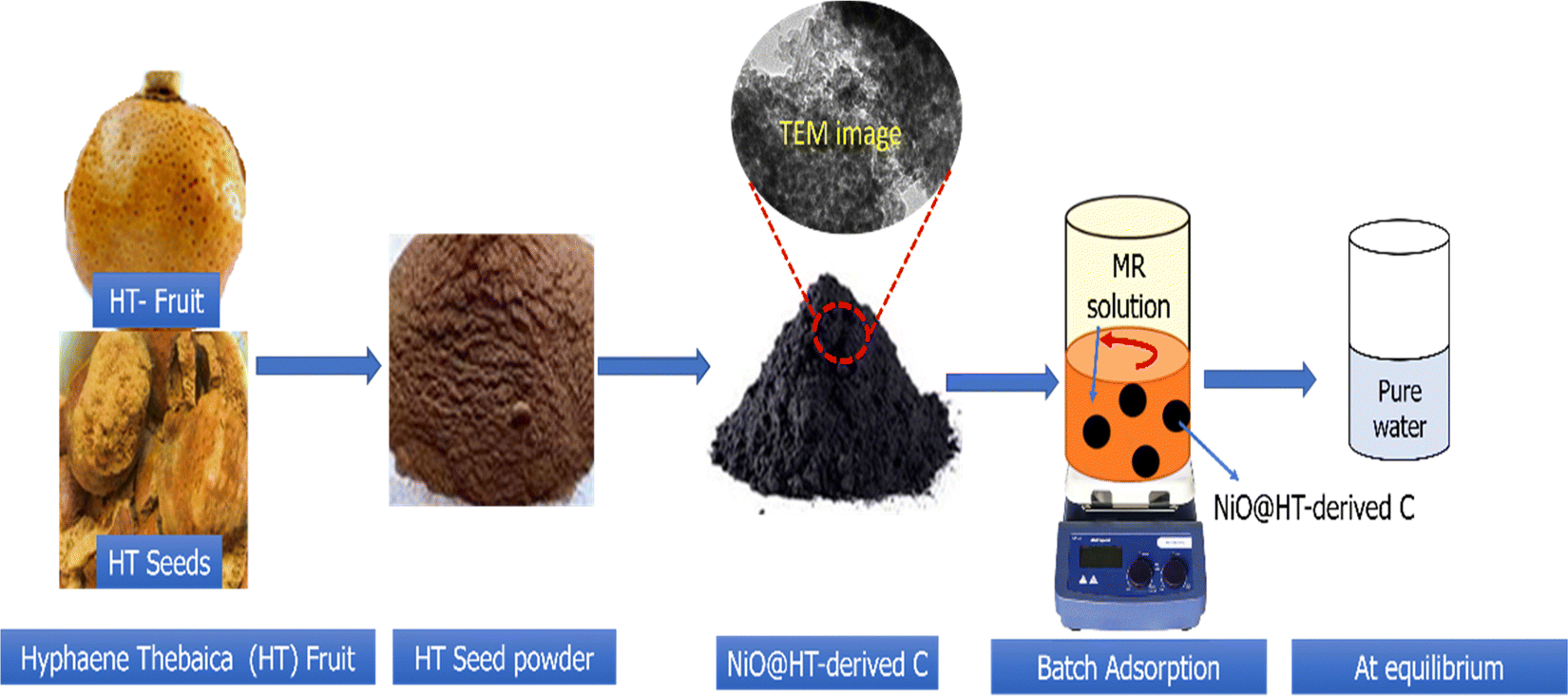 | ||
| Fig. 1 Scheme for the preparation of the NiO@HT-derived C composite. | ||
2.2. Batch adsorption experiments
Batch adsorption of MR dye was performed in a 50 mL dark bottle with a magnetic stirrer. For all tests, 0.03 g of adsorbent material was mixed with 25 mL of MR dye solution at an initial concentration of 50 mg L−1. The solution pH was adjusted by hydrochloric acid (HCl) and sodium hydroxide (NaOH) solutions with concentrations of 0.1 M. The influence of pH, adsorbent dose, contact time, and the initial dye concentration on MR dye removal was examined. The remaining concentration of the MR dye was measured using a UV-visible spectrophotometer at a wavelength of 520 nm. Each adsorption experiment was carried out in triplicate, and the average values with the related standard deviations (SD) were plotted. The percent removal (R%) of the MR dye was determined using eqn (1).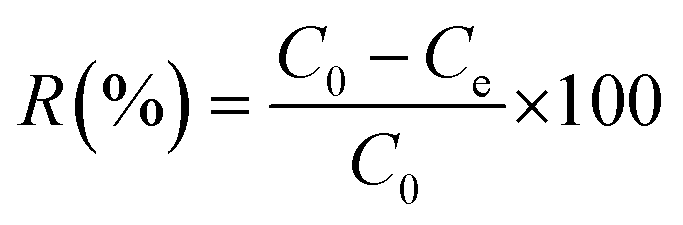 | (1) |
The adsorption capacity (qe) was measured using eqn (2).
