
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bridging the gap between tumor-on-chip and clinics: a systematic review of 15 years of studies†
Charlotte
Bouquerel
 abc,
Anastasiia
Dubrova
a,
Isabella
Hofer
b,
Duc T. T.
Phan
d,
Moencopi
Bernheim
a,
Ségolène
Ladaigue
b,
Charles
Cavaniol
a,
Danilo
Maddalo
e,
Luc
Cabel
f,
Fatima
Mechta-Grigoriou
b,
Claire
Wilhelm
a,
Gérard
Zalcman
bg,
Maria Carla
Parrini
b and
Stéphanie
Descroix
*a
abc,
Anastasiia
Dubrova
a,
Isabella
Hofer
b,
Duc T. T.
Phan
d,
Moencopi
Bernheim
a,
Ségolène
Ladaigue
b,
Charles
Cavaniol
a,
Danilo
Maddalo
e,
Luc
Cabel
f,
Fatima
Mechta-Grigoriou
b,
Claire
Wilhelm
a,
Gérard
Zalcman
bg,
Maria Carla
Parrini
b and
Stéphanie
Descroix
*a
aMacromolécules et Microsystèmes en Biologie et Médecine, UMR 168, Institut Curie, Institut Pierre Gilles de Gennes, 6 rue Jean Calvin, 75005, Paris, France
bStress and Cancer Laboratory, Inserm, U830, Institut Curie, PSL Research University, 26 rue d'Ulm, 75005, Paris, France
cFluigent, 67 avenue de Fontainebleau, 94270, Le Kremlin-Bicêtre, France
dBiomedicine Design, Pfizer Inc., San Diego, CA, USA
eDepartment of Translational Oncology, Genentech, Inc., South San Francisco, CA 94080, USA
fInstitut Curie, Department of Medical Oncology, 26 rue d'Ulm, 75005, Paris, France
gUniversité Paris Cité, Thoracic Oncology Department, INSERM CIC1425, Bichat Hospital, Cancer Institute AP-HP. Nord, Paris, France. E-mail: stephanie.descroix@curie.fr
First published on 18th August 2023
Abstract
Over the past 15 years, the field of oncology research has witnessed significant progress in the development of new cell culture models, such as tumor-on-chip (ToC) systems. In this comprehensive overview, we present a multidisciplinary perspective by bringing together physicists, biologists, clinicians, and experts from pharmaceutical companies to highlight the current state of ToC research, its unique features, and the challenges it faces. To offer readers a clear and quantitative understanding of the ToC field, we conducted an extensive systematic analysis of more than 300 publications related to ToC from 2005 to 2022. ToC offer key advantages over other in vitro models by enabling precise control over various parameters. These parameters include the properties of the extracellular matrix, mechanical forces exerted on cells, the physico-chemical environment, cell composition, and the architecture of the tumor microenvironment. Such fine control allows ToC to closely replicate the complex microenvironment and interactions within tumors, facilitating the study of cancer progression and therapeutic responses in a highly representative manner. Importantly, by incorporating patient-derived cells or tumor xenografts, ToC models have demonstrated promising results in terms of clinical validation. We also examined the potential of ToC for pharmaceutical industries in which ToC adoption is expected to occur gradually. Looking ahead, given the high failure rate of clinical trials and the increasing emphasis on the 3Rs principles (replacement, reduction, refinement of animal experimentation), ToC models hold immense potential for cancer research. In the next decade, data generated from ToC models could potentially be employed for discovering new therapeutic targets, contributing to regulatory purposes, refining preclinical drug testing and reducing reliance on animal models.
1. Introduction
For almost one century, scientists have been developing and improving cell culture models to increase their resemblance to human in vivo conditions and relevance for clinical transition. In a similar vein, novel tumor-on-a-chip (ToC) technology has developed tremendously over the past decade and holds great promise for clinical applications in oncology. The present review focuses on the quantitative analysis of the tumor on chip field up to date and offers a clear outlook of the subject in terms of both, engineering and application approaches. We have brought together academic researchers (physicists and biologists), clinicians as well as pharmaceutical companies' research experts to offer a multifaceted point of view that reflects the diversity of the actors of the field.Nowadays in Europe, the yearly number of newly diagnosed people with cancer is about 3.5 million. In 2021, the EU Cancer Mission outlined that if no further action is taken, this number will dramatically increase to more than 4.3 million by 2035. In the fight against cancer, the better understanding of cancer mechanisms and the development of more effective anti-cancer drugs still remain highly challenging. Although conventional and well-established treatments, such as chemotherapies, face many failures, the development of new therapeutic strategies, such as immunotherapies and targeted therapies, raises new hopes. Since 2015, the U.S. Food and Drug Administration (FDA) has approved more than 80 novel cancer drugs, illustrating a historically high level of successful clinical trials in oncology.1 However, it is also worth mentioning that only 4 to 7% of potential anticancer drugs obtain final clinical approval2 compared to 10 to 15% for other diseases.3 Such low success rates call into question the preclinical studies whose aim is to predict the effect of therapeutic agents in terms of efficacy, safety and dosage. The analysis of clinical trial data (from 2010 to 2017) has shown that the two main reasons for these failures are the unmanageable toxicity and lack of clinical efficacy.4 Preclinical toxicity studies are mainly assessed by animal testing. However, it is now established that animals are not always suitable models for human toxicity prediction as shown in the meta-analysis of Atkins et al.5 that compares preclinical and clinical toxicity profiles of 108 anti-cancer drugs in animal models and humans. They highlighted that the main unpredictable toxicities are of neurologic/psychiatric, cutaneous, respiratory, and cardiovascular nature. Another reason for the low success rates of anti-cancer drug development is related to intrinsic drug efficacy. Recent studies have also shown that current in vitro and in vivo models are poor predictors of drug efficacy.6 The dramatic failure rate of clinical trials not only challenges our ability to design and develop new drugs, but also the use of animal models for basic research. Common in vivo models include patient-derived xenografts in mice (PDX), which share several important characteristics with human tumors (i.e., vascularization, 3D structure and metabolism). However, PDX models also lack some crucial features such as human stroma and pharmacokinetics as well as an intact autologous immune system since such models often consist of immune deficient mice to allow efficient engrafting.7 It is also worth mentioning that syngeneic animal models will be even less predictive for novel anti-cancer therapies such as biologics, gene- and cell-based therapies, since these drugs are either mainly specific to human targeting molecular sequences or involved the human immune system.8 Very recently, the FDA has removed the requirement of animal testing before human trials (“Modernization Act 2.0”). This FDA statement is an excellent opportunity as well as a strong responsibility for the scientific community to develop and share innovative in vitro models that would faithfully reproduce disease mechanisms while improving predictive power.
Within the past decades, researchers have developed more adequate 3D in vitro models such as spheroids and organoids. They can incorporate different cell types to better mimic the complex tumor microenvironment (TME) compared to conventional 2D cell cultures. While the use of spheroids for in vitro drug testing is popular due to their relative ease of handling, they still exhibit a low degree of structural complexity.9 Organoids, cultured from embryonic, adult stem cells or induced pluripotent stem cells are better in mimicking the TME10,11 and several studies have evidenced that they are relatively good predictors of chemotherapy response in patients.12 However, the establishment of patient-derived organoids generally requires 1 to 3 months,10 and thereby limits their application in clinics as a diagnostic tool to support the choice of a specific treatment. Organoids and spheroid cultures are also associated with some major limitations: (a) they are static models which can lead to the accumulation of biochemical waste within the cell aggregate, (b) they do not fully reproduce the immune response, presence of fibroblasts and vascularization, (c) they do not reproduce the mechanical properties of tumors which can influence drug response.13
New in vitro models for oncology research are required for both basic and preclinical research. These models should especially consider that tumors are complex ecosystems dynamically evolving over time. Indeed, the TME not only contains cancer cells, the surrounding extracellular matrix (ECM) with varying mechanical and physico-chemical properties, but also a variety of other cell types such as immune lymphoid and myeloid cells, cancer-associated fibroblasts (CAFs), pericytes and endothelial cells with specific spatial organization. All before mentioned TME components have been shown to play a significant role in tumor progression, metastasis development, and resistance to treatment.14 Microfluidic technologies, and organ-on-chip approaches in particular have an enormous potential for the development of a new generation of 3D in vitro tumor models. ToC contains several features which make them highly attractive for both basic and translational research: (a) the capacity to control the cellular, mechanical and physicochemical conditions on-chip, (b) the compatibility with a wide range of analytical methods including transcriptomic analysis and live imaging, (c) the possibility to produce human and/or immunocompetent models, and (d) the relatively short experimental time of several days as compared to other 3D cancer models such as PDX or organoids, allowing clinical decision.
In this review, we conducted a systematic analysis of the publications related to ToC between 2005 and 2022 to provide the reader with a clear and quantitative vision of the emerging ToC field. Altogether, over 300 publications were identified using PubMed, Google Scholar and looking directly into key journals as shown Fig. S1.† In this systematic review, we identify crucial subject parameters and extracted percentages of each occurrence. We discuss the added values of ToC as well as the scope of possible applications: drug screening, cellular mechanisms understanding and personalized- and nano-medicine. The success of ToC for clinical applications will be determined by its ability to detect and validate new therapeutic targets as well as to guide the definition of the delicate balance between clinical dose, efficacy and safety.
2. Material and methods
We used several databases (such as Pubmed, Google Scholar) and searched for combinations of the following key words in the title and abstract (Fig. S1†): “tumor”, “cancer”, “microfluidic”, “chip”. We also searched directly for relevant publications in specific journals such as Lab On Chip. Publications with only computational approaches, as well as reviews were excluded during the first screen (titles and abstract). We screened references of more than 10 reviews to list all the ToC publications. During the second step, a qualitative analysis of all the publications results was performed and only studies including cell culture on chip were included (for example CTC sorting methods were excluded). For the purpose of the analysis, we created a table with information related to the most relevant aspects of OoC devices: a) cellular components: type (cell line, primary cells, freshly resected tumor, IPS cells, organoids), organ (breast, lung, pancreas, prostate, ovary/uterus, colorectal/intestine, liver, brain), spheroids (yes/no); b) physico-chemical control: flow, oxygen control, extracellular matrix; (c) applications: anti-cancer drug, readouts. Excel was used to count occurrence of these key points in all the publications. Over 300 publications were analysed.3. Tumor-on-chip allows to control the reconstituted tumor micro-environment
Cancer is not defined solely by cancer cells but by the whole TME, including cellular and molecular components, ECM, as well as their complex interplay. Tumors have many biomechanical abnormalities such as elevated solid stress, interstitial pressure, and stiffness.13 One beauty of the ToC approach is the possibility to finely control and thus dissect the role of each parameter of the reconstituted TME. Particularly, this includes the different cell types and their spatial organization, the ECM properties, the on-chip generation of biochemical gradients, the control of the gaseous environment or even the different mechanical forces at play.3.1. Controlling physico-chemical properties of the TME
Given the complexity and dynamic alterations of the ECM, one of the key points of ToC is the ability to properly select and design biomaterials to reconstitute the extracellular matrix in vitro. Here, we focused on ToC with extracellular matrix, but excluded articles dealing with hydrogel coating for 2D cell monolayers. Collagen I is the main ECM component in vivo and is by far the most used hydrogel in ToC being used in half of the ToC publications) (Fig. 1). Collagen I contains the tripeptide RGD (Arg–Gly–Asp) which is a very common motif in humans and animals responsible for cell adhesion.22 Another important advantage of collagen is its stiffness, which can be adjusted easily through its concentration or by covalent crosslinking via non-enzymatic glycation.23 The second most common ToC hydrogel is Matrigel which is a solubilized basement membrane matrix secreted by Engelbreth–Holm–Swarm mouse sarcoma cells. Nevertheless, it has been suggested that results based on Matrigel-cultured cells should be interpreted with caution24 due to its influence on gene expression25 as well as the lack of some human peptide motifs.22 It is worth noticing that both collagen and Matrigel can vary highly between different batches or manufacturers which can affect ToC reproducibility. Besides Matrigel and collagen, there is a wide range of hydrogels available for ToC development such as fibrin, gelatin and agarose (Fig. 1). Another less common approach relies on in vivo extracted matrix. Romero-López et al. used a decellularized ECM extracted from both healthy and cancerous colon tissues and prepared hydrogels through enzymatic digestion.26 However, variability in ECM extraction protocols could introduce further alterations in hydrogels, which may lead to inconsistent results.
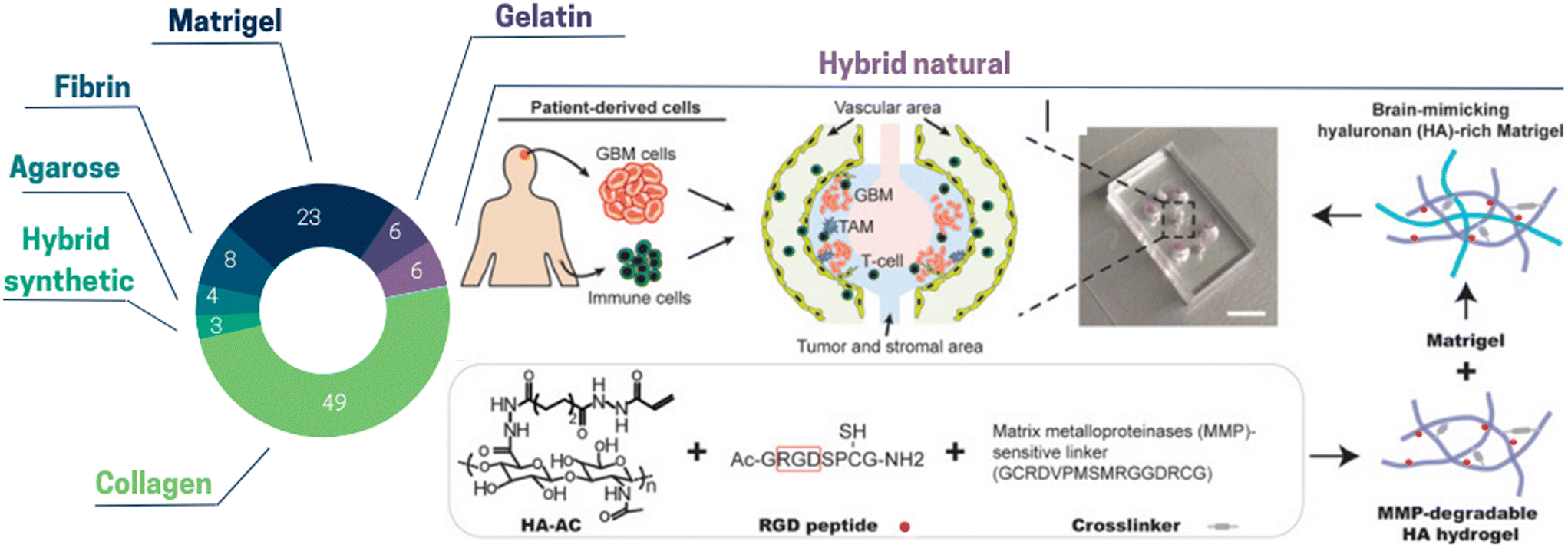 | ||
| Fig. 1 Pie chart illustrating the proportion of the different extracellular matrix types used in ToC: collagen 49%, Matrigel 23%, fibrin 8%, gelatin 6%, hybrid natural 6%, agarose 4%, hybrid synthetic 3%. (Right panel) Hybrid natural: hydrogels can be created by interpenetrating growth-factor-reduced Matrigel matrix with MMP-degradable ha hydrogel for brain tissue-mimicking extracellular matrix. RGD peptides are conjugated onto acrylated hyaluronic acid (HA–AC) and crosslinked with MMP-degradable crosslinker.27 | ||
Hybrid hydrogels are made up of building blocks that include biologically active peptides, proteins or synthetic structures. Hybrid hydrogels can be used to obtain desirable mechanical and biochemical characteristics via their functionalization with defined proteolytic sites and encapsulation of growth factors. Nevertheless, given that the ECM alone comprises more than 300 biochemical constituents,28 this remains a daunting task. Among hybrid hydrogels in ToC, natural ones comprise alginate, fibrinogen, hyaluronan (HA), chitosan, and synthetic ones include poly-ethylene glycol (PEG), poly-caprolactone (PCL) and poly(lactic-co-glycolyc) acid (PLGA). Currently, these hydrogels have not been widely adopted. Cui et al.27 used a hybrid brain tissue-mimicking hydrogel with RGD peptides conjugated onto acrylated hyaluronic acid (HA–AC) and crosslinked with MMP-degradable crosslinker (Fig. 1). With the combination of ToC and hybrid hydrogels, it is possible to not only control the ECM chemical composition and stiffness, but also to choose their spatial location. In a breast cancer model, Peela et al.29 proposed a two-step photolithography approach to create an array of cells embedded in circular constructs, with a high stiffness matrix center surrounded by low stiffness matrix. They encapsulated three cell types separately to investigate cell migratory behavior, viability, and morphology. Importantly, cells migrating through the high stiffness circular constructs exhibited different invasive behaviors compared to those migrating through the surrounding matrix. They formed morphologically accurate structures without the addition of any biochemical stimuli, illustrating the versatility of ToC in creating a biomimetic tumor microenvironment.
There are still many challenges to define and improve ECM in order to mimic accurately in vivo conditions.28 Due to the high heterogeneity between different cancer sub-types and even within the same TME, one single type of hydrogel cannot accurately recapitulate the 3D environment experienced by cells in vivo. A major challenge is still to synthesize hydrogel matrices that closely mimic the properties of the ECM components specific to each cancer subtype with properties controllable spatially and temporally. It should also be mentioned that fibroblasts and perivascular cells will be key in the future development of ToC as they also contribute to ECM production.
Recent tools have been implemented to reproduce these tumor mechanical forces in ToC. To mimic compressive stress, Onal et al.34 developed a chip with an integrated gas pressure micro-piston, applying dynamic compression on ovarian cancer cells. They studied the impact of cyclic stress on the cell nucleus, which is a mechanosensitive organelle, and demonstrated that the circularity of the cell nuclei was significantly less in compressed cells than in control. So far only a few ToC studies integrate controllable compressive stress, but this work highlighted that ToC constitutes a promising tool for studies of cell-mechanical force interaction.
In order to reproduce the interstitial fluid pressure and shear stress, fluidic control in ToC is made possible thanks to conventional fluid controllers also used for seeding cells, refreshing and controlling the cell culture media composition over time. This fluidic control is mostly performed with syringe pumps35–37 (half of ToC publications), peristaltic pumps38–44 or tilted rocking platform45 while the second half of the ToC studies does not report any flow control (Fig. S2†). Kocal et al.35 investigated how shear stress affects the epithelial to mesenchymal transition of oesophageal cancer cells in a 2D ToC. From the third day of culture under flow, cancer cells experienced a phenotypic switch with a significant decrease in cell–cell adhesion (decrease in E-cadherin expression) as well as an increase of transendothelial migration capacity (increased expression of N-cadherin). Moreover, some ToC studies showed that shear stress can also affect cancer stem cell states. Ip et al.46 found that spheroids grown under shear stress exhibited higher expression of stem cell markers Oct-4, c-Kit (CD117), efflux pumps ABCG2 and P-gp in contrast to static conditions. In clinical settings, a poor prognosis is associated with these factors, highlighting the link between shear stress applied on cancer cells and chemoresistance.47
Apart from cancer cells, shear stress can also have a high impact on normal endothelial cells. Several ToC studies showed that shear stress induced an elongated endothelial cell morphology as well as modified junctional protein expression48 which can facilitate the extravasation of cancer cells. Kim et al.49 demonstrated that the interstitial flow direction can also regulate the direction of capillary sprouting, suggesting angiogenesis occurs in the opposite direction of flow.
Among the mechanical forces experienced by cells in vivo is also peristalsis, which is the progression of coordinated muscle contractions. These forces are at play in the gut, oesophagi, uterus and many other organs. Only a few ToC devices have implemented physiological mechanical tissue deformation. Fang et al.50 developed a microfluidic chip allowing high-throughput culture under peristalsis of human colon tumoroids to screen nanomedicines. They observed an increase of stem cell markers (Lgr5) and proliferation markers (Ki67) which could be linked to a peristalsis-induced high interstitial fluid pressure and suggested that peristalsis is also involved in the reduced nanoparticle internalization via clathrin-dependent endocytosis. In a recent study, Strelez et al. studied colon metastatic spreading in a peristalsis-tunable chip using colorectal cancer (CRC) cells from patients and showed a peristalsis-induced increase of tumor cell invasion.51 Ao et al.52 evidenced that mechanical stretching of prostatic normal tissue-associated fibroblasts (NAFs) alters the structure of secreted fibronectin. They suggested that mechanical stress is one of the critical factors in NAF activation into cancer associated fibroblasts (CAFs).
There is growing evidence that a wide range of mechanical stresses can alter tumor and stromal cells behavior and ToC appears to be a powerful and innovative technological tool to decipher the role of these various mechanical forces.
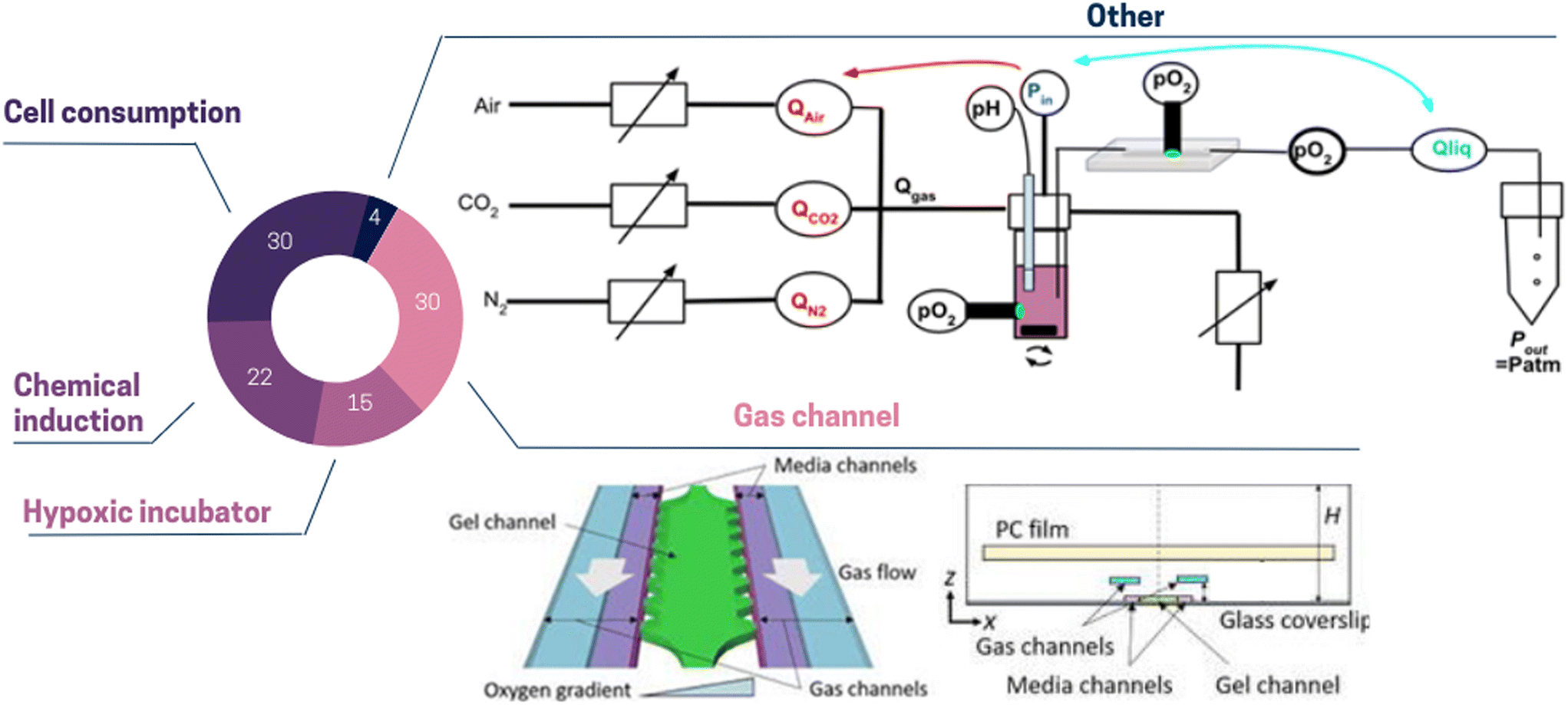 | ||
| Fig. 2 Pie chart illustrating the proportion of different oxygen control strategies used in ToC: cell consumption 30%, gas channel 30%, chemical induction 22%, hypoxic incubator 15%, other 4%. (Top panel) Other: the dissolved oxygen level can be regulated by modulating pneumatic valves opening. This method simultaneously changes the gas composition and the pressure-driven flow.62 (Bottom panel) Gas channel: to control dissolved oxygen levels in the media and gel channels, gas mixtures can be supplied to the two separated gas channels. To prevent from atmospheric oxygen diffusion, a polycarbonate film is embedded on top of the gas channels.58 | ||
Reproducing the intricate gaseous environment of the tumor in vitro presents considerable challenges, but it holds immense importance as it offers the opportunity to capture spatial metabolic and phenotypic heterogeneity. Significantly, Ayuso et al.63 demonstrated that cells situated farther from the lumen, where nutrients and oxygen originate, displayed upregulated genes associated with apoptosis resistance (e.g., BIRC3), DNA damage induced by starvation (e.g., GADD45G), and stress response (e.g., ADM).
3.2. Controlling the cellular complexity on ToC
Cancer cell lines remain the primary tool for studying biological processes since decades due to their cost-effectiveness, ease of use, and ability to provide an unlimited number of consistent samples. As shown in Fig. 3, about 70% of ToC studies use predominantly cell lines. Most of these cell lines are part of the US National Cancer Institute (NCI) 60 human tumor cell lines anticancer drug screen (NCI60), which have been extensively characterized on a molecular level: exome sequence, DNA methylation, mRNA expression, protein levels and modifications, enzyme activity, and metabolomic profiling.68 These cell lines theoretically allow an easy comparison between the results obtained inside ToC versus other in vitro models. However, it is also well-known and accepted that cell lines do not completely represent relevant models of in vivo tumors, since indeterminate transcriptomic, epigenetic, genetic and phenotype changes may occur during cell immortalization. Moreover, cells that have been cultured for several years and across different laboratories can present major differences as compared to primary cells and even to the initial source of such cell lines.69 The use of cell lines therefore raises important questions. Is it essential for the scientific community to agree on the use of specific cell lines for every clinical cancer subtype? Which specific and standardized characterization methods should be performed to confirm genomic or phenotypic drifts or cell–cell contaminations? And at last, to which extent should cell-based models represent the in vivo pathology in terms of underlying biological mechanisms and response to treatment?
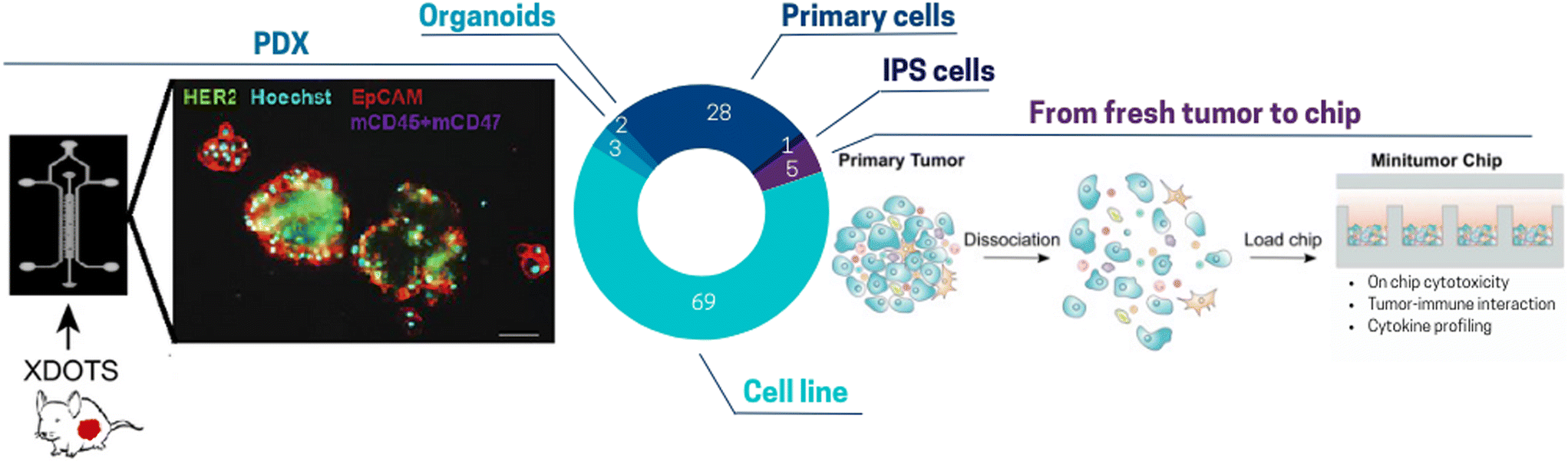 | ||
| Fig. 3 Pie chart illustrating the proportion of different cell sources used for ToC: cell line 69%, primary cells (HUVECs, PBMCs, primary fibroblasts) 28%, PDX 3%, from fresh tumor to chip (freshly resected tumor, CTCs), IPS cells 1%, organoids 2%. (Left panel) Tumors are harvested from PDXs. After dissociation and filtration isolation, spheroids are produced in ultra-low attachment plate and seeded in the toc.85 (Right panel) Freshly resected primary tumors can be subjected to tumor digestion using a human primary tumor dissociation kit and then seeded in a micro-well array ToC.89 | ||
A more faithful approach to reconstitute the in vivo TME includes the use of primary cells directly extracted from fresh tissues or fluids with subsequent ex vivo culture. Since primary cells are only cultivated for a low number of passages, they are thought to display most of the differentiated properties of their tissue of origin.69 Only one third of ToC incorporate primary cells mainly human peripheral blood mononuclear cells (PBMC), CAFs or endothelial cells (e.g. HUVECs). PBMCs include lymphocytes mostly T-cells, B cells including rare plasmocytes, NK cells, monocytes, rare dendritic cells and basophils. Numerous ToC studies use PBMCs, such as Boussomier-Calleja et al.70 who studied the effect of monocytes on cancer cell extravasation at different stages of their life cycle. However, since PBMCs are extracted from healthy donors, they cannot be fully considered to perfectly mimic tumor immune cell infiltration. Here, the use of autologous cells offers the possibility to avoid recognition of non-self cells. This is especially crucial for testing immuno-oncology drugs, such as immune checkpoint inhibitors (ICI) that function by unleashing the cytotoxic activity of T-cells.
Among the endothelial cells, HUVECs were by far the most well-represented source of ECs in vascularized ToC models. Only a few studies used micro-vascular ECs from specialized tissues in ToC.71,72 One major limitation of HUVECs and of most ECs is their non-tumoral origin. On the other hand, tumoral endothelial cells (TECs) are constrained by the isolation, availability, and viability challenges. Matsuda et al.48 estimated that TECs represent around 2% of the overall cells in the tumor. As such, novel and more efficient isolation protocols need to be established to design fully patient-derived vascularized ToC models. Isolation of ECs from peri-tumor areas could consist of a reasonable compromise allowing isolation of tumor cells, immune cells, fibroblasts, and ECs from the same patient. Notably, some ToC integrate human induced pluripotent stem cell-derived endothelial cells (iPSC-EC).73 In contrast to cell lines, iPSCs achieve immortality by inducing pluripotency rather than by transformation. For ToC development, iPSCs offer interesting advantages due to their ability to be reprogrammed into different types of tissues but their use in ToC remains scarce (only 1% of ToC publications). Lee et al.74 integrated iPSC-derived cardiac cells in a heart-breast ToC. Fibrotic stages of iPSC-derived cardiac tissues have been promoted to assess the differential functionality in healthy and fibrotic cardiac tissues after treatment with chemotherapy. iPSCs also offer the possibility to design multi-organ platforms composed of various tissues from the same donor. For example, cardiomyocytes derived from iPSCs from patients with breast cancer were shown to model at the cellular level the doxorubicin-induced cardiotoxicity observed in some patients.75
Other studies used more complex cellular models (although it is still a minority among ToC publications) such as organoids,76–79 PDX80–88 or fresh surgical tumor sample.82–87,90–96 The category “fresh surgical tumor sample” encompasses various form since there is diversity in patient tumor samples including slices, micro-dissected tumor tissues and even single cells. For instance, organotypic slices of PDX breast and prostate tumors were successfully cultured in chips with 6-well plate design for up to 14 days with an accurate prediction of cell death with conventional treatments.86 Ivanova et al.85 established a PDX ToC system for drug screening (Fig. 3 left panel). Several studies have also demonstrated the feasibility of growing circulating tumor cells (CTCs) isolated from blood samples.96–98 Often proliferation of primary cells was limited to a few days or weeks but was sufficient for drug screenings.96 ToC also indicated promising results for the maintenance of fresh surgical tumor samples in culture. Dorrigiv et al.82 demonstrated that oxygen-permeability of microfluidic devices reduces the extent of hypoxia in tissue slices in comparison to 96-well plates. In addition, Chakrabarty et al.86 reported maintaining proliferation in tissue slices for 14 days in ToC versus only 7 days with standard plates. Apart from tissue slices, diverse solid tumor types can be included on chip such as primary lesions, lymphadenectomy specimens, pleural effusions, ascites fluid, or resected metastases. Parsian et al.95 fabricated a device with three PDMS layers, where the middle layer accommodates the tissue slices, and the top/bottom layers are perfused with media. Similarly, a study by Ao et al.89 highlights the feasibility of integrating fresh tumor cells directly into microfluidic chips (Fig. 3 right panel). However, this model still lacks several features such as tumor-matching extracellular matrix, which has been demonstrated to affect several key processes, including tumor growth, immune infiltration and drug responses.15 Alternatively, it is also possible to include micro-dissections of tumors.81 Growing fresh tumor samples in 3D on ToC including its ECM could pave the way to a closer reflection of the in vivo tumor therapeutic response. However, standardized procedures still need to be developed to integrate cell populations isolated from fresh tumors.
In ToC, cellular architecture can be controlled using various microfabrication approaches. We classified ToC designs, inspired by Sleeboom et al.100 according to five categories (Fig. 4): (a) compartmentalized chip, (b) micro-wells array, (c) 2D chip, (d) membrane chip, (e) lumen chip, and (f) others. Such classification does not cover the whole spectrum of published studies, but highlights the predominant strategies currently employed in the field of tumor-on-chip research. In turn, this classification does not completely capture the diversity and complexity of ToC approaches and some configurations can be a combination of several before mentioned categories.
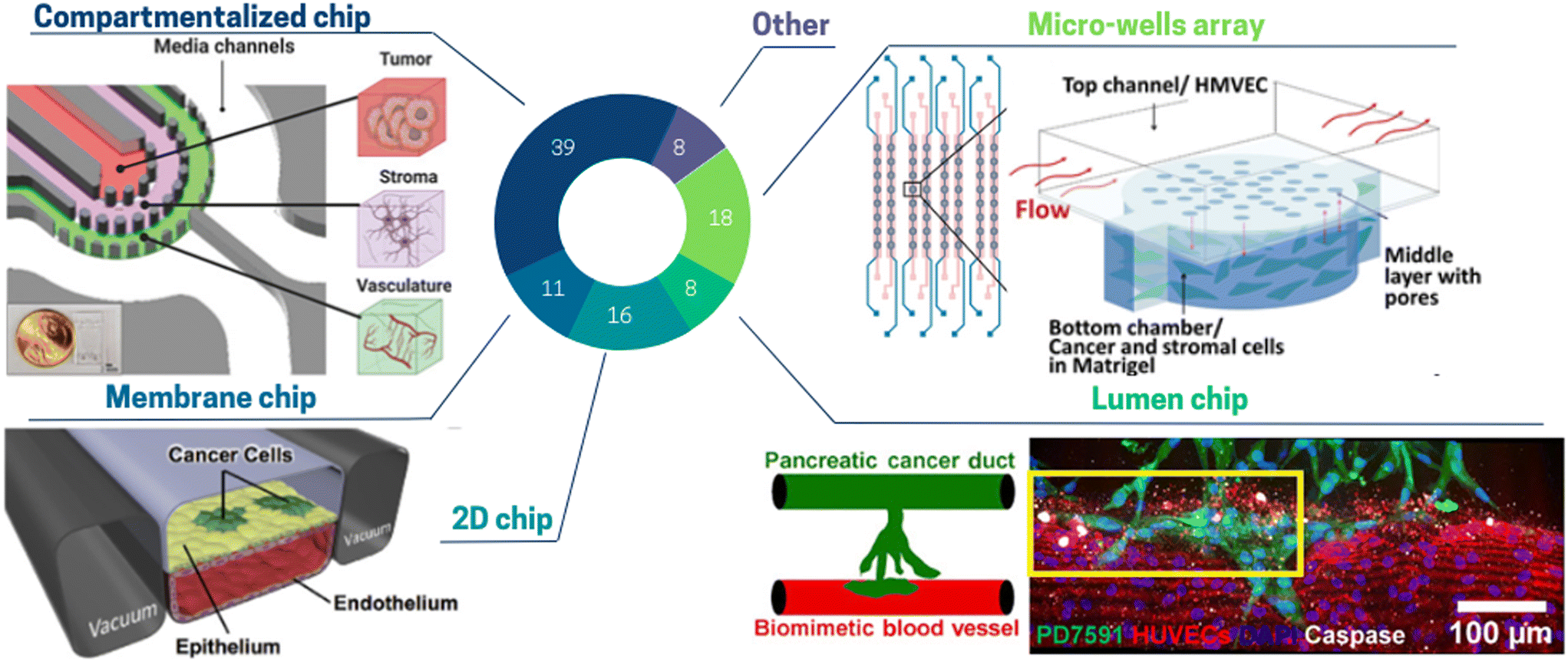 | ||
| Fig. 4 Pie chart illustrating the proportion of different toc designs used to mimic cell architecture: compartmentalized chip 39%, micro-wells array 18%, 2D chip 16%, membrane chip 11%, lumen chip 8%, other 8%. (Top left panel) The glioblastoma toc model is a compartmentalized chip with three concentric cell culture regions separated by pillars: namely, the vasculature, stroma, and tumor regions surrounded by media channels.109 (Top right) In this micro-well array ToC, cancer and stromal cells are encapsulated in Matrigel and seeded into the bottom layer, while flow is applied in the top layer.116 (Bottom left panel) The 2-channel membrane toc contains human lung epithelial cells and a low density of lung cancer cells cultured on the upper surface of a porous ECM-coated membrane. Human lung microvascular endothelial cells are cultured on all four walls of the lower channel, forming a hollow vascular lumen.71 (Bottom right panel) The lumen ToC is composed of two cylindrical channels embedded within a 3D collagen matrix. One channel is covered with endothelial cells to form a perfusable biomimetic blood vessel, and the other channel is covered with pancreatic cancer cells to form a pancreatic cancer duct.129 | ||
Almost half of the ToC belong to the “compartmentalized chips” category.76,101–109 Compartmentalization of cells embedded in hydrogels can be achieved by various physical means. Several studies use co-flow patterning flowing side by side to two different hydrogel solutions. Using this method, Jeong et al.110 co-cultured mammary epithelial cells with human mammary fibroblasts and studied in vitro the transition from ductal carcinoma to invasive ductal carcinoma. Compartmentalization can also be created in a more reproducible way by playing with capillary forces.108 Properly designed pillars induce surface tension to hold the hydrogels in a given area of the chip, while nutrients are supplied through media in adjacent channels. Adjei-Sowah et al.109 (Fig. 4 top left panel) developed a tri-culture of endothelial cells, astrocytes and glioma stem cells utilizing a pillar compartmentalized chip and focused on the identification of ligand-receptor pairs. Compartmentalized chips are very versatile as different types of hydrogel can be arranged in a controlled manner. However, some applications require a fully free interface, while pillars can hinder homogeneous diffusion of the solute. Venzac et al. described a new method of compartmentalized chip, where rigid or semi-rigid structures – sliding walls – were inserted into a guiding channel open in the PDMS microfluidic chip sides.111 The advantage of this method is the possibility to create channels of any size, independently of the hydrogel solutions properties.
Another category is the “micro-wells array” which allows studying multiple and/or replicated conditions but often at the price of a limited cell complexity. This category addresses the need for multiplexing ToC studies.89,112–115 Chi et al. (Fig. 4 top right panel) achieved an elevated reconstruction of the in vivo TME combined with high throughput screening.116 They developed a three-layered ToC containing a tumor microvasculature and tumor–stromal microenvironment, along with high throughput screening capability (8 lines of 8 wells). This platform allowed studying the function of the endothelial barrier in drug response and resistance mediated by CAFs.
The “2D chip” approach includes all different designs containing cell cultures as monolayers These 2D models do not display in vivo stromal characteristics because their key point is to focus on technological advances, for instance, automation,117 control of oxygen62 or shear stress.35 Kamei et al.117 developed an integrated heart ToC containing three sets of artificial blood circulation loops thanks to the integration of pneumatic valves and a peristaltic pump. This chip allows to automatically perform the following sequence: Matrigel coating, cell introduction, flow circulation and cell staining. 2D chips can also be used to study cell migration. Agliari et al.118 reproduced the interactions between cancer and immune cells and investigated the motility of spleen cells.
“Membrane chips”, are composed of microchannels separated by a porous membrane.41,52,71,79,86,119–121 Although cells are cultured as a monolayer on the membrane, such chips are not to be categorized as 2D chips as cells can migrate through the membrane pores. Membrane chips were originally developed by Huh et al.121 A key feature of this device is the possibility to obtain an air–liquid interface (allowing respiratory epithelial cell differentiation) as well as to apply both controlled peristalsis and shear stress (respiratory lung motion). Although membrane chips were not often used for cancer studies, they are extensively in use for other physiological and pathological models (e.g. infection-induced recruitment of immune cells, breathing-induced absorption of nanoparticles).122 In 2017, Hassell et al.71 (Fig. 4 bottom left panel) created a ToC such membrane-based device. They seeded human lung cancer cells on a monolayer of primary alveolar cells on the same membrane chip, thus recapitulating a lung adenocarcinoma growth.
The category “lumen chip” consists of ToC in which a critical element is used to form lumen in hydrogels.36,39,49,73,94,123–132 This design is typically used to model blood vessels in tumors or to tightly pack cells in a cylindrical compartment. Nguyen et al.129 (Fig. 4 bottom right panel) describes a chip in which a biomimetic ductal channel containing pancreatic cancer cells is juxtaposed to a blood vessel consisting of an endothelialized perfused lumen. To build these lumen channels, acupuncture needles were withdrawn after collagen polymerization and endothelial cells or pancreatic cancer cells were seeded into each channel. They observed that pancreatic tumor cells invaded and occupied the lumen of the biomimetic blood vessel, resulting in apoptotic endothelial cells in proximity to cancer cells. On the contrary, endothelial cells in the biomimetic blood vessels in absence of tumor cells invasion did not exhibit apoptotic activity. These data supported the notion that the “lumen chip” model, although being rather simple, allows for complex phenomena to be modelled and studied.
The incorporation of these diverse spatial TME architectures on chip has already provided valuable opportunities to investigate various tumoral mechanisms. Nevertheless, considering the advancements of biological technologies and knowledge, it is obvious that our technologies must evolve accordingly. The emergence of spatial transcriptomics has offered new and insightful perspectives into the architecture and functions of the TME. Consequently, there arises a necessity to further enhance the reconstituted TME on chip to mimic the in vivo spatial organization more faithfully. Compared to other cell types found in the TME, the 3D architecture of endothelial cells has been more deeply reproduced in ToC due to its specific organization of perfusable networks. Several in vitro approaches exist to replicate the vascular compartment, offering various levels of complexity of ECs arrangement (Fig. 5): patterned microchannel, sacrificial mold and self-assembling. The most straightforward method involves creating a 2D patterned microchannel lined with a monolayer of endothelial cells. This can be achieved by utilizing a membrane configuration, wherein the endothelial cells are seeded onto a porous membrane. The membrane serves to separate the main chamber into two compartments, while additional side channels allow for cyclic stretching of the porous membrane. This configuration is very well suited to study drug penetration of cancer cells extravasation under mechanical stresses. This configuration has been exploited by, Hassell et al.71 to generate a lung ToC model with an air/liquid interface using a 3D endothelial vessel in the basal compartment seeded with lung microvascular ECs. Despite interesting features especially regarding the application of mechanical forces, this approach remains a simplified model of vascularization: geometries are square-shaped and sizes are highly different from tumor vessel networks in vivo. 3D blood vessel models with tubular structure have been created via several approaches such as bioprinting or guiding needles, called here as “sacrificial molds” and associated with the category of “lumen chips”. In this configuration, the endothelial channel can be made in a wide range of hydrogels.133,134 Miller et al.128 used a commercial chip to incorporate patient derived tumor clusters into a 3D matrix crossed by a vessel mimicking lumen channel (Fig. 5 left panel). Key features include a controlled rate of directional media flow through the cellularized lumen. Although this strategy does not always produce complex geometries, the advantage lies in its full tunability in terms of cell input, perfusion, and 3D matrix. Finally, the most complex ECs organization consists of perfusable and self-assembling microvascular networks (MVNs),101,135,136 which in vivo require interactions with pericytes or fibroblasts. Chen et al.138 studied self-organizing perfusable human microvascular networks (Fig. 5 right panel). Self-assembly is usually restricted to the use of fibrin hydrogel, the only matrix allowing endothelial vessel sprouting, ramification and self-organization.138 Recently this approach has been exploited by the group of R. Kamm.38,108,137,139–141 They sequentially added fibroblasts to preform tumor spheroid.38 They showed that this sequential approach could enhance vascularization and that the vessels close to the tumor spheroid surrounded by fibroblasts made are more perfusable.38 All these methods have already allowed the study of several important tumoral or therapeutic mechanisms. However, other approaches are currently highly promising; especially photoablation, which could enable the generation of complex vascular networks while avoiding the use of fibrin.142,143 Besides it is also worth mentioning that there are also room for improvement on the biological aspects of the reconstituted vascular compartment of the TME. In vitro replication of the vessel structure remains limited as tumor vascularization is known to be unorganized and tortuous, with a lack of pericyte coverage and fenestrated ECs, while most current models mainly focus on accurate perfusion and structural stability. Thus, the development of more accurate in vivo mimicking vascularized TME on a chip model remains a challenge.
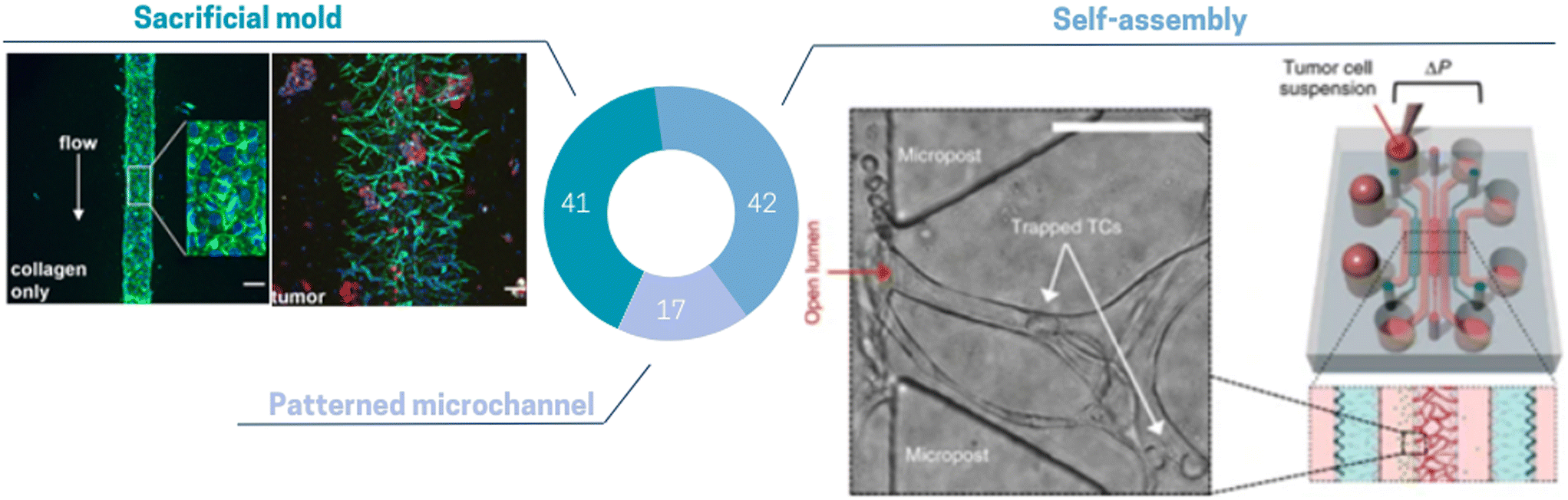 | ||
| Fig. 5 Pie chart illustrating the proportion of different vascularization methods used in ToC: self-assembly 42%, sacrificial mold 41%, patterned microchannel 17%. (Left panel) Lumen can be formed by removing a retaining rod from collagen and further cellularizing with endothelial cells.128 (Right panel) Endothelial cells elongate, form vacuoles and subsequently self-assemble into interconnected perfusable vascular networks.137 | ||
Tumor-on-chip platforms offer a promising approach to replicate the complexity of tumors in vivo within controlled in vitro settings. These models provide a unique opportunity to mimic the intricate tumor microenvironment, encompassing factors like extracellular matrix, oxygen gradients, nutrient supply, cell architecture and cell–cell interactions.
4. Applications of ToC for pre-clinical studies for cancer treatment
4.1. A wealth of new information can be extracted from tumor-on-chip
There is a large variety of information which can be extracted from ToC studies. Considering ToC studies of the past 15 years, the most common readouts used in ToC are drug-induced cell death (52% of ToC publications), followed by proteomic (23%), transcriptomic (14%) and secretome (8%) analysis (Fig. 6). Additionally, more readouts such as imaging of hydrogel fibers, diffusion of a compound through the matrix, vessel permeability,141 cell migration can also be obtained from ToC studies. This classification does not cover the whole spectrum but focuses on the most represented analytical readouts used on ToC; the analytical potential of ToC being continuously expanding.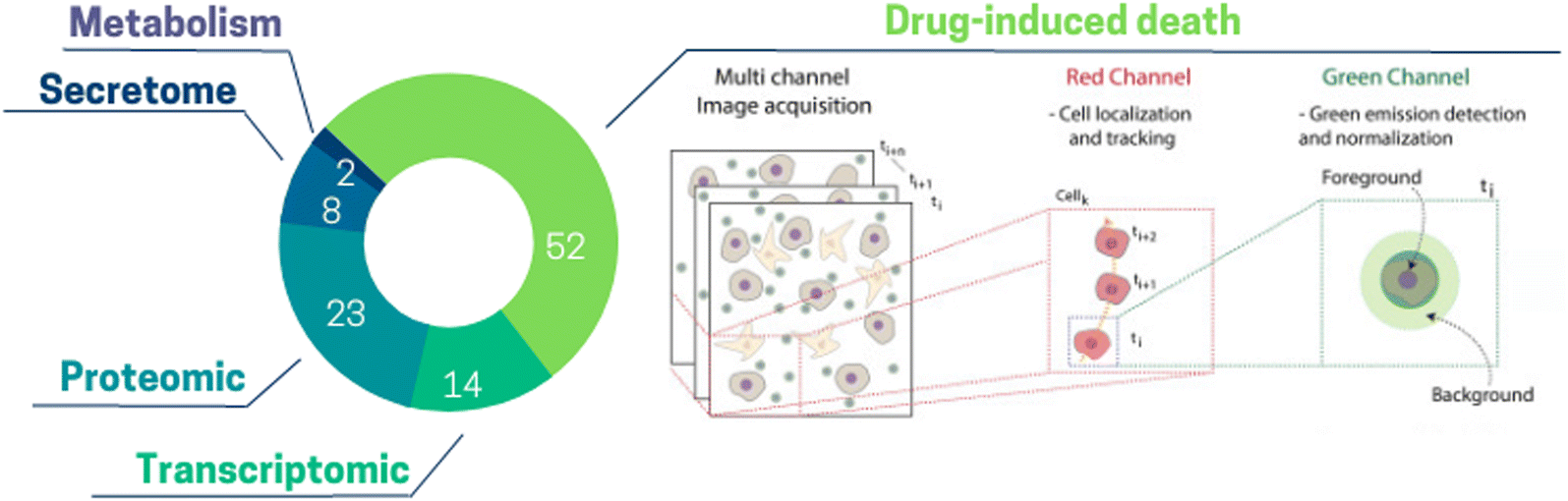 | ||
| Fig. 6 Pie chart illustrating the proportion of different information extracted from ToC: drug-induced death 52%, proteomic 23%, transcriptomic 14%, secretome 8%, metabolism 2%. (Right panel) Drug-induced death: open-source computational method can be used to extract the temporal kinetics and the spatial maps of cancer death.150 | ||
The high proportion of ToC studies measuring drug-induced death emphasizes the potential of on-chip applications for drug screening. Very recently, Jun Ye Ong et al., proposed an array device to culture PDX-derived spheroids combined with a drug concentration gradient to generate high throughput dose–response.80 This work was the first to demonstrate a quantitative correlation between drug efficacies estimated on ToC with in vivo data. Protein-based studies are conducted in about one fourth of ToC studies and mostly rely on flow cytometry, western blot and immunostaining as for example the work Boussomier-Calleja et al., which analyzed in ToC the expression of macrophage-like markers to characterize monocyte differentiation.70 Transcriptomics performed on ToC have been used to decipher the impact of several TME parameters, such as oxygen levels, matrix stiffness, cell subsets identity or gene expression. The majority of these studies used RT-qPCR, although some studies performed bulk RNA-seq. Ayuso et al., analyzed a panel of selected immunity-related genes to study stress-induced exhaustion of NK cells in co-culture with breast cancer spheroids.123 They demonstrated that NK cells in bi-culture exhibited a different transcriptomic profile, dominated by exhaustion markers, as compared to naïve NK cells. Recent studies emphasized the feasibility of single cell RNA-sequencing (scRNA-seq) of ToC derived cells. In the work of Shirure et al., several cell types (cancer cells, endothelial cells, fibroblasts, macrophages) were successfully recovered from chip devices and analyzed by scRNA-seq.104 The authors identified macrophages to dramatically impact gene expression in both endothelial cells and fibroblasts. Importantly, they were able to identify two distinct endothelial cell phenotypes as a consequence of M1/M2 macrophages and tumor cells co-culture. Several clinical applications could emerge from integrating fresh patient derived tumors on chip in combination with scRNA-seq. For example, this approach can enable the detection of rare malignant and/or chemo-resistant cancer cells, and provide supporting evidence for further suitable treatment approaches. Analysis of secreted molecules by cells – secretome – can also offer crucial information about the functional responses of the tumor ecosystem. Ligand binding assays (LBA), such as ELISA or Meso Scale Discovery (MSD) platforms can be used to measure such secreted molecules. In Jenkins et al., profiles of secreted cytokines from 3D microfluidic cultures of tumor spheroids were used to screen for the response of murine models and patient tumor samples to immune checkpoint blockade therapy (anti PD-1).144 Although cytokine analysis is fairly simple and very classical in conventional in vitro models, it is still not often used in ToC experiments.145 Recently, metabolic alterations have been shown to play a role in the sensitivity of cancer cells to drug and rewired metabolism is linked to rapid tumor growth and proliferation.146 Only a few ToC studies (2%) focused on cancer cells metabolism highlighting that this field remains to be further explored. Hou et al.147 used ultra-performance liquid chromatography coupled with mass spectrometry (UPLC-MS) to detect metabolites such as 5′-deoxy-5-fluorocytidine or 5-fluorouridine, to study metabolism-induced anticancer bioactivity. Additionally, only a low number of studies apply live markers of metabolism such as pH, hypoxia or ROS, providing both temporal and spatial information61 which are highly complementary to metabolic endpoint analysis. Among the new analyses that could be performed on ToC, it is worth citing the Assay-Guidance Manual published by the NIH Chemical Genomics Center.148 It describes guidelines for robust assay development and recommends cell proliferation and cytotoxicity assays including149 tetrazolium reduction, resazurin reduction, protease markers, and ATP detection, which are not commonly used on ToC up to date. Efforts could also be made to improve the compatibility of ToC with conventional analytical formats (e.g., the plate reader) to be able to use conventional kits and standardized assessment protocols.
To our best knowledge, ToC studies pursuing genomic analysis are rare (only 1 study was identified among the over 300 reviewed in the current article). Zhang et al.98 expanded circulating tumor cells (CTC) isolated from early-stage lung cancer patients with fibroblasts on a co-culture chip. Next-generation sequencing of 124 cancer-related genes revealed matched mutations between the primary tumor and cultured CTCs in 3 out of 8 paired CTC-tumor samples (CASP8, APC, TP53 and ERBB4 genes). Such studies suggest that the combination of ToC with next-generation sequencing could help revealing the genetic and epigenetic underpinnings of cancer, as well as help to redefine treatment paradigms.
In addition to the aforementioned readouts, the compatibility of ToC devices with high resolution imaging is also a major asset. ToC can be continuously imaged with automated video-microscopes which can additionally provide information about cell dynamics (e.g., motility, cell–cell interactions, etc.) and cellular activities (e.g. death, division, pathway activation, etc.), at single-cell resolution. Novel types of information can be deciphered by looking at the behaviors of dynamic 3D microenvironments. However, a major bottleneck is the conception and development of appropriate computer tools to process and extract the richness of biological information encrypted in on-chip images and videos. New findings can be obtained by spying tumor ecosystem dynamics within ToC in the framework of interdisciplinary collaborations between biologists and computer scientists, taking advantage also of the great power of artificial intelligence (AI) tools. For instance, a computational method, named SpatioTemporal Apoptosis MaPper (STAMP), has been developed to extract the temporal kinetics and spatial maps of cancer cell death from ToC150 (Fig. 6 right panel). STAMP revealed that cancer cells death induced by chemotherapy is transmissible, meaning dying cells promote apoptosis of cancer cells in proximity. Recently, ToC devices are also exploited to track immune cells activity around many individual cancer spheroids simultaneously at high spatiotemporal resolution. Ronteix et al.151 combined parallel imaging of T-cells behavior with probabilistic modelling to investigate the rules governing T-cells recruitment. Additionally, imaging techniques may also be used to visualize intravasation and extravasation processes, which remains a challenge for in vitro models. ToC allows studying metastatic processes in a stepwise manner.152 It enables investigating the impact of each parameter such as the presence of a specific cell type on cancer cell migration, intravasation or extravasation. ToC has been used to study the effect of monocytes, at different stages of their life cycle, on cancer cell extravasation in a 3D vascularized microfluidic model.70 Boussommier-Calleja et al. showed that breast cancer cells are less prone to extravasation in the presence of monocytes highlighting the role of monocytes in metastatic progression.70
The next step will be to apply these analysis tools for evaluation of patient-derived ToC for clinical application such as patient stratification and drug response prediction. The use of ‘personalized’ ToC composed of primary cells isolated from fresh tumors will open the possibility to conceive novel diagnostic tools that exploit AI-based analysis on ex vivo live-cell biomarkers. Some recent proof-of-concept studies support this concept. For example, morphodynamic biomarkers of primary cancer cells cultured on chips were used to predict post-surgical risks of relapse in operated breast and prostate cancer patients with high accuracy using machine-learning algorithms.153 Another study showed that a deep learning approach was able to correctly identify responding breast ToC cancer-immune co-cultures treated with a targeted therapy drug (trastuzumab) by using an atlas of immune cell trajectories.154 This suggested the feasibility of the methodology to predict drug responses using the information hidden in cell ecosystem dynamics. In the future, it will be essential to intensively pursue and to greatly expand the interdisciplinary collaboration efforts between cancer biologists, clinicians and computer scientists in order to fully take advantage of a large number of hidden information that could be extracted from ToC experiments. The combination of ToC live imaging and advanced computational image analysis, involving AI, thus provide immense opportunities to characterize the behaviors governing tumor ecosystems and to understand their responses to anti-cancer treatments.
4.2. Testing the efficacy of chemotherapies
The TME composition has a profound effect on drug efficacy and deciphering the effect this complex environment is critical.155 Since ToC allows mimicking the TME, it is a powerful platform to study drug effect as well as the dynamics of cell death in presence of drugs (Fig. 6). According to our analysis, the main anti-cancer treatments studied in ToC were chemotherapy (63% of ToC publications), targeted therapy (23%) and immunotherapy (7%) (Fig. 7).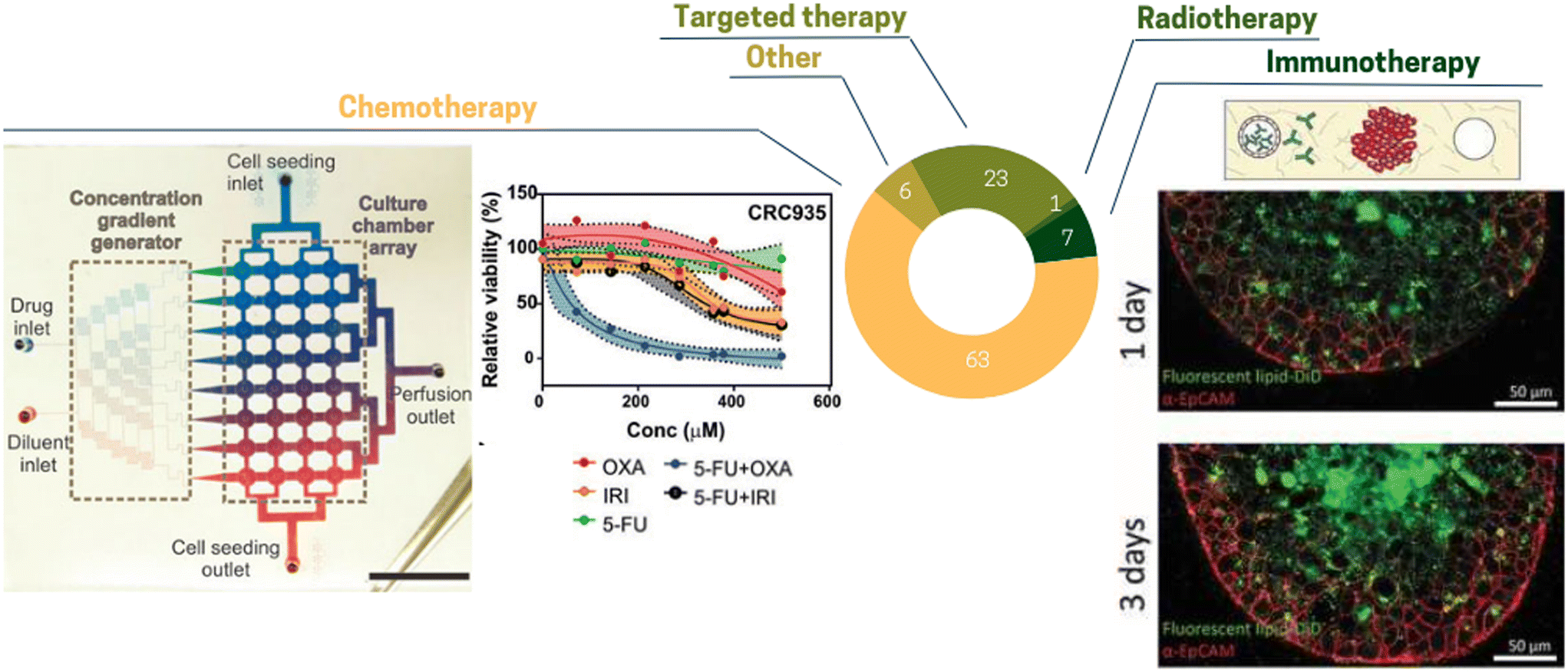 | ||
| Fig. 7 Pie chart illustrating the proportion of different treatments tested in ToC: chemotherapy 63%, targeted therapy 23%, immunotherapy 7%, other 6%, radiotherapy 1% (right panel) concentration gradient generator to test different drug combinations.80 (Left panel) Diffusion of antibody inside MCF7 spheroid.124 | ||
Chemotherapy remains the frontline treatment for advanced-stage or metastatic malignancies for which loco-regional treatments, i.e. surgery and/or radiation therapy are not useful due to the dissemination of cancer cells in the whole body.156 Chemotherapies tested in ToC are mostly taxol, doxorubicin, carboplatin or cisplatin,35,77,106,110,116,117,120,145,157–160 in agreement with clinical practice. These treatments have been mostly applied to common cell lines such as lung cancer A549, breast cancer MCF7 or MDA-MB231. Using an on-chip co-culture of cell lines (cancer cells, CAFs, stem cells and an endothelial monolayer) Chi et al.116 showed that the co-culture with CAF delayed the response of the TME to doxorubicin in comparison to the co-culture with normal fibroblasts. Apart from cell lines, ToC also enables the in vitro culture of patient samples for chemotherapy testing.160 Chakrabarty et al.86 tested the response to cisplatin using cisplatin-sensitive and cisplatin-resistant breast cancer PDX tumors. Interestingly, breast PDX cultured in the ToC platform showed a stronger response to cisplatin treatment than the conventional ex vivo culture method (6-well standard plates on an orbital shaker), suggesting that the ToC platform provides with a more optimal drug delivery into the tumor slices than the ex vivo culture method. Haque et al.79 developed a ToC incorporating patient derived organoids (PDO) and stromal cells (pancreatic stellate cells and macrophages). They showed that targeting stroma cells (stellate cells by all-trans retinoic acid or macrophages by Clodrosome®) improved therapeutic effect of gemcitabine on cancer cells. Conversely, in a ToC model of breast cancer metastasis to bone,161 including osteoblast-like cells seeded on a 3D-printed biomimetic bone scaffold, the sensitivity to cisplatin of PDX-derived triple-negative breast cancer (TNBC) cells was reduced with respect to conventional 3D cultures in Matrigel, which is consistent with the clinical observations that chemotherapy often failed to completely eliminate TNBC cells colonizing the bone.
Tumor chemosensitivity assays (TCAs) with conventional methods have been gaining attention over the past few decades. They provide a satisfactory negative predictive value (showing the possibility of drug resistance), however they only have a moderate positive predictive value (showing the possibility of drug sensitivity).156 In this context, ToC appears as a future powerful tool for TCAs with the possibility to evaluate multiple drugs and multiple doses.156
4.3. Targeted therapy developments
Over the past two decades, there has been a tremendous shift in cancer treatment from chemo- to targeted therapy in cancer patients' subsets.156 Targeted therapies often aim to deliver drugs to cells with molecular genetic alterations specific to cancer cells (gain of function, oncogene mutation or gene amplification).162 Most of these targeted drugs have a much higher affinity for altered proteins in cancer cells than their normal counterparts in normal cells, which explains their lower toxicity (favorable therapeutic index). Targeted therapies can be roughly classified into two categories: small molecules (0.1–1 kDa) and macromolecules (greater than 1 kDa, also called biologics). Biologics are relatively complex molecules derived from living cells or through biological processes, such as monoclonal antibodies or antibody–drug conjugates.163 Despite the recent interest and success of biologics, small molecules, which are made via chemical synthesis, are still in the picture of innovative drug research and development. About one fourth of drug testing in ToC focused on targeted therapy. Targeted therapies exploring a wide range of targets have been tested on chip, among them kinase inhibitors (Tarceva, erlotinib),71,164 HER2 inhibitors (trastuzumab),165 CXC chemokine receptor inhibitors,73 EGFR inhibitors (cetuximab).165Nguyen et al. studied the effect of trastuzumab, a monoclonal antibody directed against the HER2 receptor, on breast cancer cells presenting HER2 amplification.166 Trastuzumab alone or immune cells (PBMC) alone had mild effects on HER2+ cancer cells, but their combination induced a massive cancer cell apoptosis, exquisitely recapitulating on ToC a complex immune behavior, namely an anti-tumoral antibody-dependent cell cytotoxicity (ADCC). Live imaging combined with advanced cell tracking algorithms allowed to measure the number and the duration time of interactions between cancer and immune cells. In this HER2+ breast ToC, addition of trastuzumab specifically promotes long cancer–immune interactions (>50 min). Moreover, the presence of CAF (CAF-S1 sub-type) abolished this trastuzumab-dependent stimulation of cancer–immune interactions, suggesting that CAF-S1 cells may contribute to trastuzumab resistance by participating in immunomodulation.166 Other targeted therapies such as anti-angiogenic drugs have been tested in vascularized patient-derived ToC vessels.94 For different drug concentrations and different targets, the authors quantified vessel permeability as well as vessel sprouting and confluency.
Targeted therapies have already demonstrated their potential in clinics for different cancer subtypes and still holds great promise in particular with the recent emergence of the ADC drugs coupling targeting and chemotherapy precision delivery. However, small-molecule targeted anti-cancer drugs, despite high response rates and rare primary resistance, still face many challenges with the emergence of resistant clones.162 Drug resistance could be linked to several mechanisms, including gene mutation or amplification, leading to parallel signalling pathway activation, apoptosis or autophagy dysregulation, etc.167 We envision that the ability of ToC to recapitulate the cellular complexity of the tumor will be a strong asset for testing new targeted therapy testing, and we anticipate an increase of the anti-cancer drug application in ToC.
4.4. Pre-clinical studies on tumor-on-chip for immunotherapies
As ToC can include diverse immune cell types, it is emerging as a powerful model for immunotherapy testing. Immune system within the tumor microenvironment consists of adaptive and innate components168 that are interdependent. The innate system is the first defense mechanism against tumor-specific or tumor-associated antigens, and it generates short-lived responses of antigen-specific immune cells such as monocytes, macrophages, dendritic cells and natural killer (NK) cells. On the other hand, the adaptive system can generate immune memory producing long-lasting responses through T-cells and B cells.169 In turn, it has been shown that the presence of immune cells in the TME affects tumor progression, explaining why some of the on-going and very promising ToC strategies are focused on immunotherapy development.Immunotherapies are innovative anti-cancer treatments that have revolutionized anticancer therapy since the early 2000s through unleashing the immune anti-cancer response.170 Immune checkpoints are receptors expressed by immune cells that enable dynamic regulation of immune homeostasis and are particularly relevant to T-cell functionality.171 Most immune check-point inhibitors (ICI) have been focusing on reinvigorating CD8+ T-cells to target cancer cells. While their success was originally thought to be dependent on local T-cell abundance, or on the cancer cell abundance of immune checkpoint targets, favorable responses to these therapies are still largely variable, particularly in solid tumors, where T-cell infiltration is highly variable and does not necessarily correlate with therapeutic efficacy.172 Recent evidence suggested that there is more to be understood about the immune cell component within the TME and how to exploit them for therapeutic purposes. A major challenge to study tumor–immune interactions and develop therapies is the lack of effective and representative models. It is well established that the innate and adaptive immune systems of animal models, like rodents, are different from those of humans.173 Consequently, syngeneic mouse tumor models, allowing for the participation of the native rodent immune system, rarely mimic human cancer behaviors and their applicability remains limited. PDX models are by nature immune-deficient models and are not suitable for studying immune checkpoint inhibitors targeting human T-cells. Humanized mice models could be used but such mouse humanization is still a highly variable, costly and time-consuming technology. Moreover, the approaches to investigate cancer cell escape from immune surveillance are limited to intravital microscopy in mouse models or observations of tissue slices from human tumor samples. This has motivated the development of ToC as a unique technological approach to reproduce the multiple layers of complexity of cancer–immune system crosstalk.174–176
Less than one tenth of the ToC studies focused on immunotherapies27,38,83,89,123,177–180 but interestingly, the types of immunotherapy (alone or in combination) tested are broad, including immune checkpoint inhibitors (ICI), oncolytic viruses, and T-cell therapies. Parlato et al. reported that IFN-α-conditioned dendritic cells (DCs), grown in co-cultured ToC, exhibited remarkable migration and phagocytotic activity against colorectal cancer cells pre-treated with IFN-α and romidepsin, a histone deacetylase (HDAC) inhibitor.177 In this model, DCs were cultured in the central chamber while both untreated and treated cancer cells were embedded in collagen I gel in the two adjacent chambers to evaluate DCs behavior in the extracellular matrix. An increase in DC migration toward pretreated colorectal cancer cells in this model was driven by the CXCR4/CCL12 signaling axis, consistent with the DC responses in vivo. Most interestingly, by labeling DCs and SW620 with fluorescent dyes, cancer phagocytotic activity was captured in real-time using confocal microscopy, making this model more advantageous than in vivo models, where real-time monitoring of DC phagocytotic activity is not possible. Ayuso et al. developed a 3D microfluidic model that incorporated NK cells, endothelial channels (HUVECs), and MCF7 spheroids (Fig. 7 right panel). The authors studied antibody penetration into the spheroids, NK cell migration and antibody-dependent cell cytotoxicity (ADCC).124 The dynamics of ADCC shown in this model via live imaging was remarkable. NK cells were able to directly penetrate deep into the spheroid core and destroy the cancer cells in a matter of hours, without first killing the outer layer of the tumor. Such studies highlight the power of ToC to pave the way for new studies which were not feasible with classical cell culture models. Indeed, ToC could enable researchers to focus on antibody dynamics and study the impact of various parameters separately such as endothelial permeability, tumor penetration or antibody clearance by tumor cells. A very recent study from Bi et al. interrogated the role of macrophages in tumor progression using a ToC device and downstream single cell RNA-seq. They introduced M1 or M2 macrophages into a 3D tumor-on-chip model to investigate tumor behaviors in response to these macrophage subsets.104 In this model, M1 macrophages exhibited anti-tumor properties, whereas M2 macrophages showed significant pro-tumor effects, which is consistent with the current understanding of tumor-associated macrophages. However, to date, most ToC that incorporate immune cells do not allow for high-throughput testing. To address this limitation, Ronteix et al. recently introduced a platform for the parallel formation, manipulation and multiplexed observation of hundreds of tumor spheroids within stationary microfluidic droplets, in the presence of antigen-specific cytotoxic T lymphocytes.151 Exploiting mathematical probabilistic models, the quantity and quality of spatiotemporally resolved data allowed to establish that the first recruited T-cells initiate a positive feedback loop to accelerate further recruitment to the spheroid, confirming the cooperation between T-cells in the tumor killing process.151
Injecting oncolytic vaccinia viruses (OVV) directly into the tumor microenvironment is an alternative to improve tumor antigen recognition and to strengthen T cell responses.181 In a lung ToC model, infection by OVV was shown to increase cancer–immune interaction times leading to cooperative antitumoral activity of immune cells and OVV.154 Proof-of-concept studies also illustrate the feasibility to exploit ToC models for innovative immunotherapies such as Chimeric Antigen Receptor (CAR)-T-cells. In the model developed by Pavesi et al., human T-cells engineered to express tumor-specific T cell receptors (TCR-T-cells) were added into the adjacent channels to investigate the ability of the modified immune cells to migrate and kill the tumor target and their secreted soluble factors.182 A recent work by Wan et al.38 presented a vascularized in vitro model which can be used to evaluate CAR-T cell recruitment, killing capacity, and inflammatory response. After 96 h of perfusion, higher densities of both T-cells and dead cells were found in the CAR-T cell containing ToCs as compared to the control T-cell containing ToC in both co-mixed and sequential tumor spheroids.
Although ToC models offer a powerful experimental setting to quickly test immunotherapies, the vast majority of the immune-competent ToC models are so far based on cell lines and allogeneic immune cells, i.e. immune cells not coming from the same individual (mainly PBMC), exhibiting the obstacle of human leukocyte antigen (HLA) incompatibility. Immunotherapy testing requires the use of autologous cytotoxic T-cells to avoid the risk of allogeneic reactions, which, however, would require longer time than the few days needed for ToC experiences. We envision that one of the upcoming challenges in the field will be the development of patient-derived ToC for immunotherapy testing. Some latest works have shown the feasibility of such an approach. For example, a recent work reported the use of a glioblastoma-on-a-chip model to dissect a reconstituted immunosuppressive tumor microenvironment (composed of tumor-associated macrophages (TAM) and cytotoxic CD8+ T-cells) and its response to programmed cell death protein-1 (PD-1) checkpoint blockade. Interestingly, different glioblastoma subtypes displayed distinct CD8+ T-cells behaviors (extravasation, tissue infiltration, cytotoxic activities) as well as cytokine profiles. Moreover, co-targeting of PD-1 immune checkpoint and TAM-associated CSF-1R signaling improved therapeutic efficacy on-chip. However, again, human CD8+ T-cells were allogeneic, sorted from PBMCs, limiting the clinical significance.27
Even though immunotherapies can produce impressive and long-term responses in some cancer patients (e.g., 20 to 40% of lung cancer patients), their clinical benefits remain unsatisfactory because the majority of cancer patients are non-responder.170 What remains to be explored in these tumor–immune models is the incorporation of the adaptive immune cell component, in particular, autologous T-cells, as well as the integration of an in vitro immune organ, namely bone marrow- or lymph node-on-chip, to further understand tumor immunity and develop new therapeutics. Most models utilize a variety of cell sources, both immortal and primary cells in combination, making it difficult to study the adaptive immune response. A good metric of ToC would be to evaluate how deeply they are able to reproduce the in vivo functional readouts such as cytokine release, phagocytosis, IgM/IgG class switching.
Finally, immunotherapy is a fast-evolving field, and one of the further challenges in immunotherapy is the development of bispecific antibodies (BsAbs) that can directly target two different antigens on immune cells and/or tumors (tumor-associated antigens), synergistically engaging T-cells onto cancer cells, thereby increasing cytotoxic activity.170 We anticipate that in the near future ToC technology will be able, by reproducing the complexity of cancer–immune system crosstalk, to help the development of novel therapy exhibiting multiple cell interactions such as bispecific antibodies.
4.5. Drug combination in ToC
Over the years, the concept of combination therapy, which relies on combining two or more therapeutic agents, with different modes of action, has been introduced to overcome cancer treatment resistance. Such combination is either synergistic or additive, and therefore, a lower therapeutic dosage of each individual drug can be required, which also spares the cumulative toxicity.183 Combination therapy exhibits numerous benefits, such as the ability to target multiple oncogenic pathways, to improve and prolong therapeutic responses while reducing the likelihood of therapeutic resistance.184 This can include the combination of chemo- and radiotherapy, chemo- and immunotherapy, or chemotherapy and targeted agents.In contrast to conventional cell culture, fluidic control in ToC allows for creating precise drug mixing and variations over time. Ong et al.80 introduced a ToC with a concentration gradient generator (Fig. 7 left panel). This configuration allowed assessment of 8 drug concentrations and 5 different drug combinations. They showed that tumors did not respond to single-agent Oxaliplatin (OXA) and 5-fluorouracil (5-FU) treatments, but conversely had improved sensitivities when both drugs were combined (5-FU + OXA). This work represents a great illustration of the ToC potential for drug testing. Recent publications highlighted the possibility to test combined therapies on fresh surgical tumor81 or PDX.85 For example, Ivanova et al.85 tested drug combinations in breast cancer patient xenograft-derived organotypic spheroids. They identified that neratinib and trastuzumab combination was more effective compared to each agent alone, and was associated with more robust inhibition of HER2. Importantly, Eduati et al.91 presented a plug-based microfluidics platform for functional screening of drug combinations (56 different conditions with at least 20 replicates each). They suggested a novel drug combination for pancreatic cancer cell lines: PHT-427 and MK-2206, which consist of two serine/threonine kinase AKT inhibitors acting through different sites. This study highlights that the best drug combination can be different for each patient and that high throughput ToC holds a great potential for personalized medicine.
Finally, drug combinations can also include immunotherapies. With a vascularized ToC, Humayun et al.73 demonstrated that by combining immunotherapies inhibiting IL-6, IL-8 and MMP-3, the extravasation events can be reduced. In the near future, ToC could be pivotal to decipher the intricate interplay between ADC and ICI. ADCs can selectively induce death of target-expressing tumor cells and activate tumor-specific adaptive immunity through increase of T-cells infiltration in the tumor microenvironment, whereas ICI reinvigorates exhausted T-cells, enhancing antitumor immune responses.185
4.6. Support for nanomedicine innovation
Besides these conventional anti-cancer treatments, recent advancements in nanotechnology have opened new windows for the discovery and development of anti-cancer nanomedicine strategies. Having gone through several generations of nanomedicine drug development, nowadays we are at the verge of multifunctional nanomedicine therapeutics, allowing for targeted drug delivery and stimuli-triggered nanosystems, as well as their combinations. Nanocarriers appear as powerful technologies to release anticancer drugs in a stable and controlled manner. Moreover, the use of stimuli can assist in the controlled release of the drug to ensure specific toxicity to the tumor tissue, while sparing the healthy tissue.186 A variety of nanotechnology-based treatments have already gone through clinical trials, are marketed and are being currently used for clinical cancer therapy applications.187 However, the prevailing majority of research on nanomedicine therapies for cancer care is still at the stage of preclinical studies.188 Nowadays, conventional in vitro models for nanomedicine screening as well as in vivo animal models are unable to closely replicate human in vivo tissue environment which, in turn, significantly impedes adequate nanomedicine development and evaluation. Crucial parameters such as nanoparticles' toxicity, diffusion, internalization, accumulation oftentimes cannot be adequately assessed with conventional models. In regard to these limitations, ToC are gaining interest for the development and evaluation of nano-based therapies in tumors.189 ToC may allow to finely evaluate these pivotal criteria of evolving nanotherapeutic approaches through precise control of microenvironment, phenotype, dynamic fluid flows and physiological gradients among the others – all of which affect nanomedicine's suitability and progress of preclinical studies.190,191While multiple works in ToC have been focused on the various key aspects of nanomedicine evaluation for clinical applications, such as nanoparticles (NP) toxicity,192,193 transport,194,195 uptake,193,196 accumulation,197,198 they only represent 13% of reviewed ToC-related studies. For the purposes of this review, we will focus on the investigation of the therapeutic impact of NP-based therapies on tumor tissues, their efficacy and cellular effect readouts in ToC models. Broadly, nanomedicine therapies can be subclassified into drug nanocarriers and stimuli-responsive nano objects that function as a treatment itself upon activation.187 The former has been mostly explored up to date as they allow to increase loaded conventional drug's stability, solubility, blood circulation time as well as to provide controlled drug delivery to the site of interest.199 In the scope of ToC, multiple studies have been carried out to demonstrate effective tumor cell death following the nanocarrier drug exposure. Thereby, Liu et al. have reported a ToC model of human glioma to study tumor targeting with nanomedicine. They have demonstrated remarkable tumor reduction post-treatment with paclitaxel-loaded folate-decorated NPs as well as significant cell death due to both apoptosis and necrosis.200
However, following the concerns of the high NP concentrations required and high systemic drug toxicity, a new line of research is dedicated to the stimuli-responsive nano objects that can be activated by endogenous or exogenous sources at the site of interest and operate as therapeutic agents themselves. Some of the most common non-invasive, stimulating therapies are exogenous triggering mechanisms such as photodynamic therapy (PDT) and hyperthermia-based therapies (e.g., magnetic hyperthermia, photothermal therapy (PTT), ultrasound, among others).201 PDT is based on the phototoxic reactions that stem from the photosensitizer activation by light within tumor cells.201,202 Recently, Flont et al. designed a ToC model of ovarian cancer to evaluate the effect of PDT through free vs. nanoencapsulated photosensitizer on cancer cells.203 Their results have demonstrated remarkably higher cytotoxicity of nanoencapsulated photosensitizer as compared to the free one, which was further boosted by the PDT. They also demonstrated that PDT induced ROS generation in cancer cells while having little effect on non-malignant cells. Another ToC study on externally triggered nanoparticles was conducted by Lee et al. on a co-culture of breast and glioblastoma cancer cells via gold nanorod-mediated PTT.204 They have demonstrated drastic reduction in cell viability upon PTT exposure as compared to separate conditions with either nanorods presence or PTT, indeed indicating an advantage for the stimuli-induced effect.
Stemming from the beneficial characteristics and therapeutic outcomes presented by the stimuli-responsive nanoobjects, some of the recent strategies in nanomedicine therapies development have focused on the combination of nanocarrier and stimuli-responsive nanosystems, which allow for the dual action with triggered drug release, thus limiting the systemic exposure to the toxic chemotherapy. Agarwal et al. using a vascularized ToC model of breast cancer, have shown that nano-encapsulated doxorubicin is significantly more effective for cell death induction than free doxorubicin.205 Moreover, the Dox release can be controlled via NIR irradiation thanks to the photothermal and photodynamic sensitivity of the NPs.205 Another study involving stimuli-responsive nanocarrier drug delivery was conducted by Zervantonakis et al. in which they demonstrated in a ToC model of rat glioblastoma that Dox-loaded thermosensitive liposomes, when heated induce the greatest effect on tumor cell death and proliferation inhibition as compared to the single application of either drug-loaded NPs or heating.206
These different examples evidenced that ToC modeling would allow us to better investigate the effect of the new generation of nanomedicine therapies by being able to finely assess their influence on the tumor microenvironment in a human-mimicking tissue, therefore facilitating the transition towards the clinical studies.
5. ToC paths towards pharmaceutical and clinical applications
Classically, from the conception of a new drug to the launch of the product, the drug development process can take up to 15 years (although shorter times have been recently shown for targeted therapies), and cost upwards of $1 billion.207 To date, the success rate of new anticancer drugs development is very low for two main reasons: the lack of clinical efficacy and the unmanageable toxicity of the drug.4 One third of drugs fail in clinical phases due to an insufficient therapeutic index, meaning that the efficacious dose and the toxic dose are too close.208 This number evidences the shortcomings of current preclinical models to accurately predict the right target at the right dose. In order to reduce such attrition between preclinical and clinical research, the FDA has recently requested 5 million US$ to develop a comprehensive strategy on alternative testing methods.Among the different in vitro preclinical models, patient-derived cancer organoids (PDO) appear as a powerful new model. A recent review, including 60 studies, indicated that organoid cultures are faithful predictors of patient response to chemotherapy for different types of cancer.12 However, the establishment of PDO is not equally effective for all solid cancers and cancer organoids still lack some cells component of the TME, including stromal cells, immune cells, and endothelial cells. Moreover, a big challenge of organoids is to align the timescales of model establishment (1–6 months) to those of clinical decision-making. ToC by combining a short establishment time and a high control of several TME features are expected to better predict patient's treatment response.12 ToC has the potential to complement conventional preclinical models and the global ToC market size is projected to reach 110 million dollars by 2030.209
To make ToC seamlessly integrated into the drug discovery and development process, three main aspects must be considered: standardization, reproducibility, and throughput. Since ToC are still in the early-development stage, consistency and standardization are still lacking, and there is often variability in materials, device fabrication and operating conditions.210 The US IQ MPS workshop 2022 and the European ORCHID project have clearly identified standardization as a fundamental pillar of the advancement of MPS technologies. However, until now, there are currently no specific standards that have been agreed upon. To meet these standards, ToC will have to be independently validated by third-party testing centers. In line with these requirements, the NIH/NCATS and its Tissue Chip Consortium has been working on the establishment of the Tissue Chip Testing Center program dedicated to organ on chip validation. This independent validation is a stepping stone for the MPS field to provide more confidence for industry to onboard this new technology. Beyond standardization, reproducibility is also an important challenge from industry perspective; future studies would also be needed to properly assess the ToC reproducibility.
Another important consideration is related to the ability of this new technology to achieve the level of multiplexing that would allow rapid and efficient drug discovery at a large scale. So far, only few ToC studies were able to process samples at high throughput (i.e. custom micro-well arrays). High-throughput ToC would be a real breakthrough as it would allow for testing multiple compounds, and multiple concentrations in a complex and controlled reconstituted TME. Some studies recently showed the feasibility to automate ToC operations, such as media changes, pH testing, compounds monitoring (oxygen, lactate, glucose), or drug injection.91 On chip flow control can be used to refresh cell culture medium preventing medium acidification and O2 decrease.211 Flow control of ToC should include a partial and controlled recirculation of cell medium to balance nutrients refreshing and cell secretions dilution. The automation of ToC models would considerably improve their ease of use and therefore their reproducibility then supporting regulatory and quality standards.212 We believe a final goal would be to have automated high-throughput ToC with monitoring and feedback loop for a variety of key parameters (such as pH, oxygen and key functional metabolites).
Some pharmaceutical industries have already taken the step towards ToC tests in their pre-clinical studies. A recent review reported213 collaborations between several MPS manufacturers (Mimetas, TissUse, Emulate) and pharmaceutical industries (Astrazeneca, Galapagos, Janssen, Novo Nordisk, Bayer and Roche) at different steps of drug discovery: target identification and validation, discovery, pharmacokinetics and pharmacodynamics, preclinical safety and clinical developments. These early adopter studies will provide a better understanding of how the technology is deployed. Gaining the confidence of more Research Contract Organizations (CROs) could also help increase ToC use at the target identification and validation steps.214 In this section, we aim at providing insights on how ToC can bring added values for companies in their anti-cancer drug development process. We focus on three main aspects of this process:215 (i) “trust in target” which identifies the right biological target and understand its role, (ii) “trust in targeted patient population” that defines the right patient population with any needed stratification strategies and (iii) “trust in therapeutic index” which identifies the right molecule that delivers the right exposure at the target site of action without compromising patient safety.
5.1. Trust in target
The development of a new drug starts with target identification and validation.207 During this pre-discovery phase, high-throughput screening (HTS) campaigns are initiated to screen 103 to 106 potential drugs (called “hit”) on cell lines. After screening, the “hits” identified are further evaluated through different methods (e.g. dose–response curves). Then, during the hit-to-lead stage, “lead” development candidates are synthesized and their efficacy is estimated. An increasing amount of evidence has established that while extremely informative, screens performed with 2D cell culture often do not recapitulate key information such as drug response and sensitivity. As a result, many targets have been likely missed by hit identification experiments. In this context, ToC offers a great advantage because of both increased complexity and versatility, to allow fit-for-purpose implementations for each disease. ToC can be set up with various degrees of complexity selected based on the specific need to be addressed. Moreover, ToC are compatible with a wide range of readouts such as genomics, proteomics and transcriptomics, which could allow the identification of new targets in presence of the TME. The combination of ToC with in vivo imaging technology would be tremendously useful for target identification and validation as it would allow to identify molecules capable of eliciting the desired effect with phenotypic live monitoring. Once ToC reaches higher throughput level, it will appear as a new high-content screening (HCS) tool allowing for multiparametric phenotypic outputs that can be used for more comprehensive drug screens.5.2. Trust in targeted population
Personalized medicine aims to move apart for the “one-size fits all approach”, by providing the most suitable medicine, at the right dose, for the right person, at the right time, at a reasonable cost.216 This definition encompasses two ideas: (a) finding the most effective drug for an individual patient by testing different drugs on his/her tumor sample in the lab, (b) defining subpopulations of patients based on biomarkers to anticipate the efficient drug which should be chosen for an individual patient. Several prognostic and predictive markers have been identified (e.g., somatic or germline mutations) to determine individual profile of each patient and tumor.217 ToC could be a powerful tool in this field by combining functional testing on patient sample and biomarkers discovery during pre-clinical phases.218Given the heterogeneity of primary tumor tissues, these ToC could be employed to assess the degree of heterogeneity in drug response according to various clinical characteristics, or genetically similar subgroups. The combination of ToC personalized medicine with artificial intelligence paves the way to digital/in vitro twins for the development of predictive responders and non-responders models.219 The combination of patient ToC functional cytotoxicity assays with specific mathematical models could allow to predict the clinical response. For example, using conventional cell culture methods, Silva et al. developed a novel tool capable of predicting within 5 days the clinical response of multiple myeloma patients to 31 drugs over months, using fresh bone marrow aspirates.220 They implemented a parametric model in which the likelihood of cell death depends on drug concentration and exposure time. Importantly, the algorithm output is not only limited to a dichotomized response/no-response or depth of response, but can also predict the trajectories of clinical response during the first 3 months of treatment. The combination of mathematical algorithms with ToC could provide precise clinical insights on treatment efficacy in a timely manner and assist oncologists in practicing truly personalized management. The current challenge is to provide robust clinical validations of ToC-based predictions by performing rigorous correlations between ex vivo ToC responses and in vivo patient responses. This requires close collaborations with clinicians and establishment of appropriate patient cohorts.
5.3. Trust in therapeutic index
One of the main translational goals in drug development is the prediction of adverse events. Current toxicity regulations require testing first on a rodent and then on a larger mammal (pig, rabbit, dog, monkey) in order to identify the human dose prediction (HDP). However, very often animals tolerate higher dose than humans. For example, Chou et al.221 used Emulate chips to create an in vitro preclinical model of human hematopoiesis that recapitulates many clinically relevant features of bone marrow pathophysiology in response to drugs and ionizing radiation. The bone-marrow-on-a-chip may serve as a human-specific alternative to animal testing for the study of bone-marrow pathophysiology.A major drawback of conventional in vitro cancer models is that drugs are commonly delivered in fixed concentrations. However, this does not accurately simulate the pharmacokinetic (PK) concentration changes experienced in vivo due to drug metabolism and clearance mechanisms, and therefore reduces the predictive value of the data.
To solve this problem perfusion in ToC can be employed to simulate the liver's metabolism by changing the concentration over time, i.e., a rise in blood concentration followed by a decrease. Recent work222 reproduced in vivo PK profiles of different anti-cancer drugs over multi-week experiments, both as single-agent therapy or drug combinations. Interestingly, the authors compared the effects on tumor cell efficacy in vitro with efficacy seen in in vivo xenograft models. They reported that the incorporation of PK into ToC could improve in vitro–in vivo translation understanding for early therapeutic insight. Petreus et al.223 designed a pump-driven microfluidic platform capable of mimicking in vivo anti-cancer drug pharmacokinetic profiles in ToC. Tumor xenografts were subcutaneously implanted in mice and subjected to the same treatment schedules as the spheroids in the chip and demonstrated that such microfluidic ToC platforms can successfully predict the efficacy. Incorporating flow into ToC could thus assist with the understanding of how different sequencing (continuous, intermittent, gapped schedules) of the drugs affect their efficacy, thereby guiding the design of confirmatory in vivo studies.
Physiologically based pharmacokinetic and pharmacodynamic (PK/PD) models could also be proceeded with multi-organ co-culture. Particularly, as liver and kidney play a big role in drug metabolism and clearance, there is a need to connect liver/kidney models to cancer tissue for the study of small molecules. In the case of biologics larger than 45 kDa, there is less interest in such systems because they are cleared from the system mainly by endocytosis.224 McAleer et al.225 described a multi-organ device with a PK/PD profiling of tamoxifen on MCF-7 breast cancer cells in the presence/absence of liver cells. They also simultaneously monitored the effect of tamoxifen on cardiac function. The efficacy of multi-organ MPS to predict human PK/PD was also demonstrated in other studies: for capecitabine, and its 5-fluorouracil metabolite, in a four-organ system, composed of intestine, liver, cancer, and connective tissue models;226 for cisplatin using coupled bone marrow, liver and kidney chips.227
6. Conclusion
The past 15 years have witnessed significant advancements in the field of ToC. The microfluidics community in close collaboration with cell biologists and clinicians has actively engaged in this field and achieved impressive milestones. These efforts resulted over the years in an increasing level of complexity on-chip and, consequently, in a better biomimicry of these devices (Fig. 8 and S3†). In this review, our objective was to provide a comprehensive overview of the current state of ToC research and to identify the potential next steps and challenges. To achieve this, we conducted an extensive quantitative analysis of the literature, dissecting the diversity of cell models, drug treatments and extracted information (Fig. 9). Moreover, we highlighted the current promising results of ToC clinical validation.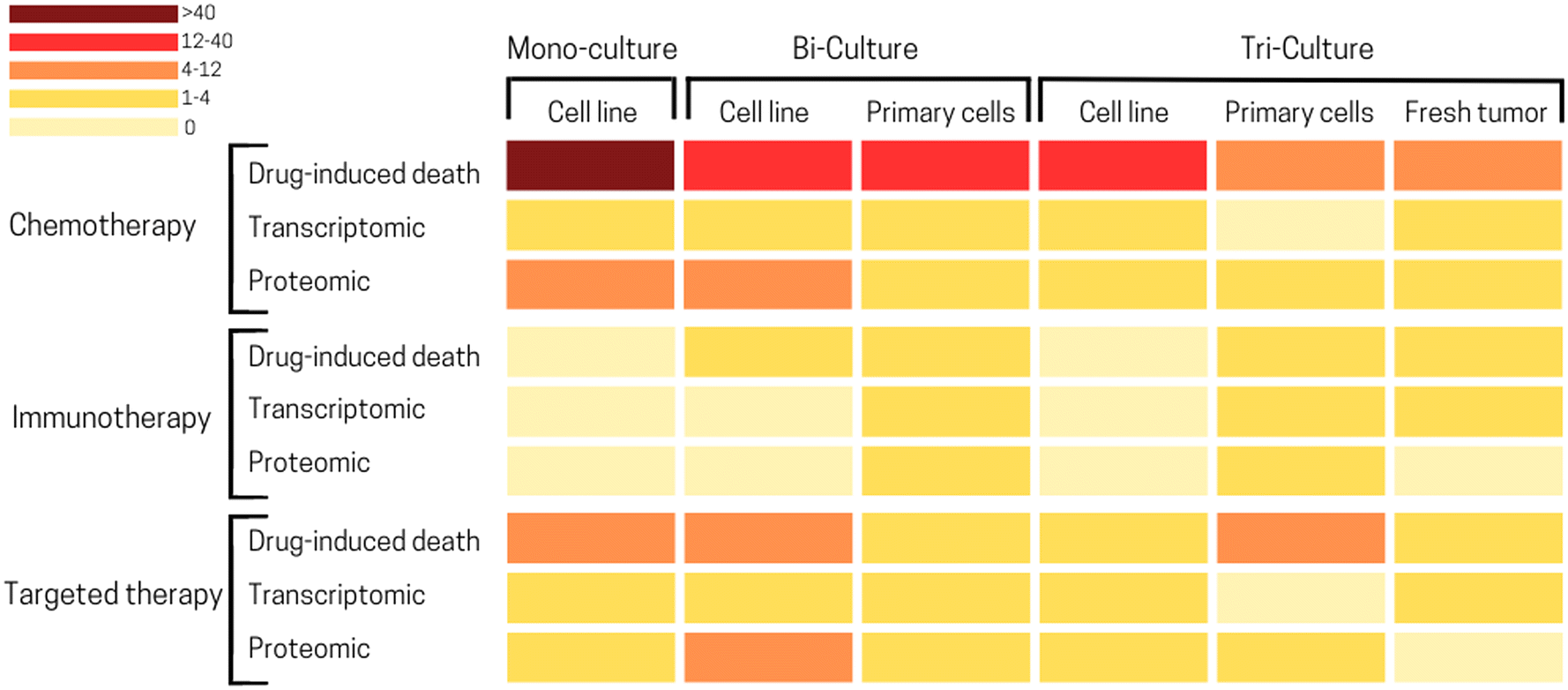 | ||
| Fig. 8 Heatmap representing the number of publications according to the cellular complexity (mono-culture, bi-culture, tri-culture) with different cell sources (cell line, primary cells or fresh tumor) for different drug treatments (chemotherapy, immunotherapy, targeted therapy) and information extracted (drug-induced death, transcriptomic, proteomic). | ||
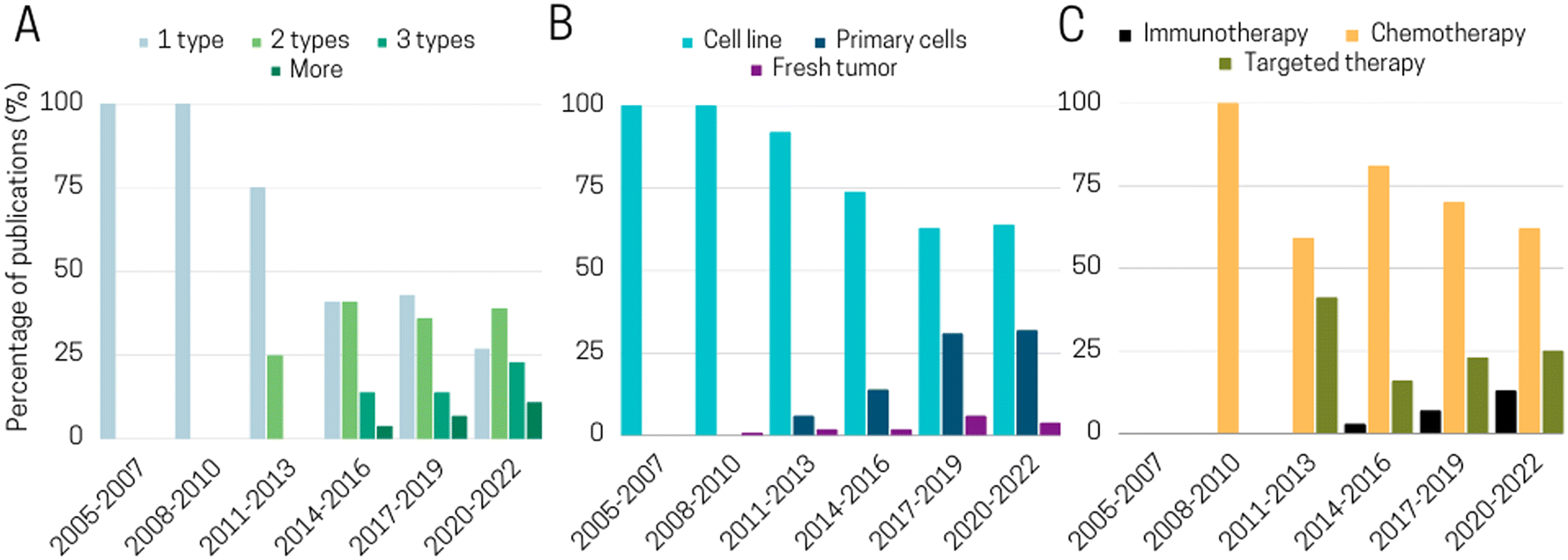 | ||
| Fig. 9 Evolution of cellular complexity and drug tested over time during the last 15 years of ToC development. Histograms illustrating evolution of A. cell complexity, B. cell source and C. tested therapy in publications. | ||
In this review, we first focused into what sets ToC apart from other in vitro models, specifically explaining the various parameters that can be finely controlled in ToC systems. These parameters include the properties of the extracellular matrix, mechanical forces exerted on cells, the physico-chemical environment, cell composition, and the architecture of the tumor microenvironment. ToC ability to precisely and individually manipulate TME parameters enables researchers to dissect the influence of these TME parameters on tumor development, growth, invasion, and response to drugs.
Nevertheless, beyond their interest for basic research, ToC have also the potential to become a transformative technology for clinical and preclinical studies. This potential relies on several factors, notably their compatibility with a wide range of analytical techniques, both conventional and state-of-the-art, such as single-cell-based technologies. Moreover, we believe that the information that can be extracted from ToC remains largely untapped, presenting an avenue for future exploration. Particularly, there is a need to exploit the hidden insights within ToC-generated data, leveraging novel approaches in data analysis. By doing so, as a community, we could be able to unlock a wealth of valuable knowledge and further enhance the capabilities of ToC platforms.
When considering ToC implementation in clinical or preclinical studies, it is also necessary to mention the demonstrated potential and compatibility of ToC devices with a wide range of therapeutic modalities. These modalities encompass chemotherapy, targeted therapy, and more recently, immunotherapy (Fig. 9C). In the field of immunotherapy, ToC hold tremendous importance due to their ability to easily include immune cells, thus, to quite faithfully recapitulate the cell composition of the tumor microenvironment. While promising results have been achieved, significant challenges remain in working with autologous patient cells or incorporating adaptive immunity mechanisms into ToC models.
We also examined the potential of ToC for pharmaceutical industries. At this stage, of course ToC cannot replace all existing in vitro and in vivo models, and the future landscape will likely involve a combination of animals, organoids, and organ-on-chip platforms. Nevertheless, considering the high failure rate of clinical trials and the increasing emphasis on the 3Rs (replacement, reduction, refinement) principles by regulatory agencies worldwide, we envision that data generated through ToC models could be employed for regulatory purposes within the next decade. While ToC models are generally more costly than conventional cultures, their adoption is expected to occur gradually by identifying applications that will overcome existing barriers in the pharmaceutical industry. A survey conducted among pharmaceutical companies estimated that microphysiological systems could potentially save the industry up to approximately 25% in research and development expenses.8
Hence, ToC has the potential to significantly enhance the efficiency and cost-effectiveness of the drug discovery process especially by defining the delicate balance between clinical dose, efficacy and safety. And the success of ToC for clinical applications will be determined by its ability to detect and validate new therapeutic targets as well as to help the clinicians to define the best therapeutic choices and sequences for patients.
Author contributions
This review was written by a consortium of engineers (CB, AD, SD, CC, CW), biologists (MB, IH, SL, MCP, FMG), clinicians (LC, GZ) and pharmaceutical industries members (DP, DM). Analysis of >300 publications were proceeded by CB and AD. Interviews of pharmaceutical industries and contract research organization were proceeded by CB.Conflicts of interest
This work was supported by Fluigent and Institut Curie.Acknowledgements
We would like to acknowledge the financial support from the CNRS, Curie Institute and Fluigent. This work was supported by ANRT, as part of a CIFRE program with the company Fluigent. This work also received the support of “Institut Pierre-Gilles de Gennes” (laboratoire d'excellence, “Investissements d'avenir” program ANR-10-IDEX-0001-02 PSL, ANR-10-EQPX-34 and ANR-10-LABX-31), Fondation ARC pour la Recherche sur le Cancer, PGA12021010002992_3578, INSERM, ITMO N°19CR046-01 and 53712INSERM/ITMO 20-DESCROIX 04/24. We would also like to extend our gratitude to the individuals who were interviewed to contribute to Section 5.Notes and references
- P. Webster, A golden era of cancer clinical trials, Nat. Med., 2022, 28, 602–605 CrossRef CAS PubMed.
- C. H. Wong, K. W. Siah and A. W. Lo, Estimation of clinical trial success rates and related parameters, Biostatistics, 2019, 20(2), 273–286, DOI:10.1093/biostatistics/kxx069.
- M. Hay, D. W. Thomas, J. L. Craighead, C. Economides and J. Rosenthal, Clinical development success rates for investigational drugs, Nat. Biotechnol., 2014, 32, 40–51 CrossRef CAS PubMed.
- D. Sun, W. Gao, H. Hu and S. Zhou, Why 90% of clinical drug development fails and how to improve it?, Acta Pharm. Sin. B, 2022, 12(7), 3049–3062, DOI:10.1016/j.apsb.2022.02.002.
- J. T. Atkins, G. C. George and K. Hess, et al., Pre-clinical animal models are poor predictors of human toxicities in phase 1 oncology clinical trials, Br. J. Cancer, 2020, 123(10), 1496–1501, DOI:10.1038/s41416-020-01033-x.
- E. Pan, D. Bogumil, V. Cortessis, S. Yu and J. Nieva, A systematic review of the efficacy of preclinical models of lung cancer drugs, Front. Oncol., 2020, 10, 591, DOI:10.3389/fonc.2020.00591.
- J. Komen, S. M. van Neerven, A. van den Berg, L. Vermeulen and A. D. van der Meer, Mimicking and surpassing the xenograft model with cancer-on-chip technology, EBioMedicine, 2021, 66, 103303, DOI:10.1016/j.ebiom.2021.103303.
- D. E. Ingber, Human organs-on-chips for disease modelling, drug development and personalized medicine, Nat. Rev. Genet., 2022, 23, 467–491, DOI:10.1038/s41576-022-00466-9.
- S. Sant and P. A. Johnston, The production of 3D tumor spheroids for cancer drug discovery, Drug Discovery Today: Technol., 2017, 23, 27–36, DOI:10.1016/j.ddtec.2017.03.002.
- S. Gunti, A. T. K. Hoke, K. P. Vu and N. R. London, Organoid and spheroid tumor models: Techniques and applications, Cancers, 2021, 13(4), 1–18, DOI:10.3390/cancers13040874.
- H. Clevers, Modeling Development and Disease with Organoids, Cell, 2016, 165(7), 1586–1597, DOI:10.1016/j.cell.2016.05.082.
- M. Verduin, A. Hoeben, D. De Ruysscher and M. Vooijs, Patient-Derived Cancer Organoids as Predictors of Treatment Response, Front. Oncol., 2021, 11, 641980, DOI:10.3389/fonc.2021.641980.
- H. T. Nia, et al., Physical traits of cancer, Science, 2020, 370(6516), eaaz0868, DOI:10.1126/science.aaz0868.
- F. R. Balkwill, M. Capasso and T. Hagemann, The tumor microenvironment at a glance, J. Cell Sci., 2012, 125(23), 5591–5596, DOI:10.1242/jcs.116392.
- S. A. Langhans, Three-dimensional in vitro cell culture models in drug discovery and drug repositioning, Front. Pharmacol., 2018, 9, 6, DOI:10.3389/fphar.2018.00006.
- B. N. Brown, C. A. Barnes, R. T. Kasick, R. Michel, T. W. Gilbert, D. Beer-Stolz, D. G. Castner, B. D. Ratner and S. F. Badylak, Surface Characterization of Extracellular Matrix Scaffolds, Biomaterials, 2010, 31(3), 428–437 CrossRef CAS PubMed.
- T. Davidov, Y. Efraim, R. Hayam, J. Oieni, L. Baruch and M. Machluf, Extracellular matrix hydrogels originated from different organs mediate tissue-specific properties and function, Int. J. Mol. Sci., 2021, 22(21), 11624, DOI:10.3390/ijms222111624.
- K. S. Kopanska, Y. Alcheikh, R. Staneva, D. Vignjevic and T. Betz, Tensile forces originating from cancer spheroids facilitate tumor invasion, PLoS One, 2016, 11(6), 1–23, DOI:10.1371/journal.pone.0156442.
- Z. L. Li, Z. J. Wang, G. H. Wei, Y. Yang and X. W. Wang, Changes in extracellular matrix in different stages of colorectal cancer and their effects on proliferation of cancer cells, World J. Gastrointest. Oncol., 2020, 12(3), 267–275, DOI:10.4251/WJGO.V12.I3.267.
- C. Bonnans, J. Chou and Z. Werb, Remodelling the extracellular matrix in development and disease, Nat. Rev. Mol. Cell Biol., 2014, 15, 786–801 CrossRef CAS PubMed.
- A. W. Holle, J. L. Young and J. P. Spatz, In vitro cancer cell-ECM interactions inform in vivo cancer treatment, Adv. Drug Delivery Rev., 2016, 97, 270–279, DOI:10.1016/j.addr.2015.10.007.
- M. R. Carvalho, D. Lima, R. L. Reis, V. M. Correlo and J. M. Oliveira, Evaluating Biomaterial- and Microfluidic-Based 3D Tumor Models, Trends Biotechnol., 2015, 33(11), 667–678, DOI:10.1016/j.tibtech.2015.09.009.
- R. Navneeta, J. Habermehl, M. F. Coté, C. J. Doillon and D. Mantovani, Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications, Nat. Protoc., 2006, 1, 2753–2758 CrossRef PubMed.
- E. A. Aisenbrey and W. L. Murphy, Synthetic alternatives to Matrigel, Nat. Rev. Mater., 2020, 5(7), 539–551, DOI:10.1038/s41578-020-0199-8.
- K. J. Price, A. Tsykin and K. M. Giles, et al., Matrigel Basement Membrane Matrix influences expression of microRNAs in cancer cell lines, Biochem. Biophys. Res. Commun., 2012, 427(2), 343–348, DOI:10.1016/j.bbrc.2012.09.059.
- M. Romero-López, A. L. Trinh and A. Sobrino, et al., Recapitulating the human tumor microenvironment: Colon tumor-derived extracellular matrix promotes angiogenesis and tumor cell growth, Biomaterials, 2017, 116, 118–129, DOI:10.1016/j.biomaterials.2016.11.034.
- X. Cui, C. Ma and V. Vasudevaraja, et al., Dissecting the immunosuppressive tumor microenvironments in glioblastoma-on-a-chip for optimized Pd-1 immunotherapy, eLife, 2020, 9, 1–21, DOI:10.7554/ELIFE.52253.
- B. Yue, Biology of the extracellular matrix: An overview, J. Glaucoma, 2014, 23(8), S20–S23, DOI:10.1097/IJG.0000000000000108.
- N. Peela, F. S. Sam and W. Christenson, et al., A three dimensional micropatterned tumor model for breast cancer cell migration studies, Biomaterials, 2016, 81, 72–83, DOI:10.1016/j.biomaterials.2015.11.039.
- F. Gong, Y. Yang, L. Wen, C. Wang, J. Li and J. Dai, An Overview of the Role of Mechanical Stretching in the Progression of Lung Cancer, Front. Cell Dev. Biol., 2021, 9, 781828, DOI:10.3389/fcell.2021.781828.
- P. Purkayastha, M. K. Jaiswal and T. P. Lele, Molecular cancer cell responses to solid compressive stress and interstitial fluid pressure, Cytoskeleton, 2021, 78(6), 312–322, DOI:10.1002/cm.21680.
- M. Delarue, F. Montel, D. Vignjevic, J. Prost, J. F. Joanny and G. Cappello, Compressive stress inhibits proliferation in tumor spheroids through a volume limitation, Biophys. J., 2014, 107(8), 1821–1828, DOI:10.1016/j.bpj.2014.08.031.
- I. F. Rizzuti, P. Mascheroni, S. Arcucci, Z. Ben-Mériem, A. Prunet, C. Barentin, C. Rivière, H. Delanoë-Ayari, H. Hatzikirou, J. Guillermet-Guibert and M. Delarue, Mechanical control of cell proliferation increases resistance to chemotherapeutic agents, Phys. Rev. Lett., 2020, 125(12), 128103, DOI:10.1103/PhysRevLett.125.128103.
- S. Onal, M. M. Alkaisi and V. Nock, A Flexible Microdevice for Mechanical Cell Stimulation and Compression in Microfluidic Settings, Front. Phys., 2021, 9 DOI:10.3389/fphy.2021.654918.
- G. Calibasi Kocal, S. Güven, K. Foygel, A. Goldman, P. Chen, S. Sengupta, R. Paulmurugan, Y. Baskin and U. Demirci, Dynamic Microenvironment Induces Phenotypic Plasticity of Esophageal Cancer Cells under Flow, Sci. Rep., 2016, 6, 38221, DOI:10.1038/srep38221.
- A. D. Wong and P. C. Searson, Live-cell imaging of invasion and intravasation in an artificial microvessel platform, Cancer Res., 2014, 74(17), 4937–4945, DOI:10.1158/0008-5472.CAN-14-1042.
- L. Zheng, B. Wang and Y. Sun, et al., An Oxygen-Concentration-Controllable Multiorgan Microfluidic Platform for Studying Hypoxia-Induced Lung Cancer-Liver Metastasis and Screening Drugs, ACS Sens., 2021, 6(3), 823–832, DOI:10.1021/acssensors.0c01846.
- Z. Wan, M. A. Floryan, M. F. Coughlin, S. Zhang, A. X. Zhong, S. E. Shelton, X. Wang, C. Xu, D. A. Barbie and R. D. Kamm, New Strategy for Promoting Vascularization in Tumor Spheroids in a Microfluidic Assay, Adv. Healthcare Mater., 2023, 12(14), e2201784, DOI:10.1002/adhm.202201784.
- X. Y. Wang, Y. Pei and M. Xie, et al., An artificial blood vessel implanted three-dimensional microsystem for modeling transvascular migration of tumor cells, Lab Chip, 2015, 15(4), 1178–1187, 10.1039/c4lc00973h.
- S. Lu, F. Cuzzucoli and J. Jiang, et al., Development of a biomimetic liver tumor-on-a-chip model based on decellularized liver matrix for toxicity testing, Lab Chip, 2018, 18(22), 3379–3392, 10.1039/C8LC00852C.
- R. Huang, W. Zheng, W. Liu, W. Zhang, Y. Long and X. Jiang, Investigation of Tumor Cell Behaviors on a Vascular Microenvironment-Mimicking Microfluidic Chip, Sci. Rep., 2015, 5, 17768, DOI:10.1038/srep17768.
- R. Fan, T. Emery, Y. Zhang, Y. Xia, J. Sun and J. Wan, Circulatory shear flow alters the viability and proliferation of circulating colon cancer cells, Sci. Rep., 2016, 6, 27073, DOI:10.1038/srep27073.
- J. Aleman and A. Skardal, A multi-site metastasis-on-a-chip microphysiological system for assessing metastatic preference of cancer cells, Biotechnol. Bioeng., 2019, 116(4), 936–944, DOI:10.1002/bit.26871.
- S. Hao, L. Ha, G. Cheng, Y. Wan, Y. Xia, D. M. Sosnoski, A. M. Mastro and S. Y. Zheng, A Spontaneous 3D Bone-On-a-Chip for Bone Metastasis Study of Breast Cancer Cells, Small, 2018, 14(12), e1702787, DOI:10.1002/smll.201702787.
- B. Kramer, L. Haan, M. Vermeer, T. Olivier, T. Hankemeier, P. Vulto, J. Joore and H. L. Lanz, Interstitial flow recapitulates gemcitabine chemoresistance in a 3D microfluidic pancreatic ductal adenocarcinoma model by induction of multidrug resistance proteins, Int. J. Mol. Sci., 2019, 20(18), 4647, DOI:10.3390/ijms20184647.
- C. K. Ip, S. S. Li, M. Y. Tang, S. K. Sy, Y. Ren, H. C. Shum and A. S. Wong, Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress, Sci. Rep., 2016, 6, 26788, DOI:10.1038/srep26788.
- S. Parvathy Pillai Babu, S. Venkatabalasubramanian, S. R. Munisankar and A. Thiyagaraj, Cancer stem cell markers interplay with chemoresistance in triple negative breast cancer: A therapeutic perspective, Bull. Cancer, 2022, 109(9), 960–971 CrossRef PubMed.
- K. Matsuda, N. Ohga and Y. Hida, et al., Isolated tumor endothelial cells maintain specific character during long-term culture, Biochem. Biophys. Res. Commun., 2010, 394(4), 947–954, DOI:10.1016/j.bbrc.2010.03.089.
- D. Kim, K. S. Hwang, E. U. Seo, S. Seo, B. C. Lee, N. Choi, J. Choi and H. N. Kim, Vascularized Lung Cancer Model for Evaluating the Promoted Transport of Anticancer Drugs and Immune Cells in an Engineered Tumor Microenvironment, Adv. Healthcare Mater., 2022, 11(12), e2102581, DOI:10.1002/adhm.202102581.
- G. Fang, H. Lu, R. Al-Nakashli, R. Chapman, Y. Zhang, L. A. Ju, G. Lin, M. H. Stenzel and D. Jin, Enabling peristalsis of human colon tumor organoids on microfluidic chips, Biofabrication, 2021, 14(1) DOI:10.1088/1758-5090/ac2ef9.
- C. Strelez, S. Chilakala and K. Ghaffarian, et al., Human colorectal cancer-on-chip model to study the microenvironmental influence on early metastatic spread, iScience, 2021, 24(5), 102509, DOI:10.1016/j.isci.2021.102509.
- M. Ao, B. M. Brewer and L. Yang, et al., Stretching fibroblasts remodels fibronectin and alters cancer cell migration, Sci. Rep., 2015, 5, 8334, DOI:10.1038/srep08334.
- S. R. McKeown, Defining normoxia, physoxia and hypoxia in tumours - Implications for treatment response, Br. J. Radiol., 2018, 87(1035), 1–12, DOI:10.1259/bjr.20130676.
- V. Petrova, M. Annicchiarico-Petruzzelli, G. Melino and I. Amelio, The hypoxic tumour microenvironment, Oncogenesis, 2018, 7, 1, DOI:10.1038/s41389-017-0011-9.
- M. D. Brennan, M. L. Rexius-Hall, L. J. Elgass and D. T. Eddington, Oxygen control with microfluidics, Lab Chip, 2014, 14(22), 4305–4318, 10.1039/c4lc00853g.
- M. B. Byrne, M. T. Leslie, H. R. Gaskins and P. J. A. Kenis, Methods to study the tumor microenvironment under controlled oxygen conditions, Trends Biotechnol., 2014, 32(11), 556–563, DOI:10.1016/j.tibtech.2014.09.006.
- P. E. Oomen, M. D. Skolimowski and E. Verpoorte, Implementing oxygen control in chip-based cell and tissue culture systems, Lab Chip, 2016, 16(18), 3394–3414, 10.1039/c6lc00772d.
- R. Koens, Y. Tabata and J. C. Serrano, et al., Microfluidic platform for three-dimensional cell culture under spatiotemporal heterogeneity of oxygen tension, APL Bioeng., 2020, 4(1), 016106, DOI:10.1063/1.5127069.
- C. W. Chang, Y. J. Cheng and M. Tu, et al., A polydimethylsiloxane-polycarbonate hybrid microfluidic device capable of generating perpendicular chemical and oxygen gradients for cell culture studies, Lab Chip, 2014, 14(19), 3762–3772, 10.1039/c4lc00732h.
- V. Palacio-Castañeda, L. Kooijman, B. Venzac, W. P. R. Verdurmen and S Le Gac, Metabolic Switching of Tumor Cells under Hypoxic Conditions in a Tumor-on-a-chip Model, Micromachines, 2020, 11(4), 382, DOI:10.3390/mi11040382.
- J. M. Ayuso, M. Virumbrales-Muñoz and A. Lacueva, et al., Development and characterization of a microfluidic model of the tumour microenvironment, Sci. Rep., 2016, 6(June), 1–16, DOI:10.1038/srep36086.
- C. Bouquerel, W. César and L. Barthod, et al., Precise and fast control of the dissolved oxygen level for tumor-on-chip, Lab Chip, 2022, 22(22), 4443–4455, 10.1039/d2lc00696k.
- J. M. Ayuso, M. Virumbrales-Munoz and P. H. McMinn, et al., Tumor-on-a-chip: A microfluidic model to study cell response to environmental gradients, Lab Chip, 2019, 19(20), 3461–3471, 10.1039/c9lc00270g.
- N. M. Anderson and M. C. Simon, The tumor microenvironment, Curr. Biol., 2020, 30(16), R921–R925, DOI:10.1016/j.cub.2020.06.081.
- M. B. Schaaf, A. D. Garg and P. Agostinis, Defining the role of the tumor vasculature in antitumor immunity and immunotherapy article, Cell Death Dis., 2018, 9(2), 115, DOI:10.1038/s41419-017-0061-0.
- A. Costa, Y. Kieffer and A. Scholer-Dahirel, et al., Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer, Cancer Cell, 2018, 33(3), 463–479.e10, DOI:10.1016/j.ccell.2018.01.011.
- F. Pelon, B. Bourachot, Y. Kieffer, I. Magagna, F. Mermet-Meillon, I. Bonnet, A. Costa, A. M. Givel, Y. Attieh, J. Barbazan, C. Bonneau, L. Fuhrmann, S. Descroix, D. Vignjevic, P. Silberzan, M. C. Parrini, A. Vincent-Salomon and F. Mechta-Grigoriou, Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms, Nat. Commun., 2020, 11(1), 404, DOI:10.1038/s41467-019-14134-w.
- D. A. Close, A. X. Wang, S. J. Kochanek, T. Shun, J. L. Eiseman and P. A. Johnston, Implementation of the NCI-60 Human Tumor Cell Line Panel to Screen 2260 Cancer Drug Combinations to Generate >3 Million Data Points Used to Populate a Large Matrix of Anti-Neoplastic Agent Combinations (ALMANAC) Database, SLAS Discovery, 2019, 24(3), 242–263, DOI:10.1177/2472555218812429.
- M. Richter, O. Piwocka, M. Musielak, I. Piotrowski, W. M. Suchorska and T. Trzeciak, From Donor to the Lab: A Fascinating Journey of Primary Cell Lines, Front. Cell Dev. Biol., 2021, 9, 711381, DOI:10.3389/fcell.2021.711381.
- A. Boussommier-Calleja, Y. Atiyas, K. Haase, M. Headley, C. Lewis and R. D. Kamm, The effects of monocytes on tumor cell extravasation in a 3D vascularized microfluidic model, Biomaterials, 2019, 198, 180–193, DOI:10.1016/j.biomaterials.2018.03.005.
- B. A. Hassell, G. Goyal and E. Lee, et al., Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro, Cell Rep., 2017, 21(2), 508–516, DOI:10.1016/j.celrep.2017.09.043.
- M. Gerigk, H. Bulstrode and H. T. H. Shi, et al., On-chip perivascular niche supporting stemness of patient-derived glioma cells in a serum-free, flowable culture, Lab Chip, 2021, 21(12), 2343–2358, 10.1039/d1lc00271f.
- M. Humayun, J. M. Ayuso, R. A. Brenneke, M. Virumbrales-Muñoz, K. Lugo-Cintrón, S. Kerr, S. M. Ponik and D. J. Beebe, Elucidating cancer-vascular paracrine signaling using a human organotypic breast cancer cell extravasation model, Biomaterials, 2021, 270, 120640, DOI:10.1016/j.biomaterials.2020.120640.
- J. Lee, S. Mehrotra, E. Zare-Eelanjegh, R. O. Rodrigues, A. Akbarinejad, D. Ge, L. Amato, K. Kiaee, Y. Fang, A. Rosenkranz, W. Keung, B. B. Mandal, R. A. Li, T. Zhang, H. Lee, M. R. Dokmeci, Y. S. Zhang, A. Khademhosseini and S. R. Shin, A Heart-Breast Cancer-on-a-Chip Platform for Disease Modeling and Monitoring of Cardiotoxicity Induced by Cancer Chemotherapy, Small, 2021, 17(15), e2004258, DOI:10.1002/smll.202004258.
- P. W. Burridge, Y. F. Li and E. Matsa, et al., Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity, Nat. Med., 2016, 22(5), 547–556, DOI:10.1038/nm.4087.
- N. Frenkel, S. Poghosyan and C. Rubio Alarcón, et al., Long-Lived Human Lymphatic Endothelial Cells to Study Lymphatic Biology and Lymphatic Vessel/Tumor Coculture in a 3D Microfluidic Model, ACS Biomater. Sci. Eng., 2021, 7(7), 3030–3042 CrossRef CAS PubMed.
- D. J. Jung, T. H. Shin, M. Kim, C. O. Sung, S. J. Jang and G. S. Jeong, A one-stop microfluidic-based lung cancer organoid culture platform for testing drug sensitivity, Lab Chip, 2019, 19(17), 2854–2865, 10.1039/c9lc00496c.
- B. Schuster, M. Junkin, S. S. Kashaf, I. Romero-Calvo, K. Kirby, J. Matthews, C. R. Weber, A. Rzhetsky, K. P. White and S. Tay, Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids, Nat. Commun., 2020, 11(1), 5271, DOI:10.1038/s41467-020-19058-4.
- M. R. Haque, C. R. Wessel, D. D. Leary, C. Wang, A. Bhushan and F. Bishehsari, Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment, Microsyst. Nanoeng., 2022, 8(1), 36, DOI:10.1038/s41378-022-00370-6.
- L. J. Y. Ong, S. Chia and S. Q. R. Wong, et al., A comparative study of tumour-on-chip models with patient-derived xenografts for predicting chemotherapy efficacy in colorectal cancer patients, Front. Bioeng. Biotechnol., 2022, 10(August), 1–14, DOI:10.3389/fbioe.2022.952726.
- M. Astolfi, B. Péant and M. A. Lateef, et al., Micro-dissected tumor tissues on chip: An ex vivo method for drug testing and personalized therapy, Lab Chip, 2016, 16(2), 312–325, 10.1039/c5lc01108f.
- D. Dorrigiv, K. Simeone, L. Communal, J. Kendall-Dupont, A. St-Georges-Robillard, B. Péant, E. Carmona, A. M. Mes-Masson and T. Gervais, Microdissected tissue vs tissue slices—a comparative study of tumor explant models cultured on-chip and off-chip, Cancers, 2021, 13(16), 4208, DOI:10.3390/cancers13164208.
- A. R. Aref and M. Campisi, 3D Microfluidic Ex Vivo Culture of Organotypic Tumor Spheroids to Model Immune Checkpoint Blockade, Physiol. Behav., 2018, 18(20), 3129–3143, DOI:10.1016/j.physbeh.2017.03.040.
- L. F. Horowitz, A. D. Rodriguez, Z. Dereli-Korkut, R. Lin, K. Castro, A. M. Mikheev, R. J. Monnat Jr, A. Folch and R. C. Rostomily, Multiplexed drug testing of tumor slices using a microfluidic platform, npj Precis. Oncol., 2020, 4, 12, DOI:10.1038/s41698-020-0117-y.
- E. Ivanova, M. Kuraguchi and M. Xu, et al., Use of Ex Vivo Patient-Derived Tumor Organotypic Spheroids to Identify Combination Therapies for HER2 Mutant Non–Small Cell Lung Cancer, Clin. Cancer Res., 2020, 26(10), 2393–2403, DOI:10.1158/1078-0432.CCR-19-1844.
- S. Chakrabarty, W. F. Quiros-Solano and M. M. P. Kuijten, et al., A Microfluidic Cancer-on-Chip Platform Predicts Drug Response Using Organotypic Tumor Slice Culture, Cancer Res., 2022, 82(3), 510–520, DOI:10.1158/0008-5472.CAN-21-0799.
- L. K. Sablatura, K. M. Bircsak, P. Shepherd, M. Bathina, K. Queiroz, M. C. Farach-Carson, R. A. Kittles, P. E. Constantinou, A. Saleh, N. M. Navone and D. A. Harrington, A 3D Perfusable Platform for In Vitro Culture of Patient Derived Xenografts, Adv. Healthcare Mater., 2023, 12(14), e2201434, DOI:10.1002/adhm.202201434.
- T. C. Chang, A. M. Mikheev, W. Huynh, R. J. Monnat, R. C. Rostomily and A. Folch, Parallel Microfluidic Chemosensitivity Testing on Individual Slice Cultures, Lab Chip, 2014, 14(23), 4540–4551, 10.1039/b000000x/NIH.
- Z. Ao, H. Cai and Z. Wu, et al., Evaluation of cancer immunotherapy using mini-tumor chips, Theranostics, 2022, 12(8), 3628–3636, DOI:10.7150/thno.71761.
- S. M. Hattersley, D. C. Sylvester, C. E. Dyer, N. D. Stafford, S. J. Haswell and J. Greenman, A microfluidic system for testing the responses of head and neck squamous cell carcinoma tissue biopsies to treatment with chemotherapy drugs, Ann. Biomed. Eng., 2012, 40(6), 1277–1288, DOI:10.1007/s10439-011-0428-9.
- F. Eduati, R. Utharala and D. Madhavan, et al., A microfluidics platform for combinatorial drug screening on cancer biopsies, Nat. Commun., 2018, 9, 2434, DOI:10.1038/s41467-018-04919-w.
- Z. P. Khin, M. L. C. Ribeiro and T. Jacobson, et al., A preclinical assay for chemosensitivity in multiple myeloma, Cancer Res., 2014, 74(1), 56–67, DOI:10.1158/0008-5472.CAN-13-2397.
- J. Sun, M. D. Masterman-Smith and N. A. Graham, et al., A microfluidic platform for systems pathology: Multiparameter single-cell signaling measurements of clinical brain tumor specimens, Cancer Res., 2010, 70(15), 6128–6138, DOI:10.1158/0008-5472.CAN-10-0076.
- M. Virumbrales-Muñoza, J. Chen, J. Ayuso, M. Lee, J. Abel and D. J. Beebe, Organotypic primary blood vessel models of clear cell renal cell carcinoma for single-patient clinical trials, Lab Chip, 2020, 20(23), 4420–4432 RSC.
- M. Parsian, P. Mutlu, E. Yildirim, C. Ildiz, C. Ozen and U. Gunduz, Development of a microfluidic platform to maintain viability of micro-dissected tumor slices in culture, Biomicrofluidics, 2022, 16(3), 034103, DOI:10.1063/5.0087532.
- F. D. Schwab, M. C. Scheidmann and L. L. Ozimski, et al., MyCTC chip: microfluidic-based drug screen with patient-derived tumour cells from liquid biopsies, Microsyst. Nanoeng., 2022, 8, 130, DOI:10.1038/s41378-022-00467-y.
- B. L. Khoo, G. Grenci, B. Y. Lim, S. C. Lee, J. Han and C. T. Lim, Expansion of patient-derived circulating tumor cells from liquid biopsies using a CTC microfluidic culture device, Nat. Protoc., 2018, 13, 34–58 CrossRef CAS PubMed.
- Z. Zhang, H. Shiratsuchi, J. Lin, G. Chen, R. M. Reddy, E. Azizi, S. Fouladdel, A. C. Chang, L. Lin, H. Jiang, M. Waghray, G. Luker, D. M. Simeone, M. S. Wicha, D. G. Beer, N. Ramnath and S. Nagrath, Expansion of CTCs from early stage lung cancer patients using a microfluidic co-culture model, Oncotarget, 2014, 5(23), 12383–12397, DOI:10.18632/oncotarget.2592.
- G. Trujillo-de Santiago, B. G. Flores-Garza, J. A. Tavares-Negrete, I. M. Lara-Mayorga, I. González-Gamboa, Y. S. Zhang, A. Rojas-Martínez, R. Ortiz-López and M. M. Álvarez, The Tumor-on-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors, Materials, 2019, 12(18), 2945, DOI:10.3390/ma12182945.
- J. J. F. Sleeboom, H. Eslami Amirabadi, P. Nair, C. M. Sahlgren and J. M. J. den Toonder, Metastasis in context: Modeling the tumor microenvironment with cancer-on-a-chip approaches, Dis. Models Mech., 2018, 11(3), dmm033100, DOI:10.1242/dmm.033100.
- S. Lee, J. Lim, J. Yu, J. Ahn, Y. Lee and N. L. Jeon, Engineering tumor vasculature on an injection-molded plastic array 3D culture (IMPACT) platform, Lab Chip, 2019, 19(12), 2071–2080, 10.1039/c9lc00148d.
- S. Bersini, J. S. Jeon and G. Dubini, et al., A microfluidic 3D invitro model for specificity of breast cancer metastasis to bone, Biomaterials, 2014, 35(8), 2454–2461, DOI:10.1016/j.biomaterials.2013.11.050.
- J. Bai, T. Y. Tu, C. Kim, J. P. Thiery and R. D. Kamm, Identification of drugs as single agents or in combination to prevent carcinoma dissemination in a microfluidic 3D environment, Oncotarget, 2015, 6(34), 36603–36614, DOI:10.18632/oncotarget.5464.
- Y. Bi, V. S. Shirure and R. Liu, et al., Tumor-on-a-chip platform to interrogate the role of macrophages in tumor progression, Integr. Biol., 2020, 12(9), 221–232, DOI:10.1093/intbio/zyaa017.
- S. Azadi, M. Tafazzoli Shadpour and M. E. Warkiani, Characterizing the effect of substrate stiffness on the extravasation potential of breast cancer cells using a 3D microfluidic model, Biotechnol. Bioeng., 2021, 118(2), 823–835, DOI:10.1002/bit.27612.
- C. R. Drifka, K. W. Eliceiri, S. M. Weber and W. J. Kao, A bioengineered heterotypic stroma-cancer microenvironment model to study pancreatic ductal adenocarcinoma, Lab Chip, 2013, 13(19), 3965–3975, 10.1039/c3lc50487e.
- W. Han, S. Chen and W. Yuan, et al., Oriented collagen fibers direct tumor cell intravasation, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(40), 11208–11213, DOI:10.1073/pnas.1610347113.
- M. Campisi, Y. Shin, T. Osaki, C. Hajal, V. Chiono and R. D. Kamm, 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes, Biomaterials, 2018, 180, 117–129, DOI:10.1016/j.biomaterials.2018.07.014.
- E. A. Adjei-Sowah, S. A. O’Connor, J. Veldhuizen, C. Lo Cascio, C. Plaisier, S. Mehta and M. Nikkhah, Investigating the Interactions of Glioma Stem Cells in the Perivascular Niche at Single-Cell Resolution using a Microfluidic Tumor Microenvironment Model, Adv. Sci., 2022, 9(21), e2201436, DOI:10.1002/advs.202201436.
- S. Y. Jeong, J. H. Lee, Y. Shin, S. Chung and H. J. Kuh, Co-Culture of Tumor Spheroids and Fibroblasts in a Collagen Matrix-Incorporated Microfluidic Chip Mimics Reciprocal Activation in Solid Tumor Microenvironment, PLoS One, 2016, 11(7), e0159013, DOI:10.1371/journal.pone.0159013.
- B. Venzac, Y. Liu, I. Ferrante, P. Vargas, A. Yamada, R. Courson, M. Verhulsel, L. Malaquin, J. L. Viovy and S. Descroix, Sliding walls: a new paradigm for fluidic actuation and protocol implementation in microfluidics, Microsyst. Nanoeng., 2020, 6, 18, DOI:10.1038/s41378-019-0125-7.
- X. Jiang, L. Ren, P. Tebon, C. Wang, X. Zhou, M. Qu, J. Zhu, H. Ling, S. Zhang, Y. Xue, Q. Wu, P. Bandaru, J. Lee, H. J. Kim, S. Ahadian, N. Ashammakhi, M. R. Dokmeci, J. Wu, Z. Gu, W. Sun and A. Khademhosseini, Cancer-on-a-Chip for Modeling Immune Checkpoint Inhibitor and Tumor Interactions, Small, 2021, 17(7), e2004282, DOI:10.1002/smll.202004282.
- D. An, K. Kim and J. Kim, Microfluidic system based high throughput drug screening system for curcumin/TRAIL combinational chemotherapy in human prostate cancer PC3 cells, Biomol. Ther., 2014, 22(4), 355–362, DOI:10.4062/biomolther.2014.078.
- N. Azizipour, R. Avazpour, M. H. Weber, M. Sawan, A. Ajji and D. H. Rosenzweig, Uniform Tumor Spheroids on Surface-Optimized Microfluidic Biochips for Reproducible Drug Screening and Personalized Medicine, Micromachines, 2022, 13(4), 587, DOI:10.3390/mi13040587.
- E. Prince, S. Kheiri, Y. Wang, F. Xu, J. Cruickshank, V. Topolskaia, H. Tao, E. W. K. Young, A. P. McGuigan, D. W. Cescon and E. Kumacheva, Microfluidic Arrays of Breast Tumor Spheroids for Drug Screening and Personalized Cancer Therapies, Adv. Healthcare Mater., 2022, 11(1), e2101085, DOI:10.1002/adhm.202101085.
- C. W. Chi, Y. H. Lao, A. H. R. Ahmed, E. C. Benoy, C. Li, Z. Dereli-Korkut, B. M. Fu, K. W. Leong and S. Wang, High-Throughput Tumor-on-a-Chip Platform to Study Tumor–Stroma Interactions and Drug Pharmacokinetics, Adv. Healthcare Mater., 2020, 9(21), e2000880, DOI:10.1002/adhm.202000880.
- K. I. Kamei, Y. Kato and Y. Hirai, et al., Integrated heart/cancer on a chip to reproduce the side effects of anti-cancer drugs: In vitro, RSC Adv., 2017, 7(58), 36777–36786, 10.1039/c7ra07716e.
- E. Agliari, E. Biselli, A. De Ninno, G. Schiavoni, L. Gabriele, A. Gerardino, F. Mattei, A. Barra and L. Businaro, Cancer-driven dynamics of immune cells in a microfluidic environment, Sci. Rep., 2014, 4, 6639, DOI:10.1038/srep06639.
- D. Gioeli, C. J. Snow and M. B. Simmers, et al., Development of a multicellular pancreatic tumor microenvironment system using patient-derived tumor cells, Lab Chip, 2019, 19(7), 1193–1204, 10.1039/c8lc00755a.
- Y. Choi, E. Hyun and J. Seo, et al., A microengineered pathophysiological model of early-stage breast cancer, Lab Chip, 2015, 15(16), 3350–3357, 10.1039/c5lc00514k.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Yuan Hsin and D. E. Ingber, Reconstituting organ-level lung functions on a chip, Science, 2010, 328(5986), 1662–1668, DOI:10.1126/science.1188302.
- A. Grassart, V. Malardé and S. Gobba, et al., Bioengineered Human Organ-on-Chip Reveals Intestinal Microenvironment and Mechanical Forces Impacting Shigella Infection, Cell Host Microbe, 2019, 26(3), 435–444.e4, DOI:10.1016/j.chom.2019.08.007.
- J. M. Ayuso, S. Rehman, M. Virumbrales-Munoz, P. H. McMinn, P. Geiger, C. Fitzgerald, T. Heaster, M. C. Skala and D. J. Beebe, Microfluidic tumor-on-a-chip model to evaluate the role of tumor environmental stress on NK cell exhaustion, Sci. Adv., 2021, 7(8), eabc2331, DOI:10.1126/sciadv.abc2331.
- J. M. Ayuso, R. Truttschel and M. M. Gong, et al., Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model, Oncoimmunology, 2019, 8(3), 1–11, DOI:10.1080/2162402X.2018.1553477.
- L. L. Bischel, D. J. Beebe and K. E. Sung, Microfluidic model of ductal carcinoma in situ with 3D, organotypic structure, BMC Cancer, 2015, 15(1), 1–10, DOI:10.1186/s12885-015-1007-5.
- X. Cao, R. Ashfaq and F. Cheng, et al., A Tumor-on-a-Chip System with Bioprinted Blood and Lymphatic Vessel Pair, Adv. Funct. Mater., 2019, 29(31), 1–13, DOI:10.1002/adfm.201807173.
- T. J. Kwak and E. Lee, Rapid multilayer microfabrication for modeling organotropic metastasis in breast cancer, Biofabrication, 2020, 13, 015002, DOI:10.1088/1758-5090/abbd28.
- C. P. Miller, C. Tsuchida, Y. Zheng, J. Himmelfarb and S. Akilesh, A 3D Human Renal Cell Carcinoma-on-a-Chip for the Study of Tumor Angiogenesis, Neoplasia, 2018, 20(6), 610–620, DOI:10.1016/j.neo.2018.02.011.
- D. T. Nguyen, E. Lee, S. Alimperti, R. J. Norgard, A. Wong, J. J. Lee, J. Eyckmans, B. Z. Stanger and C. S. Chen, A Biomimetic Pancreatic Cancer On-Chip Reveals Endothelial Ablation via ALK7 Signaling, Sci. Adv., 2019, 5(8), eaav6789, DOI:10.1126/sciadv.aav6789.
- D. Nothdurfter, C. Ploner, D. C. Coraça-Huber, D. Wilflingseder, T. Müller, M. Hermann, J. Hagenbuchner and M. J. Ausserlechner, 3D bioprinted, vascularized neuroblastoma tumor environment in fluidic chip devices for precision medicine drug testing, Biofabrication, 2022, 14(3) DOI:10.1088/1758-5090/ac5fb7.
- S. Pradhan, A. M. Smith, C. J. Garson, I. Hassani, W. J. Seeto, K. Pant, R. D. Arnold, B. Prabhakarpandian and E. A. Lipke, A Microvascularized Tumor-mimetic Platform for Assessing Anti-cancer Drug Efficacy, Sci. Rep., 2018, 8(1), 3171, DOI:10.1038/s41598-018-21075-9.
- J. Tien, J. G. Truslow and C. M. Nelson, Modulation of Invasive Phenotype by Interstitial Pressure-Driven Convection in Aggregates of Human Breast Cancer Cells, PLoS One, 2012, 7(9), e45191, DOI:10.1371/journal.pone.0045191.
- K. A. DiVito, M. A. Daniele, S. A. Roberts, F. S. Ligler and A. A. Adams, Microfabricated blood vessels undergo neoangiogenesis, Biomaterials, 2017, 138, 142–152, DOI:10.1016/j.biomaterials.2017.05.012.
- L. Yang, S. V. Shridhar, M. Gerwitz and P. Soman, An in vitro vascular chip using 3D printing-enabled hydrogel casting, Biofabrication, 2016, 8(3), 035015, DOI:10.1088/1758-5090/8/3/035015.
- J. Ko, J. Ahn and S. Kim, et al., Tumor spheroid-on-a-chip: A standardized microfluidic culture platform for investigating tumor angiogenesis, Lab Chip, 2019, 19(17), 2822–2833, 10.1039/c9lc00140a.
- Y. Kim, J. Ko and N. Shin, et al., All-in-one microfluidic design to integrate vascularized tumor spheroid into high-throughput platform, Biotechnol. Bioeng., 2022, 119(12), 3678–3693, DOI:10.1002/bit.28221.
- M. B. Chen, J. A. Whisler, J. Fröse, C. Yu, Y. Shin and R. D. Kamm, On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics, Nat. Protoc., 2017, 12(5), 865–880, DOI:10.1038/nprot.2017.018.
- D. T. T. Phan, X. Wang and B. M. Craver, et al., A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications, Lab Chip, 2017, 17(3), 511–520, 10.1039/c6lc01422d.
- C. Hajal, L. Ibrahim, J. C. Serrano, G. S. Offeddu and R. D. Kamm, The effects of luminal and trans-endothelial fluid flows on the extravasation and tissue invasion of tumor cells in a 3D in vitro microvascular platform, Biomaterials, 2021, 265, 120470, DOI:10.1016/j.biomaterials.2020.120470.
- J. Song, A. Miermont, C. T. Lim and R. D. Kamm, A 3D microvascular network model to study the impact of hypoxia on the extravasation potential of breast cell lines, Sci. Rep., 2018, 8(1), 1–11, DOI:10.1038/s41598-018-36381-5.
- K. Haase, G. S. Offeddu, M. R. Gillrie and R. D. Kamm, Endothelial Regulation of Drug Transport in a 3D Vascularized Tumor Model, Adv. Funct. Mater., 2020, 30, 2002444, DOI:10.1002/adfm.202002444.
- S. Pradhan, K. A. Keller, J. L. Sperduto and J. H. Slater, Fundamentals of Laser-Based Hydrogel Degradation and Applications in Cell and Tissue Engineering, Adv. Healthcare Mater., 2017, 6(24) DOI:10.1002/adhm.201700681.
- O. Sarig-Nadir, N. Livnat, R. Zajdman, S. Shoham and D. Seliktar, Laser photoablation of guidance microchannels into hydrogels directs cell growth in three dimensions, Biophys. J., 2009, 96(11), 4743–4752, DOI:10.1016/j.bpj.2009.03.019.
- R. W. Jenkins, A. R. Aref and P. H. Lizotte, et al., Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids, Cancer Discovery, 2018, 8(2), 196–215, DOI:10.1158/2159-8290.CD-17-0833.
- Z. Du, S. Mi, X. Yi, Y. Xu and W. Sun, Microfluidic system for modelling 3D tumour invasion into surrounding stroma and drug screening, Biofabrication, 2018, 10(3), 034102, DOI:10.1088/1758-5090/aac70c.
- E. A. Zaal and C. R. Berkers, The influence of metabolism on drug response in cancer, Front Oncol, 2018, 8, 500, DOI:10.3389/fonc.2018.00500.
- Y. Hou, X. Ai and L. Zhao, et al., An integrated biomimetic array chip for high-throughput co-culture of liver and tumor microtissues for advanced anticancer bioactivity screening, Lab Chip, 2020, 20(14), 2482–2494, 10.1039/d0lc00288g.
- M. Glicksman, Z. Li and S. Markossian, et al., In Vitro Cell Based Assays, Assay Guidance Manual, 2023 Search PubMed.
- T. L. Riss, R. A. Moravec and A. L. Niles, et al., Cell Viability Assays, Assay Guidance Manual, 2013 Search PubMed.
- I. Veith, A. Mencattini and V. Picant, et al., Apoptosis mapping in space and time of 3D tumor ecosystems reveals transmissibility of cytotoxic cancer death, PLoS Comput. Biol., 2021, 17(3), 1–23, DOI:10.1371/JOURNAL.PCBI.1008870.
- G. Ronteix, S. Jain and C. Angely, et al., High resolution microfluidic assay and probabilistic modeling reveal cooperation between T cells in tumor killing, Nat. Commun., 2022, 13(1), 25–28, DOI:10.1038/s41467-022-30575-2.
- Y. S. Zhang, Y. N. Zhang and W. Zhang, Cancer-on-a-chip systems at the frontier of nanomedicine, Drug Discovery Today, 2017, 22, 1392–1399 CrossRef CAS PubMed.
- M. S. Manak, J. S. Varsanik and B. J. Hogan, et al., Live-cell phenotypic-biomarker microfluidic assay for the risk stratification of cancer patients via machine learning, Nat. Biomed. Eng., 2018, 47(3), 549–562, DOI:10.1038/s41551-018-0285-z.Live-cell.
- A. Mencattini, D. Di Giuseppe and M. C. Comes, et al., Discovering the hidden messages within cell trajectories using a deep learning approach for in vitro evaluation of cancer drug treatments, Sci. Rep., 2020, 10(1), 1–11, DOI:10.1038/s41598-020-64246-3.
- F. Klemm and J. A. Joyce, Microenvironmental regulation of therapeutic response in cancer, Trends Cell Biol., 2015, 25(4), 198–213, DOI:10.1016/j.tcb.2014.11.006.
- E. Ulukaya, D. Karakas and K. Dimas, Tumor Chemosensitivity Assays Are Helpful for Personalized Cytotoxic Treatments in Cancer Patients, Medicina, 2021, 57(6), 636, DOI:10.3390/medicina57060636.
- I. Desyatnik, M. Krasner, L. Frolov, M. Ronen, O. Guy, D. Wasserman, A. Tzur, D. Avrahami, E. Barbiro-Michaely and D. Gerber, An Integrated Microfluidics Approach for Personalized Cancer Drug Sensitivity and Resistance Assay, Adv. Biosyst., 2019, 3(11), e1900001, DOI:10.1002/adbi.201900001.
- B. Gokce, I. Akcok, A. Cagir and D. Pesen-Okvur, A new drug testing platform based on 3D tri-culture in lab-on-a-chip devices, Eur. J. Pharm. Sci., 2020, 155, 105542, DOI:10.1016/j.ejps.2020.105542.
- Y. A. Guerrero, D. Desai, C. Sullivan, E. Kindt, M. E. Spilker, T. S. Maurer, D. E. Solomon and D. W. Bartlett, A Microfluidic Perfusion Platform for In Vitro Analysis of Drug Pharmacokinetic-Pharmacodynamic (PK-PD) Relationships, AAPS J., 2020, 22(2), 53, DOI:10.1208/s12248-020-0430-y.
- A. D. Rodriguez and L. F. Horowitz, et al., A microfluidic platform for functional testing of cancer drugs on intact tumor slices, Lab Chip, 2020, 20(9), 1658–1675 RSC.
- W. Han, R. El Botty, E. Montaudon, L. Malaquin, F. Deschaseaux, N. Espagnolle, E. Marangoni, P. Cottu, G. Zalcman, M. C. Parrini, F. Assayag, L. Sensebe, P. Silberzan, A. Vincent-Salomon, G. Dutertre, S. Roman-Roman, S. Descroix and J. Camonis, In vitro bone metastasis dwelling in a 3D bioengineered niche, Biomaterials, 2021, 269, 120624, DOI:10.1016/j.biomaterials.2020.120624.
- V. V. Padma, An overview of targeted cancer therapy, Biomedicine, 2015, 5(4), 1–6, DOI:10.7603/s40681-015-0019-4.
- F. D. Makurvet, Biologics vs. small molecules: Drug costs and patient access, Med. Drug Discovery, 2021, 9, 100075, DOI:10.1016/j.medidd.2020.100075.
- Z. Dereli-Korkut, H. D. Akaydin, A. H. R. Ahmed, X. Jiang and S. Wang, Three dimensional microfluidic cell arrays for ex vivo drug screening with mimicked vascular flow, Anal. Chem., 2014, 86(6), 2997–3004, DOI:10.1021/ac403899j.
- S. Azadi, E. Mohammadi, M. Tafazzoli-Shadpour and M. Tabatabaei, Effects of chemically EGFR targeting on non-targeted physical cell behaviors in 2D and 3D microfluidic cultures of invasive and non-invasive breast cancer cell lines, Biochem. Biophys. Res. Commun., 2022, 622, 1–7, DOI:10.1016/j.bbrc.2022.07.013.
- M. Nguyen, A. De Ninno and A. Mencattini, et al., Dissecting Effects of Anti-cancer Drugs and Cancer-Associated Fibroblasts by On-Chip Reconstitution of Immunocompetent Tumor Microenvironments, Cell Rep., 2018, 25(13), 3884–3893.e3, DOI:10.1016/j.celrep.2018.12.015.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Small molecules in targeted cancer therapy: advances, challenges, and future perspectives, Signal Transduction Targeted Ther., 2021, 6(1), 201, DOI:10.1038/s41392-021-00572-w.
- S. Akkın, G. Varan and E. Bilensoy, A review on cancer immunotherapy and applications of nanotechnology to chemoimmunotherapy of different cancers, Molecules, 2021, 26(11), 3382, DOI:10.3390/molecules26113382.
- M. Binnewies, E. W. Roberts and K. Kersten, et al., Understanding the tumor immune microenvironment (TIME) for effective therapy, Nat. Med., 2018, 24(5), 541–550, DOI:10.1038/s41591-018-0014-x.
- G. You, J. Won, Y. Lee, D. Moon, Y. Park, S. H. Lee and S. W. Lee, Bispecific antibodies: A smart arsenal for cancer immunotherapies, Vaccines, 2021, 9(7), 724, DOI:10.3390/vaccines9070724.
- D. B. Johnson, C. A. Nebhan, J. J. Moslehi and J. M. Balko, Immune-checkpoint inhibitors: long-term implications of toxicity, Nat. Rev. Clin. Oncol., 2022, 19(4), 254–267, DOI:10.1038/s41571-022-00600-w.
- L. Galluzzi, T. A. Chan, G. Kroemer, J. D. Wolchok and A. López-Soto, The hallmarks of successful anticancer immunotherapy, Sci. Transl. Med., 2018, 10(459), 1–15, DOI:10.1126/scitranslmed.aat7807.
- J. Mestas and C. C. W. Hughes, Of Mice and Not Men: Differences between Mouse and Human Immunology, J. Immunol., 2004, 172(5), 2731–2738, DOI:10.4049/jimmunol.172.5.2731.
- S. Parlato, G. Grisanti, G. Sinibaldi, G. Peruzzi, C. M. Casciola and L. Gabriele, Tumor-on-a-chip platforms to study cancer-immune system crosstalk in the era of immunotherapy, Lab Chip, 2021, 21(2), 234–253, 10.1039/d0lc00799d.
- T. I. Maulana, E. Kromidas and L. Wallstabe, et al., Immunocompetent cancer-on-chip models to assess immuno-oncology therapy, Adv. Drug Delivery Rev., 2021, 173, 281–305, DOI:10.1016/j.addr.2021.03.015.
- M. A. J. Morsink, N. G. A. Willemen, J. Leijten, R. Bansal and S. R. Shin, Immune organs and immune cells on a chip: An overview of biomedical applications, Micromachines, 2020, 11(9), 1–25, DOI:10.3390/MI11090849.
- S. Parlato, A. De Ninno and R. Molfetta, et al., 3D Microfluidic model for evaluating immunotherapy efficacy by tracking dendritic cell behaviour toward tumor cells, Sci. Rep., 2017, 7(1), 1–16, DOI:10.1038/s41598-017-01013-x.
- A. Mencattini, C. Lansche, I. Veith, P. Erbs, J. M. Balloul, E. Quemeneur, S. Descroix, F. Mechta-Grigoriou, G. Zalcman, C. Zaupa, M. C. Parrini and E. Martinelli, Direct imaging and automatic analysis in tumor-on-chip reveal cooperative antitumoral activity of immune cells and oncolytic vaccinia virus, Biosens. Bioelectron., 2022, 215, 114571, DOI:10.1016/j.bios.2022.114571.
- A. Pavesi, A. T. Tan, S. Koh, A. Chia, M. Colombo, E. Antonecchia, C. Miccolis, E. Ceccarello, G. Adriani, M. T. Raimondi, R. D. Kamm and A. Bertoletti, A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors, JCI Insight, 2017, 2(12), e89762, DOI:10.1172/jci.insight.89762.
- M. Chernyavska, C. K. J. C. Hermans and C. Chan, et al., Evaluation of immunotherapies improving macrophage anti-tumor response using a microfluidic model, Organs-on-a-Chip, 2022, 4, 100019, DOI:10.1016/j.ooc.2022.100019.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez de la Lastra, Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2022, 10(4), 1367–1401, DOI:10.1016/j.gendis.2022.02.007.
- A. Pavesi, G. Adriani, A. Tay, M. E. Warkiani, W. H. Yeap, S. C. Wong and R. D. Kamm, Engineering a 3D microfluidic culture platform for tumor-treating field application, Sci. Rep., 2016, 6, 26584, DOI:10.1038/srep26584.
- R. Bayat Mokhtari, T. S. Homayouni, N. Baluch, E. Morgatskaya, S. Kumar, B. Das and H. Yeger, Combination therapy in combating cancer, Oncotarget, 2017, 8(23), 38022–38043, DOI:10.18632/oncotarget.16723.
- W. Y. Kong, S. C. Ngai, B. H. Goh, L. H. Lee, T. T. Htar and L. H. Chuah, Is Curcumin the Answer to Future Chemotherapy Cocktail?, Molecules, 2021, 26(14), 4329, DOI:10.3390/molecules26144329.
- E. Nicolò, F. Giugliano, L. Ascione, P. Tarantino, C. Corti, S. M. Tolaney, M. Cristofanilli and G. Curigliano, Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives, Cancer Treat. Rev., 2022, 106, 102395, DOI:10.1016/j.ctrv.2022.102395.
- S. Tran, P. J. DeGiovanni, B. Piel and P. Rai, Cancer nanomedicine: a review of recent success in drug delivery, Clin. Transl. Med., 2017, 6(1), 44, DOI:10.1186/s40169-017-0175-0.
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications, J. Controlled Release, 2015, 200, 138–157, DOI:10.1016/j.jconrel.2014.12.030.
- J. K. Patra, G. Das and L. F. Fraceto, et al., Nano based drug delivery systems: Recent developments and future prospects 10 Technology 1007 Nanotechnology 03 Chemical Sciences 0306 Physical Chemistry (incl. Structural) 03 Chemical Sciences 0303 Macromolecular and Materials Chemistry 11 Medical and He, J. Nanobiotechnol., 2018, 16(1), 1–33, DOI:10.1186/s12951-018-0392-8.
- H. Zhang, Y. Zhu and Y. Shen, Microfluidics for Cancer Nanomedicine: From Fabrication to Evaluation, Small, 2018, 14(28), 1–25, DOI:10.1002/smll.201800360.
- N. S. Bhise, J. Ribas and V. Manoharan, et al., Organ-on-a-chip platforms for studying drug delivery systems, J. Controlled Release, 2014, 190, 82–93, DOI:10.1016/j.jconrel.2014.05.004.
- D. Zhu, Q. Long, Y. Xu and J. Xing, Evaluating Nanoparticles in Preclinical Research Using Microfluidic Systems, Micromachines, 2019, 10(6), 414, DOI:10.3390/mi10060414.
- O. Mitxelena-Iribarren, J. Zabalo, S. Arana and M. Mujika, Improved microfluidic platform for simultaneous multiple drug screening towards personalized treatment, Biosens. Bioelectron., 2019, 123, 237–243, DOI:10.1016/j.bios.2018.09.001.
- J. Wu, H. Li, Q. Chen, X. Lin, W. Liu and J. M. Lin, Statistical single-cell analysis of cell cycle-dependent quantum dot cytotoxicity and cellular uptake using a microfluidic system, RSC Adv., 2014, 4(47), 24929–24934, 10.1039/c4ra01665c.
- A. Albanese, A. K. Lam, E. A. Sykes, J. V. Rocheleau and W. C. W. Chan, Tumour-on-a-chip provides an optical window into nanoparticle tissue transport, Nat. Commun., 2013, 4, 1–8, DOI:10.1038/ncomms3718.
- B. Kwak, A. Ozcelikkale, C. S. Shin, K. Park and B. Han, Simulation of complex transport of nanoparticles around a tumor using tumor-microenvironment-on-chip, J. Controlled Release, 2014, 194, 157–167, DOI:10.1016/j.jconrel.2014.08.027.
- C. A. Cunha-Matos, O. R. Millington, A. W. Wark and M. Zagnoni, Real-time assessment of nanoparticle-mediated antigen delivery and cell response, Lab Chip, 2016, 16(17), 3374–3381, 10.1039/c6lc00599c.
- Y. Tang, F. Soroush, J. B. Sheffield, B. Wang, B. Prabhakarpandian and M. F. Kiani, A Biomimetic Microfluidic Tumor Microenvironment Platform Mimicking the EPR Effect for Rapid Screening of Drug Delivery Systems, Sci. Rep., 2017, 7(1), 1–14, DOI:10.1038/s41598-017-09815-9.
- K. Tokarska, M. Bułka and U. Bazylińska, et al., Evaluation of nanoencapsulated verteporfin's cytotoxicity using a microfluidic system, J. Pharm. Biomed. Anal., 2016, 127, 39–48, DOI:10.1016/j.jpba.2016.02.052.
- R. Duncan and R. Gaspar, Nanomedicine(s) under the microscope, Mol. Pharmaceutics, 2011, 8(6), 2101–2141, DOI:10.1021/mp200394t.
- W. Liu, J. C. Wang and J. Wang, Controllable organization and high throughput production of recoverable 3D tumors using pneumatic microfluidics, Lab Chip, 2015, 15(4), 1195–1204, 10.1039/c4lc01242a.
- M. Chudy, K. Tokarska and E. Jastrzębska, et al., Lab-on-a-chip systems for photodynamic therapy investigations, Biosens. Bioelectron., 2018, 101, 37–51, DOI:10.1016/j.bios.2017.10.013.
- A. Dao, R. Kushwaha, A. Kumar, H. Huang and S. Banerjee, Engineered Exosomes as a Photosensitizer Delivery Platform for Cancer Photodynamic Therapy, ChemMedChem, 2022, 17(10), e202200119, DOI:10.1002/cmdc.202200119.
- M. Flont, E. Jastrzȩbska and Z. Brzózka, A multilayered cancer-on-a-chip model to analyze the effectiveness of new-generation photosensitizers, Analyst, 2020, 145(21), 6937–6947, 10.1039/d0an00911c.
- J. M. Lee, H. I. Seo, J. H. Bae and B. G. Chung, Hydrogel microfluidic co-culture device for photothermal therapy and cancer migration, Electrophoresis, 2017, 38(9–10), 1318–1324, DOI:10.1002/elps.201600540.
- P. Agarwal, H. Wang and M. Sun, et al., Microfluidics Enabled Bottom-Up Engineering of 3D Vascularized Tumor for Drug Discovery, ACS Nano, 2017, 11(7), 6691–6702, DOI:10.1021/acsnano.7b00824.
- I. K. Zervantonakis and C. D. Arvanitis, Controlled Drug Release and Chemotherapy Response in a Novel Acoustofluidic 3D Tumor Platform, Small, 2016, 12(19), 2616–2626, DOI:10.1002/smll.201503342.
- J. P. Hughes, S. S. Rees, S. B. Kalindjian and K. L. Philpott, Principles of early drug discovery, Br. J. Pharmacol., 2011, 162(6), 1239–1249, DOI:10.1111/j.1476-5381.2010.01127.x.
- A. A. Adjei, What is the right dose? The elusive optimal biologic dose in phase I clinical trials, J. Clin. Oncol., 2006, 24(25), 4054–4055, DOI:10.1200/JCO.2006.07.4658.
- K. Sherkar, S. Choudante and O. Sumant, Organ-Tumor-on-a-Chip Market by Type (Lung Tumor-on-a-Chip, Bone Marrow Tumor-on-a-Chip, Brain Tumor-on-a-Chip, Breast Tumor-on-a-Chip, Urinary System Tumor-on-a-Chip, Intestine Tumor-on-a-Chip, and Liver Tumor-on-a-Chip): Global Opportunity Analysis and Industry Forecast, 2021–2030, 2021 Search PubMed.
- S. W. Baran, P. C. Brown, A. R. Baudy, S. C. Fitzpatrick, C. Frantz, A. Fullerton, J. Gan, R. N. Hardwick, K. M. Hillgren, A. K. Kopec, J. L. Liras, D. L. Mendrick, R. Nagao, W. R. Proctor, D. Ramsden, A. J. S. Ribeiro, D. Stresser, K. E. Sung, R. Sura, K. Tetsuka, L. Tomlinson, T. Van Vleet, M. P. Wagoner, Q. Wang, S. Y. Arslan, G. Yoder and J. E. Ekert, Perspectives on the evaluation and adoption of complex in vitro models in drug development: Workshop with the FDA and the pharmaceutical industry (IQ MPS Affiliate), ALTEX, 2022, 39(2), 297–314, DOI:10.14573/altex.2112203.
- R. Baudoin, L. Griscom, J. M. Prot, C. Legallais and E. Leclerc, Behavior of HepG2/C3A cell cultures in a microfluidic bioreactor, Biochem. Eng. J., 2011, 53(2), 172–181, DOI:10.1016/j.bej.2010.10.007.
- V. Allwardt, A. J. Ainscough and P. Viswanathan, et al., Translational roadmap for the organs-on-a-chip industry toward broad adoption, Bioengineering, 2020, 7(3), 1–27, DOI:10.3390/bioengineering7030112.
- P. Vulto and J. Joore, Adoption of organ-on-chip platforms by the pharmaceutical industry, Nat. Rev. Drug Discovery, 2021, 20(12), 961–962, DOI:10.1038/s41573-021-00323-0.
- G. K. Kiriiri, P. M. Njogu and A. N. Mwangi, Exploring different approaches to improve the success of drug discovery and development projects: a review, Future J. Pharm. Sci., 2020, 6(1), 27, DOI:10.1186/s43094-020-00047-9.
- H. Dolgos, M. Trusheim and D. Gross, et al., Translational Medicine Guide transforms drug development processes: The recent Merck experience, Drug Discovery Today, 2016, 21(3), 517–526, DOI:10.1016/j.drudis.2016.01.003.
- O. Strianese, F. Rizzo and M. Ciccarelli, et al., Precision and personalized medicine: How genomic approach improves the management of cardiovascular and neurodegenerative disease, Genes, 2020, 11(7), 1–24, DOI:10.3390/genes11070747.
- C. B. M. Du Puch, M. Vanderstraete, S. Giraud, C. Lautrette, N. Christou and M. Mathonnet, Benefits of functional assays in personalized cancer medicine: More than just a proof-of-concept, Theranostics, 2021, 11(19), 9538–9556, DOI:10.7150/THNO.55954.
- E. L. S. Fong, T. B. Toh, H. Yu and E. K. H. Chow, 3D Culture as a Clinically Relevant Model for Personalized Medicine, SLAS Technol., 2017, 22(3), 245–253, DOI:10.1177/2472630317697251.
- T. Hernandez-Boussard, P. Macklin and E. J. Greenspan, et al., Digital twins for predictive oncology will be a paradigm shift for precision cancer care, Nat. Med., 2021, 27(12), 2065–2066, DOI:10.1038/s41591-021-01558-5.
- A. Silva, M. C. Silva and P. Sudalagunta, et al., An ex vivo platform for the prediction of clinical response in multiple myeloma, Cancer Res., 2017, 77(12), 3336–3351, DOI:10.1158/0008-5472.CAN-17-0502.
- D. B. Chou, V. Frismantas and Y. Milton, et al., On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology, Nat. Biomed. Eng., 2020, 4(4), 394–406, DOI:10.1038/s41551-019-0495-z.
- D. Singh, S. P. Deosarkar, E. Cadogan, V. Flemington, A. Bray, J. Zhang, R. S. Reiserer, D. K. Schaffer, G. B. Gerken, C. M. Britt, E. M. Werner, F. D. Gibbons, T. Kostrzewski, C. E. Chambers, E. J. Davies, A. R. Montoya, J. H. L. Fok, D. Hughes, K. Fabre, M. P. Wagoner, J. P. Wikswo and C. W. Scott, A microfluidic system that replicates pharmacokinetic (PK) profiles in vitro improves prediction of in vivo efficacy in preclinical models, PLoS Biol., 2022, 20(5), e3001624, DOI:10.1371/journal.pbio.3001624.
- T. Petreus, E. Cadogan, G. Hughes, A. Smith, V. Pilla Reddy, A. Lau, M. J. O'Connor, S. Critchlow, M. Ashford and L. Oplustil O'Connor, Tumour-on-chip microfluidic platform for assessment of drug pharmacokinetics and treatment response, Commun. Biol., 2021, 4(1), 1001, DOI:10.1038/s42003-021-02526-y.
- D. Venturoli and B. Rippe, Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: effects of molecular size, shape, charge, and deformability, Am. J. Physiol. - Ren. Physiol., 2005, 288, 605–613, DOI:10.1152/ajprenal.00171.2004.-Polydisperse.
- C. W. McAleer, C. J. Long, D. Elbrecht, T. Sasserath, L. R. Bridges, J. W. Rumsey, C. Martin, M. Schnepper, Y. Wang, F. Schuler, A. B. Roth, C. Funk, M. L. Shuler and J. J. Hickman, Multi-organ system for the evaluation of efficacy and off-target toxicity of anticancer therapeutics, Sci. Transl. Med., 2019, 11(497), eaav1386, DOI:10.1126/scitranslmed.aav1386.
- T. Satoh, S. Sugiura and K. Shin, et al., A multi-throughput multi-organ-on-a-chip system on a plate formatted pneumatic pressure-driven medium circulation platform, Lab Chip, 2018, 18(1), 115–125, 10.1039/c7lc00952f.
- A. Herland, B. M. Maoz and D. Das, et al., Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips, Nat. Biomed. Eng., 2020, 4(4), 421–436, DOI:10.1038/s41551-019-0498-9.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00531c |
| This journal is © The Royal Society of Chemistry 2023 |