
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-cell patterning and characterisation of antibiotic persistent bacteria using bio-sCAPA†
Cameron
Boggon
*a,
Srikanth
Mairpady Shambat
 b,
Annelies S.
Zinkernagel
b,
Eleonora
Secchi
*c and
Lucio
Isa
*a
b,
Annelies S.
Zinkernagel
b,
Eleonora
Secchi
*c and
Lucio
Isa
*a
aLaboratory for Soft Materials and Interfaces, Department of Materials, ETH Zürich, Switzerland. E-mail: lucio.isa@mat.ethz.ch
bDepartment of Infectious Diseases and Hospital Epidemiology, University Hospital Zürich, University of Zurich, Switzerland
cInstitute of Environmental Engineering, Department of Civil, Environmental, and Geomatic Engineering, ETH Zürich, Switzerland. E-mail: secchi@ifu.baug.ethz.ch
First published on 16th October 2023
Abstract
In microbiology, accessing single-cell information within large populations is pivotal. Here we introduce bio-sCAPA, a technique for patterning bacterial cells in defined geometric arrangements and monitoring their growth in various nutrient environments. We demonstrate bio-sCAPA with a study of subpopulations of antibiotic-tolerant bacteria, known as persister cells, which can survive exposure to high doses of antibiotics despite lacking any genetic resistance to the drug. Persister cells are associated with chronic and relapsing infections, yet are difficult to study due in part to a lack of scalable, single-cell characterisation methods. As >105 cells can be patterned on each template, and multiple templates can be patterned in parallel, bio-sCAPA allows for very rare population phenotypes to be monitored with single-cell precision across various environmental conditions. Using bio-sCAPA, we analysed the phenotypic characteristics of single Staphylococcus aureus cells tolerant to flucloxacillin and rifampicin killing. We find that antibiotic-tolerant S. aureus cells do not display significant heterogeneity in growth rate and are instead characterised by prolonged lag-time phenotypes alone.
Introduction
Tracking the fate of individual bacterial cells in response to various environments is critical for studying microbial communities. Even in clonal bacterial populations, it is well established that individual cells display heterogeneous phenotypes. Some common examples include sporulation,1 antibiotic persistence2 or division of labour,3,4 and there is a growing body of literature attempting to understand the role of phenotypic heterogeneity in microbial ecology.5–7In order to study such heterogeneity, techniques for tracking large numbers of individual cells in response to varying environmental stimuli are required and a number of approaches have been previously proposed. Droplet microfluidics has been used to encapsulate single bacteria into droplets combined with defined quantities of certain chemicals or effectors, including antibiotics.8,9 While having a very high throughput, one limitation of this approach is that once the droplets are formed it is difficult to exchange the chemical environment within the droplet. ‘Mother machines’ have been widely used to monitor single-cell doubling-times in response to fluctuating environmental conditions10–14 and ‘family machines’ have been used to study the interaction distance between different species in crowded communities.15,16 While these devices have been highly successful in many applications, they offer limited control to investigate the interaction between individual organisms and require enclosed microfluidic devices, thus restricting analysis of growth to liquid culture and hampering access to individual cells of interest during or after the experiment for further analysis.
The latter obstacle can be overcome by investigating growth on solid culture media, e.g. by spotting cells on agar pads17–19 or employing a more complex centrifugation-based approach to isolate cells into specific agarose wells.20 However, the limited control in cell positioning and the close proximity of many cells impose the requirements of using high magnifications to segment individual cells, thus practically restricting the size of populations that can be monitored over time. Nonetheless, recent progress in the development of single-cell transcriptomics indicates that extremely rare population phenotypes could be common in bacterial communities.21,22 Therefore, monitoring and accessing large bacterial populations exposed to dynamically changing environments is essential for many highly relevant phenomena.
To address these challenges, in this work we demonstrate a method for patterning >105 cells in tailored geometrical configurations with single-cell precision to allow us to monitor very rare phenotypes in a bacterial population. This method is based upon sequential capillarity-assisted particle assembly (sCAPA), a technology originally designed to fabricate complex patterns of colloidal particles for a wide range of applications23 such as plasmonics24 and active matter.25–27 Capillary forces have previously been applied for the patterning of live yeast cells and fungal spores for imaging by atomic force microscopy28–32 and, more recently, a proof-of-principle application of sCAPA to model Escherichia coli strains has been demonstrated.33 The technique presented here, termed bio-sCAPA, hinges upon a key advancement: the utilization of agar pads to which the cells are exposed after patterning via a microfluidic device. The pad allows for robust cell growth and stable time-lapses over the course of up to several days. Bio-sCAPA presents a flexible and easily parallelisable platform for multiple bacterial isolates that can be exposed to time-varying stimuli by exchanging agar pads. Direct comparisons with standardized plate assays can be easily made, pads can be removed from the template to allow cells of interest to be accessed during or after the experiment, and, by taking time-lapses and using a custom image analysis script, we can measure live and dead cells, cell fluorescence intensity, growth rate, and lag-times over time on individual bacterial cells.
As a demonstration of bio-sCAPA, in this work we focus on antibiotic persistence in Staphylococcus aureus, a common cause of both hospital and community-acquired infections.34,35S. aureus isolated directly from patient-derived samples display significant colony size heterogeneity when grown on blood-agar plates.36–38 Considerable work has been performed on stable small colony variants (SCV), which have a size significantly smaller than the average colony size on the plate and are formed by bacterial cells that have developed genetic adaptations during the course of infection.37–40 However, the majority of small colonies are non-stable and revert to a normal colony size upon subculturing.36,41 It was recently demonstrated that non-stable small colonies (nsSC) are associated with prolonged lag-time and constitute a subpopulation of persister cells.42 However, it remains unclear whether lag-time heterogeneity is the only phenotypic marker of persistence. Indirect evidence from E. coli indicates that persister cells may also be characterised by slow growth rate,43,44 small cell size11 or may derive specifically from stressed and filamenting cells,45,46 suggesting that varying mechanisms may be involved in persistence, which result in varying phenotypes.47,48
We used bio-sCAPA to examine persister phenotypes in S. aureus to verify if delayed lag-time is the primary marker of persistence and gives rise to non-stable small colonies or whether growth-rate heterogeneity is also a critical factor. We challenged S. aureus with the bactericidal antibiotics flucloxacillin and rifampicin, which are commonly used to treat patients with staphylococcal infections. We showed that nutrient starvation is a simple model to stress bacteria and demonstrated that prior nutrient stress can radically alter the antibiotic tolerance profile of S. aureus. Analysing recovery after antibiotic challenge, we demonstrate that tolerant cells are characterised by prolonged lag-time alone as we observe no other phenotypic markers that correlate with antibiotic tolerance. This indicates that monitoring colony formation on agar plates to identify the presence of non-stable small colonies can be used to faithfully identify antibiotic tolerant S. aureus which, in future, could be used in clinical diagnostics to help better inform treatment strategies for chronic bacterial infections.
Materials and methods
Fabricating PDMS chips for bio-sCAPA
Two separate methods were employed for preparing the template molds for bio-sCAPA. The first method employed microcontact printing on 4-inch silicon wafers prepared by soft lithography, as detailed in ref. 27 and 49. These templates contained features, which we refer to as ‘traps’, that were 3 × 1 μm wide and 0.5 μm tall. Each template contained an array of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 traps with 10 μm spacing between traps. Before use, the silicon wafer templates were plasma treated (Zepto Plasma Unit, Diener electronic GmbH) in air for 30 s and then silanised with trichloro(1H,1H,2H,2H-perfluorooctyl) silane via vapour deposition to make the surface non-adhesive. These templates were used for all antibiotic exposure experiments.
000 traps with 10 μm spacing between traps. Before use, the silicon wafer templates were plasma treated (Zepto Plasma Unit, Diener electronic GmbH) in air for 30 s and then silanised with trichloro(1H,1H,2H,2H-perfluorooctyl) silane via vapour deposition to make the surface non-adhesive. These templates were used for all antibiotic exposure experiments.
The second fabrication method used to prepare the templates in Fig. 5 employed a high-resolution 3D printer (NanoScribe GmbH). Trap arrays were printed using commercial photo-resist (IP-dip, NanoScribe GmbH) onto fused silica glass substrates. After printing, the substrates were developed in PGMEA for 20 minutes, washed with isopropanol and cured overnight under UV. The templates were then silanised as above.
Molds for the chip roofs were 3D printed using a Prusa SL1 3D printer. After printing, the moulds were left overnight under a UV lamp and then left in the oven at 100 °C overnight to remove any non-cured resin before being silanised as above. Chip dimensions are illustrated in Fig. S1.†
Bio-sCAPA chips were prepared using polydimethylsiloxane (PDMS; Sylgard 184 silicone elastomer kit, Dow Corning, Midland, MI), mixed, and degassed in a 10:1 ratio of polymer:crosslinker using a Thinky ARE-250. For the chip roofs, 20 g of PDMS was poured onto the 3D printed mold, wrapped with aluminium to form a box around the mold. For the template, 3 g of PDMS were spin-coated onto the silica wafer mold at 500 rpm for 5 s and at 800 rpm for 10 s to obtain a layer 400 μm thick. After curing overnight at 75 °C, 1 mm diameter holes were punched into the PDMS roofs. The PDMS chips were assembled by binding the roofs to the templates using microscope spacers (Grace Bio-labs SecureSeal) designed in a cutting plotter (Silhouette Cameo 4), making it very simple to remove the roofs from the templates after deposition in order to add agar pads.
Bacteria strains
All assays performed in this work used a methicillin-resistant strain S. aureus USA300 JE2 containing a pCN56 plasmid with the GFP gene under the control of the blaZ promoter. For all experiments performed, frozen stocks kept at −80 °C were plated on tryptone soy broth (TSB) agar plates containing 10 μg ml−1 erythromycin. Liquid cultures were prepared by inoculating single colonies from plates in 50 ml falcon tubes containing 5 ml TSB and 10 μg ml−1 erythromycin. Cultures were placed in an incubator (Edmund Buehler TH30 Incubation Hood), with lids loosely closed, at 37 °C with oval shaking at 220 rpm and grown overnight.Bio-sCAPA nutrient stress and antibiotic assays
Liquid bacteria cultures 1-day, 2-day or 6-days old were washed, resuspended in TSB, and their optical density (OD) measured at 600 nm using a Vernier Go Direct SpectroVis Plus spectrometer. A bacteria suspension of OD 0.8 containing 0.1 μg ml−1 Tween20 (critical micelle concentration = 0.05 μg ml−1) was then prepared, and the PDMS chips were filled with ∼30 μl of the suspension using a pipette. The chip was then placed on a heat plate at 30 °C and left for around 1.5 hours to allow the droplet to evaporate.After deposition was complete, the bacteria-patterned templates were then cut out using a sterilised razor blade and placed into a 6-well plate. Each template was cut in half, with one half used as a control and the other used for the antibiotic exposure assay. TSB agar pads were prepared with 3 wt% agar (slightly stiffer agar was easier to handle than the conventional 1.5 wt%), 10 μg ml−1 erythromycin and 2 μg ml−1 propidium iodide, and were placed over the patterned PDMS template. For the 40× MIC antibiotic exposures, pads additionally contained either 10 μg ml−1 flucloxacillin (MIC = 0.25 μg ml−1) or 0.32 μg ml−1 rifampicin (MIC = 0.008 μg ml−1). After antibiotic exposures of a specific time, the antibiotic pad was switched with a fresh nutrient agar pad, as used in the controls. For each experiment reported, either 3 or 4 biological repeats were performed (see ESI† Fig. S6 for breakdown by experiment).
Time-lapses were taken using a Nikon Eclipse TI2-E inverted microscope with an incubator box maintained at 37 °C throughout the experiment. Up to 30 positions per sample were imaged every 20 minutes with a 20× magnification Plan Fluor objective in phase contrast, green fluorescence (470 nm excitation wavelength) to detect GFP signal and red fluorescence (555 nm excitation wavelength) to detect propidium iodide positive cells. We found the main limiting factor to the total number of cells we could image in an experiment was determined by the autofocus speed of the microscope, which limited the number of fields of view we could image.
To control for whether antibiotic-resistant mutants had formed during the experiment, 3 colonies were picked from the template at random at the end of the experiment using an inoculation loop. These were placed in a 50 ml falcon tube containing 5 ml TSB, 10 μg ml−1 erythromycin, and 40× MIC of the antibiotic and placed in the incubator overnight. If we saw growth in these tubes within 24 hours of incubation, the experiment was ignored as we could not rule out antibiotic resistance development. This was particularly crucial for rifampicin experiments as S. aureus is known to require only a single point mutation to develop resistance.50
CFU plating antibiotic assay
After washing and OD measurement of overnight liquid cultures, 2 × 5 ml cultures per biological replicate were prepared with fresh TSB media, 1 × 105 CFU ml−1S. aureus and 10 μg ml−1 erythromycin in 50 ml falcon tubes. One tube was used as control, in the second tube was added 40× MIC antibiotic concentration as above. Both tubes were then placed in an incubator at 37 °C and 220 rpm oval shaking. For each experiment, 4 biological replicates were prepared.At the same time, a small aliquot of the control was serially diluted, with 2 dilutions, and plated on TSB-agar plates to determine the CFU counts at the start of the experiment.
After a defined period of antibiotic exposure, an aliquot of the antibiotic cultures was washed 3× in PBS and serially plated on TSB-agar plates, as above, to determine CFU counts after antibiotic exposure. The rest of the antibiotic cultures were kept in the incubator for at least another 24 hours to ensure no resistant mutants emerged.
Image analysis
A custom MATLAB script was written to perform the image analysis. The analysis pipeline is summarised here.Fluorescence images were first background-subtracted on ImageJ using a Gaussian blurred image of the fluorescence profile taken on the day of the experiment, before being imported into MATLAB. Particle localisation was performed based on the Crocker and Grier algorithm51 to identify the centroid of all GFP-positive cells.
To identify growing bacteria, all background-subtracted images in the time-lapse were binarised using a fixed brightness threshold. The brightness threshold was defined such that single cells were not identified in the binarised image but were identified when they began growing as the colony became brighter. Circles were fitted to the growing bright ‘blobs’, which constituted growing colonies, and used to define the colony area and radius.
Linking colonies through time was performed as follows: 1) all images in the time-lapse were first registered based on the first frame in the timelapse to ensure all bacteria positions were in the same coordinate system. For this, we performed intensity-based image registration on the phase contrast images, which were taken concomitantly with the fluorescence images. 2) For the time update, all colony centroids and radii were identified in frame tn and tn+1 and a colony track was updated if a colony in tn+1 overlapped with one colony in tn. If no colonies in tn overlapped with those in tn+1, a new colony was defined (i.e. a new cell has started growing). If two colonies in tn overlapped with a colony in tn+1, the colonies merged. In this case, the tracking update was attributed to the larger of the two colonies, and the small colony was no longer updated.
A growing colony was used in the analysis only if it fulfilled the following criteria: 1) the colony radius increases with time, 2) the colony must be tracked for at least 5 consecutive frames, 3) the colony must not grow out of the field of view, 4) the colony must grow to a radius of at least 6.5 μm. For colonies that fulfilled these criteria, the colony lag-time was defined as the time point when the colony radius is >6.5 μm.
Results
Patterning bacterial cells with bio-sCAPA
Bio-sCAPA allows for the patterning of >105 single bacterial cells on a polydimethylsiloxane (PDMS) surface in controlled geometrical configurations with single-cell resolution. After the patterning, the location of growing cells, their growth rate, and lag time can be quantified over time. To perform the patterning, a droplet of bacterial culture is pipetted into an enclosed PDMS chip containing a template designed with an array of ‘traps’ approximately the same size as the bacteria to be patterned (see Fig. S1† for chip design). By gently heating the template from the bottom at 30 °C, the droplet begins to evaporate, generating an internal, radial flow inside the droplet, leading to what is commonly known as the coffee stain, or coffee ring, effect.52 Here, the asymmetric curvature of the droplet meniscus inside the PDMS chip leads to a higher evaporation rate at the edges of the droplet compared to the middle, which generates a flow towards the droplet edge that leads to the accumulation of bacteria at the meniscus. As the droplet evaporates, it recedes over the template with a receding contact angle between ∼30 to 60°, the bacterial cells get caught between the back wall of the trap and the moving meniscus, and they are left deposited in the trap after the meniscus passes over it (Fig. 1a and S1†). Due to the strength of the capillary forces integral to this method and the size selectivity of the traps, we can deposit single cells from bacterial populations that are highly prone to forming aggregates, such as the characteristic ‘grape-like’ structures of S. aureus.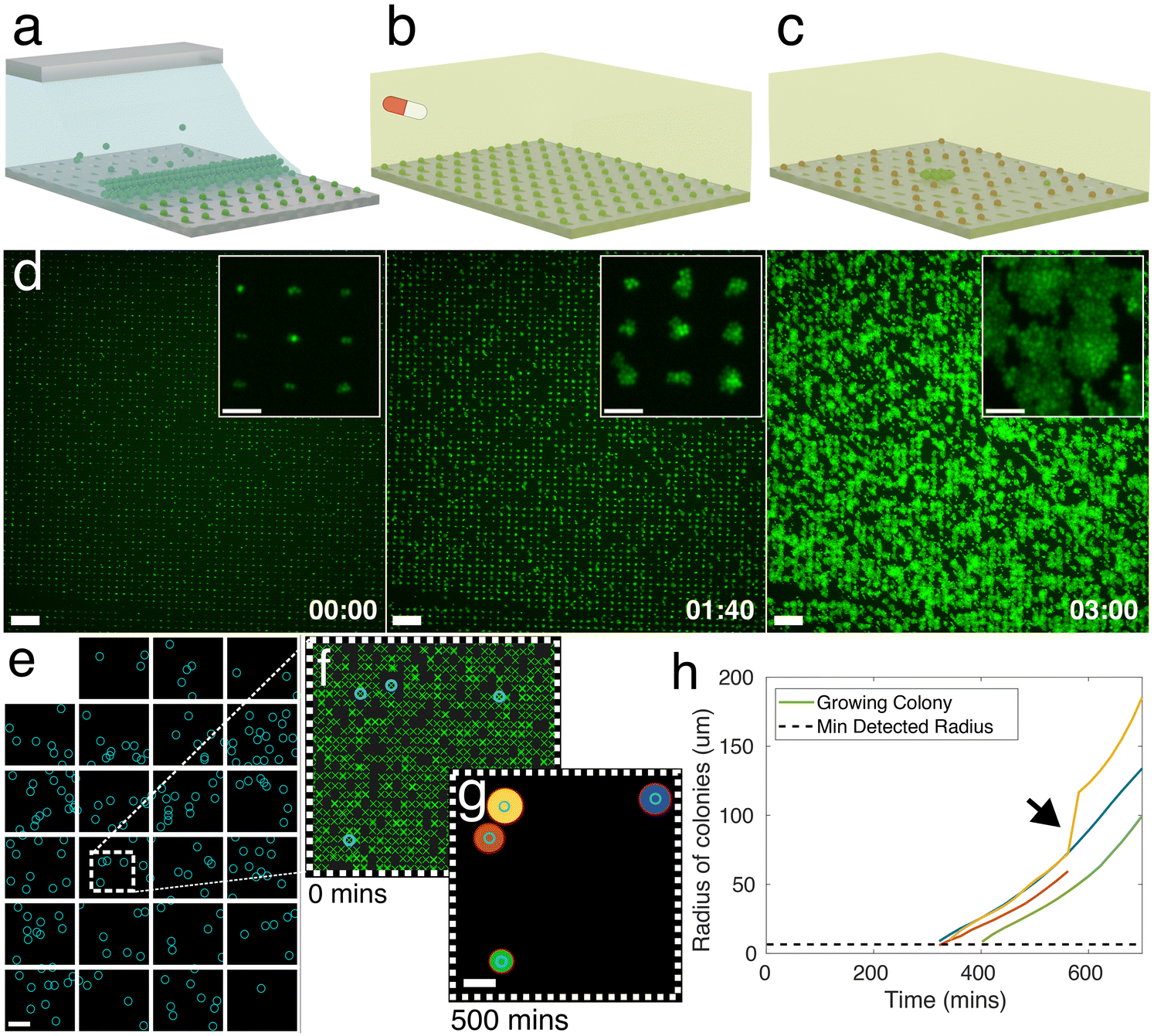 | ||
| Fig. 1 Bio-sCAPA allows for single-cell patterning and characterisation of hundreds of thousands of bacteria. a) Graphical illustration of capillarity-assisted deposition of bacteria into micron-sized traps made of polydimethylsiloxane (PDMS). b) After deposition, nutrient agar containing an antibiotic is placed on top of the patterned template, and a time-lapse is taken to observe bacterial growth. c) After a defined time, the antibiotic agar is removed and replaced with a nutrient agar pad so that surviving cells will grow and can be characterised. d) Green fluorescence images of an overnight culture of Staphylococcus aureus strain JE2 patterned and growing in 3 × 1 μm traps. Timestamp in hours: minutes. Scale bar = 50 μm, inset scale bar = 10 μm. e) Stitched image of all microscope fields of view for one culture of S. aureus JE2 during a 5 hour, 40× MIC flucloxacillin challenge. Microscope fields of view represented by white boxes. Blue circles represent the location of cells that survive and grow after exchanging the flucloxacillin pad. Dead cells are not shown. The total number of bacteria imaged for this culture = 73619, with 3 biological replicates typically measured per experiment. Scale bar = 200 μm. Dashed white box shown in f). f) Bacteria localisation. Green crosses indicate the location of all S. aureus cells at t = 0. Blue-circled cells grow after removing the antibiotic. g) Binarised image of bacteria colonies growing on the template 500 min after exchanging the antibiotic agar pad with nutrient agar without antibiotics. Scale bar = 100 μm. Red circles indicate fit to colony. Colony colour corresponds to lines reported in h), a plot of the circle radius against time. Inset arrow indicates the point when yellow and red colonies merge. | ||
The key parameters needed to achieve high yields in sCAPA are discussed in detail in ref. 53. For bio-sCAPA, we used a trap depth of 0.5 μm, because individual S. aureus cells are approximately 0.8–1 μm in diameter, and 0.1 wt% of the surfactant Tween20 to lower the droplet contact angle to around 30°. We controlled the speed of the meniscus movement simply by evaporation at 30 °C. It typically takes ∼100 minutes for a 5 × 3 mm2 area of traps to be deposited (∼150000 traps with 10 μm spacing) where >90% of traps contain cells.
After patterning the bacteria with bio-sCAPA, we placed a nutrient agar pad over the PDMS template for the controls (see Materials and methods). For antibiotic exposure assays, we added 40× MIC of the antibiotic to the agar pads and, after antibiotic exposures of a specified duration, switched the pad to nutrient agar without antibiotics, allowing the surviving cells to re-establish growth (Fig. 1b and c). We monitored growth from the single-cell using time-lapse image microscopy (Fig. 1d) where, by stitching together multiple fields of view, we monitored the full patterned area of S. aureus cells, containing >104–105 cells depending on the experiment (Fig. 1e). To analyse cell growth, we used a custom image analysis script (see Materials and methods for details). We first identified the positions of all individual patterned cells at the start of the time-lapse following standard fluorescent particle localisation techniques (Fig. 1f).51 Growing cells formed radially-expanding colonies that were identified by binarising the fluorescence image and fitting circles to all growing colonies (Fig. 1g). This allowed us to identify the locations of growing individual cells compared to non-growing cells and measure their lag-times and growth rates over time (Fig. 1h).
Nutrient starvation as a simple model to stress S. aureus
Given the importance of prior stress, such as heat shock, low pH, nutrient depletion, and oxidative stress, in altering the antibiotic tolerance profile of S. aureus,54–59 we characterized the signatures of stress in nutrient-starved S. aureus cultures both at the population and single-cell level. Stress response at the population level was analyzed using liquid culture and CFU counting assays, while the single-cell response was quantified with bio-sCAPA. The increased lag time in liquid culture and delayed colony appearance time in the plate assay for starved populations could be explained by direct observation of starvation-delayed growth onset combined with a higher fraction of dead cells using bio-sCAPA.We grew liquid cultures of S. aureus strain JE2 in tryptone soy broth (TSB) for 1, 2 or 6 days overnight. 1-day-old cultures correspond to leaving the cultures in the incubator for 16 hours, 2-day-old corresponds to 40 hours and 6-day-old, 136 hours. We performed optical density (OD) growth measurements in liquid culture by washing and diluting these cultures in fresh TSB and measuring how the time for growth to re-establish depended on the age of the culture. We observed a significant delay in lag-time between 1-day-old cultures compared to 2 and 6-day-old cultures (Fig. 2a). By fitting the OD curves to a Gompertz growth law,60,61 the lag was measured to be 217 ± 7, 283 ± 7 and 292 ± 7 minutes for 1, 2 and 6-day old cultures, respectively (see Fig. S2† for fitting details).
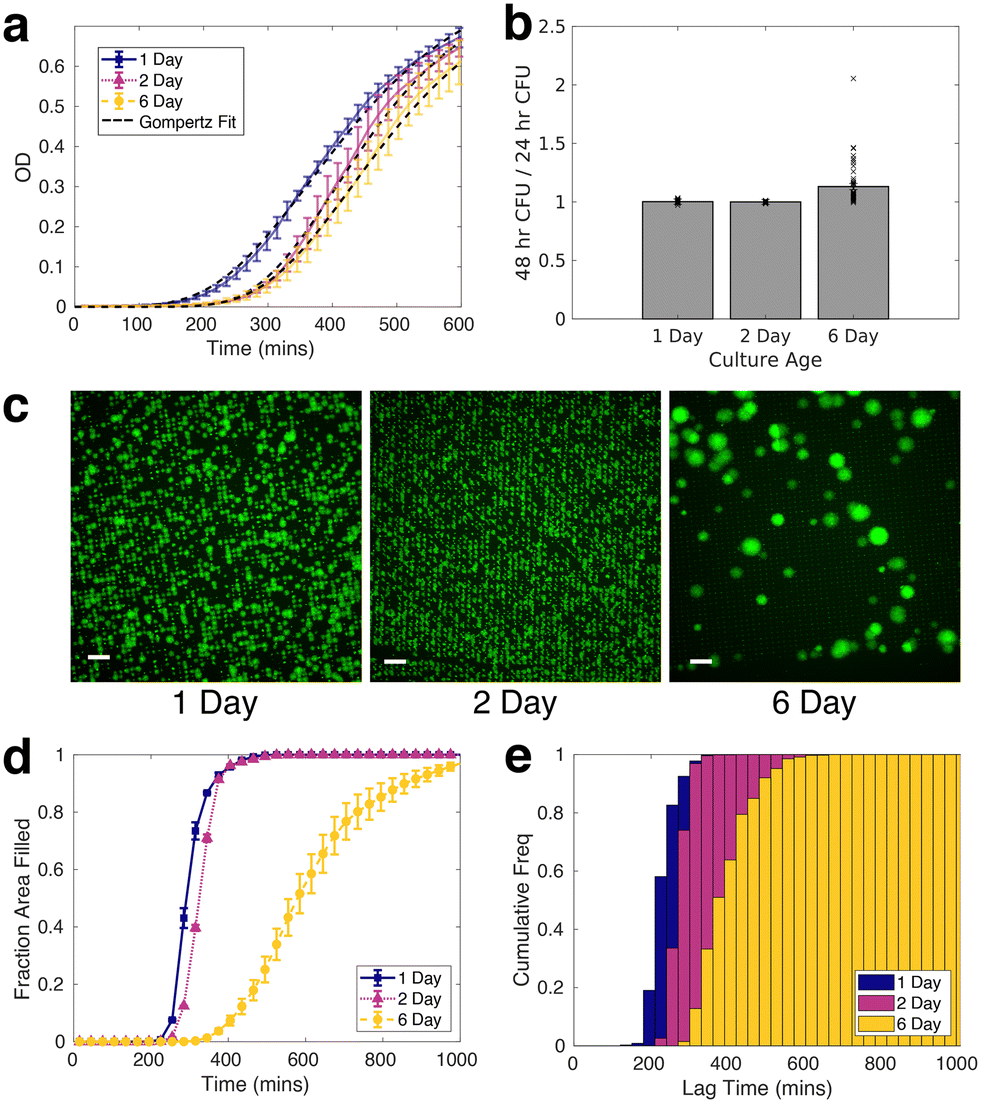 | ||
Fig. 2 Nutrient-stressed S. aureus is characterised by prolonged lag time. a) Optical density measurements of 1, 2, and 6-day overnight cultures. Coloured lines represent the mean of 8 biological replicates per nutrient starvation condition. Error bars represent standard error on the mean for 4 technical repeats. Black dashed lines represent fit to Gompertz's law. Lag-time parameter fitted are 217 ± 7, 283 ± 7, 292 ± 7 minutes for 1-day, 2-day and 6-day-old cultures, respectively. b) CFU analysis: ratio of colonies counted 48 hours after plating, to count at 24 hours after plating  for different culture age. A value of 1 corresponds to all colonies on the plate becoming visible within 24 hours after plating. c) Representative green fluorescence images of 1, 2 and 6-day-old cultures growing after sCAPA deposition. Scale bar = 50 μm. d) Area fraction of microscope field of view covered by growing cells. e) Bio-sCAPA analysis: cumulative lag-time distribution. Lag time is defined as when the bacterial colony has grown to a radius of 6.5 μm. Each distribution in d) and e) corresponds to pooled data of at least 4 independent biological replicates. for different culture age. A value of 1 corresponds to all colonies on the plate becoming visible within 24 hours after plating. c) Representative green fluorescence images of 1, 2 and 6-day-old cultures growing after sCAPA deposition. Scale bar = 50 μm. d) Area fraction of microscope field of view covered by growing cells. e) Bio-sCAPA analysis: cumulative lag-time distribution. Lag time is defined as when the bacterial colony has grown to a radius of 6.5 μm. Each distribution in d) and e) corresponds to pooled data of at least 4 independent biological replicates. | ||
On plating and counting colony-forming units (CFUs), we observed pronounced heterogeneity in colony appearance time after 6-days in the incubator, compared to those incubated for 1 and 2 days (Fig. 2b, see Fig. S3† for absolute CFU counts). This corresponded to visible colony size heterogeneity on the plate, similar to what has been documented for acid stress and oxidative stress on S. aureus.39,41,42,62,63 Whereas almost all colonies had grown to a visible size within 24 hours for 1 and 2-day-old cultures, many colonies only became visible after 24 hours in the case of 6-day-old cultures. The colonies counted 48 hours after initial plating were always smaller than the average colony size on the plate (Fig. S3e and f†).
To gain insight at the single-cell level, we observed different growth dynamics in the 6-day-old cultures compared to the 1 and 2-day-old cultures in samples prepared by bio-sCAPA (Fig. 2c). The colonies in the 6-day cultures were separated by a greater distance and appeared more heterogeneous in size than 1 and 2-day-old cultures. Using propidium iodide, a fluorescent reporter of membrane integrity commonly used as a death stain, we found a high fraction of dead cells in the 6-day cultures deposited with bio-sCAPA (see Fig. S4c and d† for propidium iodide data). One consequence of this is that the ‘effective distance’ between living cells is greater in the 6-day cultures compared to the 1 and 2-day cases, and we can track growing colonies for longer before they merge with other growing colonies. This can be seen in Fig. 2d, where we plot the area fraction of the microscope field of view filled by growing bacteria. Here we find that 6-day-old cultures take around 600 minutes longer to completely fill the microscope field of view than 1 and 2-day-old cultures.
We also observed that nutrient starvation delays growth onset, as shown by cumulative histograms of single-cell lag times. These are calculated by binning the number of cells that have begun to grow within a given time frame and normalising by the total number of cells ultimately found to grow in the whole sample (Fig. 2e). In the bio-sCAPA analysis, the lag time is defined as when the bacterial colony has grown to a radius of 6.5 μm, corresponding to ∼120 cells (i.e. approximately 7 doubling times). In Fig. 2e, almost all of the 1 and 2-day-old cultures had begun to grow by the time the first cells began growing in the 6-day-old cultures. Additionally, the shape of the cumulative histogram was much less steep in the 6-day-old culture, indicating greater lag-time heterogeneity. Characterisation at the single-cell level, therefore, indicates that the greater colony-size heterogeneity observed in the 6-day CFU plating (Fig. 2b) can be attributed to a combination of greater lag-time heterogeneity and delayed onset of growth of the entire population.
Nutrient stress increases S. aureus tolerance against flucloxacillin and rifampicin challenge
Nutrient stress increases the tolerance of S. aureus JE2 against a 40× MIC challenge of either flucloxacillin or rifampicin. These antibiotics were chosen because they are commonly used in the clinic to treat staphylococcal infections but both kill S. aureus via different functional mechanisms: flucloxacillin is a cell-wall-targeting beta-lactam antibiotic,64 while rifampicin targets RNA polymerase and shuts down RNA metabolism.65 The increase in tolerance for each starvation condition and antibiotic tested was quantified both at the population level with CFU counting and confirmed at the single-cell level using bio-sCAPA. For the CFU plating assays, the controls were plated immediately after removing cultures from the incubator and not exposed to antibiotics. The fraction of cells that survived the antibiotic challenge was calculated by comparing CFUs from the controls to CFUs enumerated after the antibiotic exposure. For bio-sCAPA, the fraction of tolerant cells was calculated by comparing the number of cells that grew in the bio-sCAPA controls and the number of cells that grew after exchanging the antibiotic agar pad with fresh nutrient agar. For all antibiotic assays performed with bio-sCAPA, we performed CFU-plating assays in parallel using the same biological replicates. The antibiotic assays are illustrated in Fig. 3a and detailed in the Materials and methods.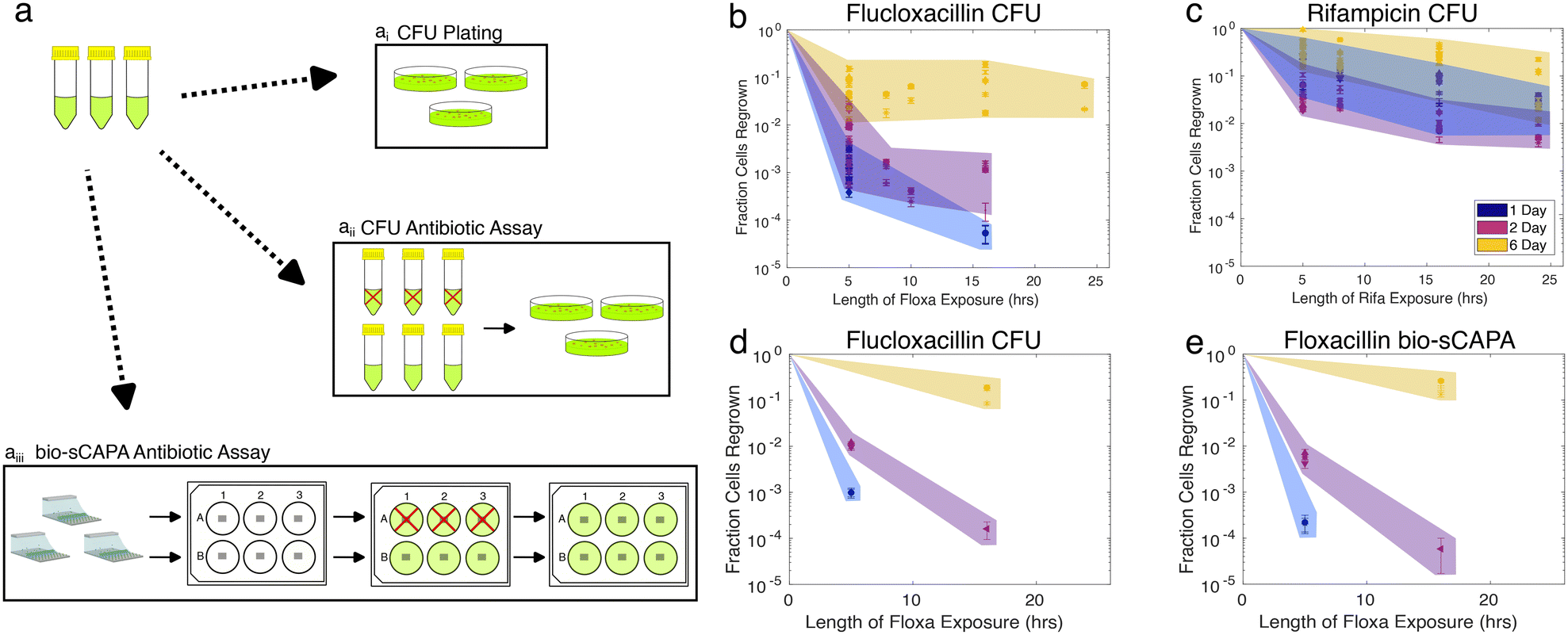 | ||
| Fig. 3 Nutrient stress increases S. aureus tolerance to flucloxacillin and rifampicin challenge. a) Graphical illustration of antibiotic tolerance assays performed in parallel. 1, 2, or 6-day overnight cultures washed, diluted, and ai) CFU plated, aii) exposed to 40× MIC antibiotic for a given antibiotic exposure time before washing and plating, aiii) deposited with bio-sCAPA, transferred to a 6-well plate, and exposed to an agar pad containing 40× MIC antibiotic before replacing it with a fresh agar pad after a given antibiotic exposure time. Red crosses indicate antibiotic exposure. b and c) S. aureus tolerance to 40× MIC flucloxacillin (b) or rifampicin (c) challenge, as measured by CFU plating. CFUs counted 48 hours after plating. d and e) Comparison of CFU assay and bio-sCAPA assays for flucloxacillin challenge. Data markers in d) and e) correspond to the same biological replicate. | ||
CFU plating revealed that nutrient stress increased tolerance to both flucloxacillin and rifampicin for any duration of the antibiotic exposure tested (Fig. 3b and c). This was very evident after flucloxacillin exposure, with 6-day-old cultures having 1000× greater tolerance compared to 1-day-old cultures. This effect was much less pronounced in the case of rifampicin, but this is, in part, due to the lower overall killing efficacy of rifampicin as compared to flucloxacillin in our assay. Though the fraction of cells killed tended to increase with longer antibiotic exposure time, the majority of cells were killed within 5 hours, as would be expected for logarithmic killing.
Fig. 3d and e show a direct comparison of measured tolerance fractions between CFU-plating and bio-sCAPA for flucloxacillin-challenged cultures (see Fig. S4a and b† for equivalent data on rifampicin). We found the tolerance profiles measured with the two methods to largely agree, confirming that the two methods can be reasonably compared and that bio-sCAPA captures the population-level behaviour we measured through CFU plating.
Prolonged lag-time rather than slow growth rate accounts for S. aureus tolerance
The single-cell characterisation performed with bio-sCAPA can explain the phenotypes driving the emergence of antibiotic tolerance and the differences in tolerance that we observed between flucloxacillin and rifampicin (Fig. 3b and c). In particular, we conclude that antibiotic-tolerant bacteria have significantly delayed lag times and growth rate heterogeneity does not play a significant role.First we analysed cell growth during antibiotic exposure using bio-sCAPA by counting the total number of bacterial cells and propidium-iodide-positive fluorescent cells during the antibiotic exposure (Fig. 4a and S5†). We found that rifampicin completely shut down all growth of S. aureus as no cells grew, in any nutrient-starved conditions, in the presence of rifampicin (green curve in Fig. 4a). This might be expected as rifampicin is an RNA-inhibitor. However, we observed no positive propidium iodide signal during antibiotic exposure either, despite the measured killing efficacy in Fig. 3c. As propidium iodide can only reach the cellular DNA if the cell membrane is compromised, this may indicate that rifampicin-challenged cells maintain a relatively intact cell membrane (see Fig. S5b and d† for propidium iodide data). This is in contrast to flucloxacillin, for which we observed cells beginning to grow even in the presence of the antibiotic, in line with the controls (blue curve in Fig. 4a). The number of cells reached a maximum 200 minutes after adding the antibiotic agar pad on the template and rapidly decreased afterwards. This was accompanied by an increase in propidium iodide positive cells, indicating that the cells were dead (Fig. S5b and c†).
 | ||
Fig. 4
S. aureus unstable small colony formation is driven by phenotypic heterogeneity in lag-time and not growth rate. a) Bio-sCAPA analysis: 1-day-old S. aureus JE2 culture growth during a 5-hour antibiotic exposure. Bars indicate the mean and standard error on the mean. b) CFU analysis: ratio of colonies counted 48 hours after plating against count at 24 hours after plating  . Cultures plated after 5-hour antibiotic exposure. A value of 1 corresponds to all colonies becoming visible within 24 hours after plating. c and d) Scatter plots of lag-time of cells that grow after a 5-hour flucloxacillin (c) or rifampicin (d) challenge. Horizontal black bars indicate mean and variance. Each condition represents the pool of at least 3 biological repeats. Black-dashed lines indicate time when 90% of the field of view is covered by growing colonies. p-Values represent 2-sample Kolmogorov–Smirnov tests comparing mean lag times between experiments. e) Representative curves demonstrating growth rate analysis. Straight lines (in red) fitted to the linear region in log radius vs. time plot. Inset arrow indicates the point when most colonies merge. The distribution of the gradient in the straight line fit is reported in f). f) Growth rate's mean and variance for all cultures growing after 5-hour antibiotic exposure. Each data point corresponds to the pool of at least 3 biological repeats. Unless marked with *, all p-values represent student t-tests comparing growth rate means between experiments. Starred p-values represent Welch's unequal variance t-test, which was performed against 6-day-old, rifampicin-challenged cultures. . Cultures plated after 5-hour antibiotic exposure. A value of 1 corresponds to all colonies becoming visible within 24 hours after plating. c and d) Scatter plots of lag-time of cells that grow after a 5-hour flucloxacillin (c) or rifampicin (d) challenge. Horizontal black bars indicate mean and variance. Each condition represents the pool of at least 3 biological repeats. Black-dashed lines indicate time when 90% of the field of view is covered by growing colonies. p-Values represent 2-sample Kolmogorov–Smirnov tests comparing mean lag times between experiments. e) Representative curves demonstrating growth rate analysis. Straight lines (in red) fitted to the linear region in log radius vs. time plot. Inset arrow indicates the point when most colonies merge. The distribution of the gradient in the straight line fit is reported in f). f) Growth rate's mean and variance for all cultures growing after 5-hour antibiotic exposure. Each data point corresponds to the pool of at least 3 biological repeats. Unless marked with *, all p-values represent student t-tests comparing growth rate means between experiments. Starred p-values represent Welch's unequal variance t-test, which was performed against 6-day-old, rifampicin-challenged cultures. | ||
Exploring how S. aureus recovered from antibiotic exposure, we initially screened lag-time at the population level via CFU plate counts and measured the number of visible colonies 24 and 48 hours after plating (Fig. 4b). For all antibiotic-challenged cultures, we observed an increase in the number of visible colonies 48 hours after plating compared to 24 hours. This effect was more pronounced in the 6-day-old cultures. However, this effect was also consistently much more pronounced in the case of rifampicin compared to flucloxacillin after both 5 hour and 16 hour antibiotic challenge (Fig. 4b and S3†).
With bio-sCAPA, we measured the distribution in single-cell lag-time of these tolerant bacterial cells and compared them against controls that had not been exposed to the antibiotics. In Fig. 4c and d, we plotted the lag-time distribution as scatter plots, where each point represents a growing cell. The corresponding grey scatter plots represent the control for each culture age. For 1 and 2-day-old flucloxacillin-challenged cultures, we observed a pronounced delay, compared to the controls, in when the first tolerant cells in the culture began to grow. For the 6-day-old cultures the time for earliest growth onset in the population was similar between the controls and flucloxacillin challenged cultures. For all flucloxacillin-challenged cultures, the earliest growth appeared to be constant at around 300 minutes regardless of nutrient stress. This indicated that flucloxacillin challenge delayed growth onset to some minimum lag-time of around 300 minutes.
For rifampicin-challenged cultures (Fig. 4d), we similarly saw a significantly delayed onset of first growth compared to the controls for all culture ages. The most prominent distinction we observed between the rifampicin and flucloxacillin was that the time for the first growth of tolerant bacterial cells was offset significantly in the rifampicin case compared to flucloxacillin. Times for first division were 339 ± 17, 288 ± 3 and 356 ± 8 minutes for 1, 2 and 6 day flucloxacillin challenged cultures, respectively. For rifampicin cultures, these were 428 ± 12, 458 ± 10, and 491 ± 6 minutes (see also Fig. S6c†). We, therefore, observed an offset of at least 90 minutes in the time for first growth after the rifampicin challenge compared to flucloxacillin for all nutrient-stressed cultures (see also Fig. S6†). This suggests that recovery from rifampicin stress takes longer than recovery from flucloxacillin stress and this is independent of how stressed the cultures are. This phenomenon, which amounts to essentially shifting the lag-time distribution by >1.5 hours, at least partly contributes to why we observed greater colony size heterogeneity on CFU plating of rifampicin-challenged cultures compared to flucloxacillin in Fig. 4b.
Turning to heterogeneity in lag-time, for all 5-hour-flucloxacillin-challenged cultures (Fig. 4c), we observed rare phenotypes with lag times of 1000 minutes (17 hours), regardless of nutrient conditions. For rifampicin-challenged cultures (Fig. 4d), we similarly saw increased lag-time heterogeneity compared to controls and we captured lag times of >1000 minutes (17 hours) for the 6-day-old cultures. However, interpreting lag-time heterogeneity in the 1 and 2-day-old cultures requires care. Though the lag-time variance in rifampicin-challenged 1 and 2-day cultures appears small (Fig. 4d blue and magenta data), this is driven by the fact that almost 2% of cells survived and grew after the rifampicin exposure. This made it difficult to capture extreme outliers in lag-time as the field of view was covered by cells that began growing earlier. This problem is less pronounced in the 6-day overnight cultures due to the previously discussed deposition of dead cells. We marked the time points where the imaging field of view was saturated using a black-dotted-bracket in Fig. 4c and d. Based on this caveat, we consider that there were cells with longer lag times that we were not able to observe in our current setup.
We analysed the growth rate of tolerant single-cells in our bio-sCAPA assays, finding that colony-size heterogeneity and the prolonged colony-appearance time on the plate observed in Fig. 4b could not be accounted for by growth-rate heterogeneity. We observed that growing single cells rapidly form spherical micro-colonies with an exponentially-growing radius as cells were just exiting lag-phase (Fig. 4e). To quantify growth-rate, we measured the radius of these colonies and then fitted straight lines to a log-linear plot of colony radius against time. By only fitting lines to colonies <100 μm in radius (red dotted curves in Fig. 4e), we ensured we were in the exponential regime and thus were measuring a good proxy for single-cell growth (colonies on agar plates ≫100 μm in radius typically grow linearly with radius42,66). We used the slope of the log-linear fit of colony radius to compare growth rates of antibiotic-tolerant cells, which were reported in Fig. 4f (see also Fig. S7†). We did not find significant differences in growth rate between culture conditions (t-test p-values reported in Fig. 4f). Furthermore, we found no correlation between growth rate and lag time (Fig. S8†), and the greatest source of variance in growth rate between culture conditions was derived from differences in clonal replicates rather than the conditions themselves (Fig. S9a†). Lastly, by modelling the extremes in growth rates that we measured for any one biological replicate, we found that the time for a colony to reach a radius of 100 μm differs by an absolute maximum of 448 minutes, i.e. 7.5 hours (Fig. S9b†). When we compared 25th and 75th percentile growth rates, this decreased to a difference of 240 minutes. When we compared this to the large differences we observed in lag-time in Fig. 4c and d of >1000 minutes, we concluded that single-cell growth rate heterogeneity had a minimal effect on the colony size heterogeneity we observed by CFU plating in Fig. 4b and could not explain the differences observed between flucloxacillin and rifampicin challenged cultures.
Discussion
In this work, we have demonstrated a novel single-cell characterisation method for analysing >105 cells in a bacterial population, allowing us to assess population-scale behaviour with single-cell resolution. In particular, the fraction of dead cells, growth rate, and lag time could be analyzed in cell populations exposed to different antibiotic treatments, highlighting the phenotypic heterogeneity in the population.As a first demonstration of bio-sCAPA, here we explored the phenotypic signatures of S. aureus tolerance to flucloxacillin and rifampicin challenge. Using a nutrient-starvation model to stress S. aureus cultures, we showed that nutrient stress drove lag-time heterogeneity, delayed the onset of first growth in the population and thus increased antibiotic tolerance. We showed that S. aureus cells tolerant to flucloxacillin and rifampicin were characterised by phenotypic heterogeneity in lag-time, which is a well-established observation of antibiotic-tolerant bacteria. However, we also found that cells tolerant to rifampicin took >90 minutes longer to recover from antibiotic exposure compared to flucloxacillin-tolerant cells, and this was independent of nutrient stress. On carefully monitoring growth of colonies forming directly from single-cells, we did not observe significant heterogeneity in growth rates across all conditions when compared to the effects of lag-time. Furthermore, we did not observe the development of resistance on subculturing tolerant bacterial cells freshly exposed to antibiotics. Together, this suggests that colony size heterogeneity observed on agar plates from nutrient-stressed cultures was driven by delayed lag time alone. If there are multiple pathways in which S. aureus can form persister cells, this does not appear to lead to significant phenotypic heterogeneity in growth rate, as has been observed for genetic variants.36,38,40 This work, therefore, provides single-cell evidence in support of the hypothesis that S. aureus non-stable small colonies constitute antibiotic-tolerant cells that are characterised by prolonged lag time and can be faithfully identified by monitoring colony growth dynamics.62,66,67 In the future this could be used in clinical diagnostics to distinguish tolerance from resistance, thus helping to better inform treatment strategies for chronic bacterial infections.
Thinking more broadly than antibiotic persistence, we believe that bio-sCAPA offers a unique platform for engineering the structure and composition of surface-attached bacterial communities (Fig. 5). We can control the number of cells deposited in any specific position down to a single cell, allowing one to define the initial community size and the distance between cells in the community. As bio-sCAPA is essentially size-selective, it offers full control in patterning multiple species of different sizes e.g. yeast and bacteria, by sequentially depositing large cells in large traps, followed by small cells in small traps. The capillary forces associated with bio-sCAPA can separate cells that are prone to aggregation (such as S. aureus), and thus enable high control on intra-cellular spacing. Furthermore, the ease of removing the agar pad from the template makes it easy to access specific cells of interest during or after the experiment. One important caveat, however, is that bio-sCAPA necessitates some degree of desiccation. We tested the effect of desiccation on S. aureus growth and lag-time and found no significant influence (Fig. S10†). However, as desiccation is a known stressor,68,69 this may prove problematic for certain species. A second limitation is that our current image analysis requires the use of fluorescent bacterial strains. For non-fluorescent cultures, however, live stains can be used or the algorithm can be extended to monitor growth under phase contrast. With these caveats in mind, we nonetheless believe that bio-sCAPA could find applications in a range of areas in microbiology.
 | ||
| Fig. 5 Outlook: engineering the structure of bacterial communities using bio-sCAPA. a) S. aureus JE2 GFP and S. aureus USA300 RFP deposited in 100:1 ratio in 3 × 1 μm traps with 10 μm spacing. ai) Graphical representation. aii) Merged green and red fluorescence image at t = 0 and aiii) t = 260 min. GFP signal is significantly stronger than RFP signal. b) Single S. aureus JE2 GFP cells deposited in 1 × 1 μm traps with 50 μm spacing. bi) Graphical representation, bii) green fluorescence image at t = 0 and biii) t = 960 min. c) S. aureus JE2 GFP deposited in traps of 3 × 3, 2 × 2 and 1 × 1 μm with 20 μm spacing. ci) Graphical representation, cii) green fluorescence image at t = 0 and ciii) t = 540 min. Scale bars = 10 μm for all images. | ||
As the vast majority of bacteria live in complex, structured, and surface-attached communities in close proximity to other bacterial species,70–72 bio-sCAPA may prove to be a powerful tool for exploring within-species communication, inter-species interactions and early-stage biofilm development in a range of organisms and environmental contexts.
Author contributions
Author contributions are defined based on the CRediT (Contributor Roles Taxonomy) and listed alphabetically. Conceptualisation: C. B., S. M. S., A. S. Z., E. S., L. I.; data curation: C. B.; formal analysis: C. B.; funding acquisition: S. M. S., A. S. Z., L. I.; investigation: C. B.; methodology: C. B., E. S.; project administration: C. B., S. M. S., L. I.; resources: C. B., S. M. S., A. S. Z.; software: C. B.; supervision: S. M. S., A. S. Z., E. S., L. I.; validation: C. B.; visualisation: C. B., E. S., L. I.; writing – original draft: C. B., E. S., L. I.; writing – review and editing: C. B., S. M. S., A. S. Z., E. S., L. I.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
E. S. acknowledges support from SNSF PRIMA grant 179834. A. S. Z. and S. M. S. acknowledge support from the Uniscientia Foundation grant and by the Swiss National Science Foundation (SNSF) project grant 310030_204343 (to A. S. Z.). Special thanks to Dr Federica Andreoni for initial training and early support in the lab. Jeruscha Baum, Dr Clement Vulin and Dr Sam Charlton contributed valuable discussions, which were highly appreciated.Notes and references
- P. J. Piggot and D. W. Hilbert, Curr. Opin. Microbiol., 2004, 7, 579–586 CrossRef CAS PubMed.
- N. Q. Balaban, S. Helaine, K. Lewis, M. Ackermann, B. Aldridge, D. I. Andersson, M. P. Brynildsen, D. Bumann, A. Camilli and J. J. Collins, et al. , Nat. Rev. Microbiol., 2019, 17, 441–448 CrossRef CAS PubMed.
- S. A. West and G. A. Cooper, Nat. Rev. Microbiol., 2016, 14, 716–723 CrossRef CAS PubMed.
- J. Van Gestel, H. Vlamakis and R. Kolter, PLoS Biol., 2015, 13, e1002141 CrossRef PubMed.
- D. A. F. Fuentes, P. Manfredi, U. Jenal and M. Zampieri, Nat. Commun., 2021, 12, 3204 CrossRef CAS PubMed.
- M. Ackermann, Nat. Rev. Microbiol., 2015, 13, 497–508 CrossRef CAS PubMed.
- M. E. Lidstrom and M. C. Konopka, Nat. Chem. Biol., 2010, 6, 705–712 CrossRef CAS PubMed.
- K. Hsieh, K. E. Mach, P. Zhang, J. C. Liao and T.-H. Wang, Acc. Chem. Res., 2021, 55, 123–133 CrossRef PubMed.
- K. Leung, H. Zahn, T. Leaver, K. M. Konwar, N. W. Hanson, A. P. Pagé, C.-C. Lo, P. S. Chain, S. J. Hallam and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7665–7670 CrossRef CAS PubMed.
- Y. Yang, O. Karin, A. Mayo, X. Song, P. Chen, A. L. Santos, A. B. Lindner and U. Alon, Nat. Commun., 2023, 14, 2209 CrossRef CAS PubMed.
- S. Bakshi, E. Leoncini, C. Baker, S. J. Cañas-Duarte, B. Okumus and J. Paulsson, Nat. Microbiol., 2021, 6, 783–791 CrossRef CAS PubMed.
- J. Ollion, M. Elez and L. Robert, Nat. Protoc., 2019, 14, 3144–3161 CrossRef CAS PubMed.
- P. Wang, L. Robert, J. Pelletier, W. L. Dang, F. Taddei, A. Wright and S. Jun, Curr. Biol., 2010, 20, 1099–1103 CrossRef CAS PubMed.
- Y. Yang, A. L. Santos, L. Xu, C. Lotton, F. Taddei and A. B. Lindner, Sci. Adv., 2019, 5, eaaw2069 CrossRef CAS PubMed.
- A. Dal Co, S. van Vliet, D. J. Kiviet, S. Schlegel and M. Ackermann, Nat. Ecol. Evol., 2020, 4, 366–375 CrossRef PubMed.
- A. Dal Co, S. van Vliet and M. Ackermann, Philos. Trans. R. Soc., B, 2019, 374, 20190080 CrossRef CAS PubMed.
- H. Jeckel and K. Drescher, FEMS Microbiol. Rev., 2021, 45, fuaa062 CrossRef CAS PubMed.
- E. Moen, D. Bannon, T. Kudo, W. Graf, M. Covert and D. Van Valen, Nat. Methods, 2019, 16, 1233–1246 CrossRef CAS PubMed.
- K. J. Cutler, C. Stringer, T. W. Lo, L. Rappez, N. Stroustrup, S. Brook Peterson, P. A. Wiggins and J. D. Mougous, Nat. Methods, 2022, 19, 1438–1448 CrossRef CAS PubMed.
- K. D. Whitley, S. Middlemiss, C. Jukes, C. Dekker and S. Holden, Nat. Protoc., 2022, 17, 847–869 CrossRef CAS PubMed.
- P. Ma, H. M. Amemiya, L. L. He, S. J. Gandhi, R. Nicol, R. P. Bhattacharyya, C. S. Smillie and D. T. Hung, Cell, 2023, 186, 877–891 CrossRef CAS PubMed.
- D. Dar, N. Dar, L. Cai and D. K. Newman, Science, 2021, 373, eabi4882 CrossRef CAS PubMed.
- S. Ni, L. Isa and H. Wolf, Soft Matter, 2018, 14, 2978–2995 RSC.
- Y. Yin, Y. Lu, B. Gates and Y. Xia, J. Am. Chem. Soc., 2001, 123, 8718–8729 CrossRef CAS PubMed.
- S. van Kesteren, X. Shen, M. Aldeghi and L. Isa, Adv. Mater., 2023, 2207101 CrossRef CAS PubMed.
- S. Ni, E. Marini, I. Buttinoni, H. Wolf and L. Isa, Soft Matter, 2017, 13, 4252–4259 RSC.
- R. Pioli, M. A. Fernandez-Rodriguez, F. Grillo, L. Alvarez, R. Stocker, L. Isa and E. Secchi, Lab Chip, 2021, 21, 888–895 RSC.
- L. Chopinet, C. Formosa, M. Rols, R. Duval and E. Dague, Micron, 2013, 48, 26–33 CrossRef CAS PubMed.
- C. Formosa, M. Schiavone, H. Martin-Yken, J. François, R. E. Duval and E. Dague, Antimicrob. Agents Chemother., 2013, 57, 3498–3506 CrossRef CAS PubMed.
- F. Pillet, S. Lemonier, M. Schiavone, C. Formosa, H. Martin-Yken, J. M. Francois and E. Dague, BMC Biol., 2014, 12, 1–11 CrossRef PubMed.
- A. Beauvais, S. Bozza, O. Kniemeyer, C. Formosa, V. Balloy, C. Henry, R. W. Roberson, E. Dague, M. Chignard and A. A. Brakhage, et al. , PLoS Pathog., 2013, 9, e1003716 CrossRef PubMed.
- C. Formosa, F. Pillet, M. Schiavone, R. E. Duval, L. Ressier and E. Dague, Nat. Protoc., 2015, 10, 199–204 CrossRef PubMed.
- R. Pioli, R. Stocker, L. Isa and E. Secchi, J. Visualized Exp., 2021,(177), e63131 CAS.
- R. Kuehl, L. Morata, S. Meylan, J. Mensa and A. Soriano, J. Antimicrob. Chemother., 2020, 75, 1071–1086 CrossRef CAS PubMed.
- S. Y. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler Jr, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- M. Huemer, S. Mairpady Shambat, S. D. Brugger and A. S. Zinkernagel, EMBO Rep., 2020, 21, e51034 CrossRef CAS PubMed.
- B. C. Kahl, K. Becker and B. Löffler, Clin. Microbiol. Rev., 2016, 29, 401–427 CrossRef CAS PubMed.
- R. A. Proctor, P. van Langevelde, M. Kristjansson, J. N. Maslow and R. D. Arbeit, Clin. Infect. Dis., 1995, 20, 95–102 CrossRef CAS PubMed.
- R. A. Proctor, C. Von Eiff, B. C. Kahl, K. Becker, P. McNamara, M. Herrmann and G. Peters, Nat. Rev. Microbiol., 2006, 4, 295–305 CrossRef CAS PubMed.
- R. Proctor, Microbiol. Spectrum, 2019, 7(3), GPP3-0069-2019 Search PubMed.
- L. Tuchscherr, E. Medina, M. Hussain, W. Völker, V. Heitmann, S. Niemann, D. Holzinger, J. Roth, R. A. Proctor and K. Becker, et al. , EMBO Mol. Med., 2011, 3, 129–141 CrossRef CAS PubMed.
- C. Vulin, N. Leimer, M. Huemer, M. Ackermann and A. S. Zinkernagel, Nat. Commun., 2018, 9, 1–8 CrossRef CAS PubMed.
- N. Kaldalu, V. Hauryliuk, K. J. Turnbull, A. La Mensa, M. Putrinš and T. Tenson, Microbiol. Mol. Biol. Rev., 2020, 84(4), e00070-20 CrossRef PubMed.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS PubMed.
- F. Goormaghtigh and L. Van Melderen, Sci. Adv., 2019, 5, eaav9462 CrossRef PubMed.
- J.-S. Kim, R. Yamasaki, S. Song, W. Zhang and T. K. Wood, Environ. Microbiol., 2018, 20, 2085–2098 CrossRef CAS PubMed.
- E. Şimşek and M. Kim, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17635–17640 CrossRef PubMed.
- R. A. Fisher, B. Gollan and S. Helaine, Nat. Rev. Microbiol., 2017, 15, 453–464 CrossRef CAS PubMed.
- M. Geissler, H. Wolf, R. Stutz, E. Delamarche, U.-W. Grummt, B. Michel and A. Bietsch, Langmuir, 2003, 19, 6301–6311 CrossRef CAS.
- M. C. Maranan, B. Moreira, S. Boyle-Vavra and R. S. Daum, Infect. Dis. Clin. North Am., 1997, 11, 813–849 CrossRef CAS PubMed.
- J. C. Crocker and D. G. Grier, J. Colloid Interface Sci., 1996, 179, 298–310 CrossRef CAS.
- R. D. Deegan, O. Bakajin, T. F. Dupont, G. Huber, S. R. Nagel and T. A. Witten, Nature, 1997, 389, 827–829 CrossRef CAS.
- S. Ni, J. Leemann, H. Wolf and L. Isa, Faraday Discuss., 2015, 181, 225–242 RSC.
- V. Leung and C. M. Lévesque, J. Bacteriol., 2012, 194, 2265–2274 CrossRef CAS PubMed.
- N. Leimer, C. Rachmühl, M. Palheiros Marques, A. S. Bahlmann, A. Furrer, F. Eichenseher, K. Seidl, U. Matt, M. J. Loessner and R. A. Schuepbach, et al. , J. Infect. Dis., 2016, 213, 305–313 CrossRef CAS PubMed.
- D. Nguyen, A. Joshi-Datar, F. Lepine, E. Bauerle, O. Olakanmi, K. Beer, G. McKay, R. Siehnel, J. Schafhauser and Y. Wang, et al. , Science, 2011, 334, 982–986 CrossRef CAS PubMed.
- S. P. Bernier, D. Lebeaux, A. S. DeFrancesco, A. Valomon, G. Soubigou, J.-Y. Coppée, J.-M. Ghigo and C. Beloin, PLoS Genet., 2013, 9, e1003144 CrossRef CAS PubMed.
- N. M. Vega, K. R. Allison, A. S. Khalil and J. J. Collins, Nat. Chem. Biol., 2012, 8, 431–433 CrossRef CAS PubMed.
- F. Peyrusson, H. Varet, T. K. Nguyen, R. Legendre, O. Sismeiro, J.-Y. Coppée, C. Wolz, T. Tenson and F. Van Bambeke, Nat. Commun., 2020, 11, 1–14 CrossRef PubMed.
- M. Zwietering, I. Jongenburger, F. Rombouts and K. Van't Riet, Appl. Environ. Microbiol., 1990, 56, 1875–1881 CrossRef CAS PubMed.
- K. M. Tjørve and E. Tjørve, PLoS One, 2017, 12, e0178691 CrossRef PubMed.
- J. Bär, M. Boumasmoud, S. M. Shambat, C. Vulin, M. Huemer, T. A. Schweizer, A. Gómez-Mejia, N. Eberhard, Y. Achermann and P. O. Zingg, et al. , Clin. Microbiol. Infect., 2022, 28, 1022 CrossRef PubMed.
- M. Huemer, S. Mairpady Shambat, J. Bergada-Pijuan, S. Söderholm, M. Boumasmoud, C. Vulin, A. Gómez-Mejia, M. Antelo Varela, V. Tripathi and S. Götschi, et al. , Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2014920118 CrossRef CAS PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- C. Wang, R. Fang, B. Zhou, X. Tian, X. Zhang, X. Zheng, S. Zhang, G. Dong, J. Cao and T. Zhou, BMC Microbiol., 2019, 19, 1–8 CrossRef PubMed.
- J. Bär, M. Boumasmoud, R. D. Kouyos, A. S. Zinkernagel and C. Vulin, Sci. Rep., 2020, 10, 1–15 CrossRef PubMed.
- I. Levin-Reisman, O. Gefen, O. Fridman, I. Ronin, D. Shwa, H. Sheftel and N. Q. Balaban, Nat. Methods, 2010, 7, 737–739 CrossRef CAS PubMed.
- J. Esbelin, T. Santos and M. Hébraud, Food Microbiol., 2018, 69, 82–88 CrossRef CAS PubMed.
- M. Potts, Microbiol. Rev., 1994, 58, 755–805 CrossRef CAS PubMed.
- H.-C. Flemming and S. Wuertz, Nat. Rev. Microbiol., 2019, 17, 247–260 CrossRef CAS PubMed.
- A. K. Wessel, L. Hmelo, M. R. Parsek and M. Whiteley, Nat. Rev. Microbiol., 2013, 11, 337–348 CrossRef CAS PubMed.
- R. Krishna Kumar and K. R. Foster, Microb. Biotechnol., 2023, 16, 489–493 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00611e |
| This journal is © The Royal Society of Chemistry 2023 |