
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe pronounced effect of cobalt oxide on the electrocatalytic activity of palladium nanoparticles anchored on CoOx/NC towards the ORR with increased MA and ECSA†
Fatima
Nasim
a,
Hassan
Ali
a and
Muhammad Arif
Nadeem
 *ab
*ab
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan. E-mail: manadeem@qau.edu.pk
bPakistan Academy of Sciences, 3-Constitution Avenue Sector G-5/2, Islamabad, Pakistan
First published on 29th November 2022
Abstract
As an electrocatalyst of the oxygen reduction reaction (ORR), palladium (Pd) has been the focus of research as an alternative to standard Pt-based catalysts. The effect on the palladium d-band center as a consequence of the electronic interaction with cobalt species has rarely been observed. Herein, we report a cost effective, highly efficient, and stable palladium-based catalyst (Pd@CoOx/NC1) with low Pd loading (ca. 5% only) over CoOx embedded nitrogen doped tubular carbon (CoOx/NC) in both 0.1 M KOH and 0.1 M HClO4. The as-synthesized catalyst (Pd@CoOx/NC1) exhibits superior activity to standard 20 wt% Pt/C in both alkaline (Eonset: 1.07 VRHE, E1/2 0.95 VRHE) and acidic (Eonset: 1.02 VRHE, E1/2 0.91 VRHE) environments. The novel support (CoOx/NC) has been derived via one step carbonization of ZIF-12. As a control catalyst, palladium nanoparticles supported over commercial multiwalled CNTs (Pd@MWCNTs) demonstrates lower E1/2 values in both alkaline (0.80 VRHE) and acidic (0.73 VRHE) environments in comparison to Pd@CoOx/NC1, strengthening the effect of cobalt on the palladium d-band center. Pd@CoOx/NC1 also exhibits increased mass activity in 0.1 M HClO4 (230 mA mgpd−1 at 0.91 VRHE) and 0.1 M KOH (280 mA mgpd−1 at 0.95 VRHE) and an increased electrochemically active surface area (233 m2 g−1). Enhanced tolerance in the presence of toxic anions is also demonstrated by Pd@CoOx/NC1. This work provides a new avenue for the exploration of synergism between CoOx and noble metals to circumvent the cost problem associated with the development of highly efficient and stable cathodic ORR electrocatalysts.
Introduction
Technical challenges encountered by precious metals as catalysts for sustainable energy technologies, such as vulnerability to poisoning species during the operation of fuel cells, high cost and limited availability, inspired researchers to search for efficient, cost effective, anti-poisoning and long-term stable catalysts for proton exchange membrane fuel cells (PEMFCs).1,2 Among the different environment friendly technologies, the cathodic oxygen reduction reaction (ORR) is of particular importance due to its clean nature and abundant availability of oxygen in the environment.3 However, the sluggish reaction kinetics of the ORR is the main obstacle in their widespread applications, because it significantly limits the conversion efficiency of the above mentioned technology.4–7 Platinum (Pt) is the most efficient metal for catalysing the ORR but the high cost (approximately ½ of the automotive fuel cell stack)8 and vulnerability to small amounts of toxic species such as NOx, PO4, SOx, etc. rule out the use of Pt as a cathode catalyst for the ORR.9–11 The design of new palladium (Pd) based catalysts has been proven to be a good choice for catalysing the reduction of oxygen due to its resemblance with the intrinsic properties of benchmark metal i.e., platinum3,12 including the electronic structure and lattice parameters.13–18 The cost problem can be cracked by reducing the amount of precious metal (Pd) at the expense of a non-noble metal19 with increased specific activity (SA) and mass activity (MA).20 Moreover, the kinetically inhibited ORR due to the coverage of oxygenated intermediates on the surface of palladium13 can be optimized by a strong metal–support interaction (SMSI) through a synergistic effect.21 However, the two main areas in which palladium is an inferior candidate compared to commercial Pt/C are, its relatively rapid leaching under drastic conditions of the operating fuel cell and low activity.5,22 The readily oozing of Pd compared to Pt in the harsh environment of PEMFCs22 necessitates the encapsulation of palladium nanoparticles within the substrate material.2 The relatively low activity of Pd/C for the ORR compared to traditional Pt/C can be enhanced through alloying and support modification.5 The choice of appropriate support circumvents the inherent carbon corrosion problems, and can help to stabilize the loaded metal particles and prevent aggregation.23 Carbon based materials, especially nitrogen doped carbon materials, have been used enormously as a support due to their unique features like higher conductivities and surface area, thus enhancing ORR activity through the interaction of loaded particles and support material. However, the oxidation of carbon during the operation of fuel cells significantly hampers their performance. To address this issue, there is a recent surge in using transition metal oxides as support, which changes the electronic properties of loaded nanoparticles through SMSI.5 The SMSI minimizes the surface energy by modulating the properties of the supported noble metal particles, leading to enhancement in electrocatalytic ability and durability.2 Ignacio et al. synthesized Pt/Ta/SnO2 as a highly efficient catalyst for the ORR.24 Likewise Fuma et al. observed an enhancement in the oxygen reduction reaction ability of the Pt NPs in 0.1 M HClO4 by depositing platinum nanoparticles on transition metal oxides formed on cup-stacked carbon nanotubes. The low electrical conductivity of transition metal oxides was enhanced by cup stacked carbon nanotubes to enable the use of transition metal oxides.25 Felipe reported Pt/MOx/C and observed the changes induced via the interaction of transition metal oxides/carbon supports and Pt NPs.26 In view of the above discussion, transition metal oxide embedded nitrogen doped carbon-based materials can be a good choice as cathode catalysts for the ORR, benefiting from the useful properties of both transition metal oxides and heteroatom doped carbon based materials.In our present report, we describe a simple, facile, and advantageous method without the use of capping agents9 and oily solvents27 for the synthesis of novel, highly stable, anti-poisoning and highly dispersed 5% Pd nanoparticles supported over transition metal oxide (CoOx) nanoparticles encapsulated by nitrogen doped tubular carbon derived from ZIF-12 as a highly efficient material for catalysing the oxygen reduction reaction in fuel cells. The method presented here can be used for the production scale-up. Nitrogen-doping provides anchoring sites for the dispersion of palladium nanoparticles and adsorbed intermediates respectively and at the same time tunes the electronic structure of dispersed nanoparticles28 together with embedded CoOx nanoparticles. The SMSI greatly improved the catalytic activity and stability. The concentration effect was examined by synthesizing different materials by varying the concentration of the precursor metal salt (Palladium diacetate). The enhanced electrocatalytic ability is attributed to the outstanding properties of one-dimensional nitrogen-doped tubular carbon (NC) encapsulating CoOx nanoparticles. The high mass activity and high electrochemical surface area (ECSA) are the result of a minimal amount of palladium loading and SMSI.
Experimental
Synthesis of ZIF-12
Cobalt nitrate hexahydrate (1.3 mmol) and benzimidazole (6 mmol) were dissolved in N,N-dimethylformamide (DMF) and transferred to a Teflon lined stainless steel autoclave. The autoclave was kept in a furnace at 150 °C for 48 h; the purple product obtained was centrifuged and washed with DMF and distilled water.Synthesis of the CoOx/NC support material
The support material (CoOx/NC) was synthesized via carbonization of ZIF-12 at elevated temperature under argon atmosphere. For this purpose, ZIF-12 (200 mg) was transferred to a quartz boat and placed in a tube furnace. The tube furnace was degassed by flowing Ar for 2 h to eliminate oxygen gas. The system was then heated to 850 °C for 8 h at a rate of 5 °C per min. After natural cooling to room temperature, the product was collected and further treated with analytical grade HCl to remove excess cobalt species adhered to the surface and to functionalize the surface so that the adsorption capacity and dispensability can be increased. After acid leaching, the material still retained ca. 2.2 wt% of cobalt as indicated by XPS and AAS analyses.Synthesis of catalysts
The catalyst was synthesized by dispersing CoOx/NC (70 mg) in DMF and sonicated for 60 min. Afterward, 5% palladium diacetate (20 mg) solution was added dropwise and sonicated further for 30 min followed by the dropwise addition of sodium borohydride (2 mg mg−1 of metal salt). After the addition of the reducing agent, the whole mixture was sonicated for 30 min followed by stirring at 60 °C for 30 min. After cooling, the product was filtered, washed with DMF and methanol, dried, characterized and used for electrochemical studies. By adjusting the concentration of the precursor metal salt (palladium diacetate), catalysts with different concentrations were obtained and their electrochemical activities were measured subsequently to investigate the concentration effect. For comparison, 20 wt% Pt/C supported on Vulcan XC-72 was synthesized by the polyol reduction method.Electrochemical measurements
An electrochemical workstation was used for evaluating the catalytic performance. The electrochemical activity requires the preparation of a working electrode coated with the synthesized catalyst. For the fabrication of the working electrode, 2 mg of synthesized catalyst was dispersed in 100 μL of isopropanol and 5% Nafion mixture and sonicated for 2 h. A thin layer (2.5 μL) of homogeneous slurry was then fabricated on an RDE by the drop casting technique, dried under ambient air at room temperature and ready to be used for electrochemical testing. The cathodic oxygen reduction ability was determined by performing linear sweep voltammetry in 0.1 M KOH and 0.1 M HClO4 at 10 mV s−1 and 1600 revolutions per minute in an O2 saturated environment. The electrochemical workstation was furnished with a three electrode system, in which a glassy carbon disk rotating electrode was used as the working electrode, graphite as the counter electrode and Hg/HgO as the reference electrode. All the potentials were converted to the RHE scale for comparison according to Nernst's equation (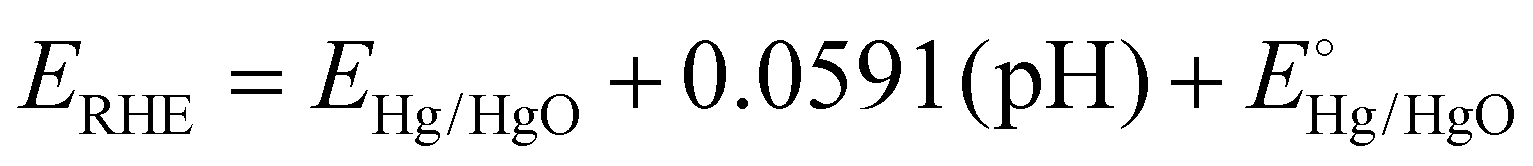 ). The current was converted to current density by dividing the obtained current over the geometric area of the electrode (0.19625 cm2). For the determination of the electrochemically active surface area (ECSA) through the palladium oxide layer reduction method, cyclic voltammetry experiments were performed at a scan rate of 50 mV s−1 in Ar-saturated 0.1 M HClO4. An accelerated durability test (ADT) was performed to check the stability of the catalyst over 10
). The current was converted to current density by dividing the obtained current over the geometric area of the electrode (0.19625 cm2). For the determination of the electrochemically active surface area (ECSA) through the palladium oxide layer reduction method, cyclic voltammetry experiments were performed at a scan rate of 50 mV s−1 in Ar-saturated 0.1 M HClO4. An accelerated durability test (ADT) was performed to check the stability of the catalyst over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. The resistance of the catalyst to small molecular poisonous species was determined by adding different concentrations (10 mM–50 mM) of NOx, PO4 and SOx species to the electrolyte (0.1 M KOH). All the obtained electrochemical results were compared to Pt/C (20 wt%), a reference and benchmark catalyst. All the measurements were carried out at room temperature. The catalyst loading was 0.01273 mgPd cm−2, 0.016815 mgPd cm−2, and 0.02242 mgPd cm−2 for Pd@CoOx/NC1, Pd@CoOx/NC2, and Pd@CoOx/NC3, respectively. Scheme 1 presents the synthetic protocol for the synthesis of Pd@CoOx/NC.
000 cycles. The resistance of the catalyst to small molecular poisonous species was determined by adding different concentrations (10 mM–50 mM) of NOx, PO4 and SOx species to the electrolyte (0.1 M KOH). All the obtained electrochemical results were compared to Pt/C (20 wt%), a reference and benchmark catalyst. All the measurements were carried out at room temperature. The catalyst loading was 0.01273 mgPd cm−2, 0.016815 mgPd cm−2, and 0.02242 mgPd cm−2 for Pd@CoOx/NC1, Pd@CoOx/NC2, and Pd@CoOx/NC3, respectively. Scheme 1 presents the synthetic protocol for the synthesis of Pd@CoOx/NC.
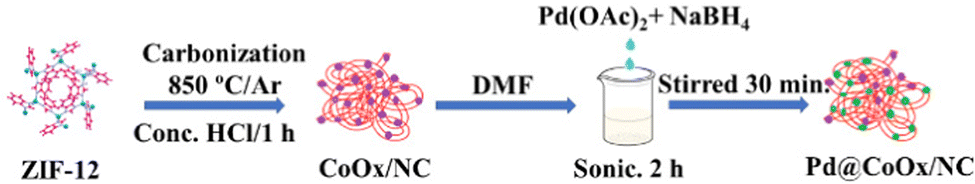 | ||
| Scheme 1 Schematic representation of the synthesis of Pd@CoOx/NCs. | ||
Results and discussion
The powder XRD pattern was investigated to determine the phase purity, crystal structure and chemical composition. Palladium metal crystallizes in the cubic system with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The graph clearly shows that after the successful formation of palladium nanoparticles, the integrity of CoOx encapsulated NC was maintained. Fig. 1(a) shows the XRD pattern of calcined ZIF-12 i.e., CoOx embedded NC and synthesized catalysts. The peak at 2θ = 25.8° corresponding to the (002) plane is assigned to the formation and perseverance of graphitic carbon in all the synthesized samples. The other two peaks located at 2θ values of ∼44.4° and ∼51.5° (Fig. 1(a)) could also be indexed to the graphitic carbon material according to the standard pattern (JCPDS card no. 00-013-0148). The peaks for CoOx nanoparticles evident from HRTEM, XPS and SAED analyses29 are not visible in the powder XRD pattern due to the very low quantity of cobalt (2 wt%). These CoOx nanoparticles are found to be encapsulated at the tip of NC. Some of the particles can also be seen inside the NC, while some are attached to the surface of nitrogen doped tubular carbon as confirmed by HRTEM analysis in our previous report (Fig. S2, ESI†).29 The very low concentration of cobalt (2 wt%) makes it difficult to determine the type of cobalt oxide by PXRD and hence more likely the mixed cobalt oxides. The peaks at ∼39.6°, ∼44.2° and ∼68.1° are the signals of the formation of palladium nanoparticles according to the standard pattern (JCPDS card no. 00-001-1201). The peak shift of Pd nanoparticles toward a lower 2θ value from the standard reference card is the result of the lattice expansion of palladium due to the interaction of the relatively larger atomic size of cobalt. The cell parameters (a) and d-spacing values calculated for Pd (111), (200) and (220) planes using Bragg's law are given in Table 1 which are in good agreement and consistent with the HRTEM results. A small increase in “a” and “d” compared to fcc-type Pd NPs is observed for the synthesized catalysts. The small deviation is the result of the electronic interaction of palladium with cobalt leading to lattice expansion. These values suggest that the metals in the synthesized electrocatalysts are under strain which slightly distorts the crystal lattice exhibiting a synergistic effect. Fig. S1 (ESI†) presents the XRD patterns of MWCNTs and Pd@MWCNTs.
m. The graph clearly shows that after the successful formation of palladium nanoparticles, the integrity of CoOx encapsulated NC was maintained. Fig. 1(a) shows the XRD pattern of calcined ZIF-12 i.e., CoOx embedded NC and synthesized catalysts. The peak at 2θ = 25.8° corresponding to the (002) plane is assigned to the formation and perseverance of graphitic carbon in all the synthesized samples. The other two peaks located at 2θ values of ∼44.4° and ∼51.5° (Fig. 1(a)) could also be indexed to the graphitic carbon material according to the standard pattern (JCPDS card no. 00-013-0148). The peaks for CoOx nanoparticles evident from HRTEM, XPS and SAED analyses29 are not visible in the powder XRD pattern due to the very low quantity of cobalt (2 wt%). These CoOx nanoparticles are found to be encapsulated at the tip of NC. Some of the particles can also be seen inside the NC, while some are attached to the surface of nitrogen doped tubular carbon as confirmed by HRTEM analysis in our previous report (Fig. S2, ESI†).29 The very low concentration of cobalt (2 wt%) makes it difficult to determine the type of cobalt oxide by PXRD and hence more likely the mixed cobalt oxides. The peaks at ∼39.6°, ∼44.2° and ∼68.1° are the signals of the formation of palladium nanoparticles according to the standard pattern (JCPDS card no. 00-001-1201). The peak shift of Pd nanoparticles toward a lower 2θ value from the standard reference card is the result of the lattice expansion of palladium due to the interaction of the relatively larger atomic size of cobalt. The cell parameters (a) and d-spacing values calculated for Pd (111), (200) and (220) planes using Bragg's law are given in Table 1 which are in good agreement and consistent with the HRTEM results. A small increase in “a” and “d” compared to fcc-type Pd NPs is observed for the synthesized catalysts. The small deviation is the result of the electronic interaction of palladium with cobalt leading to lattice expansion. These values suggest that the metals in the synthesized electrocatalysts are under strain which slightly distorts the crystal lattice exhibiting a synergistic effect. Fig. S1 (ESI†) presents the XRD patterns of MWCNTs and Pd@MWCNTs.
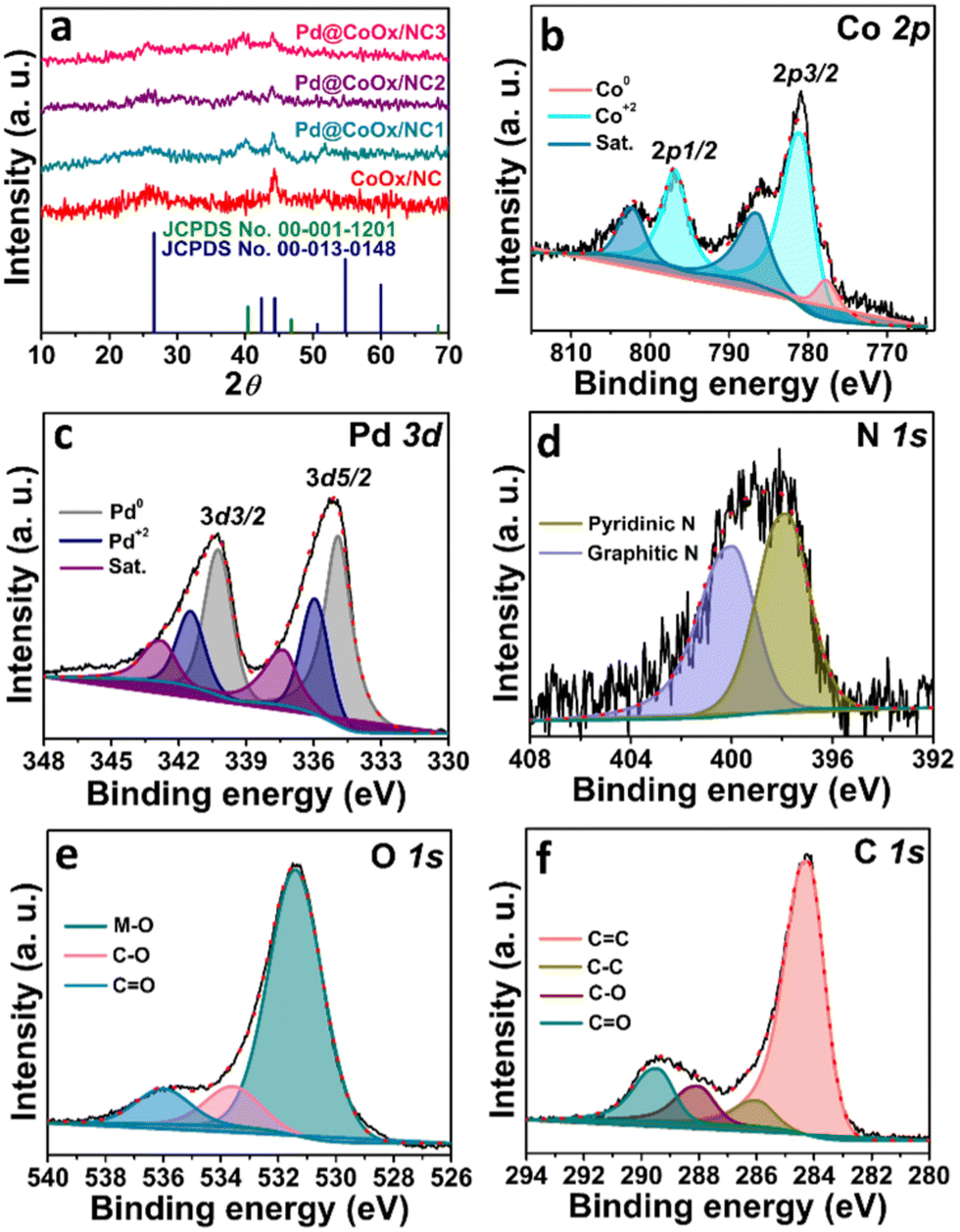 | ||
| Fig. 1 (a) PXRD patterns of synthesized electrocatalysts and comparison with standard reference cards. (b) XPS spectrum of Co 2p (c) Pd 3d (d) N 1s (e) O 1s and (f) C 1s. | ||
| Miller indices (hkl) | 2 Theta (deg) | cell parameter (a) nm | d-spacing (nm) |
|---|---|---|---|
| 111 | 39.9 | 0.39 | 0.2258 |
| 200 | 44.2 | 0.40 | 0.204 |
| 220 | 68.10 | 0.39 | 0.1379 |
For the determination of the oxidation state, surface elemental composition and electronic properties of the electrocatalyst, X-ray photoelectron spectroscopy was performed (Fig. 1). The surface analysis confirms the presence of cobalt, palladium, nitrogen, oxygen, and carbon in the synthesized electrocatalyst. The Co 2p spectrum is deconvoluted into two doublets corresponding to Co 2p3/2 and Co 2p1/2. The peak at 777.8 eV corresponds to metallic cobalt. The peak shift from 778.2 eV (standard binding energy of cobalt metal) towards a higher binding energy side, i.e., 781.2 eV and 796.9 eV corresponding to Co 2p3/2 and Co 2p1/2, respectively, is the result of oxidation of cobalt in the support material.29Fig. 1(c) presents the core level XPS spectrum of Pd nanoparticles. The two peaks at 334.8 eV and 340.2 eV can be assigned to Pd 3d5/2 (lower-energy band) and Pd 3d3/2 (high-energy band) spin orbit states, respectively. The binding energy values suggest that palladium is mostly in the metallic state. The binding energy of the palladium species is shifted negatively from 335 eV, suggesting the charge transfer from the cobalt species to the palladium species and formulating a different electronic environment around Pd which results in a downshift of the palladium d-band centre from the Fermi level. The higher electronegativity of palladium (2.20) relative to cobalt (1.88) causes the extraction of electrons from cobalt and dictates the decrease in the energy of the palladium d-band centre. The chemical shift from the standard binding energy of the palladium metal towards the lower side is due to the increased valence charge at the palladium center.30 These results are in agreement with the d-band centre model since the Pd metal shifted its d-band downward to compensate for the distortion caused as a result of the electronic interaction with cobalt. The electronic interaction of cobalt and palladium is responsible for the enhanced electrocatalytic activity of palladium species due to the weakening of the Pd–O bond of the adsorbed oxygenated intermediates. In addition to the palladium nanoparticles, the formation of surface bound PdO can also be observed in the fitted region of Pd 3d. The high resolution XPS spectrum of the N 1s core line consists of two components. The co-existence of two peaks at a binding energy of 397.8 eV and 400 eV is due to the pyridinic nitrogen and graphitic nitrogen, respectively (Fig. 1(d)).29 The high-resolution profile of the O 1s spectrum comprises three peaks at binding energies of ∼531.3 eV, ∼533.6 eV and ∼535.9 eV. These peaks represent the oxygen bonded to the metal and surface bound oxygen, respectively (Fig. 1(e)).29 The core level spectrum of C 1s is deconvoluted into four peaks. The peaks at binding energies of ∼284.2 eV and ∼286 eV can be ascribed to the sp2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and sp3 (C–C) hybridized carbon, respectively. The other two peaks at ∼288.1 eV and ∼289.4 eV are the result of surface bound oxygen groups (Fig. 1(f)).29,31 Fig. S4 (ESI†) shows the XPS analysis of CoOx/NC. The metal contents of the synthesized catalysts were also determined using an AAS analyser. The palladium metal contents of the synthesized catalysts were found to be 5.19%, 6.81%, and 8.90% for Pd@CoOx/NC1, Pd@CoOx/NC2 and Pd@CoOx/NC3, respectively.
C) and sp3 (C–C) hybridized carbon, respectively. The other two peaks at ∼288.1 eV and ∼289.4 eV are the result of surface bound oxygen groups (Fig. 1(f)).29,31 Fig. S4 (ESI†) shows the XPS analysis of CoOx/NC. The metal contents of the synthesized catalysts were also determined using an AAS analyser. The palladium metal contents of the synthesized catalysts were found to be 5.19%, 6.81%, and 8.90% for Pd@CoOx/NC1, Pd@CoOx/NC2 and Pd@CoOx/NC3, respectively.
The morphological and structural characterization was further analysed by HRTEM measurements (Fig. 2). Higher resolution transmission electron microscopy reveals the presence of well-defined cylindrical tubes with the encapsulation of cobalt oxide (CoOx) nanoparticles. The size of CoOx nanoparticles is around 50 nm (Fig. S2, ESI†) occupying the ends of nitrogen doped tubular carbon (NC). The higher magnification of the NC exposed the encapsulation of the nanoparticles within layers of tubular carbon. Curved growth lines are prominent within the tubes, curved in a single direction relative to one another. The growth lines form in semi-circular layers following from the surface of the nanoparticles and they are not concentric (Fig. S2, ESI†) The presence of heteroatoms i.e., oxygen and nitrogen, in the preparatory constituents encourage the condensation of carbon at high temperatures resulting in the formation of an entangled mat of tubular carbon with interconnected nodes and internodes. These interconnected nodes are believed to provide anchoring sites for the loaded palladium metal nanoparticles (Fig. S2, ESI†). Elemental mapping results confirm the presence of cobalt, palladium, nitrogen, carbon, and oxygen. Palladium nanoparticles with a sphere like shape are homogenously dispersed around CoOx embedded nitrogen doped tubular carbon. These results are an indication of the successful loading of palladium nanoparticles over the CoOx/NC skeleton. HRTEM results clearly reveal the appearance of fringes of loaded nanoparticles. These fringes of the d-spacing of ∼0.204 nm and ∼0.138 nm can be allocated to the (200) and (220) planes of palladium nanoparticles, respectively. The findings of HRTEM analysis are in accordance with the XRD results (Table 1). The EDX pattern of Pd@CoOx/NC1 has been shown in Fig. S3 (ESI†).
 | ||
| Fig. 2 (a) HRTEM analysis of Pd@CoOx/NC1 at 100 nm; the inset in (a) presents the HRTEM analysis of CoOx/NC at 50 nm, (b) HRTEM image of Pd@CoOx/NC1 at 50 nm (c) and (d) HRTEM image of Pd@CoOx/NC1 at 10 nm, (e) elemental mapping of Pd@CoOx/NC1, (f) nitrogen, (g) carbon, (h) oxygen, (i) palladium and (j) cobalt. | ||
Electrochemical studies
ORR electrocatalytic activity evaluation was done by performing cyclic voltammetry and linear sweep voltammetry in 0.1 M KOH and 0.1 M HClO4 in O2 and Ar saturated environments. The motivation of the study was to enhance the catalytic activity by virtue of the SMSI between palladium and the support material (CoOx/NC). Our material with the least amount of Pd metal exhibited unexpectedly higher activity than the materials with a higher loading of palladium metal. From the XPS and AAS data of our present work, the contents of cobalt and Pd were found to be 2.2%29 and 5.16%, respectively, in Pd@CoOx/NC1. Therefore, the cobalt to palladium percentage ratio in the synthesized sample (Pd@CoOx/NC1) is 2:5. The detailed mechanism of transition metal oxide (TMO) based catalysts for the ORR is still under investigation. The decrease in activity with the increase in palladium concentration could be attributed to the electronic interaction between the optimum concentration of cobalt and palladium. The charge transfer between the loaded nanoparticles and the substrate is due to the SMSI resulting in a noteworthy variation in the electronic distribution. This re-distribution of electronic densities modulates the chemisorption energies of adsorbed intermediates, facilitating the ORR kinetics.32,33 As we increase the concentration of palladium, keeping the concentration of cobalt constant, the activity decreases. From this we assume that the ideal ratio of cobalt and palladium, at which the synergistic interaction is maximum, leading to an enhancement in activity, is 2%:5%, respectively. All the results obtained were compared to commercial Pt/C under the same experimental conditions to govern the efficiency of catalysts.
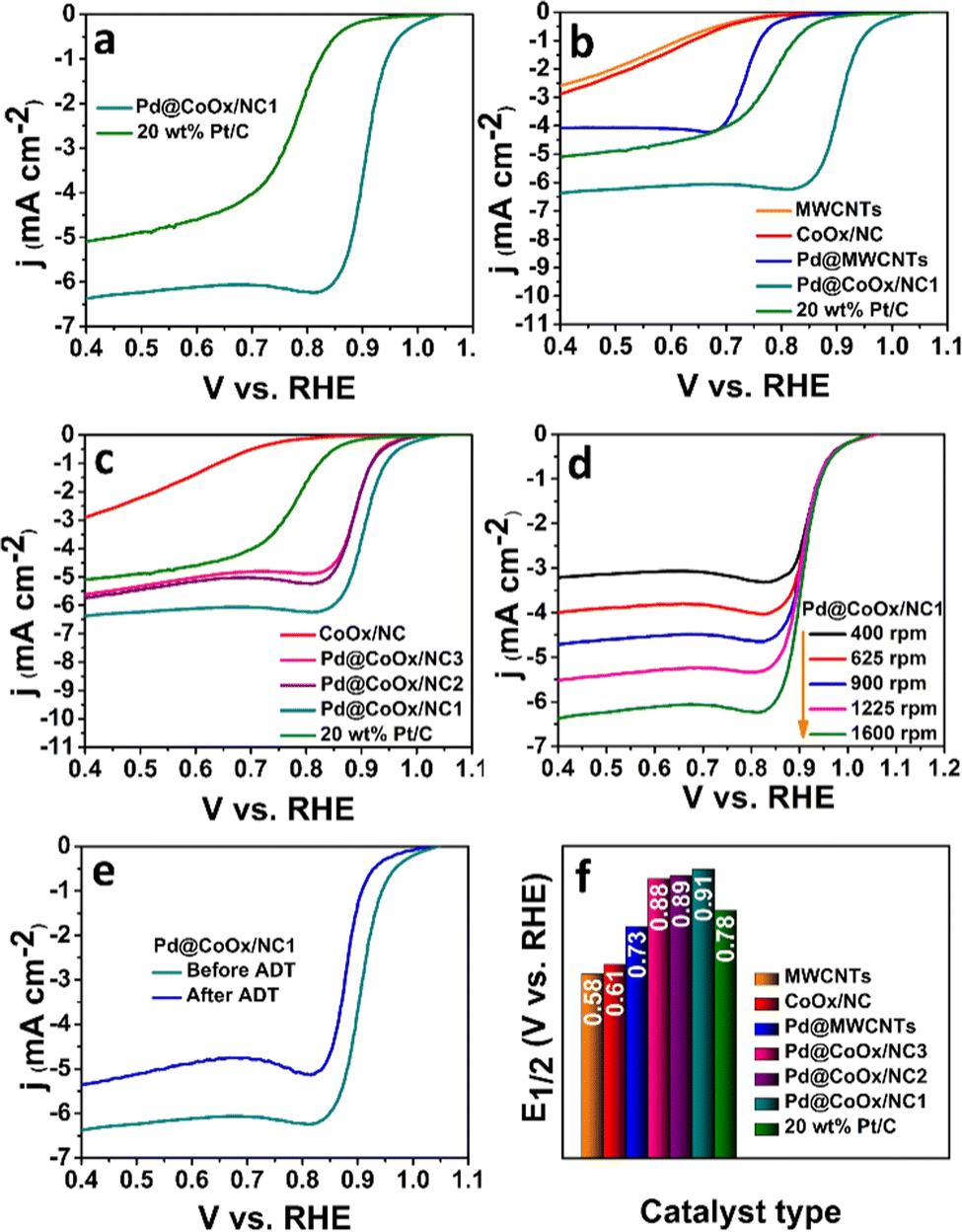
The sluggish ORR generally proceeds through two mechanisms in both acidic and alkaline media i.e., direct (four electron) and indirect (two electron) pathways. The direct pathway is preferable over the indirect pathway. The intermediate binding strength with the electrocatalyst is also a contributing factor towards the effectiveness of the catalyst.27,31,34 The significant catalytic role played by Pd based bimetallic catalysts have been proven from the reported literature.35,36 Hence the design of extremely low cost noble metal based electrocatalysts, exhibiting enhanced synergism with cobalt with a minimal amount of palladium in this work, is noteworthy. The ORR activities of all the synthesized electrocatalysts were investigated by rotating disk electrode (RDE) measurements in oxygen saturated alkaline (0.1 M KOH) and acidic (0.1 M HClO4) environments by recording linear sweep voltammetry curves (LSV) at different rotations. The ORR activities of all catalysts in basic and acidic media are reflected by bar graphs in Fig. 3(f) and 4(f), respectively. The representative LSV curves of Pd@CoOx/NC1 and all other electrocatalysts in basic and acidic media are shown in Fig. 3(b) and 4(b). Among all the presented catalysts, the Pd@CoOx/NC1 demonstrated superior ORR catalytic activity in both basic and acidic media which is far more superior than 20 wt% Pt/C in terms of mass activity (Fig. 5(d) and (e)) and the electrochemically active surface area (ECSA) (Fig. 5(f)). The enhanced ORR activity is due to a reduction in oxide coverage on the surface of the synthesized catalysts. Appropriate encapsulation of palladium nanoparticles within the NC and the electronic interaction with cobalt weaken the chemisorption of oxygenated intermediates due to a downshift in the palladium d-band centre, resulting in the improvement of ORR electrocatalytic parameters. As a control, palladium nanoparticles were deposited on commercial multiwalled carbon nanotubes. The catalyst (Pd@MWCNTs) demonstrated E1/2 values of 0.80 V in 0.1 M KOH and 0.73 V in 0.1 M HClO4 which are considerably lower than those of our synthesized catalysts, confirming the contribution of cobalt, embedded in NC towards enhanced ORR activity. The onset potential was recorded when the current density reached to 0.1 mA cm−2. The synthesized final catalyst greatly shows a positive shift in 0.1 M KOH (Eonset: 1.07 VRHE, E1/2: 0.95 VRHE) and 0.1 M HClO4 environments (Eonset: 1.02 VRHE, E1/2: 0.91 VRHE). Koutechy–Levich plots were drawn in a wide potential window ranging from 0.3 VRHE to 0.7 VRHE to determine the reaction kinetics. The electron transfer number involved in the direct pathway of Pd@CoOx/NC1 was calculated based on different rotating speeds of LSV curves which turns out to be 4.02 (Fig. S5a, ESI†) and 3.7 (Fig. S6a, ESI†) in alkaline and acidic environments respectively. These values are close to 4, suggesting four electron reduction pathways. The value of n does not remain constant in the potential range studied. It is obvious from the LSV curves of Pd@CoOx/NC1 in 0.1 M KOH (Fig. 3(d)) that the current density gradually increases and no diffusion limiting current density is reached, so the average value of electrons transferred per O2 molecule on the surface of Pd@CoOx/NC1 was calculated. The smaller Tafel slope of Pd@CoOx/NC1 in 0.1 M KOH (64 mV dec−1) among all the synthesized electrocatalysts further validates the better ORR kinetics (Fig. 5(b)). For comparison, the electrocatalytic activity of 20 wt% Pt/C towards the ORR was also recorded by linear sweep voltammetry at 1600 rpm. It was observed that the E1/2 values of the synthesized catalysts, which is the main determining parameter for evaluating the catalytic activity towards the ORR, shifted positively by 0.085 V in alkaline (Fig. 3(f)) and 0.08 V in acidic media (Fig. 4(f)) in comparison to 20 wt% Pt/C, while for Pd@MWCNTs it shifted negatively which further strengthens the idea of synergism between palladium and cobalt. These results suggest that the as-synthesized catalysts (Pd@CoOx/NC1) can effectively reduce oxygen molecules with low overpotentials.
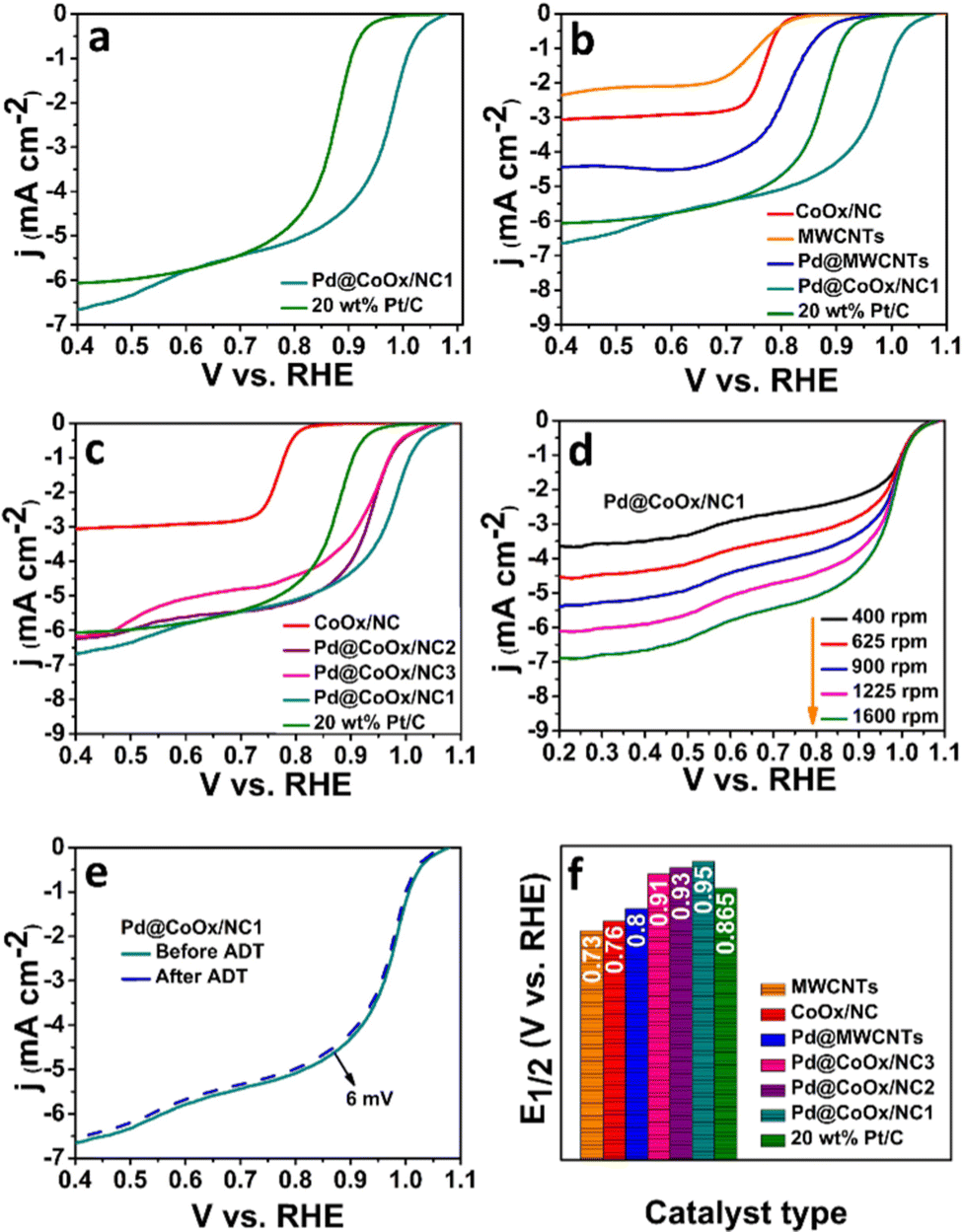 | ||
| Fig. 3 Electrochemical performance of the synthesized catalysts in 0.1 M KOH at 1600 rpm (scan rate 10 mV s−1) in an oxygen saturated environment: (a) comparison of the ORR performances of Pd@CoOx/NC1 and 20 wt% Pt/C, (b) controlled experiment and comparison of the ORR performances of the synthesized catalysts and 20 wt% Pt/C, (c) concentration effect of the synthesized catalysts, (d) rotation effect of Pd@CoOx/NC1, (e) ADT curves of Pd@CoOx/NC1 and (f) comparison of the E1/2 values of the synthesized catalysts and 20 wt% Pt/C. | ||
 | ||
| Fig. 4 Electrochemical performance of the synthesized catalysts in 0.1 M HClO4 at 1600 rpm (scan rate 10 mV s−1) in an oxygen saturated environment: (a) comparison of the ORR performances of Pd@CoOx/NC1 and 20 wt% Pt/C, (b) controlled experiment and comparison of the ORR performances with those of the synthesized catalysts and 20 wt% Pt/C (c) concentration effect of the synthesized catalysts, (d) rotation effect of Pd@CoOx/NC1 (e) ADT curves of Pd@CoOx/NC1, (f) comparison of the E1/2 values of the synthesized catalysts and 20 wt% Pt/C. | ||
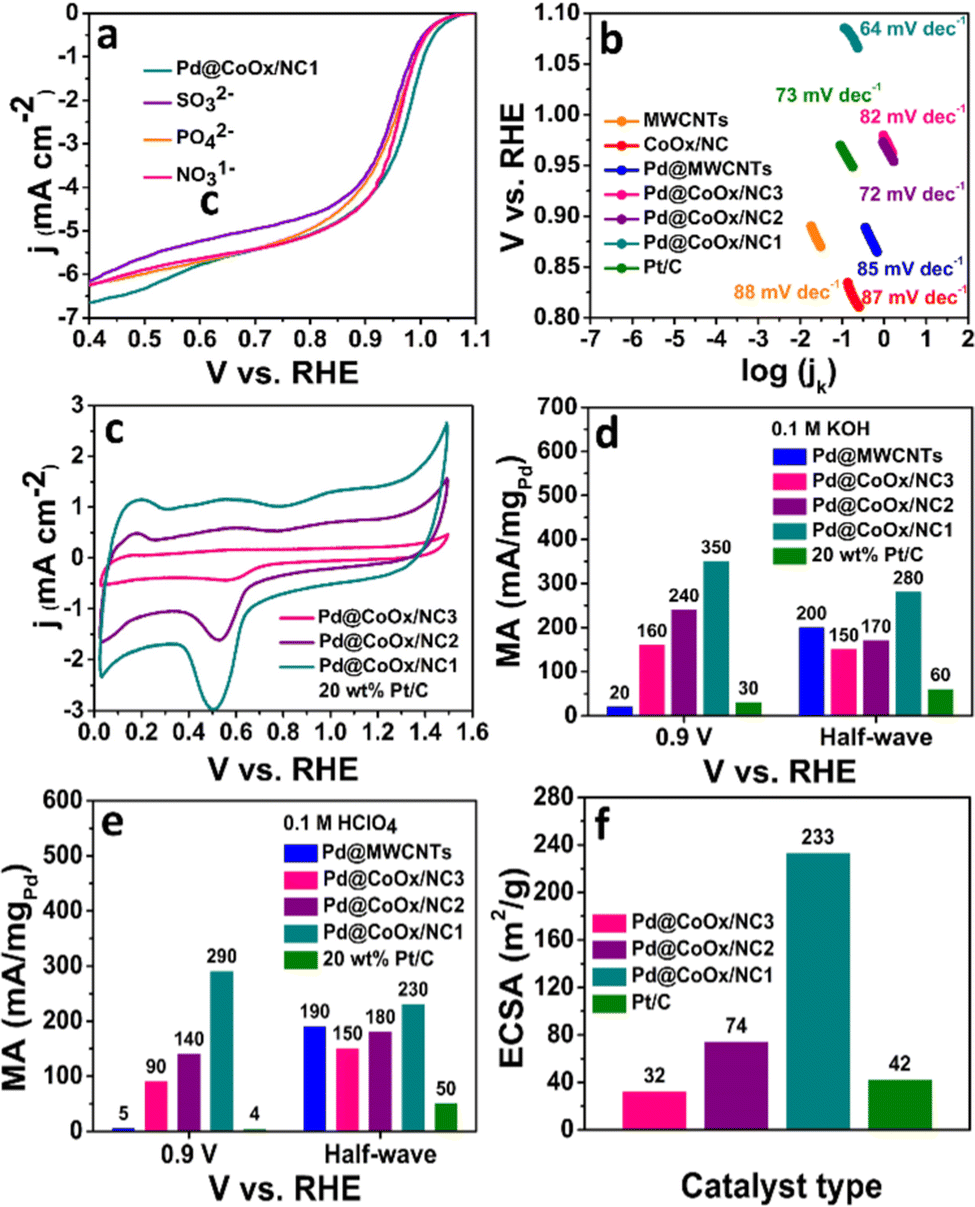 | ||
| Fig. 5 (a) ORR electrocatalytic performance of Pd@CoOx/NC1 in the presence of SO32−, NO31−, HPO42− anions in O2 saturated 0.1 M KOH at 10 mV s−1. (b) Tafel plots of all the synthesized electrocatalysts in 0.1 M KOH. (c) CV curves for oxide layer reduction in 0.1 M HClO4 as a function of the upper potential limit in an Ar saturated environment at 50 mV s−1, (d) comparison of the MA of the synthesized electrocatalysts in 0.1 M KOH at 0.9 VRHE and E1/2VRHE, (e) comparison of the MA of the synthesized electrocatalysts in 0.1 M HClO4 at 0.9 VRHE and E1/2 VRHE, (f) comparison of the ECSAs of the synthesized electrocatalysts and 20 wt% Pt/C. | ||
The mass activities of the synthesized catalysts (Pd@CoOx/NC1, Pd@CoOx/NC2, Pd@CoOx/NC3, Pd@MWCNTs) and 20 wt% Pt/C were also calculated at 0.9 VRHE (standard value) and half wave potential in both acidic and basic medium. The mass activities calculated were in the order of Pd@CoOx/NC1 > Pd@CoOx/NC2 > Pd@CoOx/NC3 (Fig. 5(e)). The electrochemical surface area was calculated from the palladium oxide layer (Pd–O) reduction method by recording cycling voltammetry curves at 1.4 VRHE as a function of the upper potential limit (Fig. 5(c)). Among all the synthesized catalysts, the Pd@CoOx/NC1 exhibited the highest electrochemical surface area i.e.; 233 m2 g−1. Fig. 5(f) shows the comparison of the electrochemically active surface area of the as-synthesized catalysts. The electrochemical surface area of all the catalysts, i.e. Pd@CoOx/NC1, Pd@CoOx/NC2, and Pd@CoOx/NC3, is superior to that of 20 wt% Pt/C. The acid leaching of the CoOx/NC could probably lead to the creation of defects, which provide anchoring sites for the uniform distribution of palladium nanocrystals and increase the electrochemically active surface area. The stability of the synthesized final material i.e. Pd@CoOx/NC1 was also evaluated by the accelerated durability test (ADT) over 10000 cycles in basic (0.1 M KOH) and 500 cycles in acidic (0.1 M HClO4) environments (Fig. 3(e) and 4(e), respectively). The catalyst demonstrated excellent cycling stability with only 6 mV decay in the alkaline environment and moderate stability with 30 mV decay under harsh acidic conditions. The high stability can be attributed to the appropriate encapsulation of Pd nanoparticles within the carbon matrix, preventing the irremediable deterioration of synthesized material in harsh reaction conditions. The E1/2 of the Pd@CoOx/NC1 was shifted negatively after ADT in 0.1 M HClO4 as well as its mass activity was decreased. Nevertheless, the ORR evaluating parameters were shifted negatively after ADT, the catalyst still retained 40% of its mass activity after 500 cycles. Fig. S5 and S6 (ESI†) show the electron transfer numbers, comparison of onset potentials and current densities in 0.1 M KOH and 0.1 M HClO4, respectively.
The ORR electrocatalytic activity of the as-synthesized catalysts and 20 wt% Pt/C was also tested in the presence of various concentrations of SO32−, NO31−, HPO42− ions in 0.1 M KOH. The onset potential, half wave potential and limiting current densities were decreased by a negligible amount for Pd@CoOx/NC1 in the presence of poisonous species. However, the SO3−2 ion strongly poisoned the 20 wt% Pt/C after the addition of 20 mM sodium sulphite. The Eonset and E1/2 were shifted negatively to a large degree for 20 wt% Pt/C in the presence of sulphite ions (Fig. S7, ESI†). The decrease in activity is probably due to the inactivation of Pt nanocrystal active sites and alteration in the electronic arrangement of the constituent materials. The adsorption of poisonous anions blocks the active sites of the commercial catalyst and consequently lowers the ORR performance. The small decrease in the E1/2 value of Pd@CoOx/NC1 is an indication of the chemisorption of poisonous anions with optimum binding strength. The large decrease in the E1/2 value of 20 wt% Pt/C in the presence of SO32− suggests the strong adsorption of anionic species, consequently shifting the ORR evaluating parameters towards the negative side. The synergistic effect of palladium and cobalt seems to be a major contributing factor towards the enhanced stability of Pd@CoOx/NC1 in the presence of poisonous species. The structural modification and rearrangement of electrons as a result of the synergistic effect of the incorporated metals (cobalt and palladium) could inhibit the loss of active sites in the presence of toxic species, resulting in pronounced stability of the synthesized material. The shift in ORR evaluating parameters in the presence of poisonous species (SO32−, NO31−, HPO42−) for Pd@CoOx/NC1 and 20 wt% Pt/C is presented in Table S1 (ESI†).
Tables 2 and 3 present the comparison of ORR evaluating parameters for palladium-based catalysts with the current work in 0.1 M HClO4 and 0.1 M KOH, respectively.
Conclusion
A series of highly efficient Pd based electro-catalysts (Pd@CoOx/NC1, Pd@CoOx/NC2, and Pd@CoOx/NC3) having different compositions were successfully synthesized. Superior ORR activity in terms of half-wave potential, current density, and mass activity has been investigated for all the synthesized catalysts in 0.1 M KOH and 0.1 M HClO4. The optimized Pd@CoOx/NC1 also exhibited an increased electrochemically active surface area calculated by the Pd–O layer reduction method. The electron transfer numbers were calculated to be 4.02 and 3.7 in alkaline and acidic environments, respectively, suggesting the direct and preferable four electron reduction process. The effect of poisoning species on the ORR activity of Pd@CoOx/NC1 was observed in 0.1 M KOH. The pronounced activities of all the as-synthesized catalysts are mainly attributed to the homogenous dispersion of the loaded nanocrystals and downshifting of the palladium d-band centre because of the synergistic effect with cobalt species. Thus, the present study provides a cost-effective, simple, and environmentally sound approach for the development of a highly active and stable oxygen reduction reaction catalyst with a minimal amount of palladium loading (5.16%). The highest activity and stability of Pd@CoOx/NC1 present a new avenue for the use of CoOx nanoparticles in combination with other noble transition metals towards electrochemical reduction of a variety of materials in fuel cell technologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Pakistan Academy of Sciences (PAS) and the Higher Education Commission (HEC) of Pakistan (No. 8400/Federal/NRPU/R&D/HEC/2017).Notes and references
- C. Wang, T. Qu, D. Wang and Z. Kang, Appl. Catal., A, 2018, 560, 103–110 CrossRef.
- J. Zhang, J. Ma, T. S. Choksi, D. Zhou, S. Han, Y.-F. Liao, H. B. Yang, D. Liu, Z. Zeng, W. Liu, X. Sun, T. Zhang and B. Liu, J. Am. Chem. Soc., 2022, 144, 2255–2263 CrossRef CAS.
- W. Jiao, C. Chen, W. You, J. Zhang, J. Liu and R. Che, Small, 2019, 15, 1805032 CrossRef PubMed.
- J. E. Choe, M. S. Ahmed and S. Jeon, J. Power Sources, 2015, 281, 211–218 CrossRef CAS.
- J. Li, H. Zhou, H. Zhuo, Z. Wei, G. Zhuang, X. Zhong, S. Deng, X. Li and J. Wang, J. Mater. Chem. A, 2018, 6, 2264–2272 RSC.
- X. Wang, Z. Chen, S. Chen, H. Wang and M. Huang, Eur. J. Chem., 2020, 26, 12589–12595 CrossRef CAS PubMed.
- J. M. Linge, D. Kozhemyakin, H. Erikson, S. Vlassov, N. Kongi and K. Tammeveski, ChemCatChem, 2021, 13(20), 4364–4371 CrossRef CAS.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-J. Liu and L. Zhuang, Nat. Commun., 2017, 8, 1–10 CrossRef.
- H. Ali, H. Lin, Q. Lu and X. Wang, J. Phys. Chem. C, 2021, 125, 14646–14655 CrossRef.
- M. Lüsi, H. Erikson, M. Merisalu, M. Rähn, V. Sammelselg and K. Tammeveski, J. Electroanal. Chem., 2019, 834, 223–232 CrossRef.
- K. Jukk, N. Alexeyeva, A. Sarapuu, P. Ritslaid, J. Kozlova, V. Sammelselg and K. Tammeveski, Int. J. Hydrogen Energy, 2013, 38(9), 3614–3620 CrossRef.
- X. Huang, A. J. Shumski, X. Zhang and C. W. Li, J. Am. Chem. Soc., 2018, 140, 8918–8923 CrossRef.
- Y. Lu, J. Wang, Y. Peng, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 7, 1700919 CrossRef.
- H. Erikson, A. Sarapuu, J. Solla-Gullón and K. Tammeveski, J. Electroanal. Chem., 2016, 780, 327–336 CrossRef CAS.
- M. Lüsi, H. Erikson, K. Tammeveski, A. Treshchalov, A. Kikas, H.-M. Piirsoo, V. Kisand, A. Tamm, J. Aruväli and J. Solla-Gullon, Electrochim. Acta, 2021, 394, 139132 CrossRef.
- K. Jukk, N. Kongi, L. Matisen, T. Kallio, K. Kontturi and K. Tammeveski, Electrochim. Acta, 2014, 137, 206–212 CrossRef CAS.
- M. Lüsi, H. Erikson, A. Sarapuu, K. Tammeveski, J. Solla-Gullón and J. M. Feliu, Electrochem. Commun., 2016, 64, 9–13 CrossRef.
- H. Erikson, M. Lüsi, A. Sarapuu, K. Tammeveski, J. Solla-Gullón and J. M. Feliu, Electrochim. Acta, 2016, 188, 301–308 CrossRef CAS.
- Y. Ge, X. Wang, B. Huang, Z. Huang, B. Chen, C. Ling, J. Liu, G. Liu, J. Zhang, G. Wang, Y. Chen, L. Li, L. Liao, L. Wang, Q. Yun, Z. Lai, S. Lu, Q. Luo, J. Wang, Z. Zheng and H. Zhang, J. Am. Chem. Soc., 2021, 143, 17292–17299 CrossRef CAS PubMed.
- G. W. Sievers, A. W. Jensen, J. Quinson, A. Zana, F. Bizzotto, M. Oezaslan, A. Dworzak, J. J. Kirkensgaard, T. E. Smitshuysen and S. Kadkhodazadeh, Nat. Mater., 2021, 20, 208–213 CrossRef CAS.
- X. Yuan, X. Wang, X. Liu, H. Ge, G. Yin, C. Dong and F. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 27654–27660 CrossRef CAS.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Möller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef.
- H. E. Kim, I. H. Lee, J. Cho, S. Shin, H. C. Ham, J. Y. Kim and H. Lee, ChemElectroChem, 2019, 6, 4757–4764 CrossRef.
- I. Jiménez-Morales, F. Haidar, S. Cavaliere, D. Jones and J. Roziere, ACS Catal., 2020, 10, 10399–10411 CrossRef.
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, H. D. Abruna, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317–9332 CrossRef.
- F. B. Ometto, E. A. Carbonio, E. Teixeira-Neto and H. M. Villullas, J. Mater. Chem. A, 2019, 7, 2075–2086 RSC.
- I. A. Khan, Y. Qian, A. Badshah, M. A. Nadeem and D. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 17268–17275 CrossRef CAS PubMed.
- Q. Xiang, W. Yin, Y. Liu, D. Yu, X. Wang, S. Li and C. Chen, J. Mater. Chem. A, 2017, 5, 24314–24320 RSC.
- I. Khan, F. Nasim, M. Choucair, S. Ullah, A. Badshah and M. Nadeem, RSC Adv., 2016, 6, 1129–1135 RSC.
- S. Mondal and C. R. Raj, ACS Appl. Mater. Interfaces, 2019, 11, 14110–14119 CrossRef.
- C. Goswami, H. Saikia, K. Tada, S. Tanaka, P. Sudarsanam, S. K. Bhargava and P. Bharali, ACS Appl. Energy Mater., 2020, 3, 9285–9295 CrossRef.
- K. Maiti, N. H. Kim and J. H. Lee, Chem. Eng. J., 2021, 410, 128358 CrossRef.
- L. Wei, H. E. Karahan, S. Zhai, H. Liu, X. Chen, Z. Zhou, Y. Lei, Z. Liu and Y. Chen, Adv. Mater., 2017, 29(38), 1701410 CrossRef.
- H. Erikson, A. Sarapuu and K. Tammeveski, ChemElectroChem, 2019, 6(1), 73–86 CrossRef.
- H. Yang, K. Wang, Z. Tang, Z. Liu and S. Chen, J. Catal., 2020, 382, 181–191 CrossRef.
- G. Bampos, S. Bebelis, D. I. Kondarides and X. Verykios, Top. Catal., 2017, 60, 1260–1273 CrossRef.
- C. Lo Vecchio, C. Alegre, D. Sebastián, A. Stassi, A. S. Aricò and V. Baglio, Mater., 2015, 8, 7997–8008 CrossRef PubMed.
- S. H. Park, C. H. Choi, J. K. Koh, C. Pak, S.-A. Jin and S. I. Woo, ACS Comb. Sci., 2013, 15, 572–579 CrossRef.
- M. Wang, X. Qin, K. Jiang, Y. Dong, M. Shao and W.-B. Cai, J. Phys. Chem. C, 2017, 121, 3416–3423 CrossRef.
- G. Bampos, L. Sygellou and S. Bebelis, Catal., 2020, 355, 685–697 Search PubMed.
- Q. Liu, Y. Peng, Q. Li, T. He, D. Morris, F. Nichols, R. Mercado, P. Zhang and S. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 17641–17650 CrossRef.
- X. Lu, M. Ahmadi, F. J. DiSalvo and H. D. Abruna, ACS Catal., 2020, 10, 5891–5898 CrossRef.
- J. Li, J. Chen, Q. Wang, W.-B. Cai and S. Chen, Chem. Mater., 2017, 29, 10060–10067 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Determination of the electron number, ECSA measurement, XRD patterns, HRTEM, EDX, XPS, K–L plots and comparison of ORR parameters, the CV curve for ECSA measurements, and anti-poisoning results. See DOI: https://doi.org/10.1039/d2ma00871h |
| This journal is © The Royal Society of Chemistry 2023 |