
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyacrylonitrile-grafted lignin copolymer derived carbon nanofibers as a flexible electrode for high-performance capacitive-energy storage†
Da-Young
Kim
a,
Sivaprakasam
Radhakrishnan
b,
Seungmin
Yu
a and
Byoung-Suhk
Kim
 *ab
*ab
aDepartment of Carbon Composites Convergence Materials Engineering, Jeonbuk National University, 567 Baekje-daero, Deokjin-gu, Jeonju-si, Jeollabuk-do 54896, Korea. E-mail: kbsuhk@jbnu.ac.kr
bDepartment of Organic Materials & Textile Engineering, Jeonbuk National University, 567, Baekje-daero, Deokjin-gu, Jeonju-si, Jeollabuk-do 54896, Korea
First published on 27th June 2023
Abstract
Fabrication of flexible, low-cost, lightweight supercapacitors is in great demand for flexible electronics used in various applications. In this work, highly flexible carbon nanofibers (CNFs) were prepared using polyacrylonitrile grafted lignin (PAN-g-lignin) copolymers by electrospinning and consecutive stabilization/carbonization. The as-prepared PAN-g-lignin copolymer-derived CNFs were characterized by spectroscopy (FT-IR, NMR, FE-SEM, XRD), TGA-DSC, electrical conductivity, bending tests, and contact angles. The PAN-g-lignin-derived CNFs showed high carbon content, a uniform fiber structure, excellent flexibility for 5000 cycles at a bending radius of 10 mm, and remarkable retention (∼99.9%). A fabricated solid-state symmetric supercapacitor (SSC) device based on flexible CNFs possessed a high specific capacitance of 93.8 F g−1 at 1 A g−1 and excellent cyclic stability with 92% capacitance retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge–discharge cycles. This fabricated SSC device delivered a high energy density of 33 W h kg−1 at a power density of 800 W kg−1, thereby demonstrating potential use for high-performance flexible energy-storage devices.
000 charge–discharge cycles. This fabricated SSC device delivered a high energy density of 33 W h kg−1 at a power density of 800 W kg−1, thereby demonstrating potential use for high-performance flexible energy-storage devices.
1. Introduction
Compared with batteries and fuel cells, electrochemical supercapacitors are important energy-storage products that work through an adsorption/desorption mechanism of electrolyte ions.1–9 In recent years, they have garnered more attention due to their excellent cycle-lifetime, superior power density, and rapid charge–discharge.5–17 These peculiar advantages of supercapacitors are highly useful for real-world applications in electric automobiles and microelectronic instruments. Different types of carbon materials (carbon nanotubes, graphenes, carbon nanofibers (CNFs), activated carbons) have been broadly studied as electrode materials for supercapacitor applications.18–21 Among these carbon materials, CNFs are an important class of materials for energy-related applications due to their remarkable physiochemical properties, such as high specific surface area, high mechanical strength, light weight, and rich morphology.22,23 However, most CNFs are mainly derived from polyacrylonitrile (PAN) as a precursor due to a high carbon yield, good mechanical properties, and good electrospinnability.24 Therefore, the high cost (>50% of the manufacturing cost of carbon fibers) of PAN-based precursors has limited their utilization in various types of markets.25–27 Hence, it is important for research communities to develop high-quality CNFs through a cost-effective approach. Hence, lignin (the second most abundant biopolymer with high carbon content and aromatic groups) has attracted considerable interest as a promising candidate to make a low-cost renewable carbon fiber.28–30 Devadas et al. prepared poly(acrylonitrile-co-methyl acrylate)/lignin nanofibers by electrospinning and studied the effects of lignin on their physiochemical properties.30 Awal et al. reported soda hardwood lignin-based fibers prepared by melt-spinning, and studied their structural and thermal properties in detail.31 Francisco et al. proposed a new method to obtain one-dimensional (1-D) porous CNFs from lignin and used them for supercapacitor applications: a good performance was obtained.32 Those studies clearly showed that lignin was a suitable material for the fabrication of low-cost CNFs. Furthermore, graft polymerization was the most effective method to increase the high level of lignin content in a PAN solution to achieve good-quality CNFs. However, the flexibility of the lignin-based CNFs was limited.33,34 Hence, it is highly important to develop flexible CNFs with a cost-effective and “green” approach due to their potential use in various applications.35–37 In particular, CNF-based materials have been broadly used as flexible electrodes in wearable electronics and flexible supercapacitors.35 Nevertheless, CNFs as flexible electrodes have often shown poor performance for the fabrication of flexible supercapacitor devices with long cycle life.36,37 Few reports are available for the fabrication of flexible CNFs because of various limitations, such as complicated experimental procedure, time-consuming process, and low electrochemical performance.37Herein, we report remarkably flexible CNFs derived from polyacrylonitrile-grafted lignin (PAN-g-lignin) copolymers. For the first time, the obtained PAN-g-lignin CNFs were used as electrode materials for flexible-supercapacitor applications. The CNFs-based electrode exhibited good electrochemical and energy-storage performances when compared with recently reported CNFs-based electrodes.
2. Experimental
2.1. Materials
Alkali lignin (AL) powder (low sulfonate content, molecular weight = 10 kDa), acrylonitrile (AN; purity >99%), and dimethylformamide (DMF) were obtained from MilliporeSigma. Dimethyl sulfoxide (DMSO), hydrogen peroxide (H2O2; 30% aqueous solution), 2,2′-azoisobutryonitrile (AIBN; purity = 98%, recrystallized), calcium chloride (CaCl2), and hydrochloric acid (HCl) were received from Junsei Chemicals.2.2. Synthesis of lignin-g-PAN copolymer
First, 8 g of AL was dissolved in 80 mL of distilled (DI) water and stirred well for 3 h to obtain a uniform AL solution. Then, 5 wt% aqueous HCl solution (∼400 g) was added to the AL solution, and stirred for 3 h. Afterwards, precipitated AL was centrifuged at 8000 rpm for 10 min and then dialyzed (molecular weight cutoff = 10000) until pH = 7. Finally, the purified AL was freeze-dried.
