
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA low-cost, paper-based hybrid capture assay to detect high-risk HPV DNA for cervical cancer screening in low-resource settings†
Chelsey A.
Smith‡
 a,
Megan M.
Chang‡
a,
Kathryn A.
Kundrod
a,
Emilie N.
Novak
a,
Sonia G.
Parra
a,
Leticia
López
b,
Celda
Mavume
c,
Cesaltina
Lorenzoni
cd,
Mauricio
Maza
b,
Mila P.
Salcedo
e,
Jennifer L.
Carns
a,
Ellen
Baker
e,
Jane
Montealegre
f,
Michael
Scheurer
f,
Philip E.
Castle
g,
Kathleen M.
Schmeler
e and
Rebecca R.
Richards-Kortum
*a
a,
Megan M.
Chang‡
a,
Kathryn A.
Kundrod
a,
Emilie N.
Novak
a,
Sonia G.
Parra
a,
Leticia
López
b,
Celda
Mavume
c,
Cesaltina
Lorenzoni
cd,
Mauricio
Maza
b,
Mila P.
Salcedo
e,
Jennifer L.
Carns
a,
Ellen
Baker
e,
Jane
Montealegre
f,
Michael
Scheurer
f,
Philip E.
Castle
g,
Kathleen M.
Schmeler
e and
Rebecca R.
Richards-Kortum
*a
aDepartment of Bioengineering, Rice University, Houston, TX, USA. E-mail: rkortum@rice.edu
bBasic Health International, San Salvador, El Salvador
cHospital Central de Maputo, Maputo, Mozambique
dMinisterio da Saude de Moçambique (MISAU), Maputo, Mozambique
eDepartment of Gynecologic Oncology & Reproductive Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA
fDepartment of Pediatrics-Hematology/Oncology, Baylor College of Medicine, Houston, TX, USA
gDivisions of Cancer Prevention and Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, MD, USA
First published on 22nd December 2022
Abstract
Cervical cancer is a leading cause of cancer death for women in low-resource settings. The World Health Organization recommends that cervical cancer screening programs incorporate HPV DNA testing, but available tests are expensive, require laboratory infrastructure, and cannot be performed at the point-of-care. We developed a two-dimensional paper network (2DPN), hybrid-capture, signal amplification assay and a point-of-care sample preparation protocol to detect high-risk HPV DNA from exfoliated cervical cells within an hour. The test does not require expensive equipment and has an estimated cost of <$3 per test without the need for batching. We evaluated performance of the paper HPV DNA assay with short synthetic and genomic HPV DNA targets, HPV positive and negative cellular samples, and two sets of clinical samples. The first set of clinical samples consisted of 16 biobanked, provider-collected cervical samples from a study in El Salvador previously tested with careHPV and subsequently tested in a controlled laboratory environment. The paper HPV DNA test correctly identified eight of eight HPV-negative clinical samples and seven of eight HPV-positive clinical samples. We then performed a field evaluation of the paper HPV DNA test in a hospital laboratory in Mozambique. Cellular controls generated expected results throughout field testing with fully lyophilized sample preparation and 2DPN reagents. When evaluated with 16 residual self-collected cervicovaginal samples previously tested by the GeneXpert HPV assay (“Xpert”), the accuracy of the HPV DNA paper test in the field was reduced compared to testing in the controlled laboratory environment, with positive results obtained for all eight HPV-positive samples as well as seven of eight HPV-negative samples. Further evaluation showed reduction in performance was likely due in part to increased concentration of exfoliated cells in the self-collected clinical samples from Mozambique compared with provider-collected samples from El Salvador. Finally, a formal usability assessment was conducted with users in El Salvador and Mozambique; the assay was rated as acceptable to perform after minimal training. With additional optimization for higher cell concentrations and inclusion of an internal cellular control, the paper HPV DNA assay offers promise as a low-cost, point-of-care cervical cancer screening test in low-resource settings.
Introduction
Cervical cancer is preventable, yet 604![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases and 342000 deaths due to cervical cancer are reported annually.1 Countries with accessible prophylactic human papillomavirus (HPV) vaccination and large-scale programs to screen for cervical cancer and its precursors have dramatically reduced the incidence and mortality of cervical cancer.2 However, women living in low-resource settings often lack access to these preventive services; as a result, they bear the majority of the global burden of cervical cancer.3,4 Although HPV vaccination is the ultimate cervical cancer prevention strategy, global HPV vaccination coverage remains low.5,6 In addition, HPV vaccines do not treat pre-existing infections and related abnormalities, and millions of women who did not receive the vaccine at an early age remain in need of screening.7,8
000 new cases and 342000 deaths due to cervical cancer are reported annually.1 Countries with accessible prophylactic human papillomavirus (HPV) vaccination and large-scale programs to screen for cervical cancer and its precursors have dramatically reduced the incidence and mortality of cervical cancer.2 However, women living in low-resource settings often lack access to these preventive services; as a result, they bear the majority of the global burden of cervical cancer.3,4 Although HPV vaccination is the ultimate cervical cancer prevention strategy, global HPV vaccination coverage remains low.5,6 In addition, HPV vaccines do not treat pre-existing infections and related abnormalities, and millions of women who did not receive the vaccine at an early age remain in need of screening.7,8
The most sensitive screening method for cervical cancer and its precursors is high-risk HPV DNA testing, which has negative predictive values over 99%.9–11 One study reported that a single screen using HPV DNA testing is effective at reducing up to 50% of advanced cervical cancers and related deaths over an 8-year period.12 Additionally, effective HPV DNA testing can be performed with self-collected cervical samples, which may increase access to cervical cancer screening for many women.13–15 However, commercially available HPV DNA tests are often not appropriate for use in low-resource settings.16,17 Initial HPV DNA tests were based on the principle of hybrid capture, in which RNA probes are used to capture target HPV DNA, followed by antibody labeling and ELISA-based signal amplification and detection. The industry standard hybrid capture test, digene HC2 HPV DNA Test (Qiagen, Germantown, MD, USA), requires highly trained personnel and significant laboratory infrastructure, and has a high per-test cost.18,19 careHPV (Qiagen), a hybrid-capture based HPV DNA test developed specifically for use in lower-resource settings, also requires expensive equipment and trained laboratory technicians, and samples must be run in batches of 90 to achieve the target per-test cost of $5.20,21 The need for batching specimens for careHPV testing can delay test results substantially and increases the likelihood that some patients will be lost to follow-up. A low-cost, sensitive HPV DNA test that can be performed at the point-of-care is needed to support global implementation and scale of cervical cancer prevention programs.
To meet this need, we developed a low-cost, paper-based HPV DNA assay that can be performed at the point-of-care. The assay uses a highly sensitive two-dimensional paper network (2DPN) to perform hybrid capture and detect high-risk HPV DNA (Fig. 1). In parallel, we developed a sample preparation method that can be used at the point-of-care to process cervical swabs for direct input to the assay. Together, the sample-to-answer workflow includes seven user steps, can be performed in one hour, and the only ancillary equipment required is a benchtop heater (<$300) (Fig. 2).
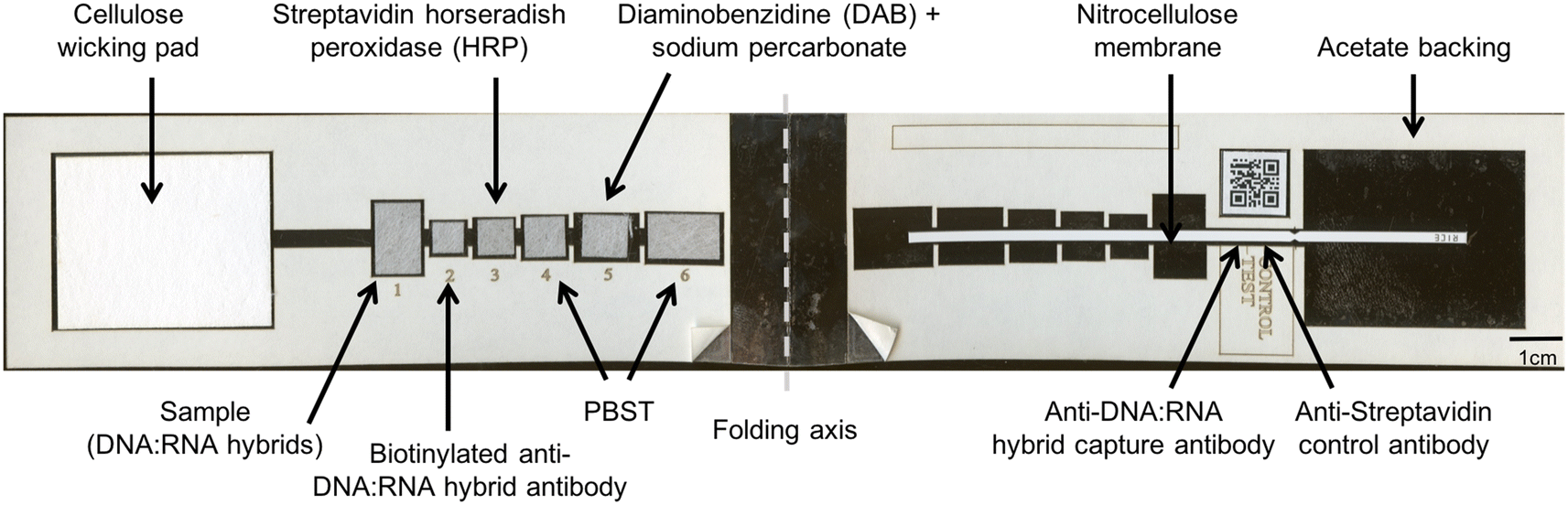 | ||
| Fig. 1 Paper HPV DNA assay components. The paper assay includes a wicking pad, six glass fiber pads with lyophilized detection reagents to perform the hybrid-capture reaction, and a nitrocellulose membrane with capture antibodies spotted in designated test and control line zones, all placed atop an adhesive acetate backing. A QR code adjacent to the test and control lines facilitates use with low-cost readers. A 1 cm scale bar is shown. | ||
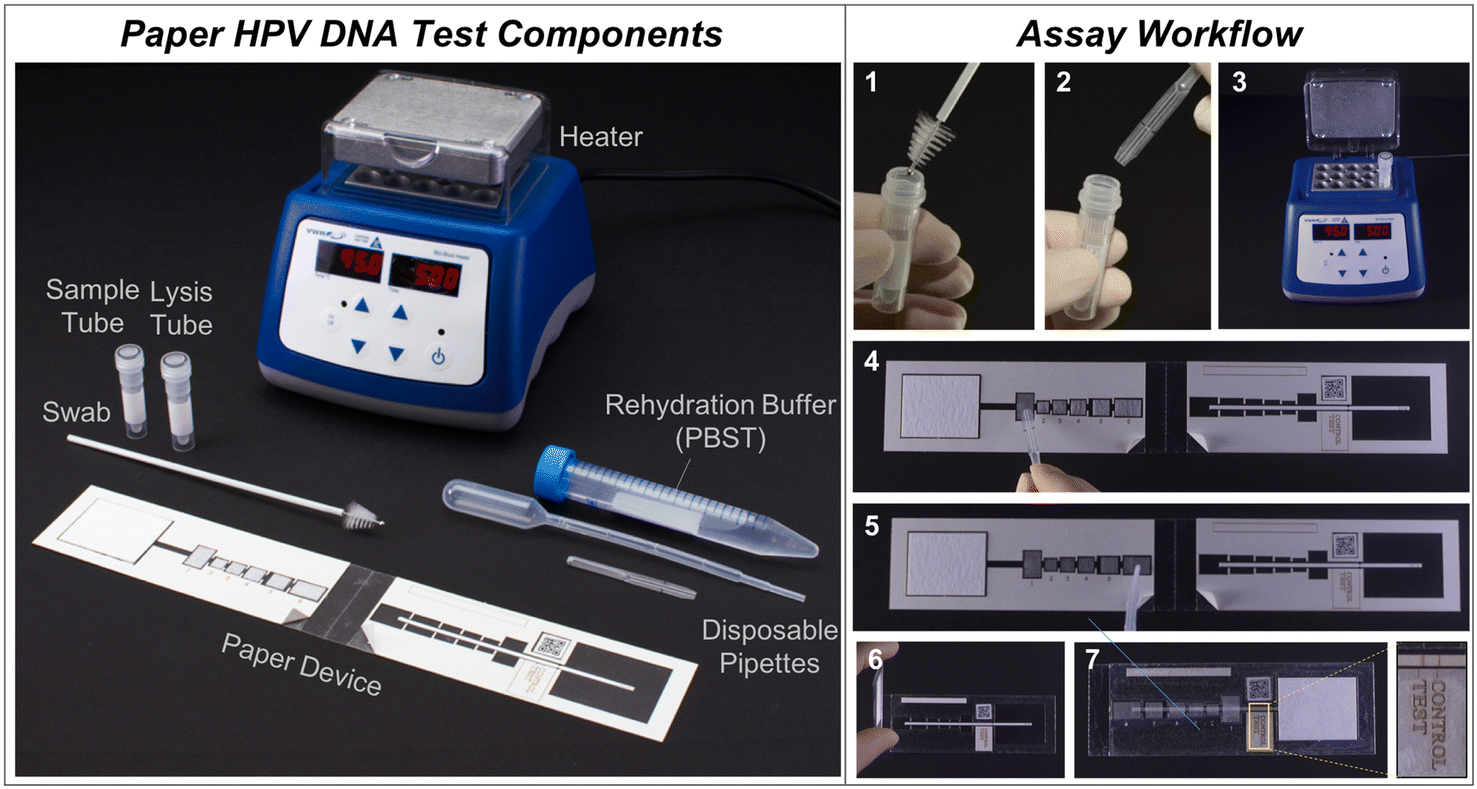 | ||
| Fig. 2 Point-of-care paper HPV DNA assay workflow. (Left) All necessary components for the assay, including the sample collection swab, sample tube, lysis tube, disposable pipettes, paper HPV DNA test, rehydration buffer, and heater. (Right) The workflow involves seven user steps. 1) Swab the cervix with a brush and place into the sample tube. 2) Using an exact volume disposable pipette, add sample into a vial with lyophilized high-risk HPV RNA and achromopeptidase (ACP). Mix and incubate for 5 minutes at room temperature. 3) Heat at 95 °C for 5 minutes. 4) Add sample to the first pad on the paper device. 5) Rehydrate lyophilized pads 2–6 with PBST (phosphate-buffered saline with 0.05% Tween20) rehydration buffer. 6) Peel paper backing to reveal sticky acetate and fold assay in half to initiate fluid flow, and 7) after 45 minutes observe signal visually or with a low-cost, automated reader. For visual interpretation, two visible lines indicate a positive result. For automated interpretation, a portable reader can be used.46 | ||
Experimental
We characterized the performance of the 2DPN hybrid capture-based assay and optimized a sample preparation protocol using samples of progressively increasing biological complexity, including short synthetic DNA targets, genomic DNA targets, cellular samples, and finally cervicovaginal swab samples. Using synthetic targets, we compared the limit of detection (LoD) of the paper HPV DNA assay to that of the gold standard technology for hybrid capture, the digene Hybrid Capture 2 assay. We incorporated lyophilized sample preparation and lyophilized 2DPN reagents into the assay workflow and evaluated performance using HPV-positive and HPV-negative cell lines to assess the LoD with more complex samples. We then performed two clinical pilot studies with 16 samples each to compare performance of the paper-based assay to a commercially available reference standard. The first clinical study was in a controlled laboratory environment where we tested 16 provider collected cervical samples from El Salvador and compared the results of the paper-based assay to prior testing by careHPV. The second was a field evaluation of the paper-based assay in Mozambique to evaluate feasibility in a point-of-care setting and compared the results to prior testing by the Xpert HPV Test (Cepheid, Sunnyvale, CA, USA). Finally, we report results of a study to assess usability of the point-of-care assay by target users (n = 44) in El Salvador and Mozambique.Paper HPV DNA assay components
The paper HPV DNA assay was assembled using a laser cutter (Universal Laser Systems, Scottsdale, AZ) to create device components, including adhesive plastic (5 mm Dura-Lar, Blick Art Supplies, Galesburg, IL), membrane backed CN95 nitrocellulose (Sartorius, Goettingen, Germany), glass fiber pads (grade 8951, Ahlstrom, Helsinki, Finland), and a wicking pad (C083, Millipore, Billerica, MA). The device components are shown in Fig. 1.Briefly, the 2DPN designed builds on a previous two-dimensional paper immunoassay for ultrasensitive detection of the malaria protein plasmodium falciparum HRP2;22 here, we adapted this design to perform a hybrid-capture reaction with visual detection of high-risk HPV DNA in a processed sample. The paper network consists of a sample pad that accepts a processed sample containing RNA–DNA hybrids, six reagent pads containing detection reagents or wash buffer, and a nitrocellulose strip with capture antibodies at a test and control line. To perform the test, the user adds a processed sample containing RNA–DNA hybrids to the sample pad. The user adds a drop of buffer to rehydrate each pad in the device and folds the device shut to initiate fluid flow down the strip. The test runs without further user interaction, and delivers the following reagents sequentially to the test and control lines to perform the hybrid capture reaction: (i) RNA–DNA hybrids; (ii) biotinylated monoclonal anti-RNA:DNA hybrid antibodies; (iii) streptavidin with poly-HRP; (iv) wash buffer (v) the colorimetric reagent diaminobenzidine (DAB) and (vi) a final wash buffer. The device is read visually.
A sciFLEXARRAYER S3 machine was used to print antibodies at the control and test lines on the nitrocellulose membrane. At the control line, the printer deposited 80 nL of streptavidin monoclonal antibody (S10D4, Thermo Fisher Scientific, Waltham, MA) prepared at 250 μg mL−1 in 1× PBST buffer containing 1% BSA, 5% sucrose and 5% trehalose. At the test line, the printer deposited 400 nL of anti-DNA–RNA hybrid antibody (MABE1095, Millipore, Billerica, MA) at a concentration of 1 mg mL−1. Once antibodies were printed, the nitrocellulose membranes were dried at 37 °C for 60 minutes, blocked for 30 minutes with 0.5% BSA (Sigma-Aldrich Inc, St. Louis, MO), 4% trehalose (Thermo Fisher Scientific, Waltham, MA), and 1% sucrose (Thermo Fisher Scientific, Waltham, MA) in PBST, and dried for an additional 90 minutes at 37 °C before being stored at 4 °C in a foil pouch with desiccant.
Enzyme-linked immunoassay (ELISA) reagents to perform the hybrid capture reaction included: 16 μg mL−1 biotinylated anti-DNA–RNA-hybrid detection antibody (ENH001, Kerafast, Boston, MA) applied to pad 2; 15 μg mL−1 streptavidin poly-HRP80 applied to pad 3; and 1 mg mL−1 diaminobenzidine (DAB, Sigma-Aldrich, St. Louis, MO) and sodium percarbonate (Sigma-Aldrich, St. Louis, Missouri) applied to pad 5. The sodium percarbonate was added to diaminobenzidine directly before running the assay with fresh reagents at 0.5% w/v or lyophilized onto a glass fiber pad and stacked with lyophilized DAB pad for all lyophilization experiments. A solution of 1% BSA in PBST was used as the wash buffer at pads 4 and 6 to separate the poly-HRP enzyme and colorimetric reagents while flowing down the nitrocellulose membrane. Volumes of reagents were: 1) 50 μL sample, 2) 15 μL detection antibody, 3) 20 μL streptavidin poly-HRP80, 4) 25 μL wash buffer, 5) 30 μL colorimetric reagents, and 6) 50 μL final wash buffer.
Point-of-care sample preparation protocol
ACP was evaluated for use in a buffer to fragment DNA and RNA and to lyse cells; the proteolytic enzyme has previously been shown to effectively lyse cellular samples in a point-of-care friendly format.23–25 ACP (MilliPore Sigma A3547, Burlington, MA) was reconstituted into 10 mM Tris (pH 8.0) with 5% trehalose at 20 U μL−1. For the point-of-care sample preparation protocol, 0.5 μL of high-risk HPV RNA Probe Cocktail (digene HC2, Qiagen, Germantown, MD), 18.25 μL of nuclease-free water with 5% trehalose, and 1.25 μL of 20 U μL−1 ACP were mixed together, incubated for 5 minutes at room temperature, and heated at 95 °C for 10 minutes to fragment RNA. After removal from the heater, 5 μL of 10× STE (Thermo Fisher Scientific, Waltham, MA) was added as a source of EDTA. At this point, the pre-treated RNA and lysis solution was either combined with 25 μL of sample immediately or lyophilized prior to sample addition. Lyophilization of RNA and ACP occurred in a PCR tube to create pellets. After sample addition, the solution was incubated for 5 minutes at room temperature, followed by a heating step at 95 °C for 5 minutes to denature and fragment the DNA. After removal from the heater, the tube was placed on ice until it could be tested. Upon cooling, the DNA hybridized to RNA in the solution, and the resulting solution containing DNA–RNA hybrids was added to the sample pad on the paper assay.Reagent lyophilization
Before lyophilization, detection antibody and streptavidin poly-HRP 80 were reconstituted in 1% BSA, 5% trehalose, and 5% sucrose in 1× PBST. DAB and sodium percarbonate were reconstituted in nuclease-free water with 5% trehalose. Wash pads were prepared using 1% BSA in PBST. Both reagent pads and RNA with ACP solutions were flash-frozen in liquid nitrogen for 20 seconds before lyophilizing for a minimum of 24 hours. Lyophilized reagents were stored with desiccant at −20 °C until use.Point-of-care workflow
To run the assay, the sample preparation protocol was performed to lyse cells, fragment DNA, and form DNA–RNA hybrids. The sample was placed on ice, and ELISA reagents were added directly to pads 2–6 for fresh reactions or pads were rehydrated using 1× PBST for assays assembled with lyophilized reagents. Then, an aliquot of the prepared sample was added to pad 1. Once the paper backing was removed to expose adhesive, each assay was folded in half to initiate fluid flow. After 45 minutes, signal formation within each test was analyzed visually, and all assays were imaged at 600 dots-per-inch (DPI) with a flatbed color scanner. The signal-to-background ratio was calculated as described below (see Signal-to-background analysis) at the test and control lines and compared to a preset threshold. If the signal-to-background ratio exceeded the threshold at the test and control lines, the assay was deemed positive for high-risk HPV DNA; if the threshold was exceeded only at the control line, the assay was deemed negative for high-risk HPV DNA. If the threshold was not exceeded at the control line, the assay was deemed invalid. In addition, visual interpretation of signal at the test and control lines was used to interpret results. The complete workflow is shown in Fig. 2.Signal-to-background analysis
Signal-to-background ratios at the test and control lines of the paper HPV DNA assay were calculated using a custom MATLAB code.22 A fixed-size region-of-interest ROI was placed over the test and control zones (signal ROI) and another ROI was placed over the adjacent nitrocellulose (background ROI). To compute intensity in each ROI, the maximum pixel value in each row of the ROI was averaged. The intensity of the signal ROI was divided by intensity of the corresponding background ROI to calculate the signal-to-background ratio.Statistical analysis
Probit analysis was used to determine reported limits of detection. First, a positivity threshold was determined as the average negative signal from three samples plus three standard deviations. Then, test results were binarized as positive or negative compared to that positivity threshold, and probit analysis was used to calculate the limit of detection with a probability value of 0.95 (XLSTAT, Addinsoft, Paris, France). All other statistical tests were conducted in GraphPad Prism.Digene HC2 reference standard
The digene HC2 test was performed on HPV calibrators and quality control solutions from the digene HC2 kit, including 5.0 × 105, 2.5 × 105, and 1.0 × 105 copies mL−1 of HPV16 DNA (High-Risk HPV Quality Control Solution), 5.0 × 105 copies mL−1 of HPV6 DNA (Low-Risk HPV Quality Control Solution), and a negative calibrator consisting of carrier DNA. The digene HC2 test was performed according to kit instructions. Briefly, DNA was denatured using a sodium hydroxide-based denaturant for one hour at 65 °C, followed by addition of probe RNA and annealing at 65 °C for 45 minutes. 100 μL of the hybrid solution were added to the HC2 capture plate and incubated at room temperature on a shaker at 1100 RPM for 60 minutes. Next, 75 μL of Detection Agent 1 was added and incubated for 45 minutes at room temperature. Wells were washed 6× with the Wash Buffer before 75 μL of Detection Agent 2 were added and incubated for 15 minutes at room temperature. Finally, chemiluminescence was measured using a plate reader (Tecan, Zürich, Switzerland).Evaluation of paper HPV DNA assay with short HPV synthetic DNA targets
Short, synthetic targets of HPV16 and HPV6 DNA containing 36 base pairs of type-specific sequences (Table S1†) were checked in NCBI Blast for specificity and ordered from Integrated DNA Technologies (Coralville, IA). They were hybridized to short synthetic HPV16 RNA probes (36 bases, Table S1;† IDT) and tested with both digene HC2 and the paper HPV DNA assay. HPV16 and HPV6 sequences are shown in Table S1.† Briefly, a linear dilution of HPV16 short synthetic targets was created from 5 × 1013 copies mL−1 to 5 × 109 copies mL−1. These standards, along with a buffer control and 5 × 1013 copies mL−1 of low-risk HPV6 DNA, were each combined with 10 μM complementary HPV16 RNA in 1× STE solution and heated for 0.5 minutes at 95 °C to denature DNA and create DNA–RNA hybrids. The resultant hybrids were tested in both digene Hybrid Capture 2 and on the paper HPV DNA assays as described in the workflow above.Evaluation of paper HPV DNA assay with genomic HPV DNA controls
Genomic HPV controls from the digene HC2 kit, including 5.0 × 105, 2.5 × 105, and 1.0 × 105 copies mL−1 of HPV16 DNA (High-Risk HPV Quality Control Solution), 5.0 × 105 copies mL−1 of HPV6 DNA (Low-Risk HPV Quality Control Solution), and a negative calibrator were evaluated per manufacturer's instruments in the digene HC2 kit; samples were also evaluated using the paper HPV DNA assay with the following three sample preparation and sample delivery protocols. In the first protocol, samples processed following digene HC2 instructions were added to paper assays held atop an orbital shaker at 220 rpm to enhance fluidic flow of genomic DNA through the nitrocellulose membrane. Orbital shaking assisted delivery of unfragmented nucleic acid through the nitrocellulose. In the second approach, DNA–RNA hybrids were incubated with ACP for 5 minutes at room temperature, heated for 30 seconds at 95 °C, followed by EDTA addition. These paper assays were also added to an orbital shaker at 220 RPM to enhance fluidic flow. In the third approach, RNA was incubated with ACP for 5 minutes at room temperature and pre-treated by heating for 10 minutes at 95 °C, followed by EDTA addition. Pre-treated RNA was combined with DNA and heated for an additional 5 minutes at 95 °C. Samples were run in triplicate on the paper HPV DNA assays without orbital shaking.Assessment of DNA and RNA fragmentation and development of sample preparation protocol
Theorizing that secondary structure from full genome HPV DNA and RNA could cause false-positive results in the paper assay, an experiment was performed to assess the size distribution of HPV DNA and HPV RNA fragments created by heating with ACP at 95 °C for various times.DNA was extracted from SiHa cells using the DNeasy®Blood & Tissue Handbook (Qiagen, Germantown, MD), and then added to a solution containing 1× STE and ACP to a final concentration of 0.5 U μL−1. Samples were incubated at room temperature for 5 minutes and then heated for 0, 0.5, 1, 2, 5, 10, or 30 minutes at 95 °C. Products were run on a 2% agarose gel at 140 V for 1.5 hours.
RNA was extracted from SiHa cells using the GeneJET RNA Purification Kit (Thermo Fisher Scientific, Waltham, MA), including performing Genomic DNA Removal and RNA Cleanup. RNA was added to a solution containing either 1) ACP to a final concentration of 0.5 U μL−1 without EDTA, or 2) a solution of 1× STE with ACP to a final concentration of 0.5 U μL−1. Samples without EDTA were incubated at room temperature for 5 minutes and then heated for 0, 0.5, 1, 2, 5, 10, or 30 minutes at 95 °C; samples with EDTA were similarly incubated at room temperature for five minutes, then heated at 95 °C for 0, 5, 10, 15, or 30 minutes. Products were run on a 1% agarose gel at 70 V for 2 hours.
After fragmentation, both a high-risk genomic HPV DNA control (5.0 × 105 copies mL−1 HPV16, digene HC2 high-risk quality control standard, Qiagen) and a low-risk genomic HPV DNA control (5.0 × 105 copies mL−1 HPV6, digene HC2 low-risk quality control standard, Qiagen) were tested in duplicate using the following two protocols. In the first, DNA and RNA were heated for 30 seconds at 95 °C after the addition of ACP and 5 minute room temperature incubation, followed by EDTA addition. In the second, RNA was first heated for 0.5, 5, or 10 minutes with only ACP. After heating, EDTA in the form of 1× STE was added to the vial along with the DNA. Samples were mixed, incubated for 5 minutes at room temperature, and then heated a second time for 0.5, 5, or 10 minutes at 95 °C. Hybrids resulting from these fragmentation profiles were tested on the paper HPV DNA assay.
Evaluation of paper HPV DNA assay with lyophilized reagents and cellular samples
To assess assay performance with cellular samples, a set of samples was assembled by combining an increasing number of HPV-positive SiHa (HPV16) or HeLa (HPV18) cells together with HPV-negative C33A cells. The total cell count was kept consistent at 1 million cells mL−1. Contrived cellular samples were processed using the point-of-care sample preparation protocol with lyophilized reagents. Samples were run in triplicate on the paper HPV DNA assays and imaged after an hour.Collection buffer assessment
HPV-positive HeLa and HPV-negative C33A cells were reconstituted in two sample collection buffers commonly used for cervical cytology specimens, namely SurePath preservation buffer (Becton Dickinson, Franklin Lakes, NJ) and PreservCyt (Hologic, Marlborough, MA). Prior to sample preparation, preserved cells were converted to a Tris-based buffer. For samples preserved in SurePath, 1 mL of sample was centrifuged at 4000 RPM for 10 minutes, samples were resuspended in 10 mM Tris, and heated at 120 °C for 20 minutes to reverse formalin-induced crosslinking. For samples preserved in PreservCyt, samples were converted to 10 mM Tris using the Sample Conversion Kit (Qiagen, Germantown, MD) per kit instructions. After conversion, positive (HeLa) and negative (C33A) controls were tested on the HPV DNA paper assay to determine buffer compatibility (Fig. S2†).Evaluation of paper HPV DNA assay with clinical samples in a controlled laboratory environment
Banked samples were collected from a screening study at Basic Health International in El Salvador. Women were eligible to participate if they: 1) were over the age of 30; 2) had a negative pregnancy test; 3) had an intact cervix; 4) had no history of invasive cervical cancer; and 5) were able and willing to provide written informed consent. Participants provided written informed consent, and the protocol was reviewed and approved by the Rice University Institutional Review Board (IRB), the University of Texas MD Anderson Cancer Center IRB, and the Comité Nacional de Ética de El Salvador. A standard-of-care cervicovaginal swab was collected and tested using careHPV to determine the participant's HPV status. A second cervicovagnial swab was collected and placed into PreservCyt buffer for testing with the paper HPV DNA test. For women with colposcopic lesions, a cervical biopsy was also obtained according to standard-of-care clinical protocols and histopathologic diagnosis was performed using standard criteria.Before clinical assessment with the point-of-care HPV DNA paper assay, 4 mL of clinical samples collected into PreservCyt buffer were converted to 150 μL of 10 mM Tris using the Qiagen Sample Conversion Kit. Sample conversion was only necessary because the paper assay was evaluated using banked preserved specimens. Sensitivity, specificity, and accuracy of the paper HPV DNA assay were determined using careHPV results as the reference standard.
Field evaluation of paper HPV DNA assay in Mozambique
2DPNs were prepared as described in Reagent lyophilization with two modifications: streptavidin poly-HRP80 was lyophilized at a concentration of 7.5 μg mL−1, and colorimetric reagents DAB and sodium percarbonate were reconstituted in nuclease-free water with 1% pullulan and 5% trehalose before lyophilization. Assembled 2DPNs with lyophilized reagent pads, nitrocellulose test membranes, and lyophilized sample preparation reagents were stored in foil bags with desiccant and vacuum-sealed before transport at ambient temperature.To assess reagent stability following transport and storage, HPV-positive (HeLa) and HPV-negative (C33A) cellular samples prepared at 1 million cells mL−1 in 10 mM Tris buffer were tested periodically with the procedure described in Point-of-care workflow using lyophilized sample preparation reagents and rehydrated 2DPN with two exceptions: 1) samples were not cooled on ice prior to being added to pad 1, and 2) devices were imaged after two hours.
Banked samples were collected from a cervical screening study conducted by the Mozambique Ministry of Health, Population Services International, MD Anderson and Rice University in Mozambique. Women were eligible to participate if they: 1) were between the ages of 30–49; 2) were not pregnant; 3) had an intact cervix; 4) lived in Maputo or Gaza; 5) were willing and able to provide informed consent. Participants provided written informed consent, and the protocol was reviewed and approved by the Rice University Institutional Review Board (IRB), the University of Texas MD Anderson Cancer Center IRB, the Population Services International IRB, and the Comité Nacional de Bioética Para Saúde de Mozambique. A cervicovaginal swab, either self-collected (95%) or provider-collected (5%), was placed into PreservCyt buffer and tested using Xpert HPV Test HPV to determine the participant's HPV status. The remaining volume of sample was stored at −20 °C for later evaluation with the paper HPV DNA test. Samples were converted from PreservCyt buffer to 10 mM Tris using the Qiagen Sample Conversion Kit prior to being assessed on the paper HPV DNA test using the Point-of-care workflow without ice, unless otherwise noted.
Assessment of clinical sample cellularity
DNA content of clinical samples was quantified with the Qubit™ 1× dsDNA High Sensitivity Assay Kit (Thermo Fisher Q33230) according to manufacturer's instructions. Briefly, 10 μL of Qubit™ standard, 10 μL of HPV-positive or 20 μL of HPV-negative sample was combined with Qubit™ 1× dsDNA HS Working Solution to reach a final volume of 200 μL. Samples were vortexed for 3 seconds, then incubated at room temperature for 2 minutes. dsDNA concentration was measured with the Qubit™ 3 Fluorometer following standards calibration.RNAse treatment of cellular samples
HPV-negative C33A cells were reconstituted in 10 mM Tris buffer at a concentration of 10 million cells mL−1. 25 μL of sample were combined with 1.25 μL of ACP. For RNase-positive samples, 6 μL of RNase A (Thermo Fisher EN0531) was also included. Samples were incubated at room temperature for 5 minutes, then heated at 95 °C for 5 minutes. Samples were immediately added to the sample pad of 2DPNs containing freshly prepared ELISA reagents, and devices were folded to initiate fluid flow. Samples were run in duplicate, and devices were scanned after 1 hour.Usability studies
The usability of the paper HPV DNA assay was assessed via two usability studies: one in El Salvador and one in Mozambique. Institutional Review Board approval was obtained from all participating institutions and informed consent was obtained from all participants. In El Salvador, participants were able to perform the usability study if: 1) they worked as a nurse, physician, research scientist, or laboratory technician associated with Basic Health International; 2) they were over the age of 18; and 3) they were willing and able to provide informed consent. In Mozambique, participants were able to perform the usability study if they were: 1) part of an existing colposcopy and Loop Electrosurgical Excision Procedure (LEEP) course held by The University of Texas MD Anderson Cancer Center; and 2) willing and able to provide informed consent.For each study, participants were provided with a 30 minute training course and demonstration on how to perform the paper HPV DNA test. They were then asked to perform the test independently with mock samples with the assistance of a one-page job aid that illustrated the test workflow (Fig. S3†). Positive mock samples included short synthetic DNA. After running the paper HPV DNA assay, participants filled out a written System Usability Scale (SUS) assessment. In addition, they had the option to provide verbal feedback noting any perceived challenges and suggested improvements.
Results
Paper HPV DNA assay performance with short HPV synthetic DNA targets
The paper HPV DNA assay showed sensitive and specific detection of short synthetic targets of high-risk HPV16 DNA hybridized to complementary short high-risk RNA fragments. Probit analysis showed that the paper-based assay could detect samples containing at least 3.4 × 1011 copies mL−1 of short synthetic high-risk HPV16 DNA (Fig. 3), approximately one order of magnitude higher than the LoD observed using digene HC2. In both assays, no signal was observed for samples containing low-risk, HPV6 DNA or high-risk RNA fragments.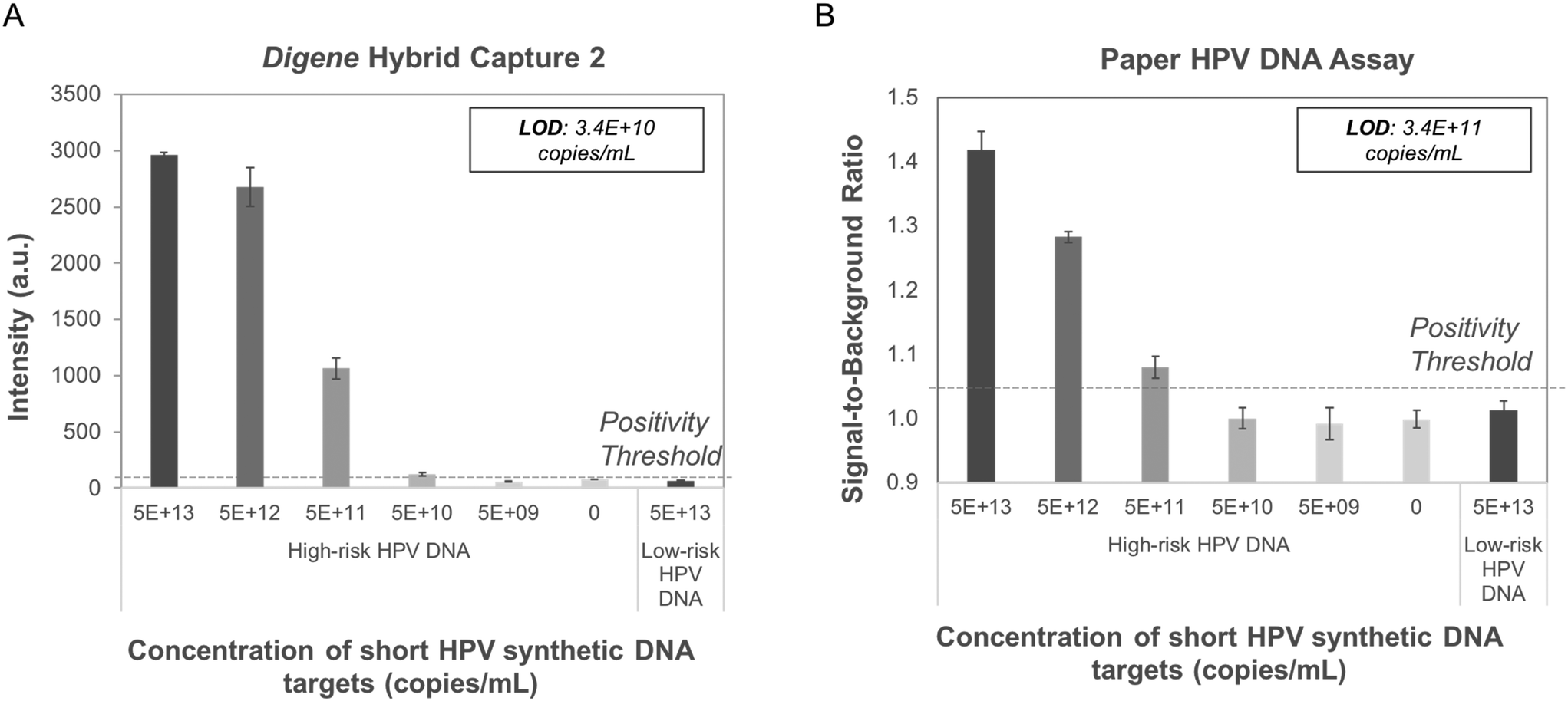 | ||
| Fig. 3 Performance of digene HC2 and paper HPV DNA assay with short HPV synthetic DNA targets (n = 3 for each condition). Signal intensity vs. target concentration for short high- and low-risk HPV synthetic DNA and complementary short high-risk synthetic RNA tested in (A) the digene HC2 assay and (B) the paper HPV DNA assay. Probit analysis of high-risk DNA showed the LoD for digene HC2 is 3.4 × 1010 copies mL−1 while the LoD for the paper HPV DNA assay is 3.4 × 1011 copies mL−1. No positive signal was observed for low-risk HPV DNA (5.0 × 1013 copies mL−1) in either assay. High-risk HPV DNA: HPV16 target; low-risk HPV DNA: HPV6 target; dashed line = positivity threshold determined as average negative signal + three standard deviations. A.u.: Arbitrary units. | ||
Paper HPV DNA assay performance with genomic HPV DNA controls
Performance of the paper HPV DNA assay was evaluated with full genome HPV16 and HPV6 DNA sequences hybridized to full genome high-risk RNA templates and processed with three different sample preparation strategies. High-risk HPV16 DNA samples processed with the digene HC2 sample preparation method produced positive signal at low DNA concentrations; however, false-positive results were observed when HPV6 DNA was processed and tested with the same method (Fig. 4A). A second sample processing strategy that included achromopeptidase (ACP) treatment of hybrids for 5 minutes at room temperature followed by heating for 30 seconds at 95 °C and subsequent addition of EDTA also yielded false-positive results for low-risk HPV DNA (Fig. 4B).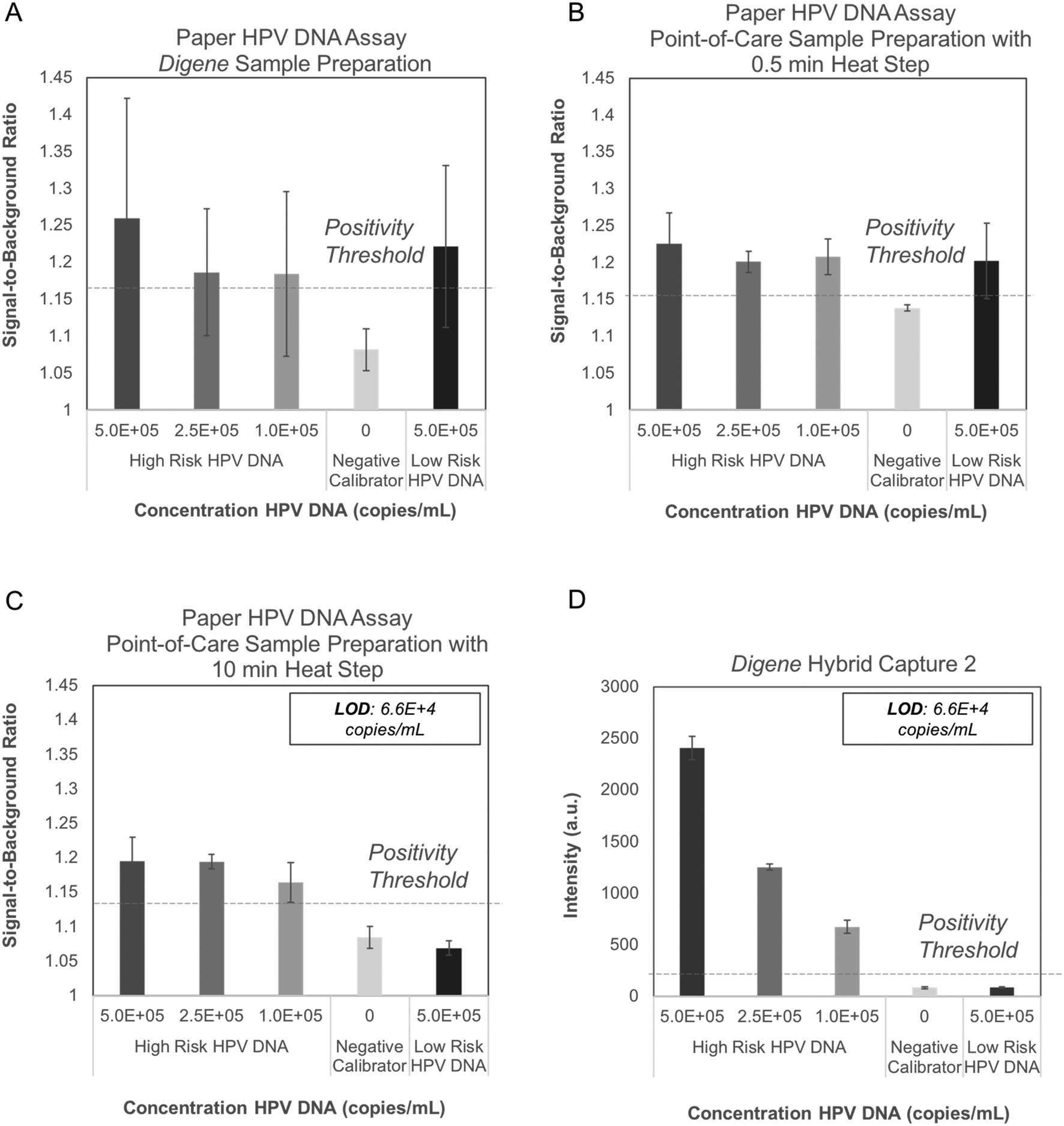 | ||
| Fig. 4 Performance of digene HC2 and paper HPV DNA assay with genomic HPV DNA standards. The paper HPV DNA assay was tested with three sample preparation methods (n = 3 for each condition): (A) digene HC2 sample preparation instructions, and (B) a sample preparation method including treatment with ACP for 5 min at room temperature and heating at 95 °C for 30 seconds. Both sample preparation methods yielded false positive results when the low-risk HPV6 DNA control was evaluated with the paper device. (C) Pre-treatment of RNA probes with ACP and heat followed by a five-minute point-of-care sample preparation method yielded minimal signal for the negative calibrator and low-risk HPV6 DNA controls. The limit of detection for high-risk HPV16 DNA using the paper HPV DNA assay is 6.6 × 104 copies mL−1, equivalent to that of the digene HC2 assay shown in (D). High-risk HPV DNA: HPV16 target (digene high-risk HPV quality control solution); low-risk HPV DNA: HPV6 target (digene low-risk HPV quality control solution); dashed line: positivity threshold determined as average signal of the negative calibrator + three standard deviations. | ||
We theorized that the false-positive signal was due to DNA and RNA forming secondary structures and sterically binding to the anti-DNA–RNA hybrid capture antibody immobilized in the paper assay.26 An experiment to evaluate assay performance following various DNA and RNA fragmentation strategies to reduce secondary structure supported this theory (Fig. S1†).
To avoid false positives due to RNA secondary structure, we pre-treated RNA probes by mixing with ACP, incubating for 5 minutes at room temperature, heating for 10 minutes at 95 °C, and adding EDTA. To avoid false-positive signals due to DNA secondary structure, we added sample DNA to pre-treated RNA probes, incubated for 5 minutes at room temperature, and then heated for 5 minutes at 95 °C. To improve ease-of-use, the pre-treated RNA and ACP were lyophilized after EDTA addition, requiring just one user step of adding sample DNA to lyophilized, pre-treated RNA and heating for 5 minutes at 95 °C. With this point-of-care sample preparation protocol, no false-positive signal was observed with low-risk HPV DNA using the paper HPV DNA assay (Fig. 4C).
The LoD for the paper HPV DNA assay using the developed point-of-care sample preparation method was found to be 6.6 × 104 copies mL−1 (Fig. 4C), equivalent to that found for the digene HC2 assay with the same input samples (Fig. 4D). In all cases, the positivity threshold was defined as the average signal of the negative calibrator supplied with the digene HC2 kit plus three standard deviations (n = 3).
Paper HPV DNA assay performance with lyophilized reagents and cellular samples
To evaluate performance of the paper assay using cellular samples, increasing amounts of high-risk HPV-positive cells, including SiHa (HPV16-positive) and HeLa (HPV18-positive) cells, were spiked into HPV-negative cells (C33A), maintaining the same total cell count in each sample. Linear ranges of high-risk HPV-positive cellular samples were run in triplicate on the paper HPV DNA assay with all assay reagents lyophilized. Cellular samples were added to lyophilized pre-treated RNA and ACP, and subsequently treated using the point-of-care sample preparation protocol to lyse cells and create DNA–RNA hybrids. Samples were run on the paper HPV DNA assay assembled with fully lyophilized reagent pads. The LoD for SiHa cells was 1.43 × 105 cells mL−1 (Fig. 5A) and for HeLa cells was 6.87 × 104 cells mL−1 (Fig. 5B). A lower LoD is expected for HeLa cells since HeLa cells have 10–50 copies of HPV18 per cell, whereas SiHa cells have approximately 1–2 copies of HPV16 per cell.27 No false-positive signal was observed for controls containing only C33A HPV-negative cells.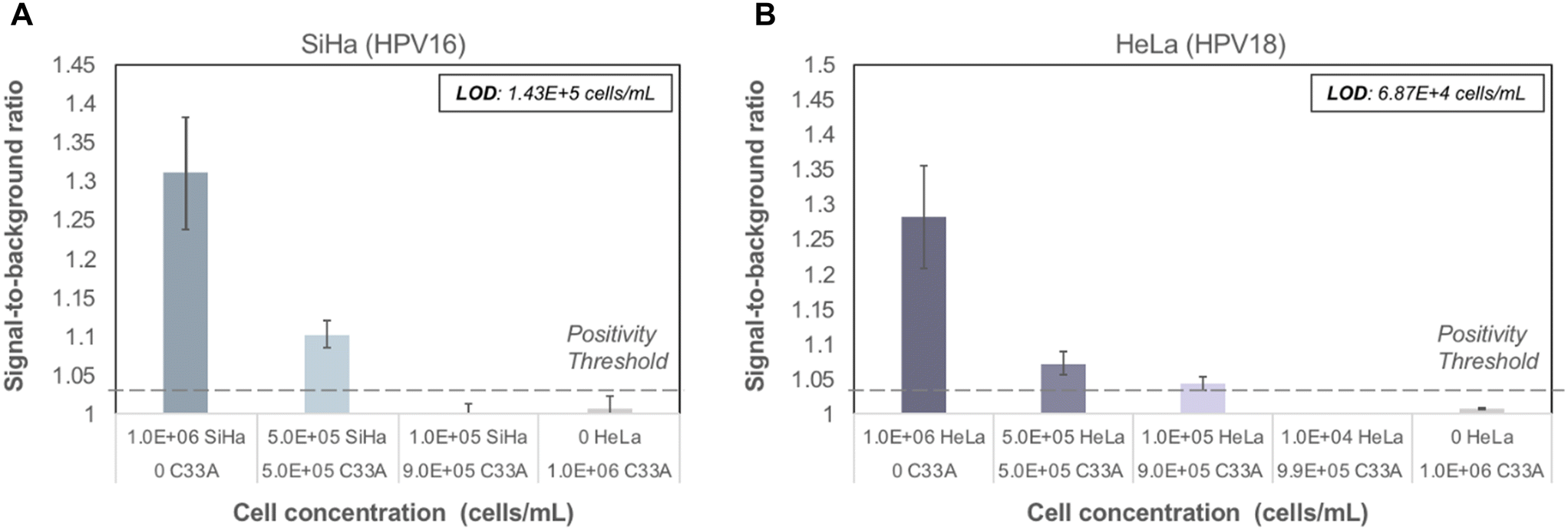 | ||
| Fig. 5 Performance of paper HPV DNA assay with lyophilized reagents for cellular samples containing a range of HPV-positive cells. Contrived cellular samples were prepared containing a decreasing number of HPV-positive cells (SiHa, HPV16; HeLa, HPV18) combined with HPV-negative cells (C33A) to maintain a constant number of cells. Cellular samples were tested using the seven-step assay workflow described in Fig. 2. (A) The limit of detection for SiHa cells was 1.43 × 105 cells mL−1; (B) the limit of detection for HeLa cells was 6.87 × 104 cells mL−1. Dashed line: positivity threshold determined as average signal of C33A cells + three standard deviations. | ||
Cervical cytology samples are typically collected into commercial sample collection buffers. To assess whether the paper HPV DNA assay was compatible with collection buffers used to preserve cervical cytology samples, we assessed assay performance for HPV-positive and -negative cells stored in PreservCyt and SurePath, two commonly used collection buffers. Preserved cellular samples were converted to a Tris-based solution and then prepared using the point-of-care sample processing method before testing on the paper HPV DNA assay (Fig. S2†). Following buffer conversion, results obtained with the paper HPV DNA assay were comparable to those obtained for unpreserved cellular samples suspended in Tris buffer.
Paper HPV DNA assay performance with clinical samples
Sixteen biobanked cervical cytology specimens collected into PreservCyt buffer were obtained from women participating in a cervical cancer screening study in El Salvador and evaluated in a high-resourced laboratory in Houston, Texas, USA. Samples included eight that tested positive and eight that tested negative for high-risk HPV by careHPV, the clinical reference standard used in the original study.28 Following buffer conversion, samples were tested using the paper HPV DNA test and results were compared to the reference standard. The threshold for test positivity was set as the average signal measured for HPV-negative cells stored in PreservCyt buffer plus three standard deviations (n = 3, Fig. S2;† positivity threshold = 1.14). The results of clinical testing are shown in Fig. 6. The mean signal-to-background ratio for high-risk HPV-positive clinical samples was significantly greater than the mean signal-to-background ratio of high-risk HPV-negative clinical samples (p = 0.001). When compared to the careHPV reference standard, the paper HPV DNA test correctly identified eight of eight HPV-negative clinical samples and seven of eight HPV-positive clinical samples (100% specificity, 87.5% sensitivity, and 93.8% accuracy). Results were the same using positivity thresholds set based on HPV-negative cells suspended in Tris buffer (Fig. S2;† positivity threshold = 1.14) or the average negative calibrator signal plus three standard deviations (Fig. 4C; positivity threshold = 1.13).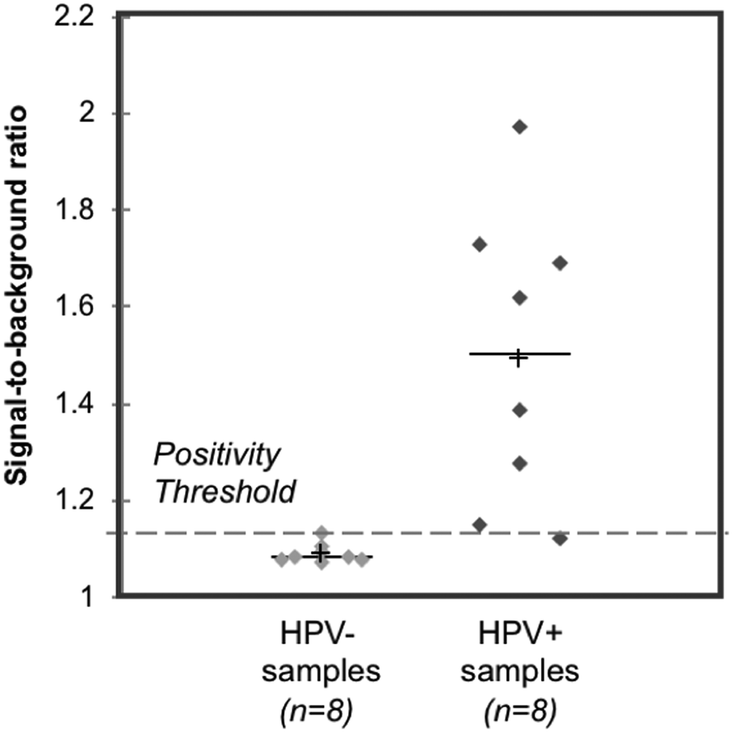 | ||
| Fig. 6 Clinical assessment of paper HPV DNA assay. Signal to background ratios of the paper HPV DNA assay when performed with clinical samples collected into PreservCyt buffer and stratified by results of the reference standard, careHPV. The positivity threshold was determined using the negative C33A signal plus three standard deviations from Fig. S2.† There was a statistically significant difference in the mean signal-to-background ratio of HPV-negative (HPV−) and positive (HPV+) clinical samples (p = 0.001, significance determined using a one-tailed unpaired t-test). Dashed line: positivity threshold determined as average negative signal + three standard deviations. +: Mean; line: median. | ||
Field evaluation of paper HPV DNA assay in Mozambique
Following promising results with clinical samples using the paper HPV DNA assay in a controlled laboratory environment, a field evaluation of the test was conducted over the course of one week in a low-resource laboratory in Mozambique. Fully lyophilized sample preparation reagents and 2DPNs were transported at ambient temperature for >50 hours in desiccated, vacuum-sealed bags, and were stored at −20 °C upon arrival.C33A (HPV-negative) and HeLa (HPV-positive) cellular controls were assessed using lyophilized sample preparation reagents and lyophilized 2DPNs periodically to determine the stability of reagents following transport and storage. Throughout the week of field testing, cellular controls yielded expected results (Fig. 7), with a statistically significant difference between the mean signal-to-background ratios of C33A and HeLa cells (p = 0.009). However, large variability was observed in the signal-to-background ratios of cellular controls, precluding the calculation of a meaningful positivity threshold.
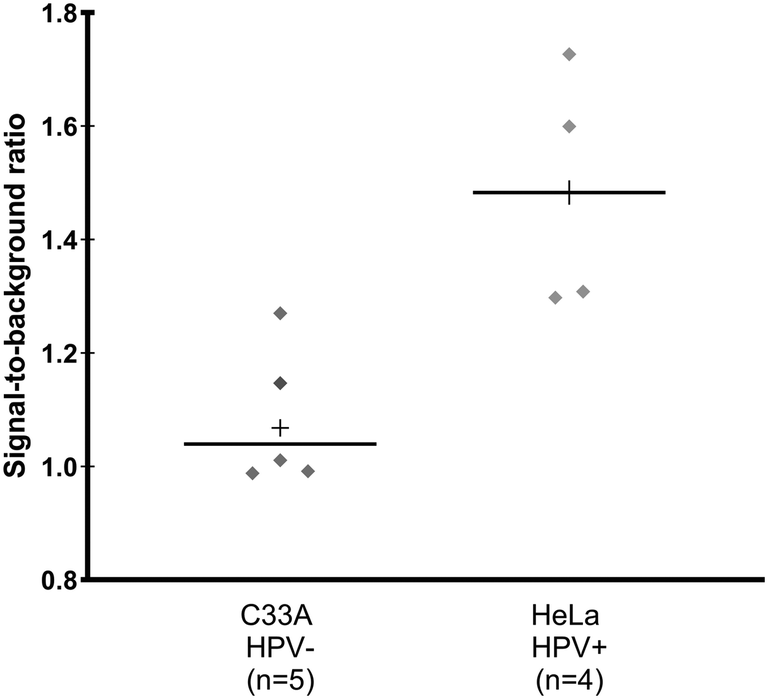 | ||
| Fig. 7 Field evaluation of fully lyophilized paper HPV DNA test in Mozambique with cellular controls. Signal-to-background ratios of the paper HPV DNA assay with lyophilized sample preparation and lyophilized 2DPN reagents when performed with cellular controls prepared at 1 million cells mL−1 in 10 mM Tris buffer throughout a week of field testing. There was a statistically significant difference in mean signal-to-background ratio of HPV-negative C33A cells and HPV18-positive HeLa cells (p = 0.005 by a one-tailed unpaired t-test). +: Mean; solid line: median. | ||
Sixteen biobanked self-collected cervicovaginal specimens collected into PreservCyt buffer were obtained from women participating in an ongoing cervical cancer screening study in Mozambique. Samples included eight that tested positive and eight that tested negative for high-risk HPV by the GeneXpert HPV Test (“Xpert”), the clinical reference standard used in the original study. Following buffer conversion, samples were tested using the paper HPV DNA test and results were compared to the reference standard. Using the positivity threshold from Fig. S2,† positive results were obtained for all eight HPV-positive samples as well as seven of eight HPV-negative samples. We hypothesized that false positive results occurred due to residual secondary structure in the cellular RNA and DNA in the clinical samples, increasing nonspecific binding to the capture antibody. Due to the finite number of lyophilized 2DPNs that were transported and the limited amount of laboratory resources, we were not able to fully test this hypothesis. However, we adapted the sample preparation protocol to try to further reduce secondary structure and improve specificity of the paper HPV DNA assay (Fig. S1†). We excluded EDTA from the 95 °C sample preparation heat step so that both cellular DNA and RNA would be fragmented, extended heating by 2 minutes, and incorporated a 10 minute cooling step in a refrigerator. Using this modified sample preparation protocol and lyophilized 2DPNs, the mean signal-to-background ratio of high-risk HPV-positive clinical samples was significantly greater than the mean signal-to-background ratio of HPV-negative clinical samples (p = 0.037) (Fig. 8A). To confirm that modifications to the sample preparation protocol were responsible for improved performance, we compared signal-to-background ratios obtained with lyophilized sample preparation reagents to that obtained with the modified sample preparation protocol (Fig. 8B); a larger decrease in signal-to-background ratio was observed for HPV-negative samples (57%) than for HPV-positive samples (34%).
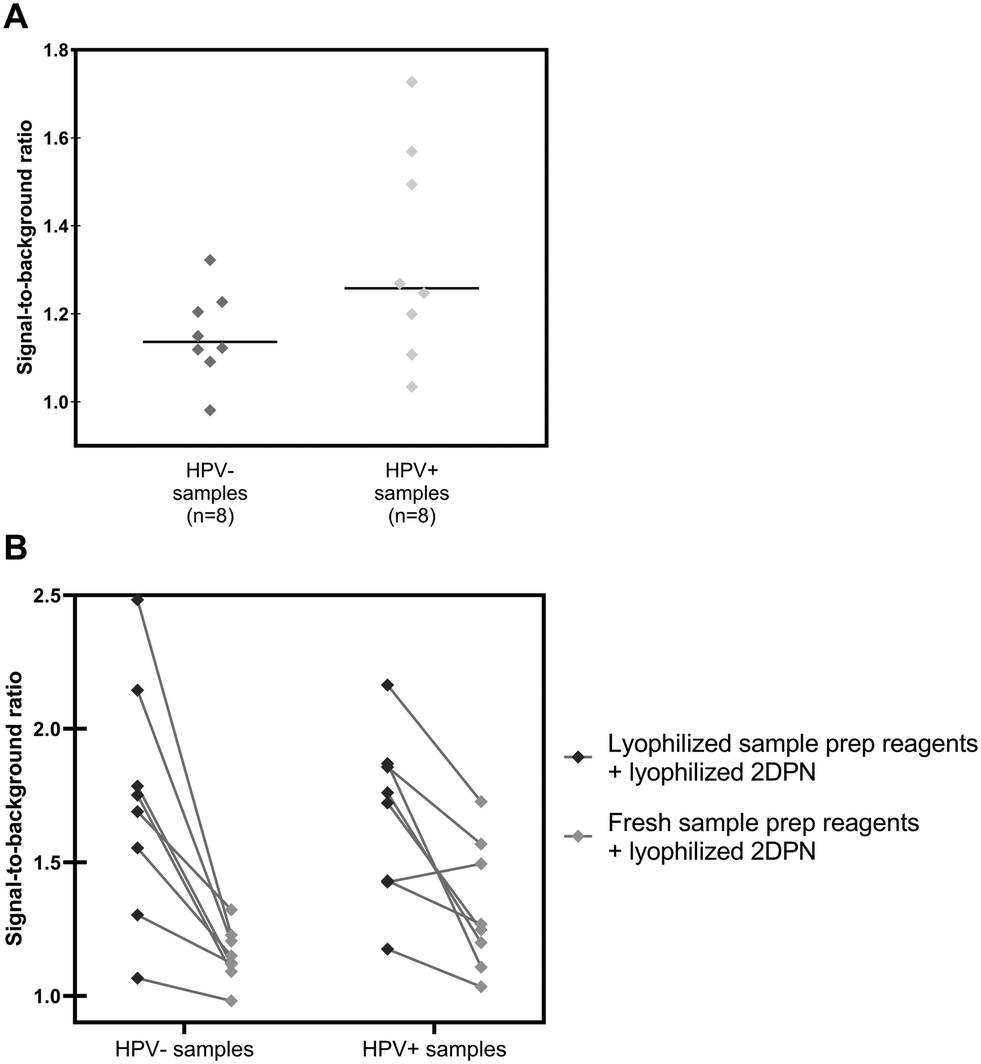 | ||
| Fig. 8 Field evaluation of fully lyophilized paper HPV DNA test in Mozambique with clinical samples. Signal to background ratios of the paper HPV DNA assay when performed with clinical samples collected into PreservCyt buffer and stratified by results of the reference standard, Xpert HPV. (A) Signal-to-background ratios of clinical samples assessed on the paper HPV DNA assay with a re-optimized sample preparation protocol and lyophilized 2DPN reagents; the mean signal-to-background ratio of HPV-positive clinical samples is significantly greater (p = 0.037) than the mean signal-to-background ratio of HPV-negative clinical samples, as determined by a one-tailed unpaired t-test. +: Mean; solid line: median. (B) Comparison of signal-to-background ratios obtained on the paper HPV DNA test using lyophilized sample preparation reagents (left) versus a field-optimized sample preparation method (right). With the field-optimized sample preparation, the signal-to-background ratio for HPV-negative samples decreased by 57%, whereas the ratio decreased by only 34% for HPV-positive samples. | ||
To confirm that cellular RNA was the source of false positives, a high concentration of HPV-negative C33A cells (10 million mL−1) was tested with the paper HPV DNA assay, with and without the addition of RNAse A; the high-risk RNA probe was excluded from the sample preparation protocol. We found that in the absence of RNase A and the high-risk RNA probe, a strong false positive signal was observed for C33A cells; in contrast, the addition of RNAse A completely eliminated this false positive signal at the test line (Fig. S6†).
The field evaluation uncovered a problem with specificity of the paper HPV DNA test that had not arisen when conducting clinical testing in a controlled laboratory environment. Samples tested in a controlled laboratory environment had a reference standard of careHPV, a hybridization-based test, whereas the reference standard for samples tested in the field was Xpert, a nucleic acid amplification test. In addition to being more analytically sensitive, Xpert also measures a cellular control to ensure that the sample contains a sufficient number of cervical cells. The careHPV test does not have an equivalent control to confirm that samples are of sufficient cellular quantity. We measured DNA content in the remaining clinical samples originally tested with the paper HPV DNA assay using a careHPV reference standard in the controlled lab environment. Samples with sufficient remaining volume were analyzed using the Qubit high-sensitivity double-stranded DNA assay to measure DNA content, and we found that samples that were HPV negative by careHPV had a lower DNA content (range: 22.9–53.5 ng mL−1, mean: 41.5 ng mL−1, n = 8) than samples that were HPV positive by careHPV (range: 51.2–908 ng mL−1, mean: 338.8 ng mL−1, n = 7; Fig. S5†); this difference in means was statistically significant (p = 0.031). Resource limitations did not allow us to test the DNA content of clinical samples evaluated in the field evaluation; however, sample turbidity for all samples evaluated during field testing was higher on visual inspection (data not shown), suggesting higher cell concentration.
Usability of paper HPV DNA assay
Forty-four participants in El Salvador and Mozambique (Table S2†) performed two tests with the paper HPV DNA assay with the assistance of a job-aid (Fig. S3†) and then completed a standard system usability scale (SUS) assessment. Results are shown in Fig. 9. In El Salvador, the average SUS score was 82.1 ± 13.3, while in Mozambique the average SUS score was 76.3 ± 14.1. SUS scores greater than 70 indicate acceptable usability,29 and three-fourths of participants scored the paper HPV DNA test 70 or higher. Participants reported the precise timing of the heating step and the use of exact volume disposable pipettes as the most difficult aspects of running the assay.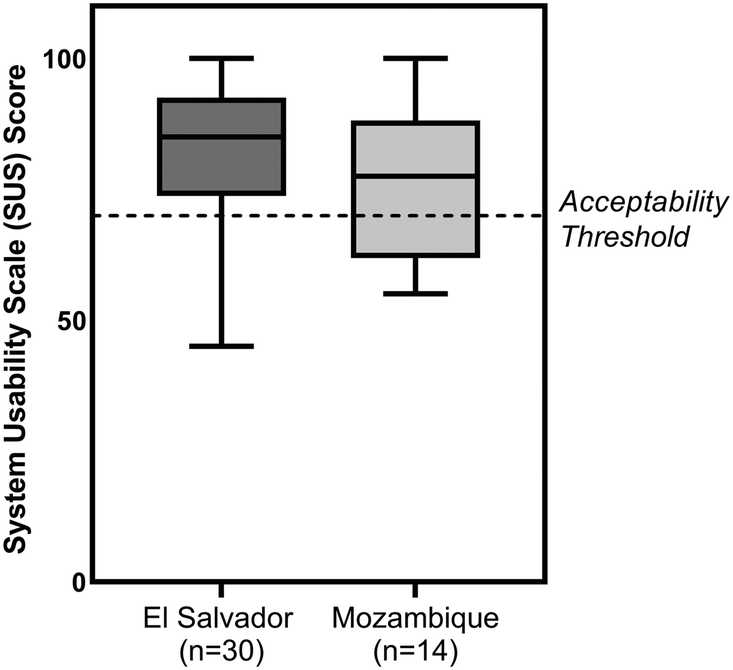 | ||
| Fig. 9 Usability testing. System usability scale (SUS) scores for the paper HPV DNA assay for users in two locations. Usability of the HPV DNA assay was assessed in El Salvador (n = 30) with physicians practicing in rural (n = 20) and urban locations (n = 8), a nurse (n = 1), and a lab technician (n = 1); and in Mozambique (n = 14) with physicians and nurses (n = 13) and a lab technician (n = 1). Participants performed two mock paper HPV DNA assays with the assistance of a job aid and subsequently filled out a usability survey. All groups rated the HPV DNA assay as acceptable to use (SUS score ≥70, indicated with dashed line). | ||
Discussion
We successfully developed a low-cost, rapid, user-friendly paper HPV DNA test for sample-to-answer testing of cervicovaginal swabs, and evaluated the assay in two studies: a small pilot study with provider-collected banked samples from El Salvador tested in a high-resourced, controlled laboratory environment in Houston, Texas, USA, and a field evaluation with self-collected banked samples tested in a lower-resourced laboratory in Mozambique. The paper HPV DNA assay builds on recent advances in 2DPNs to implement a hybrid capture reaction on paper,30–36 adds to the literature incorporating ACP into point-of-care sample preparation methods,24,37 and demonstrates the utility of fully lyophilized assay reagents following ambient temperature transport to a field setting in Mozambique. Our findings are important in furthering the development of a point-of-care HPV test with acceptable cost and usability in low- and middle-income countries, where the burden of cervical cancer is highest.In our study, the paper HPV DNA assay performed well for short, perfectly complementary sequences of DNA and RNA, however further optimization was needed to reduce background signal for longer nucleic acid sequences. Without fragmentation of sample DNA and capture RNA hybrids, steric binding of full-length DNA and RNA to the capture antibody on a paper membrane consistently led to false-positive results. Appropriately pre-fragmenting RNA sequences and fragmenting sample DNA using a combination of ACP and heat reduced nonspecific binding to the capture antibody; however, cellular RNA still posed a challenge for the assay in the field evaluation in Mozambique.
The demonstrated LoD of the developed sample-to-answer paper HPV DNA test is comparable to the gold standard digene HC2 assay for full genome high-risk HPV DNA sequences and for HPV16- and HPV18-positive cell lines. Both the paper HPV DNA assay and the digene HC2 had LoDs of 6.6 × 104 copies mL−1 when tested with full genome HPV DNA standards; this is consistent with the reported limit of detection for the digene HC2 assay but higher than the limit of detection reported for GeneXpert (2903–50493 copies mL−1).38 The paper HPV assay could detect 1.4 × 105 SiHa cells mL−1 and 6.8 × 104 HeLa cells mL−1; these limits are consistent with reports showing SiHa cells contain 1–2 copies of HPV16 DNA per cell and HeLa cells contain 10–50 copies of HPV18 per cell.27 Several studies have shown that high HPV viral load is linked to increased risk of cervical precancer and cancer, but the link between viral load and disease progression can vary by high-risk HPV genotype.39 Finally, the paper HPV DNA assay reliably produced negative results when tested with high levels of full genome low-risk HPV6 DNA sequences and high-risk HPV-negative cell lines below 1 million cells mL−1.
When evaluated with banked, provider-collected clinical samples in PreservCyt buffer, the paper HPV DNA assay agreed with the careHPV reference standard for 94% of samples tested in a high-resourced laboratory setting (Fig. 6). However, careHPV is an imperfect reference standard with no internal cellular control to ensure sample adequacy. In fact, when DNA content on this set of clinical samples was assessed, the measured DNA content of HPV-negative samples was significantly lower than the DNA content of HPV-positive samples, highlighting the need for a cellular control to ensure that samples with negative results contain adequate numbers of cells for evaluation.
When evaluated with banked self-collected clinical samples in a low-resource setting using a reference test of Xpert, a nucleic acid amplification test that contains a cellular control, the accuracy of the paper HPV DNA assay was reduced (Fig. 8), underlining the challenge of translating laboratory-developed tests to point-of-care settings. The reduction in test specificity is likely due to a higher concentration of cells, as observations in the field noted higher sample turbidity; this is consistent with other studies that found the median cellular concentration of self-collected samples to be more than five-fold higher than in provider-collected samples from the same patients (p < 0.001).40 Moreover, follow-up experiments revealed that cellular RNA from highly concentrated HPV-negative cellular samples caused false positive signal (Fig. S6†).
These results show that two improvements are needed before the developed test can be clinically useful. First, a cellular control must be incorporated to prevent false negative results that may arise simply because samples lack sufficient numbers of cells. Second, additional optimization of the sample preparation method is needed to ensure appropriate fragmentation of both cellular DNA and RNA to avoid false positive results in self-collected samples. While the addition of RNAse A successfully eliminated false positives due to cellular RNA (Fig. S6†), the addition of RNAse A would require a heat deactivation step before hybridization of the cellular sample to the high-risk RNA probe cocktail. Further work is needed to optimize a user-friendly workflow for a sample preparation strategy incorporating RNAse A.
Additionally, the results highlight limitations of assessing the performance of a new test in low-resource settings where access to gold standard testing may be limited. Here, we compared performance of the paper HPV DNA test to the clinical reference standard used in the target setting. For the first clinical evaluation, results of the paper HPV DNA assay were compared to the clinical reference standard of careHPV. Both approaches are based on hybrid capture, and although careHPV has limitations, comparative studies have shown that careHPV and digene HC2 have very good agreement for detection of high-risk HPV.41 In the second clinical evaluation, results of the paper HPV DNA assay were compared to the clinical reference standard of Xpert HPV, a PCR-based approach that has a higher sensitivity than hybrid capture-based approaches.17 Unfortunately, clinical protocol limitations did not allow sample transport outside Mozambique for additional testing, and it was not possible to access digene HC2 locally.
All of the clinical samples assessed in this work were collected into PreservCyt solution, a methanol-based preservative buffer used for the collection, transport, and storage of cervical samples, necessitating centrifugation and sample buffer conversion. With collection directly into Tris buffer, no conversion step is necessary; centrifugation and sample conversion are only necessary for processing preserved cells. Testing samples collected into Tris is necessary to identify any additional areas of test refinement that might be needed with non-preserved samples.
Users in El Salvador and Mozambique with no previous training in test operation were able to accurately perform the paper HPV DNA assay workflow and rated the test as acceptable to use. Participants reported the timing of the DNA heating step and use of exact volume disposable pipettes as the most difficult aspects of running the assay. A self-timed heater could remove the need for precise timing of sample preparation (5 minutes at 95 °C).
A comparison of the paper HPV DNA test to other commercially available HPV DNA screening tests is shown in Table 1. The digene HC2 test has a high per-test cost, requires expensive infrastructure to read the assay, and takes over four hours to produce a result.18,19 careHPV is less expensive, but requires batching in groups of 90 samples at a time to achieve a low cost, and uses expensive readout equipment. As a result of batching, the test results are not available immediately at the point-of-care and women must return for additional visits to receive HPV results, diagnostic work-up, and treatment if needed and are therefore often lost to follow-up.20 The paper HPV DNA assay eliminates the need for batching and produces results within one hour, which is appropriate for screening in a “Screen & Treat” approach.20 Additionally, the paper HPV DNA assay costs less than $3 per test (Table S4†) and requires only a heater.
| Digene hybrid capture 2 (HC2) | careHPV | GeneXpert | Paper-based HPV DNA test | |
|---|---|---|---|---|
| Commercially available? | Yes | Yes | Yes | No |
| Batching required? | No | Yes | No | No |
| Limit of detection (per literature) | 100000 copies mL−1 |
100000 copies mL−1 |
2903 to 50493 copies mL−1 |
N/A |
| Limit of detection (as evaluated in this work) | 66000 copies mL−1 |
66000 copies mL−1 |
||
| Time to result | 4.5+ hours | 3+ hours | 1 hour | 1 hour |
| Cold storage requirements | Refrigerator | Refrigerator | None | None |
| Level of lab expertise required | High | Medium | Low | Low |
To address the need for improved cervical cancer screening in low-resource settings, a number of point-of-care strategies are in development.17 Several investigators have shown promising results using strategies based on nucleic acid amplification.42,43 Rodriguez et al. demonstrated a fully integrated paperfluidic device to extract, amplify, and detect HPV16 DNA from cervical specimens using isothermal amplification with loop-mediated isothermal amplification; the limit of detection of the integrated device was 10000 copies of DNA in a 100 μL sample (100000 copies mL−1).42 Results with 10 clinical samples evaluated in a high-resource lab correctly detected all five HPV 16 positive samples but two of five HPV 16 negative samples tested falsely positive. Chen et al. demonstrated a PCR-based approach that combines a one-step lysis protocol, simultaneous amplification of HPV 16 and HPV 18 with a portable PCR thermal controller, followed by lateral flow detection.43 The limit of detection was 700 copies of HPV 16 or HPV 18 in 1 μL of sample (700000 copies mL−1). Twenty clinical samples were tested in a high-resource setting and results agreed with standard PCR. These studies based on amplification of high-risk HPV DNA showed a similar limit of detection as hybrid capture-based strategies.
The advantages of approaches based on hybrid capture include the ease of simultaneous detection of multiple high-risk genotypes and the potential to easily include detection of a cellular control to ensure that an adequate sample was collected. Unlike strategies based on nucleic acid amplification, hybrid capture-based approaches do not generate large amounts of target DNA that can lead to environmental contamination and subsequent false positive test results. However, it is known that the high-risk HPV RNA probe cocktail in the digene HC2 kit cross-reacts with some untargeted, non-carcinogenic HPV types.44,45 In this small pilot study, we were not able to assess the impact of cross-reactivity; however, in a study of 954 clinical samples, HC2 cross-reactivity resulted in minor changes in screening performance, increasing sensitivity from 84.3% to 87.9% and decreasing specificity from 89.6% to 88.1%.44 In a study of 3179 women, 7.8% of all HC2 positive results were due to cross-reactivity with untargeted, noncarcinogenic HPV genotypes.45
In conclusion, we developed a sample-to-answer screening test for high-risk HPV DNA that is sensitive, low-cost, and simple to use. The assay was equivalent in sensitivity to commercially available hybrid-capture HPV DNA tests when performed in a controlled laboratory environment, with a 94% accuracy compared to reported careHPV results in a pilot study. Furthermore, the test produces results within one hour, does not require batching, and involves only seven user steps to perform. The only instrumentation required to run the test is a low-cost, benchtop heater, reducing the level of infrastructure necessary to run the assay relative to existing tests. Together, these characteristics could prove useful for a “Screen & Treat” setting in low-resource areas with the highest burden of cervical cancer. Once the test is further optimized for low-resource settings and evaluated in a larger clinical study, the paper HPV DNA assay could serve as a rapid, point-of-care test to improve access to cervical cancer screening for women in low-resource areas.
Author contributions
CAS, MMC, KAK, PEC, KMS, and RRK conceived of the project and its scope. CAS designed and performed experiments and analyzed data leading to the initial development and optimization of the assay. MMC led the field evaluation, and with ENN, designed and performed experiments and analyzed data. CAS and KAK developed the sample preparation protocol. CAS and MMC prepared the manuscript, with input and supervision from PEC, KMS, and RRK and editing from KAK and ENN. Additional editing was provided by MPS, JLC, and JM. CAS, KAK, SGP performed the usability field testing. SGP and LL coordinated the clinical sample collection in El Salvador with supervision from MM, and all three authors edited the manuscript. CM, CL, MPS, JLC, EB and KMS coordinated the clinical sample collection in Mozambique. JM and MS provided clinical expertise. Funding for this research was acquired by KMS and RRK.Conflicts of interest
Dr. Castle has received HPV tests and assays for research at a reduced or no cost from Roche, Cepheid, Becton Dickinson, Arbor Vita Corporation, and Atila Biosystems. The other authors have no conflicts of interest to declare.Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA186132 and the National Academy of Sciences, United States Agency for International Development under the Partnerships for Enhanced Engagement in Research, Cooperative Agreement AID-OAA-A-11-00012. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, USAID, or the United States Government. The authors would like to thank all the physicians and medical personnel at Mavalane General Hospital for their invaluable help to complete the studies in this publication. Additionally, thank you to the physicians and lab technicians from the Instituto del Cáncer de El Salvador for their work to make our clinical and usability studies possible. Special thanks to Dr. Joao Salomao from Mozambique, Dr. Cristina Mendes de Oliveira and Viviane Andrade from Barretos Cancer Hospital, and Melissa Lopez Varon, Cindy Melendez, Jessica Gallegos, and Ana Lopez from The University of Texas MD Anderson Cancer Center for their assistance in data collection, coordinating studies, and ensuring each study was completed. Thank you to Dereq Ogoe for assistance assembling and testing devices. Thanks to Imran Vorha for photography of images shown in Fig. 2, and thanks to Deborah Marquez-Do and Luis Olivares for lending the Qubit™ 3.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- National Institute of Health, Cervical Cancer Fact Sheet, 2010 Search PubMed.
- WHO, Global strategy to accelerate the elimination of cervical cancer as a public health problem, World Health Organization, 2020 Search PubMed.
- G. K. Singh, R. E. Azuine and M. Siahpush, Int. j. MCH AIDS, 2012, 1, 17–30 Search PubMed.
- L. Bruni, M. Diaz, L. Barrionuevo-Rosas, R. Herrero, F. Bray, F. X. Bosch, S. de Sanjosé and X. Castellsagué, Lancet Glob. Health, 2016, 4, e453–e463 CrossRef PubMed.
- J. M. Agosti and S. J. Goldie, N. Engl. J. Med., 2007, 356, 1908–1910 CrossRef PubMed.
- J. C. Gage and P. E. Castle, J. Natl. Cancer Inst., 2010, 102, 1524–1527 CrossRef.
- M. Schiffman, N. Wentzensen, S. Wacholder, W. Kinney, J. C. Gage and P. E. Castle, J. Natl. Cancer Inst., 2011, 103, 368–383 CrossRef.
- J. C. Gage, M. Schiffman, H. A. Katki, P. E. Castle, B. Fetterman, N. Wentzensen, N. E. Poitras, T. Lorey, L. C. Cheung and W. K. Kinney, J. Natl. Cancer Inst., 2014, 106, dju153 CrossRef PubMed.
- P. E. Castle, W. K. Kinney, X. Xue, L. C. Cheung, J. C. Gage, F.-H. Zhao, B. Fetterman, N. E. Poitras, T. S. Lorey, N. Wentzensen, H. A. Katki and M. Schiffman, Ann. Intern. Med., 2018, 168, 20 CrossRef.
- G. Ronco, J. Dillner, K. M. Elfström, S. Tunesi, P. J. F. Snijders, M. Arbyn, H. Kitchener, N. Segnan, C. Gilham, P. Giorgi-Rossi, J. Berkhof, J. Peto, C. J. L. M. Meijer, J. Cuzick, M. Zappa, F. Carozzi, M. Confortini, P. D. Palma, M. Zorzi, A. Del Mistro, A. Gillio-Tos, C. Naldoni, D. Rijkaart, F. Van Kemenade, N. Bulkmans, D. Heideman, R. Rozendaal, G. Kenter, M. Almonte, C. Roberts, M. Desai, A. Sargent, W. Ryd and P. Naucler, Lancet, 2014, 383, 524–532 CrossRef PubMed.
- R. Sankaranarayanan, B. M. Nene, S. S. Shastri, K. Jayant, R. Muwonge, A. M. Budukh, S. Hingmire, S. G. Malvi, R. Thorat, A. Kothari, R. Chinoy, R. Kelkar, S. Kane, S. Desai, V. R. Keskar, R. Rajeshwarkar, N. Panse and K. A. Dinshaw, N. Engl. J. Med., 2009, 360, 1385–1394 CrossRef PubMed.
- S. Arrossi, L. Thouyaret, R. Herrero, A. Campanera, A. Magdaleno, M. Cuberli, P. Barletta, R. Laudi and L. Orellana, Lancet Glob. Health, 2015, 3, e85–e94 CrossRef PubMed.
- M. Cremer, M. Maza, K. Alfaro, M. M. Velado, J. Felix, P. E. Castle, J. Kim and J. C. Gage, J. Low. Genit. Tract Dis., 2017, 21, 26–32 CrossRef PubMed.
- M. Arbyn, S. B. Smith, S. Temin, F. Sultana, P. Castle, F. Sultana and P. Castle, BMJ, 2018, 363, k4823 CrossRef PubMed.
- J. Cuzick, L. Cadman, D. Mesher, J. Austin, L. Ashdown-Barr, L. Ho, G. Terry, S. Liddle, C. Wright, D. Lyons and A. Szarewski, Br. J. Cancer, 2013, 108, 908–913 CrossRef.
- K. A. Kundrod, C. A. Smith, B. Hunt, R. A. Schwarz, K. Schmeler and R. Richards-Kortum, Expert Rev. Mol. Diagn., 2019, 1–19 Search PubMed.
- H. Ying, F. Jing, Z. Fanghui, Q. Youlin and H. Yali, Sci. Rep., 2015, 4, 4704 CrossRef.
- S. Shah, S. Senapati, F. Klacsmann, D. Miller, J. Johnson, H.-C. Chang and M. Stack, Cancers, 2016, 8, 85 CrossRef PubMed.
- N. G. Campos, V. Tsu, J. Jeronimo, M. Mvundura and J. J. Kim, BMC Cancer, 2017, 17, 791 CrossRef.
- E. L. Vodicka, J. B. Babigumira, M. R. Mann, R. J. Kosgei, F. Lee, N. R. Mugo, T. C. Okech, S. R. Sakr, L. P. Garrison and M. H. Chung, Int. J. Gynecol. Obstet., 2017, 136, 220–228 CrossRef.
- B. D. Grant, C. A. Smith, K. A. Karvonen and R. Richards-Kortum, Anal. Chem., 2016, 88, 2553–2557 CrossRef CAS PubMed.
- J. R. Buser, X. Zhang, S. A. Byrnes, P. D. Ladd, E. K. Heiniger, M. D. Wheeler, J. D. Bishop, J. A. Englund, B. Lutz, B. H. Weigl and P. Yager, Anal. Methods, 2016, 8, 2880–2886 RSC.
- E. K. Heiniger, J. R. Buser, L. Mireles, X. Zhang, P. D. Ladd, B. R. Lutz and P. Yager, J. Microbiol. Methods, 2016, 128, 80–87 CrossRef.
- L. K. Lafleur, J. D. Bishop, E. K. Heiniger, R. P. Gallagher, M. D. Wheeler, P. Kauffman, X. Zhang, E. C. Kline, J. R. Buser, S. Kumar, S. A. Byrnes, N. M. J. J. Vermeulen, N. K. Scarr, Y. Belousov, W. Mahoney, B. J. Toley, P. D. Ladd, B. R. Lutz and P. Yager, Lab Chip, 2016, 16, 3777 RSC.
- D. D. Phillips, D. N. Garboczi, K. Singh, Z. Hu, S. H. Leppla and C. E. Leysath, J. Mol. Recognit., 2013, 26, 376–381 CrossRef PubMed.
- P.-F. Su and F.-H. Wu, Br. J. Cancer, 1996, 73, 1533–1537 CrossRef PubMed.
- S. G. Parra, L. M. López-Orellana, A. R. M. Duque, J. L. Carns, R. A. Schwarz, C. A. Smith, M. Ortiz Silvestre, S. Diaz Bazan, P. A. Escobar, J. C. Felix, P. Ramalingam, P. E. Castle, M. L. Cremer, M. Maza, K. M. Schmeler and R. R. Richards-Kortum, Int. J. Cancer, 2021, 148, 2571–2578 CrossRef PubMed.
- A. Bangor, P. T. Kortum and J. T. Miller, Int. J. Hum.-Comput. Interact., 2008, 24, 574–594 CrossRef.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- P. Yager, G. J. Domingo and J. Gerdes, Annu. Rev. Biomed. Eng., 2008, 10, 107–144 CrossRef CAS.
- E. Fu, B. Lutz, P. Kauffman and P. Yager, Lab Chip, 2010, 10, 918 RSC.
- E. Fu, P. Kauffman, B. Lutz and P. Yager, Sens. Actuators, B, 2010, 149, 325–328 CrossRef CAS PubMed.
- G. E. Fridley, H. Le and P. Yager, Anal. Chem., 2014, 86, 6447–6453 CrossRef CAS PubMed.
- E. Fu, T. Liang, P. Spicar-Mihalic, J. Houghtaling, S. Ramachandran and P. Yager, Anal. Chem., 2012, 84, 4574–4579 CrossRef CAS.
- S. Ramachandran, E. Fu, B. Lutz and P. Yager, Analyst, 2014, 139, 1456–1462 RSC.
- G. Chondrogiannis, S. Khaliliazar, A. Toldrà, P. Réu and M. M. Hamedi, Sci. Rep., 2021, 11, 6140 CrossRef.
- World Health Organization, WHO Prequalification of In Vitro Diagnostics Public Report: Xpert HPV, 2017 Search PubMed.
- M. A. Molina, L. C. Diatricch, M. C. Quintana, W. J. Melchers and K. M. Andralojc, Expert Rev. Mol. Diagn., 2020, 20, 1099–1120 CrossRef CAS PubMed.
- C. E. A. Flores, G. G. Gutierrez, J. M. O. Leon, D. C. Rodriguez and S. W. Sørbye, BMC Infect. Dis., 2021, 21, 504 CrossRef.
- W. Chen, J. Jeronimo, F.-H. Zhao, Y.-L. Qiao, M. Valdez, X. Zhang, L.-N. Kang, P. Bansil, P. Paul, P. Bai, R. Peck, J. Li, F. Chen, M. H. Stoler and P. E. Castle, J. Clin. Virol., 2014, 61, 553–557 CrossRef CAS.
- N. M. Rodriguez, W. S. Wong, L. Liu, R. Dewar and C. M. Klapperich, Lab Chip, 2016, 16, 753–763 RSC.
- Q. Chen, L. Yao, Q. Wu, J. Xu, C. Yan, C. Guo, C. Zhang, T. Xu, P. Qin and W. Chen, Microchim. Acta, 2022, 189, 350 CrossRef CAS.
- P. E. Castle, M. Schiffman, R. D. Burk, S. Wacholder, A. Hildesheim, R. Herrero, M. C. Bratti, M. E. Sherman and A. Lorincz, Cancer Epidemiol., Biomarkers Prev., 2002, 11, 1394–1399 Search PubMed.
- P. E. Castle, D. Solomon, C. M. Wheeler, P. E. Gravitt, S. Wacholder and M. Schiffman, J. Clin. Microbiol., 2008, 46, 2595–2604 CrossRef.
- S. Parra, P. Keahey, K. Schmeler, M. Maza, P. Castle and R. Richards-Kortum, in Advances in Optics for Biotechnology, Medicine and Surgery XV, 2017 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00885h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |