
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterisation of gas cell reactions for 70+ elements using N2O for ICP tandem mass spectrometry measurements†
Shaun T.
Lancaster
 *a,
Thomas
Prohaska
ab and
Johanna
Irrgeher
ab
*a,
Thomas
Prohaska
ab and
Johanna
Irrgeher
ab
aDepartment of General, Analytical and Physical Chemistry, Chair of General and Analytical Chemistry, Montanuniversität Leoben, Leoben, Austria. E-mail: shaun.lancaster@unileoben.ac.at
bDepartment of Physics and Astronomy, University of Calgary, Calgary, Canada
First published on 28th April 2023
Abstract
One widely utilised method to reduce spectral interferences for measurements using inductively coupled plasma mass spectrometry (ICP-MS) is to employ the use of a reaction cell gas. Nitrous oxide (N2O) is a highly reactive gas typically used for mass-shifting only target analytes to a higher mass-to-charge ratio with increased sensitivity (e.g. +16, +32, +48 amu for monoxide, dioxide, and trioxide product ions respectively). Traditionally, the use of N2O was limited to selected applications due to the creation of new interferences that also interfere with the detected masses of interest. However, with the advent of inductively coupled plasma tandem mass spectrometry (ICP-MS/MS), the use of N2O has gained more traction, with a growing number of publications in recent years. Here, a comprehensive study of the use of N2O for the determination of 73 elements has been conducted, with a comparison to the most widely used mass-shift method using oxygen (O2) as a reaction gas. In total, 59 elements showed improved sensitivity when performing mass-shift with N2O compared to O2, with 8 elements showing no reaction with either gas. Additionally, N2O demonstrated a collisional focusing effect for 36 elements when measuring on-mass. This effect was not observed using O2. Monitoring asymmetric charge transfer reactions with N2O highlighted 14 elements, primarily non-metals and semi-metals, that enter the gas cell as metastable ions and could be used as an alternative mass-shift option. The results from this study highlight the high versatility of N2O as a reaction cell gas for routine ICP-MS/MS measurements.
Introduction
Overcoming spectral interferences in inductively coupled plasma mass spectrometry (ICP-MS) measurements is of utmost importance when striving to achieve reliable data. One widely utilised method to obtain interference free determinations is to employ the use of a reaction cell gas. A multitude of different gases have been used, summarised in detail previously.1–4 In the cell, two possible reactions can occur: atom transfer and asymmetric charge transfer. With the advent of tandem mass spectrometry (MS/MS), only interferences with the same mass-to-charge ratio (m/z) as the target analyte remain of concern as all other ions (that can potentially become interferences depending on the reactions employed) are removed by a mass filter before entering the reaction cell.Resolution of spectral interferences by atom transfer is often achieved in two ways: atom transfer to the analyte or atom transfer to the interference. In the former case, the most common approach is to perform the reaction of the target analyte with oxygen (O2),3 such that M+ + O2 → MO+ + O. This is known as a “mass-shift” determination, where the analyte, M, is detected at a higher m/z (in this case +16 amu). Conversely, mass-shifting the interference allows the target analyte to be analysed at the same m/z as it enters the cell – known as an “on-mass” determination. Other gases used for atom transfer reactions include nitrous oxide (N2O),5–8 carbon dioxide,9 sulphur hexafluoride,10 and methyl fluoride.11,12
Asymmetric charge transfer reactions remove interferences by transferring the charge of an interference to the reaction gas (e.g. ammonia (NH3)), such that M+ + NH3 → M + NH3+. Due to the vacuum conditions of the gas cell, only exothermic reactions can proceed. Therefore, for charge transfer to occur, the ionisation energy of the reaction gas must be lower than that of the interfering ion. The use of NH3 is a widely-used option2,13 due to its low ionisation energy (10.070 eV)14 as well as its reactivity to form clusters with selected elements.4 Other gases used for charge transfer reactions include hydrogen, xenon, methane, and nitrogen.1
The application of a cell gas can also lead to a collisional focusing effect, first observed by Douglas and French.15 This effect gives an apparent increase in sensitivity with increased gas pressures due to loss of radial kinetic energy. This focuses the ions to the minimum of the effective potential of the quadrupole, allowing for increased transmission through the exit aperture.16 This, in turn, can lead to increased sensitivity of the target analyte. Gases that have previously demonstrated a collisional focusing effect include hydrogen,12,17 helium,1,18,19 and NH3.20,21
While the choice of reaction gas is an important consideration for removal of interferences, differences in the types of reaction cells used have shown to have a significant contribution to the removal of interferences. Koppenaal et al. summarized that quadrupole-based reaction cells allow for effective mass-selective ejection of unwanted precursor and product ions but lower transmission and higher scattering losses. In contrast to this, multipole-based reaction cells allow for higher transmission but lower selectivity – thus generally requiring more selective reaction gases.1 Therefore, although both cell designs give good analytical performance, it should be noted that differences may be seen between instruments with different types of gas cells.
While O2 and NH3 have been successfully used with ICP-MS/MS determinations, they are not without their disadvantages. NH3 is a corrosive and toxic gas that is not universally compatible with every gas cell and O2 only has a selective range of elements that react readily enough to benefit from mass-shift. N2O is an alternative reaction gas to form oxide species. It has a much higher reactivity than O2 and leads to increased sensitivity when used in ICP-MS.22,23 However, due to its high reactivity, a tendency to form new spectral interferences from other matrix components historically rendered N2O unfavourable for ICP-MS measurements. Nevertheless, with ICP-MS/MS, formations of new interferences are no longer as problematic. Thus, N2O becomes a more viable option and is recently gaining traction within the community.
Here, a comprehensive evaluation of N2O as a reaction cell gas for ICP-MS/MS measurements with a quadrupole-based reaction cell has been carried out for 73 elements. Mass-shift determinations have been compared against the most widely used alternative of O2 for the removal of interferences. Additionally, possibilities for on-mass determinations of these elements using N2O have been determined based on collisional focusing effects observed for the target analytes, as well as oxide and doubly-charged interferences. Finally, charge transfer reactions have been considered as possibilities for removal of interferences.
Experimental
Reagents
Nitric acid (HNO3, w = 65%, p.a. grade; Carl Roth GmbH, Karlsruhe, Germany) was purified using a sub-boiling distillation system (Savillex DST-4000, AHF Analysentechnik, Tübingen, Germany). Hydrochloric acid (HCl, w = 65%, p.a. grade; Carl Roth GmbH) was purified by sub-boiling (Savillex DST-1000, AHF Analysentechnik). Reagent grade I water (18.2 MΩ cm; MilliQ IQ 7000, Merck-Millipore, Darmstadt, Germany) was used for all acid dilutions. Vials and pipette tips were pre-cleaned by soaking overnight in diluted sub-boiled nitric acid (w = 3%) before use.Single-element standards of beryllium (Be), boron (B), cadmium (Cd), caesium (Cs), chromium (Cr), copper (Cu), gadolinium (Gd), hafnium (Hf), indium (In), lithium (Li), magnesium (Mg), molybdenum (Mo), phosphorus (P), potassium (K), rubidium (Rb), sodium (Na), and tungsten (W) (β = 1000 μg mL−1; Certipur, Merck); cerium (Ce), dysprosium (Dy), erbium (Er), europium (Eu), germanium (Ge), holmium (Ho), lanthanum (La), lutetium (Lu), neodymium (Nd), osmium (Os), praseodymium (Pr), samarium (Sm), terbium (Tb), thulium (Tm), and ytterbium (Y) (β = 1000 μg mL−1; High Purity Standards, North Charleston, SC, USA); aluminium (Al), antimony (Sb), barium (Ba), bismuth (Bi) calcium (Ca), cobalt (Co), gold (Au), iodine (I), iron (Fe), lead (Pb), manganese (Mn), niobium (Nb), palladium (Pd), platinum (Pt), rhenium (Re), rhenium (Re), scandium (Sc), strontium (Sr), sulphur (S), tantalum (Ta), thallium (Tl), tin (Sn), titanium (Ti), zinc (Zn), zirconium (Zr), selenium (Se), and vanadium (V) (β = 1000 μg mL−1; Inorganic Ventures, Christiansburg, VA, USA); selenium (Se) and vanadium (V) (β = 1000 μg mL−1; Peak Performance, CPI international, Santa Rosa, CA, USA); nickel (Ni, β = 1000 μg mL−1; Alfa Aesar, Karlsruhe, Germany); silicon (Si, β = 1000 μg mL−1; SPEX Chemicals, Metuchen, NJ, USA); gallium (Ga) and yttrium (Y) (β = 10 μg mL−1; Elemental Scientific, Omaha, NE, USA); tellurium (Te, β = 10 μg mL−1; High Purity Standards); arsenic (As), iridium (Ir), ruthenium (Ru), and silver (Ag) (β = 10 μg mL−1; Inorganic Ventures); and mercury (Hg, β = 1 μg mL−1; Inorganic Ventures) were used throughout this work. Additionally, ICP multi-element standard solution VI (Certipur, Merck), rare earth element multi-element standard AHF-CAL-7 (Inorganic Ventures), and precious metal multi-element calibration standard #2 (AccuStandard, New Haven, CT, USA) were also used.
Tetramethylammonium hydroxide (TMAH, w = 25%; Sigma-Aldrich, Steinheim, Germany) diluted to w = 1% in reagent grade I water was used for analysis of the halogens. Sodium chloride (≥99.5%, p.a. grade; Carl Roth GmbH) and potassium bromide (NORMAPUR grade; VWR, Vienna, Austria) salts were used to prepare standards for the analysis of chlorine (Cl) and bromine (Br) respectively.
Instrumentation
All work was carried out using a NexION 5000 (PerkinElmer, Waltham, MA, USA), an ICP-MS/MS system equipped with a quadrupole-based dynamic reaction cell (DRC). Here, the mass filter (prior to the reaction cell) is termed Q1 and the mass analyser (after the reaction cell) is termed Q3. Instrument parameters for the different measurement modes applied are given in Table 1. Argon (purity 5.0 (≥99.999%); Linde Gas GmbH, Stadl-Paura, Austria) was used as the plasma gas. An in-house tuning solution containing 400 pg g−1 Ce was used for tuning the oxide rate to 1.6–1.9%. The rate of Ce2+ formation was monitored (Ce2+/Ce+ of 4.5–5.0%). Nitrous oxide (medicinal grade; Linde Gas GmbH) and oxygen (purity 3.5 (≥99.95%); Linde Gas GmbH) were used as reaction gases.| Parameter | Standard mode | O2 DRC mode | N2O DRC mode |
|---|---|---|---|
| Scan mode | MS/MS | MS/MS | MS/MS |
| Cell gas | None | O2 | N2O |
| RPq | 0.25 | 0.45 | 0.45 |
| Sample introduction | Self-aspiration | Self-aspiration | Self-aspiration |
| Nebulizer | PFA MicroFlow | PFA MicroFlow | PFA MicroFlow |
| Spray chamber | Peltier cooled SilQ cyclonic spray chamber | Peltier cooled SilQ cyclonic spray chamber | Peltier cooled SilQ cyclonic spray chamber |
| Spray chamber temperature | 5 °C | 5 °C | 5 °C |
| Interface cones | Nickel | Nickel | Nickel |
| RF power | 1600 W | 1600 W | 1600 W |
| Ar nebulizer gas flow | 0.96–1.01 L min−1 | 0.96–1.01 L min−1 | 0.96–1.01 L min−1 |
| Ar auxiliary gas flow | 1.2 L min−1 | 1.2 L min−1 | 1.2 L min−1 |
| Ar plasma gas flow | 16 L min−1 | 16 L min−1 | 16 L min−1 |
| QID fixed voltage | −12 V | −12 V | −12 V |
| Hyperskimmer park voltage | 5 V | 5 V | 5 V |
| OmniRing park voltage | −185 V | −185 V | −185 V |
| Inner target lens voltage | 2 V | 2 V | 2 V |
| Outer target lens voltage | −7 V | −7 V | −7 V |
| Deflector exit voltage | −8 V | −8 V | −8 V |
| Differential aperture voltage | −3.5 V | −3.5 V | −3.5 V |
| Q1 AC rod offset | −6 V | −10 V | −7.5 V |
| Q1 rod offset | −2 V | −0 V | 0 V |
| Cell rod offset | −33 V | −5 V | −2 V |
| Axial field voltage | 0 V | 125 V | 250 V |
| Cell entrance voltage | −5 V | −8.5 V | −7.5 V |
| Cell exit voltage | −2 V | −5.5 V | −5 V |
| Q3 AC rod offset | −2.5 V | −7 V | −8 V |
| Q3 rod offset | −2 V | −10 V | −10 V |
| Dwell time | 50 ms | 50 ms | 50 ms |
| Mass range | 7–238 amu | 7–280 amu | 7–285 amu |
Analytical measurement
A qualitative profile of the on-mass interaction of Xe with N2O was carried out using the trace levels of Xe found within the Ar gas supply between 0–1 mL min−1 of N2O.
In order to determine if interfering oxide and doubly-charged ions (which are formed in the plasma) could be removed using on-mass determinations, further measurements were carried out between 0–1 mL min−1 using N2O only. The instrument was tuned to a higher oxide-rate of 200–300% in standard mode (compared to 1.6–1.9% used previously) by adjusting the z-position of the torch 1.5 mm closer to the sampler cone and increasing the nebulizer gas flow to 1.01 mL min−1. Formation of Ce2+ was monitored (Ce2+/Ce+ of 40–50%) but not further tuned. In-house multi-element standards were prepared accounting for the interferences on oxides and doubly-charged ions of analytes (ESI A; Table S1b†).
Results and discussion
General observations
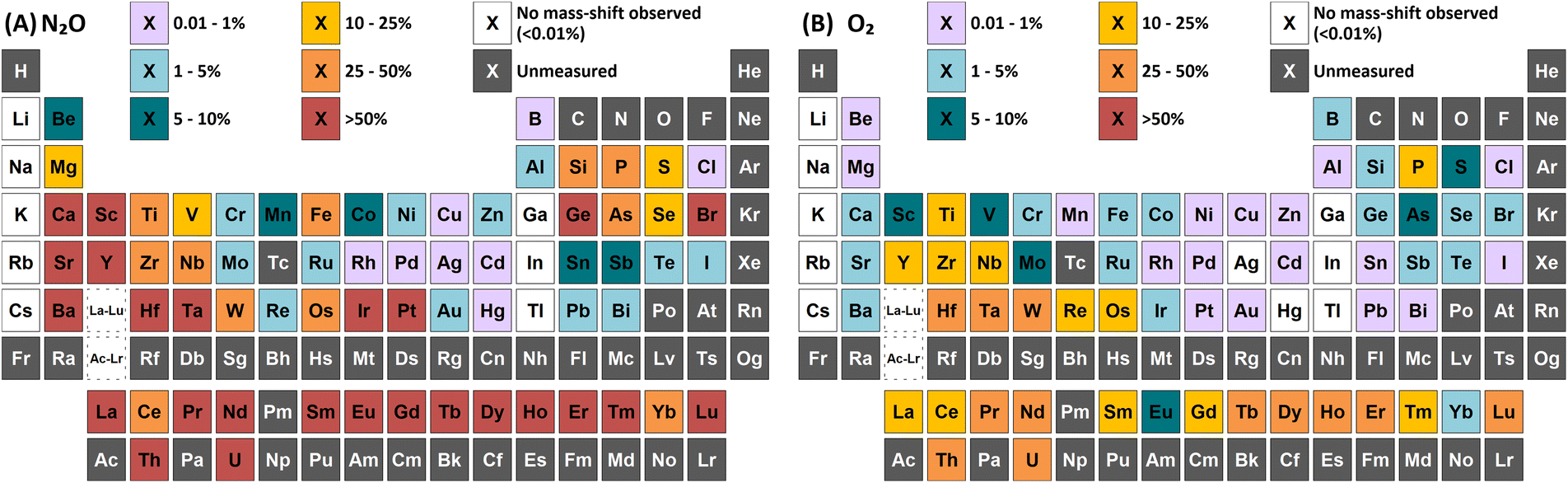 | ||
| Fig. 1 Comparison of maximum achieved sensitivity (relative to standard mode) for mass-shift determinations (by atom transfer) using (A) N2O and (B) O2 reaction gases. | ||
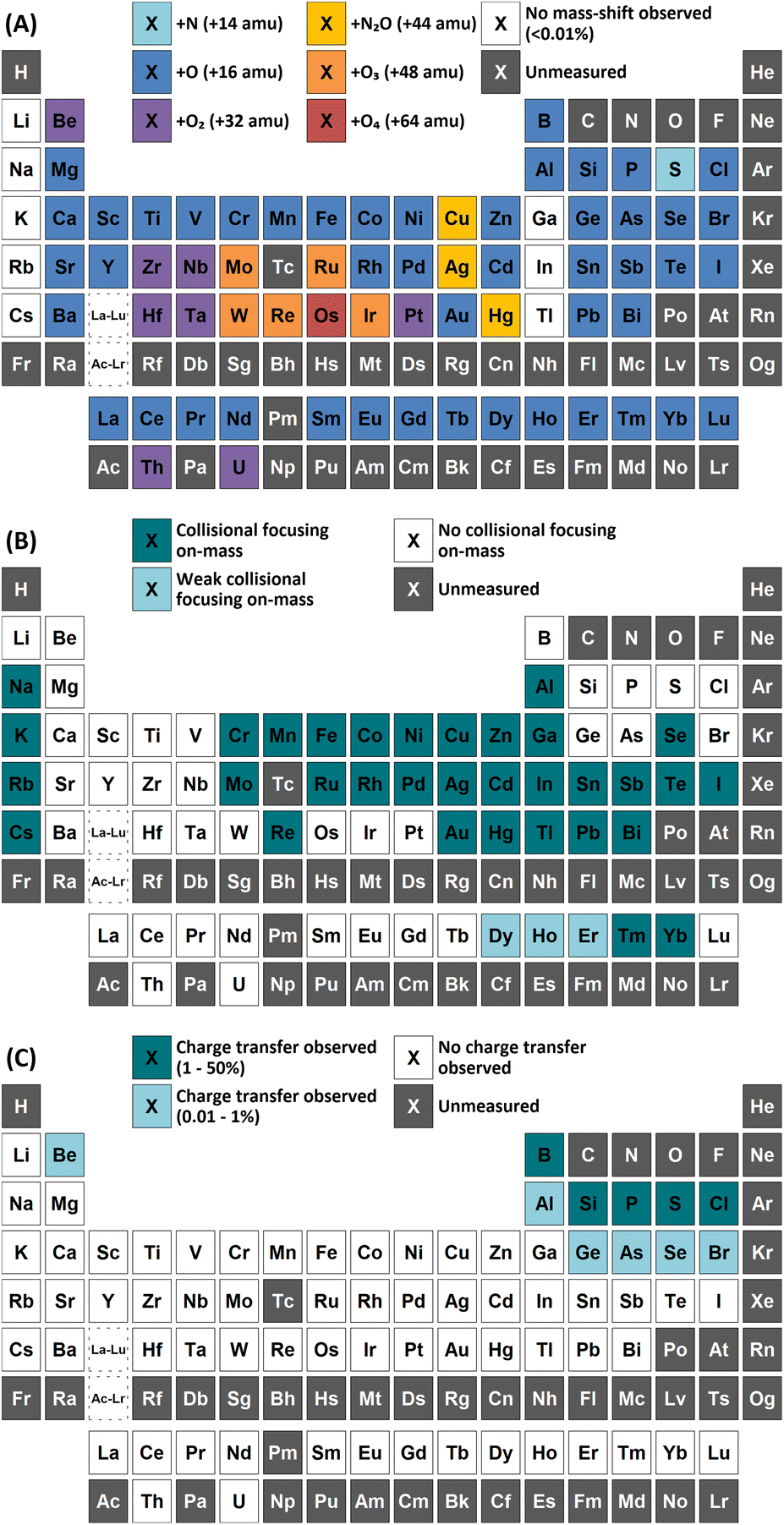 | ||
| Fig. 2 Observations of (A) atom transfer with greatest sensitivity, (B) on-mass collisional focusing, and (C) asymmetric charge transfer reactions using N2O reaction gas. | ||
Reaction with argon
Both N2O and O2 primarily react with Ar by the process of an asymmetric charge transfer reaction. For O2, the 16O2+ product was primarily observed, with an additional small formation of 38Ar16O+ (0.008%) on m/z = 54 at 0.1 mL min−1 that fell to baseline levels by 0.5 mL min−1. In the case of N2O, the primary product formed was the 14N216O+, with 14N16O+ and 16O2+ also present at lower intensities. No formation of 38Ar16O+ was observed on m/z = 54 and, as reported previously, no significant oxide formation was noted for 40Ar above 0.4 mL min−1 when considering the oxide formation from blank 40Ca.25s-Block elements
Group 1 metals do not react with either N2O or O2. However, a collisional focusing effect was observed for on-mass determinations of these elements using N2O, with the exception of Li. The magnitude of this effect was observed to increase down the group. On the other hand, group 2 metals react greatly with N2O to form the monoxide product ion, with negligible formation of the dioxide product ion (<0.1%) observed (except Be). As such, the use of N2O is a highly effective option of avoiding the isobaric interferences of 40Ar+ and 40K+ on 40Ca+ (as well as 24Mg16O+)25,26 and 87Rb+ on 87Sr+,26,27 as well as for the removal of isobaric Ba+ interferences for measurements of Cs radioisotopes.28–30 Relative sensitivities of the oxide product ion (compared to standard mode) increased down the group, from Mg (13%) to Ba (70%). In the case of Be, both the monoxide and dioxide product ions formed, with relative sensitivities of 5.5% and 7.8% compared to that of standard mode respectively. The group 2 metals did not react well with O2 (<2% formation) as it is an endothermic reaction. Be also showed evidence of charge transfer with N2O, with 0.28% formation of 14N16O+ and 0.12% formation of 14N216O+.p-Block elements
A wide variety of product ions were observed to form when using N2O. High formation of the SiO+ product ion (29%) was obtained – an improvement of 9 times greater than using O2 reaction gas. Additionally, the charge transfer reaction between 29Si(m)+ and N2O was found to give higher formations of 16O2+ (28%) compared to the formation of 14N216O+ (23%) and 14N216O+ (7.3%). While mass-shift of 28Si with O2 to 28Si16O+ has previously been demonstrated to successfully remove interferences and provide low-level Si determinations,31 the high background due to charge transfer with 14N2+ that cannot be resolved makes the same approach using N2O unfavourable. A good alternative is to mass-shift using the 28Si16O2+ product ion, which gave 11% formation. This approach allows for an increase of 3.4 times sensitivity and similar LODs and BECs compared to O2.
In addition, both P and S showed a high rate of charge transfer with N2O due to the formation of metastable ions. In both cases, formation of the 14N16O+ product ion was the greatest, at 16% and 12% for 31P(m)+ and 34S(m)+ respectively. Charge transfer of 31P(m)+ additionally formed 14N216O+ at 12% and 16O2+ at 1.9% (although this may also include partial formation of 31P1H+ from trace impurities in the N2O, such as water vapour), while charge transfer of 34S(m)+ additionally formed 14N216O+ at 5.5% and 16O2+ at 6.2%. Such a process may also be an option for removing interferences for both elements.
As and Se showed improvements in sensitivity of 4.4 times and 2.9 times respectively using N2O, while the formation rate achieved was 37% and 11% respectively. The use of N2O still allows for the effective removal of interferences of d-block element oxides and doubly-charged f-block elements, as well as interferences of ArCl+ (by charge transfer),32 while boosting sensitivity and detection limits compared to O2. 74Se+ also shares an isobaric interference with 74Ge+. The mass-shift approach only allowed for the measurement of 74Ge+ with reduced interference from 74Se+, as optimum sensitivity for 74Ge+ was obtained with a lower flow rate of N2O (0.4 mL min−1) compared to that of 74Se+ (1.3 mL min−1) but the sensitivity of 74Ge+ still remained relatively high at higher flow rates. Alternatively, Se showed collisional focusing on-mass using N2O, whereas Ge did not. Therefore, on-mass determinations of 74Se+ may be more suited to reducing the isobaric interference from 74Ge+ and 76Ge+ in addition to the aforementioned interferences, with the exception of Sm2+ and Eu2+, which also showed collisional focusing (see f-Block section).
A charge transfer reaction was also observed between Ge(m)+ and N2O, forming 14N216O+ at 0.19%. Interestingly, As and Se also showed evidence of charge transfer with N2O despite the ionisation potential of the low-lying energy levels of metastable As(m)+ (9.920–12.590 eV) and Se(m)+ (11.385–12.715 eV)24 being slightly lower than that of N2O (12.886 eV), thus leading to an endothermic reaction that should be unfavourable. 14N216O+ formations of 0.57% and 0.12% were observed for As and Se. It could be that the charge transfer reaction proceeds due to the addition of the kinetic energy component of the ions passing through the cell, or perhaps that the ions exist in an even higher energy state (induced by charge transfer reactions with metastable Ar(m)+). Further investigation is required to evaluate the reason for this anomaly.
Of these elements, Sn showed the greatest benefit from mass-shift using N2O, with 9.8% formation. This allows for the removal of the isobaric interference of 115In+ on 115Sn+, as well as the removal of interferences from oxides of d-block elements. However, 238U2+ may be an issue due to observed high UO2+ formation with N2O, which may also indicate high formation 238U16O22+ that would interfere on 119Sn16O+. Here, the collisional focusing effect is advantageous, as this offers reduction of 238U2+ as well as the aforementioned interferences. For Sb, Te, Pb, and Bi, on-mass determinations may be preferable for maintaining higher sensitivity and reducing oxide-based interferences, however the isobaric interferences between Sn, Sb, and Te cannot be resolved using N2O due to similarities in reactivity and collisional focusing effects.
Cl did not show a significant improvement in sensitivity of oxide formation, with a ClO+ formation rate of approximately 0.7% for both reaction gases. Instead, Cl primarily underwent a charge transfer reaction process, as the ionisation potential of Cl+ (12.968 eV) is greater than that of both N2O and O2. Here, the formation of 14N216O+ was 50%, with additional formations of 16O2+ and 14N16O+ at 0.76% and 0.88% respectively. Asymmetric charge transfer was also observed for Br (due to Br(m)+) but to a much lower degree, with 14N216O+ formation of 0.60% and 14N16O+ formation of 0.07% observed.
d-Block elements
Comparison of the formations of selected product ions for the d-block elements using N2O and O2 reaction gases is provided in Fig. 3.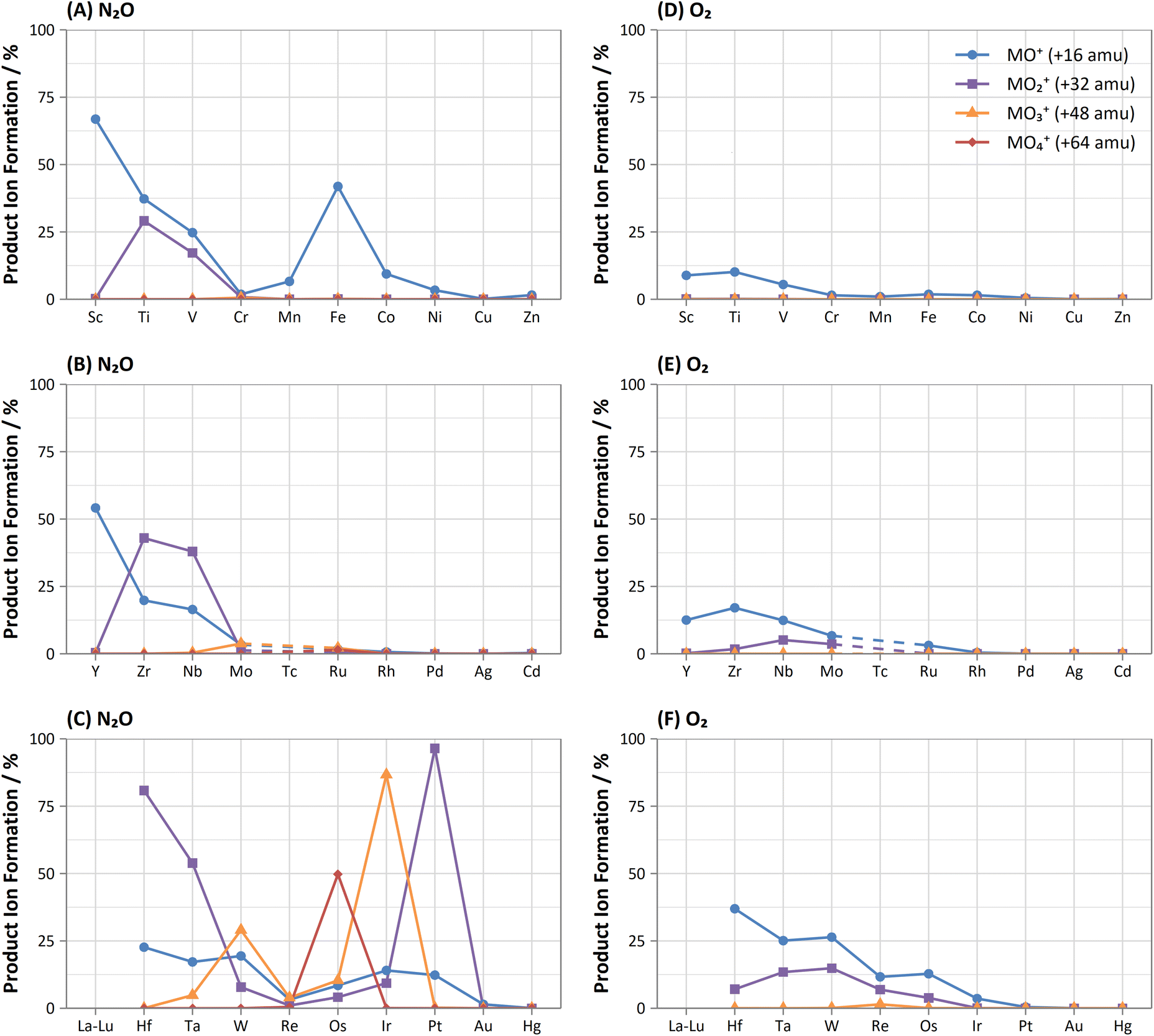 | ||
| Fig. 3 Comparison of obtained maximum formations of metal-oxide (blue, circle), -dioxide (purple, square), -trioxide (orange, triangle), and -tetroxide (red, diamond) product ions for d-block elements using (A–C) N2O and (D–F) O2 reaction gases between 0–1 mL min−1. | ||
Collisional focusing of Au and Hg, as well as removal of TaO+, HfO+, WO+, ReO+ and OsO+ with N2O, allows for on-mass determinations. Equally, the same approach could resolve the interferences of ZrO+, NbO+, and MoO+ on Ag+ and Cd+. However, Cd also suffers from isobaric interferences of Pd+, In+, and Sn+, which could not be resolved in this way due to similar collisional focusing effects of these elements. Therefore, for routine measurements, on-mass determinations of Cd with N2O on m/z = 111 would be advisable. Oxide interferences of Be- and Sc-group elements, as well as VO+, CrO+, and MnO+ demonstrated collisional focussing on-mass. Therefore, these interferences cannot be resolved for Co, Ni, and Cu, as well as Pd using N2O.
Contrary to the other elements in these groups, Pt shows markedly higher reactivity with N2O, while still producing minimal oxide formation using O2. In this case, the PtO2+ product ion gave the highest sensitivity, with 96% formation. Therefore, mass-shift of Pt to PtO2+ could be used to resolve interferences from HfO+, TaO+, and even the isobaric interferences of 196Hg+ and 198Hg+, however the observed high formation of WO3+ may indicate that the WO+ interference (on m/z = 196 and 198) may not be resolved. Additionally, isobaric interferences from 190Os+ and 192Os+ cannot be resolved due to high formation of OsO2+.
f-Block elements
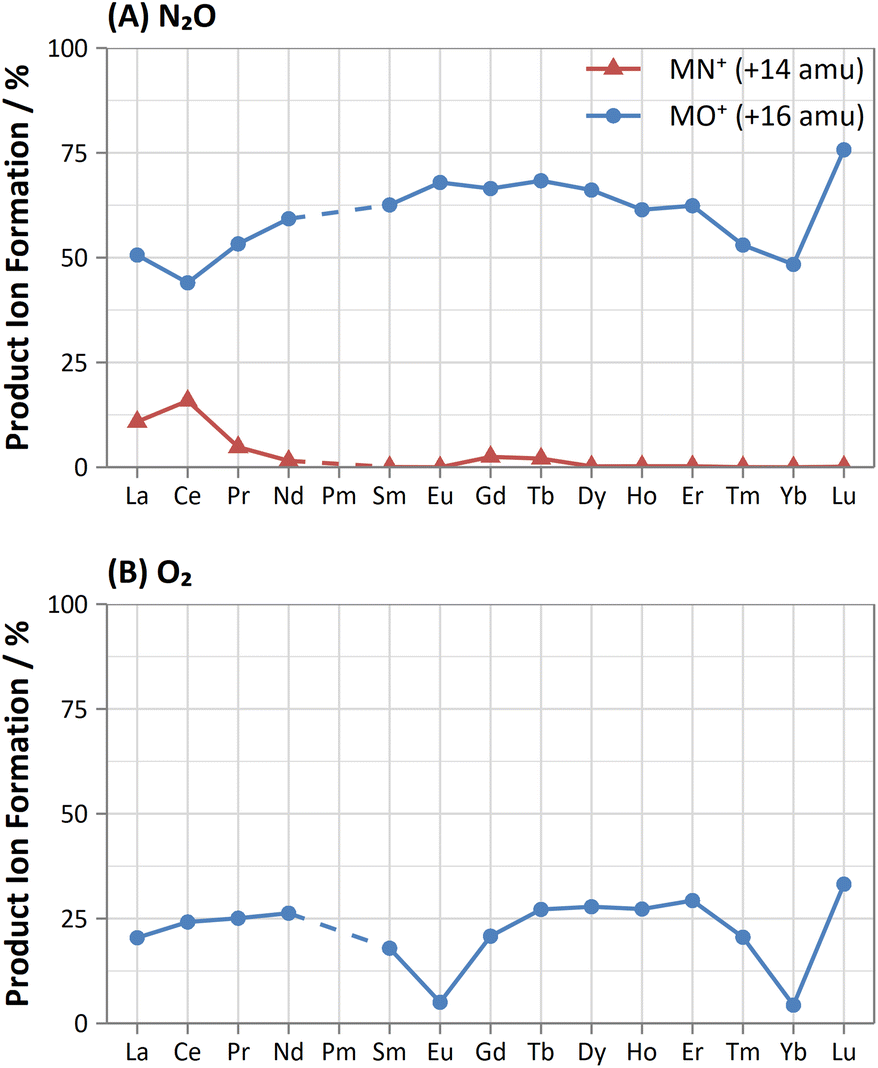 | ||
| Fig. 4 Comparison of obtained product ion formations for oxide (blue, circle) and nitride (red, triangle) product ion formation using (A) N2O and (B) O2 reaction gases. | ||
The flow rate of N2O that gave the greatest MO+ sensitivity increased across the period, from 0.5 mL min−1 for La to 0.8 mL min−1 and 0.7 mL min−1 for Yb and Lu respectively, whereas for O2 the flow rate was in most cases 0.3 mL min−1 (but slightly higher for Eu and Yb). Both N2O and O2 are capable of resolving oxide-based interferences of other lanthanides, as well as SnO+, SbO+, TeO+, CsO+, and BaO+, however the increased sensitivity offered by N2O makes it more preferable.
On-mass determinations with N2O showed evidence of collisional focusing from Dy to Yb, which became more prevalent across the period (Fig. 5). The observed strong on-mass collisional focusing effect for Yb allows for the reduction of isobaric interferences from 174Hf+, 176Hf+ and 176Lu+, as no collisional focusing effect was observed for these interfering elements. However, the reduction of isobaric interferences from 168Er+ and 170Er+ are less effective as the interference also exhibits some collisional focusing effect. Equally so, any interferences of lanthanide oxides formed in the plasma also could not be resolved on-mass due to the collisional focusing effect. Therefore, this observation may be limited to a few specific applications.
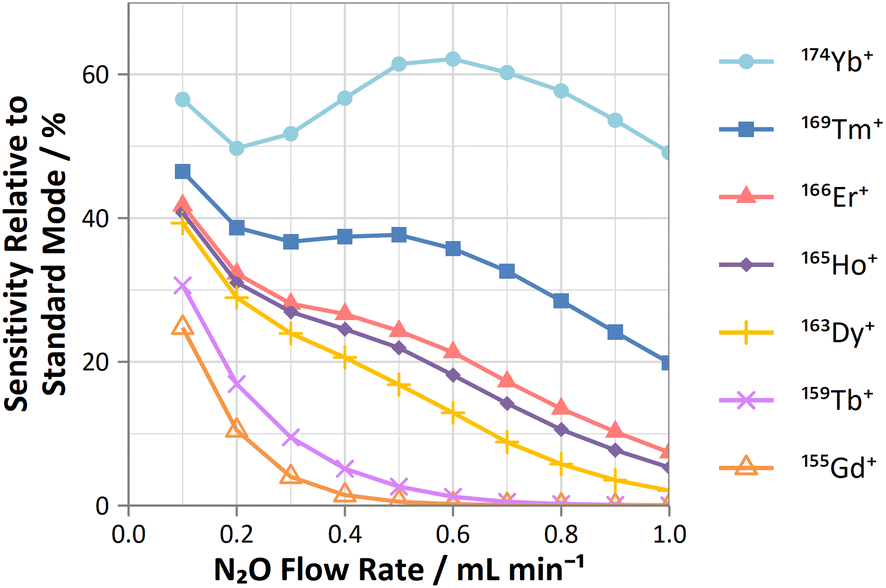 | ||
| Fig. 5 Profiles of sensitivities for Gd, Tb, Dy, Ho, Er, Tm, and Yb (relative to standard mode) determined on-mass using N2O reaction gas. | ||
Furthermore, on-mass collisional focusing of doubly-charged lanthanides with N2O was observed primarily for Sm2+, Eu2+, Tm2+ and Yb2+ (ESI B†), whereas other doubly-charged lanthanides were generally removed with the addition of N2O. This indicates that on-mass determinations of other analytes (such as As+ and Se+) will continue to suffer interferences from these four lanthanides. Product ion scans of the doubly-charged lanthanides revealed minimal product ion formation where a strong collisional focusing effect was observed, which was similarly observed for other singly-charged elements with low reactivity (Fig. 2A and B). Doubly-charged lanthanides that did not show collisional focusing were removed by mass-shifting with N2O to a different m/z. For these lanthanides, MO+ was the major product ion observed, with MO2+ and N2O+ product ions additionally present. The M+ product ion was not observed in any case. The observed product ions likely indicate an initial atom transfer reaction to form MO2+, followed by a charge transfer reaction with N2O to form MO+ and N2O+. This is supported in part by the thermodynamically unfavourable (endothermic) charge transfer reaction between N2O and doubly-charged lanthanides (second ionisation potentials between 10.55–12.18 eV for La–Yb)14 combined with the absence of M+ product ions. However, this cannot yet be stated with certainty as literature values for the second ionisation potential of gas phase lanthanide oxides is, to the authors' knowledge, not presently available.
Conclusions
This work highlights the versatility and applicability of N2O as a reaction cell gas for determinations of 73 elements. While it may not be suitable for every individual application, its high effectiveness of oxide formations compared to oxygen, as well as its ability to induce a collisional focusing effect on-mass, makes it a strong choice for routine analysis using ICP-MS/MS.While the rate of product ion formation by atom transfer may traditionally be the primary determining factor for performing mass-shift or on-mass determinations, the data provided here demonstrates considerable overlap of effectiveness of both approaches. Given this context, different interference removal approaches using N2O may be preferable depending on the level of interfering matrix elements and the concentration of the target analyte in solution.
Author contributions
Shaun T. Lancaster: conceptualization, investigation, data curation, writing–original draft preparation; Thomas Prohaska; conceptualization, supervision, writing–review and editing; Johanna Irrgeher: conceptualization, funding acquisition, supervision, writing–review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially funded by the Austrian Science Fund FWF (Fonds zur Förderung der wissenschaftlichen Forschung), grant number P 33099-N. This project (20IND01 MetroCycleEU) has received funding from the EMPIR programme co-financed by the Participating States and from the European Union's Horizon 2020 research and innovation programme. The authors would like to thank PerkinElmer for their cooperation in the study.Notes and references
- D. W. Koppenaal, G. C. Eiden and C. J. Barinaga, J. Anal. At. Spectrom., 2004, 19, 561–570 RSC.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2015, 894, 7–19 CrossRef CAS PubMed.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 1660–1679 RSC.
- Y. Zhu, T. Ariga, K. Nakano and Y. Shikamori, At. Spectrosc., 2021, 42, 299–309 CrossRef CAS.
- K. Harouaka, C. Allen, E. Bylaska, R. M. Cox, G. C. Eiden, M. L. di Vacri, E. W. Hoppe and I. J. Arnquist, Spectrochim. Acta, Part B, 2021, 186, 106309 CrossRef CAS.
- O. Klein, T. Zimmermann and D. Pröfrock, J. Anal. At. Spectrom., 2021, 36, 1524–1532 RSC.
- S. T. Lancaster, T. Prohaska and J. Irrgeher, Anal. Bioanal. Chem., 2022, 3–10 Search PubMed.
- S. Trimmel, T. C. Meisel, S. T. Lancaster, T. Prohaska and J. Irrgeher, Anal. Bioanal. Chem., 2023, 415, 1159–1172 CrossRef CAS PubMed.
- K. Harouaka, K. Melby, E. J. Bylaska, R. M. Cox, G. C. Eiden, A. French, E. W. Hoppe and I. J. Arnquist, Geostand. Geoanal. Res., 2022, 46, 387–399 CrossRef CAS.
- D. Pröfrock, P. Leonhard, S. Wilbur and A. Prange, J. Anal. At. Spectrom., 2004, 19, 623–631 RSC.
- E. Bolea-Fernandez, L. Balcaen, M. Resano and F. Vanhaecke, Anal. Chem., 2014, 86, 7969–7977 CrossRef CAS PubMed.
- E. Bolea-Fernandez, D. Leite, A. Rua-Ibarz, L. Balcaen, M. Aramendía, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2017, 32, 2140–2152 RSC.
- S. D'Ilio, N. Violante, C. Majorani and F. Petrucci, Anal. Chim. Acta, 2011, 698, 6–13 CrossRef PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 80th edn, 2000, pp. 10–175 to 10–177 Search PubMed.
- D. J. Douglas and C. J. B. French, J. Am. Soc. Mass Spectrom., 1992, 3, 398–408 CrossRef CAS PubMed.
- D. J. Douglas, J. Am. Soc. Mass Spectrom., 1998, 9, 101–113 CrossRef CAS.
- B. Russell, S. L. Goddard, H. Mohamud, O. Pearson, Y. Zhang, H. Thompkins and R. J. C. Brown, J. Anal. At. Spectrom., 2021, 36, 2704–2714 RSC.
- S. F. Boulyga and J. S. Becker, J. Anal. At. Spectrom., 2002, 17, 1202–1206 RSC.
- E. McCurdy and G. Woods, J. Anal. At. Spectrom., 2004, 19, 607–615 RSC.
- F. Ardini, E. Magi and M. Grotti, Anal. Chim. Acta, 2011, 706, 84–88 CrossRef CAS PubMed.
- C. Suárez-Oubiña, P. Herbello-Hermelo, P. Bermejo-Barrera and A. Moreda-Piñeiro, Spectrochim. Acta, Part B, 2022, 187 Search PubMed.
- V. V. Lavrov, V. Blagojevic, G. K. Koyanagi, G. Orlova and D. K. Bohme, J. Phys. Chem. A, 2004, 108, 5610–5624 CrossRef CAS.
- G. K. Koyanagi and D. K. Bohme, J. Phys. Chem. A, 2001, 105, 8964–8968 CrossRef CAS.
- K. Böting, S. Treu, P. Leonhard, C. Heiß and N. H. Bings, J. Anal. At. Spectrom., 2014, 29, 578–582 RSC.
- S. T. Lancaster, T. Prohaska and J. Irrgeher, Anal. Bioanal. Chem., 2022, 414, 7495–7502 CrossRef CAS PubMed.
- K. J. Hogmalm, T. Zack, A. K. O. Karlsson, A. S. L. Sjöqvist and D. Garbe-Schönberg, J. Anal. At. Spectrom., 2017, 32, 305–313 RSC.
- D. T. Murphy, C. M. Allen, O. Ghidan, A. Dickson, W. P. Hu, E. Briggs, P. W. Holder and K. F. Armstrong, Rapid Commun. Mass Spectrom., 2020, 34(5), e8604 CrossRef CAS PubMed.
- M. Granet, A. Nonell, G. Favre, F. Chartier, H. Isnard, J. Moureau, C. Caussignac and B. Tran, Spectrochim. Acta, Part B, 2008, 63, 1309–1314 CrossRef.
- T. Ohno and Y. Muramatsu, J. Anal. At. Spectrom., 2014, 29, 347–351 RSC.
- L. Zhu, X. Hou and J. Qiao, Anal. Chem., 2020, 92, 7884–7892 CrossRef CAS PubMed.
- R. S. Amais, C. D. B. Amaral, L. L. Fialho, D. Schiavo and J. A. Nóbrega, Anal. Methods, 2014, 6, 4516–4520 RSC.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Fresenius. J. Anal. Chem., 2001, 370, 454–470 CrossRef CAS PubMed.
- T. Ohno, Y. Muramatsu, Y. Shikamori, C. Toyama, N. Okabe and H. Matsuzaki, J. Anal. At. Spectrom., 2013, 28, 1283–1287 RSC.
- L. Fu, G. Huang, Y. Hu and F. Pan, Anal. Chem., 2022, 94, 3035–3040 CrossRef CAS PubMed.
- R. J. Ackermann, E. G. Rauh and R. J. Thorn, J. Chem. Phys., 1976, 65, 1027–1031 CrossRef CAS.
- Y. Zhu, Talanta, 2020, 209, 120536 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ja00025g |
| This journal is © The Royal Society of Chemistry 2023 |