
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum-group elements and gold in silver coinage and the issue of salt cementation†
Francis
Albarède
 *,
Chloé
Malod-Dognin
and
Philippe
Télouk
*,
Chloé
Malod-Dognin
and
Philippe
Télouk
Ecole Normale Supérieure de Lyon, Université de Lyon, France. E-mail: albarede@ens-lyon.fr
First published on 14th September 2023
Abstract
It has been proposed that gold purification by cementation could account for the low gold content of ancient Greek coinage from Attica and the Cyclades. In order to place new constraints on this suggestion, the concentrations of platinum-group elements (PGEs) and gold have been measured in 72 silver coins mostly from the Greek Archaic and Classical periods, but also from Rome, India, medieval Europe, and colonial Spanish Americas, by inductively coupled plasma mass spectrometry. A novel technique allowing these concentrations to be determined in silver coins is described. Variations are consistent with element position in the periodic table. The volatile elements Rh and Os are commonly at or below the detection level, which may reflect evaporation during smelting and cupellation. Ruthenium and Ir, which binary phase equilibrium experiments show to be insoluble in solid silver and gold, and soluble Pd and Pt, show variations in coinage consistent with these properties. The dichotomy of Ir/Au ratios is not consistent with Ir loss in gold during salt cementation (parting) and is better explained by the contrast between Au-rich and Au-poor ore districts. This contrast is suggested to reflect either regional differences or the variability of conditions during ore genesis, such as hydrothermal solution chlorinity.
1 Introduction
The distribution of platinum-group elements (PGEs, Fig. 1) in silver coins and artefacts is largely unknown, essentially on account of their very low concentrations. Gold distribution, in contrast, is better understood. Historically, it has been suggested that the main sources of Greek silver, a metal that has been minted since the last quarter of the 6th century BCE, were located in the fabled mines of Lavrion next to Athens, in Thrace, and in Macedonia (modern northern Greece) with the highly productive mines of Mount Pangaion, Thasos, and the Rhodope. The latter group of mines is often described as producing Au-rich silver with respect to Lavrion.1–3 This suggestion raises a number of questions, in particular about how the construction of the Athenian fleet that repelled the second Persian invasion in Salamis (480 BCE) was financed. Finally, gold-silver parting from natural electrum by salt cementation (admixing with NaCl, clay, and urine as a source of ammonia)4–6 is thought to have been used around the Mediterranean as a technique for retrieving silver, but this hypothesis is not generally agreed upon. Some authors consider cementation a plausible practice because the gold content of Athens' silver coinage from the Classical period (480–330 BCE) is distinctly lower than the content of the coins minted in other regions, such as Macedonia and Thrace.1–3 A difficulty with this interpretation is that producing silver from gold-silver alloys comes at a high financial and energetic cost, and that the amount of silver obtained by this process is not expected to be much greater than the overall gold output, which is certainly small. | ||
| Fig. 1 Position of platinum-group elements (in orange) and related Re and Au (in pink) in the periodic table. | ||
The abundances of one of the PGEs, iridium, are better known because Ir concentrations can be determined by instrumental neutron activation. Wood et al.7 suggested that Au and Ir concentrations in silver coins could be used, especially in conjunction with lead isotope compositions, as a provenance tool for silver coins and artefacts from the first millennium BCE and for the Sassanian and Byzantine empire. Pernicka6 nevertheless argue that gold parting confuses the Ir signal and therefore assessed that Wood et al.'s arguments are unfounded. The cementation problem therefore needs to be addressed through new observations on silver coinage. Because this dispute has important consequences in archeology, it warrants a more in-depth study of PGE and Au traces in ancient silver.
The purpose of the present work is to explore the potential of PGEs and Au as markers of silver provenance and metallurgy. The sources of silver can be related to relatively low-temperature and low-sulfur epithermal ore deposits typically associated with Au.8 In contrast Au and Cu are often precipitated from the higher-sulfur, higher-temperature hydrothermal fluids that are expelled from felsic magmas in subduction zones and continental collision sutures (porphyry coppers). The preference of the elements in question for the melt phase decreases in the order Re > Au > Pd > Pt > Rh > Ir ≥ Ru ≥ Os.9–13 Basalts and, in general, the whole oceanic and continental crust therefore are enriched in Re over Os with respect to their mantle source, which makes Os enriched in radiogenic 187Os. Platinum, Pd, and Au are enriched over the rest of the PGEs in Cu sulfides.14 Platinum-group elements and gold's boiling temperatures exceed 2800 °C, but Ru and Os are easily oxidized as RuO4 and OsO4, which evaporate at ca. 130 °C.15 The latter two elements will therefore be easily lost during silver smelting. The relative abundances of PGEs and Au hence hold some potential for identifying both silver provenance and metallurgic processes.
Overall, literature on PGEs in silver coins and artefacts is sparse and data quality is variable. Gold and iridium analysis by neutron activation analysis were successful for some coins and artefacts7,16–19 but many of them remained below detection limits.20 Early PGE analysis by LA-ICP-MS were apparently not successful enough to be reported.21 Over the last two decades, laser-ablation ICP-MS allowed some PGEs to be measured on depth profiles in silver coins22 but the detection limit in silver coins, in general on the order of a few tenths or a few hundredths of μg g−1, seems to be a limitation and only few studies, if any, have been dedicated to these elements.
Here we describe a new protocol for the measurement of Au and PGEs in silver and apply it to 72 coins of different ages (Ancient Greece, Rome, Gaul, 16–18th century AD) and from different localities (eastern and western Mediterranean Europe, South America, Mexico) with the expectation that the new data will contribute to the issue of silver produced during gold parting.
2 Material and methods
2.1 Chemicals and reagents
Water was purified with a Millipore Gradient water purification system (18.2 MΩ cm−1).The hydrochloric 37% laboratory reagent grade and nitric >68% trace element grade acids were obtained from Fischer Scientific and home-distilled. The Thermo Scientific ICAP-RQ was tuned with Tune solution, Tune B (1 ng g−1 Li, Co, In, Ba, Ce, Bi, U) in 2% HNO3 and 0.5% HCl. Single element standard solutions with 1000 μg mL−1 concentrations were obtained from Alpha Aesar. Abrasive paper was from Presi, type M, P320.
2.2 Sample preparation
Sample preparation is described in Table 1. An aliquot of 200 mg was first cut with pliers and polished with abrasive paper in order to remove impurities and metal oxidation. Fragments were cleaned with distilled water, air dried, weighed, and dissolved in PFA Savillex beakers in 5 mL distilled 8 M HNO3. The beaker was closed and heated overnight on a hotplate at 120 °C. The orange fumes of nitrous oxides disappeared within a few hours, indicating that digestion was complete, except for Pt, Au, and other impurities which occasionally partly remained insoluble in nitric acid. The beaker was finally ultrasonicated for 30 minutes to complete sample digestion. After centrifugation at 8000 rpm for 10 minutes, the supernatant was set aside and the residue dissolved in 3 mL aqua regia prepared from concentrated, distilled HNO3 and HCl, in the proportions 1/3 and 2/3, respectively. This step produced a solution referred to as ‘solution 1’ which was used for ICP-MS elemental analysis.| Step | Reagent | Volume | Time | Hotplate |
|---|---|---|---|---|
| Cutting coins (200 mg with pliers) | ||||
| Polishing | ||||
| Cleaning | Distilled water | |||
| Weighing | ||||
| Digestion | 8 M HNO3 | 5 mL | Overnight | 120 °C |
| Ultrasonic bath (optional) | 30 min | |||
| Centrifuge 8000 rpm | 10 min | |||
| Precipitate separation | ||||
| Aqua regia digestion: solution 1 | Aqua regia | 3 mL | Overnight | |
| Supernatant AgCl precipitation | 6 M HCl | 3 mL | ||
| Centrifuge 8000 rpm and separate AgCl | 10 min | |||
| Adding distilled H2O: solution 2 | Distilled water | 5 mL | ||
| Solutions 1 and 2 ICP-MS analyses | 0.5 M HNO3 with In |
The supernatant was treated with HCl to remove Ag. This step is necessary because, PGEs being ultra-traces, Ag must be removed to avoid polluting and clogging the instrument with metal and keep the beam stable. Three mL concentrated, distilled HCl were added to the nitric supernatant, which produced an abundant AgCl precipitate. The solution was centrifuged at 8000 rpm for 10 minutes and the supernatant transferred into a beaker containing 5 mL of deionized water, which brought the total volume to 12.5 mL. This second step produced ‘solution 2’. Because of the volatility of some elements, such as Ru and Os, solutions were not fully evaporated to maximize the yield of the chemistry. The yields of this procedure were determined by analyzing Alfa Aesar standard solutions combined to reproduce the composition of the samples. A full procedural yield of 100 ± 2% was achieved for Rh, Ir, Pd, and Pt. The yields for Ru and Os were smaller, 75% and 56%, respectively, and the missing fraction of these volatile elements seems to have been lost during the digestion stage. The Au yield was 61% with the missing Au likely being lost during AgCl precipitation. All the yields were reproducible within ∼10% and a correction was applied to the final results. The consistency between the 1-sigma coefficient of variation of Au concentrations (91%) is consistent with the coefficients for Pd and Pt (84 and 114%, respectively), showing that the applied correction is valid.
The total blanks of the procedure are reported in Table 2 and the instrumental noise (or background noise) is discussed below.
| Ru | Rh | Pd | Os | Ir | Pt | Au | |
|---|---|---|---|---|---|---|---|
| Chemistry blank (ng g−1) | 0.0000 | 0.0010 | 0.0015 | 0.0000 | 0.0000 | 0.0000 | 0.4209 |
| Instrument blank (ng g−1) | 0.0002 | 0.0003 | 0.0012 | 0.0039 | 0.0002 | 0.0012 | 0.0334 |
2.3 Instrumentation
Platinum-group elements and Au in both solutions 1 and 2 were analyzed by Q-ICP-MS using a Thermo Scientific ICAP-RQ quadrupole ICP-MS at the Laboratoire de Géologie de Lyon, Ecole Normale Supérieure de Lyon. The instrument was tuned using a Thermo Scientific solution and the parameters summarized in Table 3.| iCAP-RQ ICP MS operating conditions | |
|---|---|
| Instrument parameters | |
| RF power (W) | 1550 |
| Nebulizer gas flow (L min−1) | 1.05 |
| Auxiliary gas flow (L min−1) | 0.8 |
| Cooling gas flow (L min−1) | 14 |
| Hexapole bias (V) | −1.000198 |
| Collision cell gas (mL min−1) | He at 5 mL min−1 |
| CCT bias (V) | −20.49 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| Analytical parameters | |
| Acquisition mode | KED, peak jumping, simultaneous pulse count/analog detector |
| Dwell time/isotope (s) | System |
| Channel per mass | 0.01 |
| Number of repetitions/samples | 1 |
| Sample uptake/wash time (s) | 3 |
| Total acquisition time per sample | 120 |
2.4 Standard preparation
For instrument calibration, a bulk standard solution was prepared from Alfa Aesar stock solutions at 1 g l−1 for the 10 elements measured (PGEs and Au). Because some commercial standards are in HCl form, combining different standards may cause unwanted chloride precipitation. Each elemental solution therefore was evaporated and nitric acid added several times to remove any chloride traces. The solutions in this way transformed to the nitric form were then combined. The final standard solution contained all the elements in the same acid. Five solutions diluted at different concentrations were used to prepare a five-point calibration curve with the respective concentrations summarized in Table S1.†The monitored isotopes are listed in the ESI.† The kinetic-energy discrimination (KED) mode, which uses a collision cell filled with helium gas, reduces sensibility but minimizes spectral interferences. In order to reduce blanks, the mass spectrometer was first rinsed with 8 M distilled HNO3 for 20 minutes. The procedural blanks at this stage are given in Table 2. The sample/blank ratio, both for PGEs and Au, were higher than 100, which show that the blank can be neglected compared to the amounts of PGEs and Au processed.
All samples and standard solutions were spiked with a 2 ng g−1 In solution as an internal standard to correct for instrumental drift. The data were acquired and processed using the Thermo Scientific Qtegra Intelligent Scientific Data Solution Software.
Standard limits of detection (LD) are shown in Table 2. However, in order to evaluate data precision, and, critically, the effect of blank subtraction, the simple expressions derived in the ESI† were used. The error amplification factors (f) and final uncertainties were calculated for each element. Precisions better than 2% were achieved for Rh, Pd, Pt, and Au (Table 4). The relative errors on Os and Ir (53 and 33%, respectively) were rather large but, given that PGEs and Au concentrations typically vary over several orders of magnitude, the data remain informative. Finally, the relative errors on Ru and Re were on the order of 100‰ and the data only give an upper limit for the concentration (limit of detection). Adjusting sample size, as long as permitted by the separation protocol, and dilution may help minimize errors.
| Re | Ru | Os | Rh | Ir | Pd | Pt | Au | |
|---|---|---|---|---|---|---|---|---|
| a Blank signal > sample signal. b Limit of detection (in ng of element per g of silver). | ||||||||
| Blank ng g−1 | 26 | 12 | 148 | 7 | 24 | 4 | 32 | 1885 |
| Rel. error | 0.36 | 0.53 | 0.15 | 0.71 | 0.38 | 0.89 | 0.32 | 0.04 |
| Sample ng g−1 | 13 | 3.33 | 213 | 5908 | 77 | 45405 |
11462 |
1311140 |
| Rel. error | 0.5 | 1 | 0.12 | 0.024 | 0.21 | 0.01 | 0.02 | 0.002 |
|
||||||||
| Amplification factor | ||||||||
| f meas | −1.05a | −0.4a | 3.27 | 1.001 | 1.44 | 1 | 1.003 | 1.001 |
| f blk | −2.05a | −1.4a | 2.27 | 0.001 | 0.44 | 0 | 0.003 | 0.00 |
| σ n /n | 1.06 | 0.96 | 0.53 | 0.02 | 0.33 | 0.009 | 0.02 | 0.002 |
| LDb | n.d. | 0.02 | 0.02 | 0.02 | 0.04 | 0.02 | 0.05 | 1.4 |
2.5 Deconvolution
Potential isobaric interferences of Fe, Co, Ni, Cu, Zn argides (Ar compounds), and Mo on the mass spectrum of Ru, Rh, Pd, Ag, and Cd were evaluated by standard peak stripping (e.g., Albarede23 Section 5.1) using the abundance matrix shown in Table S2† and straightforward unconstrained least-square inversion. The contribution of minor Ar isotopes is ignored. The system is suitably over-determined which warrants a stable and precise inversion. When the calculated abundance of a particular argide was negative, its contributions was eliminated. The procedure was repeated for every argide and the final results suggest that the contribution of these compounds to the PGE contents can be safely neglected, as can the Mo contribution.2.6 Material
The 72 silver coins analyzed in this study were acquired on the market since 2009 by ENS collections for the purpose of technological development. Some samples were used (and previously damaged) in earlier studies by Desaulty et al.24 and for the ERC SILVER Project.25 All coins were acquired from dealers formally compliant with the code of ethics. The identifications provided by the sellers were carefully checked and verified.The coins with their type, origin, and age are listed in Tables S3 and S4† and represent a selection of coins from around the ancient Mediterranean world, a few samples from the Indian kingdoms such as Gandhara, and more modern coins from medieval and post-medieval Europe, as well as colonial Mexico and Peru. Pb and Ag isotopic data are taken from ref. 24–26.
3 Results and discussion
The PGE and Au data are reported in ESI Table S5.† In Fig. 2, the data are normalized to the PGE and Au abundances in the EH3 enstatite Sahara 97072 meteorite.27 The PGE abundances of the primitive terrestrial mantle are accounted for by a late veneer of impactors28 and we opted for a type of chondrite that has many properties in common with the Earth.29,30 With exceptions, the overall pattern is rather reproducible regardless of the origin and age of the coin. The range of variation is a factor of 14 for Ir, 8 for Ru, 5 for Pt, and about 3–4 for Rh, Pd, and Au. These variations are not obviously correlated with the analytical uncertainties. Fig. 2 shows that the EH3 chondrite-normalized PGEs and Au follow the common pattern of mantle-derived sulfides for Ru, Os, Rh, and Ir, but not for Pd, Pt, and Au. The most notable discrepancies are the normalized ratios Au/Pd > 1 and the Ru/Ir < 1 in silver coinage.
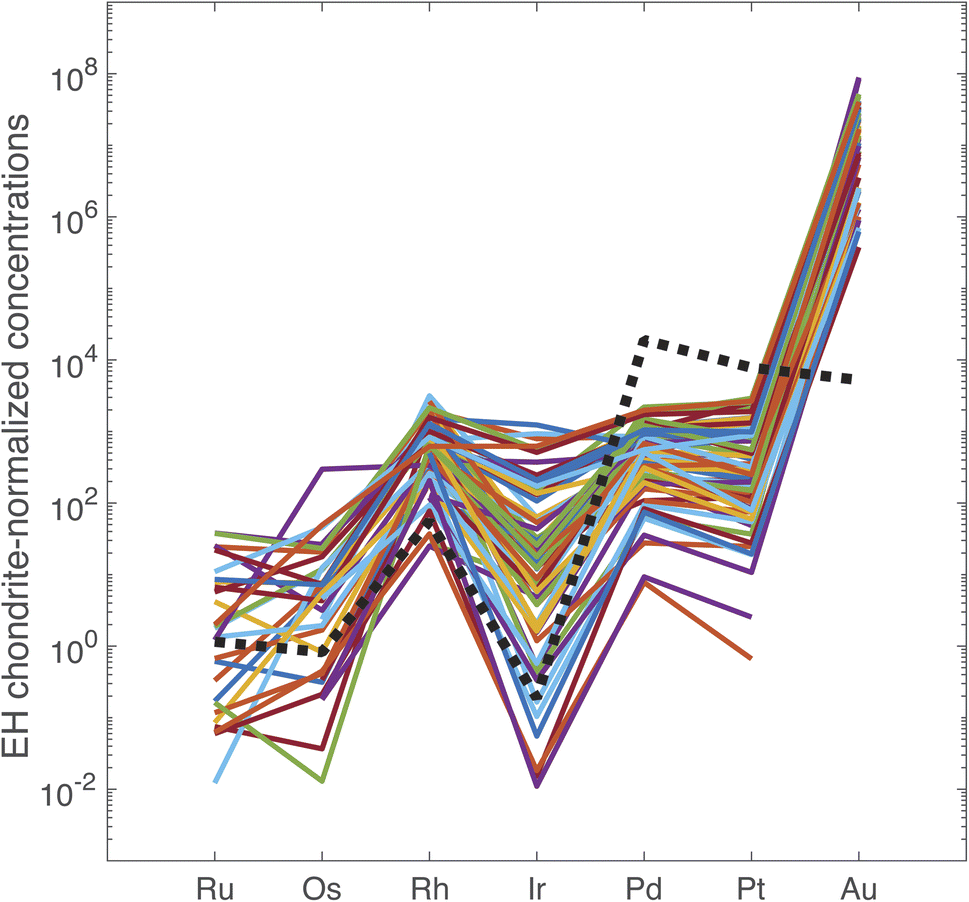 | ||
| Fig. 2 PGE concentrations of the 72 silver coins normalized to concentrations in EH chondrites. Note that the order of elements on the abscissa reflects the order of columns in the periodic table from the Fe group (Ru, Os), to the Co group (Rh, Ir), the Ni group (Pd, Pt), and the Cu group (Au). Dashed line: McCreedy Cu-rich sulfide from the Sudbury mine, Canada.14 | ||
Osmium concentrations are in general very low and often below detection limit. The perspective of using 187Os/188Os as a tracer is therefore unpromising. Osmium is most likely lost during smelting and cupellation. This is supported by the lack of a strong correlation between Os and other PGEs + Au.
Correlations between the logarithm of the different variables are summarized in Table 5 and the slopes in Table S6.† Palladium and Pt are strongly correlated (r = 0.93) (Fig. 3, bottom). Correlations between Au and Pt (0.83), Ir (0.72), and Pd (0.76) and between Ir and Pt (0.74) (Fig. 3, top), Ru (0.77), and Rh (0.71) are significantly weaker, but still significant at the 95% confidence level. Some of the correlation coefficients are high because of some data being exceptionally high or low (e.g., Ir–Rh, Au–Pt), notably those on the samples from colonial Spanish Americas which are particularly poor in Ir. Iridium seems to be constant at low Ru and only correlates with Ru when the abundance of the latter rises above the chondritic Ru abundance.
| Ru | Os | Rh | Ir | Pd | Pt | Au | |
|---|---|---|---|---|---|---|---|
| Ru | 1.00 | ||||||
| Os | 0.49 | 1.00 | |||||
| Rh | 0.45 | 0.24 | 1.00 | ||||
| Ir | 0.77 | 0.46 | 0.71 | 1.00 | |||
| Pd | 0.49 | 0.45 | 0.31 | 0.64 | 1.00 | ||
| Pt | 0.56 | 0.50 | 0.38 | 0.74 | 0.93 | 1.00 | |
| Au | 0.53 | 0.36 | 0.30 | 0.72 | 0.76 | 0.83 | 1.00 |
| CV% | 773 | 168 | 18 | 74 | 22 | 28 | 8 |
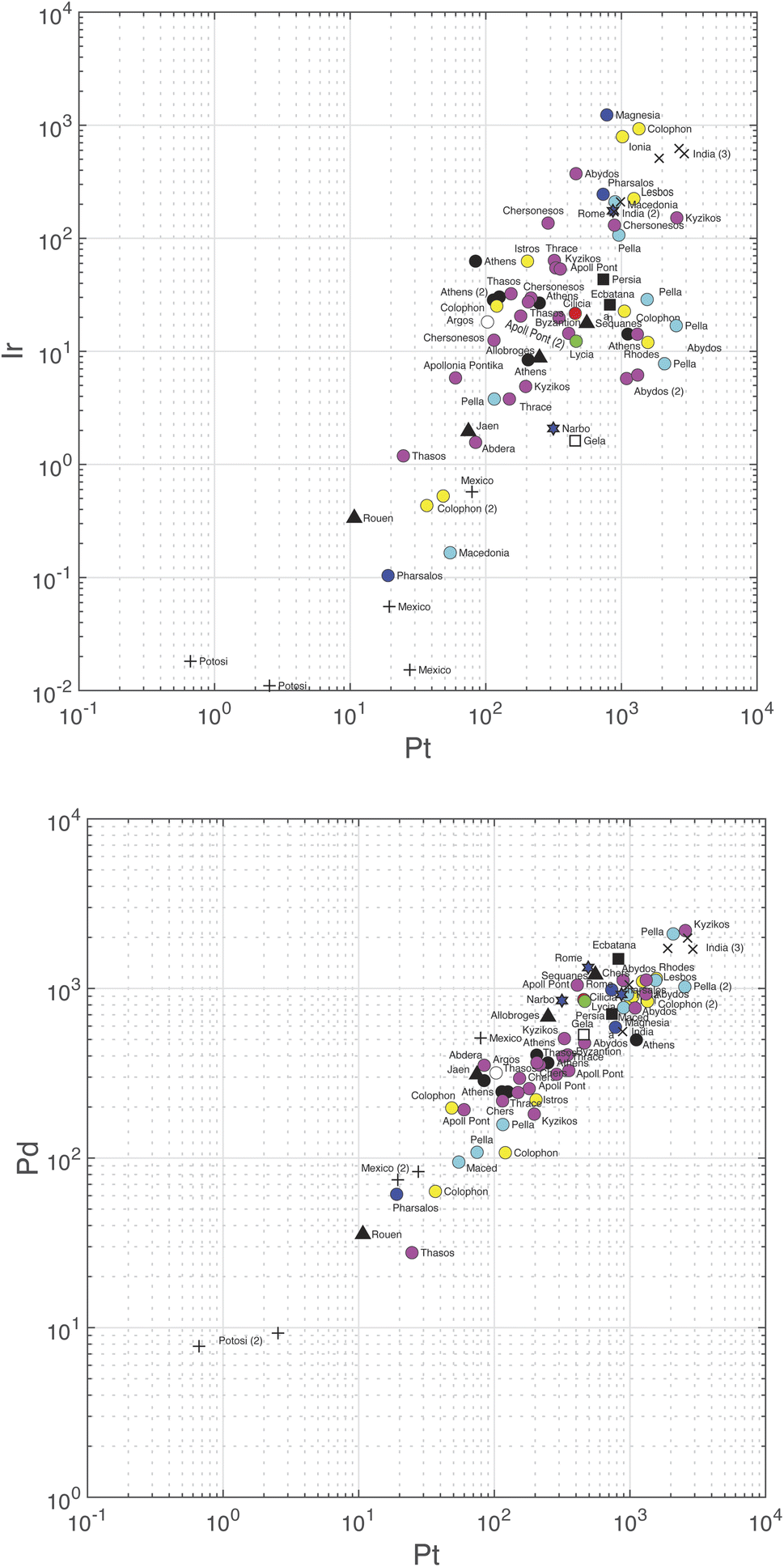 | ||
| Fig. 3 Plot of Pd (bottom) and Ir (top) concentrations vs. Pt. Note the different scales on the vertical axes. Color coding according to Table S3.† | ||
Gold is the least variable of all the elements (±100.08 or ±21%) followed by Rh (±52%), Pd, (±67%), and Pt (±89%). The strongest correlations observed, Pt vs. Pd (slope = 1.7), Pt vs. Au (slope = 1.6), and Ru vs. Ir (slope = 1.2) are defined with rather narrow uncertainties. The Pt/Pd ratios vary by a mere ∼30 percent, which shows the very coherent behavior of these two elements.
Binary phase Ag–M diagrams, where M stands for a PGE or Au, are reasonably well known from the metallurgical literature.31–39 Positive slopes of the liquidus and the solidus of Ag binary phase diagrams attest to solubility of PGEs in solid silver at high temperatures. This is the case of Au, Pd, and Pt, which, however, do not fractionate to a large extent between solid and liquid silver. In contrast, Ir and Rh form no solid solution with silver.
Binary diagrams between PGEs and Au are less well documented.40–43 As for silver, Pd and Ag do not fractionate between liquid and solid gold, while Pt is slightly more soluble in solid gold. Ir and Rh solubility has not recently been revisited but early experiments attest to the insolubility of these elements in both liquid and solid gold.44–46
Metallurgical data therefore show that PGEs react as pairs defined by their position in the periodic table: (Cu)–Ag–Au (group 11, a.k.a. coinage metals), (Ni)–Pd–Pt (group 10), and (Co)–Rh–Ir (group 9). Information on the (Fe)–Ru–Os (group 8) is scant beyond the fact that both elements are volatile. The Cu group is the least sensitive to redox conditions.
The present results are relevant to the conflicting interpretation of Ir concentrations in Sassanide and Byzantine coinage by Wood et al.,7 who support a silver source in the Taurus Mountain range of southern Anatolia, and Pernicka et al.,6 who claim that Ir variations reflect the effect of cementation. We have seen above that Pd and Pt are soluble in both silver and gold, whereas, as far as the still incomplete thermodynamics show, Ir and Rh are insoluble in either metal. Iridium should therefore be lost during gold parting and fractionated with respect to Au-soluble Pd and Pt. Given the high abundance of silver in silver coins, ratios such as Ir/Ag used by Wood et al.7 are, for all practical purposes, equivalent to Ir concentrations in weight percent. The remarkable dual trend observed in the present data when Ir/Au is plotted against Ir (Fig. 4) is essentially equivalent to Wood et al.'s7 Fig. 3 of log10(Au/Ir) vs. log10(Ag/Ir). A plot of log10(Rh/Au) vs. log10(Rh) (not shown) demonstrates similar relationships. The data show that Au-poor and Au-rich groups form at constant Ir contents. The gap visible in this plot is clearly created by a gap in Au concentrations and is independent of the Ir variations. These plots are just other pieces of evidence of the dual distribution of Au in Greek silver coinage and of a contrast between high-Au and low-Au metallogenic provinces. In these plots, Athenian coins form a well-defined Au-depleted upper group. Nevertheless, a few non-Athenian coins (Argos, Thasos, Jaen, medieval Rouen, colonial Mexico) belong to the same group of Au-poor silver as Athenian coins. Source Au depletion rather than human intervention best accounts for these relationships.
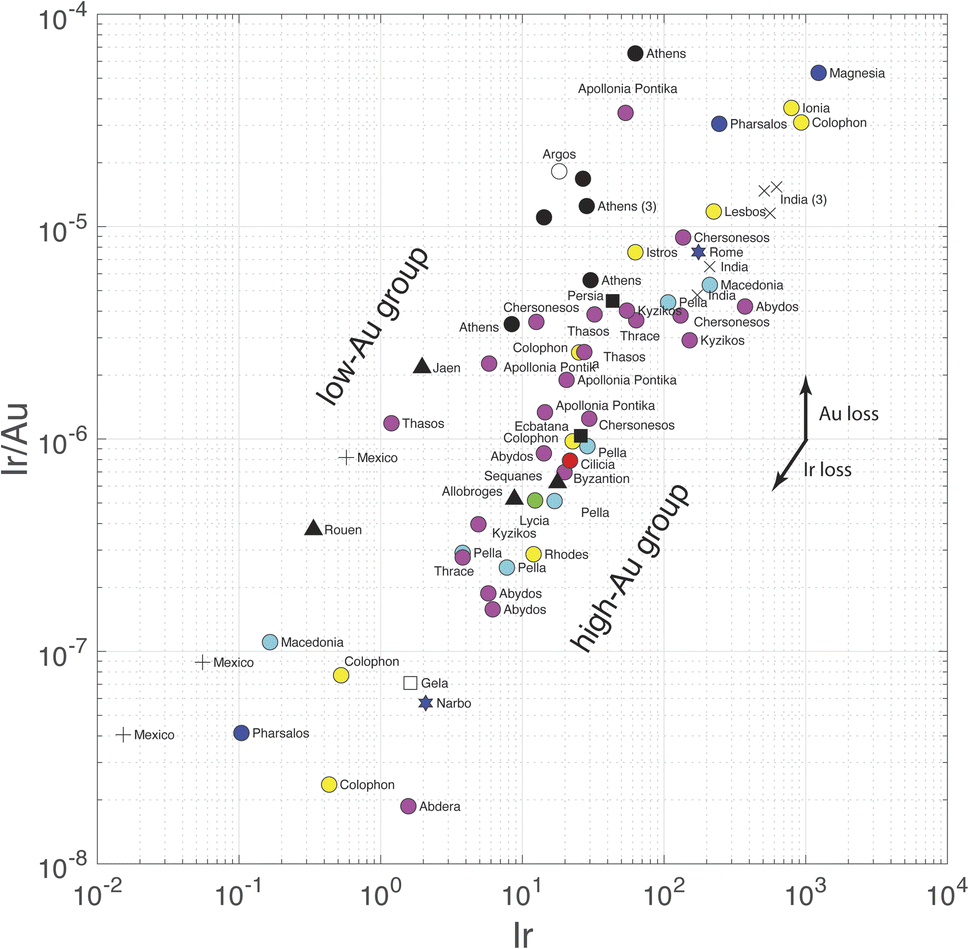 | ||
| Fig. 4 Plot of Ir/Au vs. Ir (also Ir/Ag). Note the two parallel bands spreading along the direction of Ir loss/gain. The two groups are distinct because of different Au contents, not Ir contents. Note also the presence of coins from non-Athenian origin in the low-Au group. The slope of the Ir loss/gain arrow is equal to 1. | ||
The dichotomy observed in the Ir/Au vs. Ir plot is most straightforwardly interpreted as reflecting an effect of higher Au solubility in brines of variable chlorinity.47–53 Again, Au depletion of southern Aegean ores may correspond to a contrast between southern and northern Greece. Variable conditions, such as depth of emplacement and infiltration of meteoric water during the genesis of silver ores may lead to a different role of liquid–vapor separation or to dilution of ore fluids by the water table. At this stage, we conclude that the case of salt cementation in Athenian mints6 is not supported.
Can PGEs and Au be used as a provenance tracer? In Athenian samples, Au concentrations are low and the Rh/Ir ratios vary within the rather narrow range of 12–35. As for Athens, some consistent behavior is observed but, given the destructive nature of the analytical technique, it is difficult to see a bright future for the use of PGEs and Au as a routine provenance tool. Colonial silver coins from Spanish Americas (Peru and Mexico) are also strongly depleted in Ir, Pd, and Pt, and, for Mexico, Au, which may or may not be due to losses at the amalgamation stage of the patio method. Potosi coins are depleted in Pt and Pd with respect to contemporaneous coins from Mexico, which may indicate different silver refinement procedures. For Potosi samples, whether Au contents below the detection limit are due to an analytical problem or to intentional extraction of Au from metallic silver is not known: the very small Au content of the replicated analysis of Poto D suggests that gold may have been extracted from silver.
Using the published Pb isotope data obtained on the same samples, we followed Wood et al.'s7 lead of a possible correlation between Ir contents and Pb model ages. Iridium does not correlate with Pb isotopic ratios, except weakly with 207Pb/204Pb (r = 0.58), nor with model ages or κ (232Th/238U) values. A weak correlation is observed between Ir and μ values (238U/204Pb, r = 0.53). These weak correlations suggest that high Ir contents signal a source in terranes located south of the Variscan front (see Fig. 4 in Blichert-Toft et al.54), probably in crustal slivers detached from the Gondwana continent and embedded in the recent (<100 Ma) Alpine collisions. Wood et al.7 are not incorrect when they suggest that Ir contents are potential provenance tracers, but the present work nevertheless suggests that the resolution of this tracer is only at the broad scale of tectonic provinces, not at the scale of individual mining districts.
4 Conclusions
Five of the six platinum-group elements (Pt–Pd–Rh–Ir ± Ru) and Au were successfully analyzed for the first time by Q-ICP-MS in 72 silver coins originating from the ancient Greek and Hellenistic world, Rome, medieval Europe, and Spanish Americas.The behavior of PGEs and Au define coherent groups corresponding to the four successive columns of the periodic table: (Fe)–Ru–Os (volatile), (Co)–Rh–Ir, (Ni)–Pd–Pt, and (Cu)–Ag–Au. Phase diagrams provided by metallurgy establish that elements from the Ni group are soluble in Ag and Au without major fractionation. Elements of the Co group are nearly insoluble in Ag and Au. Salt cementation of gold considered as a source of silver, notably for Athenian coinage, must be more sensitive to Ir and Rh fractionation than Pd and Pt. The data show that Au-poor and Au-rich groups form independently of Ir content variations.
It is concluded that parting does not account for the relationship between Au and Ir first demonstrated by Wood et al.7 Rather, this relationship reflects bimodal Au distribution of silver sources possibly inherited from regional conditions of ore genesis. Iridium, however, is not a helpful tracer of geographic provenance.
Author contributions
FA: project design, data validation, interpretation, writing; CMD and PT: methodology, data curation, validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council H2020 Advanced Grant 741454-SILVER-ERC-2016-ADG ‘Silver isotopes and the rise of Money’ awarded to FA. Marine Pinto is gratefully acknowledged for the measurement of limits of detection. We thank Janne Blichert-Toft and Gil Davis for comments on the manuscript. Three reviewers provided useful and courteous comments on the manuscript.References
- C. M. Kraay and V. M. Emeleus, The Composition of Greek Silver Coins: Analysis by Neutron Activation, Ashmolean Museum, Oxford, 1962 Search PubMed.
- O. Picard, Revue Belge de numismatique et de sigillographie, 2001, 147, 1–10 Search PubMed.
- G. Davis, D. B. Gore, K. A. Sheedy and F. Albarède, J. Archaeol. Sci., 2020, 114, 105068 CrossRef CAS.
- P. Meyers, Metalwork from Sardis. Harvard University Press, Cambridge, Massachusetts, 1983, pp. 185–191 Search PubMed.
- A. Ramage and P. T. Craddock, King Croesus' Gold: Excavations at Sardis and the History of Gold Refining, British Museum Publications Limited, 2000 Search PubMed.
- E. Pernicka, J. Archaeol. Sci., 2017, 86, 123–126 CrossRef CAS.
- J. R. Wood, M. F. Charlton, M. Murillo-Barroso and M. Martinón-Torres, J. Archaeol. Sci., 2017, 81, 1–12 CrossRef CAS.
- L. Robb, Introduction to Ore-Forming Processes, John Wiley & Sons, 2020 Search PubMed.
- B. Peucker-Ehrenbrink and B. M. Jahn, Geochem., Geophys., Geosyst., 2001, 2, 1061 CrossRef.
- D. Pearson, G. Irvine, D. Ionov, F. Boyd and G. Dreibus, Chem. Geol., 2004, 208, 29–59 CrossRef CAS.
- M. Becker and A. P. Le Roex, J. Petrol., 2006, 47, 673–703 CrossRef CAS.
- M. Fischer-Gödde, H. Becker and F. Wombacher, Chem. Geol., 2011, 280, 365–383 CrossRef.
- A. Gannoun, K. W. Burton, J. M. Day, J. Harvey, P. Schiano and I. Parkinson, Rev. Mineral. Geochem., 2016, 81, 651–724 CrossRef.
- S.-J. Barnes and E. M. Ripley, Rev. Mineral. Geochem., 2016, 81, 725–774 CrossRef.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2012 Search PubMed.
- P. Meyers, L. Van Zelst and E. Sayre, J. Radioanal. Nucl. Chem., 1973, 16, 67–78 CrossRef CAS.
- P. Meyers, L. Van Zelst and E. V. Sayre, Major and trace elements in sasanian silver, Archaeological chemistry - a symposium sponsored by the division of the history of chemistry at the 165th meeting of the american chemical society, 1974, vol. 138, pp. 22–33 Search PubMed.
- P. O. Harper and P. Meyers, Silver Vessels of the Sasanian Period: Royal Imagery, Metropolitan Museum of Art, 1981 Search PubMed.
- E. Le Roy Ladurie, J.-N. Barrandon, B. Collin, M.-F. Guerra and C. Morrisson, Annales ESC, 1990, 2, 483–505 Search PubMed.
- N. H. Gale, W. Gentner and G. A. Wagner, in Metallurgy in Numismatics I, ed. D. M. Metcalf and W. A. Oddy, The Royal Numismatics Society, London, 1980, pp. 3–49 Search PubMed.
- B. Gratuze, A. Giovagnoli, J.-N. Barrandon, P. Telouk and J.-L. Imbert, Revue d’Archéométrie, 1993, 17, 89–104 CrossRef.
- G. Sarah and B. Gratuze, in Recent Advances in Laser Ablation ICP-MS for Archaeology, Springer, 2016, pp. 73–87 Search PubMed.
- F. Albarède, Introduction to Geochemical Modeling, Cambridge University Press, Cambridge, 1995 Search PubMed.
- A. M. Desaulty, P. Telouk, E. Albalat and F. Albarede, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9002–9007 CrossRef CAS.
- J. Blichert-Toft, F. de Callataÿ, P. Télouk and F. Albarède, Archaeol. Anthropol. Sci., 2022, 14, 1–10 CrossRef.
- M. Vaxevanopoulos, G. Davis, J. Milot, J. Blichert-Toft, C. Malod-Dognin and F. Albarède, J. Archaeol. Sci., 2022, 145, 105645 CrossRef CAS.
- M. Fischer-Gödde, H. Becker and F. Wombacher, Geochim. Cosmochim. Acta, 2010, 74, 356–379 CrossRef.
- C. L. Chou, Proc. Lunar Planet. Sci. Conf., 1978, 9, pp. 219–230 Search PubMed.
- M. Javoy, J. Geophys. Res. Lett., 1995, 22, 2219–2222 CrossRef CAS.
- M. Javoy, E. Kaminski, F. Guyot, D. Andrault, C. Sanloup, M. Moreira, S. Labrosse, A. Jambon, P. Agrinier and A. Davaille, Earth Planet. Sci. Lett., 2010, 293, 259–268 CrossRef CAS.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1986, 7, 360–362 CrossRef CAS.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1988, 9, 243–244 CrossRef.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1988, 9, 237–243 CrossRef.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1986, 7, 362–365 CrossRef CAS.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1986, 7, 359–360 CrossRef CAS.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1987, 8, 334–340 CrossRef CAS.
- I. Karakaya and W. Thompson, Bull. Alloy Phase Diagrams, 1986, 7, 365–368 CrossRef CAS.
- H. Okamoto and T. Massalski, Bull. Alloy Phase Diagrams, 1983, 4, 30–38 CrossRef.
- P. Durussel and P. Feschotte, J. Alloys Compd., 1996, 239, 226–230 CrossRef CAS.
- H. Okamoto and T. Massalski, Bull. Alloy Phase Diagrams, 1984, 5, 384–387 CrossRef CAS.
- H. Okamoto and T. Massalski, Bull. Alloy Phase Diagrams, 1985, 6, 46–56 CrossRef CAS.
- H. Okamoto and T. B. Massalski, ASM Int., 1987, 1987, 358 Search PubMed.
- H. Okamoto and T. Massalski, Bull. Alloy Phase Diagrams, 1985, 6, 229–235 CrossRef CAS.
- A. A. Rudnitskii and V. P. Volyakova, Russ. J. Inorg. Chem., 1959, 4, 1051–1053 Search PubMed.
- A. A. Rudnitskii and A. N. Khotinskaya, Russ. J. Inorg. Chem., 1959, 4, 1160–1162 Search PubMed.
- Y. Shubin, P. Plyusnin, M. Sharafutdinov, E. Makotchenko and S. Korenev, Nanotechnology, 2017, 28, 205302 CrossRef PubMed.
- P. C. Voudouris, V. Melfos, P. G. Spry, R. Moritz, C. Papavassiliou and G. Falalakis, Mineral. Petrol., 2011, 103, 79–100 CrossRef CAS.
- P. Voudouris, V. Melfos, C. Mavrogonatos, A. Photiades, E. Moraiti, B. Rieck, U. Kolitsch, A. Tarantola, C. Scheffer and D. Morin, Minerals, 2021, 11, 76 CrossRef CAS.
- S. Kilias, S. Kalogeropoulos and J. Konnerup-Madsen, Miner. Deposita, 1996, 31, 394–406 CrossRef CAS.
- T. A. Bonsall, P. G. Spry, P. C. Voudouris, S. Tombros, K. S. Seymour and V. Melfos, Econ. Geol., 2011, 106, 619–651 CrossRef CAS.
- M. Frenzel, P. Voudouris, N. J. Cook, C. L. Ciobanu, S. Gilbert and B. P. Wade, Miner. Deposita, 2021, 1–22 Search PubMed.
- N. A. Sullivan, Z. Zajacz, J. M. Brenan and A. Tsay, Geochim. Cosmochim. Acta, 2022, 316, 253–272 CrossRef CAS.
- N. A. Sullivan, Z. Zajacz, J. M. Brenan, J. C. Hinde, A. Tsay and Y. Yin, Geochim. Cosmochim. Acta, 2022, 316, 230–252 CrossRef CAS.
- J. Blichert-Toft, H. Delile, C. T. Lee, Z. Stos-Gale, K. Billström, T. Andersen, H. Hannu and F. Albarède, Geochem., Geophys., Geosyst., 2016, 17, 3854–3864 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00112a |
| This journal is © The Royal Society of Chemistry 2023 |