
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring anodes for calcium-ion batteries
Henry R.
Tinker
a,
Christopher A.
Howard
 b,
Min
Zhou
c and
Yang
Xu
*a
b,
Min
Zhou
c and
Yang
Xu
*a
aDepartment of Chemistry, University College London, London WC1H 0AJ, UK. E-mail: y.xu.1@ucl.ac.uk
bDepartment of Physics & Astronomy, University College London, London WC1E 6BT, UK
cHefei National Laboratory for Physical Sciences at the Microscale, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 8th March 2023
Abstract
Calcium ion batteries have been increasingly explored as an alternative energy storage system as industry begins to manoeuvre towards an age of ‘Beyond lithium-ion’ research and development. However, using calcium metal as the battery's anode presents a multitude of issues, including the inability to strip ions off the metal, and the creation of an inactive passivation layer. Most research conducted around calcium ion batteries focuses on the electrolytic and cathodic study, whereas little focuses on the anode, due in part to the complexity and difficulty in resolving its challenges. Herein, this review will address the issues calcium has, including its lack of reversibility and solid electrolyte interface formation, as well as explore the alternative anode materials that have been utilised, noting their viability and future prospects.
 Henry R. Tinker | Henry Tinker is currently a postdoctoral researcher at University College London (UCL). He received his BSc in Chemistry from the University of Liverpool in 2016, before undertaking a biomedical research MRes in 2017. He completed his PhD in 2021 at UCL, focussing on combining graphitic materials with inorganic precursors to design electrochemical carbon composites for fuel cell catalysts and supercapacitor electrodes. He joined the Xu group in 2021, where he works on calcium ion battery electrode design. |
 Christopher A. Howard | Christopher A. Howard is Professor of Materials Physics at UCL. His lab focuses on creating and investigating novel nanostructured and low-dimensional materials that have interesting and useful properties, with a focus on energy applications. He enjoys teaching undergraduate and postgraduate programs and is co-director of the UK ESPRC Center for Doctoral Training in Advanced Characterisation of Materials. He is passionate about improving the mechanisms for achieving real-world impact from scientific research, which he believes is critically important for achieving carbon neutrality. To this end, he co-founded Prosemino, a clean-tech incubator that both creates and accelerates early-stage ventures in this area. |
 Min Zhou | Prof. Min Zhou received her PhD degree in the Department of Chemistry, University of Science and Technology of China (USTC). After being a postdoctoral fellow at Ilmenau University of Technology (Germany), she is currently a professor at USTC and Hefei National Research Center for Physical Sciences at the Microscale. Her research focuses on materials geometry and their applications for energy conversion and storage. |
 Yang Xu | Dr Yang Xu is a Lecturer (Assistant Professor) in Electrochemical Energy Storage in the Department of Chemistry at University College London. He received his bachelor's and PhD degrees at the University of Science and Technology of China under the supervision of Professor Yi Xie, and carried out his postdoctoral work in US, Canada, and Germany. His research focus lies in beyond-lithium battery chemistries (sodium, potassium and calcium) including intercalation chemistry of cathode materials, electrodeposition of metal anodes, and ion storage mechanism of conversion electrodes through materials synthesis, electrode and separator designs, and advanced characterization. |
Introduction
Lithium-ion batteries (LIB) are a highly commercialised method of energy storage that is fast approaching its theoretical limitations.1 This, combined with the relative terrestrial scarcity and rising cost of lithium procurement, the dendrite formation with pure lithium metal anodes, and safety issues, has pushed energy storage research to pursue alternative mechanisms and materials.2–4 This category of battery research is called ‘beyond lithium ion’. It consists of three types of candidate ions; metallic monovalent, such as sodium (Na+) and potassium (K+), metallic multivalent, which includes magnesium (Mg2+), zinc (Zn2+), aluminium (Al3+) and calcium (Ca2+), and non-metallic like H+ and NH4+.5 These charge carriers have the added benefit of being more earth abundant than Lithium (Li+) as well as in some cases having a greater charge and energy density.6Metallic multivalent ion batteries have the prospective to perform at higher capacities compared to their monovalent counterparts, due to their ability to release more electrons per atom, under the same concentrations.7 Calcium (Ca/Ca2+; −2.87 V vs. standard hydrogen electrode (SHE)) possess a slightly higher reduction potential than that of lithium (Li/Li+; −3.04 V vs. SHE) but remains much lower than comparative multivalent ions such as aluminium (Al/Al3+; −1.68 V vs. SHE) and magnesium (Mg/Mg2+; −2.36 V vs. SHE).8,9 This means that calcium can potentially perform at similar voltages as that of lithium. Calcium additionally has a theoretical volumetric capacity of 2073 mA h cm−3, similar to that of lithium but below magnesium (3832 mA h cm−3) and aluminium (8046 mA h cm−3), although their more negative reduction potentials lead to their cell voltages being lower.10–12 Calcium has a larger effective ionic radius than magnesium (Ca2+; 0.99 Å, Mg2+; 0.66 Å), while carrying the equivalent charges, which may promote a lower charge density in the electrode, but it has a comparatively a higher power density than other metal ion alternatives.13 In addition, calcium possesses a weaker charge density and weaker coordination with solvents than magnesium, further aiding in its kinetic abilities.14 Earth abundance must be taken into consideration when reviewing viable metal ion options, as it gives perspective to the longevity and availability of certain avenues of battery research. Aluminium possesses the highest abundancy in the earth's crust (8.13 wt%) of the feasible charge carriers, followed by calcium (3.63 wt%), sodium (2.83 wt%), potassium (2.59 wt%), magnesium (2.09 wt%), and lithium (0.0065 wt%).15 Calcium's comparatively high abundance compared with other metal ions makes it a strong and viable choice.
A few reasons that calcium-ion batteries (CIBs) have not seen the same recent successes as potassium and sodium-ions are due to the poor performances of the current electrolytes used, the poor intercalation of Ca2+ in cathode materials, a low working voltage (<2.0 V) and calcium metal's anodic irreversibility in standard electrolytic systems.16,17 These challenges can be simplified into three categories; the electrolyte, cathode kinetics and anode reversibility.
The general responsibility of a battery's electrolyte is to effectively shuttle ionic charge carriers between electrodes. Multivalent ion electrolytes such as Al3+ and Mg2+ require special electrolytic compositions for their hard acid–base cations to desolvate at the electrode/electrolyte boundaries, but Ca2+ electrolytes are similar to that of Li+ and Na+, i.e. a more conventional salt in solvent compositions.18–20 There are two types of electrolyte and anode interactions employed by current CIB setups; one is a passivation layer forming on the calcium metal anodes surface via the breakdowns of metastable salts/solvents.21 The main challenge with this method is the Ca-ions lack of mobility through this passivation layer. The other mechanism is one in which no a passivation layer forms, which provides stability, but as this occurs usually with non-metallic anodes, the cell will function with a lower energy density. With regards to the electrolyte–cathode relationship, the electrolytes often have a limited electrochemically stability windows, which limits cathode design and overall practicality. The remaining major challenge of Ca electrolytes is the limitation to how many electrolyte salts that are currently available, which are currently; calcium perchlorate (Ca(ClO4)2), calcium tetrafluoroborate (Ca(BF4)2), calcium borohydride (Ca(BH4)2), calcium nitrate Ca(NO3)2 and calcium (trifluoromethanesulfonyl)imide (Ca(TFSI)2).22,23
CIB cathodes can be generally categorised into four types: (i) Prussian blue analogues (PBAs), which are usually composed of iron/copper/manganese ferrocyanides with the recorded ability to store monovalent ions due to the large interstitial voids to allow the diffusion of Ca2+ in three dimensions; (ii) layered metal oxides such as CaxCoO2, Mg0.25V2O5·H2O, and V2O5, which possess large interlayer spacings to allow Ca2+ to diffuse between layers; (iii) chalcogenides such as TiS2, CuS and VS4 have demonstrated themselves to be a viable cathode for divalent storage systems, but they currently still either exist as a theoretical concept for Ca2+ or suffer from low discharge voltages and poor stability; (iv) the others are either fluoride (e.g., FeF3·0.33H2O) and polyanionic (e.g., Na2FePO4F), a branch of materials that can increase the redox potential via the induction effect. The main focus of calcium cathodes remains with PBAs and metal oxides due to their relatively low migration energy barriers of Ca diffusion. We recommend researchers to read some of the recent review articles on the cathodes of CIBs.24,25 With regards to the progress made with CIB cathodes, they still possess poor cycling stability and insufficient capacity for intercalation and conversion-based electrodes. Some of the main issues that hamper any cathode intercalation material is Ca2+'s relatively large ionic size (0.99 Å), limiting its ability to fit into conductive ionic framework of typical cathode materials and causing significant volumetric changes leading to the potential degradation of materials.26 Additionally their sluggish ion kinetics and high activation energy barrier, which originates from the divalency of the Ca2+ ions, is created via the strong electrostatic interactions within the cathode materials and themselves. To deal with these challenges, three dimensional (3D) materials designed with redox active transition metals and polyanions (P, Si, S) can proved large interstitial spaces which can allow calcium to insert and extract much more easily.27–29 The use of polyanion groups also permits for the use of higher cell voltages as the electrodes’ redox potentials become increased via the inductive effect. There have been a few materials hypothesised to be suitable, such as LiFePO4, V2O5, CaxMnO3, and CaxCoO2 (0.26 ≤ x ≤ 0.50), but none of these materials could provide decent reversible capacities and high working voltages.27,30–33 A general trend has been to follow the suitability of sodium-ion cathodes, due to Na+ (0.95 Å) having a similar ionic radius to that of Ca2+ (0.99 Å), such as a sodium (Na) super ionic conductor (NASICON) and NaV2(PO4)3.34
With regards to the progress made with calcium ion battery cathodes, they still possess poor cycling stability and insufficient capacity for intercalation and conversion-based electrodes.
Metal anodic reversibility is seen as one of the major stumbling blocks to the progression of CIBs, majorly limiting its performance and cyclability. In this review, we will explore the issues and limitations of using calcium metal as a preferred anode, examining its lack of reversibility and the formation of the solid–electrolyte interphase (SEI), as seen in Fig. 1. We then examine the alternatives to calcium, analysing their benefits and detriments, before evaluating the future prospects for the research area.
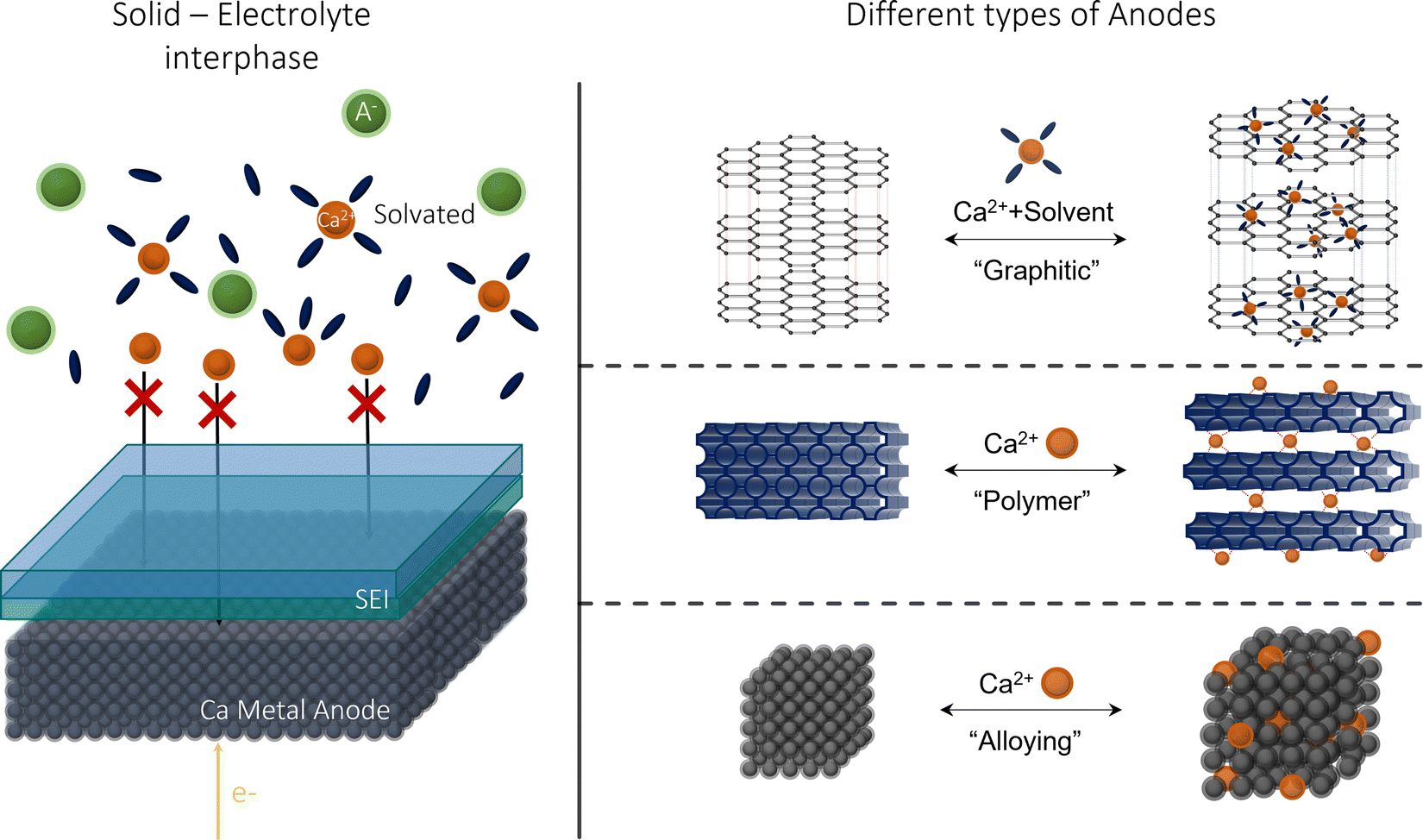 | ||
| Fig. 1 Calcium-ion anode overview: (left) an illustration of SEI at a calcium metal anode; (right) the alternative anodes suggested in this review. | ||
Understanding calcium metal anode
The use of alkali (Li, Na, K) and alkaline earth (Mg, Ca, Zn, Al) metal anodes has gained increasing attention due to their highly superior energy densities and specific capacities compared to those of non-metallic hosts such as graphite and hard carbons. The SEI on the metal anodes also plays a completely different role to that on non-metallic hosts. However, the use of metal anodes involves a more complicated electrolytic decomposition on the anode surface and yields only satisfactory performances. Na and K metal anodes are highly reactive with the electrolyte, leading to the formation of an unstable SEI, a main reason for its low coulombic efficiency (CE) and continual electrolyte degradation. Both also suffer from the similar issue Li faces, which is dendrite growth.3,35 Modifying electrolytes and cell setup can address these issues. The multivalent alternatives struggle from low ion plating and SEI formation in the electrolyte. In order to address the issues around calcium metal anodes, it is important to understand its mechanisms and its limitations. Fundamentally, using a pure metal electrode requires the calcium anode to be able to reversibly strip (discharge/oxidation) and plate (charge/reduction) Ca2+ cations in a full cell.36,37 It is common to see the decomposition of salts and solvents in the electrolyte, producing a protective layer on the surface of the chosen metal anode. To understand the issues surrounding the reversibility of calcium metal anodes, a fundamental understanding of the mechanisms of the SEI is required.21,38,39 SEI compositions will differ depending on the system they exist in and the makeup of the cell. The SEI layer is formed via the electrochemical reduction of the solvent and salt within the electrolyte into organic and inorganic compounds onto the surface of an electrode, in this case an anode.40When the Fermi energy of the anode is lower than the lowest unoccupied molecular orbital (LUMO) of the electrolyte (at the open circuit voltage in Fig. 2(a)), the battery remains stable and there is no decomposition.41 This occurs because the redox potential of the electrode is within the electrochemical window of the electrolyte, and therefore the SEI cannot form as the electrolyte cannot be reduced.42 When the anode is charged, it modifies this Fermi level which therefore allows the reduction the electrolyte, and therefore the formation of this passivation layer on the anode's surface to protect it from any further electrolytic interaction (Fig. 2(b)) consequentially limiting the continual breakdown of the electrolyte (Fig. 2(c)) by making any more SEI formation too energy intensive. The calcium SEI is considered to be a heterogeneous multi layered structure, composed of an inorganic inner layer closer to the electrode/SEI interface, which permits the movement of the cation, and an organic outer later, which has a heterogenous porous morphology.43 This outer layer is permeable to both the cations and the solvent molecules. If the SEI is ionically conductive, but also behaves as an electronic insulator, the battery can function properly. Its stability is generally key to preventing dendrite growth and any cathodic interactions.44 Over many cycles, the SEI can become thicker and begin to build resistance, which makes the battery less efficient and increases operational safety risks. There is also the issue of any reactive cations being trapped in this passivation layers, essentially reducing the capacity of the battery overtime. The existence of an electrode's SEI is key to its functionality as well as its eventual failure.45
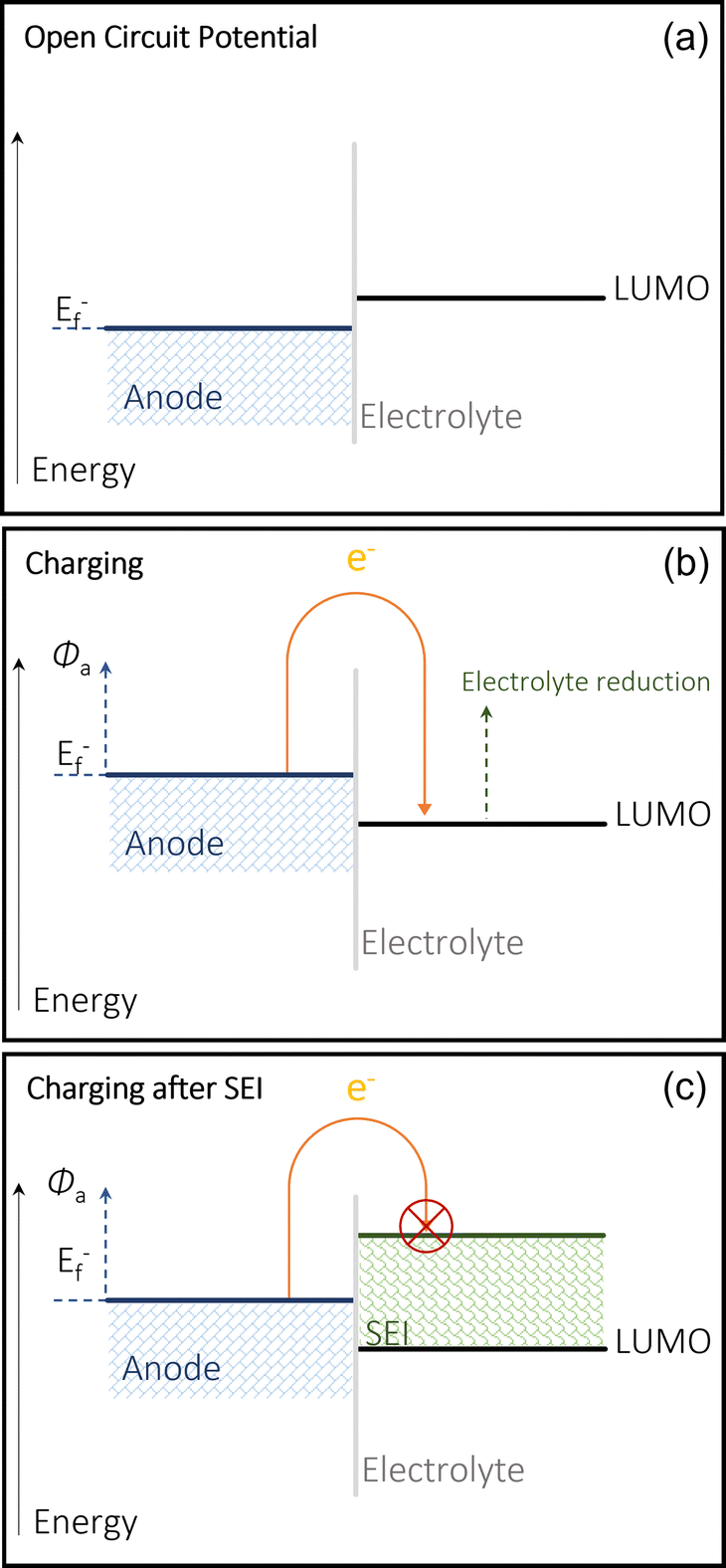 | ||
| Fig. 2 Energy diagrams of the metallic anode SEI formation. The anode's chemical potential is regarded as its Fermi energy of the anode (Ef−): (a) shows the open circuit voltage at a thermodynamic equilibrium; (b) demonstrates the shift in Fermi level when charged and (c) illustrates the energy level shift with the formation of the SEI. | ||
In the case of calcium metal anode, when the cell is charged, Ca2+ deintercalated from the cathode, immediately solvating in the electrolyte before travelling to the anode where it de-solvate and plates on the metal surface The components of the SEI, comprised of organic and inorganic compounds, due to their very strong interatomic bonding, prevents Ca2+ from moving through and therefore increases the ionic resistance of the cell, causing the circuit to break. This prevents the plating of the ions on the metal from occurring, making the process irreversible.46 The SEI is a constantly evolving layer, where its composition is key to the cell's performance. Initial observations with calcium metal anode showed that calcium can be stripped from the metal at quite high overpotentials, but it is the issue of calcium plating that presents the larger hurdles.43 Calcium plating has been extensively studied to understand the formation of non-reversible passivation layer composed of Ca(OH)2, CaCO3 and CaCl2 using the organic electrolyte salts (Ca(ClO4))2, Ca(BF4)2, and Ca(TFSI)2.47 A recent review concluded that the anode's electrochemical behaviour is dependent on the surface layer of the calcium metal, similar to lithium metal anode.48 There has been a recent study by Wang et al. utilising Ca(BH4)2/tetrahydrofuran, which during the charge–discharge cycling, eases the surface passivation, but the formation of CaH2 seen in Fig. 3 and the low anodic stability of the electrolyte (around 3 V), demonstrates there still needs to be further improvements made.49 Metal borohydrides are generally known to be strong reducing agents, and therefore are poor reversible compounds.