PAN-g-lignin copolymers were prepared in two steps. First, 6 mL of AN and 7.3 mg of AIBN were placed in a three-necked flask containing 13 mL of DMSO and stirred until AIBN was completely dissolved. Then, the mixed solution was heated and stirred for 3 h at 70 °C under a N2 atmosphere. Second, after AN polymerization, a mixture of purified AL (2 g) and CaCl2 (2 g) was dissolved in 30 mL of DMSO, added to the solution and continuously stirred. Then, 1.2 mL of H2O2 was slowly dropped into the reaction solution. Finally, the reaction was allowed to continue at 70 °C under continuous N2 flow for 6 h for the effective formation of PAN-g-lignin copolymers (CaCl2–H2O2 as a redox initiator). The final reaction solution was precipitated to DI water, and the solvent was exchanged for 1 day. The obtained products were collected by filtration and dried at 60 °C in a vacuum oven. Schematic diagrams for CNF preparation and the detailed reaction mechanism of PAN-co-lignin copolymer are given in Scheme 1 and Fig. S1 (ESI†), respectively.
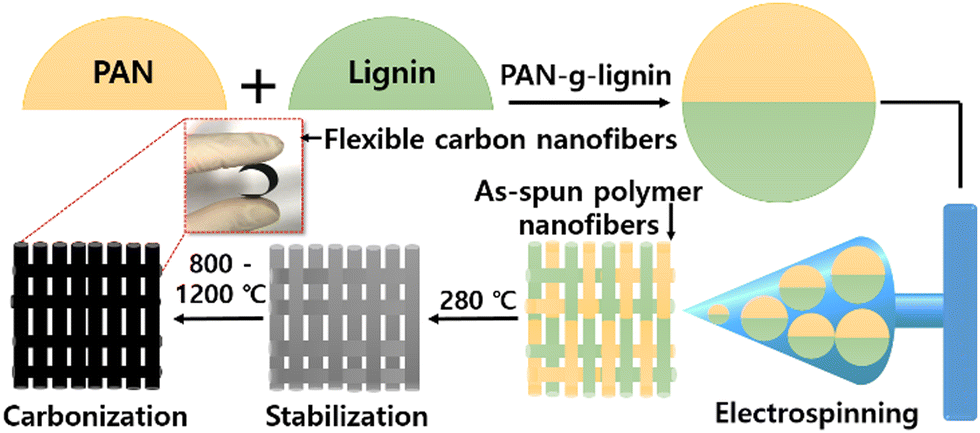 | ||
| Scheme 1 Preparation of carbon nanofibers using PAN-g-lignin copolymer (schematic). | ||
2.3. Preparation of lignin-g-PAN-derived CNFs
The PAN-g-lignin copolymer solution (10 wt%) dissolved in DMF was electrospun into nanofibers by applying a high voltage (15 kV) at a feeding rate of 1.0 mL h−1 using a 25-G metal needle. The nanofibers were collected on a drum collector for 10 h with a tip-to-collector distance of 15 cm. The obtained electrospun PAN-g-lignin nanofibers were thermally stabilized (280 °C at a heating rate of 1.0 °C min−1) in air for 1 h. The stabilized nanofibers were further carbonized at 800 °C, 1000 °C, or 1200 °C for 1 h, respectively, at a heating rate of 5 °C min−1 under a N2 atmosphere. The resultant CNFs derived from PAN-g-lignin were named C-800, C-1000, C-1200, corresponding to the carbonization temperature, respectively.2.4. Characterization
The surface morphology of samples was characterized by field-emission scanning electron microscopy (FE-SEM) using a SU-70 (Hitachi) system. Changes in functional groups were studied by Fourier transform infrared (FTIR) spectroscopy (PerkinElmer) in attenuated total reflection (ATR) mode. The chemical structure was elucidated by 1H NMR spectroscopy (Varian Unity Plus 300 spectrometer). Tetramethylsilane (TMS) was used as the internal standard. Thermal stability was studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (SDT Q600; TA Instruments) at a heating rate of 10 °C min−1 under N2. The crystallinity of CNFs was studied using X-ray diffraction (XRD; XPERT-PRO; PANalytical). A four-probe method (Keithley 2420 3A) was used for estimation of the sheet resistance of CNFs. The flexibility and bending characteristics of CNFs were studied by a Radius Bending Tester (JIRBT-610) with a DC/AC current source (6221; Keithley Instruments). The viscosity average molecular weight (Mv) of PAN-g-lignin copolymers was measured by a Ubbelohde viscometer using the following formula38| [η] = 2.33 × 10−4 × [Mv]0.75 | (1) |
2.5. Electrochemical studies
Electrochemical supercapacitor studies were undertaken using a Versastat 4 workstation composed of a free-standing CNFs mat (1.0 cm × 1.0 cm), Ag/AgCl, and platinum plate (2.5 cm × 2.5 cm) as working, reference, and counter electrodes in H2SO4 electrolyte solution (1 M), respectively. Cyclic voltammetry (CV) was undertaken in the voltage window of 0–1.0 V at a scan rate of 20 mV s−1. Galvanostatic charge–discharge (GCD) was carried out at a current density of 1 A g−1. Electrochemical impedance spectroscopy (EIS) was done in a range of 0.01–100 kHz with an amplitude of 10 mV. The true applicability of our fabricated CNFs free-standing electrode was determined by fabricating a flexible solid-state symmetric supercapacitor (SSC) device using PVA-H2SO4 as the gel-electrolyte. The specific capacitance (Cs) of the fabricated SSC device was estimated using eqn (2),| Cs = (I × t)/(m × ΔV) | (2) |
The energy density (E, W h kg−1) and power density (P, W kg−1) were derived using eqn (3) and (4)
| E = ItV/(7.2 m) | (3) |
| P = E/t × 3600 | (4) |
3. Results and discussion
3.1. Synthesis of lignin-g-PAN copolymer
The presence of functional groups and their structures was initially accessed by FT-IR spectroscopy (Fig. 1A), and 1H NMR spectroscopy (Fig. 1B). Fig. 1A shows the FT-IR spectra of PAN, lignin, and PAN-g-lignin. As seen in Fig. 1A, after the grafting of PAN with lignin, additional stretching peaks were observed at ∼2243 and ∼1452 cm−1, which were attributed to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and CH2 stretching vibrations of the incorporated PAN, respectively, thereby indicating the incorporation of the PAN copolymer into lignin. Moreover, the major functional groups in lignin were methoxy, phenolic hydroxy, and benzyl alcohol (hydroxyl group on the α-carbon) (inset in Fig. 1B). Accordingly, absorption bands at 3400 cm−1, 2900 cm−1, 1600/1450 cm−1, and 1100 cm−1 were observed, which were attributed to the OH broad band of alcoholic or hydroxyl groups, CH stretching band of methyl groups, C
N and CH2 stretching vibrations of the incorporated PAN, respectively, thereby indicating the incorporation of the PAN copolymer into lignin. Moreover, the major functional groups in lignin were methoxy, phenolic hydroxy, and benzyl alcohol (hydroxyl group on the α-carbon) (inset in Fig. 1B). Accordingly, absorption bands at 3400 cm−1, 2900 cm−1, 1600/1450 cm−1, and 1100 cm−1 were observed, which were attributed to the OH broad band of alcoholic or hydroxyl groups, CH stretching band of methyl groups, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C stretching bands in the aromatic range, and C–O stretching band of ether from lignin.39 Moreover, 1H NMR studies revealed methylene protons (2.05–2.16 ppm) and methane protons (3.13–3.18 ppm) in the PAN-g-lignin copolymer (Fig. 1B). Spectroscopy (FT-IR, NMR) studies strongly supported formation of the PAN-g-lignin copolymer.
C and C–C stretching bands in the aromatic range, and C–O stretching band of ether from lignin.39 Moreover, 1H NMR studies revealed methylene protons (2.05–2.16 ppm) and methane protons (3.13–3.18 ppm) in the PAN-g-lignin copolymer (Fig. 1B). Spectroscopy (FT-IR, NMR) studies strongly supported formation of the PAN-g-lignin copolymer.
 | ||
| Fig. 1 (A) FT-IR and (B) 1H NMR spectra of PAN, lignin, and PAN-g-lignin copolymer. | ||
The thermal stability of the precursor and copolymers was investigated by TGA (Fig. 2A) and DSC (Fig. 2B). Fig. 2A shows TGA curves of lignin, PAN homopolymer, and PAN-g-lignin copolymer. Lignin showed transition states at 100 °C, 260 °C, and 315–390 °C, which correlated to the removal of moisture, decomposition of hemicellulose, and decomposition of cellulose from lignin, respectively.40 Similar thermal degradation of the PAN-g-lignin copolymer and PAN homopolymer was also observed at second and third regions. It was quite noticeable that the thermal stability of the PAN-g-lignin copolymer (Tonset ∼300.0 °C) was largely enhanced by the incorporation of lignin, compared with that of the PAN homopolymer (Tonset ∼271.7 °C). DSC also showed the higher Tonset ∼293.9 °C of the PAN-g-lignin copolymer than that of the PAN homopolymer (Tonset ∼250.6 °C), whereas the ΔH (∼438.4 J g−1) of the PAN-g-lignin copolymer was lower than that of the PAN homopolymer (∼586.9 J g−1) (Fig. 2B), which reflected the greater cyclization during stabilization and, therefore, higher yield after carbonization, due to incorporated lignin.40,41
 | ||
| Fig. 2 (A) TGA and (B) DSC curves of PAN, lignin, and PAN-g-lignin copolymer. | ||
Fig. 3 shows the FE-SEM images of electrospun polymer nanofibers using the PAN-g-lignin copolymer. To optimize the voltage, we undertook electrospinning using PAN-g-lignin at different voltages (11, 15, 17, and 19 kV) and the corresponding FE-SEM images are displayed in Fig. 3A–D. Perfect polymeric nanofibers (920 ± 50 nm) without beads or bead-string morphologies were noticed at an applied voltage of 17 kV (Fig. 3E). Lower (<17 kV) and higher (>17 kV) applied voltages produced bead-like morphologies (due to a lower viscosity of the solution) and broader diameter distribution (due to a high viscosity of the solution), respectively. Hence, the electrospinnability of the PAN-g-lignin copolymer solution (∼10 wt%) was most stable at 17 kV. FE-SEM morphologies of stabilized (at 280 °C) (Fig. 3F) and carbonized (at 800–1200 °C) (Fig. 3G and Fig. S2, ESI†) PAN-g-lignin nanofibers were studied. Stabilized fibers were deep-brown (inset of Fig. 3F), compared with as-spun fibers (deep-yellow) (inset of Fig. 3E). Moreover, the sheet resistance and average fiber diameter decreased gradually as the carbonization temperature increased (Fig. S3, ESI†).
 | ||
| Fig. 3 SEM images of electrospun nanofibers of the PAN-g-lignin copolymer produced from 10 wt% solutions at an applied voltage of (A) 11 kV, (B) 15 kV, (C) 17 kV, and (D) 19 kV, and as-spun (at 17 kV, E), stabilized (at 280 °C, F), and carbonized (at 1000 °C, G) PAN-g-lignin nanofibers. The inset in (E, F, and G) shows real images of the corresponding stabilized and carbonized nanofibers, respectively. | ||
Fig. 4A presents the Raman spectra of CNFs carbonized at carbonization temperatures of 800 °C (a), 1000 °C (b), and 1200 °C (c). As the carbonization temperature increased, ID/IG increased from 0.99 to 1.05, indicating a decreased order of structure, probably due to the interruption caused by added lignin, though the C-800 and C-1000 samples showed the same ID/IG value.42 The nature of functional groups in as-spun (curve a), stabilized (280 °C, curve b) and CNFs carbonized at temperatures of 800 °C (C-800) (c), 1000 °C (C-1000) (d), and 1200 °C (C-1200) (e) was studied by FT-IR spectroscopy (Fig. 4B). The as-spun PAN-g-lignin nanofibers (curve a) showed a sharp band at 2250 cm−1, which was attributed to the CN stretching vibration of PAN in as-spun PAN-g-lignin. However, the same peak position was reduced significantly after stabilization (280 °C; curve b), suggesting that cyclization had occurred.43 An additional peak was observed at 1580 cm−1 (CC, and CN) for the stabilized polymer nanofibers (curve b) due to formation of a “ladder” structure.44 Carbonized (800–1200 °C) polymer nanofibers exhibited very few and weak broad bands (curves c–e) compared with those of stabilized (curve b) and as-spun PAN-g-lignin (curve a), thereby confirming removal of all organic functional groups from PAN-g-lignin at high carbonization temperatures (800–1200 °C). The microcrystalline structures of thermal-treated nanofibers were verified by XRD spectroscopy. Fig. 4C shows the XRD spectra of stabilized polymer nanofibers (curve a) and carbonized CNFs (C-800: curve b; C-1000: curve c; C-1200: curve d; C-1400: curve e).
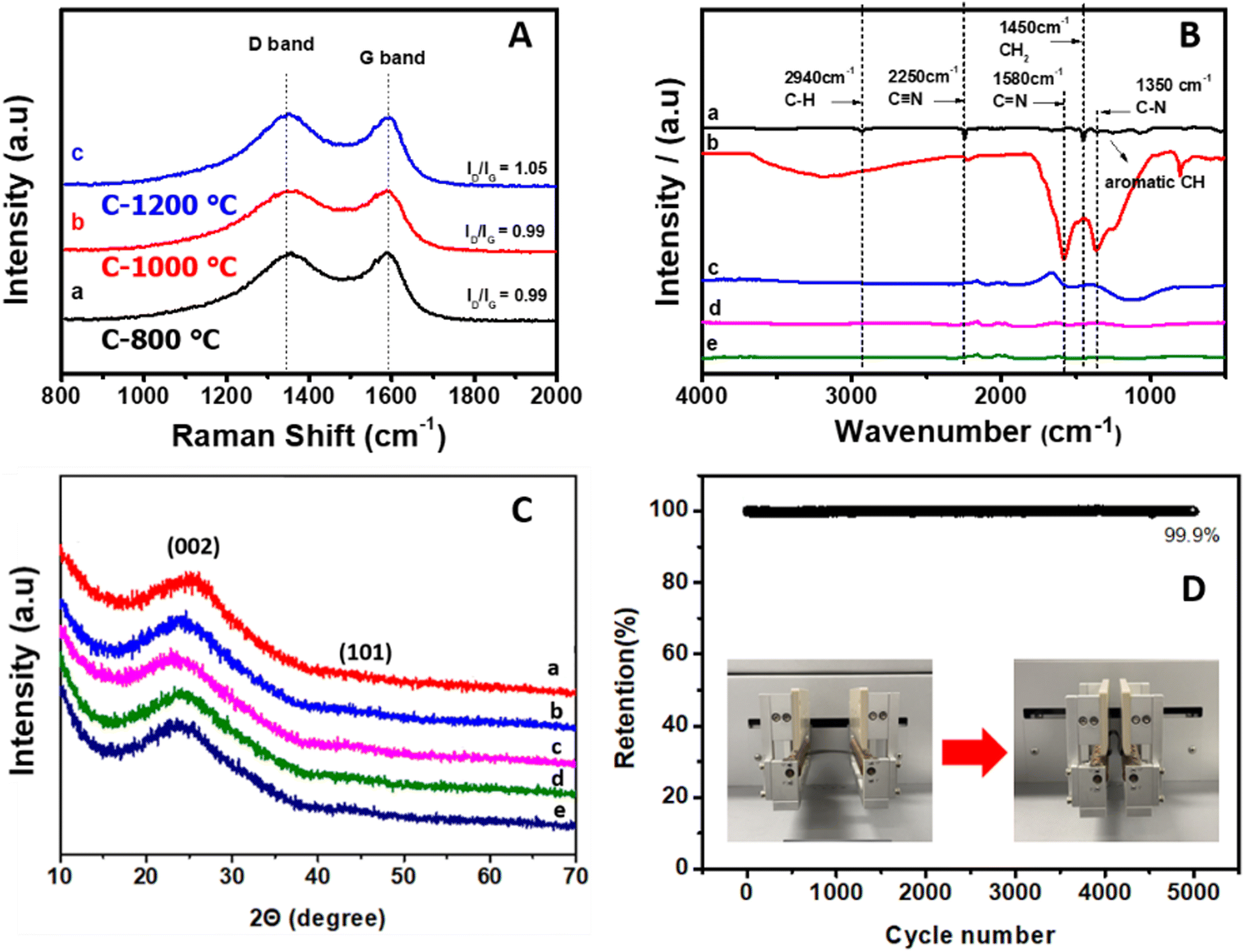 | ||
| Fig. 4 (A) Raman spectra of carbon nanofibers derived from PAN-g-lignin copolymer at carbonization temperatures of 800 °C (a), 1000 °C (b), and 1200 °C (c). (B) FT-IR spectra of (a) as-spun PAN-g-lignin polymer nanofibers, (b) stabilized PAN-g-lignin copolymer, and carbon nanofibers derived at carbonization temperatures of 800 °C (C-800) (c), 1000 °C (C-1000) (d), and 1200 °C (C-1200) (e). (C) XRD spectra of (a) stabilized PAN-g-lignin copolymer, and carbon nanofibers derived at carbonization temperatures of (b) 800 °C, (c) 1000 °C, (d) 1200 °C, and (e) 1400 °C. (D) Retention of the sheet resistance of the PAN-g-lignin carbon nanofibers for 5000 cycles at a bending radius ∼10 mm. The inset photograph shows the bending test. | ||
