A parallel-beam wavelength-dispersive X-ray emission spectrometer for high energy resolution in-air micro-PIXE analysis
Received
13th January 2023
, Accepted 11th April 2023
First published on 13th April 2023
Abstract
A new parallel-beam wavelength dispersive (PB-WDS) X-ray emission spectrometer was constructed at the external proton beamline at the Microanalytical Centre of the Jožef Stefan Institute in Ljubljana. The spectrometer combines polycapillary X-ray optics for efficient X-ray collection with diffraction on a flat crystal analyzer and achieves energy resolution in the eV range. The whole set-up is enclosed within a He bag to be able to operate in the tender X-ray energy range. The basic design is described together with the results of characterization measurements yielding the main operation characteristics. Finally, an application for the micro-PIXE analysis of biological tissue is demonstrated exploiting both spatial and energy resolution of the new set-up.
1. Introduction
Proton induced X-ray emission with a microfocused MeV proton beam (micro-PIXE) is a powerful analytical tool used to quantitatively analyze the spatial distribution of minor and trace elements in the samples with micrometer lateral resolution. The ion microprobe beamline at the Microanalytical Centre of the Jožef Stefan Institute (JSI) in Ljubljana1 is regularly used to perform micro-PIXE mapping. Due to its excellent capabilities such as micro-PIXE analysis of frozen-hydrated tissues,2 it attracts a broad user community especially from the fields of biology and medicine.3–5 The largest overall surface area analyzed at our microprobe is limited to ∼1 mm2. The latter, combined with the need for a vacuum sample environment, brings some important experimental restrictions. For that reason, we have recently upgraded our external beamline, which can now be used complementarily to the microprobe beamline to perform in-air micro-PIXE analysis of larger objects with a moderate lateral resolution (few tens of μm).6
Generally, micro-PIXE analysis is based on the energy dispersive (EDS) semiconductor X-ray detectors covering a relatively large solid angle combined with an energy resolution ΔE/E ∼ 2–3 × 10−2, which is good enough to resolve characteristic X-ray emission lines of different elements. The energy resolution can be pushed down towards the few eV range using superconducting transition-edge sensor micro calorimeters operating at cryogenic temperatures. These detectors have also been recently introduced in a high energy resolution PIXE analysis.7–9 However, such detectors are very complex, relatively difficult to operate, as well as very expensive, and have not been widely adopted. On the other hand, wavelength dispersive (WDS) Bragg crystal spectrometers have been traditionally used with X-ray fluorescence analytical techniques. WDS spectrometers are usually used in PIXE analysis to perform chemical state analysis, which is not feasible in standard PIXE analysis using EDS detectors. The main drawback of WDS spectrometers is their intrinsically low efficiency which is a consequence of a small solid angle. Curved crystal analyzers in different focusing geometries are used to enlarge the solid angle. High energy resolution PIXE spectrometers in von Hamos geometry were used to perform chemical state analysis under vacuum conditions10 and also for in-air PIXE analysis.11 In our laboratory, a crystal spectrometer in Johansson geometry has been used for high energy resolution PIXE analysis with an energy resolution below the natural core-hole broadening,12 and chemical speciation studies of low-Z elements in different materials have been performed.13–18 Apart from their moderate efficiency, such spectrometers are also quite large, and require a complex alignment procedure and a fixed beam spot, which is usually not compatible with ion microprobes. A compact flat crystal WDS spectrometer coupled to an ion microprobe has been introduced recently,19,20 mainly to perform chemical state analysis with focused ion beams and has not been used for PIXE mapping.
A promising alternative providing good collection efficiency combined with high energy resolution is a parallel-beam WDS (PB-WDS) spectrometer. Such a spectrometer employs polycapillary X-ray optics to increase the solid angle of X-ray collection and convert divergent emission into a collimated X-ray beam which is diffracted by a flat crystal analyzer to reach high energy resolution. With such a basic design, the PB-WDS spectrometer is compatible with an X-ray analysis based on a micro-focused primary beam. So far it has been used in microanalysis performed with electron microscopes21 and also for X-ray micro-fluorescence analysis.22 In the case of micro-PIXE analysis, we have temporarily installed and tested such a spectrometer at our ion microprobe demonstrating the basic principle.23 Since the spectrometer requires a fixed beam spot viewed by the polycapillary optics, it was not possible to combine it with the micro-PIXE mapping achieved by scanning the beam over the sample. However, our upgraded external beamline is perfectly suited for such a spectrometer in terms of both the fixed exit proton beam with relatively high current and beam spot size compatible with the polycapillary input field of view.
In the following sections, the design and the main operational characteristics of a new PB-WDS spectrometer installed at our external proton beamline are described. Finally, one representative application for in-air micro-PIXE mapping is presented, where the spectrometer was used to record the Cd spatial distribution within a leaf sample of a hyperaccumulating plant.
2. Spectrometer design
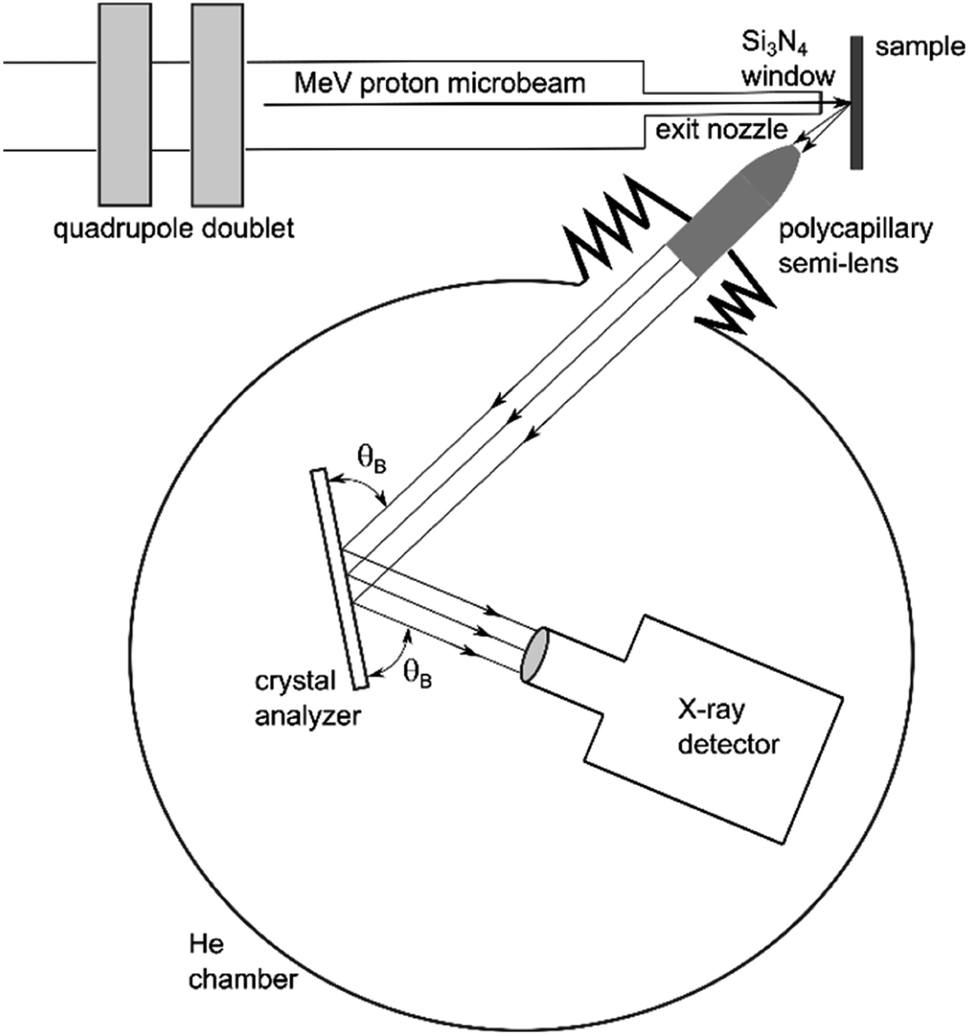
The basic operating principle of PB-WDS is shown schematically in Fig. 1. The X-ray emission is induced by irradiating the sample with a proton microbeam. The polycapillary X-ray semi-lens positioned at 45° relative to the incident beam and at a focal distance from the sample surface is used to collect the emitted X-rays and convert divergent X-ray fluorescence into an almost parallel X-ray beam. The collimated X-rays exiting the polycapillary are then further directed to a flat crystal analyzer, where they are diffracted according to Bragg's diffraction law
2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θB) = Nλ, sin(θB) = Nλ, |
where d is the crystal lattice spacing, θB is the Bragg reflection angle, λ is the X-ray wavelength and N corresponds to the diffraction order. Diffracted X-rays are collected with an X-ray detector positioned at the corresponding Bragg angle on the opposite side of the normal to the crystal surface. In order to record a wavelength dispersive spectrum, the Bragg angle is varied in a point-by-point mode by rotating the crystal over a pre-defined angular range. In the same way the detector is rotated by twice the rotation angle of the crystal. Since the X-ray beam exiting the polycapillary is almost parallel, the polycapillary to crystal and the crystal to detector distances are not crucial for the overall design. In our case, these distances are 40 cm and 5 cm, respectively, resulting in a relatively compact design. Since the spectrometer was designed to operate within the tender X-ray energy range (2–6 keV), the whole setup is enclosed within a chamber filled with He to eliminate in-air X-ray absorption. A photo of the spectrometer installed at the external proton beamline is shown in Fig. 2. The main components of the spectrometer are presented in more detail in the remaining part of this section.
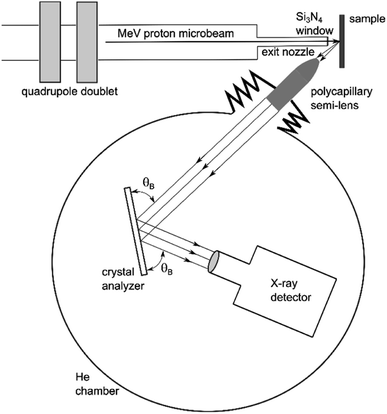 |
| | Fig. 1 A scheme of the PB-WDS setup combining polycapillary optics for efficient X-ray collection and diffraction by a flat crystal to achieve high energy resolution (objects not to scale). | |
 |
| | Fig. 2 A photo of the PB-WDS setup installed at the external proton beamline (left) and a closer view of the beam exit nozzle and polycapillary semi-lens facing the sample (right). | |
2.1 Polycapillary optics
One of the key components of the spectrometer is polycapillary optics. The input focal view of the polycapillary semi-lens was chosen to match the beam size of the focused proton beam (50–100 μm). Since we are aiming at a tender X-ray range, the optics design was optimized to maximize the transmission efficiency within this energy range. In order to remove in-air absorption, the lens is filled with He and sealed with a 12.7 μm Be window at both ends. The optics was manufactured by XOS (https://www.xos.com/), and the main specifications are given in Table 1. The lens is placed on a motorized linear micro stage MTS-65 by PImiCos (https://www.pimicos.com), with a full travel range of 13 mm and bi-directional repeatability of around 5 μm which allows for precise placement at the focal distance from the sample. Additional micro translation stages are used to align both vertical and horizontal positions of the polycapillary holder with the proton beam.
Table 1 Characteristics of polycapillary optics
| Input focal distance |
10 mm |
| Input collection angle |
65 mSr |
| Output beam diameter |
6 mm |
| Input field of view |
∼100 μm |
| Transmission efficiency |
15% at 1.5 keV, 8% at 2 keV, 11% at 3 keV, 12% at 5 keV, 5% at 8 keV |
| Output divergence |
<5 mrad |
| Enclosure length and diameter |
33 mm; 10 mm; He filled with 12.7 μm Be window at both ends |
2.2 Crystals and detector
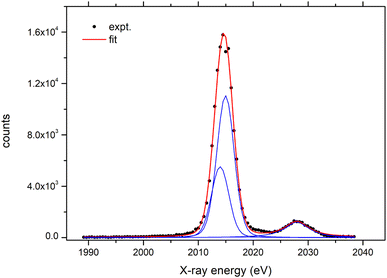
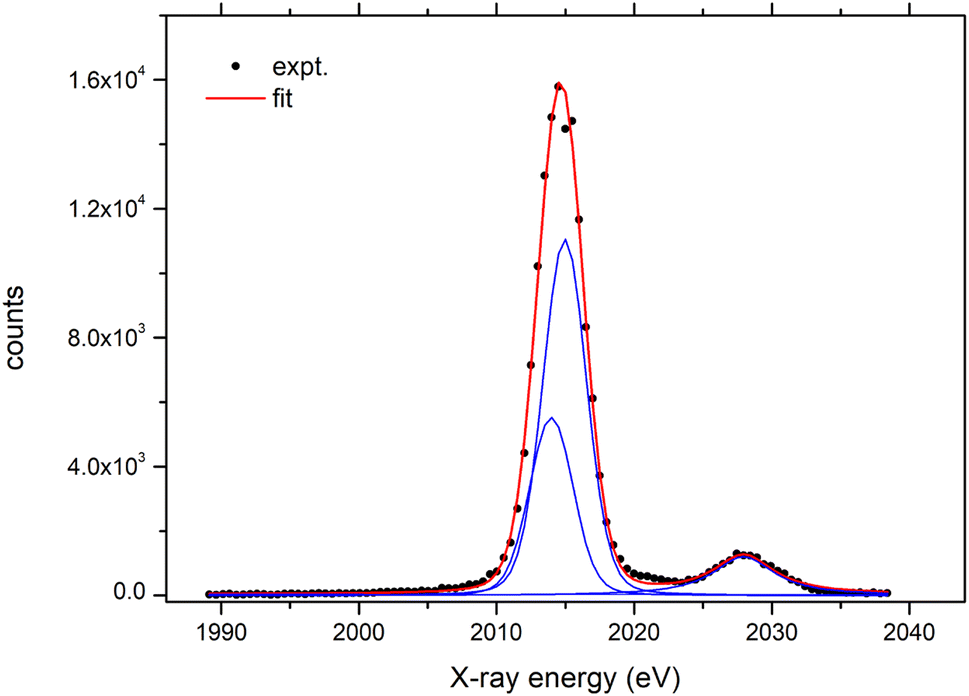
The parallel X-ray beam exiting the polycapillary optics is directed towards the flat crystal analyzer. The crystal and the detector are mounted on a goniometer, which enables precise adjustment of the angles for both the crystal and the detector. The goniometer is constructed from two separate PImiCos RS40 rotation stages providing an angular resolution of 5 mdeg with a bi-directional repeatability of ±0.04 deg. The stages are assembled together and aligned to the same rotation axis with a concentricity less than ±10 μm. The goniometer is mounted on an additional horizontal micro-translation stage to align the rotation axis with the optical axis of the polycapillary lens. Within the given mechanical arrangement of the whole setup, the Bragg angles of 35°–80° can be reached. The full working energy range is covered with three different crystal analyzers. The size of the crystals is 70 mm (width) × 30 mm (height). The list of crystals and the corresponding working energy range covered within the first order of reflection are given in Table 2. Finally, a 25 mm2 Amptek XR-100CR: Si-PIN detector with a 1.0 mil thick beryllium window is used to detect diffracted X-rays. The signal from the detector preamplifier is processed with an XIA DXP-XMAP digital pulse processor so the spectrometer can be fully integrated within the acquisition system of the external proton microbeam. As an example, the Fe signal detected by the Si-PIN detector when the spectrometer was set to the Bragg angle corresponding to the energy of the Fe Kα line is shown in Fig. 3. With the peaking time set to 25 μs, a final energy resolution of 266 eV is provided by the detector.
Table 2 List of available flat crystals with the corresponding 2d lattice spacing and covered energy range
| Crystal |
2d [Å] |
Energy range [keV] |
| Ge(111) |
6.532 |
1.9–3.3 |
| LiF(200) |
4.027 |
3.1–5.4 |
| LiF(220) |
2.848 |
4.4–7.6 |
 |
| | Fig. 3 Energy spectrum recorded with the Si-PIN detector with the spectrometer set to the Bragg angle corresponding to the energy of the Fe Kα line. | |
2.3 Spectrometer control and data acquisition
The microstep controllers driving the mechanical parts of the spectrometer and the DXP-XMAP readout are accessed by the dedicated software written in the LabVIEW programming environment. Within the acquisition procedure, the scanning energy interval, the step size and the acquisition time for a single point are defined. The spectrum is recorded in a point-by-point scanning mode according to the corresponding Bragg angles calculated from the initially given energy input values. In order to remove possible higher order reflections and/or any scattering yielding background in the measured spectra, only counts within a predefined energy window are integrated and plotted as a function of Bragg angle. Energy steps of 0.5 eV up to 2 eV are used depending on the actual energy resolution, which varies across the angular and energy range. The typical acquisition time for a single point is 1 up to 5 seconds. The total X-ray emission spectrum containing one of the main diagram lines usually consists of up to 100 points, so the total acquisition time needed to record a single spectrum is from 2 to 10 minutes.
3. Experimental results
At our external beamline, the proton beam is focused with a doublet of magnetic quadrupole lenses and exits the beamline nozzle through a 200 nm Si3N4 window yielding a beam spot of ∼50 × 50 μm2 at a working distance of few mm.6 The overall proton current on the sample can be adjusted between a few tenths of nA up to 10 nA. After installation, the separate stages of the new PB-WDS spectrometer were carefully aligned using the signal from the Fe metallic target induced with a 3 MeV proton beam. The most critical part is the alignment of the polycapillary semi-lens with the proton beam spot on the sample. This was achieved by mounting the detector directly behind the lens to achieve the maximum count rate. The focal distance from the sample was adjusted in the same way. Next, the horizontal position of the crystal stage rotation axis was aligned with the optical axis of the polycapillary optics and finally also the vertical position of the detector. The whole alignment procedure is performed in air, which allows easy access to the mechanical micro stages. At the end, a reference Fe Kα spectrum was recorded in air using a LiF(220) crystal analyzer. After that, the spectrometer was enclosed within a sealed housing filled with He. A gentle He flow through the housing was used to maintain a constant He environment during the measurements. Another control measurement of the reference Fe Kα spectrum was performed at the end within a He atmosphere. This is used to verify the removal of in-air absorption and also check the overall alignment again after enclosure within He housing.
In order to experimentally determine the main parameters of the PB-WDS set-up, Kα and Kβ X-ray spectra from P to Fe, covering the full energy range of the spectrometer, were recorded on a set of reference targets. As an example, the Kα and Kβ emission spectra of P, K and Ti recorded with Ge(111), LiF(200) and LiF(220) crystals, respectively, are shown in Fig. 4. The spectra of P and K were recorded from a KH2PO4 pressed powder pellet, while that of Ti was recorded from a thick metallic target. To reach comparable count rates, different proton currents were used in these measurements. A high proton current of 6 nA was used to measure the P spectrum with a Ge(111) crystal, and it was reduced to 2 nA to record the K spectrum with the LiF(200) crystal. For the Ti metallic target, the proton current was reduced even further down to 0.4 nA. Here, an additional scan of the region between the main Kα and Kβ diagram lines was performed to look for weak second order radiative contributions and test the sensitivity of the new setup. In this case the proton current was again increased back to 6 nA, and the acquisition time/point, from 5 to 10 seconds.
 |
| | Fig. 4 Examples of high energy resolution proton induced X-ray spectra recorded with the PB-WDS spectrometer: P (top) and K spectra (middle) of a KH2PO4 powder pellet recorded with Ge(111) and LiF(200) crystals, respectively, and the Ti spectrum (bottom) recorded with a LiF(220) crystal. | |
Due to the high energy resolution an additional peak is observed on the high-energy side of the Kα diagram lines of all three elements. This KαL satellite peak corresponds to 1s2p double ionization induced in proton-atom collisions and cannot be resolved in standard EDS PIXE spectra. The P Kβ spectrum of the KH2PO4 sample clearly exhibits a spectral structure reflecting the local chemical environment of the phosphate ion,24,25 showing a possibility to employ PB-WDS for chemical speciation studies. Aside from the high energy resolution, the Ti spectral region between both diagram lines demonstrates the high sensitivity of the setup. Despite a very low ionization cross section (σKK/σK ∼ 10−3) a Kαh hypersatellite contribution corresponding to the radiative decay of a doubly 1s ionized atom26 is clearly observed, as well as a weak K-MM radiative Auger spectral contribution27 on the low energy tail of the Ti Kβ diagram line.
3.1 Energy resolution
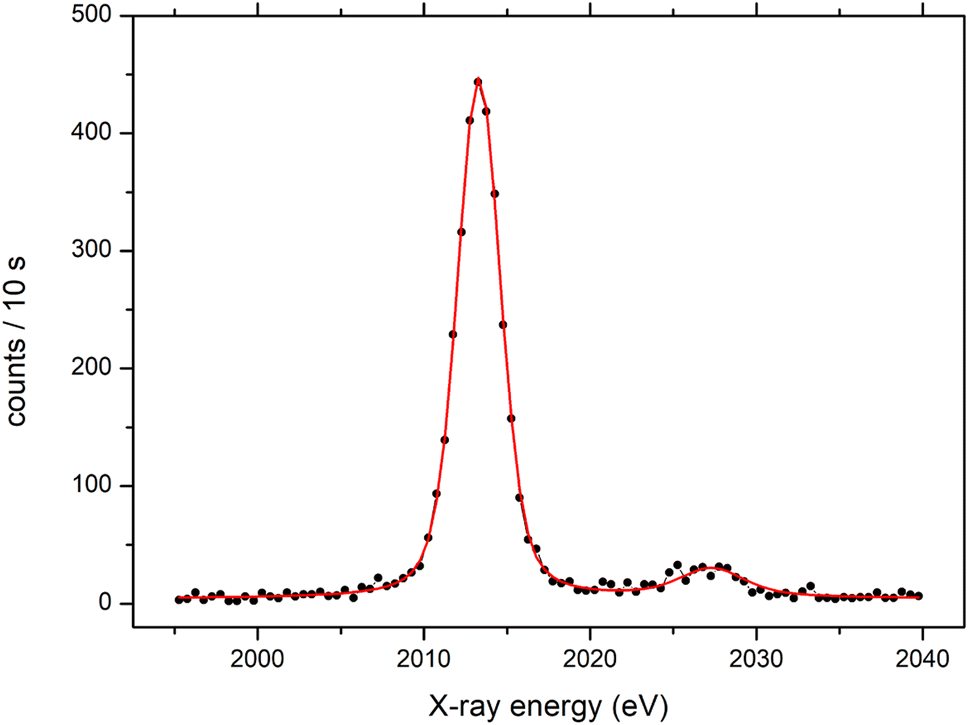
The final experimental energy resolution was determined by fitting a model spectrum to the experimental Kα spectra recorded on a set of reference targets. The model was built from two Voigt profiles with the Lorentzian width fixed to match the natural K, L atomic level widths.28 The Gaussian width of both Voigt profiles describing the spectrometer response was a free parameter in this fitting procedure, and the experimental energy resolution is given as a full width at half maximum (FWHM) value of the Gaussian component. In addition to the Kα1,2 doublet, another Voigt profile on the higher energy side of the Kα line was added to the model to account for the KαL double ionization satellite line. An example of such a fit to the P Kα spectrum measured on the KH2PO4 target is presented in Fig. 5 yielding a final energy resolution value of 3.5 eV ± 0.3 eV. In the same way, we have fitted the model spectrum to the rest of the measured Kα spectra, and the extracted energy resolution values are plotted in Fig. 6. The absolute energy resolution values are within the 3.5–23 eV range yielding resolving power ΔE/E ∼ 1.2–3.8 × 10−3 which is an order of magnitude lower than the resolution obtained with the energy dispersive detectors.
 |
| | Fig. 5 P Kα spectrum of a KH2PO4 target fitted with the model presented in the text. | |
 |
| | Fig. 6 Experimental energy resolution values determined from the fit of the model spectrum to the measured reference Kα spectra. The calculated energy resolution curves employing the divergence of the polycapillary lens are also plotted. | |
The dependence of the spectrometer energy resolution as a function of X-ray energy is obtained from a derivative of the Bragg equation
where
E is the energy of the measured X-ray line,
θB is the corresponding Bragg angle and Δ
θB represents the convolution of the beam divergence exiting the polycapillary semi-lens Δ
θpoly with the rocking curve of the analyzer crystal Δ
θcry. In our case the latter is much smaller than the divergence of the polycapillary semi-lens and

so the energy resolution is defined mainly by the divergence of the polycapillary lens. We have calculated the beam divergence for every energy resolution value obtained from the measured Kα spectra, and the energy dependence of the extracted points was fitted with the Δ
θpoly(
E) =
A/
E +
B model function where
E is the X-ray energy and
A and
B were arbitrary constants. The corresponding curves obtained with this fit are shown in
Fig. 6 for each analyzer crystal.
3.2 Detection limits
Typical X-ray spectra from reference targets presented in Fig. 4 exhibit relatively high count rates at the level of a few times 103 counts per s at the top of the Kα line. The detection limits for the new PB-WDS set-up were determined for several elements by measuring the Kα spectra from the NIST1573a standard reference material. The reported values were calculated using 3σ definition. The sigma value was determined as a square root of the measured background level. Next, the ratios of the 3σ value and the X-ray yields recorded at the top of each corresponding Kα line (see Fig. 7) were multiplied with the certified reference concentrations to reach the final numbers. Detection limits of 31 ppm ± 4 ppm for P (Kα line energy = 2013 eV), 45 ppm ± 5 ppm for K (Kα line energy = 3312 eV), 128 ppm ± 25 ppm for Ca (Kα line energy = 3690 eV), 27 ppm ± 9 ppm for Mn (Kα line energy = 5894 eV), and 37 ppm ± 8 ppm for Fe (Kα line energy = 6398 eV) were achieved at acquisition times of 10 s per point and a proton current of around 5–6 nA. As an example, the P Kα spectrum measured from the NIST1573a standard reference material is presented in Fig. 7.
 |
| | Fig. 7 P Kα spectrum of the NIST1573a reference standard containing 2161 ± 28 ppm of phosphorus. | |
3.3 PIXE mapping
With its high-energy resolution the new PB-WDS spectrometer complements the energy dispersive detectors used in a standard PIXE analysis. The main goal is to apply it for an in-air micro-PIXE mapping in parallel with the Si(Li) detector used at our external proton beamline. During mapping the spectrometer is set to the Bragg angle corresponding to the energy of the main spectral line (either Kα for low-Z or Lα for mid-Z elements), and the intensity is recorded as a function of a sample position, which is raster scanned across the proton beam to build a two-dimensional elemental map. In principle, the combination of a polycapillary half lens with the focused proton beam defines a confocal geometry and thus, a probing volume. It is therefore important that the sample surface is flat enough to keep it within this probing volume during lateral sample movement. In order to test the whole procedure, we initially mapped a test object, which was a 20 μm thick 150/inch Ti mesh (44 μm bar width and 125 μm hole width). The raster scan consisted of 50 × 100 points, with a step size of 40 μm and an acquisition time of 3 s per point. To reach comparable count rates on both detectors, a 50 μm Al absorber was placed in front of the Si(Li) detector. Both maps were recorded simultaneously with the new PB-WDS and the Si(Li) detector and are shown in Fig. 8. The excellent correspondence between them confirms the applicability of the new PB-WDS setup for PIXE mapping.
 |
| | Fig. 8 PIXE map of the 150/inch Ti grid recorded simultaneously with the Si(Li) detector (left) and with the new PB-WDS setup (right). The color scale represents measured counts. | |
4. First application in a biological study
In our laboratory, micro-PIXE mapping has been routinely used to determine the distribution of elements in biological samples, particularly in plants. Resolving element distributions within organs and tissues is essential for understanding complex processes that take place within them. Because biological samples contain many elements in different concentrations, the detection of certain elements and determination of their distribution is sometimes challenging. One such element is Cd. The first reason for this is because Cd is toxic at very low concentrations (around 10 ppm for plants), which means only low Cd concentrations are typically found in living organisms. However, extraordinary plants called hyperaccumulators can accumulate and tolerate more than 100 ppm Cd concentrations; for example, in one such plant, Noccaea (formerly Thlaspi) praecox, more than 6000 ppm Cd has been found.29 Even if this high Cd-sample is measured, the second reason, of spectroscopic origin, hinders PIXE analysis. The Cd Lα (3134 eV) and potassium Kα (3312 eV) lines overlap in a PIXE spectrum. Potassium is an essential element and is present in high concentrations (up to 20000 ppm) making its intense Kα line completely overwhelm the Cd Lα signal. When accessible, Cd K emission lines can be used, but due to the low Cd σK ionization cross section, this yields very low count rates and a poor contrast of the resulting distribution maps.30,31 Alternatively, a PB-WDS spectrometer can be used, and here, we exemplify its use for the detection and mapping of Cd distribution in N. praecox leaves.
Initially, a single PIXE spectrum from powdered leaves of N. praecox pressed into a pellet containing 1300 ppm 32 of Cd was recorded resulting in clear separation of the Cd Lα line from the major potassium and calcium K lines (Fig. 9). Secondly, the new setup was used to map the Cd distribution along the leaf of N. praecox. The leaf was removed from a flowering plant collected at a heavy metal polluted site in Žerjav, Slovenia,29 placed between Al foils, frozen in liquid nitrogen and freeze dried for three days at −97 °C and 0.001 mbar (CoolSafe, LaboGene, Allerød, Denmark). The dried leaf was sandwiched between two pieces of 1 μm Mylar foil attached to Al holders, mounted on the target goniometer and mapped using a 3 MeV focused proton beam with a beam current of 0.6 nA. The map was built from 100 × 100 points with a step size of 70 μm and a 3 seconds per point acquisition time. During mapping the spectrometer was set to the Bragg angle corresponding to the energy of the Cd Lα line. The Si(Li) detector (see Fig. 2) was used in parallel to record the standard EDS PIXE spectra. A 25 μm Kapton filter was placed in front of the Si(Li) detector to protect it from backscattered protons. Both Cd maps recorded simultaneously are presented in Fig. 10. The Cd Lα line detection provided by the PB-WDS enhanced the measured yield and improved the contrast in the Cd distribution map, compared to the one recorded with standard Si(Li) using Cd K line detection.
 |
| | Fig. 9 PIXE spectrum of a Noccaea praecox powder pellet containing around 1300 ppm32 of Cd recorded with the PB-WDS spectrometer. The spectrum was induced with 3 MeV protons; a beam current of 0.9 nA was used to scan the K and Ca energy range and it was increased to 3.8 nA during the scan over the Cd Lα range. Measured intensities of both scans were normalized to the proton dose measured with the beam chopper. | |
 |
| | Fig. 10 Cadmium distribution maps in a Noccaea praecox leaf recorded with the PB-WDS spectrometer using the Cd Lα line (left), and the Si(Li) detector using the Cd K lines (right). The color scales represent measured counts. | |
The proton beam current used in the PIXE mapping is limited by the count rate of the Si(Li) detector, which is governed by the high signal of major elements within the sample (K, Ca). When using the new PB-WDS setup, we are collecting a signal from one (minor or trace) element only and significantly higher proton current can be applied without affecting the spectrum quality. For that reason, we collected another Cd map with the PB-WDS spectrometer with an increased proton beam current of 5 nA (Fig. 11). In this case, the signal is enhanced further, yielding even higher contrast in the measured map clearly revealing the localization of Cd within small leaf regions (50–100 μm size) mainly at the tip of the leaf. A drawback in this case is increased radiation damage of the sample induced by high proton current.
 |
| | Fig. 11 Cadmium distribution within a Noccaea praecox leaf recorded with the PB-WDS spectrometer using high proton beam current. The color scale represents measured counts. | |
5. Conclusion
The parallel-beam wavelength-dispersive X-ray spectrometer developed and installed at the external beamline at the Microanalytical Centre of the Jožef Stefan Institute (JSI) in Ljubljana has been described. The spectrometer provides PIXE measurements in the tender X-ray range with energy resolution ΔE/E of ∼ 1–4 × 10−3 and sensitivity at the level of few tens of ppm. These are provided by polycapillary optics yielding high collection efficiency and diffraction by a flat crystal analyzer. A detailed description of the instrument is given with the results of characterization measurements, yielding quantitative analysis of the main operational parameters. Finally, the first example of an application for in-air PIXE mapping is given, where the capabilities of the new setup proved to be essential for successful analysis. With its high energy resolution, the spectrometer presents a novel complementary tool to the energy dispersive solid-state detectors used commonly in PIXE analysis and significantly enhances the analytical capabilities of our external proton beamline.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the RADIATE project under the Grant Agreement 824096 from the EU Research and Innovation Programme HORIZON 2020 and by the Slovenian Research Agency through the research programs P1-0112 and P1-0212, and project J4-3091. Excellent assistance of the Microanalytical Centre team with the preparation of the proton beam is acknowledged.
References
- J. Simčič, P. Pelicon, M. Budnar and Ž. Šmit, Nucl. Instrum. Methods Phys. Res., Sect. B, 2002, 190, 283–286 CrossRef.
- P. Vavpetič, K. Vogel-Mikuš, L. Jeromel, N. O. Potočnik, P. Pongrac, D. Drobne, Ž. P. Tkalec, S. Novak, M. Kos, Š. Koren, M. Regvar and P. Pelicon, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 348, 147–151 CrossRef.
- P. Pongrac, R. Tolrà, R. Hajiboland, K. Vogel-Mikuš, M. Kelemen, P. Vavpetič, P. Pelicon, J. Barceló, M. Regvar and C. Poschenrieder, Food Chem. Toxicol., 2020, 135, 110974 CrossRef CAS PubMed.
- P. Pongrac, H. Castillo-Michel, J. Reyes-Herrera, R. D. Hancock, S. Fischer, M. Kelemen, A. J. Thompson, G. Wright, M. Likar, M. R. Broadley, P. Vavpetič, P. Pelicon and P. J. White, BMC Plant Biol., 2020, 20, 368 CrossRef CAS PubMed.
- E. Punzón-Quijorna, M. Kelemen, P. Vavpetič, R. Kavalar, P. Pelicon and K. S. Fokter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2020, 462, 182–186 CrossRef.
- K. Isaković, M. Petric, Z. Rupnik, Ž. Šmit, P. Pelicon, M. Kelemen, M. Vereš, P. Pongrac, P. Vavpetič and M. Kavčič, Nucl. Instrum. Methods Phys. Res., Sect. B, 2022, 510, 69–75 CrossRef.
- M. R. J. Palosaari, K. M. Kinnunen, J. Julin, M. Laitinen, M. Napari, T. Sajavaara, W. B. Doriese, J. Fowler, C. Reintsema, D. Swetz, D. Schmidt, J. Ullom and I. J. Maasilta, J. Low Temp. Phys., 2013, 176(3–4), 285–290 Search PubMed.
- P. C. Chaves, A. Taborda, J. P. Marques and M. A. Reis, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 318, 60–64 CrossRef CAS.
- M. Käyhkö, M. Laitinen, K. Arstila, I. J. Maasilta and T. Sajavaara, Nucl. Instrum. Methods Phys. Res., Sect. B, 2019, 447, 59–67 CrossRef.
- J. Hasegawa, T. Tada, Y. Oguri, M. Hayashi, T. Toriyama, T. Kawabata and K. Masai, Rev. Sci. Instrum., 2017, 78, 073105 CrossRef PubMed.
- K. Maeda, K. Hasegawa, M. Maeda, K. Ogiwara and H. Hamanaka, X-Ray Spectrom., 2005, 34, 389–392 CrossRef CAS.
- M. Kavčič, M. Budnar, A. Mühleisen, F. Gasser, M. Žitnik, K. Bučar and R. Bohinc, Rev. Sci. Instrum., 2012, 83, 033113 CrossRef PubMed.
- M. Kavčič, A. Karydas and C. Zarkadas, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 222, 601–608 CrossRef.
- Z. Kertesz, E. Furu and M. Kavčič, Spectrochim. Acta, Part B, 2013, 79–80, 58–62 CrossRef CAS.
- M. Petric and M. Kavčič, J. Anal. At. Spectrom., 2016, 31, 450–457 RSC.
- M. Kavčič, M. Petric and K. Vogel-Mikuš, Nucl. Instrum. Methods Phys. Res., Sect. B, 2018, 417, 65–69 CrossRef.
- M. Kavčič, M. Petric, A. Rajh, K. Isaković, A. Vizintin, S. D. Talian and R. Dominko, ACS Appl. Energy Mater., 2021, 4, 2357–2364 CrossRef PubMed.
- M. Petric, A. Rajh, A. Vizintin, S. Drvarič Talian, R. Dominko and M. Kavčič, Chem. Commun., 2021, 57, 7573–7576 RSC.
- S. Fazinić, I. B. Mihalić, T. Tadić, D. Cosic, M. Jakšić and D. Mudronja, Nucl. Instrum. Methods Phys. Res., Sect. B, 2015, 363, 61–65 CrossRef.
- I. B. Mihalić, S. Fazinić, T. Tadić, D. Cosic and M. Jakšić, J. Anal. At. Spectrom., 2016, 31, 2293–2304 RSC.
- C. van Hoek and M. Koolwijk, Microchim. Acta, 2008, 161, 287–293 CrossRef CAS.
- J. Szlachetko, M. Cotte, J. Morse, M. Salomé, P. Jagodzinski, J.-C. Dousse, J. Hoszowska, Y. Kayser and J. Susini, J. Synchrotron Radiat., 2010, 17, 400–408 CrossRef CAS PubMed.
- M. Kavčič, M. Petric, F. Gasser, Z. Rupnik, B. Jenčič, M. Kelemen, P. Pelicon and P. Vavpetič, Nucl. Instrum. Methods Phys. Res., Sect. B, 2017, 404, 81–86 CrossRef.
- M. Petric, R. Bohinc, K. Bučar, M. Žitnik, J. Szlachetko and M. Kavčič, Anal. Chem., 2015, 87, 5632–5639 CrossRef CAS PubMed.
- M. Petric, R. Bohinc, K. Bučar, S. H. Nowak, M. Žitnik and M. Kavčič, Inorg. Chem., 2016, 55, 5328–5336 CrossRef CAS PubMed.
- V. Cindro, M. Budnar, M. Kregar, V. Ramšak and Ž. Šmit, J. Phys. B: At., Mol. Opt. Phys., 1989, 22, 2161–2169 CrossRef CAS.
- M. Budnar, A. Mühleisen, M. Hribar, H. Janžekovič, M. Ravnikar, Ž. Šmit and M. Žitnik, Nucl. Instrum. Methods Phys. Res., Sect. B, 1992, 4, 377–383 CrossRef.
- J. L. Campbell and T. Papp, At. Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef CAS.
- K. Vogel-Mikuš, D. Drobne and M. Regvar, Environ. Pollut., 2005, 133, 233–242 CrossRef PubMed.
- K. Vogel-Mikuš, J. Simčič, P. Pelicon, M. Budnar, P. Kump, M. Nečemer, J. Mesjasz-Przybylowicz, W. J. Przybylowicz and M. Regvar, Plant, Cell Environ., 2008, 31, 1484–1496 CrossRef PubMed.
- K. Vogel-Mikuš, M. Regvar, J. Mesjasz-Przybylowicz, W. J. Przybylowicz, J. Simčič, P. Pelicon and M. Budnar, New Phytol., 2008, 179, 712–721 CrossRef PubMed.
- M. Nečemer, P. Kump, J. Ščančar, R. Jaćimović, J. Simčič, P. Pelicon, M. Budnar, Z. Jeran, P. Pongrac, M. Regvar and K. Vogel-Mikuš, Spectrochim. Acta, Part B, 2008, 63, 1240–1247 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
A.
Rajh
bd,
Z.
Rupnik
b,
M.
Ribič
b,
K.
Bučar
bd,
P.
Pelicon
b,
P.
Pongrac
bc,
A.
Rajh
bd,
Z.
Rupnik
b,
M.
Ribič
b,
K.
Bučar
bd,
P.
Pelicon
b,
P.
Pongrac





 so the energy resolution is defined mainly by the divergence of the polycapillary lens. We have calculated the beam divergence for every energy resolution value obtained from the measured Kα spectra, and the energy dependence of the extracted points was fitted with the Δθpoly(E) = A/E + B model function where E is the X-ray energy and A and B were arbitrary constants. The corresponding curves obtained with this fit are shown in Fig. 6 for each analyzer crystal.
so the energy resolution is defined mainly by the divergence of the polycapillary lens. We have calculated the beam divergence for every energy resolution value obtained from the measured Kα spectra, and the energy dependence of the extracted points was fitted with the Δθpoly(E) = A/E + B model function where E is the X-ray energy and A and B were arbitrary constants. The corresponding curves obtained with this fit are shown in Fig. 6 for each analyzer crystal.