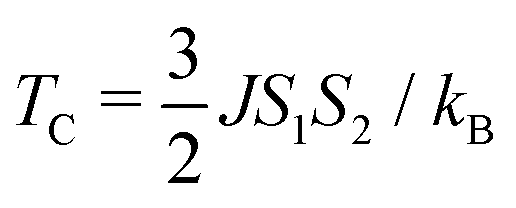DOI:
10.1039/D3MA00084B
(Paper)
Mater. Adv., 2023,
4, 2372-2378
Signature of magnetism in 2D-chromia: 2D analog of the natural α-Cr2O3 mineral and its heterostructure with graphene†
Received
23rd February 2023
, Accepted 24th April 2023
First published on 15th May 2023
Abstract
The family of intrinsic 2D magnetic materials is consistently expanding day by day due to their numerous applications and easy availability. The latest member of the family is 2D-chromia – the 2D analog of the bulk chromia mineral (α-Cr2O3). In the present work, we have thoroughly explained and investigated its structural, electronic and magnetic properties. Our first-principles calculations show that it exhibits a ferromagnetic ground state with an appreciable magnetic moment. The large value of the magnetocrystalline anisotropy energy (MAE) leads to its stability against thermal fluctuations. We also show that due to the large radius of the Cr atom, exchange interactions dominate over Cr–O–Cr antiferromagnetic superexchange interactions that result in the existence of strong magnetic ordering in contrast to bulk chromia. However, the presence of a large band gap limits its application to a great extent. We have resolved this existing ambiguity by making its suitable van der Waals (vdW) heterostructure with the graphene. Our results indicate that the resulting materials may pave the way for spintronic applications and open up a new path for research in other 2D oxides of transition metals.
1 Introduction
The latest progress in the development of two-dimensional (2D) magnets has opened up boundless opportunities for the fabrication of new devices with numerous applications, such as data storage, spintronics, magneto-optics, quantum computation and sensors.1–3 These 2D magnetic materials can be broadly classified into two categories: (i) extrinsic (e.g., graphene, transition metal dichalcogenide monolayers, etc.) and (ii) intrinsic (e.g., CrI3 monolayers, hematene, etc.). The former includes all those 2D materials that are non-magnetic in their pristine form but become magnetic upon (i) the introduction of defects (e.g., via doping with a suitable transition metal atom, creating vacancies, attaching an adatom, substitution, or forming nanoribbons), (ii) the addition of interface effects (i.e., a 2D crystal in the proximity of a magnetic substrate) or (iii) the inclusion of intercalation effects (i.e., a magnetic species is inserted in between the 2D layers).4–14 However, only localized magnetic moments can be induced through these methods, and extending this order up to a long range remains a big hurdle to overcome. In addition, if by any means the order is extended, then controlling or switching of the spin states is a significant hindrance to the process of device fabrication for these extrinsic 2D materials. Keeping this in mind, researchers nowadays rely mainly on the latter category, which includes all those 2D materials in which long-range magnetic order exists. Moreover, these intrinsic 2D magnets not only overcome the existing problem of the Mermin–Wegner theorem through MAE but also have the reliability of tuning their magnetic properties using various techniques.15 Recent advancements in these materials have boosted the intense interest of researchers to explore the use of 2D magnets in nanoscale devices. The starting members of intrinsic 2D ferromagnetic materials were monolayers of CrI3 and Cr2Ge2Te6.16 It has been observed that both of these are ferromagnets, which have out-of-plane spin orientations and magnetic ordering, are highly sensitive to the number of layers. In addition, the nature of the magnetic coupling can be easily tuned by applying strain or an electric field. Although they have emerged as exciting intrinsic magnetic materials as far as collective magnetism is concerned, their inherent low Curie temperature (TC) has limited their applications to a great extent.17–20 The next member that falls in this family is hematene (the 2D form of the iron ore hematite). Balan et al.21 have synthesized 2D hematene with a weak magnetic moment from the bulk structure of antiferromagnetic hematite. As it is less expensive, abundant and stable, even without a substrate, it has gathered the attention of many researchers. Bandyopadhyay et al.22 have reported that pristine hematene is ferrimagnetic with a weak net magnetic moment, in which the superexchange interactions (Fe–O–Fe) tend to oppose strong magnetic coupling. They also attempted to suppress this superexchange by alloying, which in turn makes the hematene ferromagnetic. Previously, our group also examined the effect of strain on the structural, electronic and magnetic properties of hematene.23 It was predicted that hematene has a very high TC (298 K) in the pristine form, which further increases with an increase in compressive strain (increasing by 21.1% at −6%). At present, van der Waals (vdW) heterostructures are emerging as a hot research topic due to their versatility of combining exciting and valuable features of two or more different materials.24–28 In another study, our group also examined the effect of the presence of graphene on the structural, electronic and magnetic properties of hematene by combining them in a vdW heterostructure.29 It was noted that the resulting heterostructure overcomes all the existing ambiguities of both graphene and hematene. Along the same lines, in the present work, we aim to study the electronic and magnetic properties of 2D-chromia (Cr2O3) in its pristine form, which is similar to 2D-hematene (Fe2O3) in every sense except that Cr is present in place of Fe, within the framework of density functional theory (DFT). Moreover, the second aim of our study is to analyze the corresponding change in these properties of 2D-chromia via modeling its suitable heterostructure with graphene. To our strongest belief, this type of heterostructure has not been targeted by any other research groups so far.
2 Computational approach
All the calculations for 2D-chromia and its corresponding heterostructure were simulated using the projector augmented wave (PAW) method as implemented in the Vienna ab initio simulation package (VASP) based on DFT.30,31 The exchange–correlation potentials were incorporated by selecting the proper Hubbard parameter (U) along with Perdew–Burke–Ernzerhof (PBE) parameterization within the generalized gradient approximation (GGA).32 We performed a series of calculations by adopting different values of U to take in to account the correct onsite Cr-d Coulomb correlations, and U = 5 eV served that purpose. The structures were relaxed over different energy cut-offs (450–520 eV) and vacuum-layer thicknesses (15–22 Å). The 15 × 15 × 1 and 9 × 9 × 1 k-meshes were used for sampling the Brillouin zone in the 2D-chromia and graphene/2D-chromia heterostructures, respectively. The DFT-D3 method with zero damping33 was employed to incorporate the vdW interactions between 2D-chromia and graphene. The electrons were relaxed under very tight convergence criteria until the energy difference between the consecutive iterations became 10−8 eV. For ionic relaxation, a conjugate-gradient algorithm was adopted to reduce the force on each atom up to 0.001 eV. The vibrational stabilities of the resulting structures were verified with the help of the phonon spectrum using the Phonopy package and the density functional perturbation theory (DFPT) approach.34 Their thermal stabilities were also checked using ab initio molecular dynamics (AIMD) simulations.35 To understand better the bonding between different atoms, Bader charge analysis was performed to study the charge transfer among them properly.36 The various exchange interactions were estimated using the Heisenberg Hamiltonian.37 In addition, the MAE was calculated in non-self-consistent calculations using the magnetic force theorem38 and incorporating spin–orbit coupling.
3 Results and discussion
Novel 2D-chromia is exfoliated from the naturally occurring bulk mineral chromia (Cr2O3) in a similar way that 2D hematene is synthesized from α-hematite. Out of the α, β and γ phases of chromia, only the α phase undergoes 2D layer formation, as heavy surface reconstruction in β and γ make these phases highly unstable when behaving as a 2D material. From the bulk α phase, 2D-chromia can be exfoliated further in two orientations, along [100] and [001]. However, experimentally, the [001] phase turns out to be more stable. That is why we have primarily focused on this phase (Fig. 1) for our detailed theoretical investigation.
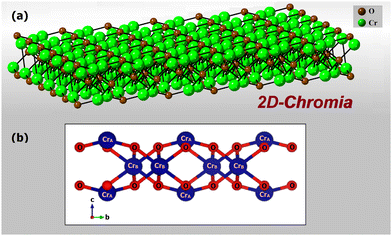 |
| | Fig. 1 (a) Schematic representation of 2D-chromia etched from the bulk α chromia along the [001] direction, and (b) optimized structure of pristine 2D-chromia, which consists of O atoms and two different types of Cr atoms. | |
The crystal structure of 2D-chromia is quite interesting as its unit cell consists of ten atoms: four Cr atoms and six O atoms. 2D-chromia as a whole has a hexagonal structure, with the lattice parameter a = 5.04 Å, and is composed of two different sublattices. Each sublattice has an occupancy of two Cr atoms. This means all four Cr atoms are not in the same environment. Two Cr atoms (i.e., CrA) are enclosed by three O atoms whereas the remaining two (CrB) are surrounded in an octahedron of six O atoms (Fig. 1). The optimized lattice parameters, unit vectors and positions of atoms in the unit cell of 2D-chromia, as obtained from relaxation calculations, are given in Table 1. Considering all the possible spin alignments within the inter- and intra-sublattices, five different magnetic configurations are possible (Table 2).
Table 1 Optimized lattice parameters, unit vectors and atomic positions for the pristine 2D-chromia unit cell
| Parameters |
a = 5.04 Å |
|
|
η = 1.48 Å |
|
|
ζ = 1.09 Å |
|
|
δ = 0.0124 |
| Unit vectors |
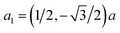
|
|
|
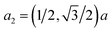
|
| Atomic positions |
CrA1 = (0, 0, η) |
|
|
CrA2 = (0.3, 0.6, 0) |
|
|
CrB1 = (0, 0, −η) |
|
|
CrB2 = (0.6, 0.3, 0) |
|
|
O1 = (0, 0.6 − δ, ζ) |
|
|
O2 = (0.3 + δ, 0.3 + δ, ζ) |
|
|
O3 = (0.6 − δ, 0, ζ) |
|
|
O4 = (0,0.3 + δ, −ζ) |
|
|
O5 = (0.3 + δ, 0, −ζ) |
|
|
O6 = (0.6 − δ, 0.6 − δ, ζ) |
Table 2 Energy (E0) and total spin magnetic moment (μs) of different possible states of 2D-chromia
| Configuration |
CrA |
CrB |
E
0 (eV) |
μ
s (μB) |
|
E
1
|
↑↑ |
↓↓ |
−73.84 |
0.13 |
|
E
2
|
↑↓ |
↑↓ |
−73.98 |
0.00 |
|
E
3
|
↑↑ |
↑↓ |
−74.03 |
5.70 |
|
E
4
|
↑↓ |
↑↑ |
−74.04 |
5.83 |
|
E
5
|
↑↑ |
↑↑ |
−74.09 |
11.81 |
Clearly, the E5 configuration in which all the Cr atoms of both sublattices are aligned parallel to each other turns out to be highly stable. This result is in complete contrast with hematene in which E1 is the most energetically favorable among its five configurations.23 We primarily computed the structural stability of this most stable configuration (E5) using the formation energy (Efor) and the following expression23,39
| | | Efor = E2D-ch − 4ECr − 6EO | (1) |
where
E2D-Ch denotes the ground state energy of 2D-chromia in the
E5 configuration, and
ECr and
EO represent the ground state energy of isolated Cr and O atoms, respectively.
Efor turns out to be negative (−1.79 eV), which indicates the structural stability of this configuration. The thermal and dynamical stability of the same configuration were also checked, and the related plots are shown in the ESI
† (Fig. S1 and S2). As is known, the primary factor in determining the magnetic state in such systems is the comparison of the anti-ferromagnetic superexchange and direct exchange interactions. The radius of the Cr atom in 2D-chromia is more than that of the Fe atom in hematene, this might be the reason for the suppression of the super exchange (Cr–O–Cr) interaction between the nearest neighboring Cr atoms and the support of the direct Cr–Cr exchange interaction. The spin magnetic moment (
μs) of the Cr
A atom is 3.31
μB and that of the Cr
B atom is 2.64
μB, both of which align in a parallel fashion with a total
μs of 11.81
μB. Since each Cr
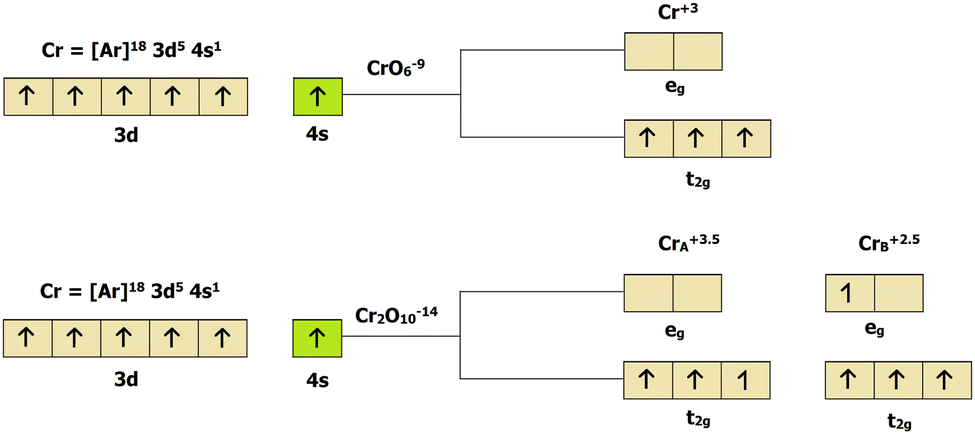
B atom in 2D-chromia is bonded to six O atoms, this octahedral crystal-field splitting uplifts the degeneracy of the d orbitals, by breaking them into their respective e
g and t
2g orbitals (
Fig. 2). If we assume that 2D-chromia exists as an isolated cluster (CrO
69−), then Cr will have a +3 oxidation state and both Cr atoms (Cr
A and Cr
B) must have an identical spin magnetic moment of 3
μB. This fact is not in agreement with our calculated results since both have different magnetic moments. This means that, instead of an isolated cluster, 2D-chromia must exist as a dimer (Cr
2O
1014−) in which the Cr atoms have mixed-valence oxidation states (+2.5 and +3.5), which is solely responsible for the different magnetic moments of the Cr atoms and the ferromagnetic nature of the material.
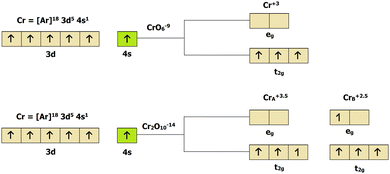 |
| | Fig. 2 Crystal field splitting of the 3d orbitals of 2D-chromia in the isolated cluster CrO69 − (top) and the dimer Cr2O1014− (bottom). | |
The concept of mixed-valence oxidation states is also verified via a study of the charge redistribution among various Cr and O atoms (Table 3). Clearly, the CrB atoms lose more charges than the CrA atoms, proving their existence in different oxidation states. Furthermore, the magnetic response is explained using spin density plots (Fig. 3). Clearly, Fig. 3 indicates that the valence charge distribution is different for both majority and minority spin channels. As each CrA atom has six CrB atoms as its first nearest neighbors, and each CrB atom has three CrA atoms as its first nearest neighbors, therefore, the CrA–CrB interactions will dominate over the CrA–CrA and CrB–CrB interactions. In addition, the CrA atom has a valencespherical valence electron cloud, whereas the electron cloud becomes deteriorated for the CrB atom. Hence, the CrB atom loses more charge than CrA, which is also evident from the amount of charge transfer as listed in Table 3.
Table 3 Calculated amount of charge transfer from CrA and CrB atoms to O atoms using Bader charge analysis
| Element |
Charge transfer (in electrons) |
| CrA |
+2.7 |
| CrB |
+3.4 |
| O |
−1.99 |
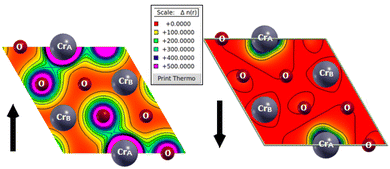 |
| | Fig. 3 Spin-resolved electronic valence charge density of pristine 2D-chromia. | |
The existence of long-range magnetic order in 2D materials is mainly due to the MAE as it can provide stability to the structure against thermal fluctuations and overcomes the problem of the Mermin–Wagner theorem.15 The physical origin of the MAE lies basically in spin–orbit coupling. When it is included, then the Hamiltonian not only depends upon the angle between the spins but also on the angle that the spin makes with respect to the crystal axis. Therefore, there will be certain special orientations of the crystal for which this anisotropy energy will be greater, and for other orientations it will be less. In this way, the MAE is determined in a non-self-consistent manner by rotating spins along all possible directions:38,40,41
Here,
E[uvw] is the sum of occupied energy eigenvalues of the respective bands for the magnetization vector aligned along different possible directions, and
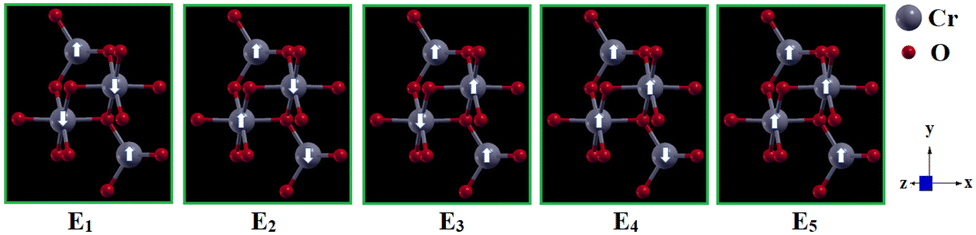
E0 is the energy of the most stable spin orientation. Thus, pristine 2D-chromia has a significant uniaxial anisotropy value (0.80 meV per Cr atom) due to its hexagonal symmetry. Since there are five possible different spin configurations (
Fig. 4) for pristine 2D-chromia due to the presence of the two sublattices, each of which contains two inequivalent atoms (Cr
A and Cr
B), so there exist three coupling constants, such that
J11/
J12/
J22 represent exchange constants due to magnetic interactions between Cr
A–Cr
A/Cr
A–Cr
B/Cr
B–Cr
B atoms, respectively.
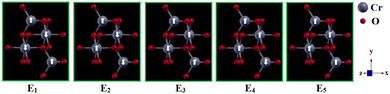 |
| | Fig. 4 Schematic representation of the five different configurations of the pristine 2D-chromia unit cell. | |
We can easily model these constants using the Heisenberg Hamiltonian:37
| | 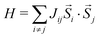 | (3) |
The eigenvalues of these five configurations can be found as:
| | J11S12 − 4J12![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) 1·2 + J22S22 = E1 1·2 + J22S22 = E1 | (4) |
| | | J11S12 + 4J121·2 + J22S22 = E5 | (8) |
These exchange interactions in 2D-chromia are only due to partially occupied d orbitals of the Cr atom, where the valence electronic configuration of Cr is 3d54s1. The electronegativity of the O atom is −2 and there are six O atoms per unit cell, which can withdraw a total charge of 12e from the Cr atoms. Since CrA/CrB exists in the +2.5/+3.5 oxidation state, thus their valence state becomes 3d3.5/3d2.5. This leads to S1 = 3.5/2 and S2 = 2.5/2. By substituting all of these values and solving eqn (4)–(8), we obtain J11 = 0.06 meV, J12 = 8.58 meV and J22 = −7.98 meV.
| | 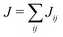 | (9) |
Now, from J, S1 and S2, TC42 can be calculated as:
| | 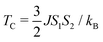 | (10) |
Here,
kB is the Boltzmann constant.
TC for pristine 2D-chromia turns out to be 22 K, which is in agreement with the experimental value of 12 K.
22 2D-chromia in the
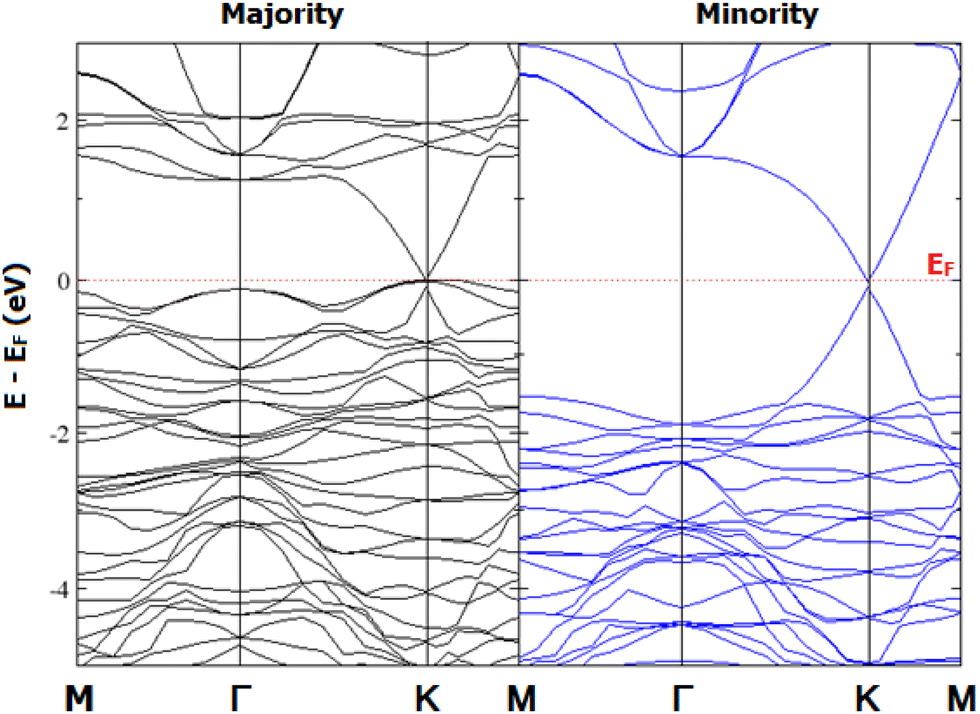
E5 configuration shows different band gaps and a total DOS for both the majority spin channel (MAC) and the minority spin channel (MIC) (
Fig. 5). The indirect gap of ∼1.12/3.76 eV along
K–
Γ/
M–
Γ occurs in the MAC/MIC, respectively. Thus, 2D-chromia behaves as a wide band gap magnetic semiconductor.
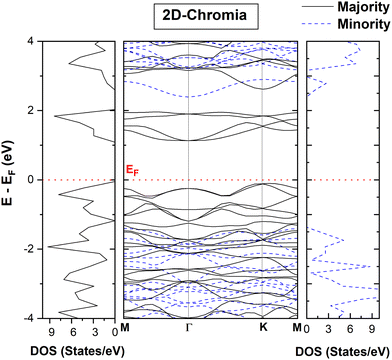 |
| | Fig. 5 Spin-resolved band structure and total density of states (DOS) of pristine 2D-chromia. | |
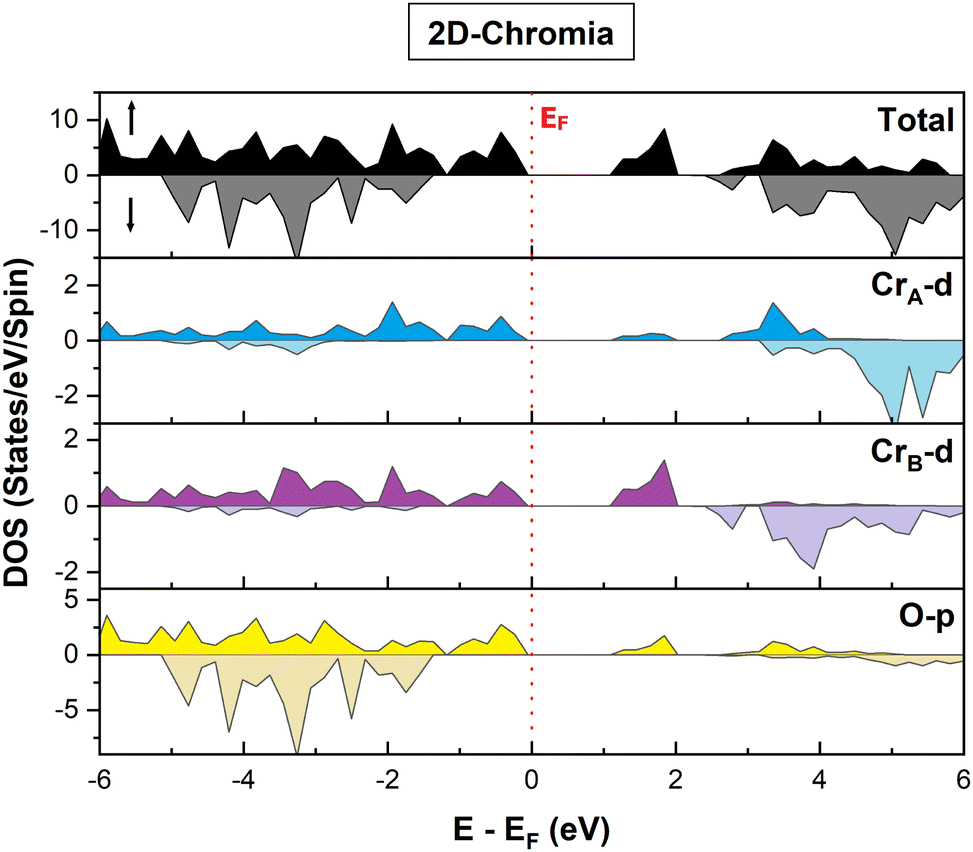
The electronic properties seem to be quite fascinating, especially in close proximity to the Fermi level (EF). The major contribution to the total DOS in the MAC arises from hybridization of the p orbital of O atoms with the d orbital of CrA and CrB (Fig. 6). However for the MIC, the maximum contribution is mainly due to the O p orbital with little contribution from the CrA and CrB d orbitals. Moreover, the gap between the d orbitals of the Cr atoms confirms the trace of crystal-field splitting among them. In addition, the significant difference in the orbital projected DOS of the CrA and CrB atoms is evident from the fact that both have different oxidation states, as explained earlier.
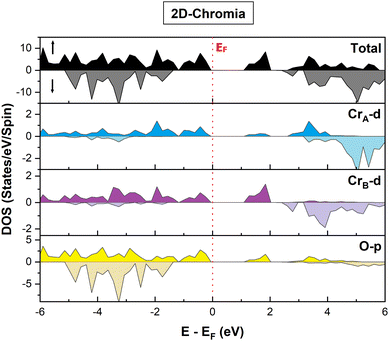 |
| | Fig. 6 Orbital projected DOS of pristine 2D-chromia in majority and minority spin channels. | |
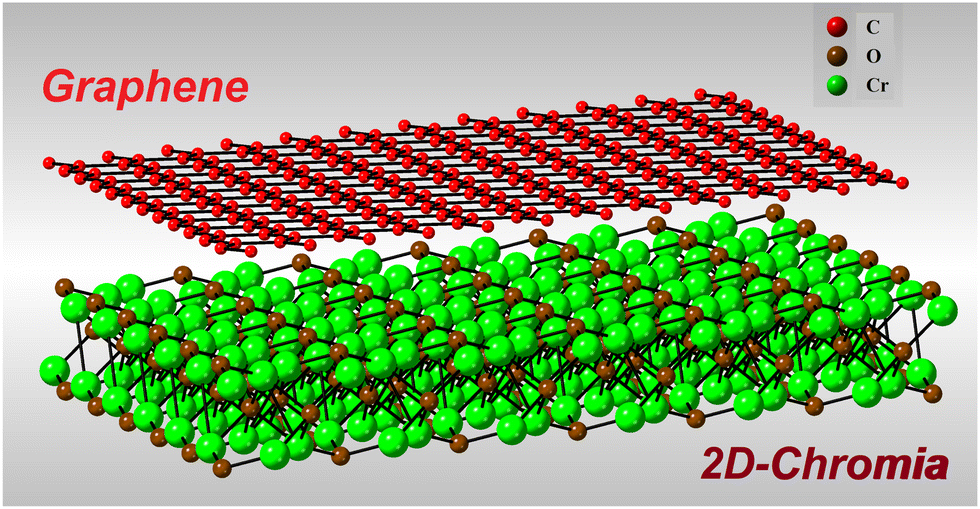
4 Graphene/2D-chromia heterostructure
After thorough checking of the magnetic, structural and electronic properties of 2D-chromia, the robustness of these properties was further analyzed by making its heterostructure with graphene. Since the lattice parameter of graphene (2.456 Å) is almost half that of 2D-chromia, thus the lattice mismatch was settled by combining a 2 × 2 × 1 supercell of graphene with a 1 × 1 × 1 primitive unit cell of 2D-chromia (Fig. 7). In doing so, the lattice mismatch between both layers turns out to be 2%. Now, there can be three different ways to achieve this lattice mismatch. The first is to compress the 2D-chromia layer by 2%, the second is to stretch the 2 × 2 × 1 supercell of graphene by 2%, and the third is to compress the 2D-chromia layer by 1% and also to stretch the 2 × 2 × 1 supercell of graphene by 1%. Out of these, we considered the third case and, after optimization, the lattice parameter of the resulting heterostructure turned out to be 5.0 Å, which indicates that the 2D-chromia monolayer is compressed by 0.7% and the graphene monolayer is stretched by 1.7%. This much strain can be easily considered for the formation of a heterostructure since we verified that, through performing calculations for (i) the 2D-chromia monolayer by adding a 1% compressive strain, and (ii) the graphene monolayer by adding a 2% tensile strain, the electronic and magnetic properties came out exactly same as those before the strain was applied.
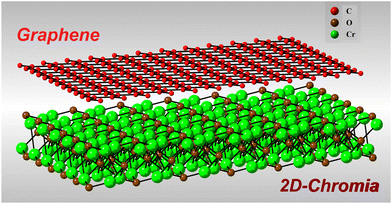 |
| | Fig. 7 Schematic representation of the graphene/2D-chromia heterostructure comprising a 2 × 2 × 1 supercell of graphene and a 1 × 1 × 1 unit cell of 2D-chromia. | |
In addition, it is very important to consider different stacking positions. For the present heterostructure, two different types of stacking exist (AA, and AB), and each stacking type consists of three different cases. The first is AA stacking in which the atoms of both layers have identical lateral coordinates, e.g., the centre of each regular carbon atom hexagon of the graphene monolayer is exactly above a CrA, CrB or O atom of the 2D-chromia monolayer. The second is AB stacking in which the centre of each regular carbon atom hexagon of the graphene monolayer is displaced by the vector equal to the edge of the hexagon, such that the hexagons are not aligned exactly above any of the CrA, CrB or O atoms of the 2D-chromia monolayer. Out of all these cases, the one with AB stacking in which the centres of the regular carbon atom hexagons of the graphene monolayer are displaced by the vector equal to the edge of the hexagon with respect to the CrA atom of the 2D-chromia monolayer turns out to be energetically favorable, and we considered the same for all the simulations of the present heterostructure. The interlayer spacing between graphene and the 2D-chromia layer turns out to be 3.41 Å. The structural stability of the resulting heterostructure is calculated using the binding energy, the related formula and the values of the binding energies in all five configurations, as given in the (Table S1, ESI†).
The presence of graphene hardly influences the magnetic properties of 2D-chromia, and the heterostructure remains in the most stable E5 configuration with an almost similar magnetic moment (Table S1, ESI†). However, there is a complete contrast in the band gap of the heterostructure from that of graphene and the 2D-chromia monolayer. Pristine graphene and the 2D-chromia monolayer have separate band gaps of 0/0 eV and 1.12/3.76 eV in the MAC/MIC, respectively, whereas their heterostructure shows half-metallic behavior (Fig. 8). This may be because when graphene and 2D-chromia are combined, the C atom p orbitals of graphene become hybridized with the d and p orbitals of the Cr and O atoms of 2D-chromia, respectively. The Dirac cone of graphene remains preserved in the heterostructure and helps to reduce the gap in both spin channels. As there is no gap in the MAC and a 0.25 eV gap in the MIC, the resulting heterostructure becomes 100% spin polarized and shows half-metallic behavior.
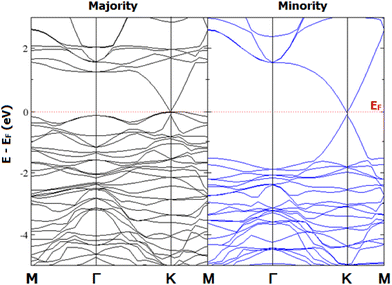 |
| | Fig. 8 Calculated spin polarized band structure of the graphene/2D-chromia heterostructure in the MAC and MIC. | |
5 Conclusion
We have studied the electronic and magnetic properties of pristine 2D-chromia theoretically via the DFT approach. Our results indicate that pristine 2D-chromia is a magnetic semiconductor. This ferromagnetic ordering in 2D-chromia is mainly because Cr–Cr exchange interactions dominate over O-mediated Cr–O–Cr superexchange interactions in contrast to its bulk analog. In addition, the dimer model was used to explain the different oxidation states of Cr atoms and their respective magnetic moments. However, like bulk chromia, the dimer acts as a semiconductor with large band gaps. This problem was overcome by forming its suitable heterostructure with graphene. In the heterostructure, all of the magnetic properties of pristine 2D-chromia remain intact, but it shows half-metallic behavior with 100% spin polarization. Thus, the graphene/2D-chromia heterostructure can play a significant role in spintronics and other memory-based storage devices. We hope that our results will help researchers to expand their research towards a microscopic understanding of the magnetism of other transition metal oxides in 2D geometry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The computations in this work were performed using the Param Shavak supercomputing machine available at the Department of Physics, Kurukshetra University, Kurukshetra (Haryana), the HPC Cluster of SPS, JNU, New Delhi and the National PARAM Supercomputing Facility (NPSF) of the Centre for the Development of Advanced Computing (C-DAC), Pune, India. R. S. would like to acknowledge the financial support from Council of Scientific & Industrial Research (CSIR), New Delhi (India) in the form of a Senior Research Fellowship (SRF).
References
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- A. S. Mayorov, R. V. Gorbachev, S. V. Morozov, L. Britnell, R. Jalil, L. A. Ponomarenko, P. Blake, K. S. Novoselov, K. Watanabe and T. Taniguchi,
et al.
, Nano Lett., 2011, 11, 2396–2399 CrossRef CAS PubMed.
- K. Wakabayashi, Mater. Nanoarchitecton., 2018, 109–123 Search PubMed.
- E. J. Santos, D. Sánchez-Portal and A. Ayuela, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125433 CrossRef.
- A. Krasheninnikov, P. Lehtinen, A. S. Foster, P. Pyykkö and R. M. Nieminen, Phys. Rev. Lett., 2009, 102, 126807 CrossRef CAS PubMed.
- X. Hu, W. Zhang, L. Sun and A. V. Krasheninnikov, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195418 CrossRef.
- X. Hu, N. Wan, L. Sun and A. V. Krasheninnikov, J. Phys. Chem. C, 2014, 118, 16133–16139 CrossRef CAS.
- J. Karthikeyan, H.-P. Komsa, M. Batzill and A. V. Krasheninnikov, Nano Lett., 2019, 19, 4581–4587 CrossRef CAS PubMed.
- G. Z. Magda, X. Jin, I. Hagymási, P. Vancsó, Z. Osváth, P. Nemes-Incze, C. Hwang, L. P. Biro and L. Tapaszto, Nature, 2014, 514, 608–611 CrossRef CAS PubMed.
- Y.-C. Lin, P.-Y. Teng, P.-W. Chiu and K. Suenaga, Phys. Rev. Lett., 2015, 115, 206803 CrossRef PubMed.
- P. M. Coelho, H.-P. Komsa, K. Lasek, V. Kalappattil, J. Karthikeyan, M.-H. Phan, A. V. Krasheninnikov and M. Batzill, Adv. Electron. Mater., 2019, 5, 1900044 CrossRef.
- S. Wang, A. Robertson and J. H. Warner, Chem. Soc. Rev., 2018, 47, 6764–6794 RSC.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao and T. Hu,
et al.
, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS PubMed.
- N. D. Mermin and H. Wagner, Phys. Rev. Lett., 1966, 17, 1133 CrossRef CAS.
- L. Ke and M. I. Katsnelson, npj Comput. Mater., 2021, 7, 4 CrossRef CAS.
- K. S. Burch, D. Mandrus and J.-G. Park, Nature, 2018, 563, 47–52 CrossRef CAS PubMed.
- C. Gong and X. Zhang, Science, 2019, 363, eaav4450 CrossRef CAS PubMed.
- D. L. Cortie, G. L. Causer, K. C. Rule, H. Fritzsche, W. Kreuzpaintner and F. Klose, Adv. Funct. Mater., 2020, 30, 1901414 CrossRef CAS.
- H. Li, S. Ruan and Y.-J. Zeng, Adv. Mater., 2019, 31, 1900065 CrossRef PubMed.
- A. P. Balan, S. Radhakrishnan, C. F. Woellner, S. K. Sinha, L. Deng, C. De Los Reyes, B. M. Rao, M. Paulose, R. Neupane and A. Apte,
et al.
, Nat. Nanotechnol., 2018, 13, 602–609 CrossRef PubMed.
- A. Bandyopadhyay, N. C. Frey, D. Jariwala and V. B. Shenoy, Nano Lett., 2019, 19, 7793–7800 CrossRef CAS PubMed.
- R. Singla, T. A. Hackett, S. Kumar, J. Sharma and M. K. Kashyap, Nanoscale Adv., 2020, 2, 5890–5896 RSC.
- C. R. Dean, A. F. Young, I. Meric, C. Lee, L. Wang, S. Sorgenfrei, K. Watanabe, T. Taniguchi, P. Kim and K. L. Shepard,
et al.
, Nat. Nanotechnol., 2010, 5, 722–726 CrossRef CAS PubMed.
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim and Y. I. Song,
et al.
, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- A. K. Geim and I. V. Grigorieva, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- R. Singla, J. Thakur, P. Rani, S. Kumar, T. A. Hackett and M. K. Kashyap, Vacuum, 2020, 182, 109685 CrossRef CAS.
- R. Singla, S. Kumar, T. A. Hackett, A. H. Reshak and M. K. Kashyap, J. Alloys Compd., 2021, 859, 157776 CrossRef CAS.
- R. Singla and M. K. Kashyap, Indian J. Phys., 2021, 1–6 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- M. Parrinello and A. Rahman, Phys. Rev. Lett., 1980, 45, 1196 CrossRef CAS.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- W.-B. Zhang, Q. Qu, P. Zhu and C.-H. Lam, J. Mater. Chem. C, 2015, 3, 12457–12468 RSC.
- R. Wu and A. J. Freeman, J. Magn. Magn. Mater., 1999, 200, 498–514 CrossRef CAS.
- P. Rani, M. K. Kashyap, R. Singla, J. Thakur and A. H. Reshak, J. Alloys Compd., 2020, 835, 155325 CrossRef CAS.
- V. Mkhitaryan and L. Ke, Phys. Rev. B, 2021, 104, 064435 CrossRef CAS.
- L. Ke and M. Van Schilfgaarde, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 014423 CrossRef.
- L. Webster and J.-A. Yan, Phys. Rev. B, 2018, 98, 144411 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bf
*bf