XRD patterns at ∼24° and ∼43° were observed, which corresponded to the (002) and (101) growth planes of graphitized carbon products, respectively.45 Such porous CNF mats with excellent graphitization aid the good electrochemical performance of an electrode by enhancing conductivity and by allowing access to many electrolyte ions. To ascertain the mechanical flexibility of as-derived CNFs (C-1000), a repeat bending test was done. Retention of the sheet resistance of PAN-g-lignin CNFs (C-1000) for 5000 cycles at a bending radius of ∼10 mm was measured (Fig. 4D). The inset photograph shows the bending test. A measurement was carried out for 5000 cycles at a bending radius of ∼5 mm (Fig. 4D). Excellent retention (99.9%) was observed, suggesting that the CNFs derived from the PAN-g-lignin copolymer had excellent mechanical flexibility and durability and, therefore, it could be suitable for the fabrication of flexible free-standing electrodes.
The type of elements and their composition in CNFs were verified by X-ray photoelectron spectroscopy (XPS). Fig. 5A shows the survey spectra of CNFs (C-800, C-1000, and C-1200) derived from PAN-g-lignin at different carbonization temperatures. All CNFs showed C, O, and N elements. C was the major content in all CNFs. The intensity of C element increased with increasing carbonization temperature from 800 °C (89.01%) to 1200 °C (92.58%). The O content decreased with increasing temperature from 800 °C (8.43%) to 1200 °C (5.15%) due to graphitization at higher temperatures (Fig. 5B). The bonding nature of C (Fig. 5C) and N (Fig. 5D) elements for C-1000 CNFs were studied in detail. In the high-resolution spectrum of C 1s, peaks at 284.8, 285.7, 287.1, and 290 eV were observed, which corresponded to the aromatic ring (C–C/CC) as well as C–N, carbonyl (CO), and ester (COOR) groups, respectively.46 Similarly, the high-resolution spectrum of N 1s exhibited peaks at 398.3, 400.2, and 400.5 eV, which were assigned to pyridinic-N, pyrrolic-N, and graphitic-N groups, respectively. Similar peak patterns were also observed for other CNFs (C-800, C-1200) derived at carbonization temperatures of 800 °C (Fig. S4A and C, ESI†) and 1200 °C (Fig. S4B and D, ESI†).
 | ||
| Fig. 5 (A) XPS survey spectra of CNFs (C-800, C-1000, and C-1200) derived at different calcination temperatures. (B) Atomic percentage of elements in CNFs. High-resolution XPS spectrum of (C) C 1s and (D) N 1s for CNFs (C-1000) derived at a calcination temperature of 1000 °C. | ||
3.2. Electrochemical supercapacitance performance of flexible free-standing CNFs
Fig. 6A shows the CV curves of as-derived CNFs (a: C-800, b: C-1000, and c: C-1200) in 1 M H2SO4. The C-1000 CNF electrode (curve b) exhibited a higher peak current with a nearly rectangular shape, compared with the CV curves of C-800 (curve a) and C-1200 (curve c). The same CNF (C-1000) electrode was studied by GCD (Fig. 6B). As expected from CV observations, the C-1000 electrode demonstrated a longer discharge time (∼135 s) compared with those of C-800 (35 s) and C-1200 (16 s) electrodes. To verify the charge-transfer resistance (RCT) of the as-derived CNFs electrode, EIS measurements were recorded for the prepared CNFs electrodes (Fig. 6C). The C-1000 electrode showed a lower RCT (∼6.98 Ω) (C-800: ∼7.26 Ω; C-1000: ∼9.44 Ω) and solution resistance (RS; ∼3.9 Ω). CV, GCD, and EIS studies clearly demonstrated that the C-1000 electrode had good electrochemical performance and energy-storage properties. | ||
| Fig. 6 (A) CV curves at a scan rate of 1 mV s−1, (B) GCD curves at 1 A g−1, and (C) EIS at OCV of (a) C-800, (b) C-1000, (c)-1200 CNFs free-standing electrodes in 1 M H2SO4. (D) CV curves of the C-1000 electrode at scan rates of 10–200 mV s−1. (E) GCD curves of the C-1000 electrode at applied current densities from 0.5 to 10 A g−1. (F) GCD profiles before and after cyclic stability at an applied current density of 5 A g−1. | ||
Fig. 6D shows the CV profiles of the C-1000 electrode at different sweeping scan rates (10–200 mV s−1) in 1 M H2SO4. The anodic and cathodic peak currents increased with an increasing scan rate. The CV curve was nearly rectangular even at a higher scan rate (200 mV s−1), which suggested that the fabricated electrode was more suitable for supercapacitor applications. To further verify the supercapacitor performance of the C-1000 electrode, GCD was undertaken. Fig. 6E shows the GCD profiles of the C-1000 electrode at different applied current densities (0.5–10 A g−1). A longer discharge time (131 s) was obtained at a low current density (0.5 A g−1), and the GCD profile had a nearly triangular shape at all current densities. The C-1000 electrode exhibited a higher specific capacitance of 137 F g−1 (at 0.5 A g−1) with excellent capacitance retention (∼83.3%) at a current density of 10 A g−1. The cyclic stability of the C-1000 electrode was verified by continuous measurement of charge–discharge for 10000 cycles at a current density of 5 A g−1 (Fig. 6F). The decrease in capacitance was estimated to be ∼10% even after 10000 GCD cycles, thereby confirming its excellent stability. The electrical properties of CNFs after the cyclic-stability test were also checked by EIS. Fig. S6 (ESI†) shows the EIS spectra before and after measurement of cyclic stability. The RCT did not change significantly after stability measurement, which confirmed the superior stability of the free-standing CNFs electrode for real-life applications.
To investigate the wettability of fabricated C-1000 CNFs, we tested the water contact-angle properties of as-derived CNFs (Fig. S5, ESI†). Interestingly, a low contact angle (∼18°) was observed for CNFs derived at a carbonization temperature of 1000 °C (enabling access to many electrolyte ions), which was nearly 6- and 7-fold lower than that of C-800 (∼108°) and C-1200 (∼120°) electrodes. These data suggested that the carbonization temperature had a significant role on the surface properties of obtained CNFs, which aided a higher electrochemical performance. In general, the more hydrophobic properties of CNFs were due to the complete reduction of oxygen-based functional groups in CNFs at higher carbonization temperatures, which was in good agreement with previous reports.47 The C-1000 electrode was used for detailed studies on the performance of electrochemical supercapacitors.
To further show the real-life applicability of our fabricated free-standing CNFs electrode, a flexible SSC device using PVA-H2SO4 as the gel-electrolyte was fabricated. The structure of the fabricated device is shown schematically in Fig. S7 (ESI†). Initially, the electrochemical performance of SSC device was accessed by CV. Optimization of a potential window is an important parameter for the fabrication of a supercapacitor device. Hence, we recorded CV curves for different potential ranges: 0–1.0 V (curve a), 0–1.1 V (curve b), 0–1.2 V (curve c), 0–1.3 V (curve d), 0–1.4 V (curve e), 0–1.5 V (curve f), and 0–1.6 V (curve g) (Fig. 7A). The fabricated SSC device worked well up to 1.6 V without distortion of CV curves, and showed nearly rectangular CV curves due to the pure double-layer capacitive nature of the device based on CNFs electrodes. The optimized potential region was used to investigate the effect of scan rates to determine the rate capability of the fabricated device. Fig. 7B shows the CV profiles recorded at different sweeping scan rates (10–200 mV s−1). Good rectangular curves at low (10 mV s−1) and high (200 mV s−1) scan rates were observed, which suggested the superior rate capability of the electrode and ideal capacitive nature of the device. Fig. 7C shows the GCD profiles of the device at different applied current densities (1–10 A g−1) in the optimized potential window of 0–1.6 V. GCD curves were nearly triangular at all current densities without a major plateau region due to the formation of electric double-layer capacitance, which aligned with the CV studies shown in Fig. 7A. The calculated specific capacitance vs. current density was plotted (Fig. 7D). The specific capacitance for a low (1 A g−1), median (5 A g−1), and high (10 A g−1) current density was estimated to be 93.8, 56.25, and 37.5 F g−1, respectively, thereby demonstrating the good rate capability of the device. The energy density (E) and power density (P) are important to ascertain the true usability of a supercapacitor device for various applications. These values for our fabricated SSC device were estimated from GCD curves, and the obtained E and P values are presented as a Ragone plot in the inset of Fig. 7E (ii). The fabricated SSC device based on the CNFs (CNF-1000) electrode delivered an energy density of 33 W h kg−1 with a power density of 800 W kg−1. Table S1 (ESI†) shows a comparison of the supercapacitor performance of the SSC device based on CNFs against literature reports based on different devices: Table S1 (ESI†) suggested the good performance of our fabricated SSC device.
 | ||
| Fig. 7 (A) CV curves of a fabricated SSC device at potential windows of 0–1.6 V. (B) Effect of scan rates for the SSC device at scan rates of 10–200 mV s−1. (C) GCD test at various current densities (1–10 A g−1) using the SSC device. (D) Plot of specific capacitance against current density. (E) Plot of capacity retention and coulombic efficiency against the cycle number; the inset shows (i) CV curves measured at various bending radii (30°–120°) and (ii) a Ragone plot of the fabricated SSC device. | ||
In addition, the cyclic-stability test was done for our device by GCD. The fabricated device demonstrated excellent capacitance retention (∼92%) at ∼10000 continuous charge–discharge cycles at an applied current density of 5 A g−1, and obtained ∼100% coulombic efficiency (Fig. 7E). These data confirmed the outstanding stability of the device which, therefore, could be used for various industries. Finally, our key aim was to demonstrate the flexible SSC device using flexible CNFs based on PAN-g-lignin. The flexibility and stability of the fabricated SSC device at different bending angles was investigated by CV. Fig. 7E (inset i) shows the CV curves obtained at bending angles of 30°, 60°, 90°, and 120°. We observed negligible changes in the current and shape in CV curves, which confirmed the excellent flexibility and stability of the as-fabricated device. These data confirmed that the fabricated SSC device based on flexible CNFs had potential use in various flexible energy-storage applications.
Conclusions
We demonstrated the preparation of flexible CNF electrodes from a PAN-g-lignin copolymer by electrospinning and consecutive stabilization/carbonization. The as-derived CNFs were extremely flexible, as confirmed by a bending test for 5000 continuous cycles. CNFs prepared at a carbonization temperature of 1000 °C demonstrated excellent supercapacitance performance under a three-electrode set-up. A fabricated flexible SSC device based on flexible CNFs (C-1000) delivered a maximum energy density of 33 W h kg−1 with a corresponding power density of 800 W kg−1. Collectively, we demonstrated a new facile preparation process for the fabrication of a highly flexible free-standing CNFs electrode and outstanding electrochemical performance of a SSC device. This work will be highly useful for the fabrication of flexible high-energy storage devices for various applications.Author contributions
Conceptualization, investigation, and data analyses: D. Y. Kim. Investigation and writing (original draft): S. Radhakrishnan. Investigation and data analyses: S. Yu. Conceptualization, funding acquisition, supervision, and review and editing: B. S. Kim.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea grant funded by the Korea government (2020R1A2C2012356) and by the Technology Innovation Program (20016795; “Development of manufacturing technology independence of advanced activated carbons and application for high performance supercapacitors”) funded by the Ministry of Trade, Industry and Energy of Korea.References
- S. Ahankari, D. Lasrdo and R. Subramaniam, Mater. Adv., 2022, 3, 1472–1496 RSC.
- S. Radhakrishnan, H. Y. Kim and B. S. Kim, J. Mater. Chem. A, 2016, 4, 12253 RSC.
- H. Li, L. Cao, H. Zhang, Z. Tian, Q. Zhang, F. Yang, H. Yang, S. He and S. Jiang, J. Colloid Interface Sci., 2022, 609, 179–187 CrossRef CAS PubMed.
- L. Cao, H. Li, X. Liu, S. Liu, L. Zhang, W. Xu, H. Yang, H. Hou, S. He, Y. Zhao and S. Jiang, J. Colloid Interface Sci., 2021, 599, 443–452 CrossRef CAS PubMed.
- J. Qi, Y. Yan, Y. Cai, J. Cao and J. Feng, Adv. Funct. Mater., 2021, 31, 2006030 CrossRef CAS.
- X. Wen, J. Luo, K. Xiang, W. Zhou, C. Zhang and H. Chen, Chem. Eng. J., 2023, 458, 141381 CrossRef CAS.
- J. Yu, S. Liu, G. Duan, H. Fang and H. Hou, Compos. Commun., 2020, 19, 239–245 CrossRef.
- D. Li, H. Guo, S. Jiang, G. Zeng, W. Zhou and Z. Li, New J. Chem., 2021, 45, 19446 RSC.
- W. Zhou, G. Zeng, H. Jin, S. Jiang, M. Huang, C. Zhang and H. Chen, Molecules, 2023, 28, 2147 CrossRef CAS PubMed.
- J. Yang, H. Li, S. He, H. Du, K. Liu, C. Zhang and S. Jiang, Polymers, 2022, 14, 2521 CrossRef CAS PubMed.
- W. Guo, X. Guo, L. Yang, T. Wang, M. Zhang, G. Duan, X. Liu and Y. Li, Polymer, 2021, 235, 124276 CrossRef CAS.
- J. G. Seong, T. H. Ko, D. Lei, W. K. Choi, Y. S. Kuk, M. K. Seo and B. S. Kim, Green Energy Environ., 2022, 7, 1228–1240 CrossRef CAS.
- G. Duan, L. Zhao, L. Chen, F. Wang, S. He, S. Jiang and Q. Zhang, New J. Chem., 2021, 45, 22602–22609 RSC.
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- D. Lei, X. D. Li, S. Radhakrishnan and B. S. Kim, Mater. Lett., 2023, 333, 133688 CrossRef CAS.
- W. Guo, X. Guo, L. Yang, T. Wang, M. Zhang, G. Duan, X. Liu and Y. Li, Polymer, 2021, 235, 124276 CrossRef CAS.
- Y. Wang, H. Li, W. Yang, S. Jian, C. Zhang and G. Duan, Diamond Relat. Mater., 2022, 130, 109526 CrossRef CAS.
- M. Karakoti, S. Pandey, G. Tatari, P. S. Dhapola, R. Jangra, S. Dhali, M. Pathak, S. Mahendia and N. G. Sahoo, Mater. Adv., 2022, 3, 2146–2157 RSC.
- O. Sangabathula and C. S. Sharma, Mater. Adv., 2020, 1, 2763–2772 RSC.
- Y. Wang, L. Zhang, H. Hou, W. Xu, G. Duan, S. He, K. Liu and S. Jian, J. Mater. Sci., 2021, 56, 173–200 CrossRef CAS.
- J. Xiao, H. Li, H. Zhang, S. He, Q. Zhang, K. Liu, S. Jiang, G. Duan and K. Zhang, J. Bioresour. Bioprod., 2022, 7, 245–269 CrossRef CAS.
- J. Liang, H. Zhao, L. Yue, G. Fan, T. Li, S. Lu, G. Chen, S. Gao, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2020, 8, 16747 RSC.
- X. Zhou, B. Liu, Y. Chen, L. Guo and G. Wei, Mater. Adv., 2020, 1, 2163–2181 RSC.
- Y. Zhang, K. Wang, G. Duan, Y. Chen, K. Liu and H. Hou, New J. Chem., 2023, 47, 5639 RSC.
- T. C. Xu, D. H. Han, Y. M. Zhu, G. G. Duan, K. M. Liu and H. Q. Hou, Chin. J. Polym. Sci., 2021, 39, 174–180 CrossRef CAS.
- Q. Mao, S. Rajabpour, M. K. Talkhoncheh, J. Zhu, M. Kowalik and A. C. T. van Duin, Nanoscale, 2022, 14, 6357–6372 RSC.
- S. Nunna, P. Blancard, D. Bukmaster, S. Davis and M. Naebe, Heliyon, 2019, 5, e02698 CrossRef PubMed.
- G. Jia, M. T. Innocent, Y. Yu, Z. Hu, X. Wang, H. Xiang and M. Zhu, Int. J. Biol. Macromol., 2023, 226, 646 CrossRef CAS PubMed.
- M. Yao, X. Bi, Z. Wang, P. Yu, A. Dufrense and C. Jian, Int. J. Biol. Macromol., 2022, 223, 980 CrossRef CAS PubMed.
- S. Devada, S. M. N. Al-Ajrash, D. A. Klosterman, K. M. Crosson, G. S. Crosson and E. S. Vasquez, Polymers, 2021, 13, 992 CrossRef PubMed.
- A. Awal and M. Sain, J. Appl. Polym. Sci., 2013, 129, 2765 CrossRef CAS.
- J. G.-M. Francisco, R.-R. Ramiro, J. M. Rosas, E. Morallon, C.-A. Diego, R.-M. Jose and T. Cordero, Sep. Purif. Technol., 2020, 241, 116724 CrossRef.
- D. Li, X. D. Li, M. K. Seo, M. S. Khil, H. Y. Kim and B. S. Kim, Polymer, 2017, 132, 31–40 CrossRef.
- X. Li, Y. Li, X. Tian, Y. Song and Y. Cui, J. Alloys Compd., 2022, 903, 163919 CrossRef CAS.
- J. Zhu, Q. Zhang, L. Guo, Y. Zhao, R. Zhang, L. Liu and J. Yu, Chem. Eng. J., 2022, 434, 134662 CrossRef CAS.
- X. Zhou, B. Liu, Y. Chen, L. Guo and G. Wei, Mater. Adv., 2020, 1, 2163–2181 RSC.
- T. H. Ko, D. Lei, S. Balasubramaniam, M. K. Seo, Y. S. Chung, H. Y. Kim and B. S. Kim, Electrochim. Acta, 2017, 247, 524–534 CrossRef CAS.
- R. L. Cleland and W. H. Stockmayer, J. Polym. Sci., 1955, 17, 473 CrossRef CAS.
- S. Kubo and J. F. Kadla, Biomacromolecule, 2003, 4, 561 CrossRef CAS PubMed.
- G. Gao, J. I. Dallmeyer and J. F. Kadla, Biomacromolecules, 2012, 13, 3602 CrossRef CAS PubMed.
- M. Ago, K. Okajima, J. E. Jakesh, S. Park and O. J. Rojas, Biomacromolecules, 2020, 161, 359 Search PubMed.
- D. B. Schuepfer, F. Badaczewski, J. M. Guerra-Castro, D. M. Hofmann, C. Heiliger, B. Smarsly and P. J. Klar, Carbon, 2020, 161, 359 CrossRef CAS.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421 CrossRef CAS.
- R. Devasia, C. P. R. Nair, R. Sadhana, N. S. Babu and K. N. Ninan, J. Appl. Polym. Sci., 2006, 100, 3055 CrossRef CAS.
- M. Wu, Q. Wang, K. Li, Y. Wu and H. Liu, Polym. Degrad. Stab., 2012, 97, 1511 CrossRef CAS.
- T. Wang, X. He, W. Gong, K. Sun, W. Lu, Y. Yao, Z. Chen, T. Sun and M. Fan, Fuel, 2020, 278, 117985 CrossRef CAS.
- M. Moorthy, M. Karnan, S. S. Balaji, S. Gokulnath and M. Sathish, J. Electroanal. Chem., 2022, 924, 116857 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 Schematic illustration for the formation mechanism of PAN-g-lignin copolymer. Fig. S2 FE-SEM images of carbonized PAN-g-lignin nanofibers at 800 °C (a) and 1200 °C (b). Fig. S3 Plot of fiber diameter and sheet resistance against carbonization temperature. Fig. S4 High-resolution XPS spectra of C 1s and N 1s for C-800 (A and C) and C-1200 (B and D), respectively. Fig. S5 Water contact angle of CNFs derived at different carbonization temperatures. Fig. S6 EIS spectra of CNFs (C-1000) electrode before and after cyclic stability measurements. Fig. S7 Schematic illustration for the fabrication of solid-state symmetric supercapacitor (SSC) device. Table S1: Supercapacitor performance comparison with literature reports. See DOI: https://doi.org/10.1039/d3ma00194f |
| This journal is © The Royal Society of Chemistry 2023 |