
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalized cellulose monolith based affinity chromatography columns for efficient separation of protein molecules†
Yusuke
Hinamoto
a,
Akihide
Sugawara
 a,
Taka-Aki
Asoh
a,
Mahasweta
Nandi
*b and
Hiroshi
Uyama
*a
a,
Taka-Aki
Asoh
a,
Mahasweta
Nandi
*b and
Hiroshi
Uyama
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita 565-0871, Japan. E-mail: uyama@chem.eng.osaka-u.ac.jp
bIntegrated Science Education and Research Centre, Siksha Bhavana, Visva-Bharati, Santiniketan 731 235, India. E-mail: mahasweta.nandi@visva-bharati.ac.in
First published on 9th August 2023
Abstract
Cellulose monoliths are prepared from cellulose acetate (CA) monoliths by the thermally induced phase separation technique followed by hydrolysis of the acetyl groups of the side chain of CA in a basic solution of sodium hydroxide. The morphology of the cellulose monoliths is found to be dependent on the fabrication conditions such as the concentration of CA, solvent ratio and cooling temperature. A comparison with ethylene-vinyl alcohol copolymer (EVOH) and poly(methyl methacrylate) (PMMA) monoliths reveals that cellulose is a better column material in terms of strength, liquid permeability and suppression of non-specific adsorption. To design the affinity columns for selective biomolecule adsorption and separation, N-hydroxysuccinimide (NHS) esters are immobilized over the cellulose monoliths by the N,N′-disuccinimidyl carbonate (DSC) activation method in the presence of N,N-dimethyl-4-aminopyridine (DMAP). The activated cellulose monoliths are allowed to react with Protein A under optimum conditions at the NHS ester sites to obtain affinity chromatography carriers for selective adsorption of Immunoglobulin G (IgG) protein by employing the association of Protein A with the IgG antibody. The adsorption capacity and adsorption efficiency are found to improve with the activated cellulose monoliths. It is revealed that cellulose monolith exhibited unique surface morphology with deep roughness in the pore structure, contributing to the enhancement of adsorption capacity. The desorption characteristics of IgG at different flow rates are found to be almost the same demonstrating that the elution time can be shortened by using higher flow rates. The high permeability coefficient of the monolith column promotes rapid diffusion to increase the flow velocity. The columns can be regenerated and reused without appreciable loss of adsorption efficiency. Such high efficiency separation of protein molecules cannot be achieved with traditional and commercially available agarose gel beads.
Introduction
Affinity chromatography is a separation technique that involves the specific adsorption of biomolecules.1,2 Among a pair of enzymes and substrates having specific actions, the enzymes can be separated by immobilizing the substrates over a separation column. It has a great advantage that only the target substance can be selectively separated with high purity from a mixture of substances by its specific adsorption. It plays an important role in clinical practice3 and proteomics4 and, in recent years, in the development of antibody drugs which has been an active area of research conducted all over the world.5 In a typical affinity chromatography column, a molecule (ligand) having a specific interaction is chemically or physically attached to a carrier. A solution of the analyte with other non-target substances is passed through the column, the analyte is adsorbed by affinity and the non-target materials retained in the column are removed by washing. After that, the analyte is eluted from the carrier by a change in pH or adding a competing substance so that the ligand and the analyte are separated from each other and the analyte is recovered. Apart from biomolecules, molecules with mimicking interactions6,7 and modified biomolecules with improved specificity and adsorption efficiency can be used as ligands.8Carriers used in affinity chromatography are either particle-filled columns or monolith columns. Particle-filled columns have been used as chromatographic carriers for a long time. One of the most common materials for carriers is cross-linked agarose gel beads with high adsorption capacity, ease of chemical modification, stability over a wide pH range, ability to suppress non-specific adsorption and high reproducibility.2,9 However, due to their gel nature, they suffer from disadvantages such as low strength and permeability, making them difficult to handle.2 On the other hand, monoliths are porous materials with a three-dimensional network structure of voids and skeletons, used as adsorbents,10,11 flow reactors,12,13 catalysts,14,15 and battery materials.16,17 They have several advantages such as (i) freely moldable and producible in a capillary column or microchannel device, (ii) low pressure loss enabling high-speed separation by using high flow velocities, thereby shortening the analysis time, (iii) efficient recovery and separation by rapid mass transfer and (iv) possibility of modification through various reactions.18 Most of the desirable requirements of a carrier such as homogeneity, macroporosity, hydrophilicity, chemical/physical stability, suppression of non-specific adsorption, good liquid permeability, high surface area for ligand immobilization and modification and ease of handling5 can be achieved by using a monolith. Different types of functionalized monolithic columns with high separation efficiency have been reported19 of which some typical carriers include silica,20,21 polymer based monoliths22,23 and metal–organic frameworks.24 Polymer-based monoliths are relatively easy to modify with functional groups.25,26 The choice of the polymer used for the fabrication of the monolith decides the desirable qualities of the carrier and hence its selection is extremely important.
Cellulose is a naturally abundant polysaccharide available from various sources27 and is known to have hydrophilicity and chemical and physical stability. Due to its biocompatibility, it is widely used as a column material for the adsorption of biomolecules,27,28 and regenerated cellulose becomes porous with a high specific surface area showing high adsorption capacity.29–31 Thus, cellulose monoliths are excellent materials as affinity chromatography carriers.32 Monoliths can be obtained either by the thermally induced phase separation (TIPS) method33 or the non-solvent-induced phase separation (NIPS) method;34 TIPS is preferable for producing homogeneous monoliths in bulk. In the case of cellulose, due to its restricted solubility in common solvents, a soluble cellulose derivative, cellulose acetate (CA), is often used to produce monoliths followed by their deacetylation to obtain cellulose monoliths. The internal morphologies of the monoliths affect the liquid permeability, surface area and adsorption performance.35 It has been found that the morphologies can be controlled by optimization of the production conditions to improve the adsorption performance and reaction efficiency in a flow reactor.36,37 Also, cellulose monoliths have sufficient heat resistance for practical applications, and their starting temperature of thermal decomposition is ca. 220 °C.38 Moreover, the rigid structure of cellulose provides excellent mechanical stability to the monoliths both in dry and wet states, contributing to the high durability required for flow systems (compression stress is ca. 2 MPa in the dry state and 0.1 MPa in the wet state, at 20% compression).38,39
The monoliths can be transformed into efficient affinity chromatography columns for selective adsorption of proteins by immobilization of suitable ligands over the porous surface.40 Various strategies are adopted to improve the performance of protein adsorption like ligand modification,41 orientation fixation,42 increasing the surface area by loading nanoparticles,43etc. Modification of the ligand onto the protein requires genetic manipulation, which involves enormous cost and time. In this respect, morphology control can be a facile route to increase the surface area for protein immobilization and adsorption to improve the efficiency and shorten the recovery time. Under certain conditions, cellulose monoliths form an internal skeletal structure composed entirely of fibers that lead to a high surface area, porosity and liquid permeability through which proteins can diffuse, thereby providing an efficient affinity column carrier.
In the present contribution, we report the design of a high-performance cellulose monolith-based column as a carrier for affinity purification (Scheme 1). The primary focus of this study has been laid on the suppression of non-specific adsorption, high liquid permeability, improvement of adsorption performance by morphology control and shortening of the recovery time by the cellulose-based column. Its performance in terms of strength, liquid permeability and suppression of non-specific adsorption is compared with monolith columns derived from ethylene-vinyl alcohol copolymer (EVOH) and poly(methyl methacrylate) (PMMA), both of which are hydrophilic thermoplastic resins known for their application in biosensors44,45 and separation of biomolecules.37,46 The affinity chromatography column, obtained from the cellulose acetate monolith by its deacetylation and successive activation with N,N′-disuccinimidyl carbonate (DSC) followed by immobilization of Protein A, exhibited high liquid permeability and an excellent separation rate for Immunoglobulin G (IgG). Here, Protein A and IgG have been used as models to evaluate the column performance of cellulose as a carrier for affinity purification.
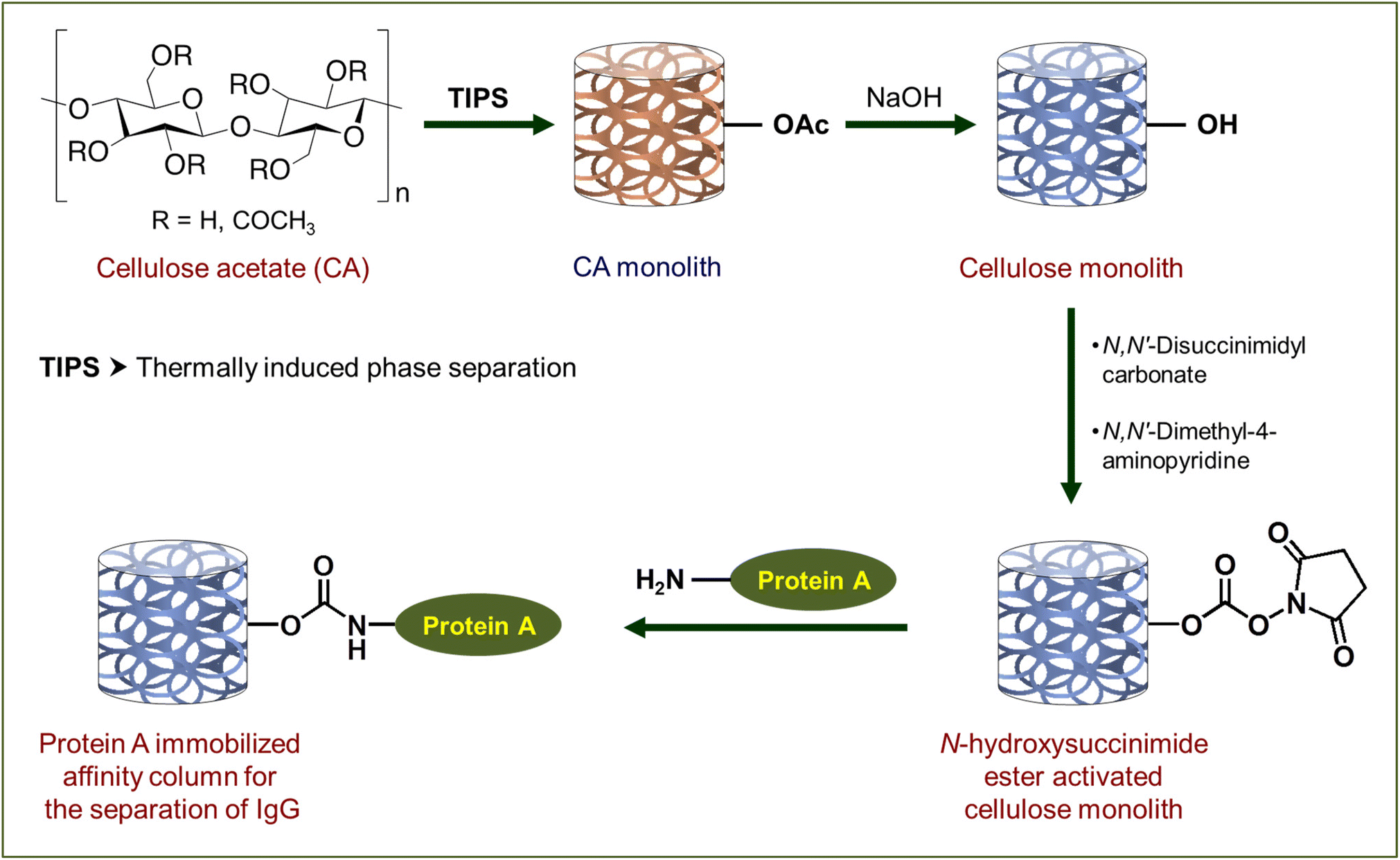 | ||
| Scheme 1 Preparation of affinity chromatography columns based on the cellulose monolith. Here functionalization of one –OH has been shown as a representative case to maintain clarity. | ||
Experimental section
Materials and methods
Potassium chloride (KCl), N,N-dimethyl-4-aminopyridine (DMAP), monoethanolamine, N,N-dimethyl formamide (DMF), dimethyl sulfoxide (DMSO) and acetonitrile were purchased from Nacalai Tesque. Sodium chloride (NaCl), potassium dihydrogen phosphate (KH2PO4), disodium hydrogen phosphate dodecahydrate (Na2HPO4·12H2O), sodium hydroxide (NaOH), glycine, methanol, ethanol and acetone were purchased from Wako Pure Chemical Industries, Ltd. The reaction solvents, namely DMF, DMSO, acetonitrile and acetone, were dehydrated using molecular sieves and others were used as received. Cellulose acetate (CA), L30, used in the synthesis was kindly provided by Daicel Co., Ltd and ethylene-vinyl alcohol (EVOH) with an ethylene content of 27% was provided by Kuraray Co., Ltd. Poly(methyl methacrylate) (PMMA) (molecular weight: 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was kindly provided by Mitsubishi Chemical. N,N′-Disuccinimidyl carbonate (DSC) was purchased from TCI Chemicals and recombinant Protein A was procured from BioVision, being used without further purification. 1-Hexanol, fluorescein isothiocyanate labelled bovine serum albumin (FITC-BSA) and Immunoglobulin G (IgG) were purchased from Sigma Aldrich and used as purchased. The pressure gauge, KDM30, was purchased from Krone Co., Ltd. For FT-IR studies, iD5 ATR and iS5 from Thermo Scientific were used. SU3500 from Hitachi was used to collect scanning electron microscopy (SEM) images and image processing was performed using ImageJ software. Heat shrink tubing (THT-6.0) was purchased from Denka Electron. A Bio-Rad BioLogic LP low-pressure chromatography system equipped with a UV spectrometer (wavelength at 280 nm) was used for biomolecule adsorption experiments. Fluorescence was observed in an OLYMPUS IX73 fluorescence microscope supported by cellSens imaging software. UV-vis spectroscopy studies were carried out using a U-2810, Hitachi spectrophotometer. Phosphate-buffered saline (PBS) was prepared by dissolving 40.0 g of NaCl, 1.0 g of KCl, 14.4 g of Na2HPO4·12H2O and 1.0 g of KH2PO4 in 900 mL of ultrapure water and adjusting the pH to 7.4, and the volume was made up to 1 L to obtain 10× PBS(−). This solution was diluted 10 times and used as 1× PBS(−). The ethanolamine buffer solution was prepared by adding 15.1 mL of monoethanolamine and 14.6 g of NaCl in 400 mL of ultrapure water and adjusting the pH to 8.3, and the volume was made up to 500 mL. The glycine buffer solution was prepared by dissolving 3.75 g of glycine in 400 mL of ultrapure water and adjusting the pH to 3.0, and the final volume was made up to 500 mL.
000) was kindly provided by Mitsubishi Chemical. N,N′-Disuccinimidyl carbonate (DSC) was purchased from TCI Chemicals and recombinant Protein A was procured from BioVision, being used without further purification. 1-Hexanol, fluorescein isothiocyanate labelled bovine serum albumin (FITC-BSA) and Immunoglobulin G (IgG) were purchased from Sigma Aldrich and used as purchased. The pressure gauge, KDM30, was purchased from Krone Co., Ltd. For FT-IR studies, iD5 ATR and iS5 from Thermo Scientific were used. SU3500 from Hitachi was used to collect scanning electron microscopy (SEM) images and image processing was performed using ImageJ software. Heat shrink tubing (THT-6.0) was purchased from Denka Electron. A Bio-Rad BioLogic LP low-pressure chromatography system equipped with a UV spectrometer (wavelength at 280 nm) was used for biomolecule adsorption experiments. Fluorescence was observed in an OLYMPUS IX73 fluorescence microscope supported by cellSens imaging software. UV-vis spectroscopy studies were carried out using a U-2810, Hitachi spectrophotometer. Phosphate-buffered saline (PBS) was prepared by dissolving 40.0 g of NaCl, 1.0 g of KCl, 14.4 g of Na2HPO4·12H2O and 1.0 g of KH2PO4 in 900 mL of ultrapure water and adjusting the pH to 7.4, and the volume was made up to 1 L to obtain 10× PBS(−). This solution was diluted 10 times and used as 1× PBS(−). The ethanolamine buffer solution was prepared by adding 15.1 mL of monoethanolamine and 14.6 g of NaCl in 400 mL of ultrapure water and adjusting the pH to 8.3, and the volume was made up to 500 mL. The glycine buffer solution was prepared by dissolving 3.75 g of glycine in 400 mL of ultrapure water and adjusting the pH to 3.0, and the final volume was made up to 500 mL.
Experimental procedure
| Sample name | CA (g) | DMF (mL) | 1-Hexanol (mL) | Cooling temperature (°C) |
|---|---|---|---|---|
| a Cellulose monoliths were prepared by their hydrolysis. | ||||
| C-90 | 2.25 | 10.0 | 15.0 | 25 |
| C-100 | 2.50 | 10.0 | 15.0 | 25 |
| C-110 | 2.75 | 10.0 | 15.0 | 25 |
| P-58 | 2.50 | 9.5 | 15.5 | 25 |
| P-60 | 2.50 | 10.0 | 15.0 | 25 |
| P-62 | 2.50 | 10.5 | 14.5 | 25 |
| T-15 | 2.50 | 10.0 | 15.0 | 15 |
| T-20 | 2.50 | 10.0 | 15.0 | 20 |
| T-25 | 2.50 | 10.0 | 15.0 | 25 |
| T-30 | 2.50 | 10.0 | 15.0 | 30 |
:7 (v/v) at a concentration of 100 mg mL−1 and dissolved by heating at 95 °C. Phase separation was induced by cooling the solution in a constant temperature bath at 25 °C. The solvent was replaced with acetone and the solid mass was dried under reduced pressure to obtain the EVOH monolith. The poly(methyl methacrylate (PMMA) monolith48,49 was prepared by adding PMMA to a mixed solvent of ethanol/water = 4:1 (v/v), maintaining the concentration at 100 mg mL−1. After PMMA was dissolved in the mixed solvent by heating at 60 °C under continuous stirring, the solution was transferred into a mold and cooled in a constant temperature bath at 25 °C to induce phase separation. Then the mold was immersed in methanol. The solid mass was taken out from the mold, and the solvent was exchanged with methanol again and dried under reduced pressure to obtain the PMMA monolith. For comparison, both PMMA and EVOH monoliths were also synthesized with 90 mg mL−1 and 110 mg mL−1 concentrations, keeping the other conditions the same.
| ΔP/L = Qμ/AB0 | (i) |
Here, L is the column length, μ is the viscosity of the solution, A is the column cross-sectional area, and B0 is the transmission coefficient. For the transmission coefficient, the average and standard error were calculated from three or more measurements. The through-pore diameters have been calculated from the transmission coefficient using Kozeny–Carman's equation50 given in eqn (ii).
| Df = 2(5B0/ε)1/2 | (ii) |
Amount of DSC charged. In the IgG adsorption experiment, a 0.2 mg mL−1 IgG solution was prepared using PBS and the breakthrough curve was measured by passing the solution through the column until adsorption reached saturation. PBS was then passed through the column and the tube to remove the residual IgG, and washed until the signal dropped to the baseline and the UV signal was stable. After that, the IgG adsorbed was eluted and recovered by passing glycine buffer solution through the solution. This experiment was carried out with all the affinity columns prepared from P-60 and activated with two-, four- and six-equivalents of DSC. The affinity columns are designated as DSC-2, DSC-4 and DSC-6. The pH for protein immobilization was maintained at 7.4 and the concentration of protein was 1.0 mg mL−1.
Immobilization reaction with different pH values. Solutions of Protein A were prepared in phosphate buffer (PB) at different pH values and the solutions were circulated through activated monolith columns separately to prepare affinity monolith columns. The pH values of PB were maintained at 7.0, 7.4 and 7.8 and accordingly the affinity columns are designated as pH-7.0, pH-7.4 and pH-7.8, respectively. In this case, the P-60 monolith column was used. Four-equivalents of DSC were charged and the concentration of protein was maintained at 1.0 mg mL−1. Using these monolith columns, the IgG adsorption experiment was performed in the same manner as in the preceding experiment and the adsorption behavior of the columns was studied.
Immobilization reaction with different concentrations of Protein A. Solutions of different concentrations of Protein A were prepared in PBS and circulated through the P-60 derived activated monolith columns (charged with four-equivalents of DSC) to create the affinity monolith columns. The concentrations of the Protein A solution were 0.5, 1.0, 1.5 and 2.0 mg mL−1 at 7.4 pH, and the affinity columns are designated as A-0.5, A-1.0, A-1.5 and A-2.0, respectively. Using these solutions, the IgG adsorption experiment was performed in the same way as that described above, and the adsorption behavior of the columns was monitored.
Differences in morphology. Activated monoliths were also prepared using cellulose monoliths, P-58, P-60 and P-62, prepared with different ratios of the anti-solvent and thereby varying in their morphologies. The DSC charging was maintained at four-equivalents, and the protein concentration was 1.0 mg mL−1 at pH 7.4. The affinity monolith columns were prepared by the same procedure in each case and the IgG adsorption experiments were performed. The affinity columns in this case are designated as P-58-A, P-60-A and P-62-A, corresponding to the cellulose monoliths P-58, P-60 and P-62, respectively.
Results and discussion
Cellulose acetate monoliths synthesized through TIPS were used to fabricate affinity chromatography columns (Scheme 1). This was carried out by dissolving the solute in a good solvent and then adding a poor solvent to the solution followed by lowering the temperature of the solution. This creates a disturbance in the balance between the solute and the good solvent, the solute is precipitated leading to phase separation thereby forming a network structure. The high molecular weight of the polymer imposes limitations on its mobility and it becomes more energetically stable to precipitate with the nearby solute molecules than to precipitate out locally. The time lag towards disruption of the interaction with the solvent results in the entanglement of the molecular chains and cross-linking due to crystallization forming a network structure by phase separation. The process occurs within a very narrow region of the concentration of the solute and solvent. When the ratio of the poor solvent is increased, the system becomes more prone to phase out and the phase separation of the system to the solid structure takes place rapidly. This results in a monolith having a fine network structure. On the other hand, when the poor solvent ratio is decreased, the opposite is true, and the phase separation progresses slowly with the growth of pores and skeletons to form a monolith with a large network structure. Also, when the polymer concentration is high and the cooling temperature is low, phase separation occurs rapidly to generate a monolith with a fine structure. Thus, by changing the monolith production conditions different morphologies can be obtained.In this study, the morphology of the cellulose monoliths was controlled by optimizing the concentration of cellulose acetate (CA), the solvent ratio and the cooling temperature. The fabrication conditions of the various CA monoliths are shown in Table 1. It is observed that the CA concentration between 90 and 110 mg mL−1, an intermediate region with a poor solvent ratio, i.e. greater than 56% and less than 64% and the phase separation temperature between 15 and 30 °C are suitable for the fabrication of the monoliths. When the concentration of CA was higher than 110 mg mL−1, the system becomes a poly(high internal phase emulsion) (HIPE) and it was difficult for any liquid to pass through it. Also, below the concentration of 90 mg mL−1, TIPS was not effective to form monolithic structures. Additionally, when the poor solvent ratio is 56%, phase separation did not occur and the solution becomes slightly cloudy. At the other end, with a poor solvent ratio of 64%, the monolith is obtained but there is severe shrinkage of the structure. At a cooling temperature of 10 °C, the diameter of the monolith shrinks from 6 mm to 4 mm. Again at a cooling temperature of 35 °C, a monolith was obtained, but with a fine network structure with fibers that shrinks drastically from 6 mm to 2 mm after drying. Thus, they could not be used as column materials. Hence, the fabrication conditions given in Table 1 are the range of various parameters under which a good monolith can be produced.
After the successful fabrication of the monoliths, the acetyl group of the CA side chains was hydrolyzed in a basic solution of sodium hydroxide. The progress of the reaction was monitored by FT-IR spectroscopy. The FT-IR spectra of CA and hydrolyzed CA for C-100, as a representative case, are shown in ESI, Fig. S1.† It is observed that the peak of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the acetyl group at ca. 1740 cm−1 disappears after hydrolysis, and on the other hand, the peak derived from the O–H stretching vibration of the hydroxyl group appears at around 3400 cm−1. This suggests that the hydrolysis proceeds sufficiently over the CA monoliths to facilitate further modification and immobilization. The morphology of the cellulose monoliths is observed using SEM and the images are shown in Fig. 1. All the monoliths prepared under the conditions given in Table 1 exhibit micrometer-order through pores. The effects of various CA concentrations, antisolvent ratios and cooling temperatures on the average size of the pores and skeletons of CA monoliths were studied. The variations are shown in Fig. 2. The through-pore and skeleton sizes tended to be larger when the concentration of CA was lesser (Fig. 2a), the antisolvent ratio was lower (Fig. 2b) and the cooling temperature of phase separation was higher (Fig. 2c). This is in line with the expected trend based on the theory of phase separation as discussed earlier. By variation of the fabrication conditions it is possible to control the through-pore sizes in the range of 5–15 μm and the skeleton sizes between 1 and 8 μm.
O stretching vibration of the acetyl group at ca. 1740 cm−1 disappears after hydrolysis, and on the other hand, the peak derived from the O–H stretching vibration of the hydroxyl group appears at around 3400 cm−1. This suggests that the hydrolysis proceeds sufficiently over the CA monoliths to facilitate further modification and immobilization. The morphology of the cellulose monoliths is observed using SEM and the images are shown in Fig. 1. All the monoliths prepared under the conditions given in Table 1 exhibit micrometer-order through pores. The effects of various CA concentrations, antisolvent ratios and cooling temperatures on the average size of the pores and skeletons of CA monoliths were studied. The variations are shown in Fig. 2. The through-pore and skeleton sizes tended to be larger when the concentration of CA was lesser (Fig. 2a), the antisolvent ratio was lower (Fig. 2b) and the cooling temperature of phase separation was higher (Fig. 2c). This is in line with the expected trend based on the theory of phase separation as discussed earlier. By variation of the fabrication conditions it is possible to control the through-pore sizes in the range of 5–15 μm and the skeleton sizes between 1 and 8 μm.
 | ||
| Fig. 1 SEM images of the fabricated cellulose monoliths. | ||
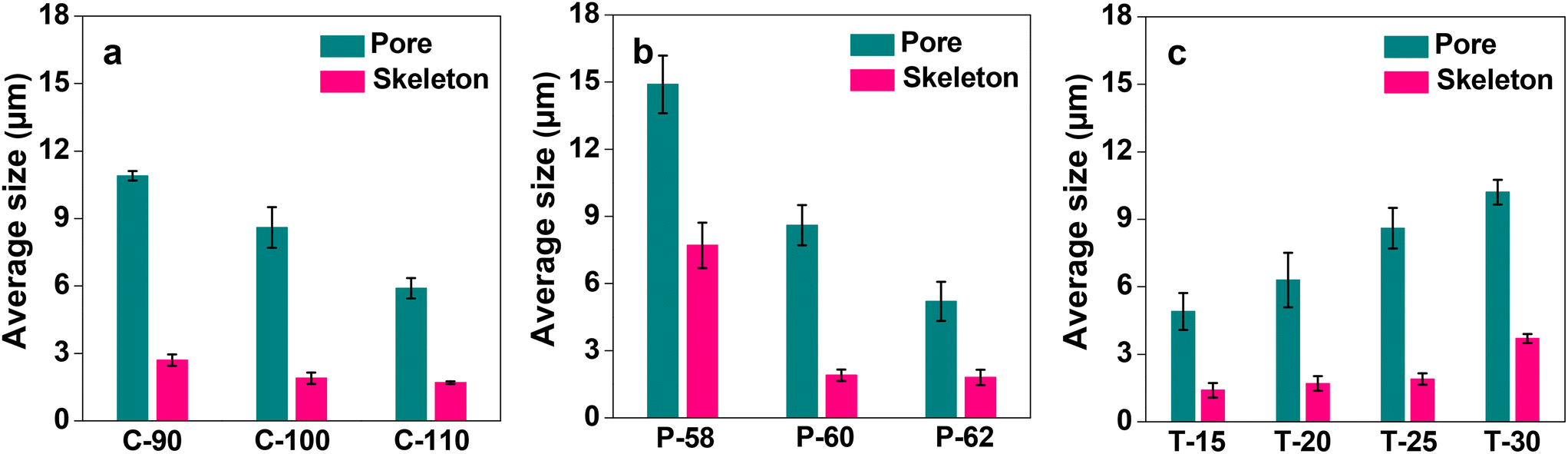 | ||
| Fig. 2 Effects of CA fabrication conditions on the average size of the pore and skeleton of the cellulose monolith: (a) CA concentration, (b) poor solvent ratio, and (c) aging temperature. Bars represent the standard error (n = 3). | ||
The microstructures of the EVOH and PMMA monolith (100 mg mL−1) were also studied by SEM. The images are shown in Fig. 3. It is evident from the images that the EVOH monolith consisted of through-pores of dimensions in the range of several tens of micrometers (Fig. 3a). A magnified view given in the inset of Fig. 3a suggests the existence of nanometer sized holes on the surface with voids inside the skeleton. The PMMA monolith, on the other hand, was composed of through-pores of nanometer-sized dimensions (Fig. 3b) and had a smooth skeletal morphology. The through-pore diameters for EVOH and PMMA monoliths were calculated to be 20.4 and 0.9 μm, and the skeletal diameters were found to be 11.1 and 0.3 μm, respectively. The variations of the average through-pore and skeleton size using the three polymers, namely cellulose (C-100), EVOH and PMMA, at a concentration of 100 mg mL−1 are shown in Fig. 4.
 | ||
| Fig. 3 SEM images of (a) EVOH monolith (inset: surface of the skeleton) and (b) PMMA monolith. | ||
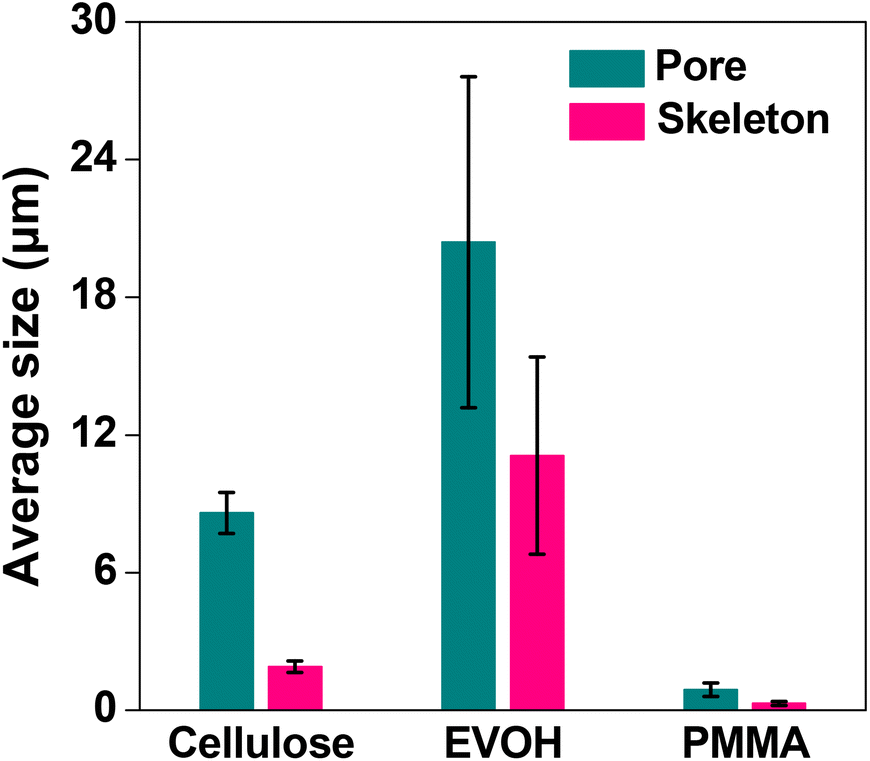 | ||
| Fig. 4 Effect of the average through-pore and skeleton size of the monoliths with different polymers. Bars represent the standard error (n = 3). | ||
Column flow experiments were carried out with the different monoliths and the permeability coefficients were measured. For this, water was passed through the monolith columns prepared and the pressure loss was measured. The generated back pressure was proportional to the linear velocity for all the columns within the range of the flow velocity used for the experiments. The effects of flow velocity on the back pressure of the cellulose monolith under different fabrication conditions are shown in Fig. 5. The permeability coefficients were measured using Darcy's equation (eqn (i)) and it is observed that the permeability coefficient tends to increase with a lower CA concentration, lower antisolvent ratio and higher cooling temperature. The effect of monolith fabrication conditions on the permeability is shown in Fig. S2.† From these results, the dependence of the permeability coefficient on the through-pore sizes, which varies with the fabrication conditions, is plotted in Fig. 6. It is evident that as the through-pore size increased, the permeability coefficient also increased. It is obvious that the larger the through-pore, the easier it was for water to flow through them. However, P-58 shows no correlation as it has a large skeleton diameter, as depicted in Fig. 1, which reduced the skeleton-water resistance and the number of through pores per cross-sectional area. For plots other than P-58, the coefficient of regression, R2 is 0.981 and the relationship is quite good with some variation. Since the permeability coefficient in a particle-filled column was inversely proportional to the square of the particle diameter, it can be considered that a similar relationship holds with respect to the through-pore diameter. The through-pore diameters have been calculated from the permeability coefficient based on Kozeny–Carman's equation (eqn (ii)).50 The plot of flow through-pore diameters calculated from Kozeny–Carman's equation versus that obtained from the SEM imaging studies is shown in Fig. S3.† From the result, it is evident that the through-pore diameter calculated from the permeability coefficient and the observed through-pore diameter had a good proportional relationship and if the result corresponding to P-58 is excluded, R2 = 0.991.
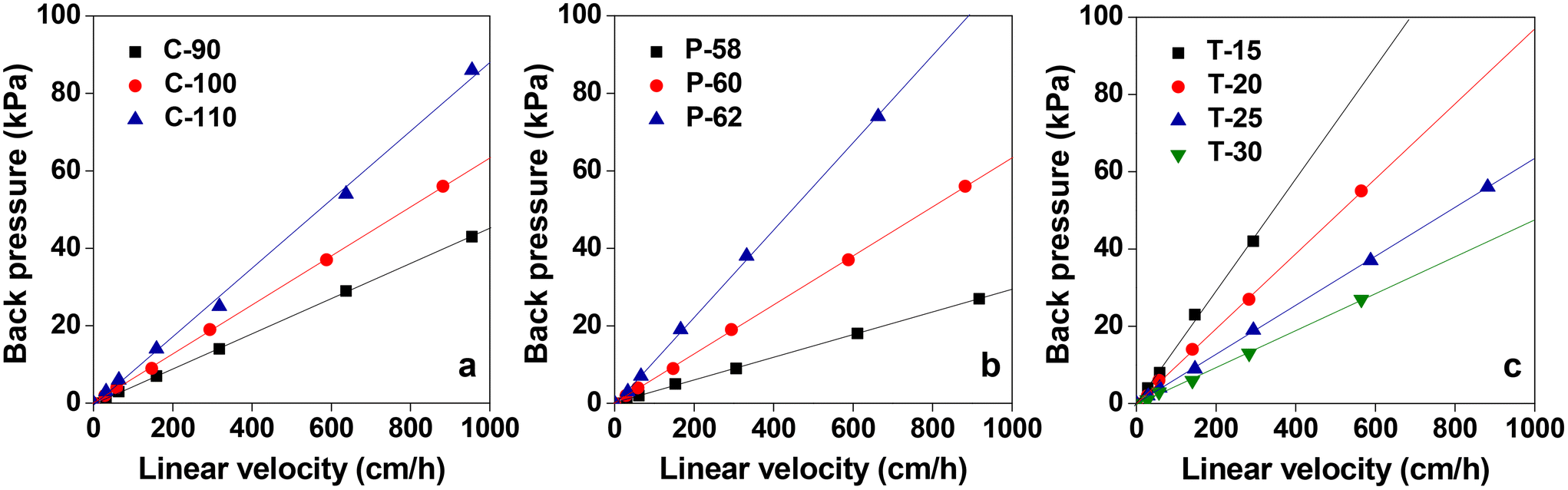 | ||
| Fig. 5 Effects of the flow velocity on the back pressure of the cellulose monolith with different fabrication conditions: (a) CA concentration, (b) poor solvent ratio and (c) aging temperature. | ||
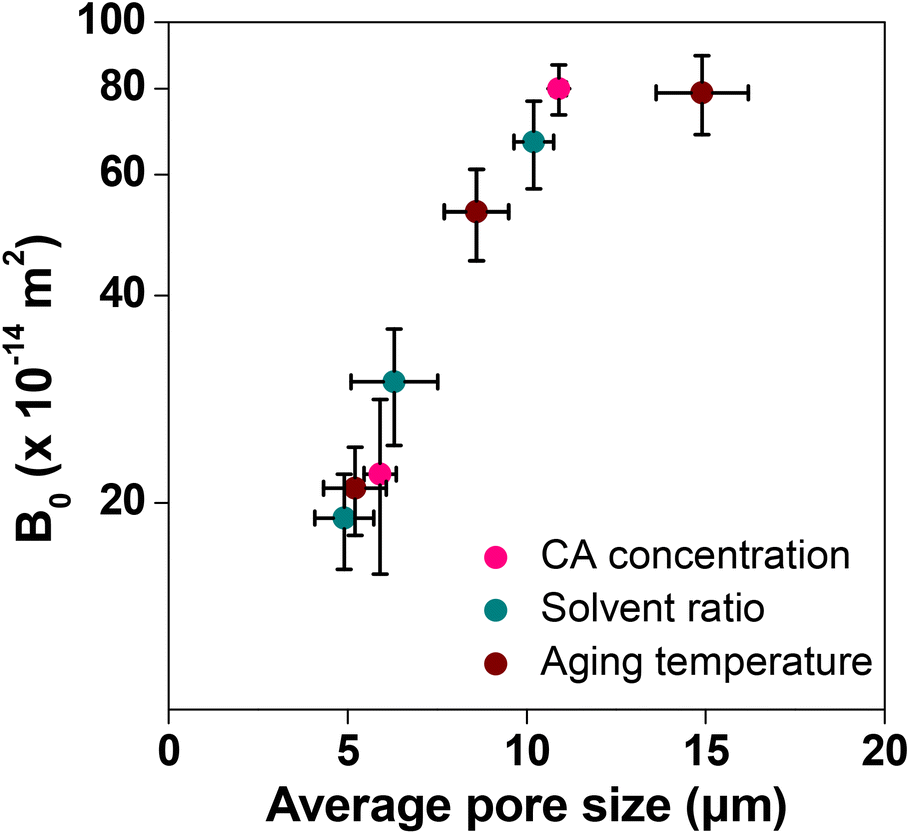 | ||
| Fig. 6 Effect of the through-pore size on the permeability of the cellulose monolith. Bars represent the standard error (n = 9). | ||
The through-pore size, skeleton size and permeability of the different samples are given in Table 2. The mismatch between the observed through-pore values from SEM images and the calculated through-pore values from the flow experiment took place because the SEM studies were carried out in the dry state which differs under the actual flow of water. Cellulose is hydrophilic, retains water well and swells. Therefore, it is expected that the through-pores will become narrow when water flows through the column. Apart from that, the high affinity between water and cellulose, the possibility of substantial narrowing of the flow path, assumptions taken in the method of measuring the average pore size, etc. also affect the results. Only P-58 does not show a relationship between the through-pore diameter and the permeability coefficient, though from the SEM image (Fig. 1) it is evident that it has a structure with voids inside the skeleton. Otherwise, the permeability coefficient is very good for the prepared monoliths and it can be anticipated that affinity monolith columns with improved adsorption performance can be designed using these samples. The permeability coefficients of EVOH and PMMA monoliths were also determined. They were in the order of 10−12 m2 and 10−15 m2, respectively. The effect of the concentration of the various polymers was compared (Fig. 7) and it is observed that the permeability coefficient did not change on the order at concentrations between 90 and 110 mg mL−1. The permeability coefficient of PMMA indicated that it is difficult for fluid to pass through it, whereas EVOH reveals excellent results in terms of the permeability coefficient.
 | ||
| Fig. 7 Effect of the polymer concentration on the permeability of CA, EVOH and PMMA. Bars represent the standard derivation (n = 3). | ||
| Sample | Through-pore size (μm) | Skeleton size (μm) | Permeability (B0) (×10−13 m2) |
|---|---|---|---|
| C-90 | 10.9 ± 0.21 | 2.67 ± 0.25 | 8.04 ± 0.67 |
| C-100 | 8.63 ± 0.90 | 1.93 ± 0.25 | 5.26 ± 0.80 |
| C-110 | 5.87 ± 0.45 | 1.73 ± 0.06 | 2.22 ± 0.63 |
| P-58 | 14.9 ± 1.29 | 7.67 ± 1.01 | 7.90 ± 1.04 |
| P-62 | 5.20 ± 0.88 | 1.75 ± 0.34 | 2.14 ± 0.31 |
| T-15 | 4.90 ± 0.82 | 1.43 ± 0.32 | 1.94 ± 0.30 |
| T-20 | 6.30 ± 1.21 | 1.73 ± 0.32 | 2.97 ± 0.58 |
| T-30 | 10.2 ± 0.55 | 3.70 ± 0.20 | 6.70 ± 0.98 |
In order to compare the adsorption in the batch method for the cellulose, EVOH and PMMA films, the fluorescence intensity of the films was observed after treating them with FITC-BSA solution to differentiate analyte adsorption from non-specific adsorption. Assuming the fluorescence intensity ratio of the cellulose film by the batch method as 1.0, the intensities for EVOH and PMMA were found to be 1.3 and 1.7, respectively (Fig. 8). Also in the flow method, PMMA exhibits the highest magnitude of non-specific adsorption, followed by EVOH and cellulose has the lowest non-specific adsorption (Fig. 9). Thus, although EVOH is found to exhibit an excellent permeability coefficient, it was unsuitable as a column material because of its extreme brittleness. EVOH is a crystalline polymer and thus the monoliths were sometimes damaged during solvent substitution due to collision between the crystallites. Therefore, it can be concluded that cellulose is an excellent column material in terms of all three qualities of strength, liquid permeability and suppression of non-specific adsorption. Thus, it is the most suitable material to be used as an affinity chromatography carrier.
 | ||
| Fig. 8 Fluorescence microscopic observations of (a) cellulose, (b) EVOH and (c) PMMA films after immersion in FITC-BSA solution and subsequent rinsing with PBS. | ||
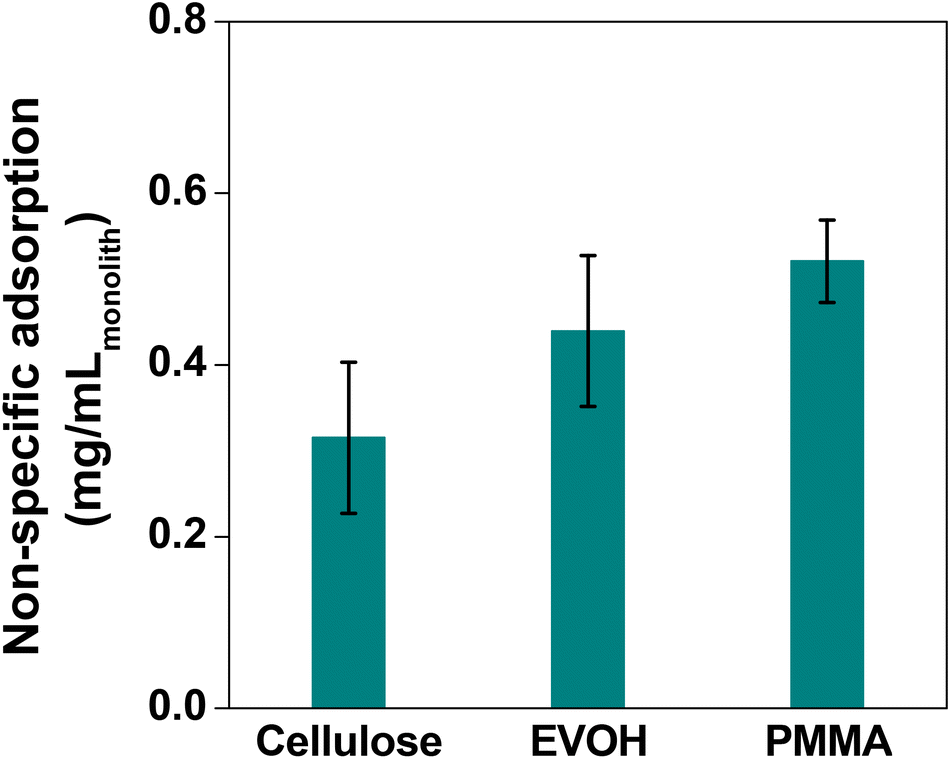 | ||
| Fig. 9 Result of the non-specific adsorption experiment with different polymer monoliths using the flow method. Bars represent the standard derivation (n = 3). | ||
Once it is established that the cellulose monolith is the most suitable column carrier for affinity chromatography, it is activated by reaction with DSC. Carrier activation and immobilization techniques are important to decide the adsorption efficiency of ligands and targets. If the immobilization amount is insufficient, the adsorption amount will decrease. On the other hand, higher ligand densities lead to lower yields and increased non-specific adsorption, and hence it is important to optimize the ligand densities.51–53 Here DSC was chosen as the activating agent for the immobilization of the NHS ester (Scheme 1) as it readily reacts with the less nucleophilic hydroxy groups of cellulose in the presence of DMAP. Due to the ease of activation, stability in water during immobilization operations and excellent reactivity with proteins, the ligand was immobilized by DSC activation.18,54 The reaction solvent as well as the charging ratio of DSC was varied and the amount of active carbonate ester introduced into the cellulose monolith was determined using the P-60 monolith as the representative sample. The results are shown in Fig. 10. Although the amount of NHS ester introduced in the monolith varies depending on the solvent, the difference is not very significant (Fig. 10a). Thus, considering the fact that the reaction product has to be washed with dehydrated ACN after its completion, ACN has been chosen as the reaction solvent for DSC activation. When the reaction was complete, the residual catalyst and the solvent were removed by washing once with ACN and drying, and finally before preparing the column, the monolith was washed with a buffer solution for more effective removal of the residual catalyst and the solvent. On the other hand, the amount of active ester introduced in the cellulose monolith was improved by increasing the amount of DSC charged in the reactions (Fig. 10b).
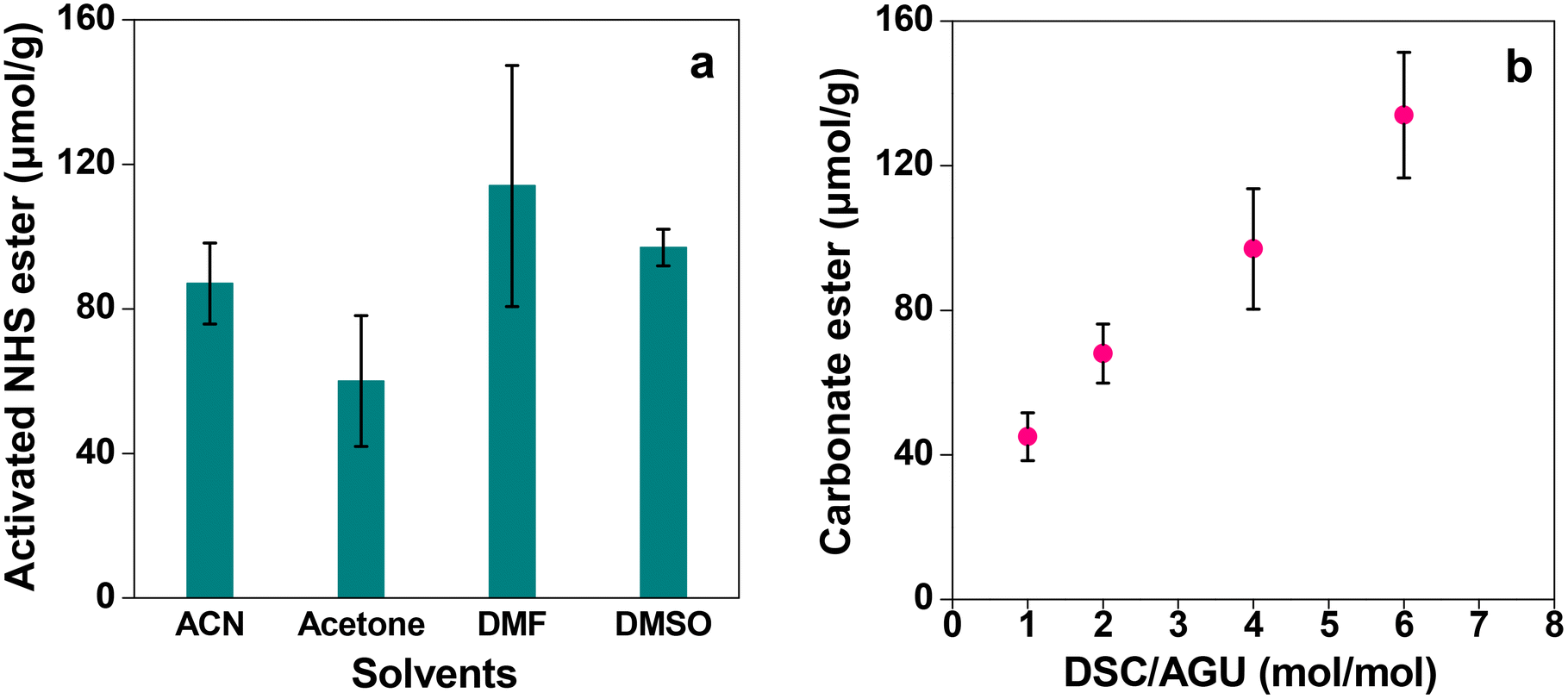 | ||
| Fig. 10 Results of the NHS ester introduced in the P-60 monolith with (a) different solvents and (b) different equivalents of DSC. Bars represent the standard error (n = 3). | ||
Finally, Protein A was immobilized onto the DSC activated monolith columns to prepare the affinity columns. Protein A is a 42 kDa protein derived from Staphylococcus aureus and composed of five IgG-adsorbing domains (E, D, A, B, C from the N-terminus).55 One domain specifically adsorbs the Fc region, which is a part of the constant region of the antibody. Each domain has three helices, one of which is considered to be contributed by hydrophobic interactions and the others by electrostatic interactions.56 With a binding dissociation constant of 10−8 M, Protein A is most widely used in antibody production because of its relatively strong affinity, rapid bond dissociation at low pH and high reusability. The affinity columns prepared by DSC activation and Protein A immobilization are used for the adsorption of antibodies, that is Immunoglobulins (Igs). Igs are proteins that are instrumental in the initial immune response in each organism's immunity. There are five classes of these antibodies, namely, IgA, IgD, IgE, IgM and IgG. Among them, 150 kDa IgG is used in ELISA, antibody drugs, etc. because it is highly reactive, more stable, has a small size and is easy to purify with Protein A.57
The affinity columns were prepared by varying the charging ratio of DSC, pH of immobilization, concentration of Protein A and ratio of the antisolvent and used for IgG antibody separation. The IgG adsorption experiments carried out over the affinity monolith columns revealed that varying the DSC charging ratio, immobilization pH and Protein A concentration has little effect on the chromatographs (Fig. 11a–c). On the other hand, when the antisolvent ratio was changed, the adsorption behavior of IgG changed only for P-58-A. Since adsorption occurs on the left side of the curve, it is considered that the adsorption amount increased only in the case of P-58-A. In Fig. 12, the chromatographs for the various monoliths obtained by changing the antisolvent ratio were compared with the commercially available agarose beads. The results of the calculation of the adsorption capacity of IgG from the breakthrough curves are given in Table 3. The bed volumes from the adsorption behavior of agarose beads and P-58-A to the leakage of IgG and saturation of adsorption are similar; however, the adsorption capacity of P-58-A is approximately twice as large as that of the commercially procured agarose beads. Thus, it can be observed that both the adsorption capacity and the adsorption efficiency were improved with the activated cellulose monoliths.
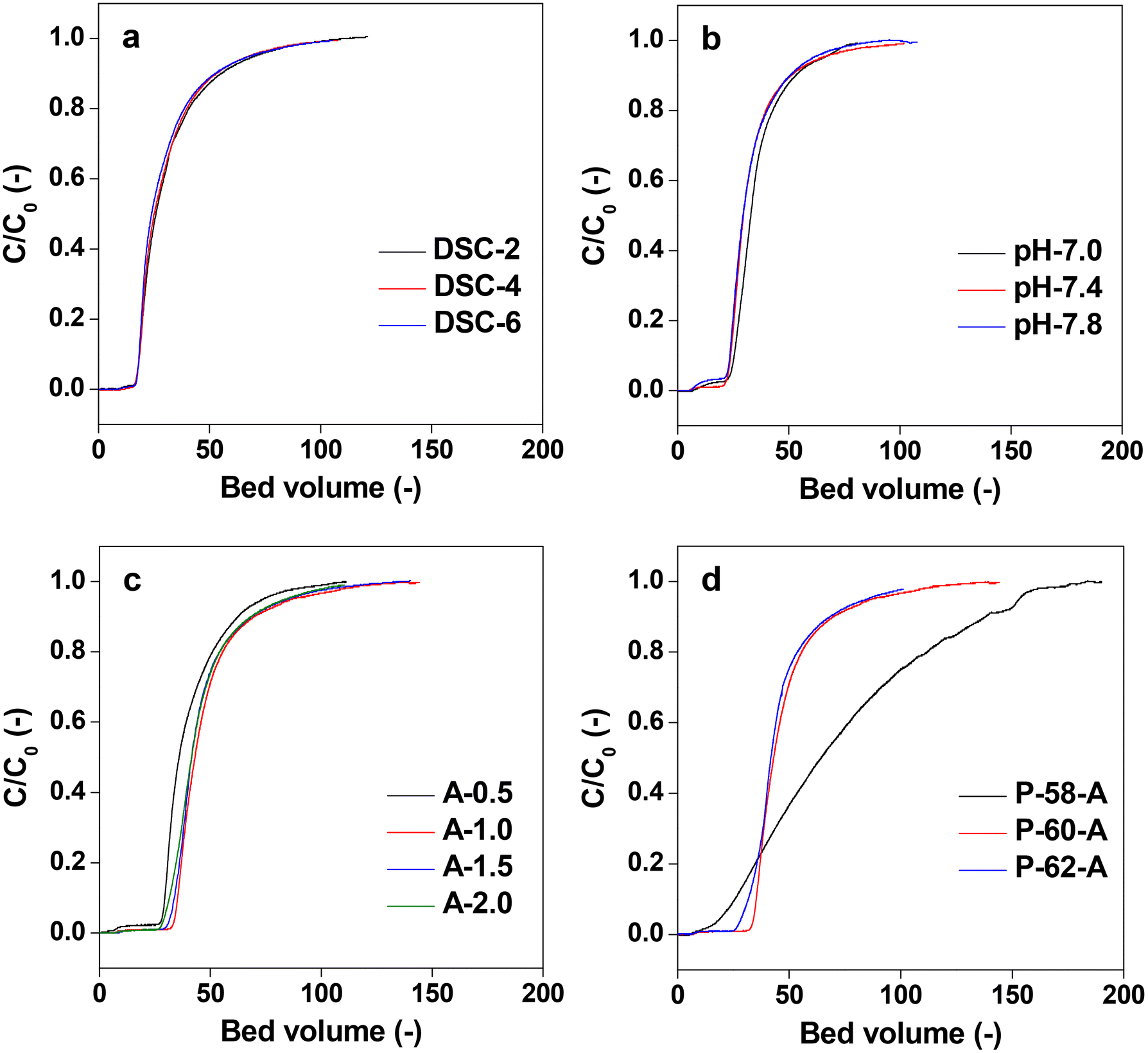 | ||
| Fig. 11 Chromatographs of varying immobilization conditions: (a) DSC stoichiometry on the anhydrous glucose unit, (b) pH from 7.0 to 7.8, (c) protein A concentration in 3 mL, and (d) different poor solvent ratio from 58 to 62. | ||
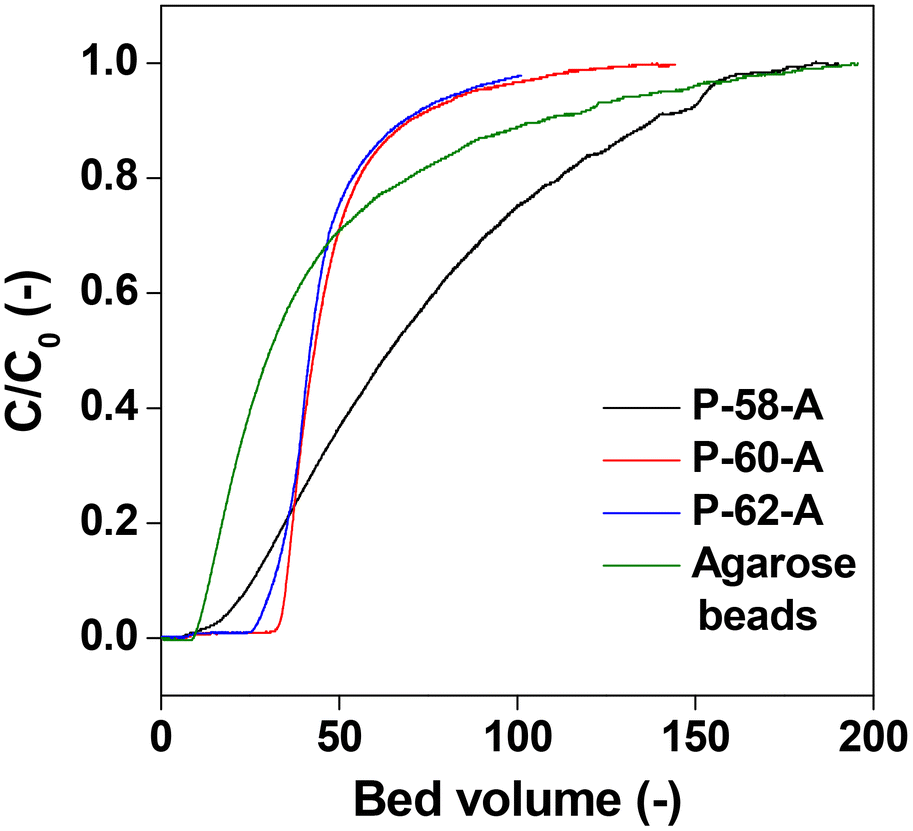 | ||
| Fig. 12 Chromatographs of Protein A immobilized commercial agarose gel beads and different poor solvent ratio monoliths. | ||
| Sample | Adsorption capacity (mg mLcolumn−1) |
|---|---|
| P-58-A | 18.3 |
| P-60-A | 7.3 |
| P-62-A | 8.2 |
| Agarose gel beads | 9.5 |
The results obtained are in line with the design guidelines for the monolith structure described earlier. It is evident that due to the presence of voids inside the skeleton, deeper unevenness of the surface and larger spacing, the surface area of the monoliths increases which facilitates the adsorption of the protein molecules. As illustrated in Fig. 13, a unique morphology of accumulated fine fibers was observed on the skeletal surface of the P-58-A monolith (Fig. 13a). This structure exhibited a deeper surface roughness compared to P-60-A or P-62-A (Fig. 13b and c) supporting that it contributed to the larger surface area in the P-58 monolith. The reason behind no significant difference in the adsorption behavior of the samples other than that for P-58-A is considered to be the high coverage of protein A on the adsorption surface area of the irregularities existing in the monoliths. The binding of protein A at multiple points of the cellulose monolith by the highly active carbonate esters reduces the degree of freedom of the affinity sites and reduces adsorption efficiency. The molecular weight of Protein A is about one-fourth of IgG and hence the bulky IgG cannot penetrate the micropores and get immobilized (Fig. 13d). Apart from that, the steric hindrance increases due to the adsorption of multiple Protein A and their interaction with adjacent Protein A or an IgG leading to a decrease in adsorption efficiency. On the other hand, in P-58-A, there was no difference in the amount of protein A immobilized per column volume and IgG that can be diffused and adsorbed. The coverage decreased due to the increase in the surface area available for adsorption (Fig. 13e); as a result, adsorption was possible more efficiently and the amount of adsorption increased.
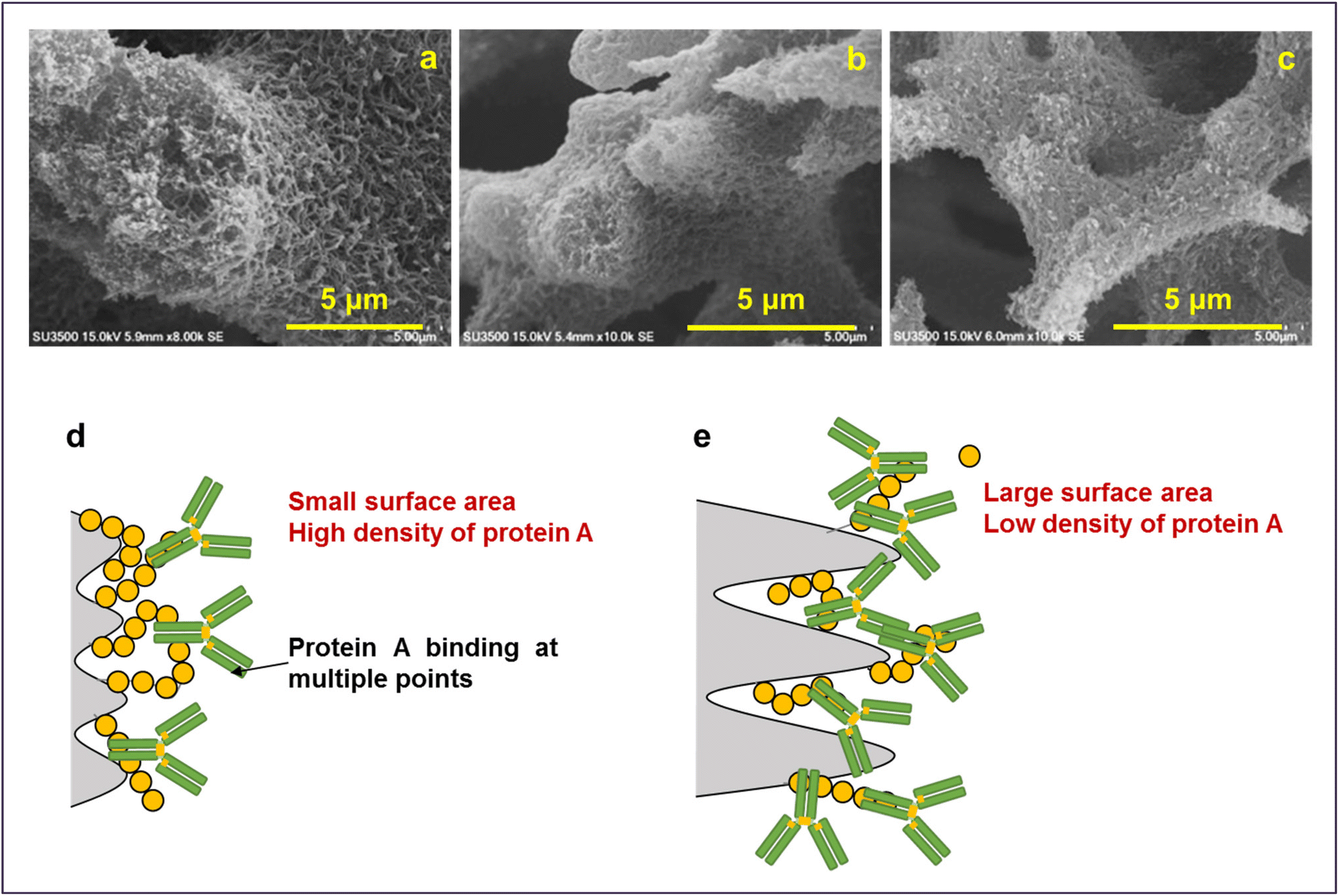 | ||
| Fig. 13 Magnified SEM images of (a) P-58, (b) P-60 and (c) P-62. Illustration of adsorption on (d) P-60-A and P-62-A affinity monolith columns and (e) P-58-A column. | ||
Fig. 14a shows the desorption behavior of IgG at different flow rates of elution with the acidic buffer (pH = 3.0) when it was passed through the P-58-A column after adsorption saturation. At all flow rates, similar desorption behavior of IgG was observed and almost all IgG was collected within 7 mL of elution, indicating that the recovery time can be shortened at high flow rates. It is attributed to the rapid diffusion through the monolith column having a high permeability coefficient with high flow velocity. Such results cannot be obtained with a particle-filled column with low strength and limited flow velocity or a monolith column with a relatively low permeability coefficient. Thus, it can be concluded that the affinity monolith column produced in this study has high performance. The recyclability of an affinity column is an important parameter to determine its practical utility. Furthermore, the recyclability was studied for the protein A immobilized column for five consecutive cycles. For this the column was subjected to the first cycle of adsorption of IgG, and the separation efficiency was determined and assigned to be 100%. Then the adsorbed IgG is eluted from the used column with buffer solution and washed thoroughly to obtain the affinity column ready for use in the next cycle. The separation efficiency for the five consecutive cycles is shown in Fig. 14b. It is evident from the plot that the separation efficiency is marginally affected after recycling the column five times. In addition to this, the mass loss from the column is also insignificant after five cycles of operation.
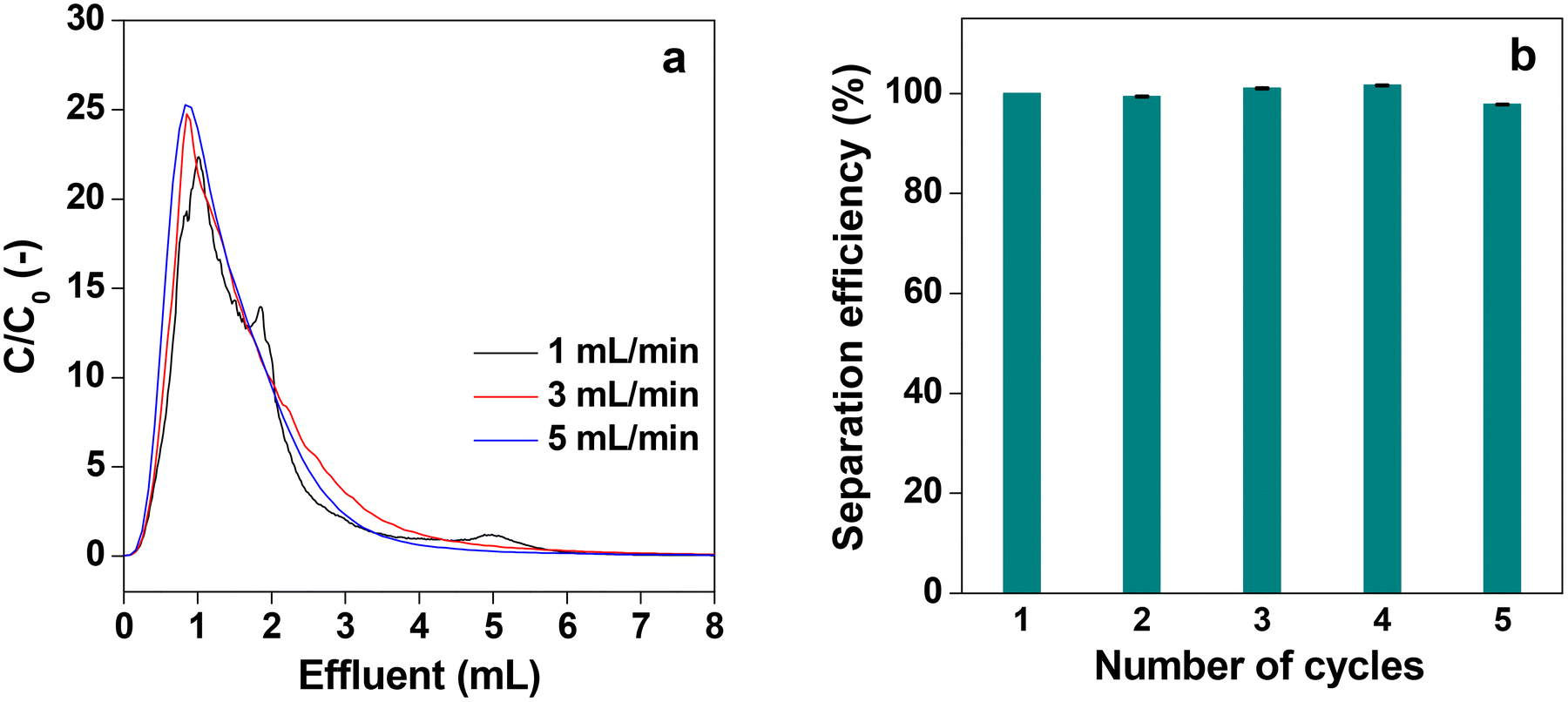 | ||
| Fig. 14 (a) Chromatographs of elution behaviors of adsorbed IgG on the P-58-A column at different flow rates and (b) recyclability of the column for five cycles. Bars represent the standard error (n = 4). | ||
Conclusions
CA monoliths were prepared and cellulose monoliths were obtained by their deacetylation. SEM observations confirm the presence of micrometer-order through-pores and fibrous skeletons in the monoliths and the morphology depended on the CA concentration, antisolvent ratio and cooling temperature. The through-pore and skeleton diameter tended to be larger when the CA concentration and antisolvent ratio were lower and the cooling temperature was higher. The permeability coefficient of the cellulose monoliths obtained from Darcy's formula shows that it changed with the morphology and increased as the through-pores increased. According to the Kozeny–Carman equation, the permeability coefficient was proportional to the square of the through-pore diameter; the value of the permeability coefficient with respect to the through-pore diameter of the cellulose monoliths prepared here is in good agreement with this. Thus, it is possible to control the through-pore, skeleton diameter and transmission coefficient by varying the monolith fabrication conditions. The non-specific adsorption experiment reveals that in the cellulose material such adsorption is small both in the batch method and the flow method. This indicates that cellulose is an excellent carrier material for affinity chromatography in terms of strength, liquid permeability and suppression of non-specific adsorption. The IgG adsorption behavior has very little dependence on the amount of DSC charged, pH and protein A concentration in the immobilization process. The small surface area and excessive Protein A closely immobilized on the cellulose monolith cannot adsorb IgG effectively due to steric effects and the adsorption capacity was limited by the amount of immobilization per surface area as well as the coverage in immobilization and adsorption. On the other hand, the cellulose monolith prepared by changing the antisolvent ratio gives an affinity monolith column with high liquid permeability and adsorption performance. It is superior both in terms of adsorption efficiency and adsorption capacity compared to commercially available products, and can withstand the use of high flow velocities and is recyclable.Conflicts of interest
There are no conflict to declare.Acknowledgements
H. U. thankfully acknowledges financial support from JSPS KAKENHI Grants (23H02024) and JST Grant Number JPMJPF2218. M. N. gratefully acknowledges financial support from the CSIR, New Delhi (Sanction Letter No. 01(3088)/21/EMR-II).References
- P. Cuatrecasas, M. Wilchek and C. B. Anfinsen, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 636–643 CrossRef CAS PubMed.
- S. Magdeldin and A. Moser, Affinity Chromatography: Principles and Applications, IntechOpen, London, 2012 Search PubMed.
- D. S. Hage, Clin. Chem., 1999, 45, 593–615 CrossRef CAS.
- M. Azarkan, J. Huet, D. B. Volant, Y. Looze and G. Vandenbussche, J. Chromatogr. B, 2007, 849, 81–90 CrossRef CAS PubMed.
- S. Arora, V. Saxena and B. V. Ayyar, Methods, 2017, 116, 84–94 CrossRef CAS PubMed.
- W. W. Zhao, F. F. Liu, Q. H. Shi, X. Y. Dong and Y. Sun, Biochem. Eng. J., 2014, 88, 1–11 CrossRef CAS.
- L. N. Lund, P.-E. Gustavsson, R. Michaela, J. Lindgren, L. N. Lauritsen, M. Lund, G. Houen, A. Staby and P. M. St. Hilaire, J. Chromatogr. A, 2012, 1225, 158–167 CrossRef CAS PubMed.
- X. Zhang, Y. Duan and X. Zeng, ACS Omega, 2017, 2, 1731–1737 CrossRef CAS PubMed.
- H. Zheng, C. Wang, T. R. Pavase and C. Xue, Colloids Surf., B, 2022, 217, 112705 CrossRef CAS PubMed.
- J. Lee, C. Y. Chuah, W. S. Tan, J. Song and T.-H. Bae, Chem. Eng. J., 2022, 427, 130883 CrossRef CAS.
- S. B. Park, T. Fujimoto, E. Mizohata, T. Inoue, M. H. Sung and H. Uyama, J. Microbiol. Biotechnol., 2013, 23, 942–952 CrossRef CAS PubMed.
- L. Zverina, M. Pinelo, J. M. Woodley and A. E. Daugaard, J. Chem. Technol. Biotechnol., 2021, 96, 2488–2495 CrossRef CAS.
- Z.-T. Xie, T.-A. Asoh and H. Uyama, Carbohydr. Polym., 2019, 214, 195–203 CrossRef CAS PubMed.
- M. Nandi and H. Uyama, RSC Adv., 2014, 4, 20847–20855 RSC.
- M. Nandi, K. Okada and H. Uyama, Funct. Mater. Lett., 2011, 4, 407–410 CrossRef CAS.
- A. Dobashi, J. Maruyama, Y. Shen, M. Nandi and H. Uyama, Carbohydr. Polym., 2018, 200, 381–390 CrossRef CAS PubMed.
- Q. Bai, Q. Xiong, C. Li, Y. Shen and H. Uyama, ACS Sustainable Chem. Eng., 2017, 5, 9390–9401 CrossRef CAS.
- R. Mallik and D. S. Hage, J. Sep. Sci., 2006, 29, 1686–1704 CrossRef CAS PubMed.
- H. Si, Q. Wang, Y. Guo, Y. Zhao, H. Li, S. Li, S. Wang and B. Zhu, Front. Chem., 2022, 10, 951649 CrossRef CAS PubMed.
- H. Kobayashi, K. Okada, S. Tokuda, E. Kanao, Y. Masuda, T. Naito, H. Takaya, M. Yan, T. Kubo and K. Otsuka, Sci. Rep., 2020, 10, 13850 CrossRef CAS PubMed.
- M. B. Espina-Benitez, J. Randon, C. Demesmay and V. Dugas, J. Chromatogr. A, 2019, 1597, 209–213 CrossRef CAS PubMed.
- Y. Mao, R. Fan, R. Li, X. Ye and U. Kulozik, Electrophoresis, 2021, 42, 2599–2614 CrossRef CAS PubMed.
- S. Mizuno, T.-A. Asoh, Y. Takashima, A. Harada and H. Uyama, Polym. Degrad. Stab., 2019, 160, 136–141 CrossRef CAS.
- R. N. Widmer, G. I. Lampronti, B. Kunz, C. Battaglia, J. H. Shepherd, S. A. T. Redfern and T. D. Bennett, ACS Appl. Nano Mater., 2018, 1, 497–500 CrossRef CAS.
- B. Liberelle, E. J. Dil, F. Sabri, B. D. Favis, G. D. Crescenzo and N. Virgilio, ACS Appl. Polym. Mater., 2021, 3, 6359–6365 CrossRef CAS.
- F. Maya and B. Paull, J. Sep. Sci., 2019, 42, 1564–1576 CrossRef CAS PubMed.
- K. Li, S. Wang, S. Koskela and Q. Zhou, Adv. Mater. Interfaces, 2021, 8, 2100787 CrossRef CAS.
- S. Rajesh, C. Crandall, S. Schneiderman and T. J. Menkhaus, ACS Appl. Nano Mater., 2018, 1, 3321–3330 CrossRef CAS.
- K.-F. Du, M. Yan, Q.-Y. Wang and H. Song, J. Chromatogr. A, 2010, 1217, 1298–1304 CrossRef CAS PubMed.
- X. Luo and L. Zhang, J. Chromatogr. A, 2010, 1217, 5922–5929 CrossRef CAS PubMed.
- D.-M. Wang, G. Hao, Q.-H. Shi and Y. Sun, J. Chromatogr. A, 2007, 1146, 32–40 CrossRef CAS PubMed.
- T. Yao, J. Song, Y. Gan, L. Qiao and K. Du, J. Chromatogr. A, 2022, 1677, 463297 CrossRef CAS PubMed.
- Q. Bai, Q. Xiong, C. Li, Y. Shen and H. Uyama, Cellulose, 2017, 24, 4275–4289 CrossRef CAS.
- X. Sun, G. Sun and X. Wang, Polymer, 2017, 108, 432–441 CrossRef CAS.
- C. Acquah, C. K. S. Moy, M. K. Danquah and C. M. Ongkudon, J. Chromatogr. B, 2016, 1015–1016, 121–134 CrossRef CAS PubMed.
- G. Wang and H. Uyama, Colloid Polym. Sci., 2015, 293, 2429–2435 CrossRef CAS.
- W. Han, M. Yamauchi, U. Hasegawa, M. Noda, K. Fukui, A. J. Vlies, S. Uchiyama and H. Uyama, J. Biosci. Bioeng., 2015, 119, 505–510 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, W. Zhang, Y. Hsu, T. Asoh, B. Qi and H. Uyama, ACS Biomater. Sci. Eng., 2022, 8, 2676–2683 CrossRef CAS PubMed.
- N. Roopsung, A. Sugawara, Y. Hsu, T. Asoh and H. Uyama, ACS Appl. Polym. Mater., 2023, 5, 4488–4497 CrossRef CAS.
- K. B. Lynch, J. Ren, M. A. Beckner, C. He and S. Liu, Anal. Chim. Acta, 2019, 1046, 48–68 CrossRef CAS PubMed.
- N. Kruljec and T. Bratkovič, Bioconjugate Chem., 2017, 28, 2009–2030 CrossRef CAS PubMed.
- M. Park, BioChip J., 2019, 13, 82–94 CrossRef CAS.
- Y. K. Baghbaderani, R. Allgayer, S. P. Schwaminger, P. F. García and S. Berensmeier, ACS Appl. Nano Mater., 2021, 4, 4956–4963 CrossRef.
- W. Wang, Q. Zhao, M. Luo, M. Li, D. Wang, Y. Wang and Q. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20046–20052 CrossRef CAS PubMed.
- S. R. Nugen, P. J. Asiello, J. T. Connelly and A. J. Baeumner, Biosens. Bioelectron., 2009, 24, 2428–2433 CrossRef CAS PubMed.
- L. Huang, H. Ye, T. Yu, X. Zhang, Y. Zhang, L. Zhao, Q. Xin, S. Wang, X. Ding and H. Li, J. Appl. Polym. Sci., 2018, 135, 46374 CrossRef.
- T. Shun, A. Sugawara, T.-A. Asoh, M. Nandi and H. Uyama, Mater. Adv., 2022, 3, 5138–5150 RSC.
- S. Yoneda, W. Han, U. Hasegawa and H. Uyama, Polymer, 2014, 55, 3212–3216 CrossRef CAS.
- H. Uyama, Kobunshi Ronbunshu, 2010, 67, 489–496 CrossRef CAS.
- R. Skudas, B. A. Grimes, M. Thommes and K. K. Unger, J. Chromatogr. A, 2009, 1216, 2625–2636 CrossRef CAS PubMed.
- A. Marechal, F. Jarrosson, J. Randon, V. Dugas and C. Demesmay, J. Chromatogr. A, 2015, 1406, 109–117 CrossRef CAS PubMed.
- W.-W. Zhao, Q.-H. Shi and Y. Sun, J. Chromatogr. A, 2014, 1355, 107–114 CrossRef CAS PubMed.
- H. S. Kim, R. Mallik and D. S. Hage, J. Chromatogr. B, 2006, 837, 138–146 CrossRef CAS PubMed.
- M. Morpurgo, E. A. Bayer and M. Wilchek, J. Biochem. Biophys. Methods, 1999, 38, 17–28 CrossRef CAS PubMed.
- K. L. Atkins, J. D. Burman, E. S. Chamberlain, J. E. Cooper, B. Poutrel, S. Bagby, A. T. A. Jenkins, E. J. Feil and J. M. H. van den Elsen, Mol. Immunol., 2008, 45, 1600–1611 CrossRef CAS PubMed.
- B. Huang, F. F. Liu, X. Y. Dong and Y. Sun, J. Phys. Chem. B, 2011, 115, 4168–4176 CrossRef CAS PubMed.
- B. V. Ayyar, S. Arora, C. Murphy and R. O'Kennedy, Methods, 2012, 56, 116–129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00041a |
| This journal is © The Royal Society of Chemistry 2023 |