
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of nitrogen on the synthesis of porous carbons by iron-catalyzed graphitization†
Robert D.
Hunter
a,
Emily C.
Hayward
a,
Glen J.
Smales
 b,
Brian R.
Pauw
b,
A.
Kulak
c,
Shaoliang
Guan
de and
Zoe
Schnepp
*a
b,
Brian R.
Pauw
b,
A.
Kulak
c,
Shaoliang
Guan
de and
Zoe
Schnepp
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: z.schnepp@bham.ac.uk
bBundesanstalt für Materialforschung und - prüfung (BAM), Unter den Eichen 87, Berlin 12205, Germany
cSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
dHarwellXPS, Research Complex at Harwell Rutherford Appleton Laboratory, Didcot, OX11 0FA, UK
eSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
First published on 22nd March 2023
Abstract
This paper reports a systematic study into the effect of nitrogen on iron-catalyzed graphitization of biomass. Chitin, chitosan, N-acetylglucosamine, gelatin and glycine were selected to represent nitrogen-rich saccharides and amino-acid/polypeptide biomass precursors. The materials were pyrolyzed with an iron catalyst to produce carbons with a wide range of chemical and structural features such as mesoporosity and nitrogen-doping. Many authors have reported the synthesis of nitrogen-doped carbons by pyrolysis and these have diverse applications. However, this is the first systematic study of how nitrogen affects pyrolysis of biomass and importantly the catalytic graphitization step. Our data demonstrates that nitrogen inhibits graphitization but that some nitrogen survives the catalytic graphitization process to become incorporated into various chemical environments in the carbon product.
Introduction
The next generation of energy storage and conversion technologies such as batteries1 and proton exchange membrane (PEM) fuel cells2 rely on low-cost, sustainable carbon materials. To function efficiently, the carbons need to have tailored porosity (for electrolyte access or flow of reactants/products) and graphitic features (for electronic conductivity). Nitrogen doping has also been shown to enhance the properties of carbons for applications such as the oxygen reduction reaction (ORR)3 or potassium ion batteries.4 There are now a plethora of examples in the literature of porous N-doped carbons for energy applications and many of these are synthesized by pyrolysis of N-rich organic precursors.5 The methods frequently use a transition metal such as iron to catalyze the formation of graphitic carbon,6 and in some cases to provide additional functionality in the resulting product.7 However, it is difficult to compare the properties of different N-doped carbons, since the precursors, iron content and heating conditions vary significantly between different reports.Systematic studies are essential for the scientific community to progress materials such as porous carbons towards applications. Even factors such as crucible size and shape have been shown to impact the formation of materials in ceramic synthesis.8 In the field of carbon synthesis from cellulose, different iron salts have been shown to produce carbons with dramatically different porosity and graphitic content.9 Raw lignocellulosic biomasses such as wood or nut shells can generate graphitic carbons with very similar porosity and graphitic content but only if they are milled to fine powders before pyrolysis with iron.10 In another example, glucose (soluble monosaccharide) and cellulose (insoluble polysaccharide) were shown to produce mesoporous carbons with very similar properties after iron-catalyzed graphitization under identical conditions.11 In contrast, starch produced microporous carbons with very little graphitic content, despite starch being a polysaccharide comprised of glucose monomers, like cellulose. The different graphitization mechanism was ascribed to the fact that starch swells in hot water and can therefore form a gel around the iron catalyst precursor. All these examples highlight the importance of studies that compare different carbon precursors in a systematic way to complement the wealth of literature that evaluates the electrocatalytic or energy storage properties of carbons produced from single organic sources.
In this paper, we report a systematic study of the effect of precursor nitrogen content and chemical structure on the formation of carbons by iron-catalyzed graphitization. To achieve this, we selected examples from the two main types of nitrogen-containing biomass: polysaccharides and polypeptides. Chitin (Fig. 1a) is an abundant polysaccharide, which is found in crustacean shells, typically as a composite with calcium carbonate. Polypeptides are a completely different class of biomass. They are polymers comprised of amino acids, where nitrogen is incorporated into the backbone of the polymer. Gelatin (a readily available derivative of collagen and abundant byproduct of the meat industry) was selected as an example of a polypeptide (Fig. 1d). Given that cellulose, starch and glucose behave so differently during graphitization, despite all being based on glucose, we also selected nitrogen-containing compounds that were chemically related to chitin and gelatin but with different physical properties. For this, we used chitosan (a soluble deacetylated derivative of chitin, Fig. 1b) and N-acetylglucosamine (GlcNAc), the water-soluble monomer of chitin (Fig. 1c). Finally, we also used glycine (Fig. 1e), the most common monomer found in gelatin. Gelatin is soluble in boiling water, while glycine is soluble in cold water. Importantly, all the above compounds display different physical properties such as solubility and thermal degradation, while representing two important chemical classes of biomass. In this paper we demonstrate that nitrogen inhibits graphitization and that the surface and structural properties of nitrogen-doped carbons vary significantly depending on the choice of organic precursor and amount of iron catalyst.
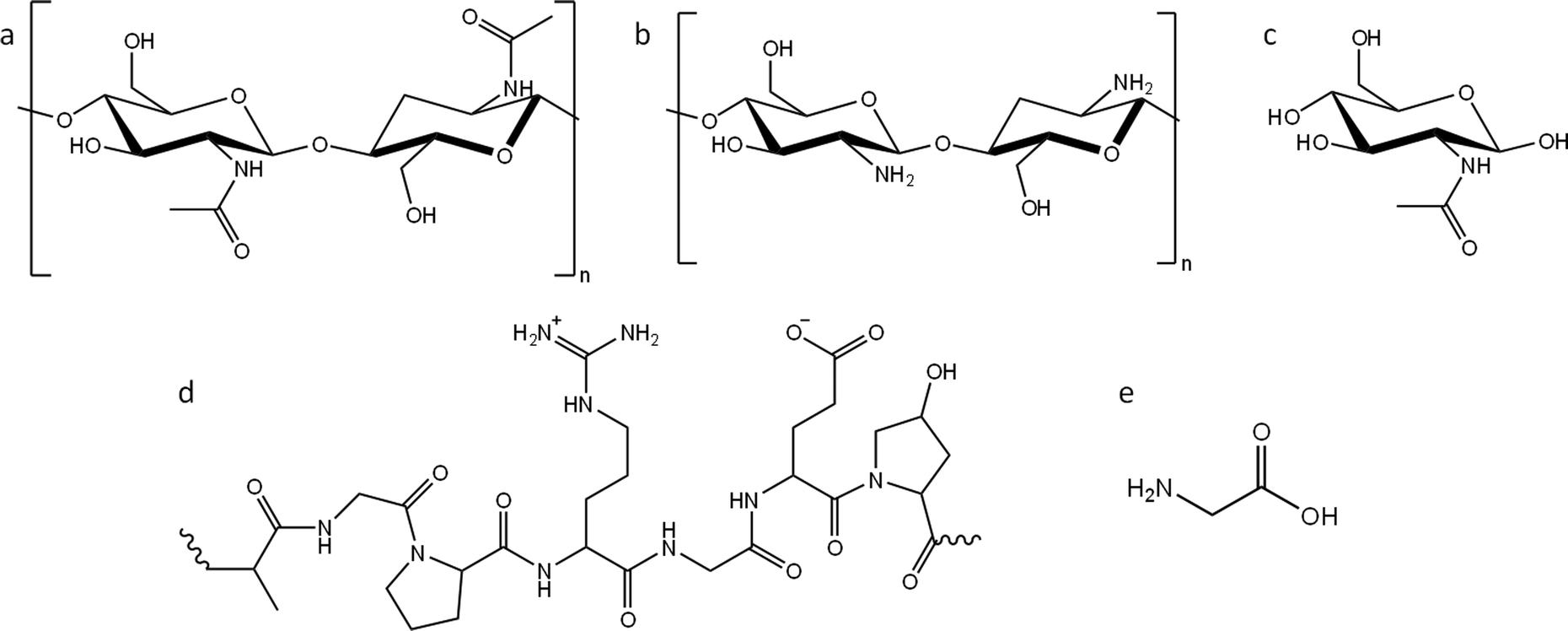 | ||
| Fig. 1 Structures of (a) chitin, (b) chitosan and (c) N-acetylglucosamine (GlcNAc), (d) representative structure of gelatin and (e) structure of glycine. | ||
Experimental
Samples were prepared at two different organic to Fe(NO3)3 ratios of 5 g to 0.67 mmol (low iron) and 5 g to 3.3 mmol (high iron). The preparation varied depending on the physical properties of the organic precursor Briefly, chitin powder (5 g) was mixed with 20 mL of deionized (DI) water containing 0.27 g (0.67 mmol) of Fe(NO3)3·9H2O until fully absorbed. The volume of water was selected to allow complete mixing of the insoluble powder with the iron nitrate solution, without pooling of excess solution. This ensured a homogeneous coating of the chitin powder with the iron salt. Chitosan (5 g) was dissolved in acetic acid (250 mL, 2 wt%) and stirred for 3 hours before addition of 20 mL of DI water containing 0.27 g of Fe(NO3)3·9H2O. N-Acetylglucosamine and glycine samples were prepared by dissolving 5 g of the organic precursor in 25 mL warm DI water then adding 4 mL of DI water containing 0.27 g Fe(NO3)3·9H2O. Gelatin powder (5 g) was dissolved in 50 mL DI water at 70 °C, followed by addition of 4 mL of DI water containing 0.27 g Fe(NO3)3·9H2O. All samples were dried at 70 °C in air before heating in a tube furnace at a rate of 5 °C min−1 under a nitrogen atmosphere with a flow rate of 1 L min−1 to 800 °C. The samples were held at 800 °C for 1 h, unless otherwise stated, before cooling completely to room temperature. Samples with a high iron content were prepared in the same way but with 1.35 g of Fe(NO3)3·9H2O. Control samples from each organic precursor were prepared using the same method but without the addition of iron nitrate solution. Full experimental details can be found in the ESI.†Results and discussion
N-doped porous carbons at low iron loading
Porous carbons were prepared by iron-catalyzed graphitization of chitin, chitosan, N-acetylglucosamine (GlcNAc), gelatin and glycine. All samples were combined with iron nitrate and pyrolyzed at 5 °C min−1 to 800 °C for 1 hour in a nitrogen atmosphere. These conditions and the organic:iron ratio are identical to those used in the study of carbonization of glucose, starch and cellulose, enabling a direct comparison of all the systems. Full details can be found in the Experimental section and ESI.†Fig. 2a shows powder X-ray diffraction (PXRD) patterns for samples of carbon prepared with 0.67 mmol of Fe(NO3)3 per 5 g of organic precursor. A diffraction pattern for cellulose prepared at this iron:biomass ratio is included in the figure as a comparison.11 All the nitrogen-containing precursors show limited graphitization compared to nitrogen-free cellulose. Chitosan and GlcNAc are the only two compounds to show a significant peak in the graphite region and these are very broad and shifted to lower 2-theta, indicative of disordered or turbostratic carbon.12 This is in stark contrast to the strong graphitic carbon peak of cellulose. The polysaccharide samples (chitin, chitosan and GlcNAc) show small peaks which correspond to Fe3C, which is believed by many to be the catalyst in the graphitization process. The very small and broad peaks in this region for glycine and gelatin indicate very small Fe3C particles. This would correlate to previous suggestions that very small Fe/Fe3C nanoparticles are unable to drive catalytic graphitization.11,13 Control samples (without iron) show only very broad peaks indicative of disordered carbon (Fig. S1, ESI†).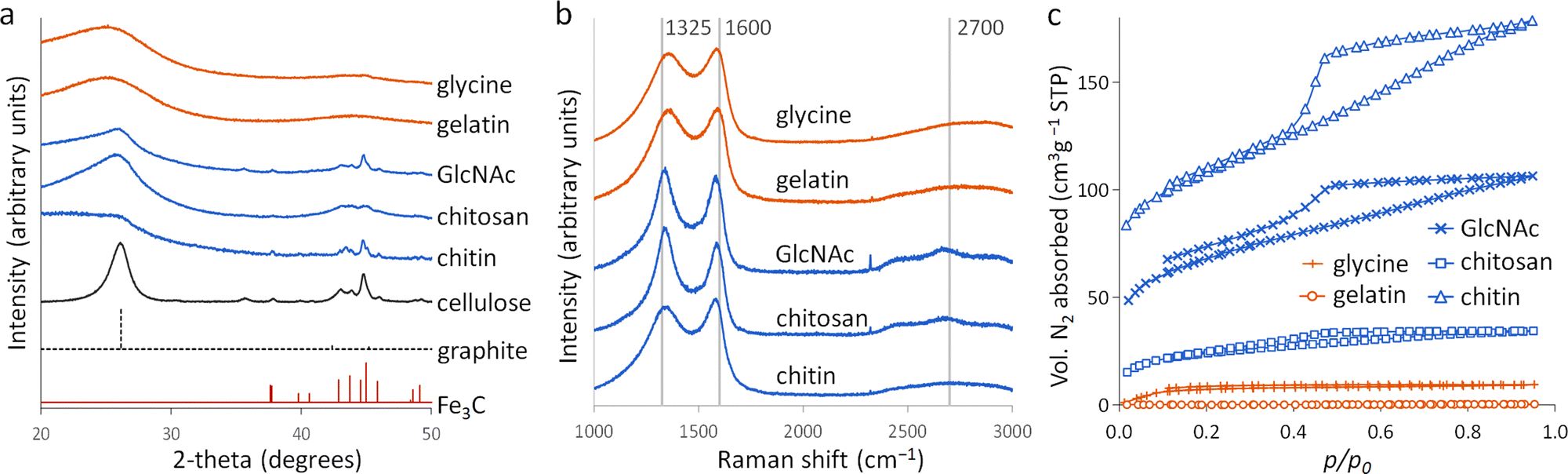 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra and (c) nitrogen sorption isotherms for carbons derived from various N-containing precursors at a ratio of 5 g of organic precursor to 0.67 mmol of Fe(NO3)3. | ||
Further characterization of the carbons with Raman spectroscopy (Fig. 2b) show peaks at approximately 1325 and 1600 cm−1, corresponding to the D and G bands respectively. The G band is present in all graphitic materials and indicates the presence of sp2 hybridised carbon within the sample. The D band is forbidden in pure graphite, so is indicative of a disordered carbon structure. All the iron-doped samples show sharper peaks than corresponding control (no iron) samples (Fig. S2, ESI†). This indicates that iron has generated local order in the form of sp2 hybridized carbon, e.g. small sheet-like structures, even if long-range graphitic order is absent. Values calculated from fitting of the spectra (Table S1, ESI†) show sharper peaks corresponding to the D1 and G bands in the chitosan- and GlcNAc-derived carbons, suggesting a greater degree of ordering than the other samples, consistent with PXRD. In all the samples, the G band is shifted from 1581 cm−1 to a value closer to 1600 cm−1, characteristic of a carbon structure composed of nanocrystalline graphitic domains.
N2 sorption measurements (Fig. 2c) were used to assess the porosity of the carbon samples. Glycine- and gelatin-derived carbons exhibit low porosity: the specific surface area of the gelatin-derived carbon is too low to calculate a meaningful value, while glycine-derived carbon has a specific surface area of just 27 m2 g−1. GlcNAc, chitosan and chitin derived carbons show greater porosity, with specific surface areas of 250, 89 and 390 m2 g−1 respectively. The isotherms for the three saccharides adopt a type IV shape with hysteresis due to capillary condensation, indicative of the presence of mesopores. This is consistent with previous work that shows iron graphitization catalysts are highly mobile and can move through an amorphous carbon matrix to produce graphitic nanotubes or capsules.14 Other calculated adsorptive properties are reported in Table 1. Interestingly, the chitin-derived carbon shows similar adsorptive properties to cellulose-derived carbon, despite much lower graphitization. This suggests that graphitization is not the only mechanism for pore development during pyrolysis of chitin. Porosimetry data for a control sample of chitin (pyrolyzed without the iron catalyst) shows a type IV isotherm and BET surface area of 290 m2 g−1 (Fig. S3 and Table S2, ESI†). This is consistent with previous reports15 and is probably because chitin exists in nature as a complex hierarchical nanocomposite with CaCO3.16 The pores left when CaCO3 was removed are likely to be maintained during the pyrolysis process. All other control samples showed minimal porosity. SEM images of the samples (Fig. S3 and S4, ESI†) show a range of structures. In chitin-derived carbon it is possible to see very small pores, as suggested by porosimetry data. Chitosan has produced a macroporous, sponge-like network, probably caused by gas evolution during the early stages of pyrolysis. Individual nanoparticles were not observable, probably due to them being too small for the resolution of the instrument. However, Fe was observable in all samples by EDXA.
| Precursor | S BET (m2 g−1) | Max Qads (cm3 g−1) | V tot (cm3 g−1) | S micro (m2 g−1) | V micro (cm3 g−1) |
|---|---|---|---|---|---|
| Cellulose | 360 | 180 | 0.27 | 140 | 0.06 |
| Glycine | 27 | 9 | 0.015 | 4 | 0.001 |
| Gelatin | — | — | — | — | — |
| GlcNAc | 250 | 110 | 0.17 | 86 | 0.035 |
| Chitosan | 89 | 34 | 0.054 | 32 | 0.012 |
| Chitin | 390 | 180 | 0.28 | 170 | 0.073 |
Elemental analysis of the carbons shows a significant amount of nitrogen remains in the system after pyrolysis (Table 2). Nitrogen content is highest in the carbons derived from gelatin/Fe(NO3)3 and glycine/Fe(NO3)3. This correlates with the higher levels of nitrogen in the polypeptide and amino acid compared to the saccharide precursors. The overall iron content (derived from residual mass, Table S3, ESI†) is similar for all 5 carbons, suggesting that iron:carbon ratio is not a factor in the different behaviour of the samples. X-Ray photoelectron spectroscopy (XPS) was used to examine the chemistry of the nitrogen in the carbons and the results can be seen in Fig. 3.
| Calculated composition of precursor | Measured composition of carbon product | |||||
|---|---|---|---|---|---|---|
| C% | H% | N% | C% | H% | N% | |
| Glycine | 56 | 12 | 33 | 82 | 0.9 | 17 |
| Gelatin (est.) | 59 | 7 | 34 | 80 | 4 | 16 |
| GlcNAc | 77 | 12 | 11 | 94 | 0.7 | 6 |
| Chitosan | 74 | 11 | 14 | 89 | 1 | 10 |
| Chitin | 78 | 11 | 11 | 92 | 0.9 | 7 |
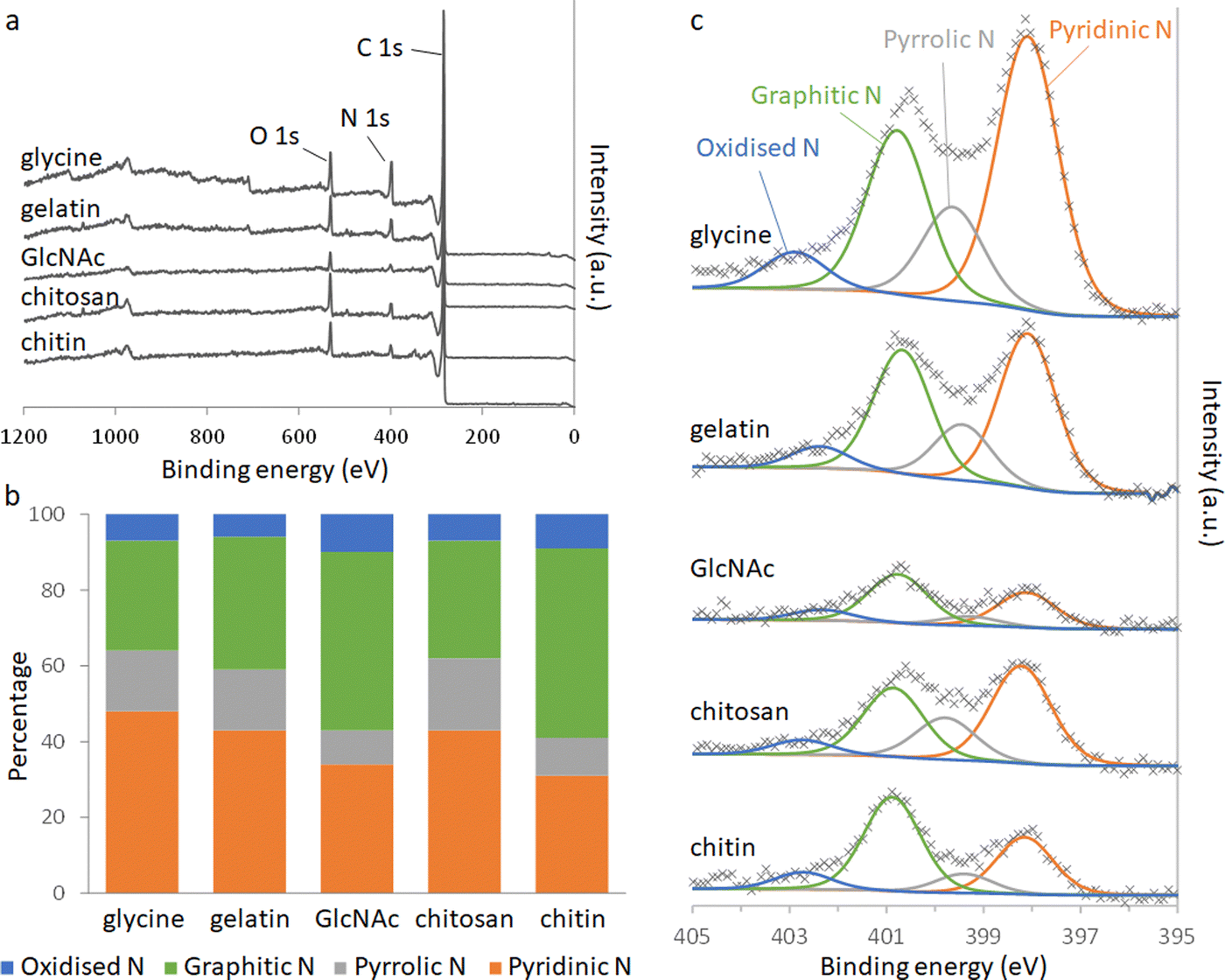 | ||
| Fig. 3 (a) XPS survey spectra and (b) relative distributions of nitrogen species calculated from deconvolution of (c) the N 1s peaks. | ||
Deconvolution of high-resolution N 1s spectra show four types of nitrogen species within the carbon samples. The peaks at approximately 398.1, 399.5, 400.8 and 402.5 eV can be ascribed to pyridinic-N, pyrrolic-N, graphitic-N and oxidised-N, respectively.17 The presence of pyridinic, pyrrolic and graphitic-N shows that nitrogen is doped into the carbon structure throughout all the systems. GlcNAc- and chitin-derived carbons show a greater proportion of graphitic-N, where the nitrogen is embedded completely within a graphitic sheet. This is consistent with the higher degree of graphitization indicated by Raman spectroscopy and suggests that some nitrogen may be incorporated into the graphitic planes during graphitization. The carbons derived from chitosan, glycine and gelatin all contain higher proportions of pyridinic and pyrrolic nitrogen. Pyrrolic nitrogen, involving a 5-membered ring, is likely to cause distortion of the carbon, consistent with a lower degree of graphitization. Pyridinic nitrogen occurs at the edge of a graphitic region, suggesting smaller regions of graphitization. These observations are both consistent with the more disordered structure indicated by PXRD and Raman spectroscopy for gelatin and glycine-derived carbons.
Small angle X-ray scattering (SAXS) was used to assess the size of the Fe/Fe3C particles within the carbon matrix. The raw SAXS data was fitted using McSAS (Fig. S6 and S7, ESI†), a Monte Carlo method to extract form-free size distributions.18 The resulting size distribution histograms for the glycine- and gelatin-derived carbons (Fig. 4) show a large population of very small scattering features (r < 1 nm). As SAXS arises from areas of varying electron density within a sample, there are various possible scattering interfaces that must be considered, such as the interface between the Fe/Fe3C particles and the surrounding carbon matrix as well as the carbon/air interface. However, the high electron density of Fe compared to carbon and air means it is reasonable to assume that a large proportion of the population comes from the Fe/C scattering interface. This small particle size may be a reason why little graphitization is observed in the glycine- and gelatin-derived carbons, as it is believed that catalyst particles must reach a certain critical size before graphitization can commence.13,19 In contrast, the size histograms for GlcNAc and chitin-derived carbons show much larger scattering features, suggesting a larger catalyst particle size. This is consistent with the high porosity of both carbons, as the process of graphitization involves movement of the catalyst through the matrix to create pores. The histogram for chitosan-derived carbon is like those of glycine and gelatin, with most particles having r < 1 nm. This pattern follows the results from XPS and XRD and supports the argument that graphitization will only proceed above a certain critical particle size. Something which is difficult to explain from the data so far in this paper is why the chitin and GlcNAc were much more resistant to graphitization than cellulose. Carbon derived from cellulose showed a very similar catalyst particle size range as carbons from chitin and GlcNAc11 but a considerably larger graphitic peak.
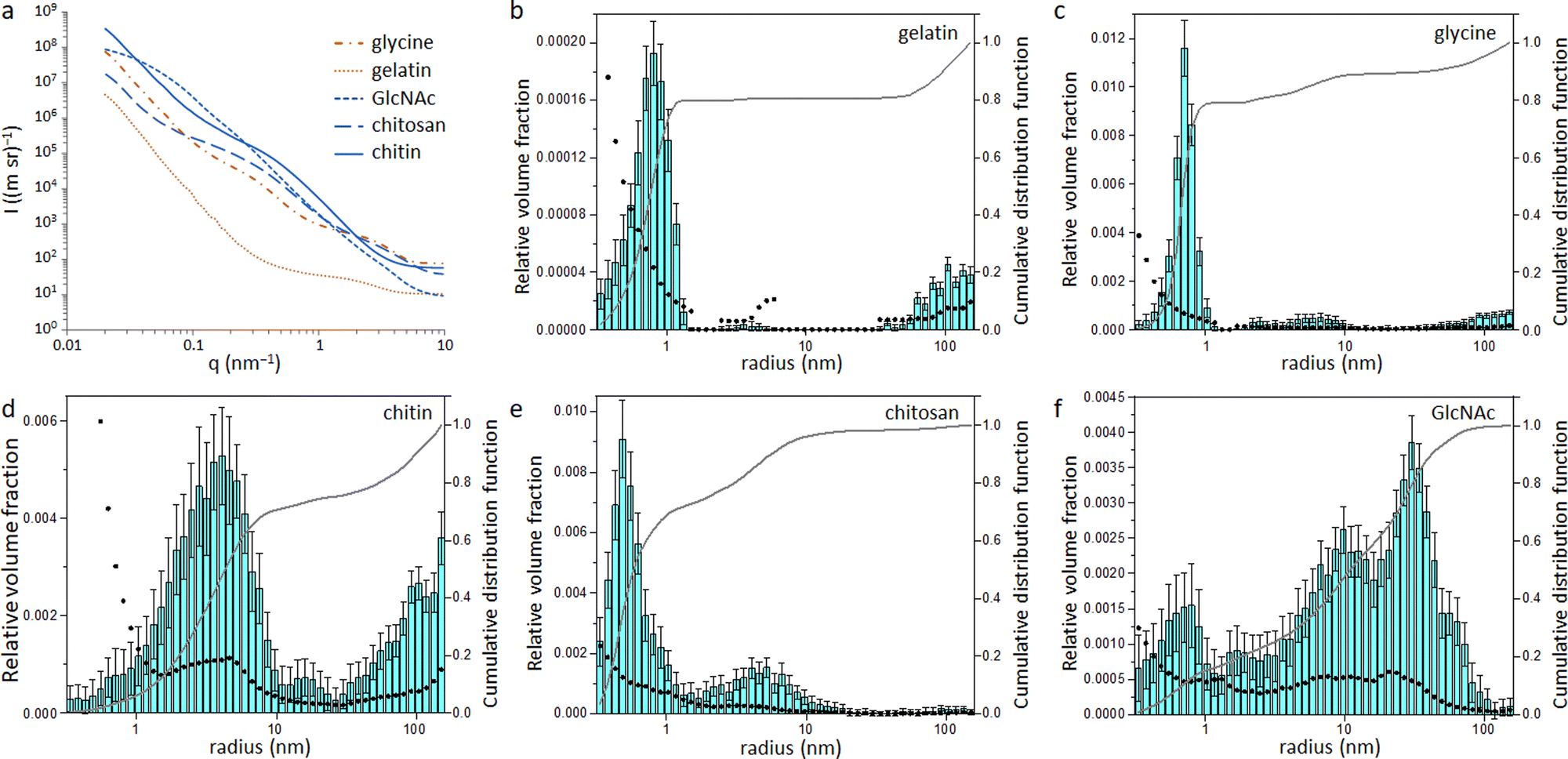 | ||
| Fig. 4 (a) Raw SAXS patterns and resulting size distribution histograms calculated from McSAS fitting of SAXS data for carbons derived from (b) gelatin, (c) glycine, (d) chitin, (e) chitosan and (f) GlcNAc. | ||
N-doped porous carbons at high iron loading
In our previous work with nitrogen-free precursors for iron-catalysed graphitization we showed that a higher iron:organic ratio produced larger catalyst particles and correspondingly higher level of graphitization. In an attempt to trigger higher levels of graphitization in the N-containing systems, we prepared samples where the iron:organic ratio was increased by a factor of 5 (3.3 mmol Fe(NO3)3 per 5 g of organic precursor). PXRD data (Fig. 5a) shows greater graphite peak intensity across all the samples. This indicates that nitrogen-rich precursors can undergo catalytic graphitization with a sufficient catalyst loading but are much harder to graphitize than analogous N-free precursors such as glucose and cellulose. Raman spectra (Fig. 5b) also show sharper D and G bands in all the samples, again indicative of a more ordered and graphitic carbon. One factor in this appears to be particle size, as the XRD data shows sharp peaks for Fe3C, indicating larger catalyst particles. These are readily observed in SEM images (Fig. S8, ESI†). Nitrogen porosimetry data (Fig. 5c) for the high-Fe samples all show a type IV isotherm, indicating the presence of mesopores. This is again consistent with greater graphitization, where the nanoparticle catalyst moves to produce hollow graphitic nanostructures, as shown in TEM (Fig. S9, ESI†). The most significant increase in porosity with higher catalyst loading comes from the gelatin and glycine systems, which suggests that the amino-acid precursors are more resistant to graphitization than the saccharides. Calculated adsorptive properties for all samples are shown in Table S4 (ESI†).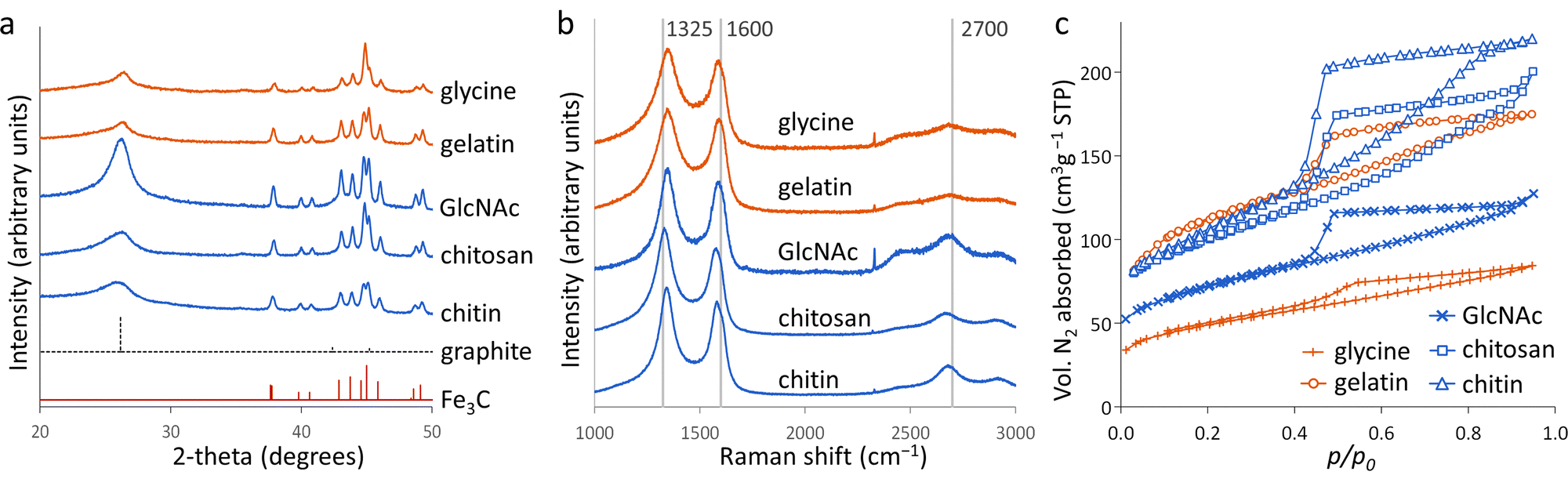 | ||
| Fig. 5 (a) XRD patterns, (b) Raman spectra and (c) nitrogen sorption isotherms for carbons derived from various N-containing precursors at a ratio of 5 g of organic precursor to 3.3 mmol of Fe(NO3)3. | ||
Elemental analysis of carbons prepared with high catalyst loading offers valuable insight into the graphitization process in these systems (Table 3). The higher catalyst loading leads to carbons with much lower nitrogen content for all 5 precursors.
| Precursor | Product composition | ||
|---|---|---|---|
| C% | H% | N% | |
| Glycine | 90 | 0.9 | 9 |
| Gelatin | 96 | 0.8 | 3 |
| GlcNAc | 98 | 0.2 | 2 |
| Chitosan | 98 | 0.5 | 2 |
| Chitin | 98 | 0.3 | 2 |
This indicates that a higher catalyst loading, and subsequently higher degree of graphitization, leads to removal of nitrogen from the system. The iron:carbon ratio (calculated from the residual mass, Table S5, ESI†) is similar for all 5 samples, again showing that it is the difference in the chemical and physical properties of the N-containing biomass that is changing the process of graphitization.
Thermogravimetric analysis (TGA) offers more insight into why graphitization is more significant at high catalyst loading. Fig. 6 shows TGA data for gelatin and chitin with a high Fe(NO3)3 loading. The degradation of chitin/Fe(NO3)3 follows a similar pattern to raw chitin,20 with a single, sharp decomposition between 300 and 400 °C. The profile for gelatin also has a major decomposition step between 300 and 400 °C, again consistent with the profile for raw gelatin.21 For the gelatin/Fe(NO3)3 mixture, however, there is also a steep mass loss at 167 °C. This probably corresponds to a rapid combustion reaction of the oxidizing nitrate with the amine-rich gelatin.22 NO, NH3 and CO2 are all released at this point (Fig. S10, ESI†), which supports this conclusion. An interesting feature in the gelatin/Fe(NO3)3 data (which is not present in data for pure gelatin) is a mass loss at ∼750 °C. This does not correlate to release of NO, NH3 or CO2. This can be explained by examining structural and compositional data for carbons prepared at high catalyst loading for various intermediate temperatures between 600 °C and 800 °C. XRD data (Fig. S11a, ESI†), shows that between 700 °C and 800 °C, strong peaks for Fe3C are generated in the gelatin-derived carbon and the graphite peak begins to emerge. The graphite peak then sharpens when the sample is held for 1 hour at 800 °C. Importantly, there is a sharp drop in nitrogen content from 12% to 3% between 700 °C and 800 °C (Table S6, ESI†). This correlates to the mass loss observed in TGA data and suggests that nitrogen is being expelled from the system in this temperature region. The lack of a peak for NH3 in the TGA-MS data at this point suggests that nitrogen leaves the system as N2 (not detectable in the N2 atmosphere of the instrument). Porosimetry data for the gelatin-derived carbons shows negligible porosity at 800 °C with 0 hour hold but significant mesoporosity (SBET = 410 m2 g−1 and Vtot = 0.27 cm3 g−1) after holding for 1 hour at 800 °C (Fig. S12, ESI†), correlating to the emergence of the graphite peak. These results together indicate that nitrogen loss occurs before graphitization takes place. Similar results are seen in the chitin system, where there are clear peaks in the XRD for Fe3C at 650 °C and a substantial peak for graphite by 700 °C (Fig. S11b, ESI†). This emergence of graphitic carbon in the XRD is again mirrored by a sharp increase in porosity (Fig. S12, ESI†). We are not able to determine whether the iron-containing catalyst is responsible for removal of nitrogen but iron is well known to catalyse reactions involving nitrogen so it seems likely.
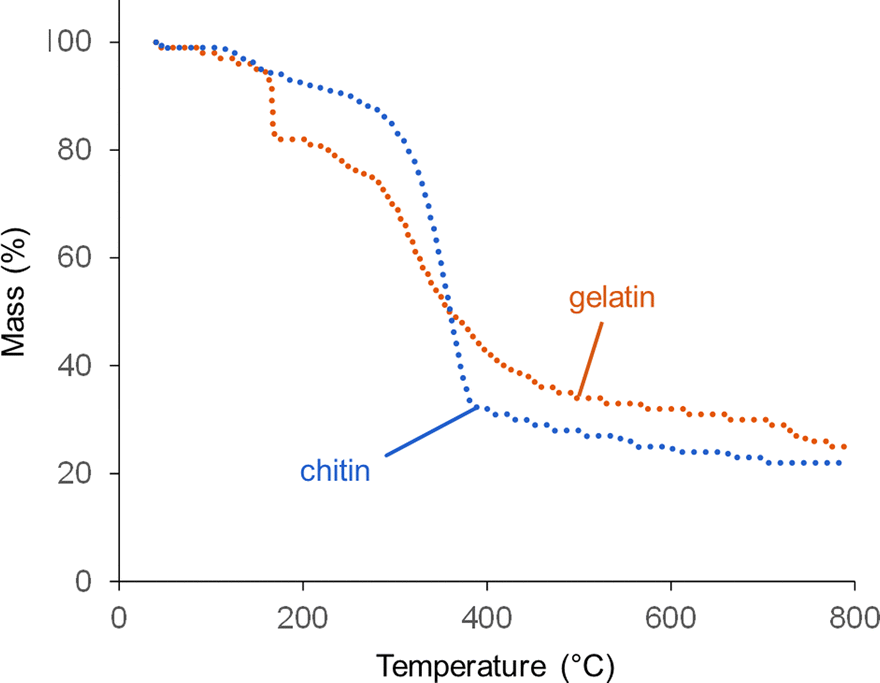 | ||
| Fig. 6 TGA data (under N2) for chitin and gelatin at organic to Fe(NO3)3 ratios of 5 g to 3.3 mmol (high Fe). | ||
Mechanism of graphitization of nitrogen-rich precursors
Two factors appear to be significant in the carbonization of nitrogen-rich organic materials. The first is the growth of the catalyst and the second is the removal of nitrogen from the system during or before the graphitization step. Catalyst size is clearly important and larger catalyst particles (produced with a higher catalyst loading) can drive more significant graphitization. However, a direct comparison of particle size data of cellulose, chitin and GlcNAc-derived carbons shows that all three have similar catalyst size distributions while only cellulose graphitizes readily at a lower catalyst loading. This clearly shows that the most significant factor in the slow graphitization of N-containing precursors is the presence of nitrogen. At present, the mechanism of iron-catalyzed graphitization is believed to be driven by dissolution of amorphous carbon in the catalyst particle, followed by precipitation of graphitic carbon. Nitrogen is poorly soluble in Fe3C and γ-Fe, which are the two possible phases for the graphitization catalyst.23 Therefore, it is possible that the nitrogen needs to be expelled from the system prior to the dissolution of amorphous carbon by the catalyst particle. Alternatively, the high temperatures of pyrolysis may naturally cause cleavage of C–N bonds, as has been proposed by other authors.24 This need to break many carbon–nitrogen bonds within the amorphous carbon during dissolution could be the reason that graphitization is slower in carbons derived from nitrogen-rich precursors. Gelatin and glycine contain the highest amount of nitrogen and are the most resistant to graphitization, which is consistent with the proposal that nitrogen inhibits graphitization.Conclusions
Nitrogen-doped carbons are increasingly important in the field of sustainable energy technologies. In this paper we have demonstrated that the synthesis of N-doped carbons by iron-catalyzed pyrolysis of nitrogen-rich precursors is influenced by multiple factors. Amine-functionalized saccharides and polysaccharides, an amino acid and a polypeptide were all used to produce nitrogen doped carbons by pyrolysis in the presence of Fe. In all cases, the samples were observed to be highly resistant to graphitization compared to glucose and cellulose. This was the case for both insoluble (i.e. chitin) and soluble biomass types. In the case of gelatin and glycine, this is partly attributed to the slow formation of the Fe3C nanoparticle catalyst. However, the fact that graphitization in amine-functionalised saccharides is slow despite the early formation of crystalline Fe3C nanoparticles proves that the nitrogen within the carbon has an inhibitory effect on the graphitization process. This is proposed to be due to the low solubility of nitrogen in the iron-rich catalyst phase and the subsequent time required for loss of nitrogen from the nitrogen rich carbon before it can undergo dissolution-reprecipitation as part of the graphitization process. Given that so many new applications for porous carbons require a high level of disorder (e.g. Na-ion batteries), the slow graphitization exhibited by these N-rich systems may be optimized for the formation of materials with lower levels of graphitization.For all the precursors, a higher level of catalyst loading was able to drive more graphitization and the introduction of graphitic mesopores in the resulting carbon. The increase in graphitization lead to a drop in the overall nitrogen content in the carbon, but some was shown to remain in all the systems. The nature of the nitrogen (pyridinic, pyrrolic, graphitic) varied significantly between the different precursors. The highest level of graphitic nitrogen was found in samples with the highest degree of graphitization, which does suggest that some nitrogen survives the graphitization step.
The literature is filled with examples of N-doped carbons that have been prepared from many different precursors under a wide range of conditions. This makes it very difficult to compare the properties of materials from different reports. This systematic study demonstrates that the structure and composition of N-doped carbons can be varied significantly by small variations in precursor. Importantly, the physical properties of the precursor seem to be less important than the chemical composition. N-Acetylglucosamine and chitin produce N-doped carbons with similar properties, despite N-acetylglucosamine being a water-soluble monomer of the insoluble chitin biomaterial. The difference in properties of the N-doped carbons from saccharide precursors and the amino-acid precursors is much more significant.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195. The authors also acknowledge the Leverhulme Trust for funding (Project RPG-2020-076).References
- W. Long, B. Fang, A. Ignaszak, Z. Wu, Y.-J. Wang and D. Wilkinson, Chem. Soc. Rev., 2017, 46, 7176–7190 RSC.
- N. Saadat, H. N. Dhakal, J. Tjong, S. Jaffer, W. Yang and M. Sain, Renewable Sustainable Energy Rev., 2021, 138, 110535 CrossRef CAS.
- H. Tan, J. Tang, J. Kim, Y. V. Kaneti, Y.-M. Kang, Y. Sugahara and Y. Yamauchi, J. Mater. Chem. A, 2019, 7, 1380–1393 RSC.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- R. D. Hunter, J. Ramírez-Rico and Z. Schnepp, J. Mater. Chem. A, 2022, 10, 4489–4516 RSC.
- D. Menga, J. L. Low, Y. Li, I. Arcon, B. Koyuturk, F. Wagner, F. Ruiz-Zepeda, M. Gaberscek, B. Paulus and T. P. Fellinger, J. Am. Chem. Soc., 2021, 143, 18010–18019 CrossRef CAS PubMed.
- X. Sun, Y. Shi, P. Zhang, C. Zheng, X. Zheng, F. Zhang, Y. Zhang, N. Guan, D. Zhao and G. D. Stucky, J. Am. Chem. Soc., 2011, 133, 14542–14545 CrossRef CAS PubMed.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, D. van de Kleut, J. M. Koelewijn, J. W. Geus and L. W. Jenneskens, Carbon, 2016, 107, 248–260 CrossRef CAS.
- R. D. Hunter, J. Davies, S. J. A. Herou, A. Kulak and Z. Schnepp, Philos. Trans. R. Soc., A, 2021, 379, 20200336 CrossRef CAS PubMed.
- R. D. Hunter, J. L. Rowlandson, G. J. Smales, B. R. Pauw, V. P. Ting, A. Kulak and Z. Schnepp, Mater. Adv., 2020, 1, 3281–3291 RSC.
- P. F. Harris, Crit. Rev. Solid State Mater. Sci., 2005, 30, 235–253 CrossRef CAS.
- K. Inomata and Y. Otake, Microporous Mesoporous Mater., 2011, 143, 60–65 CrossRef CAS.
- S. Glatzel, Z. Schnepp and C. Giordano, Angew. Chem., Int. Ed., 2013, 52, 2355–2358 CrossRef CAS PubMed.
- Y. Gao, X. Chen, J. Zhang and N. Yan, ChemPlusChem, 2015, 80, 1556–1564 CrossRef CAS PubMed.
- D. Raabe, C. Sachs and P. Romano, Acta Mater., 2005, 53, 4281–4292 CrossRef CAS.
- J. Casanovas, J. M. Ricart, J. Rubio, F. Illas and J. M. Jiménez-Mateos, J. Am. Chem. Soc., 1996, 118, 8071–8076 CrossRef CAS.
- I. Bressler, B. R. Pauw and A. F. Thunemann, J. Appl. Crystallogr., 2015, 48, 962–969 CrossRef CAS PubMed.
- C. W. Huang, L. C. Hsu and Y. Y. Li, Nanotechnology, 2006, 17, 4629–4634 CrossRef CAS PubMed.
- D. Stawski, S. Rabiej, L. Herczyńska and Z. Draczyński, J. Therm. Anal. Calorim., 2008, 93, 489–494 CrossRef CAS.
- A. Nugroho, E. B. Nursanto, C. A. Curie, H. S. Oktaviano, F. Ainurrachma and W. Trisunaryanti, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2022, 13, 035004 Search PubMed.
- L. A. Chick, L. R. Pederson, G. D. Maupin, J. L. Bates, L. E. Thomas and G. J. Exarhos, Mater. Lett., 1990, 10, 6–12 CrossRef CAS.
- M. Nikolussi, A. Leineweber and E. J. Mittemeijer, Philos. Mag., 2010, 90, 1105–1122 CrossRef CAS.
- X. Liu, L. Zhou, Y. Zhao, L. Bian, X. Feng and Q. Pu, ACS Appl. Mater. Interfaces, 2013, 5, 10280–10287 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental information and supplementary figures. See DOI: https://doi.org/10.1039/d3ma00039g |
| This journal is © The Royal Society of Chemistry 2023 |