
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microphysiological system for studying human bone biology under simultaneous control of oxygen tension and mechanical loading†
Julia
Scheinpflug
 *ab,
Chris Tina
Höfer
a,
Sarah S.
Schmerbeck‡
a,
Matthias
Steinfath
a,
Jennifer
Doka§
a,
Yonatan Afework
Tesfahunegn
e,
Norman
Violet
a,
Kostja
Renko
a,
Konrad
Gulich
a,
Thilo
John
c,
Marlon R.
Schneider¶
a,
Elisa
Wistorf
a,
Gilbert
Schönfelder
ad and
Frank
Schulze||
*a
*ab,
Chris Tina
Höfer
a,
Sarah S.
Schmerbeck‡
a,
Matthias
Steinfath
a,
Jennifer
Doka§
a,
Yonatan Afework
Tesfahunegn
e,
Norman
Violet
a,
Kostja
Renko
a,
Konrad
Gulich
a,
Thilo
John
c,
Marlon R.
Schneider¶
a,
Elisa
Wistorf
a,
Gilbert
Schönfelder
ad and
Frank
Schulze||
*a
aGerman Federal Institute for Risk Assessment, German Centre for the Protection of Laboratory Animals (Bf3R), 10589 Berlin, Germany. E-mail: julia.scheinpflug@bfr.bund.de; Frank.Schulze2@med.uni-greifswald.de
bTechnische Universität Berlin, 13355 Berlin, Germany
cDRK Kliniken Westend, 14050 Berlin, Germany
dInstitute of Clinical Pharmacology and Toxicology, Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, 10117 Berlin, Germany
eReykjavik University, 101 Reykjavik, Iceland
First published on 3rd July 2023
Abstract
Throughout life, continuous remodelling is part of human bone biology and depends on the simultaneous action of physicochemical parameters such as oxygen tension and varying mechanical load. Thus, suitable model systems are needed, which allow concomitant modulation of these factors to recapitulate in vivo bone formation. Here, we report on the development of a first microphysiological system (MPS) that enables perfusion, environment-independent regulation of the oxygen tension as well as precise quantification and control of mechanical load. To demonstrate the use of the MPS for future studies on the (patho-)biology of bone, we built a simplified 3D model for early de novo bone formation. Primary human osteoblasts (OBs), which are the key players during this process, were seeded onto type I collagen scaffolds and cultured in the MPS. We could not only monitor cell viability and metabolism of OBs under varied physicochemical conditions, but also visualise the mineralisation of the extracellular matrix. In summary, we present a MPS that uniquely combines the independent control of physicochemical parameters and allows investigation of their influence on bone biology. We consider our MPS highly valuable to gain deeper insights into (patho-)physiological processes of bone formation in the future.
Introduction
The skeleton is a highly dynamic organ, subjected to a number of internal and external stimuli that govern the process of bone remodelling necessary for tissue growth and homeostasis.1 Understanding the underlying mechanisms and the pathogenesis of bone remodelling defects2 on a molecular and cellular level is critical for the improvement of medical treatments. Common in vitro cell culture approaches often neglect physicochemical properties of the tissue of interest, and may thereby miss critical aspects for cellular function,3 such as the adequate oxygen environment.4 As an alternative to 2D cell culture, appropriate in vitro models that allow the investigation of these processes under physiological conditions are being developed. Within recent years, microphysiological systems (MPS) were established to meet these requirements and to improve in vitro protocols.5–7 Until now, a variety of elaborated MPS has been published for lung, liver or kidney models, while MPS that model bone are rather underrepresented.5,8,9 This might reflect the fact that bone is a highly intricate tissue participating in a broad range of functions such as locomotion, protection of the inner organs, mineral homeostasis and haematopoiesis, and has been considered difficult to be recreated in its entire complexity as an in vitro model.8 However, similar to any other tissue or organ, bone physiology can be reduced to key aspects which are vital to its functions and which might be selectively included in the model of choice to address specific research questions.Bone consists of a multitude of cells from different lineages such as hematopoietic stem cells, endothelial cells, mesenchymal stromal cells and their respective progeny.10 In particular, osteoblasts (OBs) are key players in bone formation and bone homeostasis, while osteocytes modulate these processes in response to external stimuli such as mechanical load.11,12 It is well established that OBs themselves are also able to sense mechanical forces and respond by a well-orchestrated sequence of cellular activities towards full maturation and subsequent formation of bone mass.13 External stimuli such as low oxygen concentrations initially lead to elevated cell proliferation in bone tissue. With increasing oxygen levels and additional mechanical stimuli, OBs secrete organic and inorganic compounds, forming the extracellular matrix (ECM).14–17 The absence of mechanical forces leads to an arrest in bone forming activity by OBs and induces degradation of bone ECM by multinucleated osteoclasts.18 Eventually, OBs facilitate bone mineralisation, thereby transforming the tissue matrix into a structure of high mass density, rigidity and stiffness. The ECM in bone is a composite material consisting of 70% organic material, mainly type I collagen, while the inorganic phase comprises calcium and phosphate as hydroxyapatite crystals.19–21 This combination grants high material flexibility while providing strong resistance to plastic deformation or fracture.14,16
To generate the required energy and molecular precursors for bone formation, OBs are able to perform aerobic glycolysis in addition to oxidative phosphorylation,22i.e. they convert glucose into lactate independently of the presence of oxygen. This process provides energy and large numbers of metabolic intermediates that are incorporated into the ECM of bone, e.g. by glycosylation of type I collagen.23,24 This ability appears even more important when considering their native environment. Oxygen tension in bone marrow was reported to be within 6.6–8.8% (physoxia) but can go down to 1% (hypoxia) and lower, e.g. in the hematopoietic stem cell niche, thereby limiting oxygen-dependent processes such as mitochondrial respiration.25,26
Till date, it is widely accepted that the tissue specific low oxygen tension in bone impacts the biology of resident cells.27 The fundamental role of (patho-)physiological oxygen levels in human bone formation, maintenance and regeneration, albeit being recognised as vital for these processes, remains poorly understood. While the effects of mechanical loading on bone formation alone were studied in more detail both in vivo and in vitro, the interplay of oxygen tension and mechanical stimulation has not been investigated yet.14,28–30
Current scientific literature indicates that, in humans, the simultaneous presence of biochemical factors such as nutrient and oxygen gradients in addition to variable mechanical loads is continuously fine-tuned to ensure bone homeostasis.31 For this reason, it is necessary to develop an appropriate in vitro model covering both aspects, modulation of oxygen tension and mechanical loading as driving factors of bone formation. Up to now, only a few published MPS enable studies on bone biology, especially mechanobiology,31 while none of them provide control over perfusion, oxygen tension and mechanical loading simultaneously.8
In this work, we present an MPS that allows studying the influence of perfusion, oxygen environment and defined longitudinal mechanical load on three-dimensional (3D) bone tissue constructs. We used primary human OBs since these cells are vital for the formation of new bone. The OBs were seeded on a type I collagen scaffold to mimic the environment found in the early phases of de novo bone formation in terms of substrate stiffness. By establishing this newly engineered MPS, we were able to demonstrate that primary human OBs exhibit distinct metabolic and osteogenic responses dependent on the respective conditions, i.e. oxygen environment and mechanical load.
Summarising, we successfully report the use of a MPS with the unique capability to combine perfusion, regulation of oxygen tension and mechanical load for the culture of bone tissue constructs under physiologic conditions. We believe that the presented MPS will provide a deeper understanding of how these parameters interact and may influence bone regeneration and development.
Results and discussion
Design and functional characteristics of the MPS
Our MPS is based on a main unit that is connected to tubing, reservoirs and pumps for perfusion and several peripheral devices that facilitate the necessary control loops. Although this set-up is inherently complex, two trained scientists can assemble all parts. Once the system is running, it only takes one person to monitor all parameters, take daily samples and occasionally remove waste. The main unit was manufactured by injection moulding using cyclic olefin copolymer (COC), a biocompatible material.32,33 The optical properties of COC allowed excitation and detection of fluorescence by optical sensors like the oxygen sensor that we integrated into the system. COC further facilitated the precise control of oxygen tension within the system since the passive exchange of most gases through this material is very low.34 The surface of the MPS main unit was made hydrophobic to prevent unwanted attachment and growth of cells outside the tissue construct. One main unit has the size of a microscopic slide (75.5 × 25.5 mm) and a maximum height of 20 mm. Our set-up enables the parallel operation of four MPS main units with independent control of oxygen levels and mechanical loading for each unit. Each main unit was divided into functional compartments: a gas exchange unit consisting of a fluidics chamber (in which the MPS culture medium circulates), a chamber containing the exchange gas (95% N2, 5% CO2) to regulate the oxygen content by oxygen depletion (Fig. 1A, blue labels), and a culture chamber for housing, perfusion and mechanical loading of the tissue construct (Fig. 1A, yellow labels). To facilitate the in- and outflux of nutrients and metabolites, the tissue construct was constantly perfused using a peristaltic pump that moves medium from the unit's reservoir RII (Fig. 1B). RII contained a fixed amount of medium of 3.5 mL and received a daily feed of fresh MPS medium from the main reservoir RI by a second peristaltic pump to simulate daily intake of nutrients. The system was thus operated in a semi-continuous feed mode under constant perfusion. Together with the volume of RII, the total volume of the system amounted to 5.8 mL. An equal amount of culture medium was added to the fluidic circuit over a time course of 4 h each day. Excess medium was removed passively through an overflow line, thereby keeping the volume in RII constant over time. The slow exchange of culture medium was intended to minimise effects of medium exchange on the accumulation of signalling factors secreted by the cells, which are necessary for efficient proliferation, but should also ensure effective removal of accumulating cellular metabolites.35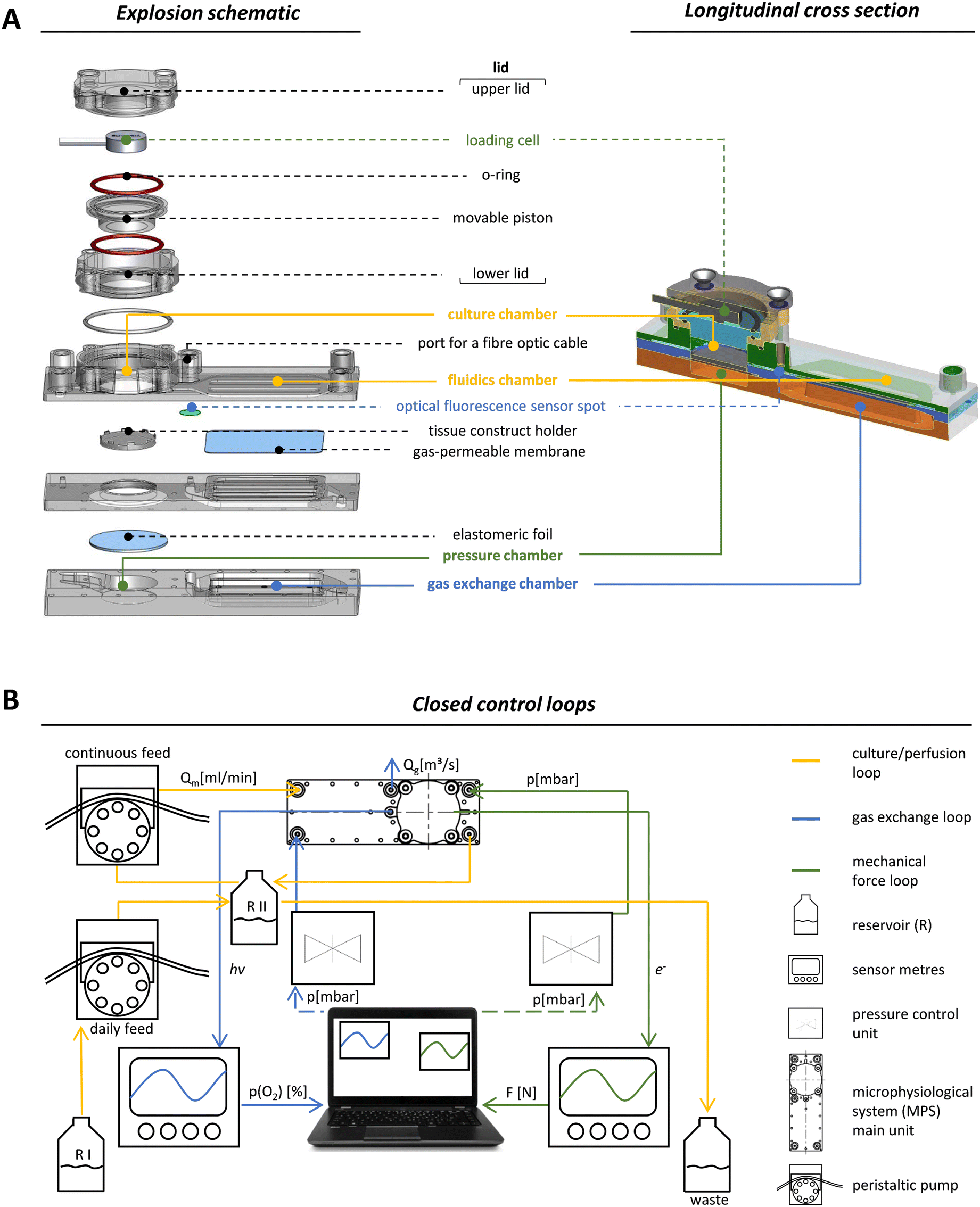 | ||
| Fig. 1 Design and control of the microphysiological system (MPS). (A) Shown is an explosion schematic (left side) and a longitudinal section (right side) of the MPS main unit. All components coloured in green are involved in the application (pressure chamber) and quantification (loading cell) of the mechanical load. The yellow colour indicates all parts of the culture unit. This includes the culture chamber, where a cylindrical tissue construct can be placed, and the fluidic chamber, where the deoxygenation of the culture medium occurs. The blue colour highlights the gas exchange unit, which includes the oxygen exchange compartment and the fluorescence optical sensor that measures the oxygen tension in the MPS medium (adapted design graphic of microfluidic ChipShop GmbH). (B) Schematic representation of the mechanical loading (green) and oxygen control (blue) circuits. The culture/perfusion loop, shown in yellow, includes a closed circuit for continuous perfusion of the MPS, which is connected to an open circuit for daily medium exchange. | ||
Volumes of the fluidic circuit were modelled according to physiologic parameters to achieve a volume ratio between RII and the culture chamber, which corresponds to the ratio of blood to bone volume in the human body. This allows for better approximation of in vivo dosages when e.g. a drug or chemical is applied in the MPS.36 The flow rate of medium was set to 0.5 mL min−1, which was balanced for efficient nutrient supply and oxygen exchange.
To implement mechanical loading, we used a pneumatic approach where variable and precisely controlled air pressure from a pressure chamber exerted mechanical forces on the tissue construct via an elastomeric COC foil that covers the pressure chamber below the culture unit (Fig. 1A, green labels). Pressure was regulated by an air pressure control unit that is able to operate in the mbar range with a response time of 100 ms (Fig. 1B, green circuit). The mechanical load that was effectively transmitted by applying air pressure was measured by a loading cell integrated in the MPS's lid opposite to the pressure chamber (Fig. 1A, green labels). When pressure was applied, the elastomeric foil deformed and pushed the tissue construct towards the movable piston resulting in longitudinal compression of the tissue construct. The movable piston was mounted, centred and sealed by two O-rings situated on its upper and lower site (Fig. 1A), which protected the entire upper lid against MPS medium leakage while allowing for longitudinal movement of the piston in a range of 180 μm. Furthermore, a plate covered the elastomeric foil to ensure that forces were transmitted evenly throughout the scaffold, which is essential for accurate force measurement. By monitoring applied forces in each MPS main unit, closed control loops for pneumatic compressive stimuli enabled individual control of the mechanical load (Fig. 1B, green circuit). In our system, frequency and amplitude of the mechanical load can be modulated dynamically throughout (and among) experiments, to study the effects of different loading scenarios on bone formation.
Closed-loop control of oxygen tension in the MPS
For monitoring oxygen tension, sensor patches were integrated into the fluidic channels of the MPS main units in between the gas exchange unit and the culture chamber (Fig. 1A, blue label). Influx of oxygen into the system occurred passively by diffusion from surrounding air into the 3D printed polypropylene container RII or across the silicone tubing. In the presented system, oxygen depletion was realised as a function of the oxygen gradient between the cell culture medium compartment and the adjacent chamber for gas exchange. Once the exchange gas is pumped into the lower compartment, the diffusion of oxygen starts from the liquid phase (MPS medium) into the gas phase (exchange gas) until an equilibrium between the two phases is reached. Use of an oxygen depletion gas instead of a gas mixture with a mixing unit offered the advantage to set and adjust oxygen tension independently for each MPS main unit. While the flow rate of oxygen-free exchange gas was kept constant, the time of the gas pulses applied was varied to control the oxygen gradient and, thus, the depletion rate in the MPS medium. To find a suitable approach for the regulation of oxygen levels, we characterised the system kinetics of oxygen depletion and parameters such as response time and system inertia, but also analysed the proportionality between pulse length and depleted oxygen. We observed an asymptotic relationship between the length of applied gas pulses and the extent of oxygen depletion (Fig. 2A).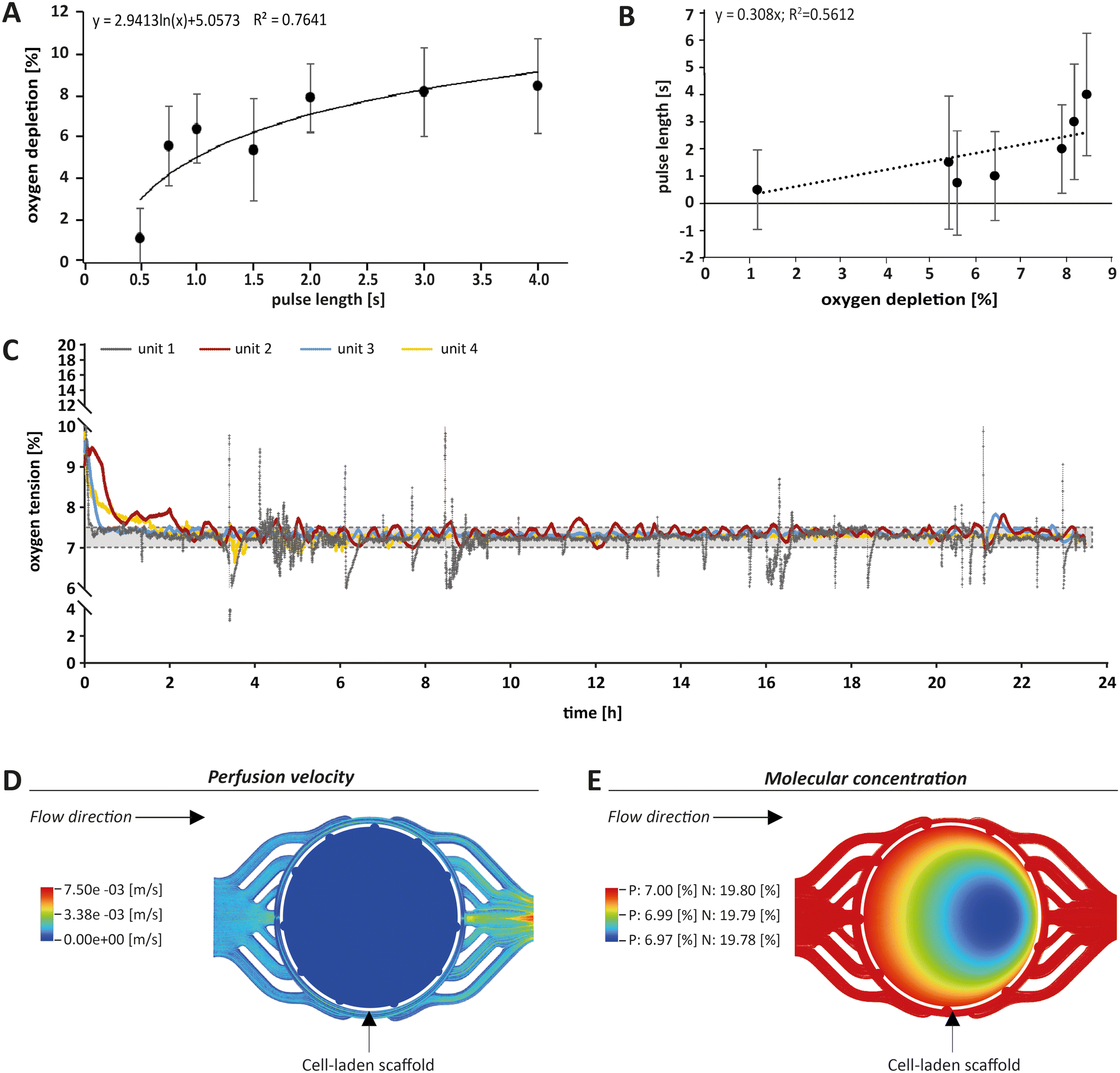 | ||
| Fig. 2 Monitoring and control of oxygen tension within the MPS. (A) The pulse length of the exchange gas was plotted against oxygen depletion measured in the MPS medium. A representative graph is shown. Pulses longer than 3 s became increasingly inefficient in reducing the oxygen concentration, which is due to technical reasons. (B) Based on the control circuit shown in (A), a proportionality factor (Kp), representing the relationship between the applied pulse length of the exchange gas and the decay of oxygen tension in the MPS medium was determined. This constant corresponds to the slope of the linear approximation calculated after changing the axes for pulse length and oxygen depletion. Standard deviation was calculated from three measurements. (C) Example of an oxygen control measurement over 24 h, showing the four different MPS units in grey, red, blue and yellow. The black dashed lines indicate the range between 7% and 7.5% oxygen tension. (D) Computational fluid dynamics (CFD) modelling (Ansys Fluent 2021 R2®) of flow velocity was performed before, within and after the culture chamber containing the cell-laden scaffold (white dashed circle) and showed a drop in velocity within the culture chamber, indicating low shear stress. (E) CFD modelling of oxygen concentrations in the MPS revealed comparable oxygen gradients within the scaffold under physoxic (P) or normoxic conditions (N). With a total drop of less than 0.03% within the scaffold, this gradient is considered negligible. | ||
By interchanging the axes for these parameters, a proportionality factor Kp was determined by a simplified linear approximation (Fig. 2B). The Kp-value, a controller parameter, is the proportionality factor between error and controller output, in this case the pulse length. The minimal pulse length of 0.2 s was given by technical constraints. Pulse lengths of more than 3 s were found to be increasingly inefficient, which reflects the upper maximum of oxygen exchange determined by the area of the nanoporous membrane and the volume of the gas-exchange chamber (Fig. 2A). Therefore, we set the maximum pulse length to 3 s, allowing to modulate the oxygen depletion rate with pulses between 0.2 and 3 s. In between single pulses, a waiting time of 60 s was inserted. These 60 s are comprised of the system's response time and its inertia. The response time of the system, defined by the decrease of oxygen tension at the sensor spot after a pulse of exchange gas, was determined to be 6.9 s (Fig. S2A†). The system inertia was found to be comparably high with 50–60 s, which is the time that it takes for the oxygen level in the medium to reach a steady state after one single pulse (after which the influx of ambient oxygen from outside the MPS would slowly increase oxygen tension over time). Since the oxygen exchange dynamics of the system do not allow description by a simple mathematical model, a proportional (P)-element based control loop (Fig. S2B†) was chosen to minimise the error between target oxygen tension and measured values. The control loop ensures the delivery of exchange gas as required to achieve or maintain the set point oxygen tension over days or weeks with a typical maximum deviation of around 0.5% from the target value (Fig. 2C) (see Experimental section “Automation of oxygen measurement by P-element based control loop” for detailed information). The chosen approach guarantees flexibility (MPS unit-independent) by free choice of the target oxygen level using a single exchange gas for depletion. Comparable systems utilised similar two-phased approaches before, yet, they relied on a pre-mixed exchange gas with the desired oxygen concentration, aiming to establish a steady state between the liquid and the gas phase.37 Supplying exchange gas for such approaches requires either a ready-to-use mixture for each use case or a gas-mixing unit and the supply of various gases in separate containers, increasing the amount of peripheral equipment needed. Therefore, our simplified and versatile approach is another step towards the implementation of oxygen control in standard experimentation. To verify the proper technical regulation of oxygen tension in our MPS, we employed a human bone osteosarcoma reporter cell line, which stably expresses a hypoxia-responsive element (HRE) fused to a luciferase reporter construct (U2OS-HRE-LUC).38
This reporter cell line is therefore sensitive to low environmental oxygen tension. Consequently, we analysed U2OS-HRE-LUC under different oxygen conditions from 1% to 19.8%. As expected, we observed the highest luciferase activity following hypoxia that was adjusted within the parallelised MPS main units by the P-element-based control loop (for detailed information see Fig. S3†). Thus, our results confirmed that we are indeed able to individually adjust the oxygen level for each MPS main unit in the studied range, thereby triggering the expected cellular responses. In conclusion, our system allows analysis of cell differentiation under various conditions, including hypoxia.
To further estimate the actual oxygen exposure throughout the scaffold, we implemented a computational approach. While our MPS setup allowed for global control of oxygen tension within the MPS, we expected that oxygen levels within a tissue construct may vary due to oxygen consumption by cells and reduced passage of medium and molecules based on steric hindrance and hydrodynamic friction (viscosity) effects. Therefore, we initially used computational fluid dynamics (CFD) software to approximate fluid velocity within the culture chamber at the employed flow rate (Fig. 2D). The overall fluid velocity was found to be very low, indicating low shear stresses exerted on the cells within scaffolds cultured in the MPS. Thus, we modelled the oxygen tension inside the porous scaffold using CFD (Fig. 2E), taking into account the porosity and pore size of the scaffold and the oxygen consumption rate of OBs as reported in literature.39 Our simulations indeed revealed a small gradient in oxygen levels within the scaffold when perfused with medium under normoxia (atmospheric oxygen) or physoxia. However, in these simulations the difference between the highest (19.80% or 7.00%, respectively) and the lowest oxygen level (19.78% or 6.97%, respectively) was proportionally low and within the measurement and control accuracy. Thus, according to these simulations, the oxygen gradient in the scaffold did not seem to be of relevance and can be neglected. Based on the CFD simulations, we considered the oxygenation within the perfused scaffold to be sufficient. In conclusion, the oxygen tension measured by the optical sensor spot can be used as an approximation for oxygen tension throughout the scaffold.
Design, application and control of the mechanical loading regime
In this work, we applied mechanical stimulation in form of dynamic compressive loading by pulsatile pneumatic pressure. The concept of using air pressure for mechanical stimulation was implemented in an MPS before.40 While in the work by Park and co-workers, a two-dimensional cell layer was treated by hydrostatic pressure via a flexible membrane, our OBs were cultured in a dynamically compressed 3D scaffold, a situation closer to in vivo biomechanics. Our MPS also allowed quantification and control of the applied forces, which is a prerequisite for biomechanical investigations.41 We chose a mechanical loading pattern that has been reported to be osteoinductive. The pattern mimics physiologically relevant interfragmentary movements during fracture healing in humans due to locomotion, which typically result in 10% compression at a frequency of 1 Hz.28,42 We decided to apply intermittent loading alternating with periods of rest, since evidence from literature suggest that such loading intervals increase bone mass, while constant loading results in desensitisation of bone cells.43,44 Tissue constructs were mechanically loaded for a total of 8 h per day in 2 h loading periods, each followed by a 2 h break.The force required to achieve a given compression of an object – such as our tissue construct – depends on the object's stiffness described by the Young's modulus.45 In this study, commercially available type I collagen scaffolds with a known Young's modulus of 8 kPa were used that adequately model biomechanical conditions during de novo bone formation in processes such as regeneration.27 According to this stiffness, the force needed for a 10% compressive strain was 106 mN (for detailed calculation see Fig. S4†). To ensure reproducible loading conditions in every experiment, not only the loading sensors themselves had to be calibrated, but also the relationship between applied pressure and resulting mechanical load in each newly assembled MPS main unit (Fig. 3A). Thermobonding of the MPS major components introduced micrometre-range variations affecting spatial parameters such as the culture chamber height. Therefore, each of the assembled MPS main units had to be calibrated for mechanical loading by application of small pressure bursts in the range of 0–200 mbar while recording the readout from the loading sensor. A logarithmic approximation was chosen in the relevant range to describe the effective mechanical force as a function of applied pressure (Fig. 3B). Using this function, we calculated the pressure to obtain a given mechanical load, and calibrated each MPS individually to set up the desired loading protocol.
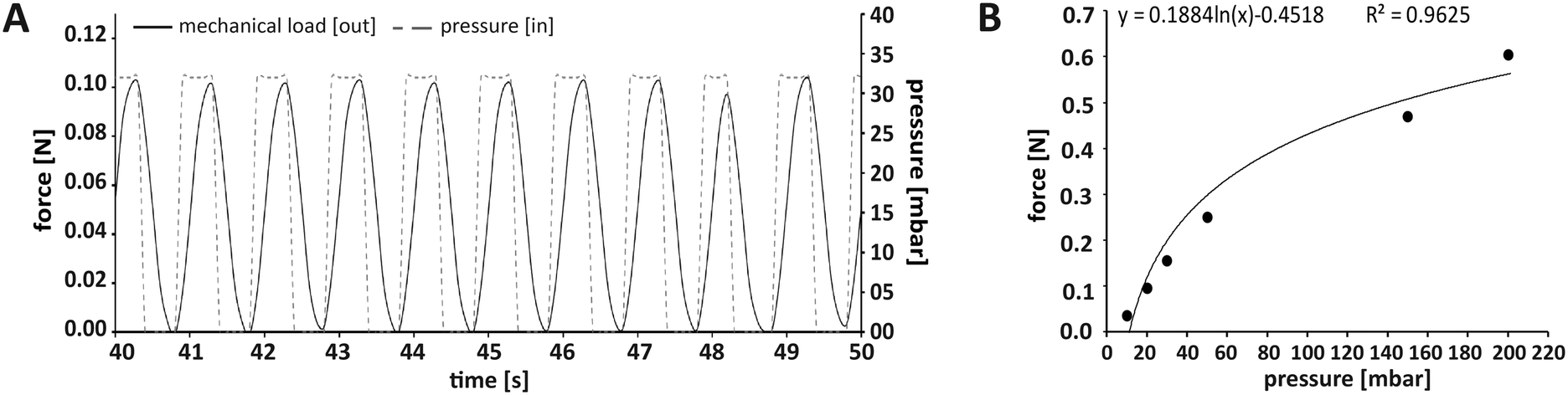 | ||
| Fig. 3 Application and control of the mechanical loading regimen within the MPS. (A) Loading protocol for 10% compression at 1 Hz. In this case, the force required for 10% compression of a scaffold with a Youngs' modulus of 8 kPa was 106 mN (see Fig. S2† for calculation). (B) Representative plot of the relationship between the applied air pressure and the mechanical loading force that was effectively transmitted through the tissue construct and measured on its surface. A logarithmic curve was fitted to the data points to determine the pressure required for a defined loading force. | ||
It is noteworthy that the maximum load that can be achieved by pneumatic compression is limited and will not be suitable to study mechanical loading of adult bone with a Young's Modulus in the range of several GPa.45 Moreover, the duration of experiments in this work was seven days, which is comparably short regarding the time line of bone formation in vivo. Taken together, our MPS is designed for, but also limited to studies in the context of de novo bone formation.
Experimental setup to test the effect of mechanical loading and oxygen tension on primary bone forming cells
Depending on the model and its purpose, different cell sources can be used for bone tissue constructs. Immortal cell lines, although convenient, lack the ability to fully recapitulate the phenotype and functional spectrum of primary cells.46,47 Primary cells from distinct donors typically cause a considerable deviation among experiments, but, at the same time, reflect the variability found among human individuals. Therefore, primary cells are thought to be of great importance as they take genetic heterogeneity into account. Further, they have the potential to differentiate, display a tissue-specific phenotype, are non-malignant, and thus often preferred over cell lines.8For initial experiments, to test the applicability of the MPS for studies on bone formation, we used a 3D model consisting of a type I collagen and 2 × 106 cells scaffold seeded with human primary OBs from four different donors (Fig. 4A, Table S1†). A representative native collagen scaffold visualised by label-free second harmonic generation (SHG) imaging prior to cell seeding is shown in Fig. 4B, and a scaffold grown with OBs in the MPS for seven days is shown in Fig. 4C. Cells, including OBs, are usually cultured and studied under normoxic conditions (standard cell culture conditions), which do not reflect the physiological oxygen tension in the body.4,48 Therefore, studies that aim to investigate the influence of oxygen tension on bone formation should consider physoxia. In this work, we first examined whether physoxic culture conditions in our MPS considerably alter the biology of OBs. Since mechanical loading is commonly described as the most important parameter in bone regeneration and new bone formation, we also investigated whether different effects of mechanical loading can be observed under physoxia as compared to normoxia. Therefore, we cultured tissue constructs in the MPS under normoxia (19.8% oxygen) or physoxia (7% oxygen) in the presence and absence of mechanical loading (Fig. 4D).
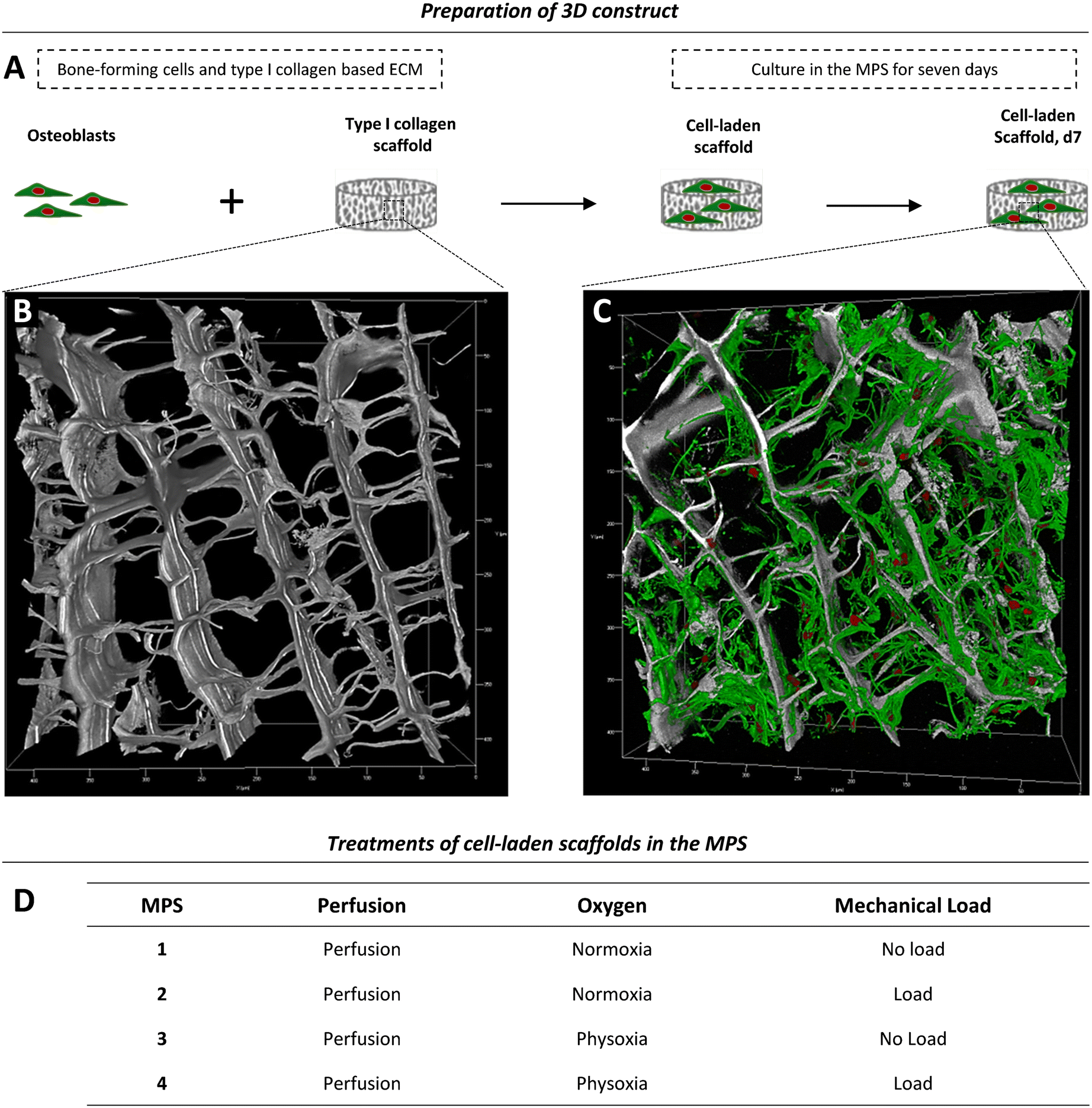 | ||
| Fig. 4 Experimental setup. (A) Primary human OBs were seeded onto a type I collagen scaffold to generate a simple 3D model for osteogenesis. (B and C) Type I collagen scaffolds were imaged by second harmonic generation (SHG) and fluorescence microscopy and are shown as 3D rendering reconstructions. (B) Native collagen scaffold. The native scaffold was soaked with medium, washed and fixed prior to imaging. The collagen-specific label-free SHG signal is shown in grey. The image stack comprises a volume of 425 × 425 × 156 μm. (C) Collagen scaffold with human OBs after seven days of incubation in the MPS under normoxia with mechanical load. Cell nuclei (red) were stained with SYTOX orange. The actin cytoskeleton (green) was stained with phalloidin-iFluor647. The image stack comprises a volume of 425 × 425 × 152 μm. (D) Parallel experimental setup for comparison of different physiological conditions in the MPS. | ||
Cell viability and aerobic glycolysis
To study if the applied culture conditions had an effect on the viability of OBs, we collected samples from circulating MPS medium daily. Cell death was examined for each of the four culture conditions over time (Fig. 5A and S5A†). Since the expression of lactate dehydrogenase (LDH) isoforms is sensitive to oxygen levels,49,50 we assessed cell viability by a protease activity assay instead. The relative protease activity in the MPS medium showed no obvious effect, indicating that culturing OBs under different test conditions did not lead to elevated cell death (Fig. 5A and S5A†). Next, we investigated cell proliferation by determining the total DNA and protein content from the cell-laden scaffolds on day seven as an approximation of cell numbers (Fig. 5B and B′). As expected, proliferation was found to vary among the donors, with donor 4 showing the overall strongest proliferation capacity. Interestingly, the application of physoxia alone did not change cell proliferation, but it tendentially increased when mechanical load was also applied (Table S2A and B†).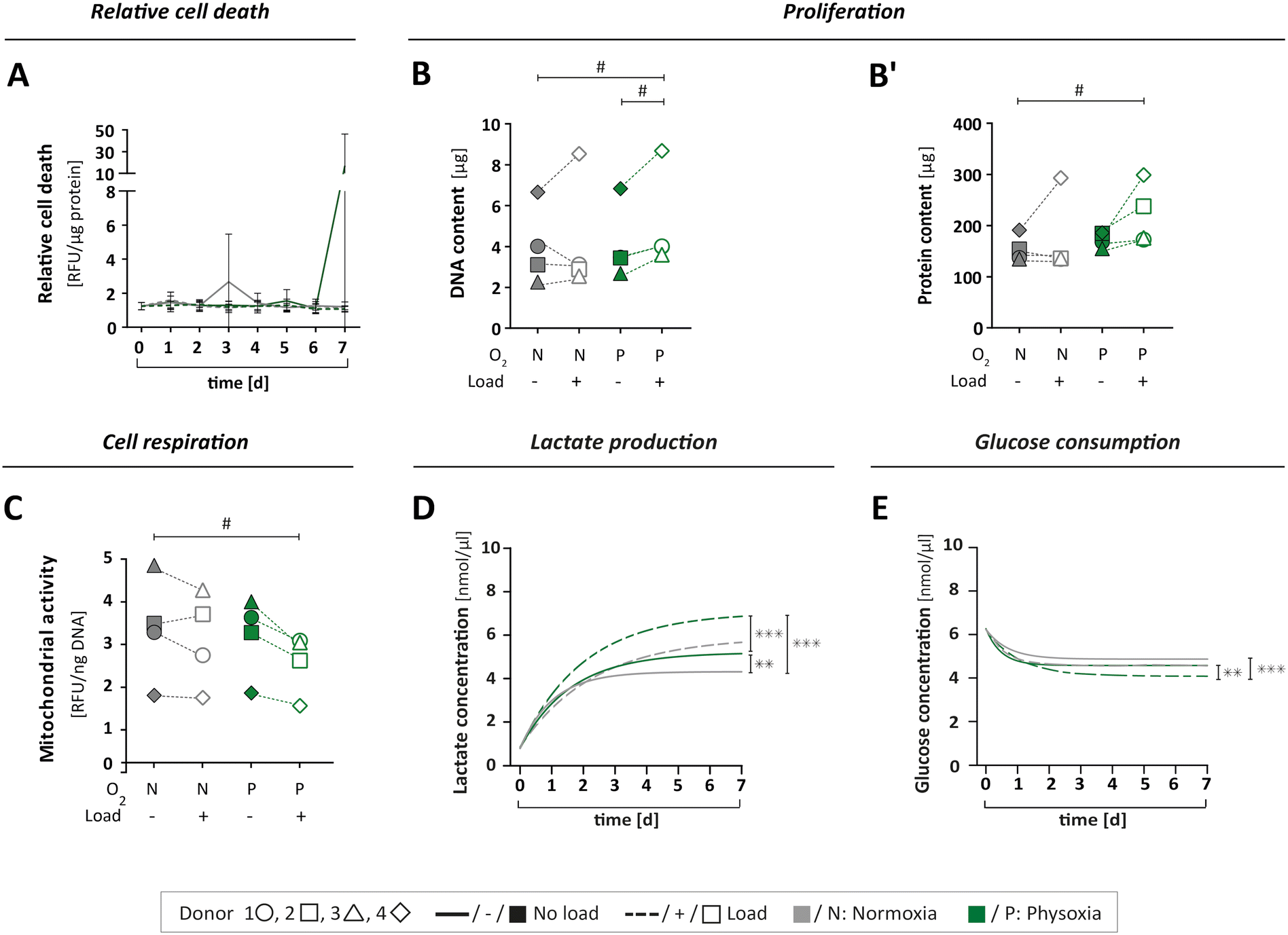 | ||
| Fig. 5 Cell viability and metabolism. (A) Relative cell death was determined by measuring specific protease activity in daily samples of MPS medium over seven days, normalised to the protein content (for single value plots see Fig. S2A†). (B and B′) The cell number of OBs on type I collagen scaffolds was indirectly determined by (B) DNA and (B′) protein content after seven days under MPS culture conditions. (C) Cell respiration was determined by mitochondrial activity in cells after seven days in MPS culture, measured as relative fluorescence units (RFU) and normalised to the DNA content. (D) Lactate production was measured in daily medium samples over seven days for all culture conditions. (E) Glucose consumption was measured in daily medium samples for all culture conditions. (D + E) Compilation of asymptotic curves based on single values plots shown inFig. 5B or D in one graph. All parameters were measured for the same four donors 1 ○, 2 □, 3 △, 4 ⋄ (n = 4) either under normoxia (N) or physoxia (P), with (+/□) or without (−/■) mechanical load. Confidence intervals and p-values obtained by nonlinear mixed model analyses are given in Table S2A–E.†P values < 0.005 were considered as statistically significant. Different significance levels are indicated as: # p < 0.05; * p < 0.005; ** p < 0.0005; *** p < 0.0001. | ||
We also analysed the levels of mitochondrial activity, glucose and lactate to characterise cell metabolism, respectively. Mitochondrial activity was not affected by physoxia (Fig. 5C, Table S2C†). However, when mechanical load was applied additionally, we could observe a decrease in mitochondrial activity.
Furthermore, metabolism of OBs was investigated by measurements of glucose consumption and lactate production over the course of seven days. When subjected to physoxia, cells produced significantly more lactate while consuming more glucose compared to normoxia (Fig. 5D and E and S5B and D†). Mechanical load under physoxic conditions had the most pronounced effect, resulting in significantly higher levels of lactate production (Fig. 5D and S5B; Table S2D†) and glucose consumption (Fig. 5E and S5D; Table S2E†). Lactate concentrations on day seven normalised to cell numbers revealed a slight increase under physoxic conditions in comparison to normoxia (Fig. S5C†). When normalised to total DNA on day seven, the glucose level seemed to be reduced under physoxia (Fig. S5E†), and under physoxia with mechanical load.
In summary, a reduction in glucose levels combined with an elevation of lactate concentration might indicate increased aerobic glycolysis,22 which is an alternative pathway in OBs for the generation of energy and production of metabolites in addition to oxidative phosphorylation. In our study, only OBs that were simultaneously cultured under physoxia and mechanical loading exhibited an apparent downregulation of mitochondrial activity. This might be due to increased aerobic glycolysis and also explain the observed increase in lactate production. A study by Zeng and colleagues supports this notion, showing that lactate levels rise when glucose is consumed in MG-63 osteoblast-like cells after mechanical stimulation.51 The current dataset, however, is not sufficient to draw definite conclusions regarding the OB's energy metabolism. The changes observed under the different culture conditions would need further investigation such as the quantification of reactive oxygen species, amount of ATP per cell and the concentrations of metabolites other than lactate.
Taken together, neither the MPS itself nor the culture conditions applied in this work had a negative effect on cell viability, but caused distinct biological responses. The combination of physoxia and mechanical loading stimulated cell proliferation while reducing mitochondrial activity. Cell culture under physoxia led to higher glucose consumption and elevated lactate levels, which further increased significantly by application of mechanical load. Our data therefore corroborates the notion that bone cell biology strongly depends on environmental factors that are missing in standard 2D cell culture.
Extracellular matrix formation and mineralisation
To investigate the progress of functional specialisation of OBs in the tissue constructs after cultivation in the MPS for seven days, we assessed clinically relevant markers for osteogenesis. However, the obtained data varied widely between cells from different donors, which might be due to numerous differences in their medical records like age, gender, comorbidities or medications (Table S1†). The N-terminal propeptide of type I collagen (P1NP) is a clinical serum marker for bone formation, and changes in serum concentrations are highly predictive for changes in bone mass.52,53 We assessed P1NP in medium samples by enzyme linked immunosorbent assay (ELISA) on day seven. Measured P1NP concentrations in the culture medium were in the range of 20 to 40 ng mL−1, which is the reported scale for this marker in human serum.54 When comparing the applied culture conditions, P1NP secretion revealed donor specific differences (Fig. 6A). It tended to increase under normoxia and mechanical load, whereby it decreased under physoxia and load, although these changes in P1NP release were not significant.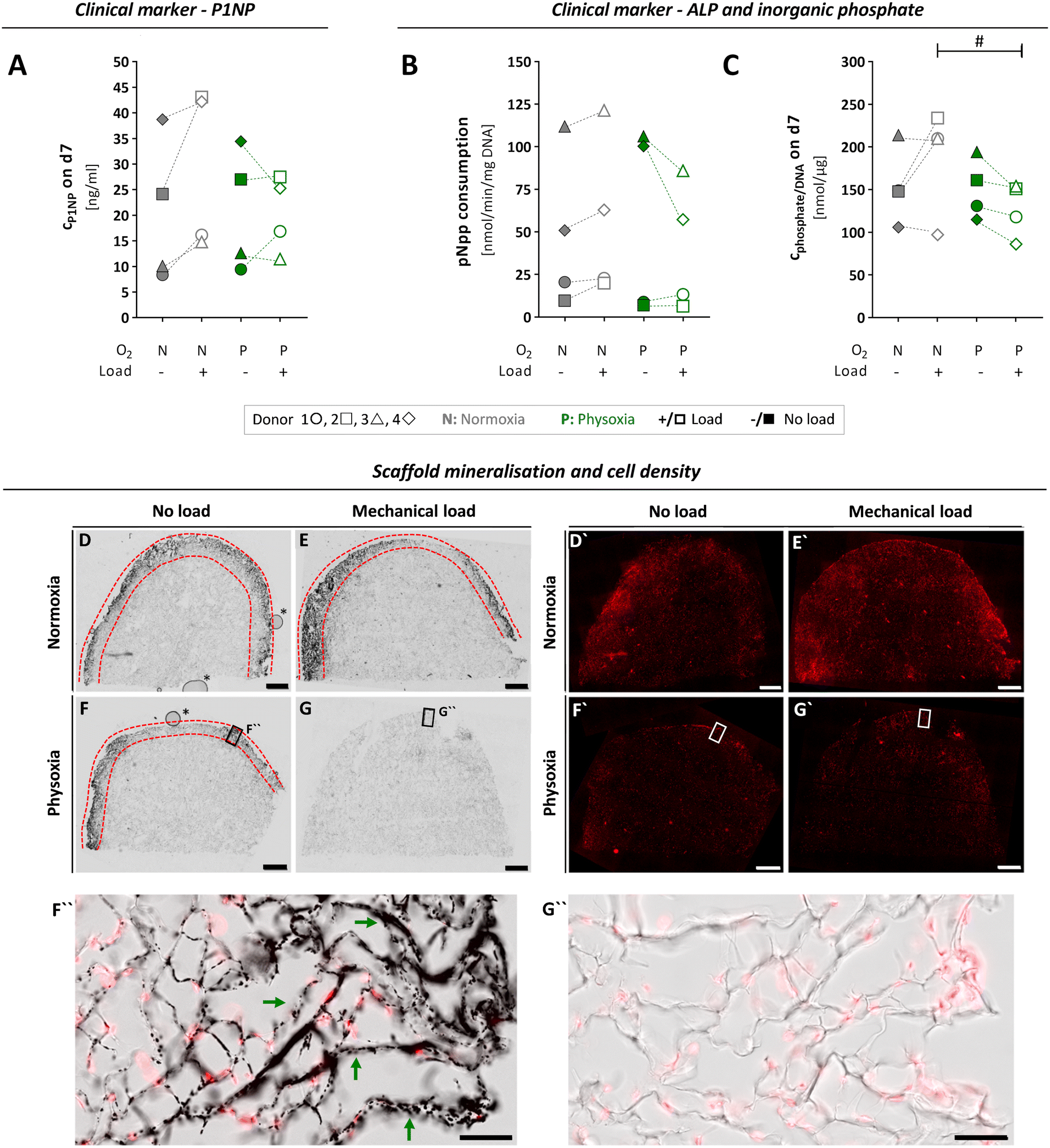 | ||
| Fig. 6 Osteogenic activity of OBs, assessed by typical clinical markers and microscopic analysis of scaffold mineralisation, after seven days of culture and differential treatment in the MPS. (A) To investigate ECM formation by OBs, P1NP concentrations in the culture medium were quantified by ELISA on day seven for each culture condition. (B) ALP activity of scaffold cells was determined based on enzymatic pNpp consumption after seven days in the MPS and normalised to the DNA content. (C) Inorganic phosphate levels measured in culture medium on day seven and normalised to DNA content. (A–C) Confidence intervals and p-values obtained by linear mixed model analyses are given in Table S3.†P values < 0.005 were considered as statistically significant. Different significance levels are indicated as: # p < 0.05; * p < 0.005; ** p < 0.0005; *** p < 0.0001. All parameters were measured for the same four donors 1 ○, 2 □, 3 △, 4 ⋄ (n = 4) either under normoxia (N) or physoxia (P), with (+/□) or without (−/■) mechanical load. (D–G) 50 μm-sections of cultivated type I collagen scaffolds were analysed comparing scaffolds grown with OBs from donor 3 under conditions of physoxia or normoxia and in the presence or absence of mechanical load. Mineralisation of type I collagen scaffolds (area between the red dashed lines) was visualised by Von Kossa staining and transmitted light microscopy, while cell nuclei (D′–G′) were detected by DRAQ5 nucleic acid fluorescence staining (scale bars, 1 mm). Higher magnification images of selected regions in (F) and (G) (black frames) are shown as overlay representations of fluorescence and transmitted light images in (F′′) and (G′′), comparing mineralised (exemplified by green arrows) and non-mineralised regions (scale bars, 100 μm). Air bubbles (*) in (D) and (F) are inclusions of the embedding medium that had no effect on samples or staining. Additional higher magnification images from D–G can be found in Fig. S6.† | ||
Next, we assessed mineralisation of ECM by OBs based on total alkaline phosphatase (ALP) activity of the cell-laden scaffold (Fig. 6B and C). Similar to P1NP, the enzyme ALP is a clinical serum marker that has a predictive value for bone formation in humans and can help to detect pathologies that affect this process. It is also used as a standard marker for osteogenic differentiation in vitro.55,56 First, we determined p-nitrophenylphosphate (pNpp) consumption and inorganic phosphate release as measures of ALP activity on day seven and normalised it to the total DNA content (Fig. 6B and C). We observed large variations of both parameters between individual donors and no significant changes when comparing normoxia vs. physoxia or mechanical load vs. no load (Table S3A and B†). However, application of mechanical load tend to increase ALP activity under normoxia, as shown in literature,57 whereas it rather decreased the enzyme activity under physoxia.
To visualise mineralisation in the scaffolds after seven days of cultivation, an extra MPS run was performed with OBs from donor 3, which allowed us to preserve the intact scaffolds for histological analysis and SHG imaging. Histological staining according to von Kossa revealed mineralisation under normoxia independent of loading conditions and under physoxia if no load was applied (Fig. 6D, F and F′′). In clear contrast, the application of mechanical load under physoxic conditions completely prevented mineralisation of the scaffold (Fig. 6G and G′′). Nuclear staining with SYTOX orange confirmed that this was not due to a lack of cells populating the scaffold (Fig. 6D′–G′). Mineralisation on day seven was limited to the outer rim close to the scaffold surface. These observations were in line with the biochemical data on ALP activity, P1NP and inorganic phosphate concentrations of donor 3, which were highest for normoxia with mechanical load and lowest for physoxia with mechanical load (Fig. 6A–C). To date, no studies have examined mechanical loading on OBs under different oxygen tensions, making it difficult to compare our initial data. However, Matziolis and colleagues demonstrated that mechanical loading at 7 kPa at 0.05 Hz for 2 weeks stimulates osteogenic differentiation of rabbit periosteal cells under standard culture conditions (21% oxygen) with increasing type I collagen synthesis, constant ALP activity, and onset of calcification of the ECM.58 Further, we noticed that mineralised regions frequently contained fewer cells compared to neighbouring non-mineralised regions, suggesting lower proliferation/altered distribution or increased cell death of OBs, which is expected to occur during bone formation (Fig. 6D–F and S6A†).59
Scaffolds cultured with OBs under the four conditions were additionally inspected by SHG and fluorescence imaging at cellular resolution. This combination of techniques allows studying both cellular organisation within scaffolds and collagen (re)organisation. Cell nuclei and the actin cytoskeleton were fluorescently labelled, while the collagen scaffolds were detected label-free (Fig. S7, Videos S1–S4†). Major remodelling of collagen scaffolds was not observed within seven days of culture in the MPS (for none of the conditions). OBs spread within the cavities of the sponge-like scaffolds similarly under all conditions. More detailed conclusions on cell densities, cell morphologies or actin organisation are, however, not possible at this point due the small sample number.
We also examined the expression of various genes involved in osteogenesis (for detailed information see Fig. S8 and Table S4†) and mostly found an upregulation of osteogenic marker genes when cells were cultured under mechanical load as compared to the corresponding non-loaded conditions. Thus, the OB phenotype seemed to establish better under mechanical loading.
In summary, we show that primary OBs in our MPS are able to produce and mineralise ECM over seven days in culture under normoxia and physoxia. When mechanical load under physoxic conditions is applied, formation and calcification of ECM seem to be reduced in favour of proliferation, according to lower ALP activity, pro-collagen production and mineralisation. Interpretation of these results should be carried out with caution. Yet, it is known from a wealth of in vivo investigations that the development of the OB phenotype during bone formation involved sequential stages of cell proliferation and differentiation, leading to ECM maturation and mineralisation.59 The initial stages of bone formation during fracture healing or development occur in an oxygen-deficient environment that gradually becomes more oxygenated as vascularisation progresses.60,61 Elevated proliferation of bone forming OBs at the beginning of these process is a very early osteogenic response to mechanical stimuli. It was previously shown that OB proliferation in mice induced by mechanical loading is a critical source of bone forming cells for subsequent maximal lamellar and woven bone formation.62 Summarising, mechanical loading seems to positively influence early osteogenesis by stimulating OB proliferation at physoxia and matrix maturation at normoxia.
We observed the expected donor-related variability of biological parameters due to the use of primary cells from donors of different age (55 to 77 years), sex, and underlying concomitant diseases (Table S1†). Albeit the apparent heterogeneity limits the scope for data interpretation, our initial experiments on early bone formation demonstrate the relevance of a MPS that independently controls physicochemical parameters. Similar to our observations regarding metabolism, we clearly demonstrated that the application of mechanical load and the control of oxygen tension had distinct effects on the bone forming ability of OBs. We therefore highlight the need to investigate cell biology under physiologic relevant conditions that are not present in standard cell culture to improve the reliability and predictive value of results. In the future, our MPS may also be suitable for a more detailed study of bone formation against the background of different disease patterns and, in particular, the influence of different treatment parameters.
Conclusions
In this work, we have developed a novel MPS that allows simultaneous control of oxygen tension and mechanical loading, and demonstrated how the complex interplay of these physicochemical parameters on bone cell biology can be studied. Our work highlights the need to integrate tissue-specific parameters into complex in vitro models to reproduce the biological and physicochemical context of cells in vivo. Albeit animal testing represents an established method in biomedical research to take the complexity of in vivo environments into account, species-specific differences hamper successful translation to human physiology.63,64 The presented MPS might pose a reasonable alternative to animal testing, as it allows the creation of a more physiologic environment for primary human bone cells. This opens up new avenues in drug discovery, for example to identify and characterise substances that have an anabolic effect on bone formation in humans to counteract degenerative diseases such as osteoporosis. Furthermore, biomaterials that exhibit biomechanical properties found in de novo bone formation might be studied in detail for their interaction with human cells using the device. In addition, the MPS might improve investigations of patient-specific biological differences in bone formation, and also allows for disease modelling by employing cells from donors that suffer from distinct pathologies.Experimental
Reagents and kits
Chemicals such as ascorbic acid (AA), beta-glycerol phosphate disodium salt pentahydrate (beta-GP), and MgCl2 × 6H2O were purchased from Merck Millipore (Burlington, MA, US). NaCl, Tris, NaOH, and ROTI Histokitt synthetic embedding medium were obtained from Carl Roth GmbH (Karlsruhe, Germany). Dulbecco's phosphate buffered saline (DPBS) was purchased from PAN-Biotech (Aidenbach, Germany). Other cell culture solutions such as penicillin/streptomycin, gentamicin, L-alanyl-L-glutamine, sodium pyruvate, TrypLE Select, HOECHST 33342, and Dulbecco's modified Eagle's medium high glucose (DMEM-HG) were received from Gibco (Grand Island, NY, US). DMEM low glucose (DMEM-LG) was purchased from Biowest (Nuaillé, France) and foetal bovine serum (FBS) from Biochrom AG (Berlin, Germany). Dexamethasone (DEXA), deferoxamine mesylate CRS (DFX), p-nitrophenylphosphate (pNpp), diethanolamine (DEA), silver nitrate, sodium thiosulfate, Triton-X 100, Tween 20, bovine serum albumin (BSA), and the assay kits for glucose, lactate, and phosphate were obtained from Sigma-Aldrich (St. Louis, MI, US). The Pierce BCA Protein Assay Kit, PrestoBlue Cell Viability Reagent, CyQUANT Cell Proliferation Assay Kit, High-Capacity cDNA Reverse Transcription Kit, DRAQ5 and SYTOX orange nucleic acid dye were purchased from Thermo Fisher Scientific (Waltham, MA, US). The CytoTox Fluor Assay Kit was received from Promega (Madison, WI, US), and the procollagen I N-terminal propeptide (P1NP) enzyme-linked immunosorbent assay (ELISA) from Elabscience (Huston, TX, US). PeqGOLD TriFAST™ for RNA purification and xylene were purchased from VWR chemicals (Radnor, PA, US). The 4% paraformaldehyde (PFA) solution (in PBS) was obtained from Alfa Aesar (part of Thermo Fisher Scientific, Lancashire, UK). Epredia type 9 paraffin was provided by Fisher Scientific (Schwerte, Germany).Fabrication and assembly of the MPS main unit
The MPS main unit was manufactured in collaboration with microfluidic ChipShop (Jena, Germany). The fabrication took place in a clean room. All parts of the MPS main unit (Fig. 1A) were made of cyclic olefin copolymers (COC) by injection moulding. All connections to the MPS main unit itself are compatible with LUER connectors. For oxygen measurements, an optical fluorescence sensor spot (OXSP5, Pyroscience, Aachen, Germany) was fixated with silicone glue right underneath the port for fibre optics (Fig. 1A) (see section “Oxygen measurement and control” for further details). For the oxygen exchange compartment, the nanoporous polytetrafluoroethylene (PTFE) membrane suitable for controlled gas exchange was laser cut to fit in between the lower gas exchange chamber and the upper fluidics chamber of the MPS main unit and fastened by thermo-bonding. An elastomeric COC foil was laser cut to seal the pressure chamber and was thermo-bonded in between the lower pressure chamber and upper the culture chamber. The lid of the MPS main unit was assembled manually under sterile conditions prior to the experiment as indicated in the explosion view.The miniature load cell (ALXLC86-050G; Althen, Kelkheim, Germany) was fixed in the upper part of the lid with silicone glue to monitor the mechanical load applied to the tissue construct. To ensure force transmission while sealing against the culture compartment, a movable piston is mounted in between the lid parts by two O-rings (silicone, 16 mm, 1 mm thick). Two self-tapping small screws (2.2 mm × 5 mm) diagonally opposed to each other held the lid together. To mount the lid to the MPS main unit, another O-ring (silicone, 19 mm, 1 mm thick) was placed in a circular notch to seal the culture chamber when closed by the lid. Then, two slightly longer self-tapping screws (2.2 mm × 9.5 mm) were used to fit into the two remaining diagonally opposite bore holes to secure the lids position. The fully assembled MPS cannot be acquired commercially. However, scientists interested in working with the MPS are welcome to contact the corresponding authors.
3D-printed peripheral components of the MPS
Some components of the MPS had to be customised and were designed using the 3D computer-aided design (CAD) software (ViaCAD11, Avanquest, Courbevoie, France). Components were printed using appropriate materials as listed in Fig. S1A† and were post-processed if necessary. We made use of two different types of printer technologies, either fused deposition modelling (FDM) or stereolithography (SLA). For FDM printing, a single print head MKII (Prusa, Prague, Czech Republic) or a double print head machine Ultimaker 3 extended (Ultimaker, Utrecht, Netherlands) were employed. For SLA printing, a Formlabs 3B was used, developed for printing biocompatible photosensitive resins such as the surgical guide SD resin (Formlabs, Somerville, MA, US). For a detailed description of the printed components with corresponding printers and materials, please refer to Fig. S1.†Set-up of the culture/perfusion circuit and integration of the MPS main units
For operating the MPS, the different compartments of the MPS had to be connected to four-channel pressure control units for subsequent deoxygenation and mechanical loading (MFCS; Fluigent, Le Kremlin-Bicêtre, France), peristaltic pumps (Ismatec; Cole-Parmer GmbH, Wertheim, Germany), miniature load cells and a four-channel fibre-optic oxygen metre (FireSting-O2; Pyroscience, Aachen, Germany) as indicated in Fig. 1B. All components of the culture/perfusion circuit, such as tubing, connectors and reservoirs, were made of autoclavable materials and were pre-packed and sterilised prior to use.The culture/perfusion circuit was assembled under sterile conditions in a laminar flow cabinet (Fig. S1A′†). The first step in the set-up of the culture/perfusion circuit resulted in a closed system without the MPS main units, which was then transferred to a hybrid incubator flow cabinet (Automated Lab Solutions, Jena, Germany) that allowed sterile working conditions at 5% CO2 and 65% humidity during cultivation, resulting in a general oxygen concentration of 19.8% in the MPS. Subsequently, the peristaltic pumps were attached and the main reservoir (RI) was filled (Fig. 1B).
The MPS consists of four individual circuits connected in parallel, which are only linked via the main reservoir (RI) and are later supplemented by the four MPS main units described above and referred to as four channels. Mechanical loading and oxygen tension in each channel can be controlled individually. Culture medium was taken by peristaltic pumps with a steady flowrate of 0.5 mL min−1 in all channels, with each channel having a total capacity of about 5.8 mL for culture medium. The circuit was flushed with at least twice the volume of a single channel (approx. 12 mL). First ethanol was used, followed by rinsing with DPBS supplemented with antibiotics penicillin (100 U mL−1), streptomycin (100 μg mL−1) and gentamicin (25 mg mL−1) to re-sterilise the entire system. Eventually, the MPS main units were inserted into the fluidic circuit, and DPBS was replaced by MPS medium (DMEM LG supplemented with 5% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin, 5 mM L-alanyl-L-glutamine, 100 nM DEXA, 50 μM AA and 10 mM beta-GP). When the entire system was equilibrated with MPS medium, the cell-laden scaffolds were inserted into the culture chambers of the MPS main units (for detailed information see Experimental section “Culture of cell-laden scaffolds in the MPS”).
Mechanical loading: calibration and application
After integration into the lid, the load cell was calibrated according to the manufacturer's instructions. In brief, since the relationship between load and voltage readout is linear in this type of load cell, a two-point calibration could be performed. The force sensing lid was connected to an appropriate sensor interface (9206 USB Multisensor Interface; Burster Präzisionsmesstechnik, Gernsbach, Germany) that was linked to a PC for data read-out. For calibration, the lid was placed in a 3D printed holder with the piston facing upwards to calibrate for zero load. The second point was calibrated by putting a 50 g weight onto the piston, which is the maximum load supported by the miniature load cell. After calibration, the whole MPS device (including the cell-laden scaffold) had to be calibrated for the relationship of applied pressure to detected mechanical load prior to every experiment. For this, the complete fluidic setup including the MPS main unit, scaffold, pumps, tubing and pressure control units had to be set up (see Experimental section “Set-up of the culture/perfusion circuit and integration of the MPS main units”). While closing the lid by tightening the longer self-tapping screws, the strain on the sensor lid had to be monitored to prevent overload that might damage the load cell. The pressure chamber was then connected to a pressure control unit via silicone tubing. The pressure control unit itself was linked to pressurised air. Once the setup was finished and sensors were connected to a PC, the calibration was started by applying four consecutive bursts at defined pressure over a range of 0–200 mbar (Fig. 2B). Higher pressures would exceed the loading cell maximum load. Minima and maxima of the monitored force were determined for every peak, averaged and plotted against the applied pressure. Data were fitted logarithmically to determine equations for each set-up describing the transmitted force at a given pressure. Every MPS unit was calibrated individually to ensure correct mechanical loading of the tissue construct according to the desired compression (Fig. S4†).Oxygen measurement and control
To quantify oxygen tension, an optical fluorescence sensor spot was used. For the contactless read-out of the oxygen sensors, a fibre-optic oxygen metre was connected to the fibre optics ports on the MPS, allowing for the simultaneous readout of up to four sensors. The calibration of the oxygen measurement within the MPS units was carried out according to the manufacturers' protocol (Pyroscience, Aachen, Germany).When operating the MPS at 19.8%, the medium was allowed to equilibrate with atmospheric oxygen tension in the hybrid incubator flow cabinet where the MPS was operated (65% humidity and 5% CO2). The use of an exchange gas (95% N2, 5% CO2) enabled oxygen depletion. However, the oxygen concentration in the circulating MPS medium slowly returns to its maximum level over time due to the gas permeability of the tubing and reservoirs and the resulting oxygen influx. After applying exchange gas, a time-delay (approx. 3 s) between the onset of gas flux and the beginning of the decrease in oxygen content was observed. In addition, changes in the exchange rate, i.e., the flux through the membrane with time and its dependence on the gas flux rate due to possible concentration gradients and dead volumes, posed a challenge for static manual control to achieve stable and reproducible conditions. Therefore, an automated control of oxygen tension was employed based on a software driven control loop. To facilitate automated control of depletion gas flow, a four-channel pressure control unit was employed. The output pressure for the pressure control unit was set to 15 mbar while the pulse lengths were varied by the control loop in response to the measured oxygen tension. This setup enabled control of oxygen tension over time.
Automation of oxygen measurement by P-element based control loop
To automate oxygen control in the MPS, a feed-back controller was implemented to achieve a sufficient accuracy (and constancy) of the oxygen. We chose a simple proportional controller (P-controller), which uses the difference between the actual and the set point value, termed error, to calculate the required pulse length proportional to the error. The pulse length is recalculated and adapted in each loop based on the feedback control algorithm (Fig. S2B′†).The P-controller was implemented in Python and ran on the same computer as other software for hardware control, e.g. the pressure control. The Python scripts are provided as ESI.†
Depending on the Kp-value, the controller can be faster or slower to prevent e.g. an overshooting (Fig. 2A). The Kp-value was determined by step attempts, i.e. oxygen levels were measured following different pulse lengths. Nonlinearity was observed between pulse length and amount of depleted oxygen, i.e. at pulse lengths of 3 s and more, the depletion rate of oxygen started to stagnate indicating that a maximum in gas exchange was reached (Fig. S2B′†). Therefore, the calculated pulse length was limited to a range between 0.2 s (due to the technical limitations of the valve) and 3 s.
Although this kind of controller leads to a permanent control offset, the resulting discrepancy was tolerable. In summary, the implemented controller achieved the goal to maintain the set oxygen level.
Modelling oxygen concentration in the scaffold using computational fluid modelling
To assess the oxygen concentrations throughout the collagen scaffold during MPS operation, a computational fluid dynamics (CFD) simulation of oxygen flow through the culture chamber of the MPS main unit containing the cell-laden scaffold was performed using the Ansys Fluent 2021 R2® software (Canonsburg, PA, US). The simulation was based on a 3D model of the culture chamber of the MPS main unit and the porous scaffold. The calculations were performed using a mesh with 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 148348 elements and 3098358 nodes that were tetraeder-shaped for both the flow across the chamber and the porous scaffold. The flow medium, defined as MPS medium with dissolved oxygen, was imported into the Ansys species model as a fluid–gas mixture (viscosity mixture: 0.006933 Pa s). The flow was defined to show a laminar behaviour with a constant volume flow rate of 0.5 mL min−1. Using Darcy's law, the permeability of the porous collagen scaffold with a diameter of 13 mm, a height of 3 mm and a porosity of 97% was derived to be 5.2 × 10−10 m2. Using an oxygen consumption rate of OBs with a value of 1.48 × 10−9 kg s−1 m−3 (ref. 39) and a mass fraction of oxygen of about 6.38 × 10−6 for normoxic (19.8%) and 2.26 × 10−6 for physoxic conditions (7%), the oxygen content in the porous cell-laden scaffold could be modelled for both oxygen levels.
148348 elements and 3098358 nodes that were tetraeder-shaped for both the flow across the chamber and the porous scaffold. The flow medium, defined as MPS medium with dissolved oxygen, was imported into the Ansys species model as a fluid–gas mixture (viscosity mixture: 0.006933 Pa s). The flow was defined to show a laminar behaviour with a constant volume flow rate of 0.5 mL min−1. Using Darcy's law, the permeability of the porous collagen scaffold with a diameter of 13 mm, a height of 3 mm and a porosity of 97% was derived to be 5.2 × 10−10 m2. Using an oxygen consumption rate of OBs with a value of 1.48 × 10−9 kg s−1 m−3 (ref. 39) and a mass fraction of oxygen of about 6.38 × 10−6 for normoxic (19.8%) and 2.26 × 10−6 for physoxic conditions (7%), the oxygen content in the porous cell-laden scaffold could be modelled for both oxygen levels.
Isolation, expansion and seeding of primary osteoblasts on type I collagen scaffolds
This study was approved by the Ethics Committee of the Berlin Medical Chamber (Eth-11/17), and all primary cell donors provided written informed consent. Primary human OBs from n = 4 donors (Table S1†) were isolated by outgrowth from cancellous bone. Hematopoietic (red) cells from freshly removed bone tissue were washed out with DPBS until the remaining tissue appeared bright white. Subsequently, the specimen was cut into small pieces with a sterile scalpel, and OBs were isolated by incubating portions of ∼5 g in polystyrene cell culture flasks of 300 cm2 (T300). For both the purification and subsequent expansion, OBs were maintained in DMEM LG supplemented with 10% FBS, 5 mM L-alanyl-L-glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. When reaching 90% confluence after 30 to 40 days, cells were trypsinised with TrypLE Select, and 2 × 106 cells were seeded onto a type I collagen scaffold (size: 13 mm in diameter × 3 mm in height; porosity: 97%), using the drop-in method described in the manufacturer's instructions (Optimaix 3D; Matricel, Herzogenrath, Germany).Culture of cell-laden scaffolds in the MPS
Cell-laden scaffolds, which were placed on holder plates, to ensure optimal positioning throughout the experiment, suitable for evenly distribution of forces, were transferred into the MPS culture chambers for seven days under the specified conditions (Fig. 4A). All scaffolds were perfused at a rate of 0.5 mL min−1. Medium exchange of the fluidic circuit was performed once a day by pumping 5.8 mL MPS medium at a flow rate of 15.7 μL min−1 into each of the unit reservoirs (RII) (Fig. 1B and S1A′†). Excess medium was removed by outflow into 3D printed waste containers. Sampling of MPS medium was performed daily prior to exchange of medium, slowly collecting 1 mL through a silicone septum (IBIDI, Gräfelfing, Germany) integrated into the fluidic circuit using a sterile syringe and needle. Samples were stored at −80 °C until further processing.If applied, mechanical loading with 10% compression at 1 Hz was performed at regular intervals for 2 h each time alternating with a 2 h resting period during the seven days of operation (the loading script is provided as ESI†). The maximum loading time did not exceed 8 h per day. Oxygen levels in the system were constantly monitored and adjusted to either 19.8% or 7% oxygen during the entire experiment. For oxygen control, see section “Oxygen measurement and control”.
Analysis of metabolism and osteogenesis in MPS culture medium
All colorimetric and fluorimetric assays described in the following section were analysed on a Multimode microplate reader (M200 PRO; Tecan, Männedorf, Switzerland). If not stated otherwise, measurements were carried out in triplicates. To determine total protein concentration and a specific protease activity as an indicator of cell death, cryopreserved samples from the MPS culture medium were thawed on ice. Total protein concentrations were determined using the Pierce BCA Protein Assay Kit. Cell death was investigated fluorometrically using the CytoTox-Fluor Assay Kit according to the manufacturer's instructions. The fluorescence signal, as a measure for protease activity, was normalised to the total amount of protein. For analysis of metabolic and osteogenic markers, medium samples were further processed using Amicon Ultra-0.5 centrifugal filters (0.5 mL; cut-off: 10 kDA) (Merck Millipore, Burlington, MA, US). The flow-through was analysed for lactate, glucose and phosphate concentrations. The retentate was used for quantification of the amount of osteogenic marker protein P1NP. For glucose, lactate and phosphate concentrations, commercially available glucose, lactate and phosphate assay kits were employed according to the manufacturer's protocols. For the assessment of extracellular P1NP formation, human P1NP ELISA Kit was utilised according to the manufacturer's protocol.Processing of the cell-laden scaffolds for endpoint measurements
After seven days of culture in the MPS the cell-laden scaffolds were processed for further analyses. The scaffolds were washed at least three times in 5 mL of 37 °C-prewarmed DPBS, inverted several times until phenol red-free and cut into halves using a scalpel. One-half was used to determine mitochondrial activity, ALP activity, total DNA and total protein content; the second half was used for gene expression analysis.Quantification of mitochondrial activity, ALP activity, protein and DNA content in cell-laden scaffolds
Seven days old OB-laden scaffolds were washed as described above. Mitochondrial activity was assessed using the PrestoBlue Cell Viability Reagent according to the manufacturer's instructions with adaptations made for cell-laden scaffolds. In brief, one-half of each cell-laden scaffolds was immersed in 1 mL working reagent (0.9 mL standard expansion medium supplemented with 0.1 mL PrestoBlue reagent), followed by 60 min of incubation under standard cell culture conditions. Triplicates of 200 μL of the reacted reagent were then transferred to a 48-well plate for fluorometric reading. Subsequently, cell-laden scaffolds were washed again at least three times with DPBS prewarmed to 37 °C until the coloration was cleared. ALP activity was determined as described by Krause and co-authors.65 Briefly, the scaffold halves were equilibrated in 2 mL of assay buffer (0.1 M NaCl, 0.1 M Tris, 0.001 M MgCl2 × 6H2O in deionised water) and then plunged into 1 mL of working solution (2.7 mM pNpp and 1 M DEA in deionised water; mixed 1:1 with assay buffer), followed by 10 min incubation under standard cell culture conditions. Scaffolds were removed from working reagent and transiently stored in 2 mL of prewarmed DPBS. To stop the enzymatic reaction, 1 mL of 1 M NaOH was added to the working solution, mixed properly, and three times 100 μL of this solution were transferred to a 96-well plate for photometric reading. Both, mitochondrial activity and ALP activity were later normalised to total DNA content. For further analysis, the cell-laden scaffolds were placed into screw-cap tubes supplemented with ceramic beads. Tubes were snap-frozen in liquid nitrogen, followed by homogenisation in a bead mill (Minilys; Bertin Instruments, Montigny-le-Bretonneux, France). The whole lysate (smashed scaffold-DPBS mixture) was mixed with DPBS to a final volume of 120 μL. 3 μL of this solution were used for DNA quantification with a Cell Proliferation Assay Kit (CyQUANT) according to the manufacturer's protocol. 54 μL were mixed with 26 μL of protein extraction buffer (PEB) to prevent further degradation of proteins. PEB was prepared as described by Davis and co-authors.66 Protein concentrations were determined in duplicates employing a Protein Assay Kit (Pierce BCA) using 25 μL of sample per measurement.
Histology
After seven days of culture in the MPS, the cell-laden scaffolds were washed as described above. Scaffolds were then cut into halves and fixed using 4% PFA solution for 10 min at RT; they were embedded in paraffin and sliced into 50 μm-sections using the manual microtome (RM 2035; Leica Microsystems, Wetzlar, Germany). Scaffold sections were transferred into 40 °C-prewarmed deionised water, mounted onto HistoBond adhesive glass slides (Paul Marienfeld GmbH & Co. KG, Lauda-Koenigshofen, Germany), dried on a hot plate, and deparaffinised with xylene for 60 min at RT followed by rehydration, von Kossa and DRAQ5 staining's. Von Kossa staining for visualisation of mineralisation was performed with 1% (w/v) silver nitrate in water for 20 min under UV light followed by washing and incubation with 5% (w/v) sodium thiosulfate in water for 5 min. To counterstain cell nuclei, sections were washed again and incubated with 5 μM DRAQ5, a far-red DNA stain. After serial dehydration of sections and soaking twice with xylene, samples were embedded in ROTI Histokitt synthetic embedding medium.Images of scaffold sections were acquired with the Axio Observer.Z1 inverted epifluorescence microscope (ZEISS Microscopy, Jena, Germany) and objectives LD-A-Plan 5×/0.15 Ph1 or Plan-Apochromat 40×/0.95 Korr. Pixel sizes of acquired images are 1.25 μm and 0.158 μm, respectively. For fluorescence detection of DRAQ5, the Zeiss filter set FS50 (BP 640/30, FT 660, BP 690/50) was used. Silver staining was detected by standard brightfield transmitted light imaging. All images were acquired as tile scans with 20% overlap. Shading corrections of vignetting effects and tile stitching were performed with ZEN blue software version 3.2 (ZEISS Microscopy, Jena, Germany) with identical settings and shading reference images for all samples.
Second harmonic generation (SHG) and fluorescence imaging
Scaffolds were washed and fixed with 4% PFA solution for 1 h at 4 °C. Fixed scaffolds were stored in DPBS with 25 mg mL−1 gentamicin, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 4 °C until further use. Prior to imaging, the cells were permeabilised with 1% Triton-X-100 in DPBS for 30 min at RT. Subsequent washing and incubation steps were performed with DPBS containing 0.05% Tween 20 (DPBS-T). Specimen were blocked with 3% (w/v) BSA in DPBS-T for 1 h at RT followed by incubation at 4 °C overnight. Phalloidin-iFluor 647 reagent (ab176759, Abcam, Cambridge, United Kingdom) was diluted 1:500 in blocking buffer and applied for 4 h at RT followed by three brief washing steps. For nuclear staining, samples were incubated with 50 nM SYTOX orange nucleic acid stain in DPBS for 35 min at RT.
Fluorescence and SHG imaging were performed with an upright confocal laser scanning LSM 880 NLO multiphoton microscope (ZEISS Microscopy, Jena, Germany) with a 20× dipping objective (W Plan-Apochromat 20×/1.0 DIC VIS-IR). Scaffolds were transferred to 6 cm Petri dishes filled with 20 ml DPBS and equipped with in-house designed 3D-printed inserts as sample holders. Label-free detection of type I collagen-specific SHG signals allowed us to image distribution and architecture of OBs in the context of the type I collagen scaffold. Confocal fluorescence signals of SYTOX orange were captured by excitation with a HeNe-laser at 543 nm and detection of fluorescence emission from 548 to 630 nm. Phalloidin-iFluor 647 was excited at 633 nm and detected between 638 and 735 nm. Scaffold-specific SHG signal was excited at 880 nm with a spectrally tuneable titanium-sapphire laser Chameleon Vision II (Coherent) and was detected on a forward non-descanned detector with SP485 filter (ZEISS Microscopy, Jena, Germany). Images were acquired as z-stacks at intervals of 0.87 μm in z (approx. 50% overlap of confocal planes, pinhole 40 μm) and xy-pixel size 415 nm. Maximum intensity projections and 3D surface rendering reconstructions of image stacks were generated using ZEN black software (ZEISS Microscopy, Jena, Germany) version 2.3 SP1. The imaging set-up had to be optimized to avoid considerable scaffold autofluorescence in the blue and green spectral range. Imaging depth was limited to the first 100–200 μm underneath the scaffold surface.
Statistical analysis
Both, timeline experiments (glucose, lactate, phosphate, cytotoxicity and P1NP analyses) and endpoint assays (DNA and protein determination, gene expression, ALP and mitochondrial activity analyses) were studied on four different cell donors (n = 4) within the MPS (1 donor = 1 MPS run). In each experiment, four different conditions were compared. For this purpose, it was investigated whether the reduction of the oxygen concentration from normoxia (19.8%) to physoxia (7%) has an influence on the cell response within the type I collagen scaffolds. Furthermore, the influence of mechanical load on the behaviour of the cells within the tissue constructs was analysed. Therefore, we assumed that the amount of glucose or lactate as a function of time (y(t)) can be described by an asymptotic regression model:67| y(t) = Asym + (R0 − Asym)exp(−exp(lrc)t) + ε | (1) |
| Asym = Xβ + Z1bdon + Z2bexp + Z3btreat. | (2) |
| bdon ∼ N(0, σb), bexp ∼ N(0, σexp), btreat ∼ N(0, σbtreat), ε ∼ N(0, σε). |
No statistical model was used to evaluate cytotoxicity over culture time and P1NP on day 7.
Comparison of mitochondrial activity, total DNA content, total protein amount, ALP activity, gene expression levels and day 7 normalisations was performed using a linear mixed model.
| y = Xβ + Z1bdon + Z2bexp + Z3btreat. | (3) |
| Symbol | Quantity |
|---|---|
| t | Time |
| Asym | Asymptote |
| R 0 | Value at time point 0 |
| exp | Experimental setting |
| lrc | Slope |
| E | Technical variance |
| X | Fixed effects |
| β | Vector of fixed effect coefficient |
| Z | Random effects |
| 1, 2, 3 | Effect number |
| b | Random coefficient |
| don | Donor |
| treat | Treatment |
| N | Normal distribution |
| σ | Standard deviation |
Author contributions
Project administration FS. Funding acquisition GS, MRS and FS. Conceptualisation FS, JS, GS and TJ. Resources GS and TJ. Methodology FS, JS and JD, YAT. Investigation JS, SSS, FS, CH, KR, KG, JD and YAT. Software FS and NV. Formal analysis MS, FS, JS. Visualisation FS, JS and CH. Writing – review & editing FS, JS, EW, CH, KR, MS, MRS, NV and GS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an internal BfR research funding program (Sonderforschungsprojekt 1322-706). In addition, this project received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 812954. We gratefully acknowledge our colleagues from BfR/Bf3R for scientific input and comments on the manuscript, especially Michael Oelgeschläger, Sebastian Dunst and Aline Stolz-Ertych for constructive and scientifically valuable discussions. We thank Beate Döring, Celina Aleth, Houda Kalot and Martin Textor for excellent technical assistance. We gratefully acknowledge Prof. Margaret Ashcroft from Cambridge University, Department of Medicine, for kindly providing us the U2OS-HRE-LUC cells. We thank Jose Manuel Villarejo Ramos and Birk Urmersbach for implementing the P-element based control loop in a python-based user interface. We gratefully acknowledge microfluidic ChipShop GmbH for joint preliminary design and scientific input in the development of the MPS main unit and for providing the design drawing of the MPS main unit in Fig. 1A.References
- A. Gonciulea and S. J. de Beur, The dynamic skeleton, Rev. Endocr. Metab. Disord., 2015, 16(2), 79–91 CrossRef CAS PubMed.
- X. Feng and J. M. McDonald, Disorders of Bone Remodeling, Annu. Rev. Pathol.: Mech. Dis., 2011, 6(1), 121–145 CrossRef CAS PubMed.
- S. G. Klein, S. M. Alsolami, A. Steckbauer, S. Arossa, A. J. Parry and G. R. Mandujano, et al., A prevalent neglect of environmental control in mammalian cell culture calls for best practices, Nat. Biomed. Eng., 2021, 5(8), 787–792 CrossRef PubMed.
- V. Palacio-Castaneda, N. Velthuijs, S. Le Gac and W. P. R. Verdurmen, Oxygen control: the often overlooked but essential piece to create better in vitro systems, Lab Chip, 2022, 22(6), 1068–1092 RSC.
- F. Zheng, F. Fu, Y. Cheng, C. Wang, Y. Zhao and Z. Gu, Organ-on-a-Chip Systems: Microengineering to Biomimic Living Systems, Small, 2016, 12(17), 2253–2282 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Microfluidic organs-on-chips, Nat. Biotechnol., 2014, 32(8), 760–772 CrossRef CAS PubMed.
- R. Kodzius, F. Schulze, X. Gao and M. R. Schneider, Organ-on-Chip Technology: Current State and Future Developments, Genes, 2017, 8(10), 266 CrossRef PubMed.
- J. Scheinpflug, M. Pfeiffenberger, A. Damerau, F. Schwarz, M. Textor and A. Lang, et al., Journey into Bone Models: A Review, Genes, 2018, 9(5), 247 CrossRef PubMed.
- J. Kim, H. Lee, S. Selimovic, R. Gauvin and H. Bae, Organ-on-a-chip: development and clinical prospects toward toxicity assessment with an emphasis on bone marrow, Drug Saf., 2015, 38(5), 409–418 CrossRef CAS PubMed.
- B. Clarke, Normal bone anatomy and physiology, Clin. J. Am. Soc. Nephrol., 2008, 3(Suppl 3), S131–S139 CrossRef CAS PubMed.
- Y. Uda, E. Azab, N. Sun, C. Shi and P. D. Pajevic, Osteocyte Mechanobiology, Curr. Osteoporos. Rep., 2017, 15(4), 318–325 CrossRef PubMed.
- A. G. Robling and L. F. Bonewald, The Osteocyte: New Insights, Annu. Rev. Physiol., 2020, 82, 485–506 CrossRef CAS PubMed.
- C. Ma, T. Du, X. Niu and Y. Fan, Biomechanics and mechanobiology of the bone matrix, Bone Res., 2022, 10(1), 59 CrossRef CAS PubMed.
- P. J. Ehrlich and L. E. Lanyon, Mechanical strain and bone cell function: a review, Osteoporosis Int., 2002, 13(9), 688–700 CrossRef CAS PubMed.
- N. Matsuda, N. Morita, K. Matsuda and M. Watanabe, Proliferation and differentiation of human osteoblastic cells associated with differential activation of MAP kinases in response to epidermal growth factor, hypoxia, and mechanical stress in vitro, Biochem. Biophys. Res. Commun., 1998, 249(2), 350–354 CrossRef CAS PubMed.
- A. Rutkovskiy, K. O. Stenslokken and I. J. Vaage, Osteoblast Differentiation at a Glance, Med. Sci. Monit. Basic Res., 2016, 22, 95–106 CrossRef PubMed.
- O. C. Tuncay, D. Ho and M. K. Barker, Oxygen tension regulates osteoblast function, Am. J. Orthod., 1994, 105(5), 457–463 CrossRef CAS PubMed.
- H. K. Vaananen and T. Laitala-Leinonen, Osteoclast lineage and function, Arch. Biochem. Biophys., 2008, 473(2), 132–138 CrossRef PubMed.
- J. A. Buckwalter, M. J. Glimcher, R. R. Cooper and R. Recker, Bone biology. I: Structure, blood supply, cells, matrix, and mineralization, Instr. Course Lect., 1996, 45, 371–386 CAS.
- M. J. Glimcher, The nature of the mineral component of bone and the mechanism of calcification, Instr. Course Lect., 1987, 36, 49–69 CAS.
- H. P. Wiesmann, U. Meyer, U. Plate and H. J. Hohling, Aspects of collagen mineralization in hard tissue formation, Int. Rev. Cytol., 2005, 242, 121–156 CrossRef CAS PubMed.
- L. Shen, G. Hu and C. M. Karner, Bioenergetic Metabolism In Osteoblast Differentiation, Curr. Osteoporos. Rep., 2022, 20(1), 53–64 CrossRef PubMed.
- B. Flanagan and G. Nichols Jr., Metabolic Studies of Bone in Vitro. V. Glucose Metabolism and Collagen Biosynthesis, J. Biol. Chem., 1964, 239, 1261–1265 CrossRef CAS PubMed.
- W. C. Lee, A. R. Guntur, F. Long and C. J. Rosen, Energy Metabolism of the Osteoblast: Implications for Osteoporosis, Endocr. Rev., 2017, 38(3), 255–266 CrossRef PubMed.
- J. S. Harrison, P. Rameshwar, V. Chang and P. Bandari, Oxygen saturation in the bone marrow of healthy volunteers, Blood, 2002, 99(1), 394 CrossRef CAS PubMed.
- M. Jez, P. Rozman, Z. Ivanovic and T. Bas, Concise review: the role of oxygen in hematopoietic stem cell physiology, J. Cell. Physiol., 2015, 230(9), 1999–2005 CrossRef CAS PubMed.
- A. Petersen, A. Princ, G. Korus, A. Ellinghaus, H. Leemhuis and A. Herrera, et al., A biomaterial with a channel-like pore architecture induces endochondral healing of bone defects, Nat. Commun., 2018, 9(1), 4430 CrossRef CAS PubMed.
- P. Klein, H. Schell, F. Streitparth, M. Heller, J. P. Kassi and F. Kandziora, et al., The initial phase of fracture healing is specifically sensitive to mechanical conditions, J. Orthop. Res., 2003, 21(4), 662–669 CrossRef PubMed.
- W. Hoffmann, S. Feliciano, I. Martin, M. de Wild and D. Wendt, Novel perfused compression bioreactor system as an in vitro model to investigate fracture healing. Front Bioeng, BioTechniques, 2015, 3, 10 Search PubMed.
- M. Jagodzinski, A. Breitbart, M. Wehmeier, E. Hesse, C. Haasper and C. Krettek, et al., Influence of perfusion and cyclic compression on proliferation and differentiation of bone marrow stromal cells in 3-dimensional culture, J. Biomech., 2008, 41(9), 1885–1891 CrossRef CAS PubMed.
- M. V. Lipreri, N. Baldini, G. Graziani and S. Avnet, Perfused Platforms to Mimic Bone Microenvironment at the Macro/Milli/Microscale: Pros and Cons, Front. Cell Dev. Biol., 2022, 9, 760667 CrossRef PubMed.
- G. Mehta, J. Lee, W. Cha, Y. C. Tung, J. J. Linderman and S. Takayama, Hard top soft bottom microfluidic devices for cell culture and chemical analysis, Anal. Chem., 2009, 81(10), 3714–3722 CrossRef CAS PubMed.
- P. M. van Midwoud, A. Janse, M. T. Merema, G. M. Groothuis and E. Verpoorte, Comparison of biocompatibility and adsorption properties of different plastics for advanced microfluidic cell and tissue culture models, Anal. Chem., 2012, 84(9), 3938–3944 CrossRef CAS PubMed.
- E. Berthier, E. W. Young and D. Beebe, Engineers are from PDMS-land, Biologists are from Polystyrenia, Lab Chip, 2012, 12(7), 1224–1237 RSC.
- D. F. Gilbert, S. A. Mofrad, O. Friedrich and J. Wiest, Proliferation characteristics of cells cultured under periodic versus static conditions, Cytotechnology, 2019, 71(1), 443–452 CrossRef CAS PubMed.
- P. G. Miller and M. L. Shuler, Design and Demonstration of a Pumpless 14 Compartment Microphysiological System, Biotechnol. Bioeng., 2016, 113(10), 2213–2227 CrossRef CAS PubMed.
- H. E. Abaci, R. Devendra, Q. Smith, S. Gerecht and G. Drazer, Design and development of microbioreactors for long-term cell culture in controlled oxygen microenvironments, Biomed. Microdevices, 2012, 14(1), 145–152 CrossRef CAS PubMed.
- N. M. Chau, P. Rogers, W. Aherne, V. Carroll, I. Collins and E. McDonald, et al., Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1alpha induction in response to hypoxic stress and growth factors, Cancer Res., 2005, 65(11), 4918–4928 CrossRef CAS PubMed.
- H. Wang, K. D. Song, L. Wang, Y. Liu, Y. C. Liu and R. P. Li, et al., Measurement of oxygen consumption rate of osteoblasts from Sprague-Dawley rat calvaria in different in vitro cultures, Afr. J. Biotechnol., 2011, 10(34), 6640–6646 Search PubMed.
- S. H. Park, W. Y. Sim, B. H. Min, S. S. Yang, A. Khademhosseini and D. L. Kaplan, Chip-based comparison of the osteogenesis of human bone marrow- and adipose tissue-derived mesenchymal stem cells under mechanical stimulation, PLoS One, 2012, 7(9), e46689 CrossRef CAS PubMed.
- A. Petersen, P. Joly, C. Bergmann, G. Korus and G. N. Duda, The impact of substrate stiffness and mechanical loading on fibroblast-induced scaffold remodeling, Tissue Eng., Part A, 2012, 18(17–18), 1804–1817 CrossRef CAS PubMed.
- M. Morlock, E. Schneider, A. Bluhm, M. Vollmer, G. Bergmann and V. Muller, et al., Duration and frequency of every day activities in total hip patients, J. Biomech., 2001, 34(7), 873–881 CrossRef CAS PubMed.
- A. G. Robling, D. B. Burr and C. H. Turner, Recovery periods restore mechanosensitivity to dynamically loaded bone, J. Exp. Biol., 2001, 204(Pt 19), 3389–3399 CrossRef CAS PubMed.
- S. Srinivasan, S. C. Agans, K. A. King, N. Y. Moy, S. L. Poliachik and T. S. Gross, Enabling bone formation in the aged skeleton via rest-inserted mechanical loading, Bone, 2003, 33(6), 946–955 CrossRef PubMed.
- M. A. K. Liebschner, Biomechanical considerations of animal models used in tissue engineering of bone, Biomaterials, 2004, 25(9), 1697–1714 CrossRef CAS PubMed.
- G. Borciani, G. Montalbano, N. Baldini, C. Vitale-Brovarone and G. Ciapetti, Protocol of Co-Culture of Human Osteoblasts and Osteoclasts to Test Biomaterials for Bone Tissue Engineering, Methods Protoc., 2022, 5(1), 8 CrossRef CAS PubMed.
- E. M. Czekanska, M. J. Stoddart, J. R. Ralphs, R. G. Richards and J. S. Hayes, A phenotypic comparison of osteoblast cell lines versus human primary osteoblasts for biomaterials testing, J. Biomed. Mater. Res., 2014, 102(8), 2636–2643 CrossRef CAS PubMed.
- A. Al-Ani, D. Toms, D. Kondro, J. Thundathil, Y. Yu and M. Ungrin, Oxygenation in cell culture: Critical parameters for reproducibility are routinely not reported, PLoS One, 2018, 13(10), e0204269 CrossRef PubMed.
- A. L. Niles, R. A. Moravec, P. Eric Hesselberth, M. A. Scurria, W. J. Daily and T. L. Riss, A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers, Anal. Biochem., 2007, 366(2), 197–206 CrossRef CAS PubMed.
- J. D. Firth, B. L. Ebert and P. J. Ratcliffe, Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements, J. Biol. Chem., 1995, 270(36), 21021–21027 CrossRef CAS PubMed.
- Z. Zeng, D. Jing, X. Zhang, Y. Duan and F. Xue, Cyclic mechanical stretch promotes energy metabolism in osteoblast-like cells through an mTOR signaling-associated mechanism, Int. J. Mol. Med., 2015, 36(4), 947–956 CrossRef CAS PubMed.
- S. Vasikaran, R. Eastell, O. Bruyere, A. J. Foldes, P. Garnero and A. Griesmacher, et al., Markers of bone turnover for the prediction of fracture risk and monitoring of osteoporosis treatment: a need for international reference standards, Osteoporosis Int., 2011, 22(2), 391–420 CrossRef CAS PubMed.
- M. K. Koivula, L. Risteli and J. Risteli, Measurement of aminoterminal propeptide of type I procollagen (PINP) in serum, Clin. Biochem., 2012, 45(12), 920–927 CrossRef CAS PubMed.
- C. Williams and A. Sapra, Osteoporosis Markers, StatPearls, Treasure Island (FL), 2022 Search PubMed.
- A. Rakow, J. Schoon, A. Dienelt, T. John, M. Textor and G. Duda, et al., Influence of particulate and dissociated metal-on-metal hip endoprosthesis wear on mesenchymal stromal cells in vivo and in vitro, Biomaterials, 2016, 98, 31–40 CrossRef CAS PubMed.
- S. Shetty, N. Kapoor, J. D. Bondu, N. Thomas and T. V. Paul, Bone turnover markers: Emerging tool in the management of osteoporosis, Indian J. Endocrinol. Metab., 2016, 20(6), 846–852 CrossRef PubMed.
- N. Matsuda, N. Morita, K. Matsuda and M. Watanabe, Proliferation and Differentiation of Human Osteoblastic Cells Associated with Differential Activation of MAP Kinases in Response to Epidermal Growth Factor, Hypoxia, and Mechanical Stressin Vitro, Biochem. Biophys. Res. Commun., 1998, 249(2), 350–354 CrossRef CAS PubMed.
- G. Matziolis, J. Tuischer, G. Kasper, M. Thompson, B. Bartmeyer and D. Krocker, et al., Simulation of cell differentiation in fracture healing: mechanically loaded composite scaffolds in a novel bioreactor system, Tissue Eng., 2006, 12(1), 201–208 CrossRef CAS PubMed.
- J. B. Lian and G. S. Stein, Development of the osteoblast phenotype: molecular mechanisms mediating osteoblast growth and differentiation, Iowa Orthop. J., 1995, 15, 118–140 CAS.
- C. S. Bahney, R. L. Zondervan, P. Allison, A. Theologis, J. W. Ashley and J. Ahn, et al., Cellular biology of fracture healing, J. Orthop. Res., 2019, 37(1), 35–50 CrossRef PubMed.
- E. Araldi and E. Schipani, Hypoxia, HIFs and bone development, Bone, 2010, 47(2), 190–196 CrossRef CAS PubMed.
- H. M. Zannit, M. D. Brodt and M. J. Silva, Proliferating osteoblasts are necessary for maximal bone anabolic response to loading in mice, FASEB J., 2020, 34(9), 12739–12750 CrossRef CAS PubMed.
- A. Knight, Systematic reviews of animal experiments demonstrate poor human clinical and toxicological utility, ATLA, Altern. Lab. Anim., 2007, 35(6), 641–659 CrossRef CAS PubMed.
- L. M. Newman, E. M. Johnson and R. E. Staples, Assessment of the effectiveness of animal developmental toxicity testing for human safety, Reprod. Toxicol., 1993, 7(4), 359–390 CrossRef CAS PubMed.
- U. Krause, A. Seckinger and C. A. Gregory, Assays of osteogenic differentiation by cultured human mesenchymal stem cells, Methods Mol. Biol., 2011, 698, 215–230 CrossRef CAS PubMed.
- L. A. Davis, A. Dienelt and N. I. zur Nieden, Absorption-based assays for the analysis of osteogenic and chondrogenic yield, Methods Mol. Biol., 2011, 690, 255–272 CrossRef CAS PubMed.
- J. C. Pinheiro and D. M. Bates, Linear Mixed-Effects Models: Basic Concepts and Examples, in Mixed-Effects Models in S and S-PLUS. Statistics and Computing, ed. J. C. Pinheiro and D. M. Bates, Springer New York, New York, NY, 2000, pp. 3–56 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00154g |
| ‡ Current address: Charité – Universitätsmedizin Berlin, Department of Neurology with Experimental Neurology, Centre for Stroke Research Berlin, 10117, Berlin, Germany. |
| § Current address: Formlabs GmbH, 12459 Berlin, Germany. |
| ¶ Current address: Institute of Veterinary Physiology, Faculty of Veterinary Medicine, University of Leipzig, Leipzig, Germany. |
| || Current address: Center for Orthopaedics, Trauma Surgery and Rehabilitation Medicine, University Medicine Greifswald, 17475 Greifswald, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |