
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping and downscaling a method by HILIC coupled simultaneously to ESIMS and ICPMS to determine the affinity of lanthanide chelating molecules using specific isotope dilution
Marina
Amaral Saraiva
a,
Pascal E.
Reiller
 a,
Cécile
Marie
b and
Carole
Bresson
*a
a,
Cécile
Marie
b and
Carole
Bresson
*a
aUniversité Paris-Saclay, CEA, Service de Physico-Chimie, F-91191, Gif-sur-Yvette Cédex, France. E-mail: carole.bresson@cea.fr
bUniversité de Montpellier, CEA, DES, ISEC, DMRC, F-30207, Bagnols sur Cèze Cédex, France
First published on 31st October 2023
Abstract
Recycling minor actinides (Am, Cm and Np) from spent nuclear fuel is considered by a few countries as an important option for a future sustainable nuclear fuel cycle. For this purpose, solvent extraction processes are developed to separate minor actinides, especially from lanthanides and other fission products. The development of fast and powerful analytical methods is essential to acquire the data needed to model these processes and to improve their performances. For this purpose, this study presents the development, validation and application of a new analytical approach based on the simultaneous coupling of hydrophilic interaction chromatography (HILIC) to electrospray ionization mass spectrometry (ESIMS) and inductively coupled plasma mass spectrometry (ICPMS), using specific isotope dilution (SID) as a method of quantification for the determination of the affinity and selectivity of chelating molecules (TPAEN, NM and DTPA) towards lanthanides (Ln). The best separation conditions of natNd and natSm complexes formed with the three molecules were defined. Then, downscaling the separation was investigated, to reduce effluent volumes, the consumption of materials and the time devoted to experiments which are of concern in the nuclear field. The separation was carried out using an amide grafted stationary phase column in isocratic and gradient elution modes depending on the separation format. The separated complexes were online identified and the quantitative distribution of the Ln among the complexes was simultaneously determined owing to our quantification strategy. The results obtained were similar for the three separation formats, allowing us to validate the robustness of the method. By applying this method, the affinity of TPAEN, NM and DTPA present in competition for the complexation reaction with natNd and natSm was further determined in a single step, allowing a quick screening. Both selectivity and affinity of these molecules could be compared to select the most promising candidates. This approach can be advantageously extended to the evaluation of the affinity of various classes of chelating molecules used in very low quantities, towards elements of interest in the fields of energy, toxicology and the environment.
1. Introduction
In the energy sector, recycling recoverable materials is a societal and economic challenge. Concerning the nuclear fuel cycle, the minor actinides (An) such as americium (Am) and curium (Cm), although representing only 1% by weight of spent nuclear fuel from current light water reactors,1 are responsible for much of the radiotoxicity and long-term heat load of the final high level waste. That is why following the extraction of uranium (U) and plutonium (Pu) from spent fuel solutions, the selective recovery of Am and Cm in order to partially or totally recycle them is considered a promising strategy by a few countries.2 Hence, several studies are focused on the development of specific molecules to set up dedicated treatment processes, especially for the partitioning of Am.2–4The treatment processes developed to ensure the selective recovery of the target radionuclides are mainly based on liquid–liquid extraction. A key step is the design of the extraction/back-extraction molecules that must exhibit high selectivity and affinity towards the elements of interest, requiring the synthesis and the screening of numerous molecules. Two of the major criteria for selecting the candidate molecules are (i) the affinity for the targeted element(s); (ii) the selectivity versus impurities or secondary elements to be able to perform separation of intra-group or inter-group elements.
In general, the affinity and the selectivity of the molecules are evaluated by combining information obtained from conventional methods such as liquid–liquid extraction, giving distribution coefficients and from solution chemistry techniques, through the thermodynamic and structural characterisation of the complexes formed with the molecules, by time-resolved laser fluorescence spectroscopy (TRLFS), fluorescence spectroscopy, microcalorimetry, potentiometry, infrared and UV-visible spectroscopy, nuclear magnetic resonance (NMR), extended X-ray absorption fine structure spectroscopy (EXAFS) or electrospray ionization mass spectrometry (ESI-MS) using direct introduction of samples.5–12 In addition, these experiments are carried out in sequential mode and are often devoted to the characterisation of each complex independently, leading to significant consumption of materials and time, and preventing to assess affinity/selectivity properties when the molecules are in competing complexation reactions towards several elements.
The development of analytical methods allowing the quick determination of the selectivity and the affinity of molecules towards the targeted elements, while reducing the consumption of materials, effluent volumes and the time devoted to the screening experiments, is of great concern for an accelerated and rational research of efficient molecules. To meet this goal, the strategy of this work was based on the simultaneous coupling of hydrophilic liquid chromatography (HILIC) to electrospray ionization mass spectrometry (ESIMS) and inductively coupled plasma mass spectrometry (ICPMS) previously set up in the laboratory.13 This powerful approach allows, online and simultaneously, the structural, multi-elemental and isotopic characterization of chemical species coming from the chromatographic separation. Hence, the quantitative distribution of the elements among the complexes formed with the different chelating molecules (CMs) in a sample can be determined, allowing the selectivity and the affinity of the molecules for the elements to be reached, in a single analysis. This approach has been undertaken to determine the selectivity and affinity of two hydrophilic CMs towards natural samarium (natSm) and natural neodymium (natNd), compared to those of commercial diethylene triamine pentaacetic acid (DTPA) as a reference molecule (Fig. 1). One of the molecules, investigated as potential selective back-extraction reagents of actinides especially for Am/Cm partitioning, contains a diamine bridge and four picolinic acid arms: N,N,N′N′-tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (TPAEN)14,15 and the second new one is a nitrogenous macrocycle (NM) (Fig. 1). Sm and Nd were chosen as analogues of Am and Cm to develop the method in a conventional laboratory, because of their similar physicochemical properties16–19 such as similar ionic radius, hard acid character according to the Pearson theory,16 same +III oxidation state in solution and high coordination numbers.
 | ||
| Fig. 1 Chemical structures of DTPA (a), TPAEN (b) and NM (c). | ||
In the first step, the best HILIC separation conditions of the natSm and natNd complexes formed with the three molecules were defined. To our knowledge, only HILIC separations of gadolinium complexes containing contrast agents based on linear/cyclic polyaminocarboxylic acid derivatives are reported in the literature20–30 and our group has also developed the separation of Ln complexes formed with DTPA and EDTA ligands.13,31 However, the separation mechanism of HILIC mode is known to be very complex,32 which required developments in order to separate these new sets of complexes. To this end, several stationary phases functionalized with different polar groups were tested in the conventional chromatographic format as well as several mobile phase compositions. An important part of this work was further dedicated to the reduction of the scale of separation to the capillary and to the nano-flow formats while maintaining the performance of the analytical method. The aim was to reduce the consumption of the quantity of materials and molecules used since their synthesis is delicate, a limiting aspect leading to the availability of very few amounts of TPAEN and NM to develop our method. Another aim was to decrease the volume of effluents generated during the analysis, which is of great concern in the nuclear domain.
Then, a method of quantification by specific isotope dilution (SID) was developed and validated, to quantify online and precisely the distribution of natSm and natNd among the separated complexes simultaneously identified by ESIMS. Compared to external calibration, the SID quantification method allows the reduction of the material consumption and analysis time and makes the results more precise because the errors resulting from the analytical procedure can be corrected mathematically and quantitatively.33 Briefly, SID is based on the measurement of isotope ratios in a sample whose analyte isotopic composition is altered by the addition of a known amount of an isotopically modified analyte (spike).34 The advantage of SID is that once the isotopic equilibrium between the sample and the spike(s) is achieved, the physical effects induced by the matrix, potential species loss (e.g., incomplete extraction) or transformation (e.g. oxidation and/or reduction) during the analytical process does not influence the method accuracy, owing to the measurement of isotopic ratios rather than the absolute or relative signals as for conventional analytical methods.33 In the nuclear domain, the quantification of Ln and/or An has been widely developed using non-specific isotopic dilution, to measure elemental concentrations with low uncertainties.35–39 To our knowledge, the separation, online identification and quantification of Ln complexes by HILIC-ESIMS-SID-ICPMS has never been reported in the literature.
By applying this approach, the affinity of the three chelating molecules for natSm and natNd could be determined online and in a single analysis. For this, several stoichiometric ratios of TPAEN, NM and DTPA were added in competition for the complexation reaction of natSm and natNd in the samples, leading to the formation of the complexes in different proportions according to the selectivity and affinity of the molecules towards the Ln. The benefits provided by our approach compared to studies reported in the literature have been highlighted.
2. Experimental section
2.1. Chemicals and reagents
All the aqueous solutions and mobile phases were prepared with ultrapure water (18.2 MΩ cm, Merck Millipore, Guyancourt – France). Acetonitrile (ACN, CH3CN, LC-MS grade), formic acid (Normapur grade) and ammonia solution 22% (v/v) were purchased from VWR Prolabo (Briare-le-Canal, France). Ammonium acetate (NH4CH3CO2) was supplied by Sigma Aldrich (Saint Quentin Fallavier, France).Standard solutions (1000 mg L−1 in HNO3 2% w/w) of natSm and natNd were provided by SCP SciencePlasmaCal (Courtaboeuf, France) and standard solutions of bismuth (209Bi) and indium (115In) were from the Spex Certiprep Group (Longjumeau, France). Nitric acid (67–69% w/w, NORMATOM®, VWR, Fontenay-sous-Bois, France) was used to prepare HNO3 2% w/w in ultrapure water. The isotopically enriched oxide powders 147Sm2O3 and 145Nd2O3 were from Euriso-top (Saint-Aubin CEDEX, France) with the isotope abundance of respectively 94.0% and 87.1% as provided by the manufacturer's certificate, but without uncertainties. The standard reference materials, Sm – 3147a and Nd – 3135a (NIST, Gaithersburg, US), both at 8.52 ± 0.02 mg g−1 in HNO3 10% w/w, were used for the quantification of the spike solutions by thermal ionization mass spectrometry (TIMS).
Diethylene triamine pentaacetic acid (DTPA, C14H23O10N3, purity ≥ 98%) was provided by Sigma Aldrich (Saint Quentin Fallavier, France), N,N,N′N′-tetrakis[(6-carboxypyridin-2-yl)methyl]ethylenediamine (TPAEN)14,15 and the nitrogenous macrocycle (NM) were synthesized, characterized and supplied by the LCIS team (Université de Montpellier, DES/ISEC/DMRC, CEA Marcoule, France).
2.2. Preparation of stock, intermediate, contact and control solutions, samples and controls
The commercial standard solutions of natSm and natNd were diluted in HNO3 2% w/w to obtain individual stock solutions at 5.0 × 10−3 mol L−1.
The stock solutions of the spikes isotopically enriched in 147Sm and 145Nd (spikeLn) were prepared by dissolving the appropriate amount of the corresponding enriched Ln2O3 oxide powders (147Sm2O3 and 145Nd2O3) in HNO3 2% w/w to obtain a concentration of each isotope of 5.1 × 10−3 mol L−1.
| natLn + spikeLn + xTPAEN + yNM + zDTPA |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y:z) compared to those of the LnT which was fixed, as reported in Table 1. The pH of each contact solution was adjusted to 1 by adding around 1 µL of ammonia solution (22% v/v).
:y:z being the stoichiometric ratios of the chelating molecules compared to this of the LnT. The experimental concentrations obtained from the weight of the Ln and the CM solutions are given in brackets (mol L−1)
:y:z) compared to those of the LnT which was fixed, as reported in Table 1. The pH of each contact solution was adjusted to 1 by adding around 1 µL of ammonia solution (22% v/v).
:y:z being the stoichiometric ratios of the chelating molecules compared to this of the LnT. The experimental concentrations obtained from the weight of the Ln and the CM solutions are given in brackets (mol L−1)
| Format | LnTa | xTPAEN | yNM | zDTPA | |
|---|---|---|---|---|---|
| a LnT = natLn + spikeLn. | |||||
| Conventional | Control 1 | 4 (1.1 × 10−5 mol L−1) | 4 (1.7 × 10−5mol L−1) | 0 | 0 |
| Control 2 | 4 (1.2 × 10−5 mol L−1) | 0 | 4 (1.8 × 10−5 mol L−1) | 0 | |
| Control 3 | 4 (1.1 × 10−5 mol L−1) | 0 | 0 | 4 (1.7 × 10−5 mol L−1) | |
| Sample 1 | 4 (1.1 × 10−5 mol L−1) | 2.5 (3.0 × 10−5 mol L−1) | 1.4 (6.1 × 10−6 mol L−1) | 0.1 (4.5 × 10−7 mol L−1) | |
| Sample 2 | 4 (1.2 × 10−5 mol L−1) | 1.4 (6.5 × 10−6 mol L−1) | 2.5 (1.1 × 10−5 mol L−1) | 0.1 (4.6 × 10−7 mol L−1) | |
| Sample 3 | 4 (1.0 × 10−5 mol L−1) | 1.8 (7.4 × 10−6 mol L−1) | 1.8 (7.6 × 10−6 mol L−1) | 0.4 (1.3 × 10−6 mol L−1) | |
| Sample 4 | — | — | — | — | |
| Capillary | Control 1 | 4 (2.2 × 10−6 mol L−1) | 4 (3.6 × 10−6 mol L−1) | 0 | 0 |
| Control 2 | 4 (2.3 × 10−6 mol L−1) | 0 | 4 (3.6 × 10−6 mol L−1) | 0 | |
| Control 3 | 4 (2.2 × 10−6 mol L−1) | 0 | 0 | 4 (3.5 × 10−6 mol L−1) | |
| Sample 1 | 4 (3.1 × 10−6 mol L−1) | 2.5 (1.6 × 10−5 mol L−1) | 1.4 (1.8 × 10−6 mol L−1) | 0.1 (1.4 × 10−7 mol L−1) | |
| Sample 2 | 4 (3.6 × 10−6 mol L−1) | 1.4 (2.0 × 10−6 mol L−1) | 2.5 (3.6 × 10−6 mol L−1) | 0.1 (1.4 × 10−7 mol L−1) | |
| Sample 3 | 4 (2.9 × 10−6 mol L−1) | 1.8 (2.0 × 10−6 mol L−1) | 1.8 (2.0 × 10−6 mol L−1) | 0.4 (4.5 × 10−7 mol L−1) | |
| Sample 4 | 4 (2.6 × 10−6 mol L−1) | 1.3 (1.3 × 10−6 mol L−1) | 1.3 (1.2 × 10−6 mol L−1) | 1.3 (1.3 × 10−6 mol L−1) | |
| Nano | Control 1 | 4 (5.8 × 10−6 mol L−1) | 4 (9.4 × 10−6 mol L−1) | 0 | 0 |
| Control 2 | 4 (5.7 × 10−6 mol L−1) | 0 | 4 (9.1 × 10−6 mol L−1) | 0 | |
| Control 3 | 4 (5.9 × 10−6 mol L−1) | 0 | 0 | 4 (9.3 × 10−6 mol L−1) | |
| Sample 1 | 4 (6.0 × 10−6 mol L−1) | 2.5 (5.7 × 10−6 mol L−1) | 1.4 (3.5 × 10−6 mol L−1) | 0.1 (2.6 × 10−7 mol L−1) | |
| Sample 2 | 4 (6.0 × 10−6 mol L−1) | 1.4 (3.0 × 10−6 mol L−1) | 2.5 (6.0 × 10−6 mol L−1) | 0.1 (2.1 × 10−7 mol L−1) | |
| Sample 3 | 4 (5.9 × 10−6 mol L−1) | 1.8 (4.1 × 10−6 mol L−1) | 1.8 (4.0 × 10−6 mol L−1) | 0.4 (9.1 × 10−7 mol L−1) | |
| Sample 4 | 4 (6.1 × 10−6 mol L−1) | 1.3 (3.1 × 10−6 mol L−1) | 1.3 (3.0 × 10−6 mol L−1) | 1.3 (3.1 × 10−6 mol L−1) | |
Samples 1, 2, 3 and 4 contained different CM stoichiometric ratios compared to this of LnT, being one equivalent of each natural and enriched Ln, according to LnT:xTPAEN:yNM:zDTPA. The composition of each sample was 4:2.5:1.4:0.1 (sample 1), 4:1.4:2.5:0.1 (sample 2), 4:1.8:1.8:0.4 (sample 3) and 4:1.3:1.3:1.3 (sample 4). Controls 1, 2 and 3 contained one of each CM in equimolar stoichiometric ratios with respect to this of LnT. The composition of each control was LnT:4TPAEN (control 1), LnT:4NM (control 2), LnT:4DTPA (control 3), with LnT = 1natSm + 1natNd + 1spikeSm + 1spikeNd. The samples and controls were prepared and injected in duplicate in the column after 1 hour. The stock, intermediate and contact solutions were stored in a refrigerator at 4–5 °C for a maximum of one month.
Additional five contact solutions containing equimolar ratios of the CM regarding this of the LnT, 4:1.33:1.33:1.33 (sample 4) were prepared and the pH was adjusted between 1 and 10 (1.36, 2.85, 5.31, 6.93 and 9.56) with ammonia solution 22% (v/v). The solutions were left overnight and diluted into the working mobile phase the next day to obtain the corresponding samples.
2.3. Instrumentation
The chromatographic separations in conventional format were carried out using an ultimate 3000 UHPLC+ system (Dionex/Thermofisher Scientific, Courtaboeuf, France), consisting of a degasser, a dual RS pump, an RS autosampler, a column compartment and an RS diode array detector. The ultimate 3000 RSLCnano System (Thermofisher Scientific, Courtaboeuf, France) was composed of a pump module equipped with proflow and capillary flowmeters.A triple quadrupole TSQ Quantum Ultra™ mass spectrometer (Thermo Fisher Scientific, San Diego CA, USA) equipped with an H-ESI II ionisation probe was used for the simultaneous coupling to the separations in conventional and capillary formats. Mass spectra were registered in full scan mode (m/z 300–800) and in selected ion monitoring mode (SIM) by considering the most abundant m/z ratio of the isotopic pattern associated with the Ln-complexes, namely m/z = 743 for the Ln–TPAEN complexes, m/z = 702 for the Ln–NM complexes and m/z = 537 for the Ln–DTPA complexes, with Ln being natural and spike Sm and Nd (spectral width m/z ± 12). The data were processed with Xcalibur software.
The ICPMS instrument used for the simultaneous coupling of the separations carried out in conventional format was an XSeriesII (Thermo Electron SAS, Courtaboeuf, France) equipped with platinum skimmer and sampler cones, a MicroMist nebulizer operating at 400 µL min−1 (Thermo Scientific BRE0009386) and a quartz cyclonic spray chamber (Thermo Scientific 1317080) thermostated at 3 °C. The data were processed with PlasmaLab software.
An 8800 Triple Quadrupole ICPMS (Agilent, Courtaboeuf, France) was also used for the coupling experiments associated with the chromatographic downscaling, owing to its better sensitivity when working at micro flow rates. A total consumption micro nebulizer operating 10 µL min−1 (Agilent G3280-80602) equipped with a single pass spray chamber (Agilent G3280-80603 in quartz material) was used for the coupling experiments in capillary and nano-flow rate formats. The data were processed with Mass Hunter software.
In order to prevent any carbon deposition on the cones due to the use of acetonitrile, oxygen was injected at 8 mL min−1 in the plasma after the spray chamber when using the ICPMS XSeriesII13,31 and O2/Ar mix at 0.5 L min−1 of for the ICPMS 8800 TQ.
The operating ESI-QqQ-MS, ICP-MS XseriesII and 8800 TQ parameters are summarized in Table 2.
| ESIMS TSQ Quantum Ultra | ICPMS XSeriesII | ICPMS 8800 TQ | ||
|---|---|---|---|---|
| a Used as internal standards. | ||||
| Spray voltage | −3.7 kV | Plasma gas flow rate | 14 L min−1 | 15 L min−1 |
| Capillary transfer temperature | 250 °C | Auxiliary gas flow rate | 0.9 L min−1 | 0.9 L min−1 |
| Vaporisation temperature | 150 °C | Nebuliser gas flow rate | 0.8 L min−1 | 0.7 L min−1 |
| Sheath gas pressure | 15 a.u | Dilution gas flow rate | 8 mL min−1 (O2) | 0.5 L min−1 (O2/Ar 20%/80%) |
| Auxiliary gas pressure | 35 a.u | Nebulizer liquid flow rate | 130 µL min−1 | 0.3 µL min−1 |
| Scan width | 20 (m/z) | Tip material | Platinum | Platinum |
| Scan time | 0.7 s | Sampler cone diameter (Pt) | 1.0 mm | 1.2 mm |
| FWHM | 0.5 | Plasma power | 1550 W | 1550 W |
| Dwell time | 30 ms | 10 ms | ||
| Monitored isotopes | 142Nd, 145Nd, 147Sm, 152Sm, 209Bia | 142Nd, 145Nd, 147Sm, 152Sm, 115Ina | ||
2.4. Chromatographic conditions
The separation of the Ln complexes was developed in the conventional format and further transposed to the capillary flow format, before reaching the nano-flow format. The physicochemical properties and the dimensions of the columns used in the different formats are summarized in Table 3.| Name (manufacturer) | Dimension (mm); particle size (µm); pore size (Å) | Material support | Stationary phase functionalization | Functional group |
|---|---|---|---|---|
| a BEH – bridged ethylene hybrid; n.a — not available. | ||||
| ACQUITY BEH Amide (Waters) | 150 × 2.1; 1.7; 130 | BEH silica | Amide |

|
| InertSustain Amide (GLSiences) | 150 × 0.3; 3.0; 100 | Ultra-pure silica | Amide | |
| InertSustain Amide (GLSiences) | 150 × 0.075; 3; 100 | Ultra-pure silica | Amide |

|
| Accucore Amide (Thermofisher) | 100 × 2.1; 2.6; 150 | Ultra-pure silica | Amide | |
| TSKgel Amide-80 (Tosoh) | 150 × 2.1; 3.0; 80 | Silica | Amide |

|
| BioZen Glycan (Phenomenex) | 150 × 2.1; 2.6; 100 | n.a | Amide polyol |

|
| Triart Diol HILIC (YMC) | 150 × 2.1; 1.9; 120 | Organic/inorganic hybrid silica | Diol |

|
| Luna HILIC (Phenomenex) | 100 × 2.1; 3.0; 200 | Silica | Diol | — |
| Inertsil HILIC (GL Science) | 100 × 2.1; 3.0; 100 | Ultra-pure silica | Diol |

|
| Inertsil Diol (GL Science) | 150 × 2.1; 3.0; 100 | Ultra-pure silica | Diol | |
| Acclaim HILIC-10 (Thermofisher) | 150 × 2.1; 3.0; 120 | Ultra-pure silica | Proprietary group | Proprietary |
| Acclaim WCX-1 (Thermofisher) | 150 × 2.1; 5.0; 120 | Ultra-pure silica | Mixed mode | — |
| Acclaim Trinity P1 (Thermofisher) | 150 × 2.1; 3.0; 300 | Hybrid silica | Mixed mode | — |
| Hypersil Gold HILIC (Thermofisher) | 100 × 2.1; 1.9; 175 | Ultra-pure silica | Polyethylenimine | — |
| Hypersil Gold silica (Thermofisher) | 100 × 2.1; 1.9; 175 | Highly pure deactivated silica | Silica | — |
| Pack Sil (YMC) | 100 × 2.1; 3.0; 120 | Ultra-pure silica | Silica | — |
| Triart Sil (YMC) | 100 × 2.1; 3.0; 120 | Organic/inorganic hybrid silica | Silica | — |
| Luna silica (Phenomenex) | 100 × 2.1; 3.0; 100 | Ultra-high purity silica | Silica |

|
| ACQUITY BEH HILIC (Waters) | 150 × 2.1; 1.7; 150 | BEH silica | Silica | — |
| Syncronis HILIC (Thermofisher) | 150 × 2.1; 5.0; 100 | Ultra-pure silica | Zwitterionic (sulfobetaine) | — |
| ZIC-HILIC (Merck) | 150 × 2.1; 3.5; 100 | Polymeric | Zwitterionic (sulfobetaine) |

|
The chromatographic parameters: retention factor (k′), selectivity and resolution factors (α and Rs) were calculated to evaluate the performance of the different columns.41
The retention factor (k′) was calculated according to eqn (1):
 | (1) |
The selectivity factor (α) was calculated based on eqn (2) and the resolution factor (Rs) based on eqn (3):
 | (2) |
 | (3) |
Analyte 2 was more retained than analyte 1 and w0.5h corresponds to full-width at half-maximum of each peak.
The chromatographic conditions for each separation format are presented in Table 4.
| Separation format | Conventional | Capillary | Nano | |
|---|---|---|---|---|
| Mobile phase | 74/26 (v/v) ACN/H2O + 0.5% v/v formic acid + 15 mmol L−1 NH4CH3CO2 | 74/26 (v/v) ACN/H2O + 0.5% v/v formic acid + 10 mmol L−1 NH4CH3CO2 | Solvent A: 60/40 (v/v) ACN/H2O+ 0.5% v/v formic acid + 7.5 mmol L−1 NH4CH3CO2 | Solvent B: 90/10 (v/v) ACN/H2O + 0.5% v/v formic acid + 7.5 mmol L−1 NH4CH3CO2 |
| Elution mode | Isocratic | Isocratic | Gradient | |
| 0–0.5 min: 75% B | ||||
| 0.5–3.0 min: 70% B | ||||
| 3.0–6.5 min: 55% B | ||||
| 6.5–25.0 min: 45% B | ||||
| Injection volume | 3 µL | 1 µL | 0.050 µL | |
| Flow rate | 300 µL min−1 | 5 µL min−1 | 0.3 µL min−1 | |
| Sampler temperature | 22 ± 1 °C | |||
| Column temperature | 40 ± 1 °C | 22 ± 1 °C | 22 ± 1 °C | |
| Separation time | 20 min | 20 min | 25 min | |
2.5. Simultaneous coupling of HILIC to ESIMS and ICPMS
The instrumental configuration of the simultaneous coupling of HILIC in conventional separation format to ESIMS and the ICPMS XSeriesII was previously developed in the laboratory.13 By applying this setup, the outflow from the column was split to enter the ESIMS with a flow rate of 295 µL min−1 (98.3%) and 5 µL min−1 for the flow entering the ICPMS (1.7%). A make-up made of 2% HNO3 w/w and containing 10 ppb of 209Bi as internal standard was added to the flow entering the ICPMS, with a flow rate of 140 µL min−1. More details regarding the instrumental conditions are provided in ref. 13.For the simultaneous coupling involving the separation downscaling, the outflow from the capillary column was split to simultaneously enter the ESIMS with a flow rate of 4.70 µL min−1 (94%) and the ICPMS with a flow rate of 0.30 µL min−1 (6%). In the case of nano-flow format experiments, the capillary was only coupled to ICPMS with a flow rate of 0.30 µL min−1.
The torch position, the nebulizer and auxiliary gas as well as the optical lens system were optimized daily using a mixed solution of natSm–natNd–DTPA. 209Bi solution at 10 ppb in 2% HNO3 w/w was used as an internal standard for the conventional separation format and 115In at 50 ppb was the internal standard added in the mobile phase of the capillary and nano-flow separations, to monitor the signal stability before and during the analyses.
2.6. Development of the quantification method by specific isotope dilution (SID)
A dedicated quantification method using SID was developed and validated based on the mathematical model provided in the literature,42 to quantify precisely and online the distribution of natSm and natNd among the complexes coming from the separations.In the first step, the concentration of 145Nd and 147Sm in the stock solutions of the spikes was determined based on thermal ionization mass spectrometry (TIMS), which was used to measure isotopic ratios of mixed solutions made of the spikes and standard reference material of natSm and natNd. The precise concentration of 145Nd and 147Sm was further quantified, by applying the methods developed by the CEA-LANIE.43,44 The measured concentrations of 145Nd and 147Sm were 1067.01 µg g−1 ± 0.94 and 984.22 µg g−1 ± 0.41 respectively and were taken into account for the SID calculations.
The samples were spiked with known amounts of 145Nd and 147Sm to achieve 145Nd/142Nd and 147Sm/152Sm isotope ratios equal to one in the blend samples. For this purpose, the mass of each spike stock solution to add to the sample was calculated based on eqn (4) (example provided here for 145Nd spike solely).
 | (4) |
The natural abundances of the considered 152Sm and 142Nd isotopes were 26.749 and 27.152% as reported by the Internal Union of Pure and Applied Chemistry.45 The abundances of 145Nd and 147Sm in the spike solutions were 87.1% and 94.0% as provided by the manufacturer's certificate.
The natSm and natNd contained in the separated complexes were further online quantified based on the elution profiles registered by ICPMS, by monitoring the signal of Sm and Nd isotopes needed for this study as given in Table 2. The integration of the total area of the chromatographic peaks corresponding to the Ln complexes formed with the three chelating molecules in different proportions, combined with the application of SID allowed determination of the quantitative distribution of natSm and natNd among the different complexes.
The measured 145Nd/142Nd and 147Sm/152Sm natural isotopic ratios were corrected with the mass bias factor (k), determined daily before each analytical procedure, using eqn (5).
| k = Rr/Rm | (5) |
The concentration of natNd and natSm in each separated complex was calculated based on eqn (6).
 | (6) |
 , measured isotope ratio based on the chromatographic peak integration of the corresponding complex; MMnat, molar mass of natNd or natSm (g mol−1).
, measured isotope ratio based on the chromatographic peak integration of the corresponding complex; MMnat, molar mass of natNd or natSm (g mol−1).
The limit of detection (LOD) and limit of quantification (LOQ) of natSm and natNd–DTPA complexes were determined based on the common method using the calibration curve applying eqn (7):46,47
 | (7) |
The LOD and LOQ obtained by nano-HILIC-ICPMS were 3.6 and 12.1 µg kg−1 for the natSm complex and 4.9 and 16.6 µg kg−1 for the natNd complex, respectively.
3. Results and discussion
3.1. Definition of the conditions for separating the Ln complexes in conventional format
HILIC separations of Ln (Er, Eu, Gd, and Nd) with strong chelating molecules such as DTPA and EDTA have been previously investigated13,31 and our first tests in this work showed that the natSm/natNd–DTPA complexes were more retained than the complexes containing NM and TPAEN molecules. Hence, our efforts were first focused on the screening of the selectivity of several columns to separate natSm and natNd complexes of NM and TPAEN.
For all the columns, a reference mobile phase composed of 70/30 (v/v) ACN/H2O containing 15 mmol L−1 NH4CH3CO2 and 0.5% formic acid was initially used and eluted in isocratic mode.13 It must be noted that using Luna HILIC, Acclaim Mixed-mode and Biozen Glycan columns, NM and TPAEN complexes co-eluted or were not observed, even with different mobile phase compositions. Therefore, these columns were not selected for the rest of the study. The chromatographic parameters such as retention factor (k′), selectivity factor (α) and resolution factor (Rs) are shown in Table 5. From this table, it can be seen that the TPAEN complexes were well separated from the NM complexes, with selectivity factors between 1.55 and 11.17, except for the Acclaim Trinity column with a value of 1.08. In all cases, NM complexes were more retained than TPAEN complexes with the highest retention factors of NM complexes ranging from 13.5 to 40. The particularly high retention factors obtained with the most polar non-grafted silica columns as well as with the ZIC-HILIC, the Acclaim Trinity or the TSK Gel Amide columns, led us to also exclude these columns. In addition, most of the columns were not selective enough in these conditions to separate efficiently the complexes containing the same molecule, namely the pairs natSm–TPAEN/natNd–TPAEN and natSm–NM/natNd–NM, as for the Triart HILIC or the Acclaim HILIC-10 columns, with α around 1. Except for the ACQUITY UPLC BEH Amide, resolution factors lower than 0.1 to separate the peaks of natSm–TPAEN and natNd–TPAEN were obtained for all the columns under the reference conditions.
| Stationary phase functionalization | Amide | Diol | Exclusive | Polyethylenimine | Amide polyol | Bare silica | Hybrid silica | Zwiterionic | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Column name | ACQUITY UPLC BEH | Accucore | TSK Gel Amide-80 | Triart HILIC | Luna HILIC | Inerstil HILIC | Inerstil Diol | Acclaim HILIC-10 | Acclaim Mixed-mode | Acclaim Trinity P1 | Hypersil Gold HILIC | Biozen Glycan | Hypersil Gold | YMC Pack Sil | Luna Silica | YMC Triart Sil | ACQUITY BEH HILIC | Syncronis HILIC | ZIC-HILIC | |
| a ND: not detected; —: all peaks co-eluted. | ||||||||||||||||||||
| k′ | Sm–TPAEN | 2.93 | 0.93 | 2.89 | 2.29 | 1.24 | 0.38 | 1.01 | 4.06 | 1.98 | 9.15 | 1.39 | ND | 1.20 | 2.43 | 4.10 | 1.65 | 2.64 | 2.11 | 1.41 |
| Nd–TPAEN | 3.00 | 1.01 | 2.94 | 2.31 | 0.43 | 1.05 | 4.15 | 9.20 | 1.43 | ND | 1.21 | 2.48 | 4.15 | 1.72 | 2.71 | 2.13 | 1.45 | |||
| Sm–NM | 4.64 | 2.25 | 15.84 | 7.31 | 1.06 | 2.03 | 6.69 | 15.63 | 14.31 | 40.28 | 13.50 | 24.74 | 39.23 | 9.64 | 9.96 | 8.42 | 14.46 | |||
| Nd–NM | 4.94 | 2.48 | 16.65 | 7.54 | 1.18 | 2.11 | 6.88 | 15.81 | 15.70 | 42.43 | 14.41 | 28.87 | 41.91 | 10.07 | 10.33 | 8.53 | 15.87 | |||
| α i,j | Sm–TPAEN | |||||||||||||||||||
| 1.03 | 1.09 | 1.02 | 1.01 | — | 1.13 | 1.04 | 1.02 | — | 1.01 | 1.03 | ND | 1.01 | 1.02 | 1.01 | 1.04 | 1.03 | 1.01 | 1.03 | ||
| Nd–TPAEN | ||||||||||||||||||||
| 1.55 | 2.22 | 5.39 | 3.16 | — | 2.50 | 1.93 | 1.61 | — | 1.08 | 10.01 | ND | 11.17 | 9.98 | 9.45 | 5.60 | 3.67 | 3.95 | 9.94 | ||
| Sm–NM | ||||||||||||||||||||
| 1.20 | 1.10 | 1.05 | 1.03 | — | 1.11 | 1.04 | 1.03 | — | 1.22 | 1.10 | 1.05 | 1.07 | 1.17 | 1.07 | 1.04 | 1.04 | 1.01 | 1.10 | ||
| Nd–NM | ||||||||||||||||||||
| Rs | Sm–TPAEN | |||||||||||||||||||
| 0.15 | 0.04 | 0.01 | 0.01 | — | 0.02 | 0.02 | 0.05 | — | 0.03 | 0.01 | ND | 0.00 | 0.01 | 0.01 | 0.02 | 0.02 | 0.01 | 0.01 | ||
| Nd–TPAEN | ||||||||||||||||||||
| 1.92 | 0.54 | 2.88 | 1.72 | — | 0.25 | 0.47 | 1.38 | — | 3.41 | 2.09 | ND | 1.99 | 3.60 | 5.52 | 2.14 | 2.22 | 2.28 | 2.10 | ||
| Sm–NM | ||||||||||||||||||||
| 2.49 | 0.15 | 0.56 | 0.16 | — | 0.07 | 0.06 | 0.13 | — | 0.13 | 1.14 | 1.20 | 0.84 | 3.19 | 2.08 | 0.37 | 0.25 | 0.10 | 1.14 | ||
| Nd–NM | ||||||||||||||||||||
Finally, the ACQUITY UPLC BEH Amide (150 × 2.1 mm; 1.7 µm) column was selected since it led to the best compromise between acceptable retention factors, satisfying separation of the NM complexes and its commercial availability in capillary and nano-flow formats for downscaling of the separation.
Finally, the best separation conditions were obtained using a mobile phase made of 74/26 (v/v) ACN/H2O containing 15 mmol L−1 NH4CH3CO2 and 0.5% formic acid, eluted in isocratic mode as presented in Fig. 2.
 | ||
| Fig. 2 Chromatograms simultaneously acquired by ESIMS and ICPMS coupled to the separation of the natNd and natSm complexes previously formed with TPAEN, NM and DTPA and mixed in the working mobile phase before the injection. ESIMS spectra were recorded using SIM mode by considering the most abundant m/z ratio of the isotopic pattern of the Ln-complexes, namely m/z = 743 for the Ln–TPAEN complexes, m/z = 702 for the Ln–NM complexes and m/z = 537 for the Ln–DTPA complexes. ICPMS profiles were obtained by registering the signal of the most abundant isotope of natNd (m/z = 142) and natSm (m/z = 152) (a) Extracted ESI mass spectra of natNd and natSm–TPAEN complexes and (b) extracted ESI mass spectrum of the natNd–DTPA complex. Column: ACQUITY UPLC BEH Amide (150 × 2.1 mm; 1.7 µm). Mobile phase: 74/26 (v/v) ACN/H2O containing 15 mmol L−1 NH4CH3CO2 and 0.5% formic acid, eluted in isocratic mode. Flow rate: 300 µL min−1 and Vinj = 3 µL. Note: *NM is a natSm and natNd tertiary complex of NM. | ||
The peaks in the chromatograms registered by ICPMS could be attributed by comparison with the mass spectra extracted from the peaks of the ESIMS chromatograms, allowing the identification of the associated complexes. For all the chelating molecules, complexes of 1/1 stoichiometry were online observed in agreement with the literature, in which TPAEN complexes were characterized by spectroscopic techniques11 and DTPA complexes using HILIC-ESIMS.13,31
In this study, the HILIC conditions have been set up to separate new sets of Ln complexes formed with hydrophilic synthetic chelating molecules, thanks to an amide functionalized stationary phase. Regarding the studies of Ln species, numerous studies have reported the analysis of Gd-based contrast agents containing DTPA and linear/cyclic polyaminocarboxylic acid derivatives such as BT-DO3A, DOTA, BMA and BOPTA, by HILIC-ESIMS20 and/or HILIC-ICPMS in blood plasma and urine samples,21–23 waters23–29 and plants.30 Most of these studies involved zwitterionic-grafted and unmodified silica columns and the separations were generally run in conventional chromatographic format (100–150 mm × 2–3 mm; 2.6–5 µm).
| Format | Classical approach | Our approach | ||
|---|---|---|---|---|
| Batcha | Conventional | Capillary | Nano | |
| a Complexation of Eu by TPAEN was followed by microcalorimetric titrations.11 | ||||
| Column inner diameter (mm) | — | 2.1 | 0.3 | 0.075 |
| Flow rate (µL min−1) | — | 300 | 5 | 0.3 |
| Injection volume (µL) | 2 | 3 | 1 | 0.05 |
| natSm (ng) | — | 1.80 | 0.15 | 0.015 |
| natNd (ng) | — | 1.73 | 0.14 | 0.014 |
| Eu (ng) | 62600 |
— | — | — |
| CMtotal (ng) | 12000 |
36.00 | 2.40 | 0.24 |
| Effluent volume (mL)/1 h | — | 18 | 0.3 | 0.018 |
| Total analysis time (min) | — | 20 | 20 | 25 |
Compared to the determination of the TPAEN affinity for Eu by microcalorimetric titrations described in the literature,11 the total amount of Ln and chelating molecules in the injection volume of our nano-flow method was decreased by around 4 × 106 times and 5 × 105 times respectively, while enabling quantitative data for several molecules complexed by two Ln to be acquired.
3.2. Online determination of the affinity of TPAEN, NM and DTPA towards natSm and natNd
To meet this aim, various stoichiometric ratios of the three chelating molecules (CMs) were added to natNd and natSm in a competing complexation reaction, as indicated in Table 1. The formed complexes were further separated by HILIC, online identified by ESIMS and simultaneously quantified by ICPMS using the SID. By applying the mass balance, the quantitative distribution of natNd and natSm among the complexes of TPAEN, NM and DTPA could be deduced and the affinity of the CM for each Ln determined.The chromatograms obtained by HILIC-ICPMS for the four samples analysed in the nano-flow format are presented in Fig. 3.
 | ||
| Fig. 3 HILIC-ICPMS chromatograms for the four samples analysed in duplicate. Separations were run in nano-flow format using the conditions presented in Table 4. Composition of the samples as indicated in Table 1: LnT:xTPAEN:yNM:zDTPA with (a) 4:2.5:1.4:0.1 (sample 1), (b) 4:1.4:2.5:0.1 (sample 2), (c) 4:1.8:1.8:0.4 (sample 3) and (d) 4:1.3:1.3:1.3 (sample 4). | ||
The peaks of the Sm and Nd complexes formed with each CM were co-eluted in the nano-flow format compared to the capillary and the conventional formats, even after optimising the composition and elution mode of the mobile phase. It must however be pointed out that this was not an obstacle since ICPMS makes it possible the integration of each individual peak, allowing the quantification of each complex in the different samples.
Samples and controls were subjected to the separation run in duplicate and the complexes were online quantified based on the integration of their total peak area, allowing us to deduce for each of them the natLn/spikeLn ratio that was used in SID calculations. The measured concentrations of natSm and natNd complexes of TPAEN, NM and DTPA in the samples and controls are summarized in Table 7.
| Complex | natSm(CM) | natNd(CM) | ||||
|---|---|---|---|---|---|---|
| CM | TPAEN | NM | DTPA | TPAEN | NM | DTPA |
| Sample 1 | ||||||
| [natLn(CM)]control | 340.9 (2.3 × 10−6 mol L−1) | 323.0 (2.1 × 10−6 mol L−1) | 324.4 (2.1 × 10−6 mol L−1) | 343.6 (2.3 × 10−6 mol L−1) | 333.2 (2.3 × 10−6 mol L−1) | 325.1 (2.3 × 10−6 mol L−1) |
| [natLn(CM)]sample | 347.6 (2.3 × 10−6 mol L−1) | 0 | 0 | 376.3 (2.6 × 10−6 mol L−1) | 0 | 0 |
| ∑(sample/control) (%) | 102.0 | 109.5 | ||||
|
||||||
| Sample 2 | ||||||
| [natLn(CM)]control | 348.7 (2.3 × 10−6 mol L−1) | 325.7 (2.2 × 10−6 mol L−1) | 323.8 (2.2 × 10−6 mol L−1) | 323.7 (2.2 × 10−6 mol L−1) | 336.9 (2.3 × 10−6 mol L−1) | 325.0 (2.3 × 10−6 mol L−1) |
| [natLn(CM)]sample | 157.6 (1.1 × 10−6 mol L−1) | 193.0 (1.3 × 10−6 mol L−1) | 4.4 (2.9 × 10−8 mol L−1) | 272.2 (1.9 × 10−6 mol L−1) | 96.9 (0.9 × 10−6 mol L−1) | 0 |
| ∑(sample/control) (%) | 101.7 | 102.7 | ||||
|
||||||
| Sample 3 | ||||||
| [natLn(CM)]control | 349.4 (2.3 × 10−6 mol L−1) | 323.5 (2.2 × 10−6 mol L−1) | 348.7 (2.3 × 10−6 mol L−1) | 340.7 (2.4 × 10−6 mol L−1) | 337.6 (2.3 × 10−6 mol L−1) | 342.5 (2.4 × 10−6 mol L−1) |
| [natLn(CM)]sample | 278.4 (1.9 × 10−6 mol L−1) | 93.9 (6.2 × 10−7 mol L−1) | 21.9 (1.5 × 10−7 mol L−1) | 298.9 (2.1 × 10−6 mol L−1) | 32.8 (2.3 × 10−7 mol L−1) | 0 |
| ∑(sample/control) (%) | 105.7 | 97.4 | ||||
|
||||||
| Sample 4 | ||||||
| [natLn(CM)]control | 361.7 (2.4 × 10−6 mol L−1) | 375.6 (2.5 × 10−6 mol L−1) | 382.4 (2.5 × 10−6 mol L−1) | 345.6 (2.4 × 10−6 mol L−1) | 349.4 (2.4 × 10−6 mol L−1) | 348.7 (2.4 × 10−6 mol L−1) |
| [natLn(CM)]sample | 150.0 (9.9 × 10−7 mol L−1) | 83.9 (5.6 × 10−7 mol L−1) | 149.4 (9.9 × 10−7 mol L−1) | 261.2 (1.8 × 10−6 mol L−1) | 42.3 (2.9 × 10−7 mol L−1) | 28.7 (1.9 × 10−7 mol L−1) |
| ∑(sample/control) (%) | 100.7 | 95.9 | ||||
The mass balance (%) of natSm and natNd was calculated by summing the ratios of the complex concentrations obtained by SID in the samples and the corresponding controls, as shown in (eqn (8)).
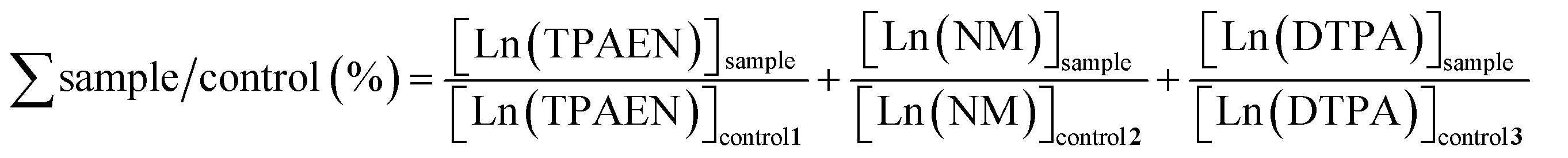 | (8) |
The quantitative distribution (%) of natSm and natNd among the separated complexes was obtained by applying eqn (9) and is presented in Fig. 4 and 5 for samples 1–4.
 | (9) |
 | ||
| Fig. 4 Quantitative distribution of natSm among the separated complexes depending on the composition of the samples. LnT:xTPAEN:yNM:zDTPA: (sample 1) 4:2.5:1.4:0.1, (sample 2) 4:1.4:2.5:0.1, (sample 3) 4:1.8:1.8:0.4 and (sample 4) 4:1.3:1.3:1.3. | ||
 | ||
| Fig. 5 Quantitative distribution of natNd among the separated complexes depending on the composition of the samples. LnT:xTPAEN:yNM:zDTPA (sample 1) 4:2.5:1.4:0.1, (sample 2) 4:1.4:2.5:0.1, (sample 3) 4:1.8:1.8:0.4 and (sample 4) 4:1.3:1.3:1.3. | ||
From Table 7, it can be seen that the mass balance of natSm and natNd ranged from 95.9 to 109.5%, which shows that all the chelating molecules fully complexed the Ln.
For samples 1, 3 and 4, when the TPAEN ratio is equal or greater than these of NM and DTPA, natSm is preferentially complexed by TPAEN than by NM, namely 102%, 74% and 39% for natSm(TPAEN) compared to 0%, 25% and 22% for natSm(NM), as can be seen in Fig. 4. In the same samples, 0%, 6% and 39% correspond to natSm(DTPA). For sample 2, when the ratio of NM is 1.8 times higher than that of TPAEN, only 55% was complexed by NM while 45% of natSm was complexed by TPAEN and the DTPA complex was negligible. Regarding sample 4 containing equimolar ratios of the three chelating molecules, natSm is equally distributed under DTPA and TPAEN complexes, being 39% while only 22% is involved in NM complexation.
For all the samples, natNd is preferentially complexed by TPAEN than NM and DTPA, as can be seen in Fig. 5. In sample 2, when the ratio of NM is 1.8 times higher than this of TPAEN, 76% of natNd remains complexed by TPAEN against 27% by NM and no DTPA complex was formed. Regarding sample 4 containing equimolar ratios of the three chelating molecules, natNd is predominantly complexed by TPAEN (76%) while only around 10% by NM and DTPA.
Overall, when the molecules are present in equimolar ratios (sample 4), TPAEN and DTPA show similar affinity towards natSm and higher than this of NM, while TPAEN exhibits much higher affinity for natNd, reflecting the selectivity of each molecule for the two Ln.
The Ln quantitative distribution among the different complexes calculated for experiments carried out in duplicate for the three separation formats was similar with an RSD < 5%, showing the robustness of the developed analytical method.
K for stepwise reactions and logβ for cumulative reactions) are expressed relative to the totally ionized form of the ligands, e.g. DTPA5− where all the carboxylic acid functions (Fig. 1a) are deprotonated. It assures that all constants can be compared. As the acidity of NM is not known, only conditional complexation constants cK, without taking into account the acidity of the functional groups, can be determined and compared to this obtained with the other molecules.
Taking into account the data determined in the previous part, the cK values were calculated for the complexes formed with the three chelating molecules at pH = 1.2 by applying eqn (10), taking natSm(CM) as an example and eqn (10.1) and (10.2) for the calculation of the free proportion of natSm and CM.
 | (10) |
| [natSmfree] = [Smtotal] − ([Sm(TPAEN)] + [Sm(NM)] + [Sm(DTPA)]) | (10.1) |
| [CMfree] = [CMtotal] − ([Sm(CM)] + [Nd(CM)]) | (10.2) |
In our case, the complexation between the Ln and CM is mostly total, rendering impossible the determination of the thermodynamic complexation constants using the usual representation, according to eqn (11).
 | (11) |
As [Ln]free tends to nil at high CM concentration, the slope of eqn (11) cannot be used.
Nevertheless, the ratio between conditional complexation constants can be used to represent the affinity of one CM towards one Ln. Affinity (A) is the degree to which a substance tends to combine with another, that is, the ratio of conditional complexation constants (cK) between complexes of elements from different groups (inter-groups) or elements within the same group (intra-group). The affinity of one molecule for natNd and natSm, as inter-group elements is represented using eqn (12).
 | (12) |
In agreement with the trends observed in the previous part, TPAEN presents the highest affinity for natSm compared to this of NM and DTPA, followed by NM having a slightly higher affinity than DTPA (ca. 0.4 times). TPAEN exhibits also the highest affinity for natNd with a factor of 25 in relation to NM and 189 when compared to the affinity of DTPA. Finally, NM shows higher affinity for natNd than DTPA (ca. 7 times). As can also be seen from the inter-group data, all the CMs exhibit higher affinity for natNd than for natSm, with a factor of 830 for TPAEN, 324 for NM and 19 for DTPA (Table 8). The selectivity and affinity of TPAEN for natSm and natNd follow the same trends observed in the literature, in which this molecule was identified as a potential back-extraction agent for the selective partitioning of Am from nuclear fuel solutions.11 Our method allowed us to determine the affinity and selectivity of NM towards both Ln simultaneously, leading to the conclusion that this latter could be less performant for selective back extraction of Am.
| Intra-group | natSm | natNd |
|---|---|---|
| A TPAEN–NM | 9.9 | 25.3 |
| A TPAEN–DTPA | 4.3 | 188.5 |
| A NM–DTPA | 0.4 | 7.4 |
| Inter-groups | natNd/natSm |
|---|---|
| A TPAEN | 830.7 |
| A NM | 324.3 |
| A DTPA | 19.0 |
A preliminary study has also been carried out to assess the complexation power of CM for natSm and natNd by increasing the pH of the contact solution, involving different degrees of CM deprotonation according to their pKas that are known for DTPA54 and TPAEN.14
Very good mass balance, between 79 and 106% for both natLn, was obtained for the different pH. The quantitative distribution of the natLn among the different complexes in samples containing an equimolar ratio of CM compared to natSm and natNd is shown in Fig. 6. The results were obtained by applying eqn (9).
 | ||
| Fig. 6 Quantitative distribution of natNd and natSm among the complexes separated by HILIC-ICPMS under nano-flow conditions reported in Table 4, for two replicates. Sample 4 (LnT:xTPAEN:yNM:zDTPA = 4:1.3:1.3:1.3) was prepared at different pH. | ||
The results obtained for pH ≈ 1 were consistent with the results presented in Fig. 4 and 5. Whatever the pH, TPAEN exhibits the highest complexation ability towards natNd, with 82% formation rate of natNd(TPAEN) at pH ≈ 1, which decreased to 65% for pH between 5 and 10. In contrast, the pH seems to have no influence on the NM complexation ability of natNd, since the proportion of natNd(NM) ranged between 15 and 19% whatever the pH. The highest formation rate of natSm(TPAEN) was 43% at pH ≈ 5 but with small variations for the other pH, between 33 and 40%. The same trend was observed for the natSm(DTPA) complex. The NM complexation ability of natSm was less efficient than those of TPAEN and DTPA whatever the pH, with 26% of natSm(NM) formation rate at pH ≈ 1, which undergoes a subsequent decrease to 9% when the pH increased up to 10.
These results show that despite the increase in the pH, involving the deprotonation of the CMs, the trends observed regarding the distribution of the complexes remain similar, with the highest affinity and selectivity of TPAEN for natNd.
5. Conclusion
The objective of the present work was to develop a dedicated method to determine on-line and in a single step the selectivity and the affinity of two hydrophilic synthesized molecules towards natNd and natSm used as analogues of Am and Cm, in comparison with DTPA as a reference molecule. The analytical strategy was based on the simultaneous coupling of HILIC to ESIMS and ICPMS combined with the online quantification of natNd and natSm among the separated complexes by specific isotopic dilution.The selectivity of several polar stationary phases of commercial columns in conventional format was evaluated and the composition of the mobile phase was adjusted to separate the Ln complexes formed with the three chelating molecules, leading to the selection of the ACQUITY UPLC BEH amide column and a mobile phase made of 74/26 (v/v) ACN/H2O, containing 0.5% (v/v) formic acid and 15 mmol L−1 NH4CH3CO2. The separation downscaling from conventional to nano-flow format allowed us to reduce 120 times the mass of Ln and 150 times the chelating molecules involved in one analysis, as well as a reduction of the effluent volumes by a factor of 1000. This latter format also allowed a significant reduction of chelating molecules and Ln involved in the analysis compared to classical solution chemistry approaches.
The quantification method developed and validated by SID allowed us to quantify online the separated Ln(CM) complexes simultaneously identified by ESIMS, in a single analysis. By applying our approach, the selectivity and the affinity of each chelating molecule for natNd and natSm were determined based on the sample containing stoichiometric ratios of TPAEN, NM and DTPA in competition towards the Ln complexation. In particular, TPAEN exhibits affinity for natNd 830 times higher than for natSm, which is consistent with the selection of TPAEN as a potential selective back-extraction agent in treatment processes dedicated to selective Am partitioning. We could also determine in the same analysis the selectivity and the affinity of NM for natNd and natSm.
This approach can be extended to the online evaluation of the affinity/selectivity of these chelating molecules for the actinides (Pu, Am, Cm…) because of similar physicochemical properties of Ln and An, and by means of an instrumental platform integrated in glove boxes. Moreover, it can be implemented for the screening of the affinity/selectivity properties of various classes of chelating molecules towards elements of interest in the fields of energy, toxicology and environment.
Author contributions
Marina Amaral Saraiva: conceptualization, methodology, investigation, validation, writing – original draft, visualization. Pascal E. Reiller: methodology, validation, writing – review & editing. Cécile Marie: validation, resources – review & editing. Carole Bresson: conceptualization, methodology, validation, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to acknowledge the Nuclear Cycle program (DES-DPE-CYN/PRATA) from the Energy Division of the CEA, the French Alternative Energies and Atomic Energy Commission, for financial support. The authors thank also Hélène Isnard and Sébastien Mialle (DES/ISAS/DRMP/SPC/LANIE) for their help in the qualification and quantification of spike solutions by TIMS.References
- C. Poinssot, C. Rostaing, S. Greandjean and B. Boullis, Recycling the actinides, the cornerstone of any sustainable nuclear fuel cycles, Procedia Chem., 2012, 7, 349–357, DOI:10.1016/j.proche.2012.10.055.
- P. Matveev, P. K. Mohapatra, S. N. Kalmykov and V. Petrov, Solvent extraction systems for mutual separation of Am(III) and Cm(III) from nitric acid solutions. A review of recent state-of-the-art, Solvent Extr. Ion Exch., 2020, 39(7), 679–713, DOI:10.1080/07366299.2020.1856998.
- S. Gracia, G. Arrachart, C. Marie, S. Chapron, M. Miguirditchian and S. Pellet-Rostaing, Separation of Am(III) by solvent extraction using water-soluble H4tpaen derivatives, Tetrahedron, 2015, 71(33), 5321–5336, DOI:10.1016/j.tet.2015.06.015.
- P. Weßling, M. Maag, G. Baruth, T. Sittel, F. S. Sauerwein, A. Wilden, G. Modolo, A. Geist and P. J. Panak, Complexation and Extraction Studies of Trivalent Actinides and Lanthanides with Water-Soluble and CHON-Compatible Ligands for the Selective Extraction of Americium, Inorg. Chem., 2022, 61(44), 17719–17729, DOI:10.1021/acs.inorgchem.2c02871.
- Y. Wang, G. J.-P. Deblonde and R. J. Abergel, Hydroxypyridinone Derivatives: A Low-pH Alternative to Polyaminocarboxylates for TALSPEAK-like Separation of Trivalent Actinides from Lanthanides, ACS Omega, 2020, 5, 12996–13005, DOI:10.1021/acsomega.0c00873.
- E. Macerata, E. Mossini, S. Scaravaggi, M. Mariani, A. Mele, W. Panzeri, N. Boubals, L. Berthon, M.-C. Charbonnel, F. Sansone, A. Arduini and A. Casnati, Hydrophilic Clicked 2,6-Bis-triazolyl-pyridines Endowed with High Actinide Selectivity and Radiochemical Stability: Toward a Closed Nuclear Fuel Cycle, J. Am. Chem. Soc., 2016, 138, 7232–7235, DOI:10.1021/jacs.6b03106.
- C. Adam, B. B. Beele, A. Geist, U. Müllich, P. Kaden and P. J. Panak, NMR and TRLFS studies of Ln(iii) and An(iii) C5-BPP complexes, Chem. Sci., 2015, 6(2), 1548–1561, 10.1039/C4SC03103B.
- T. S. Grimes, C. R. Heathman, S. J. Popova, A. S. Ivanov, S. Roy, V. S. Bryantsev and P. R. Zalupski, Influence of a Heterocyclic Nitrogen-Donor Group on the Coordination of Trivalent Actinides and Lanthanides by Aminopolycarboxylate Complexants, Inorg. Chem., 2018, 57(3), 1373–1385, DOI:10.1021/acs.inorgchem.7b02792.
- M. Audras, L. Berthon, C. Berthon, D. Guillaumont, T. Dumas, M.-C. Illy, N. Martin, I. Zilbermann, Y. Moiseev, Y. B. Eliyahu, A. Bettelheim, S. Cammelli, C. Hennig and P. Moisy, Structural Characterization of Am(III) and Pu(III)-DOTA Complexes, Inorg. Chem., 2017, 56, 12248–12259, DOI:10.1021/acs.inorgchem.7b01666.
- C. Moulin, B. Amekraz, S. Colette, D. Doizi, C. Jacopin, C. Lamouroux and G. Plancque, Electrospray mass spectrometry for actinides and lanthanide speciation, J. Alloys Compd., 2006, 408–412, 1242–1245, DOI:10.1016/j.jallcom.2005.04.177.
- N. Boubals, C. Wagner, T. Dumas, L. Chanèac, G. Manie, P. Kaufholz, C. Marie, P. J. Panak, G. Modolo, A. Geist and P. Guilbaud, Complexation of Actinide(III) and Lanthanide(III) with H4TPAEN for a Separation of Americium from Curium and Lanthanides, Inorg. Chem., 2017, 56(14), 7861–7869, DOI:10.1021/acs.inorgchem.7b00603.
- C. Rostaing, C. Poinssot, D. Warin, P. Baron and B. Lorraina, Development and Validation of the EXAm Separation Process for Single Am Recycling, Procedia Chem., 2012, 7, 367–373, DOI:10.1016/j.proche.2012.10.057.
- E. Blanchard, A. Nonell, F. Chartier, A. Rincel and C. Bresson, Evaluation of superficially and fully porous particles for HILIC separation of lanthanide–polyaminocarboxylic species and simultaneous coupling to ESIMS and ICPMS, RSC Adv., 2018, 8(44), 24760–24772, 10.1039/C8RA02961J.
- N. Chatterton, Y. Bretonnière, J. Pécaut and M. Mazzanti, An Efficient Design for the Rigid Assembly of Four Bidentate Chromophores in Water-Stable Highly Luminescent Lanthanide Complexes, Angew. Chem., Int. Ed., 2005, 44(46), 7595–7598, DOI:10.1002/anie.200502231.
- J. Borrini, A. F. Reguillon, M. Lemaire, S. Gracia, G. Arrachart, G. Bernier, X. Hérès, C. Hill and S. Pellet-Rostaing, Water Soluble PDCA Derivatives for Selective Ln(III)/An(III) and Am(III)/Cm(III) Separation, Solvent Extr. Ion Exch., 2015, 33(3), 224–235, DOI:10.1080/07366299.2014.974449.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85(22), 3533–3539, DOI:10.1021/ja00905a001.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Study of Inter Atomic Distances in Halides and Chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767, DOI:10.1107/S0567739476001551.
- K. L. Nash, A review of the basic chemistry and recent developments in trivalent f–elements separation, Solvent Extr. Ion Exch., 1993, 729–768, DOI:10.1080/07366299308918184.
- G. T. Seaborg, Overview of the Actinide and Lanthanide (the f) Elements, Radiochim. Acta, 1993, 61, 115–122, DOI:10.1524/ract.1993.61.34.115.
- J. Künnemeyer, L. Terborg, S. Nowak, A. Scheffer, L. Telgmann, F. Tokmak, A. Günsel, G. Wiesmüller, S. Reichelt and U. Karst, Speciation Analysis of Gadolinium-Based MRI Contrast Agents in Blood Plasma by Hydrophilic Interaction Chromatography/Electrospray Mass Spectrometry, Anal. Chem., 2008, 80(21), 8163–8170, DOI:10.1021/ac801264j.
- A. Maia, P. Silva, C. Fernandes, A. Silva, A. L. Barros, D. C. Soares and G. Ramaldes, Chemometric-Assisted Hydrophilic Interaction Chromatographic Method for the Determination of Gadolinium-Based Magnetic Resonance Imaging Contrast Agent in Liposomes, J. Braz. Chem. Soc., 2018, 29(11), 2426–2440, DOI:10.21577/0103-5053.20180120.
- L. Schlatt, A. Köhrer, C. Factor, P. Robert, M. Rasschaert, M. Sperling and U. K. Mild, Dissolution/Recomplexation Strategy for Speciation Analysis of Gadolinium from MR Contrast Agents in Bone Tissues by Means of HPLC-ICP-MS, Anal. Chem., 2021, 93(33), 11398–11405, DOI:10.1021/acs.analchem.1c01100.
- S. Feng, S. Shen, Y. Yao, M. Liang, Y. Chen and H. Liu, Comparison of different analytical methods for speciation of seven gadolinium‐based magnetic resonance imaging contrast agents and the applications in wastewater and whole blood, J. Sep. Sci., 2022, 46(4), 2200575, DOI:10.1002/jssc.202200575.
- C. S. Kesava Raju, A. Cossmer, H. Scharf, U. Panne and D. Lück, Speciation of gadolinium based MRI contrast agents in environmental water samples using hydrophilic interaction chromatography hyphenated with inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2010, 25(1), 55–56, 10.1039/B919959D.
- M. Horstmann, R. Gonzalez de Vega, D. P. Bishop, U. Karst, P. A. Doble and D. Clases, Determination of gadolinium MRI contrast agents in fresh and oceanic waters of Australia employing micro-solid phase extraction, HILIC-ICP-MS and bandpass mass filtering, J. Anal. At. Spectrom., 2021, 36(4), 767–775, 10.1039/D0JA00493F.
- S. Okabayashi, L. Kawane, N. Y. Mrabawani, T. Iwai, T. Narukawa, M. Tsuboi and K. Chiba, Speciation analysis of Gadolinium-based contrast agents using aqueous eluent-hydrophilic interaction liquid chromatography hyphenated with inductively coupled plasma-mass spectrometry, Talanta, 2021, 222, 121531, DOI:10.1016/j.talanta.2020.121531.
- M. Birka, C. A. Wehe, L. Telgmann, M. Sperling and U. Karst, Sensitive quantification of gadolinium-based magnetic resonance imaging contrast agents in surface waters using hydrophilic interaction liquid chromatography and inductively coupled plasma sector field mass spectrometry, J. Chromatogr. A, 2013, 1308, 125–131, DOI:10.1016/j.chroma.2013.08.017.
- M. Birka, C. A. Wehe, O. Hachmöller, M. Sperling and U. Karst, Tracing gadolinium-based contrast agents from surface water to drinking water by means of speciation analysis, J. Chromatogr. A, 2016, 1440, 105–111, DOI:10.1016/j.chroma.2016.02.050.
- L. Telgmann, C. A. Wehe, M. Birka, J. Künnemeyer, S. Nowak, M. Sperling and U. Karst, Speciation and Isotope Dilution Analysis of Gadolinium-Based Contrast Agents in Wastewater, Environ. Sci. Technol., 2012, 46(21), 11929–11936, DOI:10.1021/es301981z.
- U. Lindner, J. Lingott, S. Richter, N. Jakubowski and U. Panne, Speciation of gadolinium in surface water samples and plants by hydrophilic interaction chromatography hyphenated with inductively coupled plasma mass spectrometry, Anal. Bioanal. Chem., 2013, 405(6), 1865–1873, DOI:10.1007/s00216-012-6643-x.
- L. Beuvier, C. Bresson, A. Nonell, L. Vio, N. Henry, V. Pichon and F. Chartier, Simple separation and characterization of lanthanide–polyaminocarboxylic acid complexes by HILIC ESI-MS, RSC Adv., 2015, 5(113), 92858–92868, 10.1039/C5RA16078B.
- B. Buszewski and S. Noga, Hydrophilic interaction liquid chromatography (HILIC)—a powerful separation technique, Anal. Bioanal. Chem., 2012, 402(1), 231–247, DOI:10.1007/s00216-011-5308-5.
- J. I. G. Alonso and P. Rodriguez-Gonzalez, Isotope dilution mass spectrometry, Royal Society of Chemistry, Cambridge, 2013, ch. 1, pp. 1–38 Search PubMed.
- P. Rodríguez-González and J. I. García Alonso, Isotope Dilution Mass Spectrometry, Encyclopedia of Analytical Science, Elsevier, 3rd edn, 2019, pp. 411–420, DOI:10.1016/B978-0-12-409547-2.14387-2.
- S. Schuhmann and J. A. Philpotts, Chapter 37G Mass-spectrometric stable-isotope-dilution analysis for lanthanides in geochemical materials, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1979, vol. 4, pp. 471–481, DOI:10.1016/S0168-1273(79)04016-2.
- J. G. Alonso, Determination of fission products and actinides by inductively coupled plasma-mass spectrometry using isotope dilution analysis: A study of random and systematic errors, Anal. Chim. Acta, 1995, 312(1), 57–78, DOI:10.1016/0003-2670(95)00199-A.
- F. Chartier, M. Aubert, M. Salmon, M. Tabarant and B. H. Tran, Determination of erbium in nuclear fuels by isotope dilution thermal ionization mass spectrometry and glow discharge mass spectrometry, J. Anal. At. Spectrom., 1999, 14(9), 1461–1465, 10.1039/a902318f.
- M. Bourgeois, H. Isnard, A. Gourgiotis, G. Stadelmann, C. Gautier, S. Mialle, A. Nonell and F. Chartier, Sm isotope composition and Sm/Eu ratio determination in an irradiated 153Eu sample by ion exchange chromatography-quadrupole inductively coupled plasma mass spectrometry combined with double spike isotope dilution technique, J. Anal. At. Spectrom., 2011, 26(8), 1660, 10.1039/c1ja10070j.
- A. Beaumais, A. Nonell, C. Caussignac, S. Mialle, G. Stadelmann, M. Janin, H. Isnard, M. Aubert, T. Vercouter and F. Chartier, Determination of the 144Ce/238U atomic ratio in spent nuclear fuel using double spike isotope dilution mass spectrometry, J. Anal. At. Spectrom., 2022, 37(6), 1288–1297, 10.1039/D2JA00052K.
- A. Gaffney, Guideline on Isotope Dilution Mass Spectrometry, Lawrence Livermore National Laboratory, United States, 2017, DOI:10.2172/1358328.
- L. R. Snyder, J. J. Kirkland, and J. W. Dolan, Introduction to Modern Liquid Chromatography, John Wiley & Sons, Inc., 2009, p. 1, DOI:10.1002/9780470508183.
- USEPA, U.S, Environmental Protection Agency, Method 6800: Elemental and Speciated Isotope Dilution Mass Spectrometry, Washington, DC, 2014, https://www.epa.gov/sites/production/files/2015-12/documents/6800.pdf, accessed: 2 August 2023 Search PubMed.
- H. Isnard, R. Brennetot, C. Caussignac, N. Caussignac and F. Chartier, Investigations for determination of Gd and Sm isotopic compositions in spent nuclear fuels samples by MC ICPMS, Int. J. Mass Spectrom., 2005, 246(1–3), 66–73, DOI:10.1016/j.ijms.2005.08.008.
- F. Chartier, H. Isnard, J. P. Degros, A. L. Faure and C. Fréchou, Application of the isotope dilution technique for 93Zr determination in an irradiated cladding material by multiple collector-inductively coupled plasma mass spectrometry, Int. J. Mass Spectrom., 2008, 270(3), 127–133, DOI:10.1016/j.ijms.2007.12.005.
- J. Meija, T. B. Coplen, M. Berglund, W. A. Brand, P. D. Bièvre, M. Gröning, N. E. Holden, J. Irrgeher, R. D. Loss, T. Walczyk and T. Prohaska, IUPAC - Isotopic compositions of the elements 2013 (IUPAC Technical Report), Pure Appl. Chem., 2016, 88(3), 293–306, DOI:10.1515/pac-2015-0503.
- M. Thompson, S. L. R. Ellison and R. Wood, Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report), Pure Appl. Chem., 2002, 74(5), 835–855, DOI:10.1351/pac200274050835.
- NF V03-110-National standards and national normative documents, AFNOR, 2010, https://www.afnor.org/standard/nfv03-110, accessed: 2 August 2023 Search PubMed.
- K. A. Gschneidner, L. Eyring, G. H. Lander and G. R. Choppin, Handbook on the Physics and Chemistry of Rare Earths, Chemistry Elsevier, 1994, vol. 18 Search PubMed.
- P. Hemström and K. Irgum, Hydrophilic interaction chromatography, J. Sep. Sci., 2006, 29(12), 1784–1821, DOI:10.1002/jssc.200600199.
- P. Jandera, Stationary and mobile phases in hydrophilic interaction chromatography: a review, Anal. Chim. Acta, 2011, 692(1–2), 1–25, DOI:10.1016/j.aca.2011.02.047.
- A. Leclercq, A. Nonell, J. L. T. Torró, C. Bresson, L. Vio, T. Vercouter and F. Chartier, Introduction of organic/hydro-organic matrices in inductively coupled plasma optical emission spectrometry and mass spectrometry: A tutorial review. Part I. Theoretical considerations, Anal. Chim. Acta, 2015, 885, 33–56, DOI:10.1016/j.aca.2015.03.049.
- A. Leclercq, A. Nonell, J. L. T. Torró, C. Bresson, L. Vio, T. Vercouter and F. Chartier, Introduction of organic/hydro-organic matrices in inductively coupled plasma optical emission spectrometry and mass spectrometry: A tutorial review. Part II. Practical considerations, Anal. Chim. Acta, 2015, 885, 57–91, DOI:10.1016/j.aca.2015.04.039.
- H. P. Nguyen and K. A. Schug, The advantages of ESI-MS detection in conjunction with HILIC mode separations: Fundamentals and applications, J. Sep. Sci., 2008, 31(9), 1465–1480, DOI:10.1002/jssc.200700630.
- R. Smith, A. Martell and R. Motekaitis, NIST Standard Reference database 46, NIST Critically Selected Stability Constants of Metal Complexes Database Version 8.0, U.S. Department of Commerce, 2004 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |