
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum nanoclusters made by gas-diffusion electrocrystallization (GDEx) as electrocatalysts for methanol oxidation†
Omar
Martinez-Mora
 a,
Luis F.
Leon-Fernandez
b,
Milica
Velimirovic
b,
Frank
Vanhaecke
c,
Kristof
Tirez
b,
Jan
Fransaer
a and
Xochitl
Dominguez-Benetton
*b
a,
Luis F.
Leon-Fernandez
b,
Milica
Velimirovic
b,
Frank
Vanhaecke
c,
Kristof
Tirez
b,
Jan
Fransaer
a and
Xochitl
Dominguez-Benetton
*b
aDepartment of Materials Engineering, Surface and Interface Engineered Materials, Katholieke Universiteit Leuven, Kasteelpark Arenberg 44 – box 2450, 3001 Leuven, Belgium
bSeparation and Conversion Technologies, VITO, Flemish Institute for Technological Research, Boeretang 200, 2400, Mol, Belgium. E-mail: xoch@vito.be
cAtomic & Mass Spectrometry—A&MS Research Group, Department of Chemistry, Ghent University, Campus Sterre, Krijgslaan 281-S12, 9000 Ghent, Belgium
First published on 11th September 2023
Abstract
The development of high-performance electrocatalysts is critical for enhancing the performance and commercial viability of direct methanol fuel cells (DMFCs), which hold the potential to transform the way we power portable electronics and off-grid systems. In this study, we have employed the gas-diffusion electrocrystallization process (GDEx) at room temperature to synthesize platinum nanoclusters (NCs), using different concentrations of polyvinylpyrrolidone (PVP) to stabilize the NPs. The morphology, structure, and composition of the Pt NCs were characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), Single-particle inductively coupled plasma-sector field mass spectrometry (spICP-SFMS) and X-ray diffraction (XRD). Moreover, we assessed the electrocatalytic activity of the Pt NCs for methanol oxidation in both acidic and alkaline media. TEM and SEM analyses revealed Pt NCs of 30 nm–60, composed of much smaller primary nanoparticles with a diameter ranging from 2–4 nm. PVP played a crucial role in preventing diffusion limited aggregation of the Pt NCs. PVP-stabilized GDEx-made Pt NCs demonstrated superior electrocatalytic activity for methanol oxidation compared to aggregated Pt NCs and commercial Pt/C, which can be attributed to the porous structure of the Pt NCs, resulting in a high effective surface area. This study underscores the potential of the GDEx process as a simple and efficient strategy for synthesizing nanomaterials with remarkable catalytic activity and stability for electrochemical energy applications such as direct methanol fuel cells.
Introduction
The growing demand for energy production and the need to replace fossil fuels as a primary source of electric power have prompted the scientific community to explore renewable and green alternatives.1 In this context, fuel cells have emerged as promising technologies for energy generation, offering high energy conversion efficiency while remaining environmentally friendly.2 Direct methanol fuel cells (DMFCs) stand out as a promising alternative to their counterparts (i.e., proton exchange membrane fuel cells, PEMFCs) due to their cost-effectiveness, simplified systems and high energy density.3The core concept underlying DMFCs is centred on situating the methanol oxidation reaction (MOR) at the anode and the oxygen reduction reaction (ORR) at the cathode. At 25 °C the thermodynamic potential of a DMFC is 1.21 V and the theoretical thermodynamic efficiency of converting methanol could potentially reach up to 96.4%, this efficiency greatly surpasses that of internal combustion engines, for example.4 The biggest promise of DMFCs lies in their potential to serve as efficient, compact, and environmentally friendly energy sources for a variety of portable and remote applications. Their high energy density, low operating temperature, and ability to directly utilize liquid methanol as fuel make them particularly suitable for powering portable electronic devices, remote power generation, and backup power systems.5 DMFCs have the potential to revolutionize the way we power portable electronics and off-grid systems, offering longer runtimes, quick refuelling, and reduced environmental impact compared to conventional batteries.5 Additionally, their quiet operation and ease of use make them attractive for military and emergency response applications. The use of methanol as a liquid fuel, which facilitates their transportation and storage, and their low temperatures operation (i.e., 40–80 °C), make them particularly attractive for these applications.3
However, to fully realize the potential of DMFCs several technological challenges need to be addressed, including methanol crossover and, especially, the performance of the electrocatalysts due to their high production costs and the sluggish kinetics of the methanol oxidation reaction (MOR).6 The MOR involves a six-electron transfer and follow different pathways in acidic and alkaline electrolytes. Ideally, six electrons are exchanged, encompassing steps such as methanol adsorption, dehydrogenation of carbon-containing intermediates (i.e., COH, CHO), and the generation of CO2. However, the formation of CO2 involves intricate processes, with various pathways leading to the generation of multiple intermediaries. Notably, one of the intermediaries, CO, restricts the rate of methanol oxidation at the anode due to its great stability. CO formation arises from an indirect mechanism where COH and CHO are directly dehydrogenated.7,8 For DMFCs utilizing acid-based electrolytes, the reaction are as follows ((1) and (2)):8
| At the anode: CH3OH + H2O → CO2 + 6H+ + 6e− | (1) |
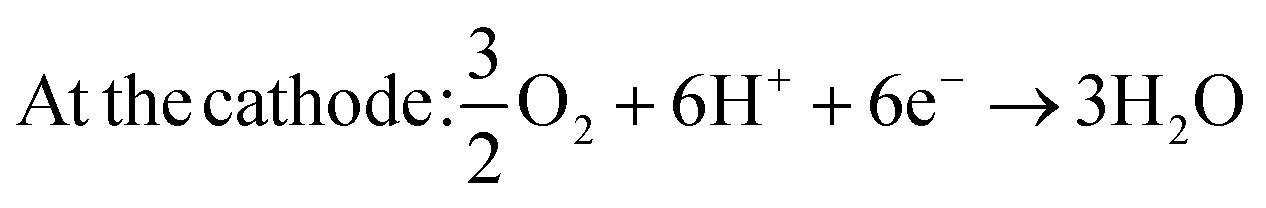 | (2) |
And for DMFCs utilizing alkaline-based electrolytes (reactions (3) and (4)):
| At the anode: CH3OH + 6OH− → CO2 + 5H2O + 6e− | (3) |
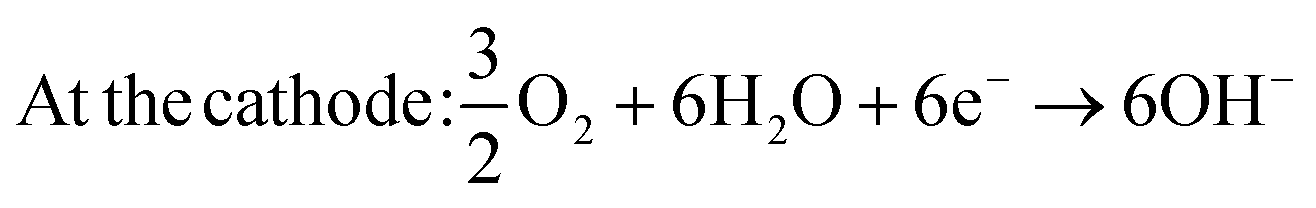 | (4) |
In both scenarios, the overall chemical process can be summarized as (reaction (5)):
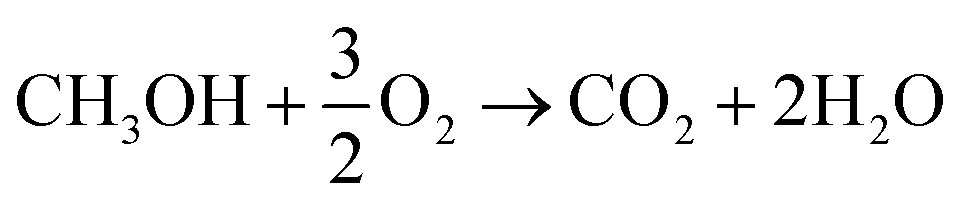 | (5) |
Despite its cost, Pt is widely recognized as the most effective catalyst for DMFCs. However, the CO intermediate formed during the methanol oxidation reaction (MOR) strongly adsorbs onto the Pt surface, blocking active sites and consequently causing a significant decrease in activity.6 In addition to poisoning, typical Pt electrocatalysts may also experience ripening, sintering or dissolution, which contribute to a lack of long-term stability. Furthermore, the use of carbon-based ESI† can lead to severe corrosion under the typical operation conditions of DMFCs, exacerbating stability problems.9 Due to these concerns, there has been a resurgence of interest in using unsupported Pt-based catalysts in recent years.9,10 Unsupported nanostructured Pt catalysts address the issues related to carbon corrosion, enhancing long-term stability. Moreover, unsupported Pt catalysts can form very thin catalytic layers even with significant loadings (i.e., 2 mgPt cm−2), which reduces the ohmic drop while providing sufficient accessible active sites and high electrochemical surface areas (ECSAs).9,10 Moreover, the dimensional and crystalline nature of unsupported Pt catalyst result in excellent chemical and electrochemical stability, making them less susceptible to aggregation and dissolution.11 Consequently, synthesis protocols have shifted focus from reducing particle size to developing nanostructures with specific geometries, shapes and compositions that boast high surface area and/or specific activity, ultimately increasing catalyst utilization.1 Different Pt nanostructures with a variety of shapes and morphologies have been explored as efficient electrocatalysts for MOR, such as nanotubes,11,12 nanowires,13,14 nanodendrites,15,16 nanoflowers,17 nanoporous structures,18 or nanocubes.19
In line with these developments, we have recently reported a method for synthesizing platinum group metal (PGM, i.e., Pt, Pd, Rh) nanoparticles (NPs), using a process called Gas Diffusion Electrocrystallization (GDEx).20 In this process, the simultaneous electrochemical reduction of CO2 and water occurs in uncatalyzed gas-diffusion electrodes, producing H2 and CO. Both gases, particularly H2, act as reducing agents for water-soluble noble metal ions, leading to the formation of small metal nanoclusters.21 CO can also serve as a capping agent.22 Furthermore, the presence of CO2 stabilizes the pH, avoiding the formation of metal (hydr)oxides.
In this work, we have used the GDEx process to synthesize unsupported Pt nanoclusters using polyvinylpyrrolidone (PVP, 55 kDa) as a stabilizer and evaluated their electrocatalytic activity for methanol oxidation. The synthesis was conducted at −30 mA cm−2, using 3.0 mM Pt4+ (as H2PtCl4) as the metal precursor and different concentrations of PVP (i.e., 0.0, 0.01, 0.1 and 1.0 g L−1). We opted for low concentrations of stabilizer to facilitate its removal after synthesis, as clean catalysts are necessary for electrocatalytic applications.23 The resulting catalysts were Pt NCs measuring 30–60 nm in size, composed of aggregates of smaller NPs of 2–5 nm in diameter. The electrochemical activity of the synthesized Pt NCs for methanol oxidation was investigated in both acidic and alkaline media. The GDEx-made Pt nanomaterials exhibited excellent electrocatalytic activity for the MOR in both media and remarkable stability in acidic media, suggesting that the Pt NCs synthesized with the GDEx process hold potential for applications as catalysts for DMFCs.
Experimental
Materials
Hexachloroplatinic acid (H2PtCl6) solution was supplied by Johnson-Matthey Co. Polyvinylpyrrolidone (PVP, 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mm), sodium hydroxide (NaOH), potassium hydroxide (KOH), and sulfuric acid (H2SO4) were obtained from Sigma-Aldrich. Nafion solution (5 wt%), sodium chloride (NaCl), and methanol (MeOH) were purchased from VWR. Carbon dioxide (CO2) and Argon (Ar) were obtained from Air Liquide. All chemicals were of analytical grade and used without further purification.
000 Mm), sodium hydroxide (NaOH), potassium hydroxide (KOH), and sulfuric acid (H2SO4) were obtained from Sigma-Aldrich. Nafion solution (5 wt%), sodium chloride (NaCl), and methanol (MeOH) were purchased from VWR. Carbon dioxide (CO2) and Argon (Ar) were obtained from Air Liquide. All chemicals were of analytical grade and used without further purification.
Electrochemical setup
The electrochemical setup employed in the GDEx process has been previously described.24 In brief, the reactor consisted of an EC Micro Flow Cell (ElectroCell), using a three-compartment configuration. A PTFE-bonded cold-rolled gas diffusion electrode (GDE) (VITO CORE®) was used as a cathode supported in a titanium frame acting as a current collector.25 A leak-free Ag/AgCl reference electrode (eDAQ) was placed in the cathodic chamber near the GDE. A platinum-coated tantalum plate electrode (Pt 10 μm thickness) was used as an anode. This anode generated mostly Cl2 and some O2, which had a negligible influence on the electrochemical (cathodic) process and products of interest. Both, the cathodic and anodic chambers had an exposed area of 10 cm2, a thickness of 0.4 cm, and a total volume of 4 mL. Both compartments were separated by an anion exchange membrane (FUMASEP® FAP-4130-PK). The supporting electrolyte in all cases was 0.5 M NaCl. CO2 was supplied at the hydrophobic (back) side of the GDE at a flow rate of 200 mL min−1 with an overpressure of 20 mbar(g).Synthesis of Pt nanoparticles using the GDEx process
A background electrolyte of 0.5 M NaCl was used to prepare all solutions. The catholyte was prepared by mixing PVP (0 g L−1, 0.01 g L−1, 0.1 g L−1 or 1.0 g L−1) and H2PtCl6 (3 mM) with the background electrolyte, and the pH was adjusted to 3 using concentrated HCl or NaOH. The background electrolyte was used as the anolyte. 100 mL of the catholyte and anolyte were added to two 3-necked glass bottles and connected to the electrochemical reactor using marprene tubing (Watson-Marlow). The solutions were pumped to their respective chambers at a flow rate of 100 mL min−1 using a peristaltic pump (530, Watson-Marlow). The solutions and gases were flushed through the cell during 30 minutes before starting the experiments (without electrode polarization) to ensure sufficient wetting of the electrode. Chronopotentiometric (CP) experiments were carried out in batch mode at −30 mA cm−2 using an AUTOLAB (PGSTAT302N) potentiostat. pH, charge, and potential were monitored throughout all experiments. The pH was measured every 5 seconds with a Metrohm 781 pH per ion meter equipped with a Metrohm Unitrode pH electrode. To monitor the Pt concentration in the liquid phase, aliquots of 1 mL were taken from the catholyte at different times, centrifuged, and analyzed with an inductively coupled plasma-optical emission spectrometer (ICP-OES) (Varian 750 ES). The experiments were run until a charge of 10000 C L−1 was consumed.
The different Pt NPs suspensions were processed as follows: for material characterization, 50 mL of the Pt NCs suspensions were dialyzed using a dialysis membrane (VWR, 12–14 kD). This process was repeated until no chlorides could be detected in the solution using AgNO3 solution. For the electrocatalytic tests, 50 mL of the Pt NCs suspensions were treated with NaOH to remove PVP from the surface of the Pt NCs following the protocol of Vidal-Iglesias et al.26 In brief, NaOH pellets were added to the Pt NCs suspension until the pH was ∼14, so that PVP is desorbed from the particles. The suspensions turn then unstable and the Pt NCs naturally settle.27 The supernatant was discarded, and the Pt NCs were washed with deionized water until the pH was ∼7. Finally, the Pt NCs were resuspended in deionized water. The Pt concentration in the different Pt NCs suspensions was measured with ICP-OES after digestion with aqua regia. All Pt NPs dispersions were kept in closed glass vessels and stored in the dark until further processing.
Material characterization
The morphology of the Pt NPs was revealed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). SEM images were obtained using a Philips XL30 FEG scanning electron microscope, images were taken with secondary electrons and an acceleration voltage of 30 kV. The samples were prepared by placing two drops of the dialyzed suspensions on an aluminium foil mounted on a sample holder. TEM images were recorded using JEOL JEM-2200FS FEG high resolution transmission electron microscope (TEM/STEM) operated at a voltage of 200 kV. Samples for analyses were prepared by direct deposition on holey carbon supported copper grids (200 mesh). Single-particle inductively coupled plasma-sector field mass spectrometry (spICP-SFMS; AttoM ES, Nu Instruments Ltd) was used to study particle size distribution. Instrumental details and operating conditions are summarized in Table S1 (SI-I) (ESI†). Data acquisition and data treatment were performed using the combination of NuAttoLab and NuQuant software (Nu Instruments Ltd) X-ray diffraction analysis (XRD) was used to determine the crystal structure on a Seifert 3003 T/T diffractometer operated at a voltage of 40 kV and a current of 40 mA with Cu Kα radiation (λ = 1.5431 Å). The data were collected in the 20°–120° (2θ) range with a step size of 0.05°, spending 1 s per step. Single-particle inductively coupled plasma-sector field mass spectrometry.Electrochemical characterization
The electrocatalytic activity of the different Pt NPs was characterized using a three-electrode system on a Bio-Logic (VMP3) multichannel potentiostat. A Pt coil (∼3.6 cm2) was used as a counter electrode, a Ag/AgCl (3 M KCl) electrode was used as a reference electrode, and a glassy carbon electrode (GCE, 3.0 mm diameter) served as the working electrode. The GCE was polished with Al2O3 (0.05 μm) and rinsed with Millipore Milli-Q® deionized water. The Pt NCs suspensions (treated with NaOH) were sonicated for 30 min, and 4–10 μL of the suspensions were deposited onto the GCE and left to dry at room temperature. Following this, 5 μL of Nafion 0.5 wt% were added on top of the GDE and let dry on air overnight to ensure catalyst adhesion. The Pt loading was controlled to be ∼60 μg cm−2. Cyclic voltammograms (CV) were acquired with or without 1.0 M MeOH in a potential window from 0.0 V to 1.2 VRHE in Ar-saturated 0.5 M H2SO4 or from 0.05 V to 1.45 VRHE in Ar-saturated 1.0 M KOH at a scan rate of 50 mV s−1 until a steady behaviour was obtained. Chronoamperometry (CA) experiments were conducted by monitoring the current over a period of 3600 s while holding the potential constant at 0.9 VRHE in Ar-saturated 0.5 M H2SO4 or at 0.75 VRHE in Ar-saturated 1.0 M KOH containing 1.0 M MeOH. The accelerated durability tests (ADT) were carried out using CVs for 1000 cycles at 50 mV s−1 from 0.0 V to 1.2 VRHE in Ar-saturated 0.5 M H2SO4 and 1.0 M MeOH or from 0.05 V to 1.45 VRHE in Ar-saturated 1.0 M KOH and 1.0 M MeOH. To convert the potentials measured against the Ag/AgCl electrode (EAg/AgCl) to values against the reversible hydrogen electrode (RHE) (ERHE) eqn (6) was used.| ERHE = EAg/AgCl + 0.059pH + E0Ag/AgCl | (6) |
Results and discussion
Synthesis and characterization of Pt nanomaterials by GDEx
In previous work, the synthesis of PGMs NPs (i.e., Pt, Pd, Rh) using the CO2-GDEx process was introduced.20 In this work, we focused only on the synthesis of the Pt NCs. The GDEx process is, in general, carried out in absence of additives except for NaCl, which acts as supporting electrolyte, and the targeted metals ions. However, to obtain stable colloidal nanoparticle suspensions, we need to add a stabilizing agent (i.e., polyvinylpyrrilidone, PVP) to protect the Pt NPs from aggregation. The reasoning behind selecting PVP the stabilizer arises from several pivotal factors. First, the mechanism of the stabilization of Pt NPs by PVP is well-established. PVP binds to Pt surfaces by interacting with the carbonyl group or nitrogen atom in the pyrrolidone ring of the repeating unit, which facilitates the creation of a protective shell around the NPs. This protective layer effectively curbs aggregation tendencies mainly by steric hindrance, thus preserving the individuality of the NPs. Furthermore, PVP exhibits high solubility in aqueous media, aligning well with our synthesis conditions. Besides, unlike stabilizers such as sodium citrate or ethylene glycol, which possess both, stabilizing and reducing properties, PVP is characterized by a weak reducing capability.28 We noted that the solutions containing Pt ions and PVP remained stable before the synthesis process, thereby eliminating the possibility of PVP serving as a reducing agent. In the context of the GDEx process, where the reducing and caping agents (H2 and CO) are generated electrochemically in situ, the role of PVP is solely focused in stabilizing the Pt NPs. Our goal was to use the minimum amount of PVP required to stabilize the suspension to facilitate the cleaning process afterwards. For this reason, we synthesized different Pt NPs using concentrations of PVP of 0 g L−1 (Pt-PVP-0), 0.01 g L−1 (Pt-PVP-001), 0.1 g L−1 (Pt-PVP-01), and 1.0 g L−1 (Pt-PVP-1). The molar ratios of Pt to PVP were 30:1, 3:1, and 1:3, for Pt-PVP-001, Pt-PVP-01, and Pt-PVP-1, respectively.
In Fig. 1, the evolution of pH and Pt concentration of the different samples as a function of the volumetric charge are shown, and the chronopotentiometric curves are shown in Fig. S1 (ESI-I) (ESI†). A blank experiment using only the background electrolyte is added for comparison. For all experiments, the variation of the pH vs. charge is similar, suggesting that the addition of PVP in the solution has little impact on the electrochemical process. The reduction of CO2 to CO (reaction (7)) and of water to H2 (reaction (8)) occurring at the GDE during the synthesis process, produce OH− ions,29 leading to an increase in the catholyte pH, from pH 3 (starting conditions) to 9.0–9.5 at which the pH is buffered. Furthermore, a small plateau between pH 6 and 7 is observed when Pt ions are present in solution. The length of this plateau increases with the concentration of PVP in the solution.
| CO2 + H2O + 2e− ⇌ CO + 2OH− E0 = −0.934 VSHE | (7) |
| 2H2O + 2e− ⇌ H2 + 2OH− E0 = −0.828 VSHE | (8) |
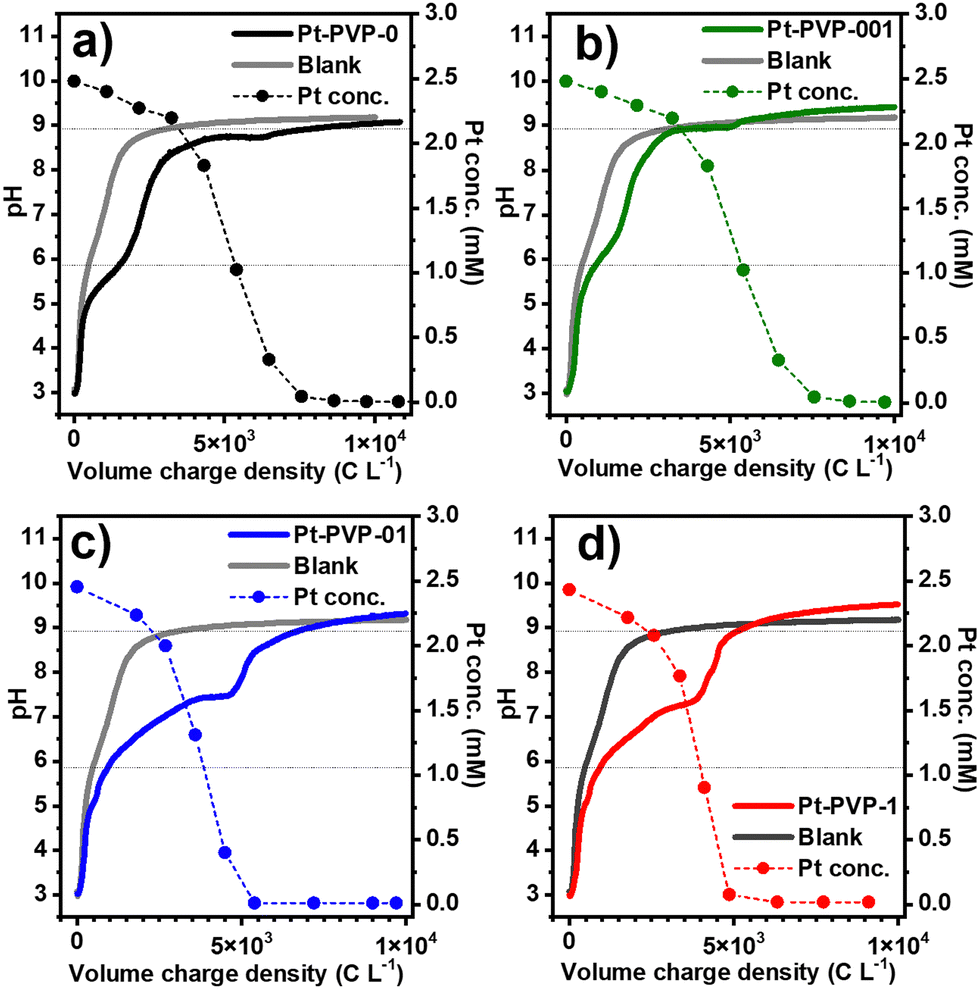 | ||
| Fig. 1 Evolution of pH and concentration of dissolved Pt ions as a function of the charge consumed per unit volume of catholyte, throughout the GDEx process with (a) Pt-PVP-0, (b) Pt-PV-001, (c) Pt-PVP-01 and (d) Pt-PVP-1. The dashed lines indicate the pKa's of CO2 equilibrium in NaCl 0.5 M. | ||
From Fig. 1, it can also be seen that the concentration of Pt in solution decreased slowly at the beginning, followed by a fast drop, while the initially yellow solutions progressively turned black. This behaviour agrees with what we previously observed20 and indicates that the reduction of Pt4+ ions to Pt0 by H2 occurs in two-steps (reactions (9) and (10)).
| [PtCl6]2− + H2 → [PtCl4]2− + 2H+ + 2Cl− | (9) |
| [PtCl4]2− + H2 → Pt0 + 2H+ + 4Cl− | (10) |
During the reduction of Pt ions, H+ is formed, which reacts with the OH− from the reduction of CO2 or H2O, explaining the small pH plateau which is not present in the blank experiment. The final pH is related to the dissolution of CO2 in the catholyte (reactions (11) and (12)) that reacts with OH−, resulting in the buffering of the pH at a value close to the pKa of HCO3−/CO32− in 0.5 M NaCl:30
CO2 + OH− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) HCO3− pKa = 5.86* HCO3− pKa = 5.86* | (11) |
| HCO3− + OH− CO32− + H2O pKa = 8.92* | (12) |
The physical appearance of the different Pt dispersions after synthesis is shown in Fig. S2 (ESI-I†). The agglomeration and sedimentation of Pt-PVP0 and Pt-PVP001 dispersions are observed after the synthesis procedure is finished. For the case of Pt-PVP-01 and Pt-PVP-1, stable dispersions were obtained, showing that a PVP concentration as low as 0.1 g L−1 is sufficient to stabilize the Pt NPs dispersions made with the GDEx process.
The morphology, structure, and composition of the Pt NCs were characterized using TEM, SEM, and XRD. TEM images are shown in Fig. 2. Nanoparticles can be formed by diffusional growth of nuclei or by a secondary growth process involving the aggregation of these nuclei.31 The TEM images indicate that all GDEx made Pt nanomaterials are nanocluster structures formed by aggregation of such primary nuclei. However, In Pt-PVP-0 and Pt-PVP-001 the nanoclusters are closely packed together in larger aggregates, while individual nanoclusters can be seen in Pt-PVP-01 and Pt-PVP-1 (Fig. 2(c) and (d)). The size of the primary NPs ranged from 2 nm to 5 nm (Fig. S3, ESI-I†). Pt nanomaterials with similar structure and composition have been synthesized using sonoelectrochemistry16 and laser pulse heating electrodeposition.18 Compared to these methods, GDEx is simpler, as it does not require specialized or costly equipment, and it is more easily scalable.
 | ||
| Fig. 2 TEM images showing the GDEx made Pt NCs. (a) Pt-PVP-0, (b) Pt-PVP-001, (c) Pt-PVP-01, and (d) Pt-PVP-1. Insets: Higher magnification images of Pt-PVP-0 and Pt-PVP-001 showing the primary Pt NPs. | ||
The formation of Pt NCs is governed by the combination of attraction and repulsion forces, and it is affected by the pH, the ionic strength, and the zeta potential of the solution.31 At low zeta potential conditions, the electrostatic repulsive force between particles is negligible and particles will aggregate once they encounter each other, leading to the formation of an open structure that falls under the diffusion-limited aggregation regime. Increasing the zeta potential leads to an increase in the repulsive force between the particles and to a reaction- controlled aggregation regime which results in the formation of porous nanoclusters.31 Under GDEx conditions, the high ionic strength of the supporting electrolyte (NaCl 0.5 M) and the local high pH (due to the formation of OH−) create the conditions enabling for the formation of the Pt NCs. However, once the Pt NCs reach the bulk, in which the zeta potential is lower due to the high ionic strength,32 they undergo diffusion-limited aggregation (as observed in Pt-PVP-0). The addition of PVP to the reaction media at a concentration of 0.1 g L−1 or higher stabilizes the Pt NCs and prevents diffusion-limited aggregation (as observed in Pt-PVP-01 and Pt-PVP-1).
The Pt NCs appeared to be spherical. To further confirm this, samples were analysed using SEM. The SEM images in Fig. S4 (ESI-I) (ESI†) confirmed the spherical nature of the nanocluster structures. However, it should be noted that these clusters are more defined when the concentration of PVP during synthesis is increased. The average diameter of the Pt NCs determined from the SEM images was 64 nm ± 22 nm, 60 nm ± 22 nm, 38 nm ± 15 nm, and 43 nm ± 18 nm, for Pt-PVP-0, Pt-PVP-001, Pt-PVP-01, and Pt-PVP-1, respectively.
The spherical equivalent diameter of the Pt NPs in the liquid was also determined using spICP-SFMS.33 In Fig. S5 (ESI-I) (ESI†) the particle size distribution of Pt-PVP01 and Pt-PVP1 as obtained by spICP-SFMS is shown. Both samples have similar spherical equivalent diameter, 41 nm ± 15 nm for Pt-PVP-01, and 40 ± 13 nm for Pt-PVP-1; it should be noted that spICP-SFMS cannot distinguish primary Pt NPs from individual Pt NCs. The agglomeration observed in Pt-PVP-0 and Pt-PVP-001 made them unsuitable for spICP-SFMS analysis.
The XRD pattern of the Pt NCs (Fig. S6, ESI-I†) revealed that they are crystalline in nature. The four major peaks at 39.9°, 46.3°, 67.7°, and 81.1° correspond to the (110), (200), (220), and (311) planes of fcc Pt (PDF 00-001-1194). The broad diffraction peaks are consistent with the nanoscale feature of the primary Pt NPs. The average crystallite sizes were calculated using Scherrer's equation,34 without subtracting the instrumental broadening. The values thus obtained were 6.7 nm, 5.6 nm, 4.4 nm, and 4.3 nm for Pt-PVP-0, Pt-PVP-001, Pt-PVP-01, and Pt-PVP-1, respectively, which are in good agreement with the TEM measurements for the primary NPs.
We have previously demonstrated that CO in the GDEx process is essential for the formation of small NPs.20 Additionally, the results of this work, suggests that the role of PVP is stabilization of the Pt NCs and avoidance of their diffusion-limited aggregation, but it does not influence the size of the Pt NCs. To confirm this, a couple of experiments were performed, using solutions under the same conditions as Pt-PVP-1. In the first, the GDEx process was carried out replacing CO2 with Ar (hence only H2 is formed). In the second experiment, H2 was bubbled through the solution for 30 min. The SEM images of the respective products (Fig. S7, ESI-I†) showed the formation of much larger and more disperse particles, confirming that PVP does not play a role in controlling the size of the Pt.
Electrocatalytic performance
To study the electrochemical properties of the Pt NPs, cyclic voltammograms (CVs) were obtained in Ar-saturated 0.5 M H2SO4 at room temperature between −0.2 V and 1.0 V vs. Ag/AgCl at a scan rate of 50 mV s−1. Prior to the electrocatalytic study the Pt NCs were subjected to a cleaning treatment to remove PVP from the surface (Fig. S8, ESI-II†). The CVs are shown in Fig. 3(a). For all Pt NPs a well-defined hydrogen adsorption–desorption region from 0.05 V to 0.350 VRHE is observed, followed by the double layer region and the hydroxyl species (OHad) adsorption region beyond 0.7 VRHE and a OHad reduction peak at about 0.8 VRHE in the reverse scan. The voltammetric features confirm the successful removal of PVP. For comparison, CVs of uncleaned Pt-PVP-01 and Pt-PVP-1 are shown in Fig. S9 (ESI-II†). The two well delineated peaks in the hydrogen adsorption region and the three peaks in the hydrogen desorption region agree with the data previously reported in the literature35 and correspond to the surface adsorption characteristics of Pt (111), Pt (100) and Pt (110) facets revealing the polycrystalline nature of the Pt NPs.17,36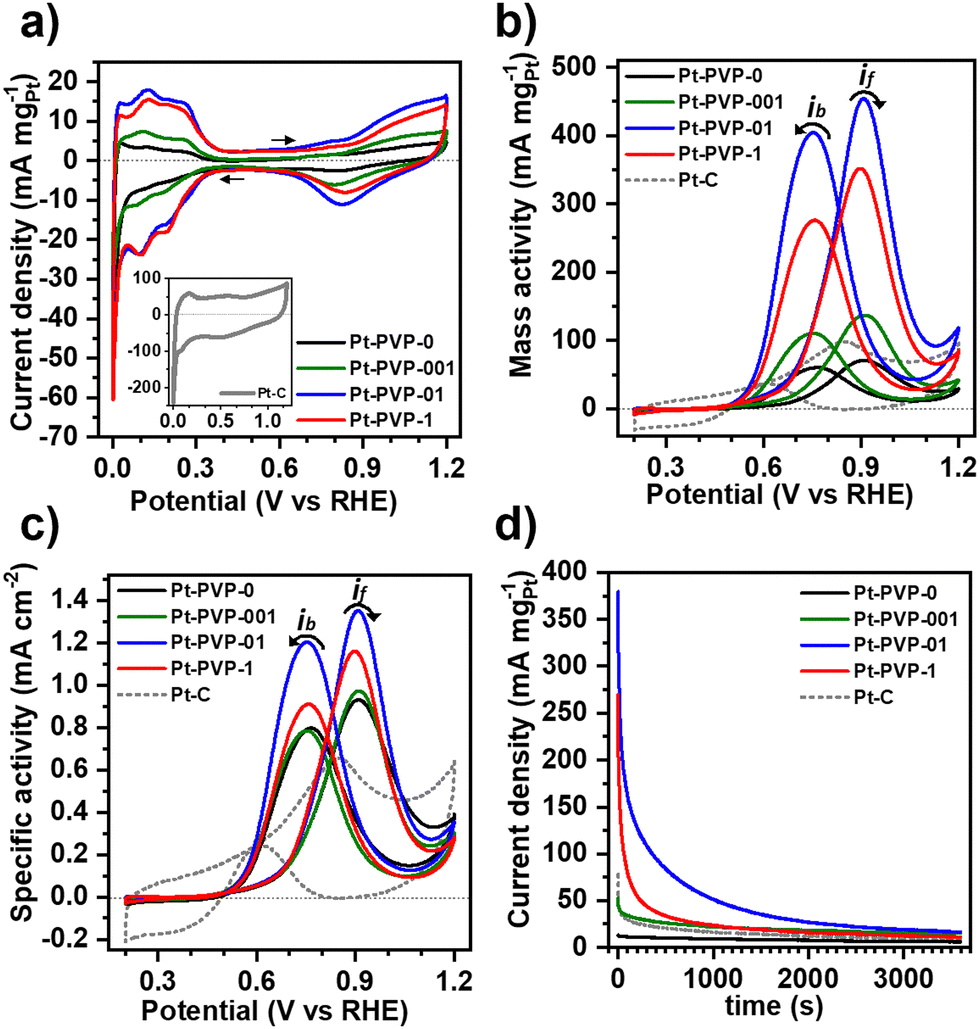 | ||
| Fig. 3 (a) CVs of the different Pt NCs in 0.5 M H2SO4 solution at 50 mV s−1, inset: CV of Pt/C. (b) and (c) mass activity and specific activity of the Pt NCs and Pt-C recorded in 0.5 M H2SO4 + 1.0 M MeOH solution at 50 mV s−1. (d) Chronoamperograms of the different materials in in 0.5 M H2SO4 + 1.0 M MeOH solution at 0.9 VRHE. Arrows indicate the direction of the scan. | ||
The electrochemically active surface area (ECSA) of the Pt NPs was calculated by integrating the charge in the hydrogen desorption region, after deduction of the double layer region. The ECSA (m2 gPt−1) was estimated according to eqn (13):37
| ECSA = QH/210 LPt | (13) |
| Material | ECSA (m2 g−1) | Mass activity (mA mg−1) | Specific activity (mA cm−2) |
|---|---|---|---|
| Acid media (0.5 M H2SO4) | |||
| a The values reported are the average and standard deviation of at least three different measurements. | |||
| Pt-PVP-0 | 7.6 ± 0.7 | 71 ± 2 | 0.94 ± 0.02 |
| Pt-PVP-001 | 14.1 ± 1.2 | 136 ± 7 | 0.97 ± 0.05 |
| Pt-PVP-01 | 33.6 ± 1.2 | 463 ± 28 | 1.38 ± 0.08 |
| Pt-PVP-1 | 30.3 ± 1.0 | 341 ± 25 | 1.12 ± 0.08 |
| Pt/C | 15.0 ± 1.3 | 103 ± 6 | 0.69 ± 0.04 |
| Alkaline media (1.0 M KOH) | |||
| Pt-PVP-0 | 3.6 ± 0.5 | 382 ± 18 | 10.8 ± 0.5 |
| Pt-PVP-001 | 5.9 ± 1.3 | 453 ± 32 | 7.7 ± 0.5 |
| Pt-PVP-01 | 16.7 ± 0.8 | 1269 ± 169 | 7.7 ± 1.0 |
| Pt-PVP-1 | 18.6 ± 0.4 | 1292 ± 150 | 6,8 ± 0.7 |
| Pt/C | 18.5 ± 3.0 | 620 ± 22 | 2.8 ± 0.1 |
The electrocatalytic activity of Pt NCs for MeOH oxidation was evaluated by cyclic voltammetry in Ar-saturated 0.5 M H2SO4 + 1.0 M MeOH solution at 50 mV s−1 between 0 V to 1.2 VRHE. The results were normalized to Pt loading and ECSA, which were used to determine the mass activity (MA) (Fig. 3(a)) and specific activity (SA) (Fig. 3(b)), respectively. Two characteristic oxidation peaks are present in the CVs. The first one at ∼0.9 VRHE in the forward scan (if) is associated with the oxidation of freshly chemisorbed MeOH on the Pt surface, while the second oxidation peak at ∼0.7 VRHE in the backward scan (ib) has been traditionally associated with the oxidation of carbonaceous species (i.e., CO) that are not completely oxidized.38 However, recent studies suggest that ib does not reflect the CO adsorption and poisoning, but is related to the oxidation of MeOH on Pt–Ox surfaces formed during the forward scan.39,40
The MA and SA values at the if are listed in Table 1. The MA and SA values scale with the calculated ECSA. It can be observed that employing PVP at concentrations exceeding 0.1 g L−1, notably enhanced the mass activity, as observed with the ECSA, attributed to the reduced agglomeration. However, normalizing the catalytic activity with ECSA (SA) showed minimal disparities, as the available surface area is the key factor for the catalysis of MOR. Moreover, the SA of all GDEx-made NCs was higher than the commercial Pt/C indicating a higher utilization of a large surface area due to the high proportion of edges and corners of the clustered porous structure.41 The results highlight that the key determinant for the catalytic activity of the Pt NCs is their stabilization with an appropriate PVP concentration, which is achieved as from 0.1 g L−1 with limited improvement on activity beyond this point. During synthesis, the PVP polymer chains are adsorbed into the Pt NCs providing stabilization through steric hindrance.28 Once enough PVP is enough to stabilize the NPs, increasing the PVP concentration might impact the stability of the Pt NCs but does not affect the properties or the catalytic activity of the Pt NCs.42 Table S2 (SI-III) (ESI†) shows the electrocatalytic activity for MeOH oxidation of unsupported Pt nanomaterials reported in literature. As can be seen, the activity of Pt-PVP-01 and Pt-PVP-1 generally matches or sometimes outperforms what has been reported in literature.
The ratio if/ib is commonly used to characterize the tolerance to CO poisoning.38 The calculated values were 1.1, 1.3, 1.1, and 1.3 for Pt-PVP-0, Pt-PVP-001, Pt-PVP-01, and Pt-PVP-1, respectively. However, as ib is related to the oxidation of MeOH on Pt–Ox rather than the CO adsorption the ratio if/ib results inadequate to assess CO tolerance.39,40 To evaluate short-term stability, chronoamperometric curves (Fig. 3(d)) were recorded at 0.9 VRHE for 3600 s. A progressive decay in the initial current with time was observed for all materials, which is characteristic for Pt and it is related to the accumulation of CO-like intermediate species on the surface of the Pt NCs.38 We used accelerated durability tests (ADT) to characterize the stability of the Pt-PVP-01 and Pt-PVP-1 for MeOH oxidation. These experiments were conducted by doing 1000 CV cycles in the same electrolyte at 50 mV s−1 between 0 V to 1.2 VRHE. Fig. 4(a) shows the change of the MA at if during cycling and Fig. 4(b) shows the normalized data with respect to the highest value of MA. Pt-PVP-01 displays high current densities and better durability by holding ∼50% of the maximum activity after 1000 cycles, while Pt-PVP-1 held ∼30% of the maximum activity. The CVs in H2SO4 0.5 M before and after the test (Fig. S10, ESI-III†) show a similar loss of the area in the hydrogen adsorption region (∼60% for Pt-PVP-01 and ∼85% for Pt-PVP-1). This behaviour is remarkable for pure Pt catalysts as the CO binding is inherent to Pt and the blocking of active sites and hence poisoning cannot be avoided.10 For comparison, a bimetallic Pt–Ru nanoflower catalyst held a similar activity (∼50%) after 900 CV cycles.43 SEM images taken of the electrode preparation before and after the test (for Pt-PVP-01, Fig. S11, ESI-III†) revealed no significant differences. This suggests that the decline in activity is likely attributed to CO poisoning. Nevertheless, the possibilities of Pt dissolution and agglomeration of Pt NCs cannot be ruled out.
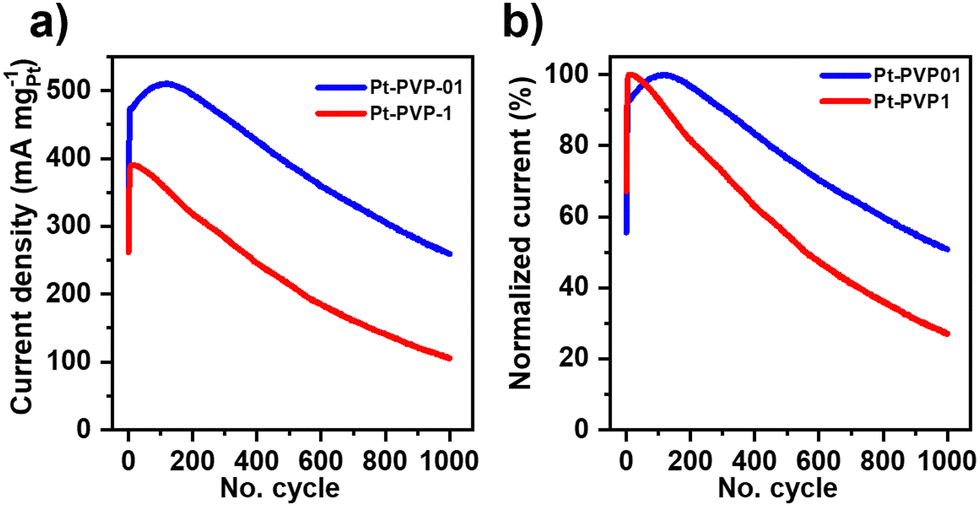 | ||
| Fig. 4 Accelerated stability test (ADT) of Pt-PVP-01 and Pt-PVP-1 in 0.5 M H2SO4 + 1.0 M MeOH solution at 50 mV s−1. (a) MA value at the if, (b) normalized data to the highest value. | ||
Based on these results, it is clear that GDEx-made Pt NCs exhibit superior catalytic properties for MeOH oxidation compared to commercial Pt/C and related materials reported in the literature, especially when their diffusion-limited aggregation is avoided using PVP during synthesis. The aggregation of the primary NPs into the Pt NCs provides a porous structure with a high surface area,31 increasing the number of active sites and, hence, enhancing their catalytic activity.16,17
The MeOH oxidation activity of the Pt NCs was also characterized in 1.0 M KOH, as the kinetics of the MOR are more favourable in alkaline media.44 The results are presented in ESI† (Fig. S12 and S13, ESI-III) and the ECSA, MA, and SA values are reported in Table 1. All materials had higher values of MA and SA for MeOH oxidation than in acid media, however the calculated ECSA were smaller. It is reported than in alkaline media, the kinetics of the hydrogen oxidation and evolution reactions are much slower and, non-specific adsorption of alkali metals onto Pt stepped surfaces can take place.45,46 This results in a smaller area of the hydrogen desorption region than in acidic media, hence the smaller ECSA values. Furthermore, the Pt NCs had poor stability in alkaline media, especially during accelerated stability tests (Fig. S13, ESI-III†). The instability is caused by the dissolution of Pt during cycling (Fig. S14, ESI-III†). It has been reported that Pt is highly susceptible to dissolution in alkaline media as the presence of OH− increases the Pt corrosion due to bulk complexation effects. However, the dissolution rate can be controlled via non-covalent effects.47 Hence, more work is necessary to improve the stability of GDEx-made Pt NPs in alkaline media.
Conclusions
Gas-diffusion electrocrystallization (GDEx) is a simple, fast, and environmentally friendly method to synthesize Pt NCs, as the reducing agents are electrochemically produced in situ via the reduction of CO2 and H2O. The Pt NCs can be stabilized by adding PVP in amounts as low as 0.1 g L−1. The GDEx-made Pt NCs exhibit good activity for the MeOH oxidation reaction in acidic and alkaline media, with improved stability observed in acidic conditions. This performance makes them promising candidates as catalysts for direct methanol fuel cells, providing a potentially more efficient alternative to traditional catalysts. The GDEx process can also be a useful method for making Pt nanomaterials in combination with other noble metals (i.e., Au, Pd, Rh, Ru), expanding its potential application areas and enhancing its versatility. The scalability and cost-effectiveness of the GDEx method make it an attractive approach for both research and industrial applications, contributing to the advancement of sustainable energy solutions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Union's Horizon 2020 research and innovation program, under grant agreement 730224 (PLATIRUS) and no. 958302 (PEACOC). The authors thank the funding from the European Union's Horizon Europe research and innovation programme, under grant agreement 101091715 (FIREFLY). OMM thanks to CONACYT-Mexico for the doctoral scholarship no. 766618. MV acknowledges Research Foundation—Flanders for the senior postdoctoral fellowship (no. 12ZD120N).References
- A. Yuda, A. Ashok and A. Kumar, Catal. Rev., 2020, 1–103 Search PubMed.
- X. Ren, Q. Lv, L. Liu, B. Liu, Y. Wang, A. Liu and G. Wu, Sustainable Energy Fuels, 2020, 4, 15–30 RSC.
- C. Koenigsmann and S. S. Wong, Energy Environ. Sci., 2011, 4, 1161–1176 RSC.
- Z. Xia, X. Zhang, H. Sun, S. Wang and G. Sun, Nano Energy, 2019, 65, 104048 CrossRef CAS.
- T. Wilberforce, A. Alaswad, A. Palumbo, M. Dassisti and A. G. Olabi, Int. J. Hydrogen Energy, 2016, 41, 16509–16522 CrossRef CAS.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 RSC.
- P. Ferrin, A. U. Nilekar, J. Greeley, M. Mavrikakis and J. Rossmeisl, Surf. Sci., 2008, 602, 3424–3431 CrossRef CAS.
- Y. Zuo, W. Sheng, W. Tao and Z. Li, J. Mater. Sci. Technol., 2022, 114, 29–41 CrossRef CAS.
- E. Antolini and J. Perez, J. Mater. Sci., 2011, 46, 4435–4457 CrossRef CAS.
- X. L. Tian, L. Wang, P. Deng, Y. Chen and B. Y. Xia, J. Energy Chem., 2017, 26, 1067–1076 CrossRef.
- S. M. Alia, G. Zhang, D. Kisailus, D. Li, S. Gu, K. Jensen and Y. Yan, Adv. Funct. Mater., 2010, 20, 3742–3746 CrossRef CAS.
- Y. Lou, C. Li, X. Gao, T. Bai, C. Chen, H. Huang, C. Liang, Z. Shi and S. Feng, ACS Appl. Mater. Interfaces, 2016, 8, 16147–16153 CrossRef CAS PubMed.
- S. M. Choi, J. H. Kim, J. Y. Jung, E. Y. Yoon and W. B. Kim, Electrochim. Acta, 2008, 53, 5804–5811 CrossRef CAS.
- L. Zhang, N. Li, F. Gao, L. Hou and Z. Xu, J. Am. Chem. Soc., 2012, 134, 11326–11329 CrossRef CAS PubMed.
- J. Wang, X.-B. Zhang, Z.-L. Wang, L.-M. Wang, W. Xing and X. Liu, Nanoscale, 2012, 4, 1549–1552 RSC.
- Q. Shen, L. Jiang, H. Zhang, Q. Min, W. Hou and J.-J. Zhu, J. Phys. Chem. C, 2008, 112, 16385–16392 CrossRef CAS.
- R. Ojani, E. Hasheminejad and J. B. Raoof, Energy, 2015, 90, 1122–1131 CrossRef CAS.
- H. E. M. Hussein, H. Amari and J. V. Macpherson, ACS Catal., 2017, 7, 7388–7398 CrossRef CAS.
- X. Sun, X. Zhu, N. Zhang, J. Guo, S. Guo and X. Huang, Chem. Commun., 2015, 51, 3529–3532 RSC.
- O. Martinez-Mora, G. Pozo, L. F. Leon-Fernandez, J. Fransaer and X. Dominguez-Benetton, RSC Sustain., 2023, 1, 454–458 RSC.
- L. D. Rampino and F. F. Nord, J. Am. Chem. Soc., 1941, 63, 2745–2749 CrossRef CAS.
- S.-H. Chang, M.-H. Yeh, C.-J. Pan, K.-J. Chen, H. Ishii, D.-G. Liu, J.-F. Lee, C.-C. Liu, J. Rick, M.-Y. Cheng and B.-J. Hwang, Chem. Commun., 2011, 47, 3864–3866 RSC.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- R. A. Prato M, V. Van Vught, K. Chayambuka, G. Pozo, S. Eggermont, J. Fransaer and X. Dominguez-Benetton, J. Mater. Chem. A, 2020, 8, 11674–11686 RSC.
- Y. Alvarez-Gallego, X. Dominguez-Benetton, D. Pant, L. Diels, K. Vanbroekhoven, I. Genné and P. Vermeiren, Electrochim. Acta, 2012, 82, 415–426 CrossRef CAS.
- F. J. Vidal-Iglesias, R. M. Arán-Ais, J. Solla-Gullón, E. Herrero and J. M. Feliu, ACS Catal., 2012, 2, 901–910 CrossRef CAS.
- A. Zalineeva, S. Baranton, C. Coutanceau and G. Jerkiewicz, Langmuir, 2015, 31, 1605–1609 CrossRef CAS PubMed.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- R. E. Zeebe and D. Wolf-Gladrow, CO2 in seawater: equilibrium, kinetics, isotopes, Gulf Professional Publishing, 2001 Search PubMed.
- B. Viswanath, S. Patra, N. Munichandraiah and N. Ravishankar, Langmuir, 2009, 25, 3115–3121 CrossRef CAS PubMed.
- J. Gustafsson, P. Mikkola, M. Jokinen and J. B. Rosenholm, Colloids Surf., A, 2000, 175, 349–359 CrossRef CAS.
- R. P. Lamsal, A. Hineman, C. Stephan, S. Tahmasebi, S. Baranton, C. Coutanceau, G. Jerkiewicz and D. Beauchemin, Anal. Chim. Acta, 2020, 1139, 36–41 CrossRef CAS PubMed.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- B. Geboes, J. Ustarroz, K. Sentosun, H. Vanrompay, A. Hubin, S. Bals and T. Breugelmans, ACS Catal., 2016, 6, 5856–5864 CrossRef CAS.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354–2358 CrossRef CAS.
- D. Z. Mezalira and M. Bron, J. Power Sources, 2013, 231, 113–121 CrossRef CAS.
- Y.-T. Liu, Q.-B. Yuan, D.-H. Duan, Z.-L. Zhang, X.-G. Hao, G.-Q. Wei and S.-B. Liu, J. Power Sources, 2013, 243, 622–629 CrossRef CAS.
- A. M. Hofstead-Duffy, D.-J. Chen, S.-G. Sun and Y. J. Tong, J. Mater. Chem., 2012, 22, 5205–5208 RSC.
- D. Y. Chung, K.-J. Lee and Y.-E. Sung, J. Phys. Chem. C, 2016, 120, 9028–9035 CrossRef CAS.
- Y. Zuo, K. Cai, L. Wu, T. Li, Z. Lv, J. Liu, K. Shao and H. Han, J. Mater. Chem. A, 2015, 3, 1388–1391 RSC.
- A. M. Levendorf, S.-G. Sun and Y. J. Tong, Electrocatalysis, 2014, 5, 248–255 CrossRef CAS.
- M. Li, H. Zheng, G. Han, Y. Xiao and Y. Li, Catal. Commun., 2017, 92, 95–99 CrossRef CAS.
- E. H. Yu, U. Krewer and K. Scott, Energies, 2010, 3, 1499–1528 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- X. Chen, I. T. McCrum, K. A. Schwarz, M. J. Janik and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 15025–15029 CrossRef CAS PubMed.
- P. P. Lopes, D. Strmcnik, D. Tripkovic, J. G. Connell, V. Stamenkovic and N. M. Markovic, ACS Catal., 2016, 6, 2536–2544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00209h |
| This journal is © The Royal Society of Chemistry 2023 |