 | ||
| Fig. 3 Cross-section of Au electrodes during calcium plating/stripping in Ca(BH4)2/tetrahydrofuran. (a) First plating. (b) First stripping. (c) Tenth plating. (d) Tenth stripping. Reproduced with permission from ref. 49, copyright 2017 Nature Springer. | ||
The electrolyte's solvation structure is how the solute molecules coordinate to the dissolved ions effectively. The Ca2+ solvation can affect the speed of diffusion of the anion in bulk electrolyte and the strong binding energy of the calcium compounds in the SEI impacts the metal ion deposition on the anode. Therefore, the solvent is also crucial in how the SEI forms, as the SEI is composed of not only the cation but the decomposition of the electrolyte's anion and solvent. The products of the electrolytic decomposition migrate to the anode, where they further inhibit the plating of calcium, and passivate the surface.23 It seems then that the issues that occur on the surface of the anode can be addressed by either changing the electrolytic composition, the calcium metal anode, or both. Some studies have noted that reversible calcium electrolyte can be achieved with use of molten salts, but this requires an extremely high working temperature of around 500–700 °C, which is not viably commercially or in a broader research scale.50
It was not until 2015, when Ponrouch et al., reopened the previously forgotten multivalent systems and demonstrated the feasible plating and stripping of calcium metal, with the use of aprotic organic electrolytes (0.3 M Ca(BF4)2 in ethylene carbonate and propylene carbonate (EC:PC)).51 Although the performance was not brilliant, the simple fact that it demonstrated some level of cycling with metallic plating and stripping, reviving interest for CIB research. They listed the following perquisites for the functionality of CIBs; solvated Mχ+ can readily diffuse in the chosen organic electrolyte, the desolvation barrier at the electrolyte SEI layer is low, the Mχ+ can pass through the SEI/passivation layer and the energy barrier for the nucleation and development of M at the electrode/substrate interface is also low. Ponrouch et al., expanded upon this initial research by adding a boron based additive, in this case borontrifluoride diethyletherate (2 wt%) to Ca(TFSI)2 (0.4 M) at 100 °C.52 Although they demonstrated a low CE of >30%, they noted the crucial role of boron in the reversibility of Ca plating/striping and its passivation layer.53 These conventional Ca2+ salts in aprotic solvents are known to be easily reduced on the calcium anode's surface, and the salt/solvent combination will determine the chemical composition of the SEI. Therefore, by using the same salt but with alternative aprotic solvents will lead to an alternative passivation layer composition on the calcium anode. An optimal ion-conductive salt should be able to demonstrate reversible plating/stripping at room temperature, while being compatible with Ca metal anodes, have a wide potential window and an efficient ion transfer mechanism. Fluorinated alkoxyaluminate ([Al(hfip)4]−) and alkoxyborate ([(B(hfip))4]−) anions (hfip = –OCH(CF3)2) have been shown to have high ionic conductivity and anodic stability due to the electronegativity of the fluorine atom, producing a strong C–F bond and the charge of the alkoxy groups being delocalised which in turn leads to weak cation–anion interactions. Li et al. compared the use of Ca[B(hfip)4]2 in 1,2-dimethoxyethane (DME) to the two most proven reversible electrolytes at room temperature, Ca(BF6)2 in EC:PC (Wang et al.) and Ca(BH4)2/THF (Ponrouch et al.)49,51,54 They found that the B(hfip)4 anion possesses higher ionic conductivity of ∼8.3 mS cm−1 compared to Ca(BF6)2 (∼5.5 mS cm−1) and higher anodic stability of >4.5 V on Al (compared to Ca(BF6)2 ∼ 3.0 V (Al) and Ca(BH4)2 ∼ 3.0 V (Au)). They additionally observed the electrolytic decomposition on the Ca anode is around 7% CaF2, which was not enough to prevent the plating/stripping of Ca2+ like with the other two electrolytes, and therefore allowing for reversibility. They stated that the coulombic efficiency was low (80%), which could be attributed to electrolytic decomposition and Ca deposits detachment from the working electrode.
One of the most recent studies that has achieved room temperature calcium plating and stripping was explored by Liao et al., where they synthesised and utilised an uncommon electrolyte, calcium tetrakis(perfluoro-tert-butoxy) aluminate (Ca(TPFA)2) in DME.55 They observed how weakly coordinated but highly stable the sterically bulk ligands were with the calcium cation. They noted a relatively low CE of 55% on a gold electrode but emphasised the reductive stability of the Ca(TPFA)2 salt. They saw that the CE and overpotentials could be due to the reductive decomposition at the anode interface or the Ca2+ post surface stripping. Although the performance of the salt was not very notable, their focus on the chemical understanding of the salt stability and ionic anodic interfaces demonstrates the direction of future electrolyte-based research.
Current research still demonstrates limited plating/stripping of Ca on calcium metal surface with the alteration of calcium ion electrolytes. The following sections will investigate the exploration of the anode materials, as opposed to the electrolyte, noting their different mechanisms, how they may relate to work conducted with other metal ions and the future scope of their research.
Graphitic carbon-based anodes
Graphitic carbon materials have been commonly practiced in organic electrolyte metal ion-based batteries due to their affordability, inert nature, cyclability and their ability to form active passivation layers on their surface.56,57 Graphitic carbon is one of the most widely used electrode materials for ion battery storage because of its simplicity, affordability, and its suitability as an anode, especially in the case of lithium cells. It consists of sp2 hybridised graphene layers, bonded by interlayer weak van der Waals (VdW) forces with the pi bands on the graphene sheets which makes graphite a very good conductor.58,59 Ions can be hosted within these graphene layers to form graphite intercalation compounds (GICs).60 Graphite has always been regarded as one of the most reliable electrode/anode materials, predominantly being used in commercial lithium batteries, but only has a maximum capacity of 372 mA h g−1.61,62 Graphite possesses the benefit of not producing an inactive passivation layer due to its co solvent based intercalation-based mechanism, removing the issue of poor Ca kinetics that usually occur when cations interact with ionically insulating inorganic and organic SEI compounds. Additionally, graphite can readily host K+ ions, for example to form KC8.63 On the other hand, when Na+ ions are inserted, the overall free energy and the binding energy between the ions and the graphene sheets are so small that it becomes difficult to form the Na+ intercalated molecules (NaC64).64 The complex balance of competing energetics is evidenced by the fact K+ intercalated into graphite and Na+ does not despite being smaller (1.33 Å and 0.95 Å, respectively).65 Pyo et al. studied the Ca2+ co-intercalation of Ca(TFSI)2 with tetraglyme (G4) into graphite. They noted reversibility beyond 2000 cycles with no degradation, with a reversible capacity of 62 mA h g−1 at 0.05 A g−1 against an activated carbon (AC) counter electrode.66 When implemented in a full cell with a perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) cathode, it achieved an 80 mA h g−1 for 100 cycles with a reversible potential of around 1.6 V versus graphite. They saw the formation of a ternary intercalation compound named Ca–G4·C72, which undergoes no calcium plating, and a large volume change during the charge–discharge cycle, with an increase of over 210% from standard graphite, which can be seen in Fig. 4(a) and (c). The situ X-ray diffraction (XRD) patterns, in Fig. 4(b), demonstrate the shifts in diffraction peaks during charge–discharge, leading to the assumption of Ca intercalation and deintercalation. The original intensity of graphite at 26.5° began to decrease with the initiation of charge, and two new peaks were observed at 24.8° (d00n = 0.362 nm) and 28.5° (d00n+1 = 0.302 nm) at −2.10 V. These peaks can be noted to shift to 24.4° and 30.0° respectively at −2.90 V, the completion of charge. They further calculated the gallery height of graphite at different stages of charge, observing its maximum d-spacing to be 1.211 nm at −2.6 V. Park et al. additionally investigated reversible intercalation into graphite, specifically at room temperature and focussing on the electrolyte salt and solvent combinations.67 They were able to cycle the graphite anode over 200 cycles with a capacity of 85 mA h g−1 and very little capacity drop, as well as maintain a high rate of 2 A g−1, using a half cell with natural graphite as the working electrode, Ca metal as a counter and Ca(BH4)2 (0.5 M) in dimethylacetamide (DMAc). They explained that the intercalation mechanism was feasible because of the strength of the solvation with the electrolytic salt. They noticed the presence of their glyme solvent in the calciated graphite, through the use of solid state 13C-NMR (Fig. 4(c)). The solvent peaks locating at 20.5, 33.6, 36.7 and 168.9 ppm correspond to the four carbon atoms of DMAc, with the latter three peaks moving to a higher frequency due to their deshielding, attributed to the strong interaction between the electronegative oxygen and nitrogen when introduced to the electrolytic Ca salt. They additionally observed broadened peaks in similar positions, shifted further upfield, which they attribute to the interaction between DMAc and graphite and the randomness of the orientations within the structure. This showed that Ca2+cointercalates with the solvent in a configuration that differed from what was observed in the electrolyte. It is interesting to observe that between these two studies conducted by Pyo and Park, they used completely different electrochemical cell setups, from the counter/cathode to the salt/solvent, but they delivered a similar solvent cointercalation mechanism and comparable performances.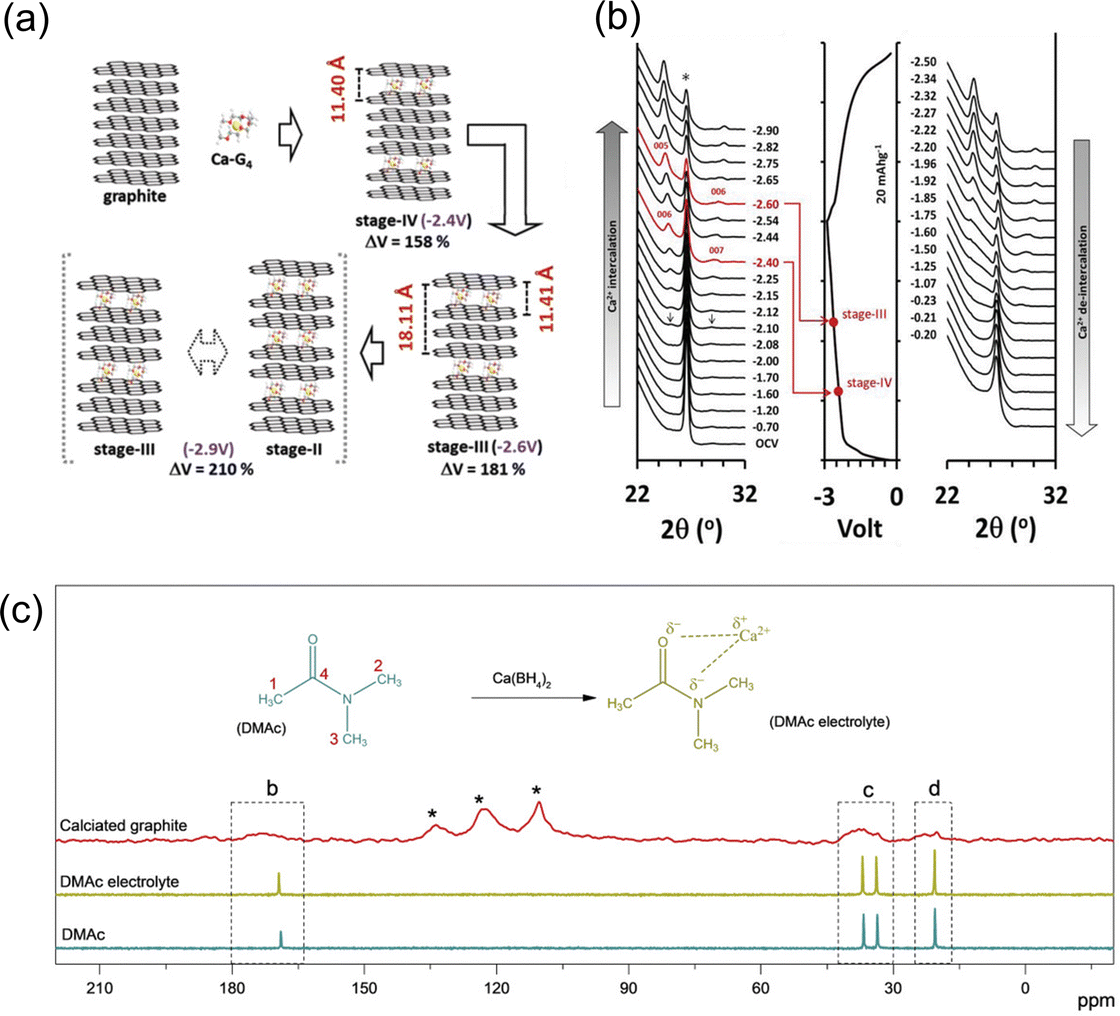 | ||
| Fig. 4 (a) An illustration of the various processes for Ca–G4 intercalation into graphite; (b) in situ XRD during charge discharge and correspond to their voltage profiles; reproduced with permission from ref. 66, copyright 2019 Wiley-VCH; (c) 13C-NMR characterisation; DMAc-based electrolyte, fully discharged graphite with chemical structures of DMAc molecule and potential interaction between Ca2+ and DMAc molecule in electrolyte, the four carbon atoms in DMAc molecule are designed as 1, 2, 3, and 4. Reproduced with permission from ref. 67, copyright 2020 Wiley-VCH. | ||
Another paper by Tang et al. observed the successful design of a dual graphitic carbon CIB operating with a carbonate calcium hexafluorophasphate (Ca(PF6)2) electrolyte.68 They used a mesocarbon microbead (MCMB) as the anode, which possesses an isotropic graphitic-type layered structure. The idea was that this material would be advantageous for Ca2+ ion intercalation at ambient temperatures. The Ca2+ ions intercalate between these layers to form a CaCx intercalation compound, while the PF6− anions move between the sheets of the expanded graphite cathode to form a Cy(PF6) intercalation compound as seen in Fig. 5(c). The Ca2+ intercalation was confirmed with ex situ XRD, seen in Fig. 5(a). It can be observed that there is a sharp diffraction peak (002) at 26.3°, with a d002 of 0.339 nm initially. When the anode is fully charged to 5.2 V, the (002) peak decreases and shifts to 25.9° and d002 value increases to 0.344 nm, which they claim is an indication of the intercalation of Ca2+. When the cell discharges, it returns to its diffraction peak returns to its initial position, demonstrating the deintercalation of the cation. This overall intercalation and deintercalation can be observed in the schematic in Fig. 5(b). They noted a discharge capacity of around 66 mA h g−1 at a current rate of 2 C with a working potential of 4.6 V, seen in Fig. 5(b). Its cycling stability remained very high, with a discharge capacity of 62 mA h g−1 over 300 cycles, with a capacity retention of 94%.
