
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-based stimuli-responsive materials for biomedical applications
Wenjing
Ma
,
Dawei
Hua
 ,
Ranhua
Xiong†
* and
Chaobo
Huang†
*
,
Ranhua
Xiong†
* and
Chaobo
Huang†
*
Joint Laboratory of Advanced Biomedical Materials (NFU-UGent), Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing 210037, P. R. China. E-mail: ranhua.xiong@njfu.edu.cn; Chaobo.Huang@njfu.edu.cn
First published on 21st December 2022
Abstract
Bio-based stimuli responsive materials have been widely studied as highly versatile materials in biomedical applications. In this review, different types of bio-based stimuli-responsive materials are introduced (most of them are carbohydrate polymers). The response mechanisms are explained and the advantages/drawbacks of the materials are discussed. Particularly in bio-based stimuli-responsive drug delivery and in bio-based drug anticounterfeiting fields, the response mechanisms and advantages are discussed, and the shortcomings are also mentioned.
1. Introduction
Bio-based materials refer to a material prepared by using biomass as raw materials. It is considered environmentally friendly and is suitable for sustainable development to replace oil-based products. Among the bio-based materials, the polymer materials encompass a large proportion, and include bioplastics, water-soluble polymers and other carbon-based lubricants (Fig. 1). These materials play an important role in the bio-carbon cycle, which is important for circular economy.1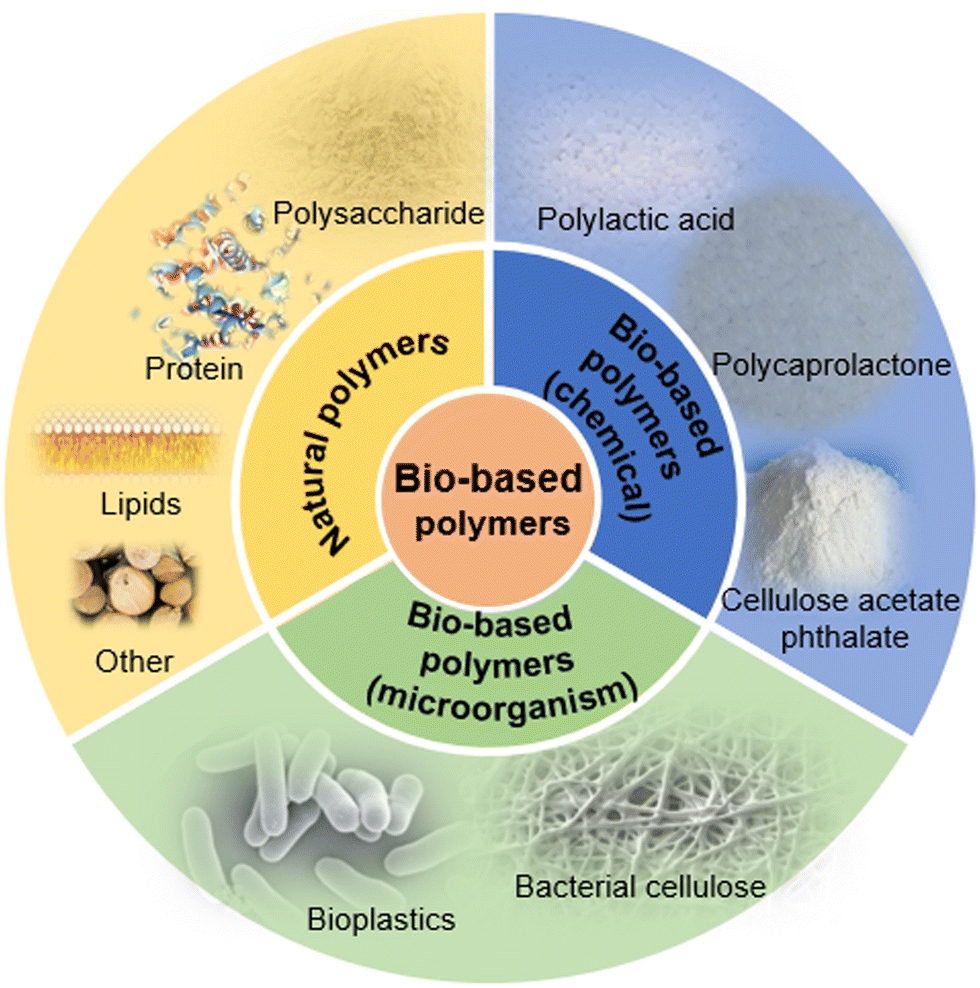 | ||
| Fig. 1 Different types of bio-based polymers. | ||
Bio-based polymer materials can be classified into three types: natural polymers, synthetic polymers and composite polymers (a mixture of multiple bio-based polymers). Among them, natural polymers mainly include natural polysaccharides, such as xylan-containing hemicellulose from wood, chitin and chitosan obtained from crabs and shrimps, as well as curdlan from the physiological activities of microorganisms. The synthetic polymers include polylactic acid (PLA), which can be synthesized by lactic acid (fermented corn starch) through chemical reaction,2 and polycaprolactone (PCL), which can be formed by the ring-opening polymerization of a caprolactone monomer in the presence of gas atmosphere and catalyst.3 Due to its excellent biological/sustained drug release properties, PCL is widely used in tissue engineering and biomedical fields. In addition, PCL can combine with a variety of natural fillers to prepare biodegradable composite materials. For example, it can be mixed with starch to prepare composite biodegradable materials with lower cost.4
With the development of science and technology, it is increasingly difficult for traditional bio-based materials to fulfill the needs of people. For this reason, many researchers have paid increasing attention to explore bio-based polymer materials, which is stimuli-responsive. A stimuli-responsive material refers to a type of material, of which physical and chemical properties can change upon exposure to external signals.5–7 It exhibits a response to stimuli, such as pH,8 temperature,9 mechanical force,10 small molecules,11 and electric/magnetic field.12–14 In order to obtain specific functions, a variety of bio-based stimuli-responsive materials are prepared. For example, chitosan is pH-responsive due to protonation and deprotonation of its amino terminal. Chitosan hydrogels could swell or collapse when the pH and ionic intensity of the external environment change. The hydrogels have tunable physicochemical properties, especially for mechanical properties (e.g., elasticity and viscoelasticity) that can provide specific functions (e.g., guiding cell behavior and fate).15,16 Furthermore, the drug molecules contained in the hydrogel could be triggered to release due to the volume change of the hydrogel. Compared with traditional stimuli responsive materials, the main advantage of a bio-based stimuli responsive material is that it can simulate natural physical and chemical conditions, and endow the materials with excellent biocompatibility. Therefore, it is widely used in food, medicine, tissue engineering and other fields (Table 1).
| Types | Strategies | Applications | Ref. |
|---|---|---|---|
| pH response | Protonation and aprotonation; chemical cleavage bonds | Biodegradable oral drug delivery system; inhibition of bacterial growth | 21 and 24 |
| Temperature response | Phase change | Drug-controlled release system; injectable gel | 33 and 37 |
| Magnetic response | Incorporated with magnetic-responsive materials, such as Fe3O4, Ni | Implantation and positioning of the material in the body; cell engineering | 42 and 48 |
| Electric response | Undergoes a reversible redox reaction with voltage | Drug release system | 50 and 54 |
| Ultrasound response | Affects material directly; secondary effect of ultrasound irradiation | Drug release system | 56,58 and 59 |
| Light response | Photoisomerization; photodegradation of polymer; photothermal and photochemical effect | Drug release system; wound dressing | 64 and 68 |
| Mechanical force response | The materials have a response to compression, tension, and shear | Sensors; electronic devices; medicine | 80–83 |
| Ion response | Forms crosslinking; affects the osmotic pressure | Self-healing; to detect heavy metal pollutants | 89 and 92 |
| Enzyme response | Incorporation with substrate molecules that have specific domains | Drug release system; cell engineering | 97 and 101 |
2. Different types of bio-based stimuli responsive materials
2.1 pH-responsive bio-based materials
pH-responsive bio-based materials have attracted increasing attention in the field of medicine. For example, due to the higher aerobic and anaerobic glycolysis efficiency, the pH value in the tumor extracellular environment is lower than that in the extracellular environment of a healthy cell. The difference of pH value between the tumor cell and healthy cell is the basis for the preparation of tumor-targeted pH-responsive drug delivery systems.17–19 The drug contained in the materials can be released with the change of pH value, thus achieving a therapeutic effect.20 Such materials are usually prepared from polymers that containing carboxylic acid groups or amine groups. Due to the protonation and aprotonation effects of these functional groups, the polymer structure can change upon pH change. In addition, there might be acid cleavage bonds in such materials, such as acetal groups or hydrazone groups, so that the structure can cleave in a low pH environment and the drug molecules contained within the materials can be released. For example, alginate is an anionic copolymer that is composed of mannuronic acid and guluronic acid residues. It is non-toxic and biodegradable, so it is often used in the preparation of oral drugs. In an acidic environment, the carboxylic acid group in the structure converts to –COOH and a hydrogen bond can form between a hydroxyl group and –COOH. The formation of a hydrogen bond limits the swelling of the hydrogel structure, and then the polymer structure shrinks.21 Researchers22 mixed polyvinyl alcohol and sodium alginate to prepare an ionic crosslinked alginate porous film. The immiscibility caused the alginate and polyvinyl alcohol phase separation to occur. Calcium chloride was used to dissolve the polyvinyl alcohol phase, leaving the alginate phase, and the alginate was crosslinked with calcium ions to form a porous alginate film. The pore size of the porous membrane could be controlled upon pH change. It showed that the porous membrane was pH responsive. The study also showed that these porous hydrogel membranes could be transferred to conventional commercial filtration substrates without damage to the membrane. This enables the porous membrane to have the potential for use as a separation membrane or a controlled drug release system.Due to the high water content of conventional hydrogels, the mechanical strength is generally weak and the burst release often occurs when the hydrogel is applied as a drug release system. Some researchers tried to enhance the mechanical strength of pH-responsive bio-based hydrogels by increasing the degree of crosslinking. However, the crosslinking agents are sometimes toxic, which limits the application of the hydrogel. Other researchers introduced different materials into stimuli responsive materials to prepare composite materials, so that the materials are not only stimuli-responsive, but also have a certain mechanical strength, which is convenient for practical application. For example, some researchers mixed calcium hydrogen phosphate and alginate to prepare calcium hydrogen phosphate-alginate hydrogels.23 The dibasic calcium phosphate could form hydrogen bonds with alginate. Furthermore, the hydrogen bonds could make the composite hydrogel structure more compact, which enhances the mechanical strength of the hydrogel to a certain extent. Since alginate is pH-responsive, when the hydrogel is in an acidic environment, the structure shrinks (drug cannot release), and the hydrogel swells under alkaline conditions (drug release). Due to the existence of hydrogen bonds in the structure, the swelling of the hydrogel is limited. In this way, the burst release could be avoided during the swelling, and an ideal drug release profile could be obtained.
Different from a passive diffusion-based drug release system, the release profile of a pH-responsive drug release system is more controllable. A biodegradable pH-responsive drug release system was prepared by the researchers,24 and aminoglycoside was applied as crosslinking agent to connect with oxidized polysaccharides (such as dextran, carboxymethyl cellulose, alginate, and chondroitin). By changing the concentration of aminoglycoside during gel preparation, the degradation rate and the release kinetics of the material could be tunable. The growth of bacteria may induce an acid environment. Due to the existence of acid cleavage bonds, the acid environment could promote the bond cleavage. The drug (such as an antibacterial drug) encapsulated in the material could then be released, and show therapeutic effects (Fig. 2(A)).
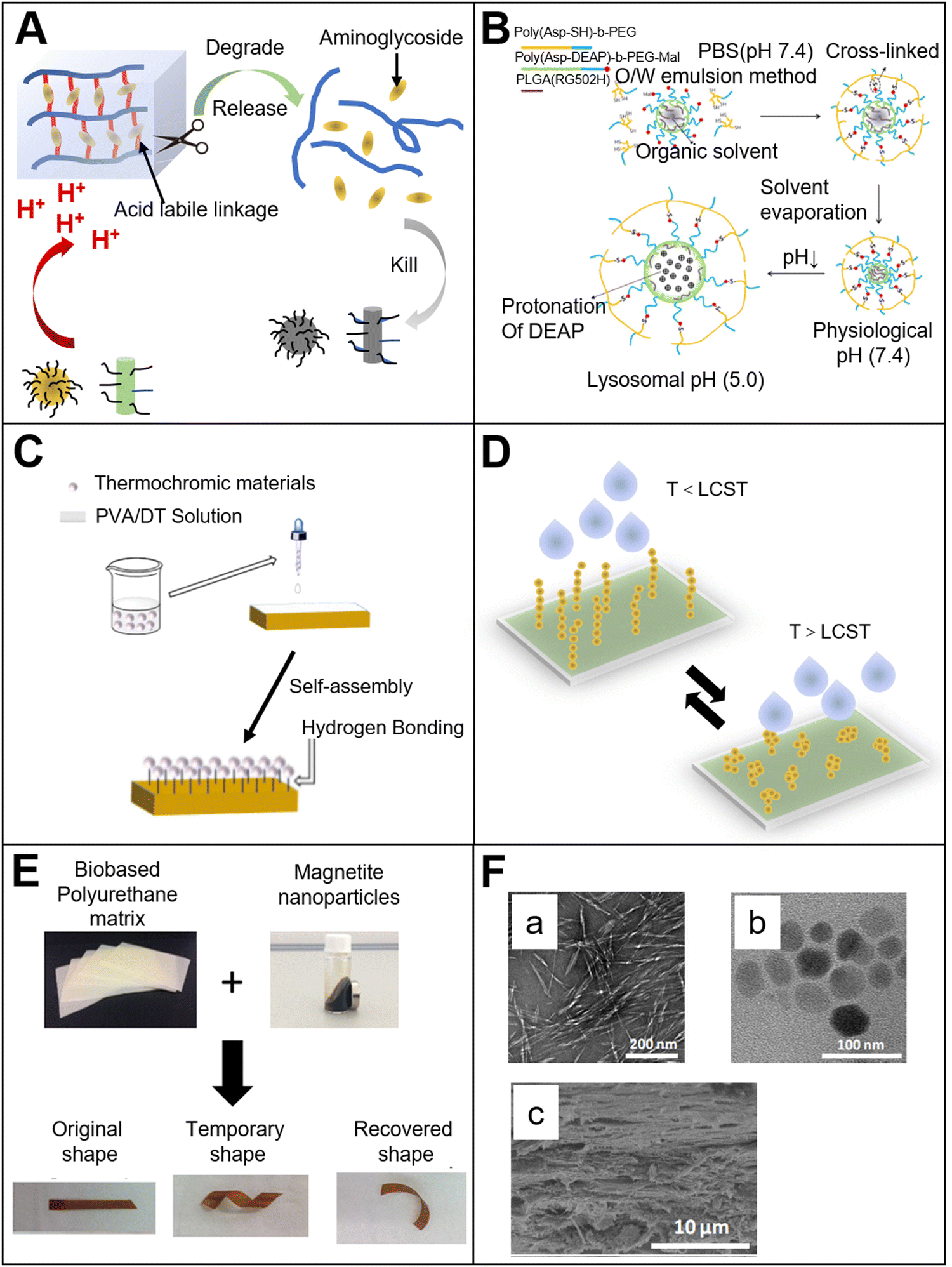 | ||
| Fig. 2 (A) pH-responsive bio-based materials. Mechanism of the pH-responsive aminoglycoside hydrogel24 (reproduced with permission24 Copyright 2017 Elsevier B.V. All rights reserved); (B) pH-responsive co-polymer micelles32 (reproduced with permission32 Copyright 2012 Elsevier B.V. All rights reserved); (C) temperature-responsive bio-based materials made by surface modification38 (reproduced with permission38 from the Royal Society of Chemistry); (D) water permeability of the temperature-responsive cellulose nanofibril film39 (reproduced with permission39 Copyright 2016 American Chemical Society); (E) bio-based magnetic-responsive materials. Bio-based polyurethane material with both magnetic response and shape memory.44 (Reproduced with permission44 Copyright 2018 Published by Elsevier B.V.); (F) magnetically responsive cobalt ferrite–cellulose nanocrystal composite film. a: cellulose nanocrystals; b: cobalt ferrite; c: ferrite–cellulose nanocrystal composite film46 (reproduced with permission46 Copyright 2017 Elsevier, Ltd. All rights reserved). | ||
In the field of stimuli-responsive materials, after the material is exposed to an external stimulus, the response efficiency of the material usually depends on the rate at which the stimulus is transmitted into the material, and the feedback to the response mainly depends on the diffusion process of the stimulus within the material. The response efficiency is usually inversely proportional to the thickness of the material.25 It is logical that a response that is too slow will limit the practical application of materials. Therefore, compared with thick hydrogels, many researchers have paid increasing attention to the fiber structure, which is pH-responsive. Poly(styrene-co-maleic anhydride) and cellulose acetate were used to prepare composite fibers by electrospinning. The composite fiber has good mechanical strength and pH-responsiveness. The composite fiber could absorb a large amount of water and swell at pH values above 8. This is because the –COOH group in maleic acid is ionized into a –COO group in an alkaline environment, which makes the fiber generate a higher density negative charge. The higher negative charge density makes the fiber form a greater electrostatic repulsion. This increases the size of the fiber, making the polymer chain more relaxed to promote the absorption of water. When the fibers were added to a high Na+ concentration environment, sufficient electrostatic repulsion could not be generated due to the neutralization of the charge, which weakens the water absorption capacity of the fiber. Similar to the hydrogels mentioned above, some researchers introduced acid-labile acetal groups into the main chain of a bio-based block copolymer composed of polylactic acid and polyethylene glycol.26 The acid-labile polymer fibers were obtained by electrospinning. When the fibers were immersed in a neutral solution, the morphology did not change. However, the acetal group could be cleaved when the fibers were added to an acidic environment, which led to the disintegration of the polymer backbone. Thus, the drugs and other substances contained within the fibers could be released. In our previous work,27 cellulose acetate phthalate (CAP) was used to prepare CAP/PU coaxial fibers by electrospinning. When in an acidic environment, the fiber mainly contains hydrophobic groups, which can maintain a hydrophobic state, and the fiber structure is stable. However, when the fiber is placed in an alkaline environment, the functional group of CAP becomes hydrophilic. The fiber is dissolved immediately, and the drug molecules can be released. In this way, the pH-responsive drug release system can be obtained.
In addition to micro/nano fibers, micro/nano particles with pH-responsiveness are also notable. The basic mechanism is similar to that of pH-responsive hydrogels. The structure of the micro/nano particles expands or collapses during the deprotonation or protonation of anions or cations. For anionic micro/nano particles, when the pKa value of the material is higher than that of an external environment, the electrostatic repulsion in the internal grid structure of the material will decrease, and then the micro/nano particles will collapse. Meanwhile, for cationic micro-nano particles, if the pKa of the material is higher than that of the external environment, the –NH2 group can be converted to –NH3+, thereby increasing the hydrophilicity of the material and causing the material to swell. To a certain extent, we could say that this pH-responsiveness includes hydrophilic–hydrophobic phase transition.28–31 Aspartic acid, which is a bio-based polymer derivative, was also applied to prepare pH stimuli-responsive materials (Fig. 2(B)). The material made by the polyaspartic acid block polymer could be protonated when the pH value was below 7.32 The material will swell due to the protonation. When the particles are taken up by lysosome, the material will swell immediately because the pH of lysosome is lower than 5. The change in the volume of the particles can damage the lysosomal membrane and make the enzymes (such as phosphatase, protease and nuclease) leak from lysosome. These enzymes can kill surrounding host cells, and thus play an important role in killing tumor cells. Meanwhile, small-molecule drugs or proteins/genes could be encapsulated in the pH-responsive particles, and lysosome escape could be achieved to avoid the degradation of the bioactive compounds by lysosome. By changing the content of the block polymer, the particles could swell to a certain level in an acidic environment, resulting in the instability of the lysosomal membrane and enhancement of the drug release rate.
2.2 Temperature-responsive bio-based materials
Temperature is one of the most studied factors in the field of stimuli-responsive materials. Researchers have prepared a variety of bio-based temperature-responsive materials by doping or chemical modification. The researchers33 selected natural fatty acids to prepare a temperature-responsive drug-controlled release system. Natural fatty acid is a type of phase change material. The phase change means that the material can undergo a reversible transition between liquid and solid under external stimulation (temperature). By changing the ratio of different types of natural fatty acids, the phase transition temperature could be tuned to 37 °C,34–36 which has great potential in the field of biomedical application. For example, lauric acid and stearic acid were mixed in a certain ratio to prepare a phase transition material. The phase transition temperature of the eutectic mixture was 39 °C. Then, the nerve growth factor was packaged in the phase transition mixture by electrospray technology, and the mixture was combined with an electrospinning polycaprolactone fiber. The nerve cells were seeded onto the composite fiber membrane. At room temperature, nerve growth factor was encapsulated in the solid mixture, and no obvious growth of the nerve protrusion was observed. When the temperature exceeded the melt point of the phase-change material, the nerve growth factor was released from the liquid mixture, resulting in obvious growth and differentiation of nerve synapses. In addition, the researchers37 prepared temperature-responsive nanoparticles made by bio-based polylactic acid (PLA) and polyethylene glycol (PEG) block copolymers. The experimental results showed that when the temperature was 20 °C, the mixture presented a sol state. When the temperature increased to 37 °C, the mixture turned into a gel state, and the mechanical strength of the material could be significantly enhanced. The temperature response mechanism of the nanoparticle can be described as follows. At low temperatures, the hydrophobic PLA segment is wrapped by the hydrophilic PEG segment, and the PLA and PLA segments are isolated from each other. With increasing temperature, the hydrophobicity of the PLA segment is weakened. Then, the segment can mix with the outside hydrophilic layer, so that the PLA chains are intertwined and mixed, and the degree of crystallinity is increased. Thus, the material is transformed from the sol state to the gel state, and the results showed that the phase transition material could be used to prepare an injectable gel.Bio-based temperature-responsive materials are not only determined by the structural properties of the bio-based material itself. It could also be obtained by surface modification of the bio-based material. The researchers38 poured thermochromic materials, polyvinyl alcohol and cyclodextrin onto the surface of wood to prepare thermochromic wood (Fig. 2(C)). The natural porous structure of wood provides many anchor points for the thermochromic material. Polyvinyl alcohol and dextrin make it possible to form more hydrogen bonds within the structure, so that the thermochromic material can be closely attached to the wood surface. At room temperature, the wood shows its initial texture color. When the temperature increase, the wood surface gradually shows a red color. The mechanism of color change is because of the organic dyes anchored on the wood surface. When the temperature exceeds the transformation temperature, the crystal phase and structure of the dyes will undergo a reversible transformation, resulting in a transformation of their color. As mentioned above, the researchers39 grafted thermo-responsive poly(N-isopropylacrylamide) onto the surface of cellulose nanofibrils (CNF) (Fig. 2(D)). As we know, natural CNF is hydrophilic. In order to facilitate grafting, a layer of polyvinyl alcohol film was coated onto the CNF surface as a crosslinking agent, and poly(N-isopropylacrylamide) was subsequently grafted onto it. The results showed that with increasing temperature, the efficiency of water permeation significantly increased. The mechanism of its temperature response is that when the temperature is lower than the phase transition temperature of poly(N-isopropylacrylamide), the polymer chains are in a stretched state and form a physical protective layer on the CNF film, which hinders the permeation of moisture. When the temperature is higher than the phase transition temperature, the polymer chains will fold, and the physical barrier hindering the water permeation disappears. Therefore, the water permeability will be significantly enhanced. In addition to hydrogels, there are many fibers with stimuli response. For example, researchers40 presented a strategy to produce an electrospinning smart fiber based on a copolymer with a cellulose acetate (CA) backbone containing grafted temperature-responsive polymer chains of MEO2MA and OEGMA. The stimuli-responsive polymers with a temperature-triggered volume phase transition (VPT)41 could transform from a hydrophilic to hydrophobic state. The study showed that the copolymer of 2-(2-methoxyethoxy) ethyl methacrylate and oligo (ethylene glycol) methacrylate, p(MEO2MA-co-OEGMA), also exhibited a VPT. Thus, if the temperature is above LCST, the temperature-responsive polymer grafted CA fibers can transform from a hydrophilic to hydrophobic state.
In addition to surface modification,42 bio-based temperature-responsive materials can be prepared by doping. Researchers43 dissolved thermal responsive methyl cellulose (MC) in an aqueous dispersion of cellulose nanocrystals (CNC) to prepare a viscoelastic dispersion at room temperature. The composite material is temperature-responsive due to the existence of MC. When the temperature rises to 60 °C, its mechanical strength is significantly enhanced. The mechanism of its temperature response is because of the reversible gelation behavior of MC. With increasing temperature, the single MC chain loses part of its affinity with the surrounding water. Then, the MC chain shrinks into a fibril structure with the size of 14 nm. The physical cross-grid within the fibril structure and the rod-shaped CNC enhance the mechanical strength of the material. This method provides a new idea for the preparation of CNC hydrogel materials by doping.
2.3 Magnetic-responsive bio-based materials
Magnetic-responsive materials have attracted increasing attention due to their non-contact response and low costs. They have been widely used in the medical imaging and industrial fields. In order to obtain ideal magnetic responsiveness during the preparation of bio-based magnetic-responsive materials, the bio-based materials are usually incorporated with magnetic-responsive materials. For example, Fe3O4 nanoparticles are most commonly used because of their low cost, non-toxicity, and excellent magnetic responsiveness. Fe3O4 nanoparticles were doped with hyperbranched polyurethane (PU), which was modified by sunflower oil (Fig. 2(E)).42 The results showed that the Fe3O4 nanoparticles could be uniformly distributed in the hyperbranched PU. Because of the magnetic Fe3O4, the thermomechanical property and shape memory property of the material were improved. Furthermore, the material showed magnetic responsiveness. The magnetic response mechanism is because of the uniform distribution of Fe3O4 nanoparticles in the material, allowing for more hydrogen bonds to be introduced into the material. It might be due to the carbamate complex, which was formed between the oxygen in Fe3O4 and the –NH group, or the interaction between the carbonyl group of the carbamate and the trace hydroxyl groups on the surface of the nanoparticles. Introducing hydrogen bonds affects the phase separation in the material segment.The magnetic responsiveness provides a certain convenience for the implantation and positioning of the material in the body. Similarly, some researchers44 mixed bio-based polyurethane with magnetic Fe3O4 nanoparticles to prepare bio-based magnetic-responsive materials. The Fe3O4 nanoparticles impart magnetic responsiveness to the material. Meanwhile, the Fe3O4 nanoparticles could reduce the crystallinity of the polymer, and increase the shape fixity of the material due to its interaction with the polymer hydroxyl groups. Also, Fe3O4 nanoparticles could heat the material to soften the polymer segment in the presence of a magnetic field. It shows the function of shape memory.
Other metal ions are also used in combination with Fe3O4 nanoparticles to give the material additional properties (not only magnetic response). For example, researchers45 mixed cellulose nanocrystals (CNC) and Fe3O4 nanoparticles to prepare composite materials. The copper ions were also added to form complexes, so that the composite showed responsiveness to external magnetic fields. Meanwhile, the formation of complexes also provided attachment sites for proteins. Thus, the adsorption and separation of proteins could be achieved. Other researchers46 doped cobalt ferrite nanoparticles with CNC, as well as glucose to prepare a homogeneous and crack-free CNC film (Fig. 2(F)). The film showed excellent magnetic response, and the incorporation of cobalt ferrite further increased the dielectric constant of the film. The results indicated that the material had the potential to be applied as a green bio-based functional material. The cobalt ferrite was chosen because of its high magnetostriction (≈200 ppm) and high magnetization compared with ordinary ferrite.
Magnetic nanoparticles could be coated onto the surface of bio-based materials to prepare bio-based magnetic responsive materials. The bio-based material could also be coated onto the surface of magnetic nanoparticles. The researchers47 anchored the lignin and its derivatives onto the surface of Fe3O4 nanoparticles to prepare bio-based magnetic-responsive materials. Different from the doping method mentioned above, the researchers mixed iron salt with lignin here, which has amine groups. The lignin was anchored onto the surface of the Fe3O4 nanoparticles after the reaction. The prepared material showed a strong adsorption capacity for dyes in sewage. Because of its magnetic responsiveness, the dyes absorbed by the nanoparticles could be easily separated from the solution. Due to the protonation and deprotonation of the material, the prepared magnetic material also had pH responsiveness, so the adsorption and desorption between lignin and dyes could be realized by tuning the pH of the solution.
In addition to Fe3O4, other metal elements were used to prepare magnetic-responsive materials. Researchers48 designed bio-based magnetic responsive materials prepared by collagen hydrogel, GelMA hydrogel and nickel beads. For cell engineering, the synergy between the extracellular matrix and the mechanical stimulation is important. A 3D magnetically actuated collagen hydrogel platform was developed and could realize combined control of the ECM architecture and mechanical stimulation, which shows great promise for regulating cell behavior and fate.
2.4 Electric-responsive bio-based materials
Compared with pH and temperature response, electric response has its unique advantages. The electric field could be precisely controlled by tuning the voltage, current or time. This is convenient for the development of a high-precision portable device. As we know, the main component of an electric stimuli-responsive material is a conductive polymer. The conductive mechanism is that the alternating single and double bonds of the polymer chain could generate π-bonded delocalized electrons in the polymer, and the electrons can move freely on the polymer chain, so that the transfer of electric charge could be achieved.49 When a voltage is applied, the polymer undergoes a reversible redox reaction, causing the conformational change of the polymer. Currently, the electric stimuli-responsive materials in the medical field mainly include: electric stimuli-responsive hydrogels, electric stimuli-responsive layer-by-layer self-assembled films, and others. The electric response mechanism is that the hydrogel undergoes a series of structural changes, such as swelling, bending, and contraction when the external electric field is applied. Due to the structure change, drug molecules could be released from the hydrogel. In addition to the simple electric stimuli response, some more complicate electric stimuli responsive bio-based materials have been prepared. For example, researchers50 have encapsulated a drug in the copolymer of methyl methacrylate and methacrylic acid (Fig. 3(A)). The copolymer is insoluble in water when the pH value is below 7, and is soluble when the pH is above 7. Chitosan was cast onto the copolymer as a protective layer. When an electric field is applied with the protonation of the copolymer, the pH value increases above 7 and the copolymer becomes water-soluble. Then, the drug encapsulated in the copolymer could be released. Interestingly, the release amount of drug is positively correlated with the intensity of the electric field. Without an electric field stimulation, the drug is only released with a small amount, which means that the electric stimuli responsive system can realize the pulse release of drugs. Another researcher51 also used chitosan as a cationic material, and combined it with ionic liquids to prepare a drug delivery film with electric stimuli responsiveness. The results showed that the incorporation of the ionic liquid significantly improved the conductivity of the film, and the sensitivity to electric stimulation under a weak electric field was enhanced. This indicates that the material can be used in drug delivery and other fields.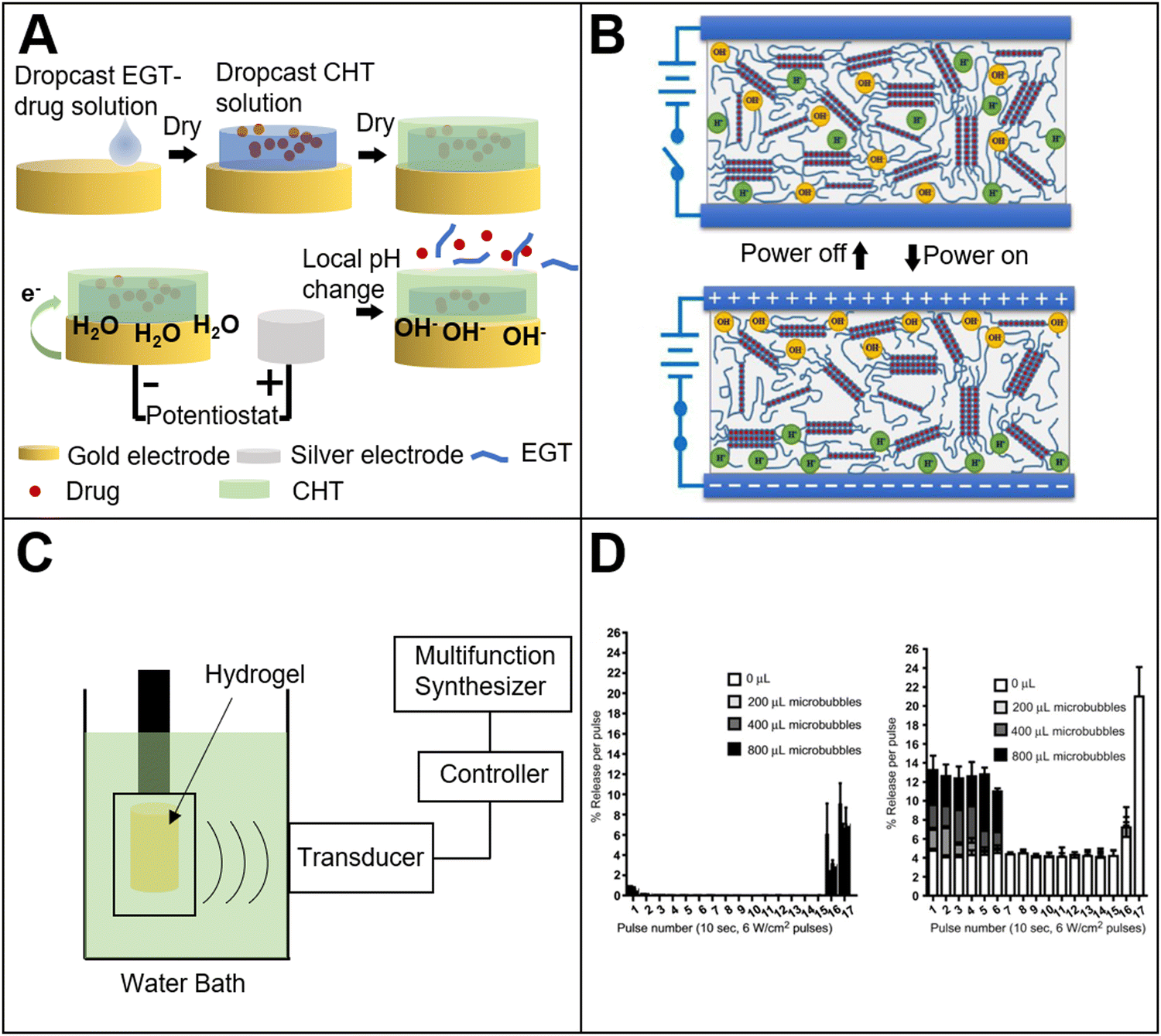 | ||
| Fig. 3 (A) Electrical field response bio-based materials. Electrically responsive methacrylic acid–chitosan composite materials for controlled drug release50 (reproduced with permission50 from the Royal Society of Chemistry); (B) electro-responsive agarose hydrogel52 (reproduced with permission52 Copyright 2020 Elsevier, Ltd. All rights reserved); (C) ultrasound-responsive bio-based materials. Schematic diagram of ultrasonic-responsive cellulose-based hydrogel film experiment56 (reproduced with permission56 Copyright 2016 Elsevier B.V. All rights reserved); (D) ultrasound-responsive hydrogel for pulsed drug delivery.59 Dye release before (left) and after (right) ultrasound pulse (reproduced with permission59 Copyright 2010 Elsevier, Ltd. All rights reserved). | ||
In addition to chitosan, some researchers52 have used other materials such as an agarose hydrogel to prepare electric stimuli responsive bio-based materials. The agarose hydrogel was prepared by solvent casting, and the internal physical cross-linking of hydrogen bonds formed a 3D hydrogel network structure (Fig. 3(B)). The experiment showed that with the electric stimulation, the polarizable molecules in the agarose structure were aligned to form a dipole moment. The hydrogel was conductive and the dielectric constant of the hydrogel increased with increasing hydrogel concentration. The large amount of hydroxyl groups in the hydrogel structure could promote the responsiveness to the electric field, and make it easier to expand and contract. Under electric stimulation, the dielectrophoresis force generated by the non-uniform electric field and non-uniform distribution of the electric dipoles inside the agarose hydrogel bends the hydrogel. This electric stimulation response has broad application prospects in biological actuators and other fields. Some other researchers53 used bio-based polyurethane blends (PCL/PU) and graphene nanosheets to prepare bio-based actuators with electric stimulation response and self-driving capabilities. The graphene nanosheets have a certain impact on the crystal structure of the polyurethane blends, and give the material a certain infrared light responsiveness. Under the infrared light irradiation, the graphene absorbs the light energy and converts it into heat. With the increasing local temperature, the mobility of the polymer segment is promoted, thereby achieving bending. Agarose and alginate are also often used to prepare electric stimuli responsive bio-based materials. The researchers54 combined agarose/alginate with an aniline tetramer to prepare a hydrogel with an electric stimulation response for drug release system. Since the mechanical strength of the aniline tetramer alone is weak, the agarose and alginate could improve its mechanical strength. In this research study, bio-based materials were used as a scaffold of the hydrogel system. It provided a certain amount of mechanical strength for the hydrogel, and made it more suitable for practical application. Meanwhile, it also improved the biocompatibility of the hydrogel system.
2.5 Ultrasound responsive bio-based materials
Bio-based ultrasound-responsive materials refer to materials, in which the structure or physical and chemical properties could change and exhibit specific functions under ultrasound irradiation. It is widely used in the medical field. There are mainly two types of ultrasound response mechanism. One is that the ultrasound could directly affect the material. For example, ultrasound could destroy the hydrogen bonds within the material, and change the structure of the material to release drugs encapsulated in the material or enable the material to achieve specific functions. Another response mechanism is the secondary effect of ultrasound irradiation. It means that the material could convert the energy into heat under ultrasound irradiation, so that the local temperature of the material will increase, and then the material could exhibit some specific functions with increasing temperature. One of the most important secondary effects of ultrasound response is called cavitation.55 It includes bubble nucleation, growth and cavitation. In the process of bubble generation or rupture, the bubble will produce corresponding changes to the structure of the material. In this way, the material could exhibit certain specific functions.Due to its good mechanical strength, suitable viscoelasticity and excellent biocompatibility, the hydrogel is also often used to prepare bio-based ultrasound responsive materials. For example, researchers prepared a cellulose-based hydrogel film, which exhibited a response to ultrasound irradiation, and the ultrasound-triggered drug release56 was obtained (Fig. 3(C)). Results showed that the hydrogen bonds between the cellulose and the drug molecule in the hydrogel were broken. Therefore, it achieved the drug release from the hydrogel. The drug release efficiency of the hydrogel with ultrasound treatment was six times higher than that of the hydrogel without ultrasound treatment. Furthermore, with increasing ultrasonic power, more drug was released. Like cellulose, researchers57 prepared a chitin hydrogel loaded with gallic acid. Under ultrasound irradiation, the hydrogen bonds between chitin and gallic acid were broken so that the drug release was accelerated. In addition to the breakage of hydrogen bonds, ultrasound can facilitate the breakage of ionic bonds in the material. For example, the researchers58 prepared a chemotherapeutic loaded sodium alginate hydrogel. The experiment showed that pulse chemotherapeutic drug delivery could be achieved by tuning the ultrasound on and off. The efficacy of therapy with ultrasound treatment was much better than without the ultrasound treatment group. The ultrasound response mechanism of this system is that the calcium ion cross-linking structure in the hydrogel is broken. The breakage could destroy the grid structure within the hydrogel, causing the small molecules of chemotherapeutic release from the hydrogel. When the ultrasound is turned off, the free calcium ions in the hydrogel will spontaneously form crosslinking, and the crosslinking could repair the hydrogel grid structure to trap the drug molecules inside the hydrogel.
In addition to forming hydrogen bonds or other groups in bio-based materials, bio-based ultrasound responsive materials could be obtained by the combination of bio-based materials and ultrasound-responsive materials. The researchers59 combined dextran hydrogels with gas microbubbles to prepare ultrasound responsive bio-based hydrogels (Fig. 3(D)). The dextran could protect the microbubbles from collapse. When ultrasound is applied to the hydrogel, the structure of the hydrogel changes as the microbubbles collapses under the shock of ultrasound, so that the drug is released and the controlled release could be realized. In addition, the drug release efficiency of the hydrogel could be improved by increasing the drug loading or microbubbles concentration.
As mentioned above, bio-base materials include natural bio-based and synthetic bio-based materials. In addition to natural bio-based materials such as sodium alginate and dextran, bio-based raw materials were used as monomers to synthesize bio-based polyamides.60 The polymer chain changed from a crosslinked structure to a non-crosslinked structure. Then, the aromatized structure was transformed into a linear structure, and the glass transition temperature of the material decreased due to the polymer free volume increasing after decrosslinking. The method could provide a new idea for the preparation of bio-based stimuli-responsive materials.
2.6 Light-responsive bio-based materials
In a stimulus response system, another kind of material that has attracted increasing attention is the light-response material. When stimulated by an external light source, the structure of these materials may undergo a certain change in conformation, polarity, amphiphilicity, electric charge, optical chirality or conjugation. This mainly depends on specific groups that are responsive to light, such as spiropyran, azobenzene, diarylethane and coumarin, which can realize reversible isomerization under light irradiation. There are also irreversibly transformed o-nitrobenzyl photo-unstable protective groups, of which the groups can be dissociated with polymer chains after light illumination. Compared with the pH response, temperature response, electrical response and magnetic response systems, the advantage of light responsiveness is its non-contact response, and the long-term or long-distance response could be realized. Meanwhile, it can achieve the required response at a low dose level. The light response system can precisely control the light intensity, wavelength, irradiation time and irradiation area to achieve specific functions, such as changing the color of the material appearance or pulsed drug release. Many different types of light irradiation have a broad application prospect in the field of non-invasive surgery.61The response mechanisms of light-responsive materials can be classified as follows: (1) molecular structure transformation by photoisomerization effect under light irradiation; (2) photodegradation of the polymer main chain; (3) damage to the material structure due to the photothermal effect; (4) chemical degradation through photochemical effects. Among these, photoisomerization is related to the chemical bonds of limited rotation, mainly double bonds. This is a reversible change (intramolecular rearrangement), such as the trans double bond in azobenzene, which has a reversible cis–trans isomerization to the light irradiation. In addition, photoisomerization can be accompanied by the cleavage of the chromophore, which is attributed to the light-induced structural transformation, and the transformation is irreversible. The photodegradation of the polymer backbone causes the release of the drug molecules contained within the structure. The polymer structure is locally destroyed by heat through the photothermal effect, so that the molecules loaded in the polymer matrix could be released. When the polymer containing the o-nitrobenzyl group is exposed to light, the group can be cleaved, thereby releasing the drug from the polymer.62
The chromophore plays an important role in the light response system.63 These groups are designed to respond to a variety of light sources, such as UV light, visible light, near-infrared and even laser. UV light, due to its high energy, easily breaks chemical bonds. Thus, UV light is applied as the main response stimulation in light response systems.64,65 Due to the advantages of UV light, many photo-crosslinkable materials have been developed to prepare light responsive systems for tissue engineering. The researchers66,67 used photo-crosslinkable gelatin methacrylate (GelMA) to prepare hydrogels. Periodontal ligament stem cells (PDLSCs) were encapsulated in the hydrogel. The designed hydrogel systems could promote regeneration of functional tissue, and it could also be a good platform to study the extracellular matrix. For wound management and repairing, an in situ imine crosslinking-based photoresponsive chitosan hydrogel was prepared by the researchers.68 Upon UV exposure, o-nitrobenzene could be converted to o-nitrosobenzaldehyde groups, which could form crosslinking on the tissue surface. Compared with recently developed hydrogel-based tissue adhesives, the photoresponsive chitosan hydrogel possessed a more effective, suitable, and promising property for future trauma emergency treatment.
However, there are some drawbacks to using UV light. Tissue permeability that is too low will limit the application of UV light.69 It only penetrates the shallow tissue and cannot reach the deep tissue. In addition, UV light is highly toxic, and free radicals are produced during UV light irradiation. The free radicals may cause progressive damage to human skin,70 destroy the structure of vitamin A71 and vitamin C,72 damage the structure of collagen,73 and even lead to more serious genetic damages.74 There is no doubt that visible light is less toxic than UV light,75 and the visible light can penetrate deeper tissue. Therefore, researchers are trying to use visible light to replace UV light as the response stimulation source in the light response material field. The researchers64 prepared a nano-drug carrier system, which is sensitive to singlet oxygen. The system could release the loaded drug under visible light irradiation. The nano-structure carrier nanoparticles are composed of porous silica nanoparticles, and a porphyrin cap that is sensitive to reactive oxygen species (ROS) is grafted onto the silica pore. With visible light irradiation, active oxygen is generated by the porphyrin structure. The active oxygen could break the chemical bonds, which is sensitive to active oxygen. The porphyrin cap on the pore could then be damaged, and the drug molecule loaded inside the nanoparticles is exposed to the surrounding environment. Then, the light responsive drug release could be realized.
In addition to visible light, another stimulation source is the two-photon near-infrared light. There are many advantages to this system, including the high anti-interference ability, low energy, deep penetration and high 3D spatial resolution.76 A composite hydrogel with polydopamine nanoparticles and polyethylene glycol was prepared by researchers.77 Since polyethylene glycol is a synthetic bio-based material with excellent biocompatibility and hydrophilicity, it is widely used to prepare bio-based hydrogels. It has been reported that the molecules containing sulfhydryl or amine groups could bind with polydopamine molecules through π–π bonds or hydrogen bonds. Due to the binding, the molecules could be trapped in the hydrogel. Under infrared light irradiation, polydopamine could absorb near-infrared light and convert its energy into heat. The heat can destroy the chemical bond interaction between the target molecule and the polydopamine molecule. Thus, the target molecules could be released. The near-infrared responsiveness of the bio-based material depends on the stacking of π–π bonds and the strength of hydrogen bonds.
2.7 Mechanical stimulus response bio-based material
Mechanical response bio-based material refers to bio-based materials that can give feedback to external mechanical stimuli and show specific functions. The external mechanical stimuli can be divided into three types: compression, tension, and shear. There are a variety of specific materials corresponding to each stimulus. The researchers78 prepared a mechanical stimuli response hydrogel, and investigated the application for controlled drug release (Fig. 4(A)). Sodium alginate-bio-based material was applied as a raw material, and was mixed with the target molecules to form a hydrogel. Two different types of hydrogels were prepared. One only contained free target molecules, while the other one contained both free target molecules and the molecules that were anchored to the hydrogel network structure. The results showed that when the hydrogel was compressed, the free target molecules could be released from the squeezed hydrogel, but the release profile was not controllable. Meanwhile, in the presence of the anchored molecules group, under the periodic compression, controlled drug release could be realized. A plausible release mechanism is described as follows. Without compression, the target molecules remained anchored inside the hydrogel. When a compression force was applied, the force damaged the anchoring structure between the hydrogel and the target molecule. Then, the pulse release profile could be achieved. β-Cyclodextrin was also combined with sodium alginate to prepare a compression response hydrogel.79 The results showed that sodium alginate hydrogel alone had no response to compression force. The reason might be that with calcium ion crosslinking, the hydrogel could be stable even under the compression. When β-cyclodextrin was incorporated, the mechanical strength of the hydrogel mesh was decreased, so that the hydrogel mesh structure could be deformed under the compression force, resulting in the release of drug molecules from the hydrogel.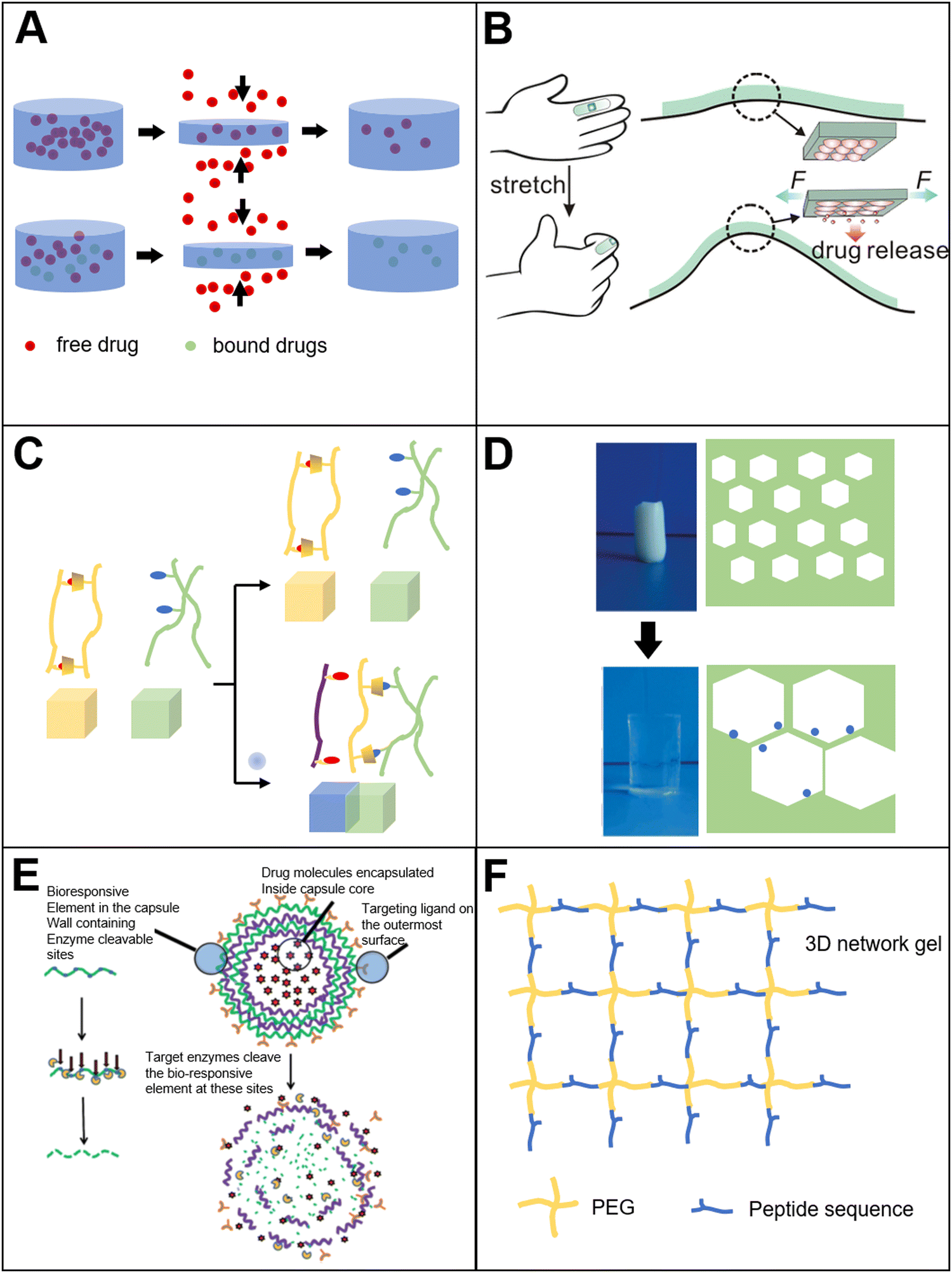 | ||
| Fig. 4 (A) Tensile-responsive bio-based materials. Pressure-responsive hydrogel for drug release78 (reproduced with permission78 Copyright 2001 WILEY-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany); (B) wearable tensile-responsive sodium alginate microsphere for drug delivery86 (reproduced with permission86 Copyright 2015 American Chemical Society); (C) ion-responsive bio-based materials. Metal ions-responsive adhesive material89 (reproduced with permission89 from the Royal Society of Chemistry); (D) ion-responsive cellulose-based hydrogel90 (reproduced with permission90 Copyright 2011 American Chemical Society); (E) enzyme-responsive bio-based materials. Enzyme-responsive layer-by-layer self-assembled microcapsules97 (reproduced with permission97 from the Royal Society of Chemistry); (F) enzyme-responsive polyethylene glycol–peptide hydrogel101 (Reproduced with permission101 Copyright 2009, Nature Publishing Group). | ||
Bio-based tension response materials are widely used in sensors,80,81 electronic devices,82,83 medicine and other fields.84 Usually, the tension response material contains elastomers. For example, the researchers85 prepared a composite hydrogel containing sodium alginate, acrylamide and enzyme. The enzyme molecules were uniformly distributed in the hydrogel. When the hydrogel was stretched, the tension could increase the surface area of the hydrogel structure and reduce the thickness of the hydrogel. More enzymes could be exposed onto the surface of the hydrogel, which made it easier for the enzyme to make contact with the substrate. In this way, the catalytic efficiency could be accelerated. Furthermore, the increase in the specific surface area of the hydrogel was positively correlated with increasing enzyme activity. The increase in the specific surface area of the hydrogel under the tension could also be used to control the release profile of the drug molecules. The researchers86 placed drug-loaded sodium alginate microspheres in an elastomer substrate (Fig. 4(B)). When an external tension stimulus was applied, the surface area of the sodium alginate microspheres was changed. This change could promote the release and diffusion of the loaded drug, and the system could be reused several times.
The response to shear force depends on the reversible deformation and disaggregation of the material since the narrowing of blood vessels increases the shear force of blood flow by more than ten times.87,88 Therefore, the shear force response materials have been widely used in cardiovascular system therapy. The researchers prepared a bio-based drug-loaded microaggregate composed of polylactic acid and glycolic acid nanoparticles. The aggregates remained stable under the condition of normal blood flow shearing force. In an embolized blood vessel, however, the increased shear force could cause microaggregate disaggregation. The concentration of free nanoparticles would then increase. With the increasing surface area of the drug-loaded nanoparticles, the drug utilization rate could be improved.
Although mechanical stimulus response bio-based materials are easy to prepare and can be used in a wide range of applications, it still faces the following problems during the late development process. For example, the mechanical stimulus needs to reach a certain range to achieve the feedback of the material. Thus, the sensitivity of the response should be improved. After the stimulation is given, it also takes a certain period for the material to recover. This means that even if the stimulation is removed, the material could still show certain functions, such as the release of drugs. For a biomedical system, a complete controllable response is necessary. Otherwise, the leakage of drug molecules contained within the material may occur. These problems mentioned above need to be further developed and improved.
2.8 Ion-responsive bio-based materials
As we mentioned above, a variety of kinds of stimuli-responsive materials are prepared, and many of them are hydrogels. For these hydrogels, metal ions is very important. Because the metal ions could form a crosslinking structure in the hydrogel. For example, one of the most widely used bio-based hydrogels is the sodium alginate hydrogel. The calcium ions are added during the hydrogel preparation process to form crosslinking with the polymer network. In addition to the crosslinking during hydrogel preparation, metal ions can be used as a source of stimulation. In order to prove that the metal ions could be used as a stimulation source (Fig. 4(C)), the researchers89 prepared two kinds of hydrogels: one containing N,N-methylenebisacrylamide/polyacrylamide (modified by β-cyclodextrin and metal ion stimuli-responsive ligand); another type is methylene bisacrylamide/tert-butyl modified polyacrylamide. In the absence of metal ions, cyclodextrin binds to the ligand, which is metal ion-responsive. The ligand is inserted into the cyclodextrin cavity, and it causes the stickiness of the hydrogel to disappear. Then, the hydrogels cannot be adherent with each other. Once the metal ions are added, the metal ions release the ligand from the cyclodextrin cavity, and the exposed cyclodextrin cavity can be combined with the tert-butyl group of polyacrylamide. Subsequently, the adhesion of the hydrogel materials is recovered and the self-healing is achieved.Metal ions not only combine with the corresponding groups to realize the stimuli response, but also affects the osmotic pressure to achieve the response. As shown in the literature,90 quaternized cellulose (QC) and carboxymethyl cellulose (CMC) were crosslinked to prepare composite hydrogels (Fig. 4(D)). When the hydrogel was placed in the solution containing metal ions, the swell occurred. The swelling mechanism is because the hydrogel absorbs a large amount of water to increase its volume. During swelling, metal ions can affect the osmotic pressure inside the hydrogel and in the solution. Thus, the change of osmotic pressure makes the hydrogel expand and shrink. For instance, in the presence of a high concentration of metal ions, the hydrogel shrinks due to the osmotic pressure, resulting in a low swelling rate. In addition, by tuning the ratio of cellulose to carboxymethyl cellulose, the swellable rate of the hydrogel can be controlled.
Biological materials, such as DNA, are also widely applied in the preparation of ion-responsive hydrogels. A Ag+ responsive hydrogel containing DNA was prepared by the researchers.91 Specifically, the hydrogel contains Y-type nucleic acid subunits or acrylamide chains, which is functionalized with nucleic acids. In the presence of silver ions, the hydrogel undergoes a reversible transition between gel and liquid. The response mechanism is that silver ions can combine with cytosine bases to form a polymer grid crosslinking structure. Cysteine can form complexes with the silver ions. Thus, when cysteine is added into the hydrogel, the silver ions used as the crosslinking agents would be taken away by cysteine due to the complex formation. With the disappearance of the crosslinking agents, the hydrogel structure collapses. Therefore, the material converts from a solid to a liquid.
Some researchers have combined ion-responsive materials with non-ion-responsive cellulose to prepare ion-responsive cellulose films. The researchers92 coated the surface of the cellulose fiber membrane with titanium dioxide so that the surface of the cellulose fiber was evenly coated with a titanium dioxide film. The ruthenium dye molecules were then anchored on the titanium dioxide film. When the cellulose film is placed in a solution containing mercury ions, the mercury ions combine with the isothiocyanate group in the ruthenium dye to form complexes. The formation of complexes makes the fiber membrane color significantly change. This could be applied to detect heavy metal pollutants in water. The high surface area of the cellulose film improves the efficiency and sensitivity of the test.
2.9 Enzyme-responsive bio-based materials
Enzymes are biomolecules that act as catalysts for chemical reactions, and participate in almost all intracellular metabolic processes. It can selectively incorporate with substrate molecules that have specific domains.93 This highly selective and specific enzyme-substrate interaction is the basis of enzyme-responsive materials. Based on this interaction, a series of applications have been developed.94 There are two main routes to prepare enzyme-responsive bio-based materials. One route is to use enzyme-responsive materials as substrates, such as peptides or sugars. These molecules could be cleaved under the enzyme interaction specifically.95 Another route is to modify the surface of non-enzyme-responsive materials with enzyme-responsive groups. Due to the existence of enzymatic reaction groups, the enzyme-responsive materials could be obtained.96A layer-by-layer self-assembled method was applied by the researchers97 to prepare hollow nanocapsules with dual enzyme response. Doxorubicin was loaded in the cavity of the nanocapsules (Fig. 4(E)). The results showed that there was no drug release from the nanocavity in the absence of enzymes. Meanwhile, if the trypsin or hyaluronidase was added, the concentration of free drug molecules in the solution increased significantly. This is because the main component of the nanocapsule wall is protamine. If the enzyme is applied, the protamine will be catalyzed by trypsin or hyaluronidase, leading to degradation. Furthermore, because of the protamine degradation, the nanocapsule wall is cracked. The degraded nanocapsules can release drug molecules from the cavity. The results also showed that if the nanocapsules surface is modified by the folate receptor, then the nanocapsules can be selectively internalized by cancer cells, thereby achieving the targeted drug delivery. In addition to using a material that is directly responsive to enzymes, the researchers98 synthesized a polymer-peptide hybrid block amphiphilic copolymer by combining N-carboxy anhydride ring-opening polymerization technology and controlled peptide synthesis technology. The copolymer also showed an enzyme response. The hydrophobic segment of the copolymer is polystyrene chain, and the hydrophilic segment is a polyglutamic acid polypeptide chain, which contains alanine. The amphiphilic copolymer can form self-assembled nanoparticles and is stable in solution. When the elastase or thermophilic protease is added to the solution, the polypeptide chain segment is degraded. This results in the disintegration of the copolymer and the performance of the corresponding functions. Similar to the previous literature, the researchers99 prepared a triblock peptide-containing composite polymer micelle composed of poly(oligoethylene glycol monomethyl ether methacrylate) (POEGMA), peptides and temperature-responsive poly(N-isopropylacrylamide) (PNIPAM). The micelle is composed of PNIPAM as the core and POEGMA/peptides as the outer layer. The structure has a dual response of temperature and enzyme. When the metalloprotease is added, the specific sequence in the peptide chain could be cleaved. As a result, the shell layer of the micelles will be removed and the micelles collapse.
From the above, we already know that polymer nanoparticles could be degraded by enzymes. However, the polymer could also be crosslinked under catalysis of the enzyme. The researchers100 prepared an enzyme-responsive tyrosinated dextran hydrogel. The swelling and degradation occur under enzyme catalysis. In the presence of horseradish peroxidase, the modified glucan chains could be quickly crosslinked to form a hydrogel. The crosslinking makes the polymer structure transfer from liquid to solid. The response mechanism is that under the catalysis of horseradish peroxidase, the carbon–carbon bond of the modified glucan chain, or the carbon–oxygen bond between the carbon and the phenoxy group is coupled. Then, the hydrogel is formed by polymer grid crosslinking. In the presence of enzymes, the hydrogel could be crosslinked and is transformed from sol to gel. Meanwhile, due to the existence of enzymes, the hydrogel could be depolymerized, and the hydrogel is then transformed from gel to sol. For example, Huisgen cycloaddition reaction was applied by the researchers101 to prepare a polyethylene glycol–peptide composite hydrogel (Fig. 4(F)). The peptide chains in the structure could be used to tune the internal crosslinking structure of the hydrogel. When the hydrogel is placed in the solution with metalloprotease, the peptide chains are degraded, thereby changing the internal grid structure of the hydrogel from compact to loose. Then, the cells could diffuse and migrate within the hydrogels.
3. Advanced technologies for preparing bio-based stimuli responsive materials
Electrospinning is a simple and versatile technique to prepare nanofibers from multifarious polymers and composite materials.102 It is rather attractive in biomedical fields due to its high loading efficiency, flexibility in surface functionalities, versatility of drug incorporation, simple procedure, feasible mass production and low cost.103 Many bio-based polymers are suitable for electrospinning, such as PLA, PCL, and chitosan.104 Connecting electrospinning technology with stimuli-responsive materials can control and manage the release of drugs from nanofibers.105 It is well known that electrospinning fibers have a high surface area-to-volume ratio. Thus, its structures mimicking the ECM have shown great potential for tissue ingrowth and cell migration.106 Microfluidic chip precisely controls and manipulates micro scale fluids, especially for submicron structures, and is also known as Lab-on-a-Chip technology. The technology is suitable to prepare nanoparticles. For the biomedical field, these drug-loading nanoparticles could achieve targeted delivery107 and controlled drug release.108 The procedure has been used to synthesize monodisperse core–shell chitosan based mesoporous silica nanoparticles with pH-sensitive characteristics for drug release.108 The physicochemical property of the nanoparticles has been well studied. Compared with electrospinning, it could produce more uniform products. However, the drawback of the microfluidic method is its low efficiency and high cost. Due to the use of microfluidic chips, channel blockage could often occur during the preparation process, which limits its further application. Bioprinting is an advanced technology for constructing three-dimensional multicellular system in vitro. This technology is a combination of rapid prototyping technology and biological manufacturing technology, which can solve the problems that are difficult to solve in traditional tissue engineering. Compared with electrospinning and microfluidic methods, the bioprinting method shows greater advantages especially in tissue engineering and cell delivery fields66,109 because bioprinting technology could realize complex and organized structures in 3D.109–111 Meanwhile, for electrospinning, it fails to recapitulate the realistic 3D microenvironment of the native system.484. Emerging biomedical applications of bio-based stimuli-responsive materials
4.1 Drug delivery
Drug delivery refers to the technology in which specific drug molecules are transferred to target locations in the organism.112 It is a comprehensive system that could control the spatial and temporal distribution of specific drugs in the organism. The concentration of the drug is maintained at a certain level by the system, so that the drug could have specific effects on the organisms and achieve therapeutic goals in related diseases. Moreover, since the drug delivery system is controllable and tunable, the therapeutic efficiency of the drugs could be effectively promoted. Meanwhile, the system can reduce the cost and side effects of the drugs.113Although the drug delivery systems have the abovementioned advantages, the current traditional drug delivery systems still face many problems. Sometimes, the release profile of the drug is less than satisfactory. Some drugs are released too quickly after entering the body and fail to realize the therapeutic effects, or some drugs are not effectively delivered to the target site of the organism. For example, after entering the body, some drugs are enriched and are decomposed in the liver before reaching the target site without any therapeutic effects. In addition, some drugs have a certain potential side effect. For this reason, the spatial and temporal controlled drug release is one of the most critical properties of drug delivery systems. Researchers are working on how to achieve a personalized and customized drug delivery. The side effects of the drugs could be further reduced through more precise and targeted administration.114
However, conventional drug delivery systems such as micelles115 and nanoparticles116 mainly rely on the circulation of body fluids, in which the drug-loaded polymer matrix is “passively” transported to the target site and release drugs to achieve therapeutic effects. Generally, the release mechanisms of drugs from the polymer matrix mainly includes two types: (1) release drugs with the polymer matrix degradation, where the rate of drug release depends on the rate of polymer degradation; (2) release drugs from the polymer matrix by diffusion, where the release rate depends on the structure of the polymer matrix and the mobility of the polymer chains. Based on the mechanisms mentioned above, the release of drugs would be very slow and the programmed release cannot be achieved.
With the development of this research field, the stimuli-responsive drug delivery system has attracted increasing attention. The system can release drugs at a specific target location based on the specific disease of patients or chemical reactions of the body.117 The goal for the stimuli-responsive drug delivery system is to build a real-time linkage between the location of the drug delivery (or the rate of drug release) and the physiological activities of patients (or other disease-related conditions). In contrast to no response drug delivery system, a stimuli-responsive drug delivery system could provide each patient with the optimal individual drug delivery strategy based on their specific physiological activity. As is known to all, the preparation of stimuli-responsive drug delivery systems relies on the polymers that have specific responses to relevant stimuli. The drug release could be achieved through related reactions. Furthermore, the response to the stimulation is reversible. Therefore, controlled drug release is obtained. Compared with the traditional drug delivery system mentioned above, the stimuli-responsive drug delivery system has many advantages. The system maintains the blood concentration in a relatively constant level. The dose of drug administration could also be reduced to improve the curative effect. The targeted delivery of drugs reduces the side effects of drugs as much as possible. Furthermore, the duration of administration is shortened, and the patient compliance is improved.
A bio-based pH-responsive drug loaded hydrogel was prepared using chitosan and acrylic acid through free radical polymerization.118 The system was then applied for cancer treatment (Fig. 5(A)). The results showed that the controlled release of 5-fluorouracil loaded in the hydrogel could be achieved by tuning the environmental pH. This system, to a certain extent, could reduce the toxic and side effects of the drugs. In addition to chitosan, carboxymethyl cellulose was used to prepare a stimuli-responsive drug delivery system.119 The hydrogel combined carboxymethyl cellulose with gelatin. The results showed that a gel–sol transition could be realized near the temperature of the human body condition. Thus, the controlled release was obtained. In addition, the researchers120 developed a pH-responsive drug delivery system by crosslinking cyclodextrin-grafted gelatin with oxidized dextran. The controlled release of 5-fluorouracil was also achieved (Fig. 5(B)).
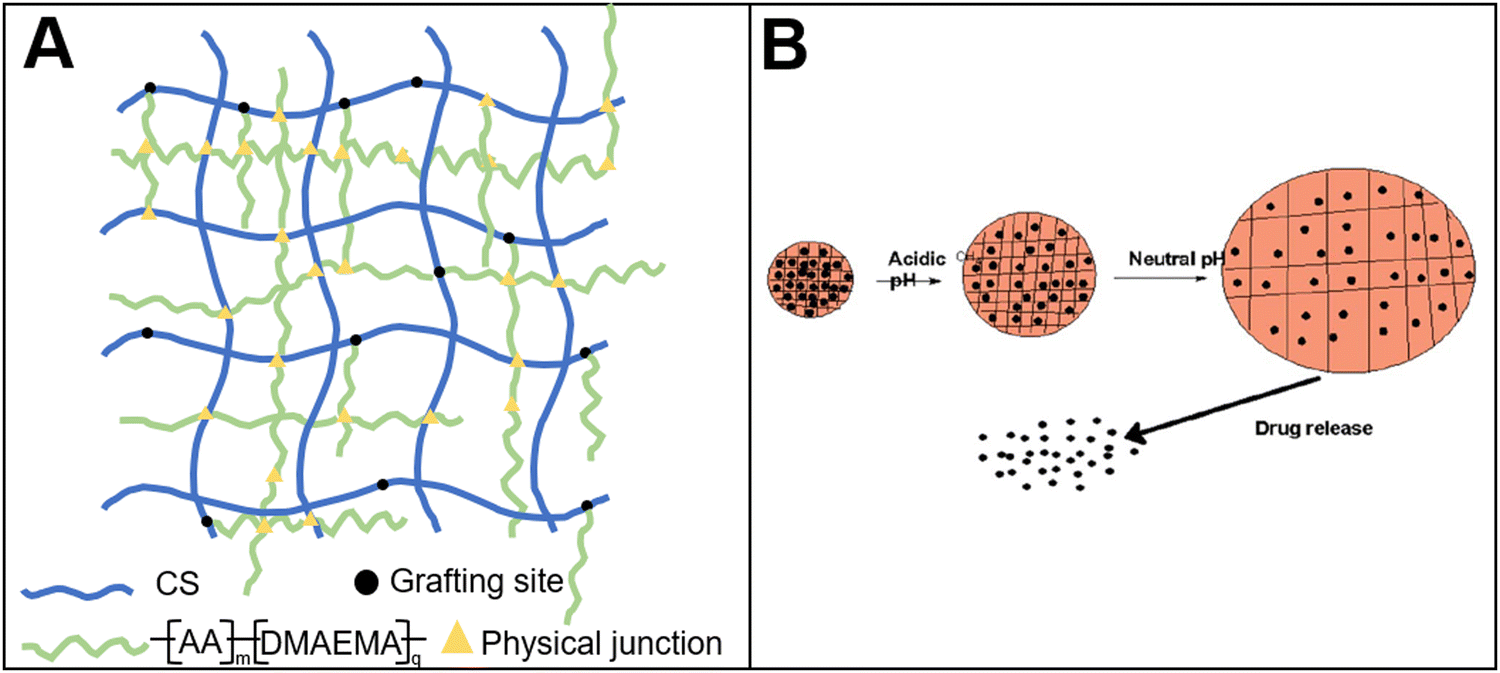 | ||
| Fig. 5 Bio-based responsive materials for drug release. (A) Chitosan-based pH-responsive drug release hydrogel118 (reproduced with permission118 from the Royal Society of Chemistry); (B) drug release from pH-responsive hydrogel120 (reproduced with permission120 from the Royal Society of Chemistry). | ||
In addition to a small molecule drug delivery, bio-based materials could be applied to macromolecular drugs such as mRNA. Cholesterol is primarily vital for maintaining the cell membrane integrity, and it could be prepared from nature. For example, a mRNA drug carrier was prepared by using ionizable lipid, cholesterol, DSPC and DMG-PEG.121 The delivery system was pH responsive. The drug carrier could maintain a neutral charge while in systemic circulation (pH 7.0), and it could become positively charged in the endosome (pH 5.0), facilitating the membrane fusion and subsequent cytosolic release. The results showed that the modified cholesterol could improve the intracellular uptake and retention of the drug carrier, resulting in a dramatic improvement in efficacy.
4.2 Drug anticounterfeiting
Drug counterfeiting is an increasingly serious problem, which has also attracted increasing attention.122 Currently, the methods to prevent drug counterfeiting mainly focus on the packaging of drugs (such as bar codes, fluorescent trademark.). However, the repackaging of drug packaging by distributors may invalidate the security marks on the packaging, thus resulting in the circulation of counterfeit drugs in the market. Based on this situation, in situ labeling technology has been developed to label the tablet itself.Cellulose acetate phthalate and polylactic acid fibers were obtained by electrospinning (Fig. 6(A)).122 The fibers were applied to prepare in situ anti-counterfeiting labels of drugs. By doping the fluorescent molecules in the polymer, the fibers exhibited strong green fluorescence. The fibers were cut into micron-sized fragments by laser. Then, a specific area on the fiber fragments was bleached by laser irradiation. During laser irradiation, the fluorescence molecules in the irradiation area would be quenched. The pattern of the quenched fluorescence molecules and fluorescence molecules could form bar codes. These fiber fragments (containing the bar codes) were coated onto the tablet surface to achieve in situ drug anti-counterfeiting. The drug information could be decoded from the bar codes by fluorescence microscope, enabling the identification of drug authenticity.
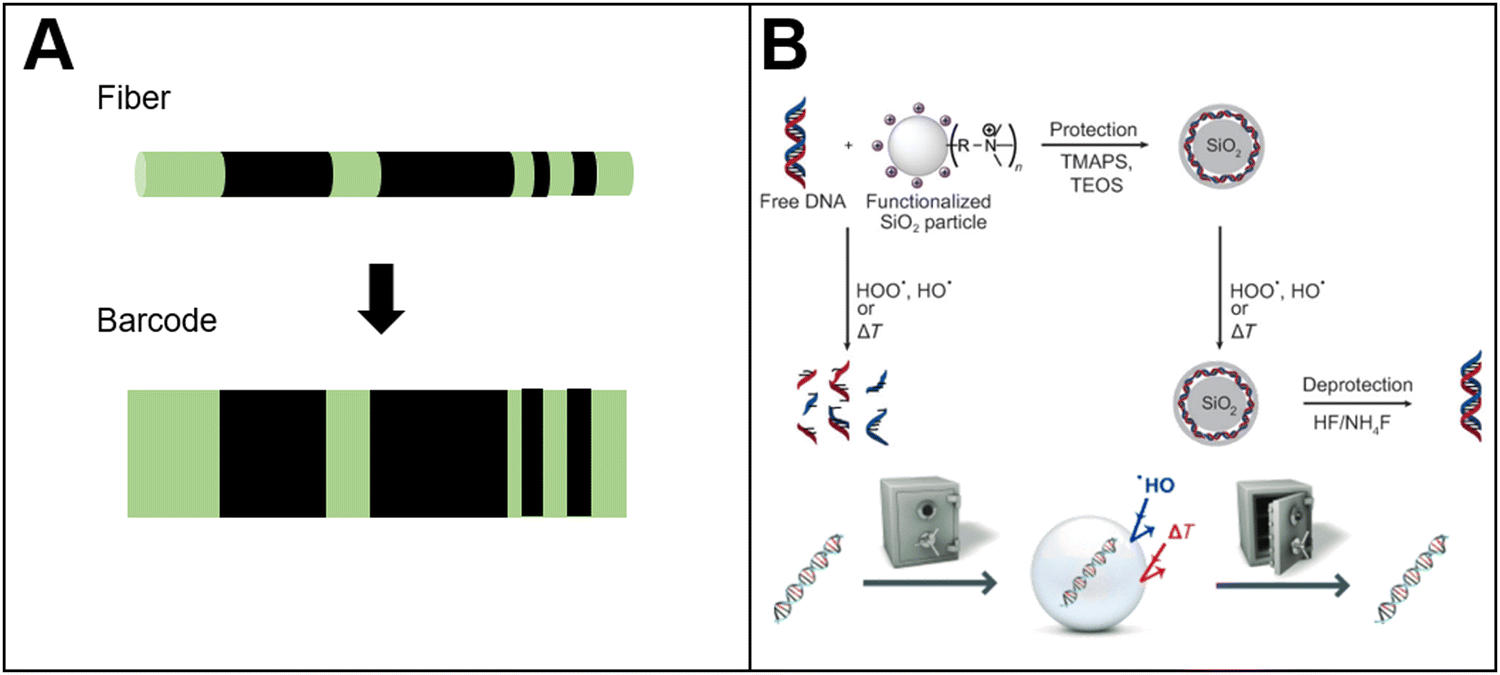 | ||
| Fig. 6 Bio-based responsive materials for drug anticounterfeiting. (A) Photobleaching PLA fluorescent barcode for anticounterfeiting122 (reproduced with permission122 Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim); (B) schematic diagram of DNA sequence to storage information for tablet anti-counterfeiting127 (reproduced with permission127 Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim). | ||
In addition to conventional polymers, DNA as a bio-based stimuli-responsive material has been applied for drug anti-counterfeiting. Due to its strong coding capacity and security (only in the presence of correct enzymes and other conditions, the sequence of the base-pairs could be decoded), the oligonucleotide is applied to prepare anti-counterfeiting labels suitable for solid and liquid formulation.123–126 For example, plant-based DNA has been applied to prepare anti-counterfeiting labels.127 The labels were mixed with tablet coating materials to track a single tablet. By using a portable DNA sequence reader, the DNA sequence can be decoded, and then people could identify the authenticity of the tablet (Fig. 6(B)). Researchers128 also combined light-responsive molecules with DNA to prepare drug anti-counterfeiting labels. The light-responsive molecules could be detected by an optical device, and then the DNA strands combined with light-responsive molecules could be extracted from the surface of the tablet or from the solution. The drug information stored inside the DNA sequence could be decoded by DNA polymerase chain reaction (PCR). However, PCR technology is complicated and not easily popularized. For this reason, a more convenient DNA-based drug anti-counterfeiting system was prepared.129 There are two parts in the system: drugs labeled by DNA, and an ink containing the complementary DNA sequence. If the tablet contains the correct DNA label, the ink will show fluorescence when it comes in contact with drugs labeled by the correct DNA sequence. No fluorescence would be observed if the ink encounters the wrong DNA label. Similarly, a DNA-based molecular beacon was prepared. Once the molecular beacons encounter the complementary DNA sequence, the fluorescence signal could be detected and the signal can be presented by secondary photoelectric tubes.
With the development of anti-counterfeiting technology, drug anti-counterfeiting technology has been gradually transitioned from the packaging of drugs to the drugs themselves. At present, most of the in situ anti-counterfeiting technologies of drugs are mainly concentrated on solid preparation. However, the injectable formulation also deserves attention. In many areas, the proliferation of counterfeit and inferior injectable formulation has attracted increasing attention. For example, it has been reported130 that in 2007, someone used fake and shoddy heparin on dialysis, resulting in an allergic reaction. In addition, the WHO reported131 that 21 patients in Iranian hospitals were injected with fake and inferior bevacizumab, causing serious vision problems. Other examples, including counterfeit vaccines, fake insulin and other counterfeit injectable formulations, have been recently reported in China.
5. Conclusion and outlook
Bio-based stimuli-responsive materials offer many advantages for a wide range of applications in a variety of fields. The mainly advantage of bio-based stimuli-responsive materials is their biodegradability and biocompatibility. Especially for biomedical applications, this feature could enhance the safety of biomedical applications and reduce the cost. Bio-based stimuli-responsive materials also show more environmental-friendly properties compared with oil-based stimuli-responsive materials. Herein, we introduce the types of bio-based stimuli-responsive materials and their preparation methods, as well as the response mechanisms. In this chapter, we mainly focus on the applications in drug delivery and drug anti-counterfeiting. We explain the release mechanism of a traditional drug delivery system. We also show the defects of the system. Compared with a traditional drug delivery system, the advantage of the bio-based stimuli-responsive material is significant. Some hydrogel-based stimuli-responsive drug delivery systems are introduced.However, we still need to face the problems of bio-based stimuli-responsive materials for drug delivery and for drug anticounterfeiting. It is not easy to prepare stimuli-responsive drug delivery systems. The preparation usually needs to go through a complex design, which makes potential drugs more difficult to develop, especially in terms of the quality control and repeatability of drugs. Meanwhile, it is necessary to improve and optimize the bio-based stimuli-responsive materials in order to make them transfer from an experimental model to clinical application. In particular, the preparation of systems that have a response to endogenous stimuli is more difficult to control because the in vivo environment is different from patient to patient (such as the pH of tumor cells or metabolites in the blood circulation). Despite the great potential of drug delivery systems that respond to exogenous stimuli, there are still some problems that need to be solved: (1) the penetration depth to the organism needs to be enhanced; (2) the sensitivity of the response needs to be improved (to avoid healthy tissues damage). At present, there are two types of stimuli-responsive drug delivery systems that have been applied at the clinical stage, temperature response system and magnetic response system. The commodity names are thermoDox and NanoTherm, respectively. The former is being used in clinical trials for stage 2 breast cancer and stage 3 liver cancer, while the latter has been approved for the treatment of glioblastoma. The development of materials, especially for the bio-based materials, greatly promotes the design of stimuli-responsive drug delivery systems. Nowadays, the design of stimuli-responsive drug delivery systems tends to focus on how to prepare a system that is suitable for clinical application. In the future, a more sensitive stimuli-responsive system that has a stronger response to the stimuli is expected.
For drug anticounterfeiting, there is no doubt that more advanced and more powerful technologies are always needed to prevent drug counterfeiting. Due to the lack of security of package labeling for drug anti-counterfeiting, in-tablet and in-capsule labeling, instead of drug packaging labeling, is becoming a powerful tool to protect drugs from counterfeiting. During the preparation, the toxicological safety and the compatibility of the anti-counterfeiting label should be the first factor to be considered. Owing to the advantages in biocompatibility and safety, bio-based stimuli-responsive materials are expected to have wide potential applications in the field of in situ drug anti-counterfeiting. In addition to the drug, closer cooperation and supervision among various agencies are of great significance to combat counterfeit and substandard drugs. At the same time, we should not overlook that patients should be fully aware of the threats by the black drug market.
Owing to all these features and many applications, we could expect that bio-based stimuli-responsive materials will become increasingly important tools that can open a new area to benefit mankind.
Conflicts of interest
There are no conflicts to declare.References
- S. RameshKumar, P. Shaiju and K. E. O’Connor, Curr. Opin. Green Sustainable Chem., 2020, 21, 75–81 CrossRef.
- T. Ahmed, M. Shahid, F. Azeem, I. Rasul, A. A. Shah, M. Noman, A. Hameed, N. Manzoor, I. Manzoor and S. Muhammad, Environ. Sci. Pollut. Res., 2018, 25, 7287–7298 CrossRef CAS PubMed.
- W. Punyodom, W. Limwanich and P. Meepowpan, Thermochim. Acta, 2017, 655, 337–343 CrossRef CAS.
- J. J. Koh, X. Zhang and C. He, Int. J. Biol. Macromol., 2018, 109, 99–113 CrossRef CAS PubMed.
- L. Jingcheng, V. S. Reddy, W. A. Jayathilaka, A. Chinnappan, S. Ramakrishna and R. Ghosh, Polymers, 2021, 13, 1427 CrossRef CAS.
- J. Park, Y. Oh, S. Jeong, H.-W. Song, E. Choi and H. Kim, Chem. Mater., 2021, 33, 8124–8132 CrossRef CAS.
- C. Kaewsaneha and P. Opaprakasit, Sci. Inno. Adv. Mater., 2021, 1, 64001 Search PubMed.
- H. Tang, W. Zhao, J. Yu, Y. Li and C. Zhao, Molecules, 2019, 24, 4 CrossRef.
- O. Werzer, S. Tumphart, R. Keimel, P. Christian and A. M. Coclite, Soft Matter, 2019, 15, 1853–1859 RSC.
- Z. Liu, Y. Faraj, X. J. Ju, W. Wang, R. Xie and L. Y. Chu, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 1306–1313 CrossRef CAS.
- Y. L. Colson and M. W. Grinstaff, Adv. Mater., 2012, 24, 3878–3886 CrossRef CAS.
- T. Tanaka, I. Nishio, S.-T. Sun and S. Ueno-Nishio, Science, 1982, 218, 467–469 CrossRef CAS PubMed.
- J. Thévenot, H. Oliveira, O. Sandre and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 7099–7116 RSC.
- M. Irie, Pure Appl. Chem., 1990, 62, 1495–1502 CrossRef CAS.
- Y. Ma, M. Lin, G. Huang, Y. Li, S. Wang, G. Bai, T. J. Lu and F. Xu, Adv. Mater., 2018, 30, 1705911 CrossRef PubMed.
- Y. Ma, T. Han, Q. Yang, J. Wang, B. Feng, Y. Jia, Z. Wei and F. Xu, Adv. Funct. Mater., 2021, 31, 2100848 CrossRef CAS.
- P. Vaupel, F. Kallinowski and P. Okunieff, Cancer Res., 1989, 49, 6449–6465 CAS.
- M. Stubbs, P. M. McSheehy, J. R. Griffiths and C. L. Bashford, Mol. Med. Today, 2000, 6, 15–19 CrossRef CAS PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- C.-L. Lo, C.-K. Huang, K.-M. Lin and G.-H. Hsiue, Biomaterials, 2007, 28, 1225–1235 CrossRef CAS.
- J. Zhao, X. Zhao, B. Guo and P. X. Ma, Biomacromolecules, 2014, 15, 3246–3252 CrossRef CAS PubMed.
- V. Gopishetty, I. Tokarev and S. Minko, J. Mater. Chem., 2012, 22, 19482–19487 RSC.
- S. M. H. Dabiri, A. Lagazzo, F. Barberis, A. Shayganpour, E. Finocchio and L. Pastorino, Carbohydr. Polym., 2017, 177, 324–333 CrossRef CAS PubMed.
- J. Hu, Y. Quan, Y. Lai, Z. Zheng, Z. Hu, X. Wang, T. Dai, Q. Zhang and Y. Cheng, J. Controlled Release, 2017, 247, 145–152 CrossRef CAS PubMed.
- S. Cao, B. Hu and H. Liu, Polym. Int., 2009, 58, 545–551 CrossRef CAS.
- W. Cui, M. Qi, X. Li, S. Huang, S. Zhou and J. Weng, Int. J. Pharm., 2008, 361, 47–55 CrossRef CAS PubMed.
- D. Hua, Z. Liu, F. Wang, B. Gao, F. Chen, Q. Zhang, R. Xiong, J. Han, S. K. Samal and S. C. De Smedt, Carbohydr. Polym., 2016, 151, 1240–1244 CrossRef CAS PubMed.
- N. Leber, L. Nuhn and R. Zentel, Macromol. Biosci., 2017, 17, 1700092 CrossRef PubMed.
- S. V. Vinogradov, T. K. Bronich and A. V. Kabanov, Adv. Drug Delivery Rev., 2002, 54, 135–147 CrossRef CAS PubMed.
- M. Miyake, K. Ogawa and E. Kokufuta, Langmuir, 2006, 22, 7335–7341 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- N. M. Oh, K. T. Oh, Y. S. Youn, D.-K. Lee, K.-H. Cha, D. H. Lee and E. S. Lee, Colloids Surf., B, 2013, 101, 298–306 CrossRef CAS.
- J. Xue, C. Zhu, J. Li, H. Li and Y. Xia, Adv. Funct. Mater., 2018, 28, 1705563 CrossRef.
- Y. J. Zeng, D. Wu, X. H. Cao, W. X. Zhou, L. M. Tang and K. Q. Chen, Adv. Funct. Mater., 2020, 30, 1903873 CrossRef CAS.
- Z. Zhang, Y. Yuan, N. Zhang and X. Cao, J. Chem. Eng. Data, 2015, 60, 2495–2501 CrossRef CAS.
- P. Zhao, Q. Yue, H. He, B. Gao, Y. Wang and Q. Li, Appl. Energy, 2014, 115, 483–490 CrossRef CAS.
- S. Somekawa, A. Mahara, K. Masutani, Y. Kimura, H. Urakawa and T. Yamaoka, Tissue Eng. Regener. Med., 2017, 14, 507–516 CrossRef CAS.
- Y. Li, B. Hui, G. Li and J. Li, J. Mater. Sci., 2017, 52, 7688–7697 CrossRef CAS.
- M. Hakalahti, A. Mautner, L.-S. Johansson, T. Hänninen, H. Setälä, E. Kontturi, A. Bismarck and T. Tammelin, ACS Appl. Mater. Interfaces, 2016, 8, 2923–2927 CrossRef CAS.
- A. C. Santos, S. Alves, M. H. Godinho, C. Baleizão and J. P. S. Farinha, Polym. Chem., 2018, 9, 3615–3623 RSC.
- B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS PubMed.
- B. Das, M. Mandal, A. Upadhyay, P. Chattopadhyay and N. Karak, Biomed. Mater., 2013, 8, 035003 CrossRef CAS PubMed.
- J. R. McKee, S. Hietala, J. Seitsonen, J. Laine, E. Kontturi and O. Ikkala, ACS Macro Lett., 2014, 3, 266–270 CrossRef CAS PubMed.
- T. Calvo-Correas, A. Shirole, F. Crippa, A. Fink, C. Weder, M. A. Corcuera and A. Eceiza, Mater. Sci. Eng., C, 2019, 97, 658–668 CrossRef CAS.
- J. Guo, I. Filpponen, L.-S. Johansson, P. Mohammadi, M. Latikka, M. B. Linder, R. H. Ras and O. J. Rojas, Biomacromolecules, 2017, 18, 898–905 CrossRef CAS.
- E. Lizundia, A. Maceiras, J. Vilas, P. Martins and S. Lanceros-Mendez, Carbohydr. Polym., 2017, 175, 425–432 CrossRef CAS PubMed.
- X. Li, Y. He, H. Sui and L. He, Nanomaterials, 2018, 8, 162 CrossRef PubMed.
- N. Shi, Y. Li, L. Chang, G. Zhao, G. Jin, Y. Lyu, G. M. Genin, Y. Ma and F. Xu, Small Methods, 2021, 5, 2100276 CrossRef CAS PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS.
- D. Samanta, R. Mehrotra, K. Margulis and R. N. Zare, Nanoscale, 2017, 9, 16429–16436 RSC.
- A. Dias, A. Cortez, M. Barsan, J. Santos, C. Brett and H. De Sousa, ACS Sustainable Chem. Eng., 2013, 1, 1480–1492 CrossRef CAS.
- K. Rotjanasuworapong, N. Thummarungsan, W. Lerdwijitjarud and A. Sirivat, Carbohydr. Polym., 2020, 247, 116709 CrossRef CAS.
- Z.-x Zhang, J.-x Dou, J.-h He, C.-x Xiao, L.-y Shen, J.-h Yang, Y. Wang and Z.-w Zhou, J. Mater. Chem. C, 2017, 5, 4145–4158 RSC.
- Z. Atoufi, P. Zarrintaj, G. H. Motlagh, A. Amiri, Z. Bagher and S. K. Kamrava, J. Biomater. Sci., Polym. Ed., 2017, 28, 1617–1638 CrossRef CAS PubMed.
- K. S. Suslick and W. L. Nyborg, J. Acoust. Soc. Am., 1990, 87, 919–920 CrossRef.
- H. Jiang, K. Tovar-Carrillo and T. Kobayashi, Ultrason. Sonochem., 2016, 32, 398–406 CrossRef CAS.
- H. Jiang and T. Kobayashi, Mater. Sci. Eng., C, 2017, 75, 478–486 CrossRef CAS PubMed.
- N. Huebsch, C. J. Kearney, X. Zhao, J. Kim, C. A. Cezar, Z. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS.
- H. Epstein-Barash, G. Orbey, B. E. Polat, R. H. Ewoldt, J. Feshitan, R. Langer, M. A. Borden and D. S. Kohane, Biomaterials, 2010, 31, 5208–5217 CrossRef CAS PubMed.
- W. Huang, J. Zhai, C. Zhang, X. Hu, N. Zhu, K. Chen and K. Guo, Ind. Eng. Chem. Res., 2020, 59, 13588–13594 CrossRef CAS.
- M. Alatorre-Meda, C. Alvarez-Lorenzo, A. Concheiro and P. Taboada, Smart Mater. Drug Deliv., 2013, 1, 304–348 Search PubMed.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- C. Alvarez-Lorenzo and A. Concheiro, Chem. Commun., 2014, 50, 7743–7765 RSC.
- M. Martínez-Carmona, D. Lozano, A. Baeza, M. Colilla and M. Vallet-Regí, Nanoscale, 2017, 9, 15967–15973 RSC.
- Q. Jin, F. Mitschang and S. Agarwal, Biomacromolecules, 2011, 12, 3684 CrossRef CAS.
- Y. Ma, Y. Ji, G. Huang, K. Ling, X. Zhang and F. Xu, Biofabrication, 2015, 7, 044105 CrossRef.
- Y. Ma, Y. Ji, T. Zhong, W. Wan, Q. Yang, A. Li, X. Zhang and M. Lin, ACS Biomater. Sci. Eng., 2017, 3, 3534–3545 CrossRef CAS.
- Y. Ma, J. Yao, Q. Liu, T. Han, J. Zhao, X. Ma, Y. Tong, G. Jin, K. Qu and B. Li, Adv. Funct. Mater., 2020, 30, 2001820 CrossRef CAS.
- S. w. group, Brussels, Belgium: European Commission. http://ec.europa.eu/health/archive/ph_risk/committees/04_scenihr/docs/scenihr_o_010.pdf, 2007.
- B. A. Jurkiewicz and G. R. Buettner, Photochem. Photobiol., 1994, 59, 1–4 CrossRef CAS.
- Z. Wang, M. Boudjelal, S. Kang, J. J. Voorhees and G. J. Fisher, Nat. Med., 1999, 5, 418–422 CrossRef CAS PubMed.
- M. Podda, M. G. Traber, C. Weber, L.-J. Yan and L. Packer, Free Radical Biol. Med., 1998, 24, 55–65 CrossRef CAS.
- K. Jariashvili, B. Madhan, B. Brodsky, A. Kuchava, L. Namicheishvili and N. Metreveli, Biopolymers, 2012, 97, 189–198 CrossRef CAS.
- R. P. Sinha and D.-P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 CrossRef CAS.
- J. Olejniczak, C.-J. Carling and A. Almutairi, J. Controlled Release, 2015, 219, 18–30 CrossRef CAS PubMed.
- M. Gary-Bobo, Y. Mir, C. Rouxel, D. Brevet, I. Basile, M. Maynadier, O. Vaillant, O. Mongin, M. Blanchard-Desce and A. Morère, Angew. Chem., Int. Ed., 2011, 123, 11627–11631 CrossRef.
- X. Wang, C. Wang, X. Wang, Y. Wang, Q. Zhang and Y. Cheng, Chem. Mater., 2017, 29, 1370–1376 CrossRef CAS.
- K. Y. Lee, M. C. Peters and D. J. Mooney, Adv. Mater., 2001, 13, 837–839 CrossRef CAS.
- L. Tan, J. Li, Y. Liu, H. Zhou, Z. Zhang and L. Deng, J. Bioact. Compat. Polym., 2015, 30, 584–599 CrossRef CAS.
- D. R. Stabley, C. Jurchenko, S. S. Marshall and K. S. Salaita, Nat. Methods, 2012, 9, 64–67 CrossRef CAS PubMed.
- W. Jiang, B. Kim, J. T. Rutka and W. C. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- C. Larson, B. Peele, S. Li, S. Robinson, M. Totaro, L. Beccai, B. Mazzolai and R. Shepherd, Science, 2016, 351, 1071–1074 CrossRef CAS PubMed.
- R. F. Riley, C. W. Don, W. Powell, C. Maynard and L. S. Dean, Circ. Cardiovasc. Qual. Outcomes., 2011, 4, 193–197 CrossRef.
- Y. Zhang, Q. Chen, J. Ge and Z. Liu, Chem. Commun., 2013, 49, 9815–9817 RSC.
- J. Di, S. Yao, Y. Ye, Z. Cui, J. Yu, T. K. Ghosh, Y. Zhu and Z. Gu, ACS Nano, 2015, 9, 9407–9415 CrossRef CAS PubMed.
- T. Saxer, A. Zumbuehl and B. Müller, Cardiovasc. Res., 2013, 99, 328–333 CrossRef CAS.
- N. Korin, M. J. Gounis, A. K. Wakhloo and D. E. Ingber, JAMA Neurol., 2015, 72, 119–122 CrossRef PubMed.
- T. Nakamura, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- C. Chang, M. He, J. Zhou and L. Zhang, Macromolecules, 2011, 44, 1642–1648 CrossRef CAS.
- W. Guo, X.-J. Qi, R. Orbach, C.-H. Lu, L. Freage, I. Mironi-Harpaz, D. Seliktar, H.-H. Yang and I. Willner, Chem. Commun., 2014, 50, 4065–4068 RSC.
- X. Zhang and J. Huang, Chem. Commun., 2010, 46, 6042–6044 RSC.
- Q. Hu, P. S. Katti and Z. Gu, Nanoscale, 2014, 6, 12273–12286 RSC.
- C. E. Callmann, C. V. Barback, M. P. Thompson, D. J. Hall, R. F. Mattrey and N. C. Gianneschi, Adv. Mater., 2015, 27, 4611–4615 CrossRef CAS.
- D. A. Bedoya, F. N. Figueroa, M. A. Macchione and M. C. Strumia, Stimuli-Responsive Polymeric Systems for Smart Drug Delivery, Advanced Biopolymeric Systems for Drug Delivery, 2020 Search PubMed.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani and M. Bozorgomid, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- K. Radhakrishnan, J. Tripathy, D. P. Gnanadhas, D. Chakravortty and A. M. Raichur, RSC Adv., 2014, 4, 45961–45968 RSC.
- G. J. Habraken, M. Peeters, P. D. Thornton, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 3761–3769 CrossRef CAS PubMed.
- A. J. de Graaf, I. I. A. P. dos Santos, E. H. Pieters, D. T. Rijkers, C. F. van Nostrum, T. Vermonden, R. J. Kok, W. E. Hennink and E. Mastrobattista, J. Controlled Release, 2012, 162, 582–590 CrossRef CAS PubMed.
- R. Jin, C. Hiemstra, Z. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS PubMed.
- D. Han, X. Yu, Q. Chai, N. Ayres and A. J. Steckl, ACS Appl. Mater. Interfaces, 2017, 9, 11858–11865 CrossRef CAS.
- J. Jiang, J. Xie, B. Ma, D. E. Bartlett, A. Xu and C.-H. Wang, Acta Biomater., 2014, 10, 1324–1332 CrossRef CAS.
- Z. Chen, X. Mo, C. He and H. Wang, Carbohydr. Polym., 2008, 72, 410–418 CrossRef CAS.
- S. Chen, S. K. Boda, S. K. Batra, X. Li and J. Xie, Adv. Healthcare Mater., 2018, 7, 1701024 CrossRef.
- W. He, Z. Ma, T. Yong, W. E. Teo and S. Ramakrishna, Biomaterials, 2005, 26, 7606–7615 CrossRef CAS.
- G. Unsoy, S. Yalcin, R. Khodadust, P. Mutlu, O. Onguru and U. Gunduz, Biomed. Pharmacother., 2014, 68, 641–648 CrossRef CAS PubMed.
- M. M. Hasani-Sadrabadi, S. Taranejoo, E. Dashtimoghadam, G. Bahlakeh, F. S. Majedi, J. J. VanDersarl, M. Janmaleki, F. Sharifi, A. Bertsch and K. Hourigan, Adv. Mater., 2016, 28, 4134–4141 CrossRef CAS PubMed.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- R. Lozano, L. Stevens, B. C. Thompson, K. J. Gilmore, R. Gorkin III, E. M. Stewart, M. in het Panhuis, M. Romero-Ortega and G. G. Wallace, Biomaterials, 2015, 67, 264–273 CrossRef CAS PubMed.
- M. Antman-Passig, S. Levy, C. Gartenberg, H. Schori and O. Shefi, Tissue Eng., Part A, 2017, 23, 403–414 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS.
- S. E. Neumann, C. F. Chamberlayne and R. N. Zare, Nanoscale, 2018, 10, 10087–10093 RSC.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- S. Gao, G. Tang, D. Hua, R. Xiong, J. Han, S. Jiang, Q. Zhang and C. Huang, J. Mater. Chem. B, 2019, 7, 709–729 RSC.
- Y. Che, D. Li, Y. Liu, Q. Ma, Y. Tan, Q. Yue and F. Meng, RSC Adv., 2016, 6, 106035 RSC.
- A. Nayak, H. Babla, T. Han and D. B. Das, Drug Delivery, 2016, 23, 658–669 CrossRef CAS PubMed.
- T. Anirudhan and A. M. Mohan, RSC Adv., 2014, 4, 12109–12118 RSC.
- S. Patel, N. Ashwanikumar, E. Robinson, Y. Xia, C. Mihai, J. P. Griffith, S. Hou, A. A. Esposito, T. Ketova and K. Welsher, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed.
- C. Huang, B. Lucas, C. Vervaet, K. Braeckmans, S. Van Calenbergh, I. Karalic, M. Vandewoestyne, D. Deforce, J. Demeester and S. C. De Smedt, Adv. Mater., 2010, 22, 2657–2662 CrossRef CAS PubMed.
- B. Popping, J. Biotechnol., 2002, 98, 107–112 CrossRef CAS PubMed.
- J. A. Hayward, M. E. Hogan, M. B. Liang and L. Jung, U.S. Pat. Search PubMed 15/800, 768, 2018.
- H. Zhang, D. Hua, C. Huang, S. K. Samal, R. Xiong, F. Sauvage, K. Braeckmans, K. Remaut and S. C. De Smedt, Adv. Mater., 2020, 32, 1905486 CrossRef CAS PubMed.
- G. M. George, U.S. Pat., 12/084, 512, 2009 Search PubMed.
- D. Paunescu, R. Fuhrer and R. N. Grass, Angew. Chem., Int. Ed., 2013, 52, 4269–4272 CrossRef CAS PubMed.
- J. A. Hayward, M. H. Liang and S. Shu-Kin-So, U.S. Pat., 8420400B2, 2013 Search PubMed.
- S. R. Owens, EMBO Rep., 2003, 4, 741–743 CrossRef CAS PubMed.
- G. Cosel, Pew Health Group, Philadelphia, PA, 2011 Search PubMed.
- M. Entezari, S. Karimi, H. Ahmadieh, A. H. Mahmoudi, H. Parhizgar and M. Yaseri, Graefe's Archive for Clinical and Experimental Ophthalmology, 2016, vol. 254, pp. 1851–1856 Search PubMed.
Footnote |
| † Senior authors who equally contributed. |
| This journal is © The Royal Society of Chemistry 2023 |