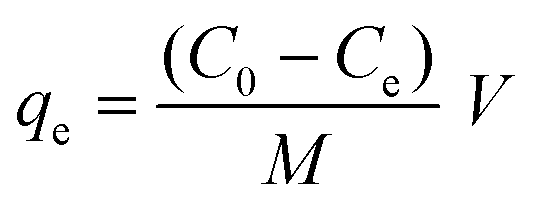 | (2) |
2.3. Adsorption kinetics and isotherms
The adsorption kinetics of the MR dye were performed by mixing 0.03 g of NiO@HT-derived C composite with 25 mL of dye solution (50 mg L−1, pH = 2.5, room temperature); the mixtures were kept under a magnetic stirrer for various contact times (5–120 min). The adsorption isotherms were investigated by using 0.03 g of NiO@HT-derived C composite mixed with 25 mL of MR dye solution (10–200 mg L−1) at pH 2.5 and a contact time of 2 h at room temperature.3. Results and discussion
3.1. Materials characterization
X-ray diffraction (XRD) was used to investigate the structure of the prepared materials. As shown in Fig. 2a, the XRD pattern of the NiO@HT-derived C composite showed diffraction peaks at 2θ of 37.1°, 43.2°, 63°, 73.5°, and 79.6°, all assigned to cubic-centered NiO (JCPDS 47-1049). In addition, two diffraction peaks for carbon were observed at around 22.5° and 40°. This indicates that NiO nanoparticles were successfully doped onto the carbon phase.34,35 | ||
| Fig. 2 (a) XRD pattern and (b) FT-IR spectra of the NiO@HT-derived C composite. (c–f) XPS spectra of the NiO@HT-derived C composite [(c) survey scan, (d) Ni2p, (e) C1s, and (f) O1s]. | ||
The FT-IR spectra of the HT powder and NiO@HT derived C samples are presented in Fig. 2b. The HT spectrum showed a broad band at 3405 cm−1 for the OH groups, which can be assigned by absorbing water molecules from the atmosphere. The small band located at 2935 cm−1 is for the asymmetric and symmetric stretching vibration of C–H bonds. However, the bands at 1740 and 1625 cm−1 are attributed to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the CC groups, respectively.36 The symmetric stretching of the carboxylate group (COO–) is assigned to the next small band at 1370 cm−1. The peak at 1255 cm−1 is assigned to the deformation vibration of CO and the stretching formation of OH of carboxylic acids and phenols, while the peak at 1050 cm−1 corresponds to the C–O band in the –OCH3 groups of lignin. For the NiO@HT-derived C composite, all peaks of the HT have been observed and the peak at 1050 cm−1 becomes much stronger, which indicates the bonding between Ni2+ ions and the functional groups of lignin present in the HT.37,38
O) and the CC groups, respectively.36 The symmetric stretching of the carboxylate group (COO–) is assigned to the next small band at 1370 cm−1. The peak at 1255 cm−1 is assigned to the deformation vibration of CO and the stretching formation of OH of carboxylic acids and phenols, while the peak at 1050 cm−1 corresponds to the C–O band in the –OCH3 groups of lignin. For the NiO@HT-derived C composite, all peaks of the HT have been observed and the peak at 1050 cm−1 becomes much stronger, which indicates the bonding between Ni2+ ions and the functional groups of lignin present in the HT.37,38
The XPS survey scan of the NiO@HT derived C composite in Fig. 3c demonstrates the presence of Ni, O, and C elements, which confirms the formation of the NiO@HT-derived C composite. Fig. 3d–f shows the Ni 2p, O 1s, and C 1s peaks, which were evaluated using XPS peak version 4.1 software.
 | ||
| Fig. 3 (a and b) TEM, (c) HRTEM images, and (d) SAED pattern of the NiO@HT-derived C composite. | ||
As shown in Fig. 2d, the Ni 2p spectrum shows five fitted peaks. These peaks are 2p3/2 (856.9 eV) and its satellites (858.3 and 862.2 eV), and 2p1/2 (874.5 eV) and its satellite (880.2 eV). The difference in binding energy between Ni 2p3/2 and Ni 2p1/2 is 17.6 eV. All these peaks are characteristic of Ni2+, revealing the presence of NiO.39,40 The C1s spectrum in Fig. 2e has four distinct peaks at 284.6, 285.5, 286.8 and 289.2 eV for nonoxygenated carbon atoms (C–C), carbon atoms in C–OH or C–O–Ni bands, carbon in carboxyl groups (CO) and carbon in carboxyl hyaluronic acid (HO–CO), respectively. The XPS profile of O1s (Fig. 2f) displayed two deconvoluted peaks. The one located at lower binding energy (531.2 eV) is assigned to the oxygen bonded with the O–Ni band, showing that oxygen bridges should be C–O–Ni bands, while the higher one at 532.4 eV is for the physically adsorbed oxygen.
The morphology of the NiO@HT-derived C composite was investigated by TEM/HR TEM and SAED measurements. As shown in TEM images (Fig. 3a and b), small dark dots can be visible, revealing that the NiO nanoparticles were equally distributed on the surface of the carbon matrix.41 The HR TEM image of the NiO@HT-derived C composite (Fig. 3c) shows a lattice fringe with an interplanar spacing of 0.24 nm, which matches the (111) planes of NiO. The SAED profile in Fig. 3d displays several distinct diffraction rings that correspond to the (111), (200), (220), (311), and (222) planes, with interplanar spacings that match the JCPDS 47-1049 (NiO) card data.41
The surface area and porous structure are important factors for providing more active sites, which promote the adsorption process.42 The nitrogen adsorption–desorption isotherm of the NiO@HT-derived C composite is presented in Fig. S1a (ESI†). Based on the IUPAC classifications, the NiO@HT-derived C composite exhibits a type-II isotherm with an H3 hysteresis loop. This is typical of micro/mesoporous materials composed of aggregates of particles creating slit-shaped pores.43 Micropore adsorption is responsible for the rapid increase in nitrogen absorption capacity at low pressure (P/P0 = 0–0.01).43 The presence of mesoporous structures is demonstrated by an increase in nitrogen absorption capability. The specific surface area (SBET) of the NiO@HT-derived C composite was estimated to be 141 m2 g−1 using the BET-equation in the pressure range of applicability (P/P0 = 0.05–0.30). The large surface area provides more adsorption sites and large contact area with the solution, resulting in improved overall adsorption efficiency.42 The Barrett–Joyner–Halenda (BJH) method pore size distribution shown in Fig. S1b (ESI†). The pore size distribution curve showed that the NiO@HT-derived C composite is mesoporous, which is ideal for dye adsorption.12
3.2. Adsorption studies
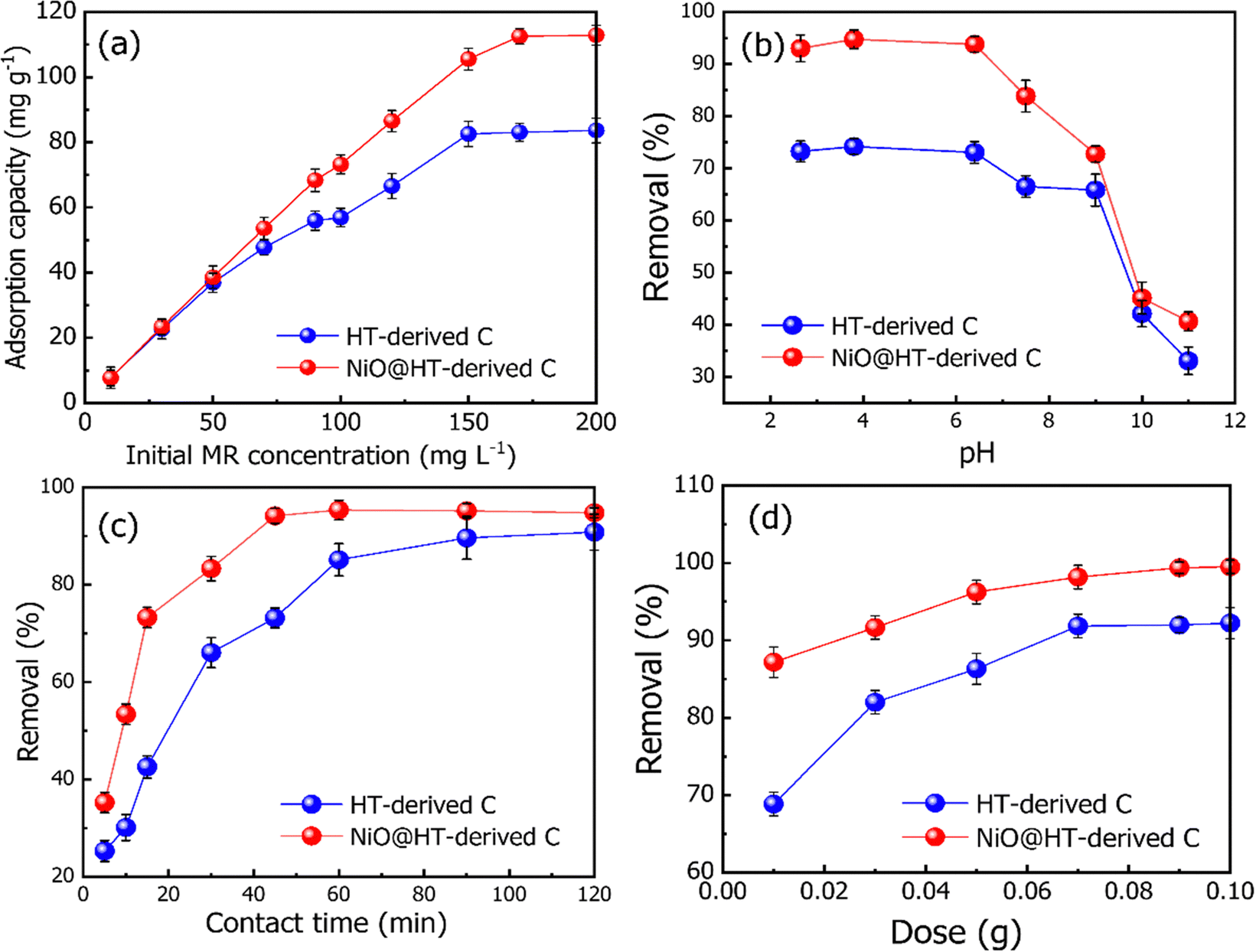 | ||
| Fig. 4 Effect of (a) initial dye concentration [M = 0.03 g, t = 60 min, pH = 2.5], (b) solution pH [C0 = 50 mg L−1, M = 0.03 g, t = 45 min, pH = 2.5–11], (c) contact time [M = 0.03 g, C0 = 50 mg L−1, pH = 2.5] and (b) adsorbent dosage [M = 0.01–0.1 g, C0 = 50 mg L−1, pH = 2.5] on the removal of the MR dye from aqueous solution. | ||
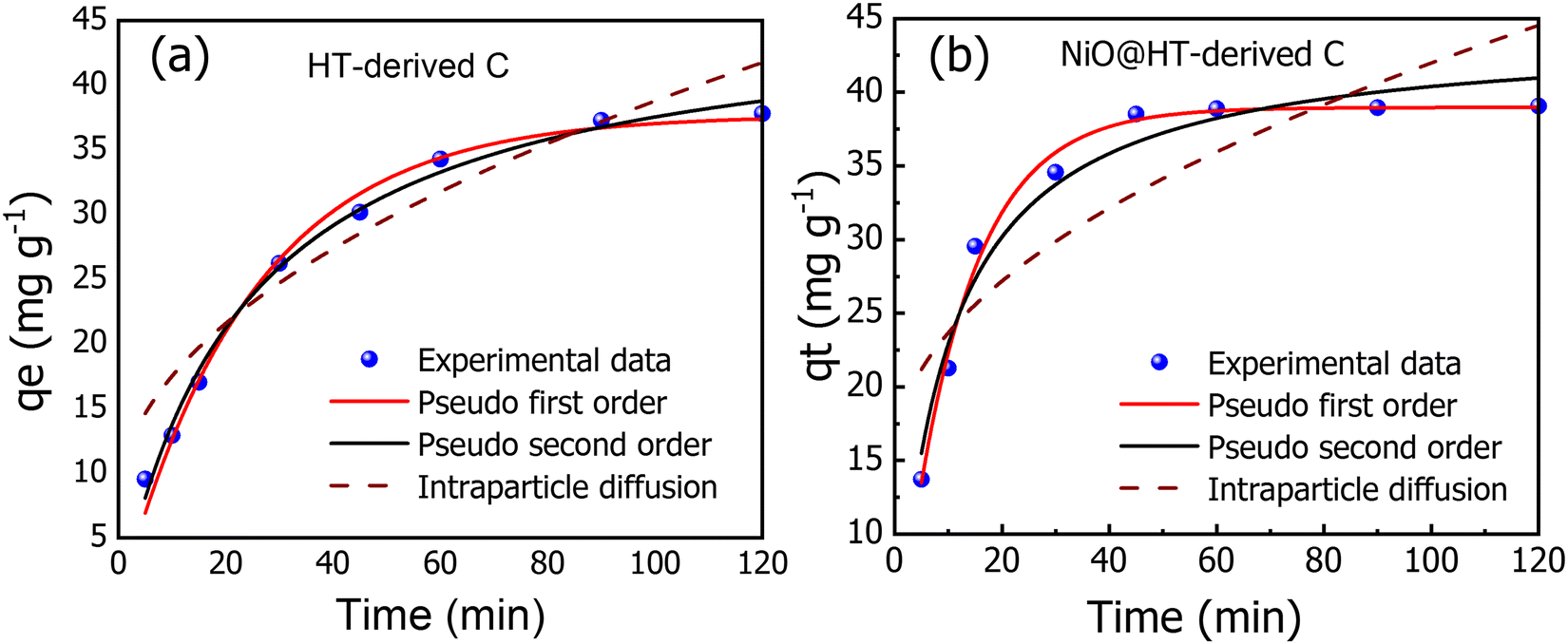 | ||
| Fig. 5 The experimental and kinetic adsorption models for the adsorption of MR onto (a) HT-derived C and (b) NiO@HT-derived C adsorbents [C0 = 50 mg L−1, M = 0.03 g, pH = 3, t = 5–120 min]. | ||
The pseudo-first-order kinetic model is described by eqn (3).53
| qt = qe(1−e−k1t) | (3) |
The pseudo-second-order kinetic model is given by eqn (4).
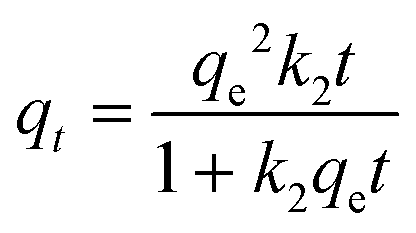 | (4) |
Neither the pseudo-first order nor the second-order model could identify the rate-limiting step. Therefore, an intra-particle mass transfer diffusion model proposed by Weber and Morris was used (eqn (5)).54
| qt = kdifft0.5 + C | (5) |
The related various kinetic parameters and their correlation coefficients are listed in Table 1. It is observed that the experimental data of the adsorbed MR on HT-derived C and NiO@HT-derived C materials were fitted by the pseudo second-order model than the first order kinetic model as indicated by higher R2 values. Significantly, the predicted and experimental adsorption capabilities have a larger affinity for the pseudo second order model.
| Kinetic model | Parameter | HT-derived C | NiO@HT-derived C |
|---|---|---|---|
| Pseudo-first order | q e (mg g−1) | 37.692 | 38.970 |
| K 1 (min−1) | 0.041 | 0.085 | |
| R 2 | 0.987 | 0.9905 | |
| Pseudo-second order | q e(mg g−1) | 46.477 | 44.123 |
| K 2 (g mg−1 min−1) | 0.00091 | 0.00245 | |
| R 2 | 0.932 | 0.969 | |
| Intra-particle diffusion | K diff (mg g−1 min−0.5) | 3.113 | 2.679 |
| C (mg g−1) | 3.666 | 15.194 | |
| R 2 | 0.881 | 0.793 |
The estimated qe values for the pseudo-second-order model were 46.477 and 44.123 mg g−1, for HT-derived C and NiO@HT-derived C, respectively, which are higher than those estimated for the pseudo-first order model as shown in Table 1. It is clearly observed from the fitted data in Fig. 5, and related parameters in Table 1, that the R2 values of the pseudo-first- and the pseudo second-order models displays good correlation with the experimental data, which indicate the presence of both physical adsorption and chemical adsorption. However, the higher R2 values confirmed that the adsorption process predominantly followed the pseudo-second-order kinetic model. It is significant to note that both physisorption and chemisorption may take place concurrently on the surface of an adsorbent, with one layer of molecules being physically adsorbed on top of the other.55
Adsorption processes are often governed by a variety of mechanisms, the most limiting of which are diffusion mechanisms, such as external diffusion and intra-particle diffusion.56 As a result, the intra-particle diffusion model was investigated to estimate the rate-controlling step of the adsorption process. The intraparticle diffusion model for MR onto HT-derived C and NiO@HT-derived C adsorbents demonstrated the least fitting to experimental kinetic data. However, the plots did not also pass through the origin, implying that the adsorption process involves intra-particle diffusion rather than a sole rate-controlling process.12 Thus external mass transfer and intra-particle diffusion can be used to manage the entire adsorption process. From the above discussion, it can be concluded that the pseudo-second-order model is the best model to fit the experimental data for adsorption of MR onto both materials for HT-derived C and NiO@HT-derived C adsorbents. This indicates that the rate limiting step for MR is probably the chemisorption process, thus, involving exchange of electrons between the absorbent and adsorbate.55
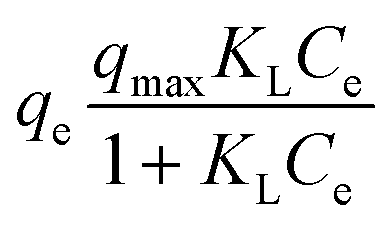 | (6) |
The Freundlich model is another model that can be employed to describe heterogeneous surfaces and multilayer adsorption systems, and its linearized form can be given by eqn (7).54
| qe = KfCe1/n | (7) |
As shown in Fig. 6(a and b), and Table 2, the experimental data of MR adsorbed onto HT-derived C and NiO@HT-derived C composite materials can be fitted by Langmuir and Freundlich isotherms. However, the experimental data were best described by the Langmuir isotherm model as indicated by higher R2 and lower χ2 values. This suggests that an MR dye monolayer has formed on a surface that is uniformly covered with localized adsorption sites, which was also reported in red 14 adsorption onto activated carbon derived from cherry tree waste.58
 | ||
| Fig. 6 The experimental and isotherm adsorption isotherm models (Langmuir and Freundlich) for adsorption of MR onto (a) HT-derived C and (b) NiO@HT-derived C materials [C0 = 10–200 mg L−1, M = 0.03 g, pH = 3, time =45 min]. | ||
| Isotherm model | Parameter | HT-derived C | NiO@HT-derived C |
|---|---|---|---|
| Langmuir | q m (mg g−1) | 101.321 | 134.075 |
| K L (L mg−1) | 0.0654 | 0.118 | |
| Adj. R2 | 0.988 | 0.989 | |
| χ 2 | 9.10 | 14.586 | |
| Freundlich | K f (L g−1) | 16.975 | 28.300 |
| N | 2.652 | 2.724 | |
| Adj. R2 | 0.935 | 0.898 | |
| χ 2 | 48.873 | 140.243 |
The Langmuir constant KL is usually used to describe the interaction between the adsorbate and absorbent. As listed in Table 2, the KL value for MR adsorption onto NiO@HT-derived C is 0.118 L mg−1, which is much higher than that for MR onto HT-derived C (0.065 L mg−1). This indicates that the interactions between MR and NiO@HT-derived C are more stable than the interactions between MR and the HT-derived C. In other words, the sorbent NiO@HT-derived C has a greater affinity for MR than HT-derived C.
The estimated maximum adsorption capacities (mg g−1) of MR onto HT-derived C and NiO@HT-derived C composites are 101.321 and 134.075 mg g−1, respectively. The higher value of the maximum adsorption capacity for MR onto the NiO@HT-derived C composite than that for MR onto HT-derived C can be attributed to the higher surface area, and due to the presence of NiO nanoparticles which act as active centers for adsorption.
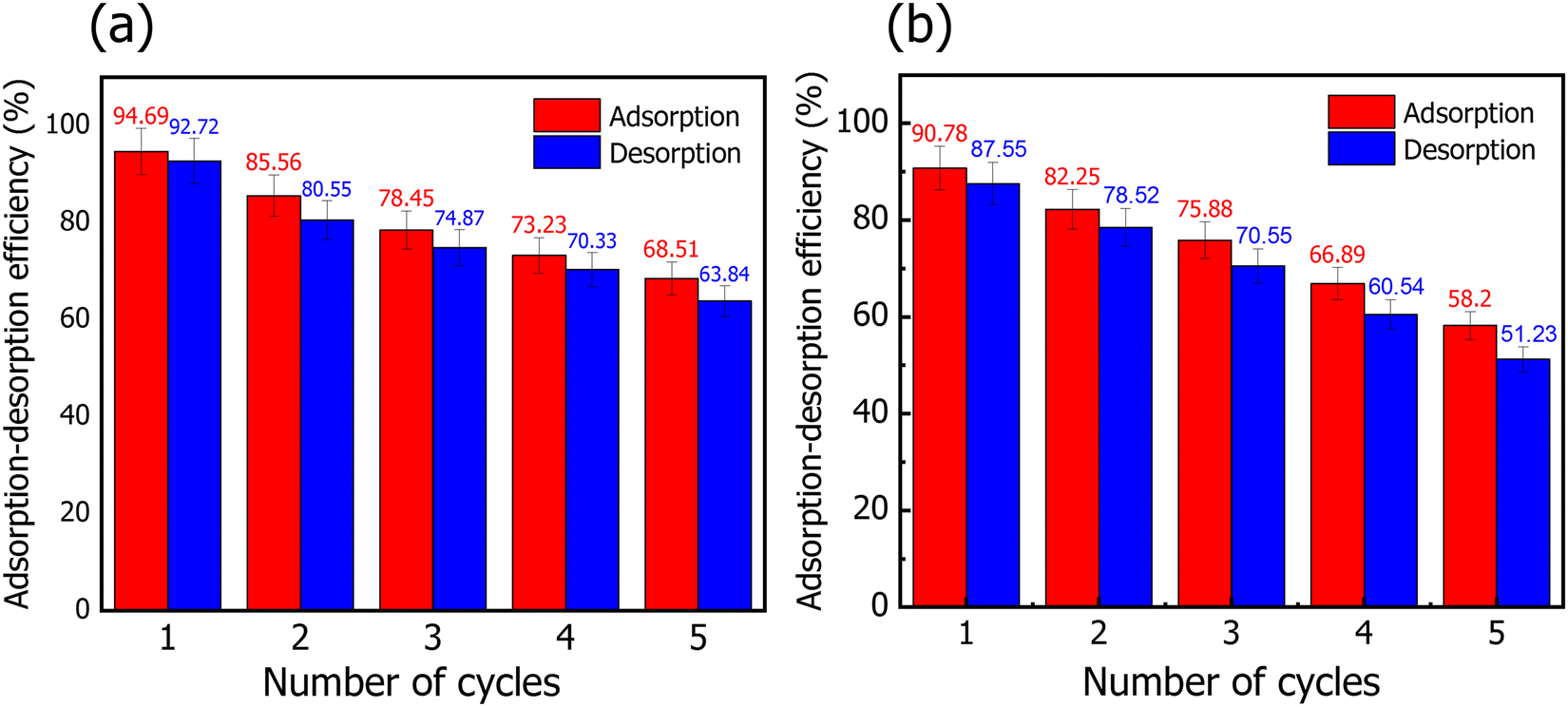 | ||
| Fig. 7 Adsorption–desorption cycles of the MR dye onto (a) NiO@HT-derived C and (b) HT-derived C adsorbents. | ||
4. Conclusions
In conclusion, a series of experiments for the absorption of MR onto HT-derived C and NiO@HT-derived C have been reported. It should be noted that the maximum adsorption capacity for MR onto NiO@HT-derived C is found to be 129.87 mg g−1 at pH 3. This value is much higher than that achieved by HT-derived C (82.82 mg g−1). The equilibrium data are studied using the Langmuir and Freundlich isotherm models. Although both the Langmuir and Freundlich isotherms could be used to describe the adsorption behaviour with a high correlation coefficient, the Langmuir model fits the experimental data better. In addition, the kinetic mechanism is studied by using the pseudo first order, pseudo second order, and intra-particle diffusion models. The obtained results reveal good agreement between the experimental data and the pseudo second order kinetic model. This reveals a chemical adsorption process in which the MR adsorption over NiO@HT-derived C and HT-derived C can be explained to be through electrostatic attraction and π–π interactions. Furthermore, the recyclability tests indicate a good capability of reusing NiO@HT-derived C and HT-derived C for many cycles with no significant reduction in the adsorption capacity, revealing high ability for practical applications.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- C. Grégorio, Bioresour. Technol., 2006, 97, 1061–1085 CrossRef.
- Z. Yanbo, L. Jian, Z. Yi and L. Yongdi, Environ. Pollut., 2019, 252, 352–365 CrossRef PubMed.
- A. J. Sunil Kumar Bajpai, Water, 2012, 4, 52–57 Search PubMed.
- K. Sushila, A. Sharma, S. C. Sahoo, G. Kumar, S. K. Mehta and R. Kataria, J. Mol. Struct., 2021, 1226, 129327 CrossRef.
- C. I. Pearce, J. R. Lloyd and J. T. Guthrie, Dyes Pigm., 2003, 58, 179–196 CrossRef CAS.
- Y. Zhou, J. Wang, Q. Zhao, H. Cai and H. Zhang, ChemistrySelect, 2022, 7, e202203280 CAS.
- R. Sunil, B. Swapnil and S. Virendra Kumar, Ultrason. Sonochem., 2017, 34, 183–194 CrossRef PubMed.
- V. Selvaraj, T. Swarna Karthika, C. Mansiya and M. Alagar, J. Mol. Struct., 2021, 1224, 129195, DOI:10.1016/j.molstruc.2020.129195.
- A. Mohd Azmier, A. Nur‘Adilah Binti, A. Kayode Adesina and B. Olugbenga Solomon, Chem. Data Collect., 2019, 22, 100249 CrossRef.
- M. A. Ahmad, N. Ahmad and O. S. Bello, Appl. Water Sci., 2014, 5, 407–423 CrossRef.
- Y. Badr, M. G. Abd El-Wahed and M. A. Mahmoud, J Hazard. Mater., 2008, 154, 245–253 CrossRef CAS PubMed.
- R. Sunil, S. Virendra Kumar, P. Avdesh Singh, N. Mohit and R. Kuldeep, Curr. Res. Green and Sustainable Chem., 2021, 4, 100180 CrossRef.
- M. Riera-Torres, C. Gutiérrez-Bouzán and M. Crespi, Desalination, 2010, 252, 53–59 CrossRef CAS.
- G.-M. Julia, R. Nilbia, M. Iván, D. Xavier, A. G.-H. José, T. Francesc and P. José, J. Hazard. Mater., 2006, 138, 218–225 CrossRef PubMed.
- W. Jiangning, A. E. Mark and S. E. Law, J. Environ. Eng., 1998, 124, 272–277 CrossRef.
- S. Venkata Mohan, P. Sailaja, M. Srimurali and J. Karthikeyan, Environ. Eng. Policy, 1998, 1, 149–154 CrossRef.
- C.-H. Wu and J.-M. Chern, Indust. Eng. Chem. Res., 2006, 45, 6450–6457 CrossRef CAS.
- K. Equbal Ahmad Shahjahan and K. Tabrez Alam, J. Mol. Liq., 2018, 249, 1195–1211 CrossRef.
- K. Amel, B. Esma Mahfouf, D. Kerroum, B. Souheila, C. Bouchra and B. Raouf, Cleaner Chem. Eng., 2022, 4, 100069 CrossRef.
- S. Gul, M. Kanwal, R. A. Qazi, H. Gul, R. Khattak, M. S. Khan, F. Khitab and A. E. Krauklis, Water, 2022, 14, 2831 CrossRef CAS.
- N. M. Hosny, I. Gomaa and M. G. Elmahgary, Appl. Surf. Sci. Adv., 2023, 15, 100395 CrossRef.
- Z. Muhammad Nadeem, D. Qamar, N. Faisal, Z. Muhammad Naveed, I. Munawar and N. Muhammad Faizan, J. Mater. Res. Technol., 2019, 8, 713–725 CrossRef.
- A. S. A. Ahmed, W. Xiang, M. Abdelmotalleib and X. Zhao, ACS Appl. Electron. Mater., 2022, 4, 1063–1071 CrossRef CAS.
- N. Arailym, C. Moon Ho, K. Jik-Soo, M. Seung-Taek, K. Sung-Soo and S. Yang-Kook, J. Power Sources, 2015, 299, 425–433 CrossRef.
- Y. Abdelbaki, A. de Arriba, R. Issaadi, R. Sánchez-Tovar, B. Solsona and J. M. López Nieto, Fuel Process. Technol., 2022, 229, 107182 CrossRef CAS.
- Q. Riaz, M. Ahmed, M. N. Zafar, M. Zubair, M. F. Nazar, S. H. Sumrra, I. Ahmad and A. Hosseini-Bandegharaei, Int. J. Environ. Anal. Chem., 2020, 102, 84–103 CrossRef.
- T. Zhu, J. S. Chen and X. W. Lou, J. Phys. Chem. C, 2012, 116, 6873–6878 CrossRef CAS.
- M. A. El-Ghobashy, H. Hashim, M. A. Darwish, M. U. Khandaker, A. Sulieman, N. Tamam, S. V. Trukhanov, A. V. Trukhanov and M. A. Salem, Nanomaterials, 2022, 12 Search PubMed.
- M. R. Al-Bahrani, W. Ahmad, H. F. Mehnane, Y. Chen, Z. Cheng and Y. Gao, Nanomicro Lett., 2015, 7, 298–306 CAS.
- S. Joshi, S. Shrestha and B. Shrestha, Chem. Eng. Technol., 2021, 44, 2327–2332 CrossRef CAS.
- H. N. Hamad and S. Idrus, Polymers, 2022, 14 Search PubMed.
- A. S. Al-Wasidi, I. I. S. AlZahrani, H. I. Thawibaraka, A. M. Naglah, M. G. El-Desouky and M. A. El-Bindary, J. Mol. Struct., 2022, 1250, 131736 CrossRef CAS.
- E. G. Magdi, Z. Nana, Y. Yanbin, L. Pansheng, W. Shoujuan, T. Tamrat and K. Fangong, J. Cleaner Prod., 2020, 267, 121949 CrossRef.
- M. Patange, S. Biswas, A. K. Yadav, S. N. Jha and D. Bhattacharyya, Phys. Chem. Chem. Phys., 2015, 17, 32398–32412 RSC.
- H. Wang, Y. Zhang, Q. Wang, C. Jia, P. Cai, G. Chen, C. Dong and H. Guan, RSC Adv., 2019, 9, 9126–9135 RSC.
- F. Yang, Y. He, S. Sun, Y. Chang, F. Zha and Z. Lei, J. Appl. Poly. Sci., 2016, 133 Search PubMed.
- D. Cholico-Gonzalez, N. Ortiz Lara, A. M. Fernandez Macedo and J. Chavez Salas, ACS Omega, 2020, 5, 3302–3314 CrossRef CAS.
- N. Azouaou, Z. Sadaoui, A. Djaafri and H. Mokaddem, J. Hazard. Mater., 2010, 184, 126–134 CrossRef CAS PubMed.
- W. Liu, C. Lu, X. Wang, K. Liang and B. K. Tay, J. Mater. Chem. A, 2015, 3, 624–633 RSC.
- N. Jayababu, M. Poloju, J. Shruthi and M. V. R. Reddy, RSC Adv., 2019, 9, 13765–13775 RSC.
- A. A. L. M. I. Chebanenko, V. N. Nevedomskiy and V. I. Popkov, Dalton Trans., 2020, 49, 12088–12097 RSC.
- H. Frost, T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2006, 110, 9565–9570 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87 Search PubMed.
- K. Mensah, H. Mahmoud, M. Fujii, M. Samy and H. Shokry, Biomass Convers. Biorefin., 2022 DOI:10.1007/s13399-022-03304-4.
- S. Hassan, E. Marwa and H. Hesham, J. Mater. Res. Technol., 2019, 8, 4477–4488 CrossRef.
- P. S. S. Venkata Mohan, M. Srimurali and J. Karthikeyan, Environ. Eng. Pol., 1999, 1, 149–154 CrossRef.
- N. Hassan, A. Shahat, A. El-Didamony, M. G. El-Desouky and A. A. El-Bindary, J. Mol. Struct., 2020, 1210, 128029 CrossRef CAS.
- C. K. Enenebeaku, N. J. Okorocha, U. E. Enenebeaku and I. C. Ukaga, Inter. Lett. Chem. Phys. Astron., 2017, 72, 52–64 Search PubMed.
- M. F. Elkady, M. I. Amal and M. M. A. El-Latif, Desalination, 2011, 278, 412–423 CrossRef CAS.
- S. Dadfarnia, A. M. S. E. Moradi and S. Emami, Appl. Surf. Sci., 2015, 330, 85–93 CrossRef CAS.
- D. Mehmet and A. Mahir, Chemosphere, 2003, 50, 517–528 CrossRef PubMed.
- Y. Liu, C. Luo, J. Sun, H. Li, Z. Sun and S. Yan, J. Mater. Chem. A, 2015, 3, 5674–5682 RSC.
- H. Y. Zhu, R. Jiang, L. Xiao and G. M. Zeng, Bioresour. Technol., 2010, 101, 5063–5069 CrossRef CAS PubMed.
- C. G. Ramos, S. A. Sousa, A. M. Grilo, J. R. Feliciano and J. H. Leitão, J. Bacteriol., 2014, 196, 3980 CrossRef CAS PubMed.
- A. Denizli, R. Say and Y. Arica, Sep. Purifi. Technol., 2000, 21, 181–190 CrossRef CAS.
- A. Ozcan, E. M. Oncü and A. S. Ozcan, J. Hazard. Mater., 2006, 129, 244–252 CrossRef PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- R. Askari, F. Mohammadi, A. Moharrami, S. Afshin, Y. Rashtbari, M. Vosoughi and A. Dargahi, Appl. Water Sci., 2023, 13, 90 CrossRef CAS.
- N. Ghosh, S. Sen, G. Biswas, A. Saxena and P. K. Haldar, Water, Air, Soil Pollut., 2023, 234, 202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00226h |
| This journal is © The Royal Society of Chemistry 2023 |