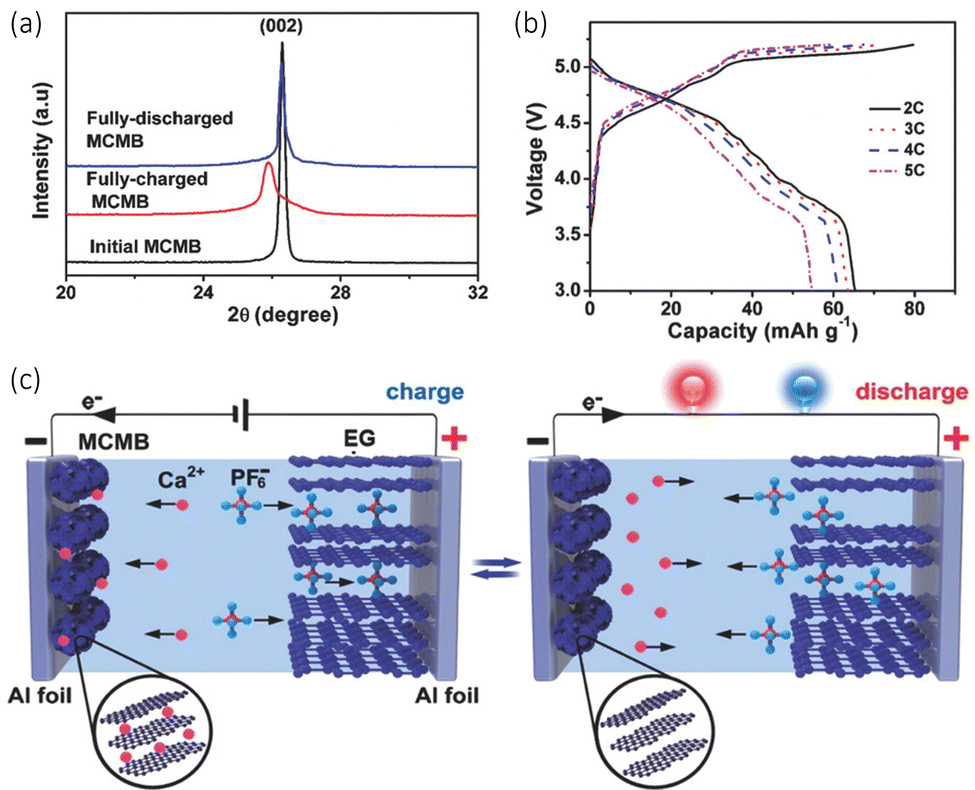 | ||
Fig. 5 (a) XRD patterns of the charge discharge initial cycle of a mesocarbon microbead (MCMB) anode in the calcium dual carbon battery (Ca-DCB). (b) Charge/discharge capacities and coulombic efficiency at different current rates. (c) A schematic configuration of the Ca-DCB, which consists of an expanded graphite (EG) cathode and a MCMB anode with Ca(PF6)2 (0.7 M, EC/DMC/EMC (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 v/v/v)) as the electrolyte Reproduced with permission from ref. 68, copyright 2018 Wiley-VCH. :3:2 v/v/v)) as the electrolyte Reproduced with permission from ref. 68, copyright 2018 Wiley-VCH. | ||
Low-dimensional carbon materials have been heavily researched over the last century, with the discovery of C60 in 1985, carbon nanotubes in 1991 through to the isolation of graphene in 2004.69–71 Graphene itself contains impurities and defects (Stone–Wales and divacancy), which improve the adsorption of Ca2+ ions onto its surface.72 Shenoy et al., compared percentage levels of defects to that of pristine graphene and graphite using simulations based on density functional theory (DFT), prediciting the improvement of charge storage with the increase of defect density and evidencing an overall higher capacity and cyclability than graphite. Their Ca2+ ion model with the maximum divancy densities predicted a capacity of 2900 mA h g−1, twice that of the Na+ ion battery. Their conclusion was pristine graphene would not allow for the adsorption of Ca2+ and requires for a reasonable level of divacancy defects (around 25% for 70% adsorption) and Stone–Wales (100% for 50% adsorption), which therefore increases the potential around the defect sites, in turn increasing the capacity. Too many defect sites and the structure will have too many dangling bonds (unstable carbon sites) and will lose adsorption ability.
2D carbon materials have been applied to many forms of technology, including that of carbon capture, catalysis and most crucially, metal ion batteries (MiB).73,74 Graphite remains the most stable and fundamental carbon anode but there have been Li+ ion batteries with carbon anode allotropes such as popgraphene, pentagraphene, ψ-graphene, phagraphene, and Θ-graphene, with capacities that range between 372 to 1489 mA h g−1.75 These allotropes have a greater success with Na+ ion batteries, with Θ-graphene and pentagraphene possessing capacities of 1489 to 1275 mA h g−1. It has been utilised for K+ ion batteries as well but does not operate with Mg2+ ion. Wang et al., proposed combining auxetic materials together with single atomic layered graphene to theorise a planar 2D carbon allotrope dubbed ‘xgraphene’.76 The conclusion was when utilising Ca2+, a capacity of 1488 mA h g−1, but more interestingly, a low energy barrier of ≤0.49 eV and an open circuit voltage of ≤0.53 V, which is an indication of an advantageous charge–discharge capability.
Organic and metal organic framework anodes
Organic and metal organic framework anodes have become an attractive proposition due to their structural diversity and resource renewability.77,78 They possess the benefit of storing electrochemical energy via the binding of ions to redox active groups within their structures, an alternative method to conventional inorganic intercalation, and therefore they are less vulnerable to temperature changes.79,80 These materials have been explored with Li-ion and Na-ion batteries, but issues have arisen with their cyclability as they suffer from a fading capacity due to their solubility in organic electrolytes.81 Li et al. explore the use of an organic anode paired with a graphite cathode in a calcium bis(fluorosulfony)imide (Ca((FSI)2) (3.5 M)) electrolyte, observing intercalation of Ca2+ into both electrodes.82 Part of their study focused on the stability and concentration of the electrolyte salts and solvents, but they had a particular interest in the use of organic materials as the anode in the cell. They used 3,4,9,10-perylenetetracarboxylic dianhydride (PTCDA), originally utilised as a cathode which displayed a discharge capacity of 75 mA h g−1 at a current density of 100 mA g−1, with an 84.7% capacity retention over 350 cycles. The reasons behind its use can be ascribed to its ability to host extraneous monovalent and multivalent cations, its high reaction kinetics and electrochemical stability, its green synthesis and low cost. They used in situ infrared spectroscopy (FT-IR) and performed a charge/discharge cycle (Fig. 6(a)) to study the change in functional groups on PTCDA during cycling (Fig. 6(b)). What they observed was when charging began, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O converted to C–O due to the insertion and chemical conversion caused by Ca2+, evidenced by the shifts of the CO peaks to lower wavelengths. During the next discharge process, the peaks shifted back to higher wavelengths, demonstrating the reversibility of the cation insertion/de-insertion. Moreover, they performed in the X-ray photoelectron spectroscopy (XPS), noting the presence of C–O bonding increasing in the charged stage of the cycle. This led to the conclusion that when Ca2+ inserts, it forms an O⋯Ca⋯O interaction, illustrated in Fig. 6(c). Jiang et al., have demonstrated an all-organic dual ion battery (ODIBs) to function with a successful calcium electrolyte.83 Their aim was to produce a low-temperature and non-dendritic cell.
O converted to C–O due to the insertion and chemical conversion caused by Ca2+, evidenced by the shifts of the CO peaks to lower wavelengths. During the next discharge process, the peaks shifted back to higher wavelengths, demonstrating the reversibility of the cation insertion/de-insertion. Moreover, they performed in the X-ray photoelectron spectroscopy (XPS), noting the presence of C–O bonding increasing in the charged stage of the cycle. This led to the conclusion that when Ca2+ inserts, it forms an O⋯Ca⋯O interaction, illustrated in Fig. 6(c). Jiang et al., have demonstrated an all-organic dual ion battery (ODIBs) to function with a successful calcium electrolyte.83 Their aim was to produce a low-temperature and non-dendritic cell.
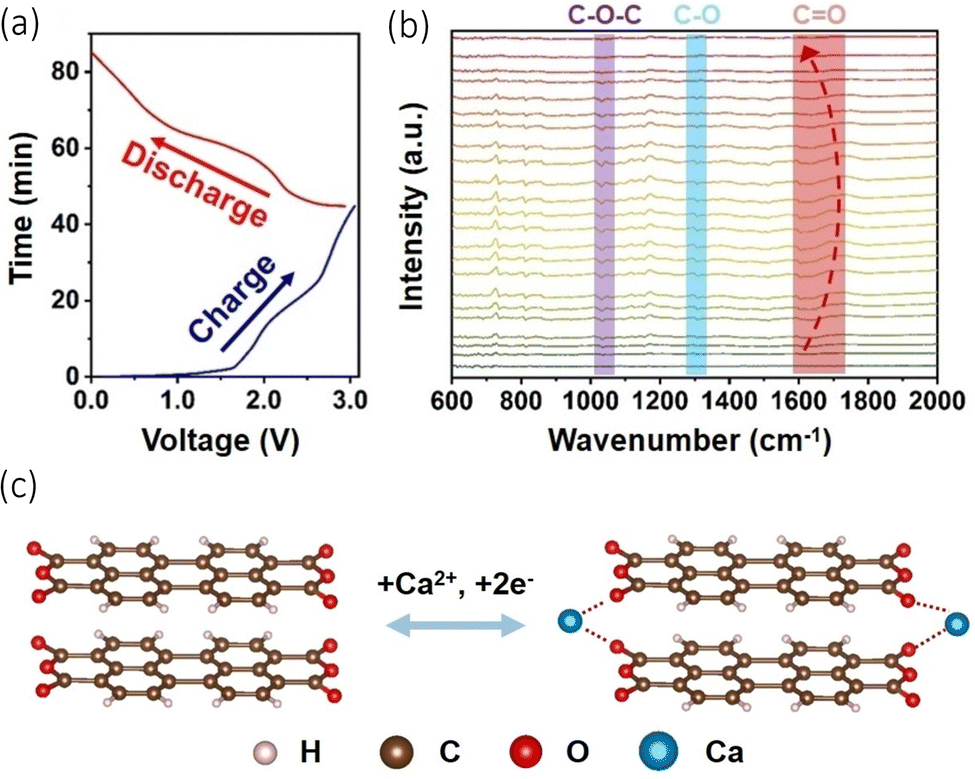 | ||
| Fig. 6 (a) Charge/discharge curves of PTCDA||graphite cathode cell during in situ test. (b) In situ FT-IR spectra for the PTCDA anode. (c) Suggested electrochemical conversion of PTCDA with Ca2+ insertion/de-insertion. Reproduced with permission from ref. 82, copyright Wiley-VCH. | ||
When analysing the electrochemical conversion of the PDI-EDA anode, the authors utilised the XPS (Fig. 7(b)) to observe the presence of Ca2+ and the change of CO and C–O ratios (Fig. 7(c)). The cell effectively limited any internal resistance of ion migration by the anions/cations associating/dissociating within the anode/cathodes. This also in turn, reduced the impact of any temperature variation on the performance of the cell. Their electrochemical setup was comprised of a polytriphenylamine (PTPAn) cathode and a perylene diimid–ethylene diamine (PDI–EDA) anode, with a calcium perchlorate (Ca(ClO4)2, 1 M) electrolyte seen in Fig. 7(a). Their choice in polyimides was from their ability to bind with metal ions in their reduction states. They managed to achieve a discharge capacity of 82.7 mA h g−1 at a 0.2 A g−1 current density, with a 100% CE. It was able to function at low temperatures, mainly due to the energy storage mechanisms of organic electrodes. While also demonstrating a highly consistent conductivity with temperature variations.
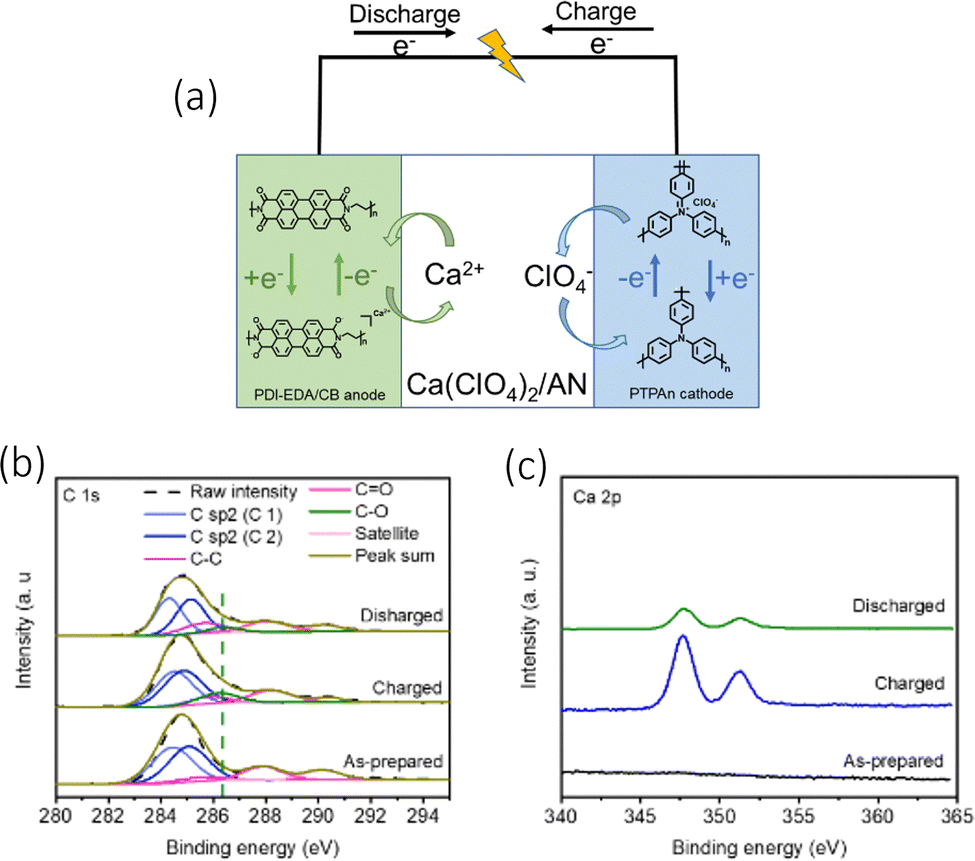 | ||
| Fig. 7 (a) The electrochemical energy storage mechanism of CAN-ODI (b) XPS spectra of PDI-EDA/CB anode C 1s and (c) Ca 2p XPS curves of the PDI-EDA/CB anode at different charging states; reproduced with permission from ref. 83, copyright 2022 Wiley-VCH. | ||
Metal organic compounds are a type of materials that have organic ligands that are coordinated around a central metal ion, and they can be classified into metal organic frameworks (MOFs) and metal organic complexes (MOCs).84 During an electrochemical energy storage process, the central metal ion can be the sites for faradaic interactions, while the ligands provide pathway of cation movements through the electrode.85 MOFs and MOCs are easily modified to improve conductivity and general morphology, through the introduction of alternative metal centres and organic ligands. MOCs that possess Co3+ and Ni2+ with 1,4-benzenedicarboxylate ligands have already been trialled successfully in Li-ion batteries, but Ni2+ based materials are observed to easily hydrolyse during synthesis, which in turn weakens ligand metal coordination.81,82 Addressing this issue, Ngoc Vo et al., added an NH2 to 1,4-benzenediacarboxylate to form 2-amino-1,4-benzenedicarboxylate (Ni[C6H4(NH2)(COO)2] or NibdcNH2), which provided greater electron density to the MOC and improved its conductivity.86 Their study, in which the MOC was in a Ca2+ cell with a Prussian blue cathode and a Ca(ClO4)2/CH3CN electrolyte (1 M), showed that the NH2 organic linker had an initial discharge capacity around 90 mA h g−1 at 100 mA g−1 with a capacity retention of 77%. They claimed that the introduction of the NH2 group to the ligand additionally acted as a conductive element to the cell due to the excess electron density provided by the nitrogen atom. This suggested that the enhancement of the conductivity improved the stability and reversibility of the cell, even though the cation diffusion coefficient was low (0.80 × 10−14 cm2 s−1), which in turn highlights the issues remaining with poor Ca2+ movement and fluctuations in the coulombic efficiency.
Alloys
Alloy electrodes are a branch of anode being widely studied in Li-ion batteries and have so far demonstrated favourable reversibility in CIBs. Other metals/metalloids that have been explored are Zn, Li, Na, Al, and Si.87–89 All except Na possess the ability to mix with calcium, in order to form a broad range of intermetallic compositions. Examples of the alloy anodes with the highest Ca-content were Ca3Zn, Ca2Sn, and CaLi2. These possessed the theoretical capacities of 1366, 903, and 3860 mA h g−1, respectively.90–92 When employed in full cells, the strong calciation of the alloying materials converts the alloys to alternative compositions, i.e., Ca7Sn6, with a theoretical capacity of 527 mA h g−1.93A pre-electrochemical calcium–tin alloy was first used by Lipson et al., who noted the calciated Sn electrode produced an initial discharge capacity of 40 mA h g−1 with a Ca(PF6)2 (0.3 M, EC:PC (3:7)) electrolyte and a Prussian blue (NaxMnFe(CN)6) cathode.88 They synthesised the alloy via a highly facile inert high temperature reaction, by simply mixing calcium and tin granules together and heating them in a tube furnace as 900 °C under argon for 1 h. The concept has further been tested in a dual ion system with a graphite electrode and Ca(PF6)2. This demonstrated a good cycling retention of 95% after 350 cycles, forming the reversible active phase anode Ca7Sn6 in the process. Zhao-Karger et al., managed to electrochemically transform an alloy (CaxSn) into a calcium rich CaSn3 alloy anode, with a 1,4 – polyanthraquinone (14PAQ) quinone polymer (COF) based anode and a Ca[B(hfip)4]2 electrolyte.94 The cell demonstrated a voltage of around 1.8 V and can be operated for 5000 cycles at a specific current of 260 mA g−1, with a capacity of 78 mA h g−1. The authors’ initial target was to design a Ca2Sn alloy, due to is higher theoretical capacity (903 mA h g−1) and low deciliation/calciation potential (0.5 vs. Ca), but they found that they produced a sample that had a mix of Ca2Sn, Ca36Sn23 and Ca32Sn21, and therefore denoted their initial anode CaxSn. They monitored the electrochemical phase progression of this anode through in situ XRD shown in Fig. 8(a). The charge–discharge cycle operated in a 0.6–2.5 V potential window. During the first discharge, there was an immediate decrease in Ca2Sn peaks due to de-calciation. The β-Sn phase appeared and became more intense over the discharge, the ‘de-alloying’ of the anode. When the cell was charged, the Ca2Sn peaks did not reappear, and were instead replaced with what was identified as CaSn3. The Sn and CaSn3 peaks increased in intensity throughout cycling. They saw the new Ca–Sn phase was more kinetically favourable than the alloys in the initial sample, and therefore designed their investigation around a CaSn3 anode. Interestingly, it was observed that the mixed alloy sample performed better than pure CaSn3 and attributed the observation to the low active surface area of Sn phase formed with the CaSn3. They electrochemically compared their CaxSn sample to that of pure CaSn3 in a full cell (CaxSn or CaSn3‖14PAQ) cells, seen in Fig. 8(b), and found that the pure phase sample exhibited the lower capacity retention and coulombic efficiency at 260 mA g−1 for 1000 cycles, which they ascribed to the lower electrochemically active surface area of the Sn phase that's found in the bulk alloy and difference in microstructure from their synthetic methods.
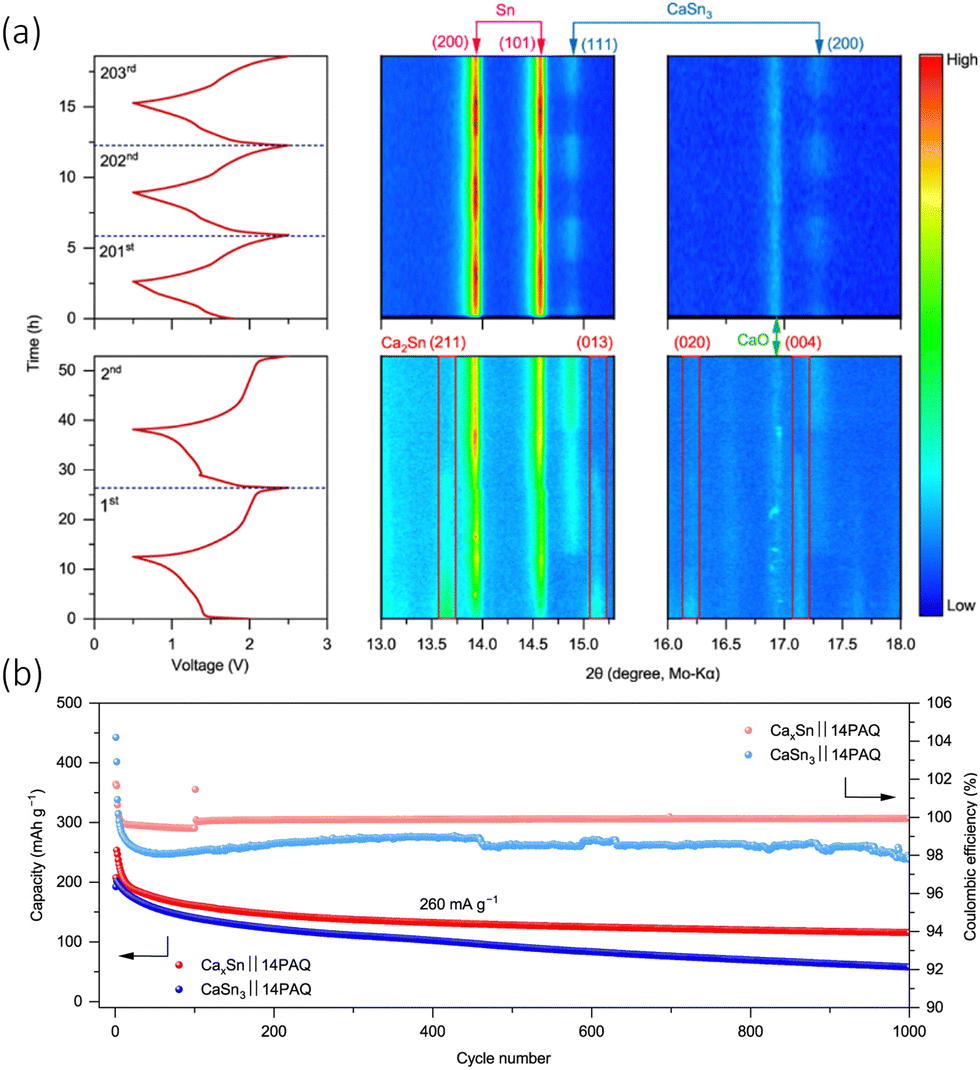 | ||
| Fig. 8 (a) Isoplots of in situ XRD scans the anode CaxSn‖14PAQ cells during the first, second and from 201st to 203rd discharge/charge cycles at specific current of 26 mA g−1 at 25 °C. The peak is observed along the top and the cell voltage profiles on the left had side. ICSD-ID: Ca2Sn 659611, β-Sn 106072, CaSn3 58934, Ca36Sn23 54619 and CaO 26959. (b) Cycling performance of CaSn3‖14PAQ compared with CaxSn‖14PAQ. Reproduced with permission from ref. 94, copyright 2022 Springer Nature. | ||
The concept of metal calcium alloys has been explored with few results, but there have been potential theoretical leads that may provide insight into alloy materials that are worth being experimentally examined. A material that has garnered some attention is a Si based anode. Ponrouch et al. used DFT-based simulations to predict the relevant Ca alloying voltage with Si at around 0.4 V, with fcc-Si producing the intermetallic CaxSi phases (0.5<x≤2), and calcium de-insertion from the electrochemically formed CaSi2 at 1.2 V.89 The authors observed a volumetric expansion of around 306%. They went on to test the concept in a two-electrode cell, employing the use of CaSi2, with a calcium tetrafluoroborate (Ca(BF4)2, 0.45 M, EC:PC (50/50 wt%)) electrolyte versus Ca2+/Ca. It was found that the capacity of 400 mA h g−1 could be achieved by charging the cell at 100 °C at a scan rate of 0.05 mV s−1. The authors noticed a change in the voltage range, moving from 1.7 to 2.3 V seen in Fig. 9(c), something usually ascribed to overpotential. To confirm this was due to the redox reaction and not any electrolytic side reactions, they characterised the pristine and oxidised samples. The pristine sample had Ca:Si ratio of 30:70 wt%, measured by EDS, but the calcium content dropped to 7 wt% (∼Ca0.15Si2), accompanied by a decrease in particle size from 7.3 to 2.6 μm. This was corroborated with the X-ray powder diffraction (SXRPD) results seen in Fig. 9(a), which shows the original Si peaks after the oxidation process, confirming the decalciation of CaSi2 (at 2.8 V vs. Ca2+/Capassivated). The authors concluded that the alloyed anode was active experimentally and computationally, but a higher specific capacity could be achieved. Alloy materials promise very good alternatives for Ca2+ incorporation but still suffer from large volume expansion and slow kinetic properties, which hugely impact their cycling performance.
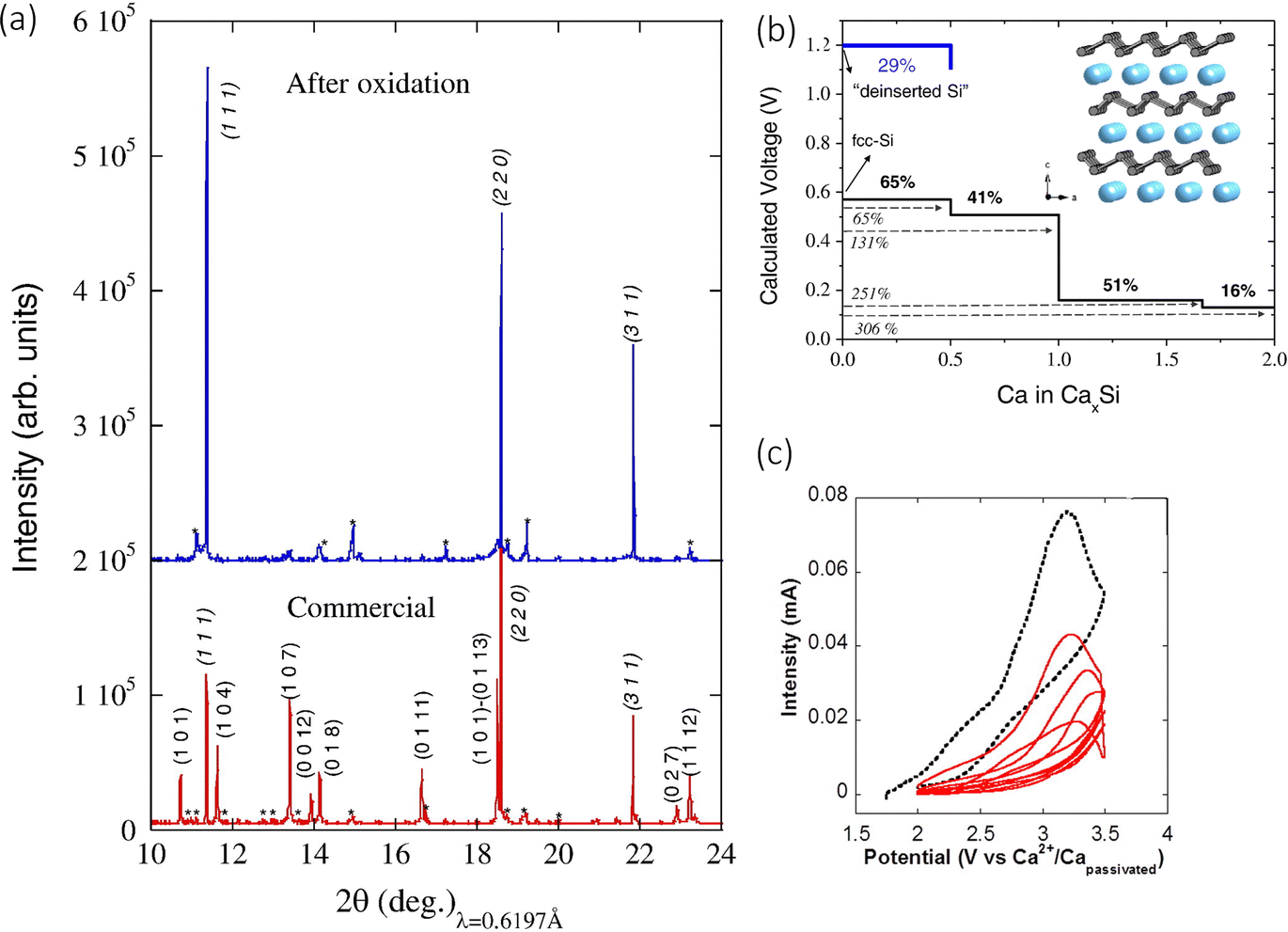 | ||
| Fig. 9 (a) SXRPD patterns; pristine anode prepared with the commercial CaSi2 before (bottom) and after oxidation to 3.5 V vs. Ca2+/Ca at 100 °C (top). Miller indices – CaSi2 (roman type) and Si Peaks (marked as *) indicate impurities present in the pristine commercial sample. The pristine powder composed of CaSi2 (70%) and Si (30%). (b) Calculated composition–voltage profiles. The black line corresponds to the calciation of fcc-Si following an alloying mechanism in which intermetallics CaxSi are formed. The blue line accounts for the deinsertion of Ca ions from the layered CaSi2. The inset shows the schematic crystal structure of CaSi2 (Si in grey, Ca in blue). (c) Cyclic voltammograms (0.05 mV s−1) of CaSi2 electrode in a 2-electrode Ca cell operating at 100 °C. Reproduced with permission from ref. 89, copyright 2016 Elsevier B.V. | ||
Although not alloys, the use of pristine gold and platinum as electrodes that can successfully strip and plate calcium has been briefly looked at.36 These are famously ideal anodes, but cost and capacitive performance would limit their commercial potential. The study by Wang et al., mainly focused on the use of a Ca(BH4)2 electrolyte in terahydrofuran, with emphasis on the ability for gold to successfully plate calcium and strip it, leaving a CaH2 passivation layer behind.
Conclusion and future outlooks
The development of viable anodes for CIBs would unlock major research in this area. The strong reducing ability of calcium metal and its high valency, mixed with the combination of available electrolytes, have inhibited the growth and development of calcium as an alternative metal ion battery to lithium, sodium, or potassium. To deal with these challenges, researchers have observed various paths, including the composition of the electrolytes, operational temperatures, and in the case of this review, utilising different anode materials. This review highlights issues around calcium metal anodes, from ineffective plating and stripping mechanism to inactive passivation layers, and finally acknowledging the anode materials required for successful reversibility and competitive capacities. It can be acknowledged that this research is still very much in its infancy, but as seen in this review, there has been significant progress made. The main material that has seen success is graphitic carbon, something that has been used throughout metal ion battery research in the past with many varying morphologies being studied, but alternatives to this have been found such as organic frameworks for electrolytic stability and alloys for attempts to achieve calcium's theoretical capacity. Organic anodes provide a promising avenue of anode research because of their adaptability physically and chemically. They possess very notable rate performances but can suffer from solubility and conductivity issues in organic electrolyte, limiting their current application for CIBs. Their affordability and adaptability have driven research in mono- and multivalent in general when the potential of these types of materials are finally being understood. With it comes a more comprehensive understanding of its potential and the future it will play in metal ion battery research.The study of alloy anodes is one of keen interest due to their high theoretical capacity, low reaction potentials, and their ability to prevent dendrite growth. There have been a lot of theoretical studies conducted, suggesting prospective metals to use, but so far only Sn has truly been tested experimentally, mainly due to large volume expansions, increasing resistances and capacitance fading. There have been studies conducted into the alloying of metals such as Ge, Sb, Bi and Mg with Ca using molten salts as the electrolyte, which could lead to favourable alloy conversion and a new avenue of research for alloying materials.95,96 Ca–Sn remains an attractive option, and the potential of exploring other alloys utilised in other multivalent metals like Mg-ion remain a strong possibility.
The development of new electrolytes and compositions hold one of the keys to the future of Ca2+-ion anodes. They would be required to form stable SEI layers that are electronically insulating but ionically conductive, which none of the salts, nor their solvents currently are currently able to address. Computational models continue to examine the issue of the current electrolyte selection, but also have begun providing alternatives such as calcium closo-monocarborane (Ca[CB11H12]2) in tetraethylene glycol dimethyl ether (G4) or exploring the optimal compositions of current electrolytes.97,98 Another electrolytic route is that of a solid electrolyte, something that could theoretically stabilise the surface of Ca metal anodes, through steady and uniform ion dispersion.99,100
Ionic liquid (IL) based electrolytes are being viewed as an alternative to solvent-based electrolytes. They are molten salts that have a melting temperature lower than 100 °C. ILs have had operational success with LIBs, due to their thermal stability, wide electrochemical stability, beneficial salt stability, and high ionic conductivity. They are generally less toxic and less volatile than their solvent-based equivalents but their main attribute in the context of CIBs is their ability to lower their operational stripping and plating potentials, something that could prove useful with calcium metal anodes, which is theoretically lower than conventional calcium electrolytes, with higher efficiencies.101 With regards to CIB ILs, the research is still in its infancy, with only a handful of publications. Stettber et al. and Pathreeker et al. have looked into using 1-butyl-1-methylpyrrolidinum bis(trifluoromethylsulfonyl) imide (Pyr14TFSI), an aprotic IL (AIL) or 1-butylpyrrolidinum bis(trifluoromethylsulfonyl) imide (PyrH4TFSI), a protic ‘free proton’ IL (PIL) with Ca(TFSI)2 as the salt.102,103 They both were able to perform cycling at room temperature and noted an improved ion transport mechanism, owing to the IL's superior ionic conductivity. Biria et al. used 1-ethyl-3-methylimidazolium trifluoromethanesulfonate ([emim][triflate]), an IL that has had success with Mg2+ systems, with a Ca(BF4)2 salt.104 They observed the stable formation of the SEI layer, while demonstrating the ability to plate and strip crystalline calcium. The SEI that forms shows Ca2+ permeability and that any passivation of the SEI later would require greater overpotentials at their current density (0.55 mA cm−2) to the plating and stripping of the Ca anode. This in turn would lead to electrolytic breakdown and a low coulombic efficiency. It would be an advantage to some extent to use ILs to form SEI, a polymeric layer instead of an inorganic one. This prevents the passivation of the surface of the anode, therefore allowing for facile Ca2+ diffusion. Some of the issues that remain are that they still have very poor power performances compared to those of commercial organic electrolytes, are high in cost and are not as environmentally friendly as once thought due to their high toxicity and poor biodegradable natures105,106
One of the issues around Ca2+ is its poor diffusion kinetics due to its large ion size and high charge density, which can lead to the instability of crystal electrode materials during intercalation/deintercalation. Looking at adapting the morphology, through doping, vacancies, disordering defects, and amorphization can lead to greater accessibility of Ca2+ to active sites through the promotion of ion diffusion, election transfer, as well as the ability to more easily intercalate/deintercalated. Defect chemistry specifically improves the intercalation capabilities of the electrode, through this improvement of the systems kinetics and thermodynamics. Defect chemistry can theoretically alleviate volume changes, while enhancing structural stability, facilitate the fast diffusion of Ca2+ ions and can form electron rich regions, through exposing more active storage sites, which all lead to better electrochemical performances. This is subject area that has been highly investigated in other multivalent battery research and could be easily applied to calcium research.24,107
The future for CIBs remains exciting, as current research is still in its infancy and if more attention is afforded to this area of study, the problems seen in this review will be addressed. Countless theoretical papers have been written about anode utilising DFT, showing that there is no lack of potential interest in Ca2+ ions, but experimental design is still lacking. The increase in computational studies for CIBs will allow for a wide and thorough screening of potential candidate anode materials with regards to their diffusion kinetics, theoretical capacities, electrochemical stabilities etc, improving the options and scope for more experimental design.
Cathodes will remain the preferred option taken when undertaking CIB research because of the more translatable design across metal ion battery research but the success of its anodes will yield more progressive results.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Z. acknowledges the support of the National Key R&D Program of China (2021YFA1501502). Y. X. acknowledges the support of the Leverhulme Trust (RPG-2021-138), Engineering and Physical Sciences Research Council (EP/V000152/1, EP/X000087/1), and Royal Society (RGS\R2\212324, SIF\R2\212002). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.References
- P. Greim, A. A. Solomon and C. Breyer, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed
.
- J. Wen, Y. Yu and C. Chen, Mater. Express, 2012, 2, 197–212 CrossRef CAS
- A. Jana, S. I. Woo, K. S. N. Vikrant and R. E. García, Energy Environ. Sci., 2019, 12, 3595–3607 RSC
- G. Berckmans, M. Messagie, J. Smekens, N. Omar, L. Vanhaverbeke and J. van Mierlo, Energies, 2017, 10, 1314 CrossRef
- J. Biemolt, P. Jungbacker, T. Van Teijlingen, N. Yan and G. Rothenberg, Materials, 2020, 1–31 Search PubMed
- X. Shen, H. Liu, X. B. Cheng, C. Yan and J. Q. Huang, Energy Storage Mater., 2018, 12, 161–175 CrossRef
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS
- A. Ponrouch, J. Bitenc, R. Dominko, N. Lindahl, P. Johansson and M. R. Palacin, Energy Storage Mater., 2019, 20, 253–262 CrossRef
- R. S. Treptow, J. Chem. Educ., 2003, 80, 1015–1020 CrossRef CAS
- L. Stievano, I. de Meatza, J. Bitenc, C. Cavallo, S. Brutti and M. A. Navarra, J. Power Sources, 2021, 482, 228875 CrossRef CAS
- D. Li, Y. Yuan, J. Liu, M. Fichtner and F. Pan, J. Magnesium Alloys, 2020, 8, 963–979 CrossRef CAS
- S. K. Das, S. Mahapatra and H. Lahan, J. Mater. Chem. A, 2017, 5, 6347–6367 RSC
- C. Ling, J. Chen and F. Mizuno, J. Phys. Chem. C, 2013, 117, 21158–21165 CrossRef CAS
- N. T. Hahn, D. M. Driscoll, Z. Yu, G. E. Sterbinsky, L. Cheng, M. Balasubramanian and K. R. Zavadil, ACS Appl. Energy Mater., 2020, 3, 8437–8447 CrossRef CAS
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J. F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS PubMed
- M. E. Arroyo-De Dompablo, A. Ponrouch, P. Johansson and M. R. Palacín, Chem. Rev., 2020, 120, 6331–6357 CrossRef CAS PubMed
- R. J. Gummow, G. Vamvounis, M. B. Kannan and Y. He, Adv. Mater., 2018, 30, 1801702 CrossRef PubMed
- N. N. Rajput, T. J. Seguin, B. M. Wood, X. Qu and K. A. Persson, Elucidating Solvation Structures for Rational Design of Multivalent Electrolytes—A Review, Top. Curr. Chem., 2018, 376, 79 CrossRef PubMed
- Z. Wang, H. Wang, S. Qi, D. Wu, J. Huang, X. Li, C. Wang and J. Ma, EcoMat, 2022, 4, 2567–3173 Search PubMed
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X. Z. Liao and Z. F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC
- Y. Zhao, A. Wang, L. Ren, X. Liu and J. Luo, J. Energy Chem., 2022, 70, 174–190 CrossRef CAS
- S. S. R. K. C. Yamijala, H. Kwon, J. Guo and B. M. Wong, ACS Appl. Mater. Interfaces, 2021, 13, 13114–13122 CrossRef CAS PubMed
- D. Aurbach, R. Skaletsky and Y. Gofer, J. Electrochem. Soc., 1991, 138, 3536–3545 CrossRef CAS
- B. Ji, H. He, W. Yao and Y. Tang, Adv. Mater., 2021, 33, 2005501 CrossRef CAS PubMed
- R. Verrelli, A. Black, R. Dugas, D. Tchitchekova, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2020, 167, 070532 CrossRef CAS
- A. Ponrouch and M. R. Palacin, Curr. Opin. Electrochem., 2018, 9, 1–7 CrossRef CAS
- S. Kim, L. Yin, M. H. Lee, P. Parajuli, L. Blanc, T. T. Fister, H. Park, B. J. Kwon, B. J. Ingram, P. Zapol, R. F. Klie, K. Kang, L. F. Nazar, S. H. Lapidus and J. T. Vaughey, ACS Energy Lett., 2020, 5, 3203–3211 CrossRef CAS
- S. Jing, J. Xiao, Y. Shen, B. Hong, D. Gu and W. Xiao, Small, 2022, 18, 2203251 CrossRef CAS PubMed
- X. Tang, D. Zhou, B. Zhang, S. Wang, P. Li, H. Liu, X. Guo, P. Jaumaux, X. Gao, Y. Fu, C. Wang, C. Wang and G. Wang, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed
- S. Pathreeker, S. Reed, P. Chando and I. D. Hosein, J. Electroanal. Chem., 2020, 874, 114453 CrossRef CAS
- M. S. Chae, J. W. Heo, J. Hyoung and S.-T. Hong, ChemNanoMat, 2020, 6, 1049–1053 CrossRef CAS
- H. Park, C. J. Bartel, G. Ceder and P. Zapol, Adv. Energy Mater., 2021, 11, 2102698 Search PubMed
- L. Wang, J. Qiu, X. Wang, L. Chen, G. Cao, J. Wang, H. Zhang and X. He, eScience, 2022, 2, 125–137 CrossRef
- B. Jeon, J. W. Heo, J. Hyoung, H. H. Kwak, D. M. Lee and S. T. Hong, Chem. Mater., 2020, 32, 8772–8780 CrossRef CAS
- Y. Guo, S. Wu, Y.-B. He, F. Kang, L. Chen, H. Li and Q.-H. Yang, eScience, 2022, 2, 138–163 CrossRef
- A. Shyamsunder, L. E. Blanc, A. Assoud and L. F. Nazar, ACS Energy Lett., 2019, 4, 2271–2276 CrossRef CAS
- S. Biria, S. Pathreeker, H. Li and I. D. Hosein, ACS Appl. Energy Mater., 2019, 2, 7738–7743 CrossRef CAS
- C. Bao, B. Wang, P. Liu, H. Wu, Y. Zhou, D. Wang, H. Liu and S. Dou, Adv. Funct. Mater., 2020, 30, 2004891 CrossRef CAS
- J. Young and M. Smeu, Adv. Theory Simul., 2021, 4, 2100018 CrossRef CAS
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446–1453 CrossRef CAS
- W. Song, S. Scholtis, P. C. Sherrell, D. K. H. Tsang, J. Ngiam, J. Lischner, S. Fearn, V. Bemmer, C. Mattevi, N. Klein and D. J. Riley, Energy Environ. Sci., 2020, 4977–4989 RSC
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4 Search PubMed
- J. Forero-Saboya, C. Davoisne, R. Dedryvère, I. Yousef, P. Canepa and A. Ponrouch, Energy Environ. Sci., 2020, 13, 3423–3431 RSC
- B. Wu, S. Wang, J. Lochala, D. Desrochers, B. Liu, W. Zhang, J. Yang and J. Xiao, Energy Environ. Sci., 2018, 11, 1803–1810 RSC
- K. Guo, R. Kumar, X. Xiao, B. W. Sheldon and H. Gao, Nano Energy, 2020, 68, 104257 CrossRef CAS
- S. Biria, S. Pathreeker, F. S. Genier, H. Li and I. D. Hosein, ACS Appl. Energy Mater., 2020, 3, 2310–2314 CrossRef CAS
- Q. Wei, L. Zhang, X. Sun and T. L. Liu, Chem. Sci., 2022, 13, 5797–5812 RSC
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Nat. Mater., 2018, 17, 16–20 CrossRef CAS PubMed
- H. Kim, D. A. Boysen, T. Ouchi and D. R. Sadoway, J. Power Sources, 2013, 241, 239–248 CrossRef CAS
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Nat. Mater., 2016, 15, 169–172 CrossRef CAS PubMed
- J. Forero-Saboya, C. Bodin and A. Ponrouch, Electrochem. Commun., 2021, 124, 106936 CrossRef CAS
- J. Forero-Saboya, C. Bodin and A. Ponrouch, Electrochem. Commun., 2021, 124, 106936 CrossRef CAS
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC
- N. J. Leon, X. Xie, M. Yang, D. M. Driscoll, J. G. Connell, S. Kim, T. Seguin, J. T. Vaughey, M. Balasubramanian, K. A. Persson and C. Liao, J. Phys. Chem. C, 2022, 126, 13579–13584 CrossRef CAS
- H. Hou, X. Qiu, W. Wei, Y. Zhang and X. Ji, Adv. Energy Mater., 2017, 7 Search PubMed
- J. Ming, Z. Cao, W. Wahyudi, M. Li, P. Kumar, Y. Wu, J. Y. Hwang, M. N. Hedhili, L. Cavallo, Y. K. Sun and L. J. Li, ACS Energy Lett., 2018, 3, 335–340 CrossRef CAS
- P. Luo, C. Zheng, J. He, X. Tu, W. Sun, H. Pan, Y. Zhou, X. Rui, B. Zhang and K. Huang, Adv. Funct. Mater., 2022, 32, 2107277 CrossRef CAS
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC
- Y. Nishimura, N. Nakatani and K. Nakagawa, J. Solid State Electrochem., 2021, 25, 2495–2501 CrossRef CAS
- L. Zhao, B. Ding, X. Y. Qin, Z. Wang, W. Lv, Y. B. He, Q. H. Yang and F. Kang, Adv. Mater., 2022, 34 Search PubMed
- J.-L. Yang, X.-X. Zhao, W.-H. Li, H.-J. Liang, Z.-Y. Gu, Y. Liu, M. Du and X.-L. Wu, eScience, 2022, 2, 95–101 CrossRef
- J. Zhao, X. Zou, Y. Zhu, Y. Xu and C. Wang, Adv. Funct. Mater., 2016, 26, 8103–8110 CrossRef CAS
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O’Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M. M. Titirici, Energy Environ. Sci., 2020, 13, 3469–3479 RSC
- Y. Li, Y. Lu, P. Adelhelm, M. M. Titirici and Y. S. Hu, Chem. Soc. Rev., 2019, 48, 4655–4687 RSC
- S. J. R. Prabakar, A. B. Ikhe, W. B. Park, K. C. Chung, H. Park, K. J. Kim, D. Ahn, J. S. Kwak, K. S. Sohn and M. Pyo, Adv. Sci, 2019, 6, 1902129 CrossRef PubMed
- J. Park, Z. L. Xu, G. Yoon, S. K. Park, J. Wang, H. Hyun, H. Park, J. Lim, Y. J. Ko, Y. S. Yun and K. Kang, Adv. Mater., 2020, 32, 1904411 CrossRef CAS PubMed
- S. Wu, F. Zhang and Y. Tang, Adv. Sci., 2018, 5, 1701082 CrossRef PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS
- D. Datta, J. Li and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 1788–1795 CrossRef CAS PubMed
- J. Wang, V. Malgras, Y. Sugahara and Y. Yamauchi, Nat. Commun., 2021, 12, 3563 CrossRef CAS PubMed
- S. Lu, F. Lou and Z. Yu, Catalysts, 2022, 12, 228 CrossRef CAS
- A. Rajkamal and R. Thapa, Adv. Mater. Technol., 2019, 4, 1900307 CrossRef CAS
- S. Wang, Y. Si, B. Yang, E. Ruckenstein and H. Chen, J. Phys. Chem. Lett., 2019, 10, 3269–3275 CrossRef CAS PubMed
- Q. Xie, H. Ou, Q. Yang, X. Lin, A. Zeb, K. Li, X. Chen and G. Ma, Dalton Trans., 2021, 50, 9669–9684 RSC
- Y. Chen, K. Fan, Y. Gao and C. Wang, Adv. Mater., 2022, 20062 Search PubMed
- Y. Liang, C. Luo, F. Wang, S. Hou, S. C. Liou, T. Qing, Q. Li, J. Zheng, C. Cui and C. Wang, Adv. Energy Mater., 2019, 9, 1802986 CrossRef
- X. Lu, D. Zhang, J. Zhong, L. Wang, L. Jiang, Q. Liu, G. Shao, D. Fu, J. Teng and W. Yang, Chem. Eng. J., 2022, 432, 134416 CrossRef CAS
- A. v Desai, R. E. Morris and A. R. Armstrong, ChemSusChem, 2020, 13, 4866–4884 CrossRef CAS PubMed
- J. Li, C. Han, X. Ou and Y. Tang, Angew. Chem., Int. Ed., 2022, 61, e202116668 CAS
- B. Jiang, Y. Su, R. Liu, Z. Sun and D. Wu, Small, 2022, 18, 2200049 CrossRef CAS PubMed
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 2, 2235–2259 CrossRef CAS
- J. Calbo, M. J. Golomb and A. Walsh, J. Mater. Chem. A, 2019, 7, 16571–16597 RSC
- T. N. Vo, J. Hur and T. il Kim, ACS Sustainable Chem. Eng., 2020, 8, 2596–2601 CrossRef CAS
- T. T. Tran and M. N. Obrovac, J. Electrochem. Soc., 2011, 158, A1411–A1416 CrossRef CAS
- A. L. Lipson, B. Pan, S. H. Lapidus, C. Liao, J. T. Vaughey and B. J. Ingram, Chem. Mater., 2015, 27, 8442–8447 CrossRef CAS
- A. Ponrouch, D. Tchitchekova, C. Frontera, F. Bardé, M. E. A. de Dompablo and M. R. Palacín, Electrochem. Commun., 2016, 66, 75–78 CrossRef CAS
- W. Jia, Z. Wang, J. Li, X. Yu, Y. Wei, Z. Yao, Y. Liu, Y. Wang, A. Zhou, W. Zou, F. Zhou and H. Li, J. Mater. Chem. A, 2019, 7, 22377–22384 RSC
- H. Okamoto, J. Phase Equilib. Diffus., 2013, 34, 171 CrossRef CAS
- A. Palenzona, P. Manfrinetti and M. L. Fornasini, J. Alloys Compd., 2000, 312, 165–171 CrossRef CAS
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H. M. Cheng, Nat. Chem., 2018, 10, 667–672 CrossRef CAS PubMed
- Z. Zhao-Karger, Y. Xiu, Z. Li, A. Reupert, T. Smok and M. Fichtner, Nat. Commun., 2022, 13, 3849 CrossRef CAS PubMed
- J. M. Newhouse, S. Poizeau, H. Kim, B. L. Spatocco and D. R. Sadoway, Electrochim. Acta, 2013, 91, 293–301 CrossRef CAS
- R. J. Gummow, G. Vamvounis, M. B. Kannan and Y. He, Adv. Mater., 2018, 30, 1801702 CrossRef PubMed
- S. S. R. K. C. Yamijala, H. Kwon, J. Guo and B. M. Wong, ACS Appl. Mater. Interfaces, 2021, 13, 13114–13122 CrossRef CAS PubMed
- N. T. Hahn, D. M. Driscoll, Z. Yu, G. E. Sterbinsky, L. Cheng, M. Balasubramanian and K. R. Zavadil, ACS Appl. Energy Mater., 2020, 3, 8437–8447 CrossRef CAS
- F. S. Genier, C. v Burdin, S. Biria and I. D. Hosein, J. Power Sources, 2019, 414, 302–307 CrossRef CAS
- S. Biria, S. Pathreeker, F. S. Genier and I. D. Hosein, ACS Appl. Polym. Mater., 2020, 2, 2111–2118 CrossRef CAS
- S. Biria, S. Pathreeker, F. S. Genier, H. Li and I. D. Hosein, ACS Appl. Energy Mater., 2020, 3, 2310–2314 CrossRef CAS
- T. Stettner, R. Dugas, A. Ponrouch and A. Balducci, J. Electrochem. Soc., 2020, 167, 100544 CrossRef CAS
- S. Pathreeker and I. D. Hosein, ACS Appl. Polym. Mater., 2022, 4, 6803–6811 CrossRef CAS PubMed
- S. Biria, S. Pathreeker, F. S. Genier, H. Li and I. D. Hosein, ACS Appl. Energy Mater., 2020, 3, 2310–2314 CrossRef CAS
- M. C. Bubalo, K. Radošević, I. R. Redovniković, I. Slivac and V. G. Srček, Arh. Hig. Rada Toksikol., 2017, 68, 171–179 CrossRef CAS PubMed
- Q. Wei, L. Zhang, X. Sun and T. L. Liu, Chem. Sci., 2022, 13, 5797–5812 RSC
- E. Uchaker and G. Cao, Chem. – Asian J., 2015, 10, 1608–1617 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |