
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the interaction mechanism of carbazole/anthracene with N,N-dimethylformamide: NMR study substantiated carbazole separation†
Hui
Cao‡
ab,
Mengyu
Dou‡
ab,
Zexiang
Lyu‡
ab,
Yingxiong
Wang
 ab,
Christian Marcus
Pedersen
c and
Yan
Qiao
*ab
ab,
Christian Marcus
Pedersen
c and
Yan
Qiao
*ab
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, 27 South Taoyuan Road, Taiyuan 030001, China. E-mail: qiaoy@sxicc.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark
First published on 16th November 2022
Abstract
Carbazole and anthracene, two aromatic hydrocarbon components contained in coal tar, are used as essential organic intermediates to synthesize various carbazole derivatives and anthraquinones. N,N-Dimethylformamide (DMF) is a commonly used solvent to extract carbazole from crude mixtures of carbazole and anthracene. However, the interaction between carbazole/anthracene and DMF in the extraction process is still to be fully understood at the molecular level. In this work, the intermolecular interaction of carbazole/anthracene with DMF was investigated using various NMR techniques, including 1H NMR titration, variable temperature NMR spectroscopy (VT-NMR), Nuclear Overhauser Effect Spectroscopy (NOESY), and diffusion-ordered spectroscopy (DOSY). The observed 1H chemical shift changes of carbazole indicated strong intermolecular hydrogen bonds between carbazole and DMF, which was further supported by the decrease in the molecular self-diffusion coefficients (D) of both carbazole and DMF according to DOSY measurements. Moreover, NOESY experiments revealed that the distance between the aldehydic hydrogen of DMF and the N–H of carbazole was smaller than 5 Å. Accordingly, an intermolecular hydrogen bond between carbazole and DMF in the form of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N was proposed. This research increases our knowledge about the separation process of carbazole and anthracene and hence helps improve the methods.
O⋯H–N was proposed. This research increases our knowledge about the separation process of carbazole and anthracene and hence helps improve the methods.
Keywords: NMR; Carbazole; Separation; Intermolecular hydrogen bonds.
1 Introduction
The by-product of coal carbonization, coal tar, adds up to an annual production of more than 20 million tons.1 Commonly, tens of thousands of organic material are contained in coal tar; however, only about 500 valuable chemical products have been identified, and these are widely used in the chemical, agricultural and pharmaceutical industries.2 Crude anthracene oil, a main fraction of coal tar, is rich in aromatic hydrocarbon components, such as anthracene, phenanthrene and carbazole. Anthracene is used as a raw material for the production of anthraquinone, which is used to prepare bio-degradable materials,3 colorants,4 anti-diabetics,5,6 and photoredox catalysts.7 Phenanthrene and carbazole are used for synthetic dyes,8 pesticides,9 pharmaceuticals10 and photoelectric materials.11 Along with the development of fine chemicals and the advancement of organic synthesis, there is an increasing demand for high purity anthracene, phenanthrene, and carbazole. At present, these three compounds are mainly obtained by separation and purification of crude anthracene oil.12Physical separation methods, such as crystallization,13 distillation, zone melting14,15 and supercritical fluid extraction,16,17 are the main ways to obtain anthracene, phenanthrene and carbazole. Due to their close boiling points, i.e. 340.7 °C (anthracene), 340.2 °C (phenanthrene) and 354.8 °C (carbazole), the separation of these compounds by distillation is a time and energy consuming process.18 A separation method based on an emulsion membrane requires high stability of the separation system.19 Zone melting technology demands complex operation steps and strict temperature control.20,21 Among these methods, crystallization is an effective method to separate anthracene, phenanthrene and carbazole from anthracene oil, and readily available solvents are the key factor for the separation. Because the solubility of phenanthrene in non-polar solvents is higher than the solubility of anthracene and carbazole, phenanthrene can be extracted from crude anthracene oil with benzene and its derivatives (e.g. benzene, toluene and xylene). For the separation of anthracene and carbazole, the difference in the solubility of carbazole and anthracene is key for the solvent selection. However, solvent screening is still lacking theoretical guidance, with most studies working on the basis of “like dissolves like”.18 Recently, DMF has been developed as an efficient solvent for carbazole and anthracene separation due to the high solubility of carbazole in DMF. Moreover, researchers have found that the separation of carbazole and anthracene benefits from an intermolecular hydrogen bond between carbazole and DMF.18,22 In most cases, hydrogen bonding can enhance the solubility greatly and research on solubilization, mediated by hydrogen bonding, has received significant attention in both academia and industry.23 Although it is well known that the hydrogen bonds between carbazole and the solvent promote the solubility of carbazole, there is no detailed study concerning the interaction mechanism between carbazole/anthracene and solvent capable of hydrogen bonding. It is important to use a versatile detection technique for analyzing hydrogen bond interactions, and hence to explain the interaction mechanism between carbazole/anthracene and DMF via hydrogen bonding on the molecular level.
NMR spectroscopy is routinely available in most chemistry research facilities, and liquid-state NMR24 is a high-resolution and non-invasive technology. NMR signals represent the different environments of atomic nuclei and provide rich information on structural details. Thus, NMR spectroscopy has developed into a powerful analytical tool for studying molecular interactions. Our research group has studied the interaction between the macromolecule PAMAM–NH2 and small guest molecules through various NMR techniques. For example, we provided a systematic NMR method monitoring the interaction between PAMAM and 6-mercaptopurine 6-MP, confirming that 6-MP enters the PAMAM–NH2 cavity and interacts with amide groups in the internal dendrimer structure via hydrophobic interactions and hydrogen bonding.25 NMR spectroscopy is very powerful in characterizing hydrogen bonds due to its multinuclear and multidimensional detection.24,26 For instance, chemical shift titration can show the signals of H-bonded protons, which move downfield because of the diamagnetic shielding effect.25 The pure shift NMR method is very effective at reducing signal overlap such as the recently reported advanced diffusion NMR method, Pure Shift Yielded by CHirp Excitation DOSY (PSYCHE-iDOSY),27 which can record a high resolution signal diffusion spectrum, facilitating composition analysis and structure interaction. Other 1D and 2D methods such as studies of the change of T1 relaxation,28 observation of intermolecular NOE,29 measurement of spin–spin coupling constants and diffusion-ordered spectroscopy (DOSY)30 are also very effective. The NOE has long been well recognized as a powerful tool for investigating hydrogen bonds.31 Recently, DOSY has been used as a qualitative probe for detecting hydrogen bond strength.30 With the aid of NMR information, we aim at a better understanding of the hydrogen bonding between DMF and carbazole leading to the solubilization of carbazole, and eventually helping in the development of methods for separating similar structures more effectively.
In this article, we study the intermolecular interaction mechanism between DMF and carbazole/anthracene by various advanced NMR techniques. Chemical shift titration and the DOSY technique provide a new insight into the interaction between DMF and carbazole and demonstrate the existence of hydrogen bonds. Pure shift NMR suppresses the J coupling of multiple structures and solves the dilemma of overlapping 1H spectra. NOESY experiments show the correlation signal between carbazole and DMF, indicating that the spatial distance of the aldehydic hydrogen and the active hydrogen is small, thus indicating hydrogen bonding between carbazole and DMF. The influence of temperature on hydrogen bonding was also investigated and finally a possible mode of interaction between carbazole and DMF is provided.
2 Results and discussion
The 1H and 13C NMR spectra of pure carbazole, anthracene and DMF are shown in Fig. S1–S4 and S7 and S8.† With the assistance of the 1H–1H COSY spectrum (Fig. S5†) and the 1H–13C HSQC spectrum (Fig. S6†), each signal of carbazole, anthracene and DMF could be assigned in Table 1.| Compound | Molecular structure | Atom number | δ C (ppm) | δ H (ppm) |
|---|---|---|---|---|
| Carbazole |

|
1, 12 | 120.7 | 7.99 |
| 3, 10 | 125.9 | 7.35 | ||
| 2, 11 | 119.6 | 7.19 | ||
| 4, 9 | 110.8 | 7.05 | ||
| N–H | — | 6.52 | ||
| Anthracene |
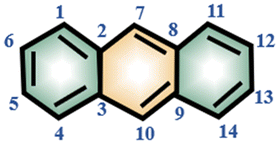
|
1, 4, 11, 14 | 128.2 | 7.79 |
| 2, 3, 8, 9 | 131.9 | — | ||
| 5, 6, 12, 13 | 125.2 | 7.24 | ||
| 7, 10 | 126.4 | 8.15 | ||
| DMF |

|
1 | 161.7 | 7.66 |
| 2 | 30.7 | 1.92 | ||
| 3 | 35.1 | 2.39 |
2.1 Chemical shift titration
In order to investigate the interaction between DMF and carbazole (or DMF and anthracene) by NMR, a small quantity of DMF was added to a mixture of carbazole and anthracene in d6-benzene. The chemical shifts of anthracene remained essentially unchanged (Fig. 1), which suggests insignificant intermolecular interactions between anthracene and DMF (showed with the orange bars). However, after adding DMF, the signal at 6.52 ppm, i.e. N–H of carbazole, clearly shifted downfield. | ||
| Fig. 1 The 1H NMR spectra of mixtures of carbazole and anthracene before and after adding DMF. 2.0 mg carbazole and 2.0 mg anthracene in 400 μL C6D6 at 25 °C. Orange bars indicate the signals of anthracene. | ||
The chemical shifts of carbazole protons 1, 3, 4, 9, 10, and 12 do also increase to varying degrees (pointed out with different colored arrows). These shift changes indicate intermolecular interactions between carbazole and DMF. Further investigation of the interaction of carbazole and DMF was conducted using chemical shift titration experiments.
Fig. 2a shows a series of chemical shift titration spectra of carbazole. With the gradual addition of DMF, the signals of carbazole move downfield. The green arrows label the variation of chemical shift of the N–H proton, from 6.52 ppm to 9.44 ppm, indicating an obvious trend of signal change to a lower field. Another distinct signal change belongs to H-4 and H-9 in the carbazole molecule, which changes from 7.05 ppm to 7.45 ppm. The resonance of protons 1 and 13 slightly shift to a lower field, from 7.99 to 8.06 ppm. The signals at 7.35 (protons 3 and 10) and 7.19 (protons 2 and 11) ppm moved about 0.02 ppm downfield, and the chemical shift changes were not obvious. The chemical shift of protons H-4 and H-9 moved slightly more downfield than those of the other protons of benzene rings (Fig. 2a). DMF (Fig. 3) contains two electronegative centers, which are negatively polarized (δ−) O and N. Because of the steric hindrance caused by the presence of the two methyl groups, hydrogen bonding between N and carbazole is much less favorable than that between O and carbazole. Furthermore, O is more electronegative and contains two free lone pairs, which are H-bond acceptors. Therefore, it is most likely that the intermolecular hydrogen bond, formed between carbazole and DMF, is CO⋯H–N, as shown in Fig. 3. The effects of hydrogen bonding on the chemical shift of the aromatic hydrogens have been mentioned in previous studies.32 The change in chemical shifts originating from the aromatic hydrogens near the hydrogen bond position is relatively obvious. It is well established that hydrogen bonding of phenolic compounds results in a change of the resonance of the shifts from para-H to a higher magnetic field than that from ortho-H, which move about 0.1–0.3 ppm downfield.33
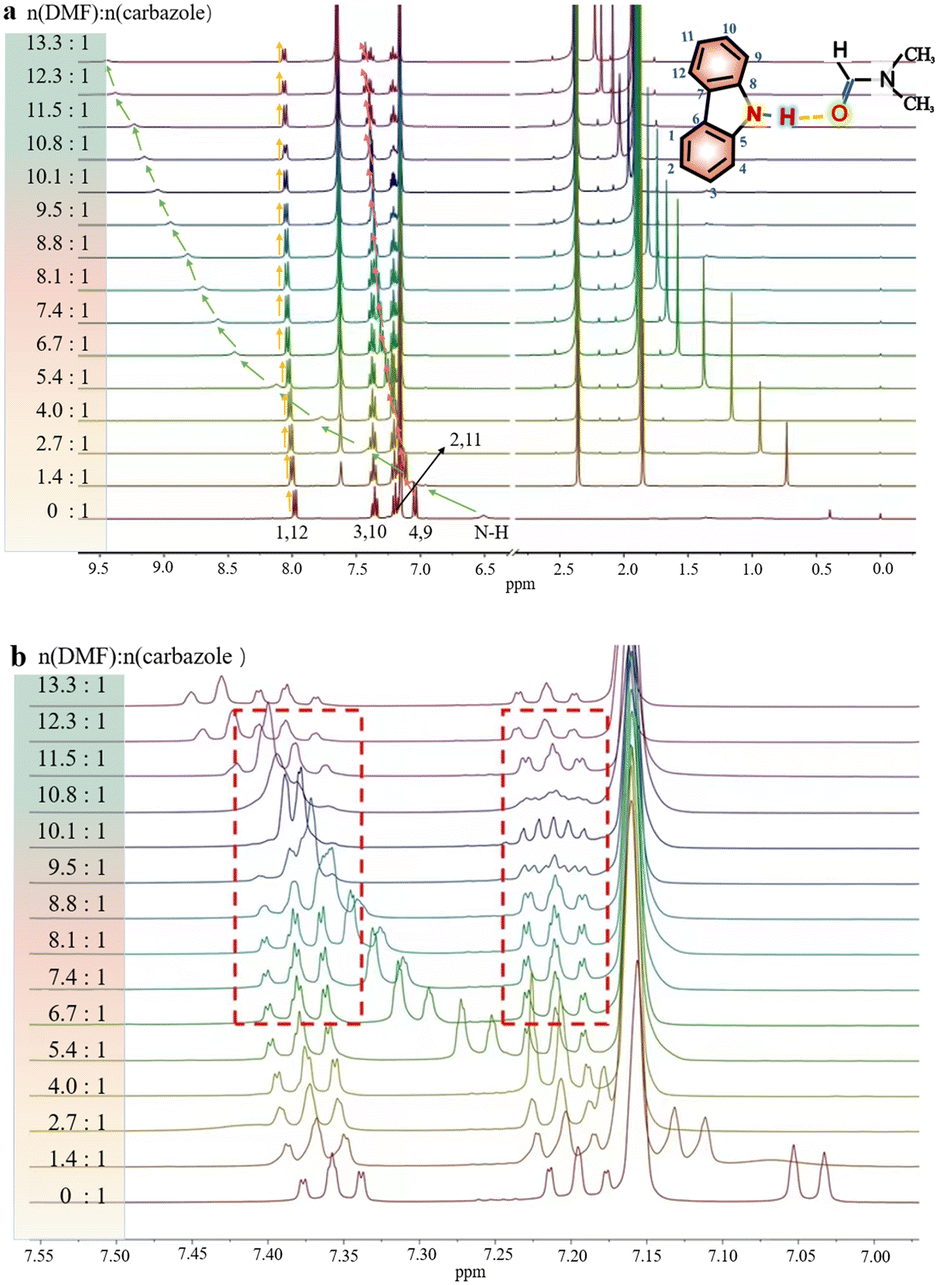 | ||
| Fig. 2 (a) The 1H NMR spectra of 2.0 mg carbazole with increasing concentration of DMF in deuterated benzene and at 25 °C. Referenced to tetramethylsilane (TMS). (b) The enlarged part between 7.15 and 7.45 ppm to show the variation of carbazole signals. | ||
 | ||
| Fig. 3 Schematic diagram of the intermolecular hydrogen bond between carbazole and DMF. | ||
Hence, upon gradual addition of DMF, the carbazole signals move downfield to different degrees, according to the π system of benzene ring conjugation with the p orbital of the N atom,34 resulting in the electron delocalization around carbazole. The hydrogen bond interactions in the system result in a decrease in diamagnetic shielding around the bridging proton, resulting in a proton shift to a lower field.35
Interestingly, the spectral splitting mode of carbazole protons between 7.15 and 7.45 ppm also clearly changed (Fig. 2b). For example, the splitting of peaks of carbazole at 7.20 ppm became very complex when 8.3 mg DMF was added to a solution containing 2.0 mg carbazole. The pure shift NMR method can diminish the influence of J-coupling.
As an example, upon the addition of 8.3 mg DMF to 2.0 mg carbazole (a molar ratio = 9.5), the pure shift spectrum compared with the traditional 1H NMR spectrum is significantly simplified, facilitating the identification of carbazole proton peaks (Fig. S9†). However, when 10.8 mg DMF was added to the solution of carbazole, the signals at 7.20 ppm seemed to change to give a triplet. The signals at 7.34 ppm also change dramatically during the addition of DMF. The change of signals in the mixture with 8.9 mg DMF and 2.0 mg carbazole (a molar ratio = 10.1) is the most obvious. The peaks at 7.20 ppm split to multiple peaks. We speculate that the underlying reason for this change is that DMF molecules construct a hydration shell around the molecule of carbazole.36 When the molar ratio of DMF to carbazole was 10.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the number of DMF molecules around each carbazole molecule reached a critical value, the hydration shell no longer expanded.
:1 and the number of DMF molecules around each carbazole molecule reached a critical value, the hydration shell no longer expanded.
Another signal change is at high field, where the signal at 0.40 ppm shifts to 2.23 ppm. This signal is assigned to the trace amount of water in the reagents, and the formation of hydrogen bonds between DMF and water makes this signal shift to a lower field.
2.2 VT-NMR experiments
Variable-temperature 1H NMR spectroscopy is often used to characterize the hydrogen bond strength as hydrogen bonding is temperature sensitive and can gradually weaken and even be broken with increasing temperature. Fig. 4 shows the VT-NMR spectra of a mixture of carbazole, anthracene and DMF. With the decrease of temperature, the signal of active hydrogen, i.e. the N–H of carbazole and the proton in H2O, moves to the downfield region, which means that the strength of hydrogen bonds between DMF and carbazole becomes stronger.37 Notably, other chemical shifts from carbazole and anthracene do not change during the cooling process.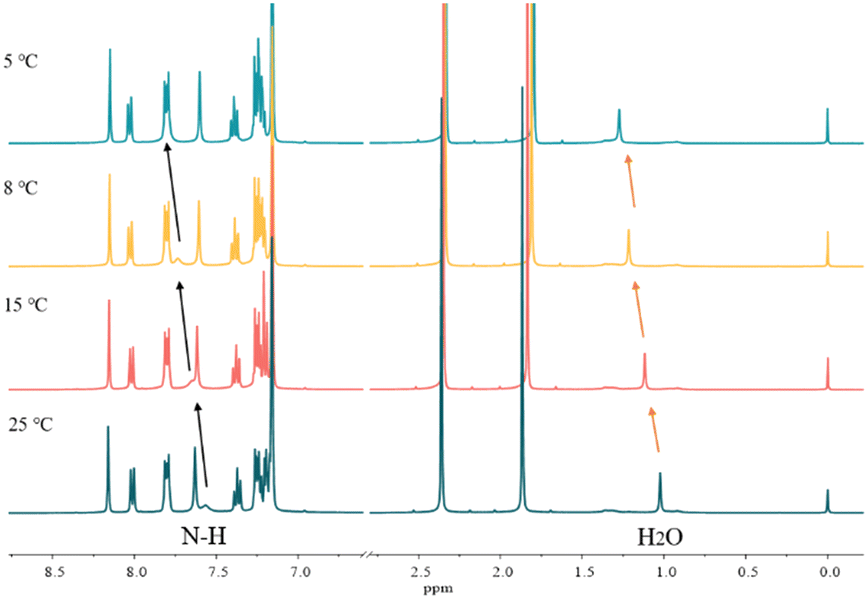 | ||
| Fig. 4 The variable temperature-NMR spectra of 0.2 mg carbazole, 0.2 mg anthracene and 4.0 mg DMF in 400 μL C6D6. | ||
2.3 NOESY measurements
NOESY relies on the spatial relationship between two nuclei. In general, the distance between protons must be <5 Å to generate NOE signals. Thus, NOE spectroscopy is a tool for studying molecular interactions and spatial arrangements, e.g. hydrogen bonds.38Fig. 5 shows the NOESY spectrum of carbazole and DMF at 8 °C. The signals in red circles are the correlation signals between the aldehyde hydrogen in DMF and N–H of carbazole, which indirectly indicate that the spatial distance between the aldehyde oxygen of DMF and N–H of carbazole is less than 5 Å and the form of hydrogen bond is shown in Fig. 3. The NOESY spectra indicate that the formation of hydrogen bonds between carbazole and DMF makes the spatial distance small between CO and N–H.
 | ||
| Fig. 5 The NOESY spectrum of 0.2 mg carbazole, 0.2 mg anthracene and 4.0 mg DMF in 400 μL C6D6 at 8 °C. | ||
2.4 DOSY experiments
DOSY experiments can provide the molecular self-diffusion coefficient (D). The D value is correlated with the fluidity and can reflect the interaction between molecules, such as hydrogen bonds. For a simple spherical molecule, the D value conforms to the Stokes–Einstein equation: D = kBT/(6πηrH), according to which hydrogen bonding can be related.To remove the influence of change in viscosity (η), tetramethylsilane (TMS) is chosen as an internal diffusion reference in our experiments. As described in the Stokes–Einstein equation, the ratio of the diffusion of a solute (D) and the reference TMS (DTMS) depends on the ratio of their hydrodynamic radius: D/DTMS = rHtms/rH. The diffusion of the reference compound in different solutions can be regarded as relatively constant because TMS does not take part in the intermolecular interactions studied, and its diffusion is hence only influenced by the viscosity of solvent. D/DTMS is the relative diffusion coefficient value of the same solute and TMS in different solutions, which is independent of the viscosity. Consequently, the changes in diffusion of hydrogen bond donor and acceptor can be presented as  . In addition, the alteration of hydrodynamic radius of the solute can be described as
. In addition, the alteration of hydrodynamic radius of the solute can be described as  because rHtms is also constant in different solutions.
because rHtms is also constant in different solutions.
With TMS as the internal reference, the diffusion coefficient values of carbazole and anthracene, before and after the addition of DMF, are listed in Table 2. The diffusion curves and DOSY spectra of carbazole and anthracene after DMF addition are shown in Fig. S10 and S11.† All of the diffusion coefficient values decrease after the addition of DMF. The values of ΔD/DTMS of DMF and carbazole are 0.05 and 0.04, respectively, indicating that the diffusion coefficients of carbazole and DMF changed after the addition of DMF.
| Compound | Before DMF addition | After DMF addition | ΔD/DTMS | ΔrH | ||
|---|---|---|---|---|---|---|
| D (10−9 m2 s−1) | D/DTMS | D′ (10−9 m2 s−1) |
|
|||
| a In different samples, the D value of TMS is slightly different. | ||||||
| TMSa | 1.93 ± 0.01 | 1 | 1.89 ± 0.02 | 1 | 1 | |
| DMF | 2.17 ± 0.02 | 1.12 | 2.02 ± 0.03 | 1.07 | 0.05 | 1.05 |
| TMSa | 1.90 ± 0.06 | 1 | 1.89 ± 0.07 | 1 | 1 | |
| Carbazole | 1.34 ± 0.02 | 0.71 | 1.26 ± 0.02 | 0.67 | 0.04 | 1.06 |
| TMSa | 1.93 ± 0.02 | 1 | 1.89 ± 0.04 | 1 | 1 | |
| DMF | 2.17 ± 0.03 | 1.12 | 2.12 ± 0.02 | 1.12 | 0.00 | 1.00 |
| TMSa | 1.91 ± 0.05 | 1 | 1.90 ± 0.03 | 1 | 1 | |
| Anthracene | 1.40 ± 0.03 | 0.73 | 1.39 ± 0.07 | 0.73 | 0.00 | 1.00 |

The diffusion coefficient value of carbazole was more precise than that of DMF and the diffusion coefficient value of carbazole changes more than that of DMF. The changes of the hydrodynamic radius of carbazole and DMF are similar because the values of ΔrH of carbazole and DMF are 1.05 and 1.06, respectively. The diffusion coefficient values obtained from a mixture of carbazole and anthracene, before and after adding DMF, were obtained by DOSY. The result is shown in Table 3. It is clear that the addition of DMF decreases the diffusion of carbazole, and the diffusion of anthracene almost stays unaffected. The values of ΔrH also indicate that hydrogen bonds are formed between carbazole and DMF, and the variation of the hydrodynamic radius of carbazole and DMF is equivalent. The DOSY results hence strongly indicate the formation of hydrogen bonds between carbazole and DMF.
| Compound | Without DMF | With DMF | ΔD/DTMS | ΔrH | ||
|---|---|---|---|---|---|---|
| D (10−9 m2 s−1) | D/DTMS | D′ (10−9 m2 s−1) |
|
|||
| TMS | 1.92 ± 0.05 | 1 | 1.81 ± 0.01 | 1 | — | 1 |
| Carbazole | 1.34 ± 0.03 | 0.70 | 1.23 ± 0.03 | 0.68 | 0.02 | 1.03 |
| Anthracene | 1.40 ± 0.02 | 0.73 | 1.32 ± 0.04 | 0.73 | 0 | 1 |
| DMF | 2.17 ± 0.01 | 1.12 | 1.96 ± 0.01 | 1.08 | 0.04 | 1.04 |

3 Conclusion
In conclusion, we have disclosed the intermolecular interaction between DMF and carbazole/anthracene. The chemical shift changes observed for the active hydrogen suggest the hydrogen bond interaction between carbazole and DMF, i.e. CO⋯H–N, and there is no hydrogen bond formation between anthracene and DMF. The NOESY spectra reveal the spatial distance between carbazole and the “aldehydic” hydrogen on DMF, which clearly suggests that the distances between the hydrogens are shorter than 5 Å, which further supports the formation of hydrogen bonds between carbazole and DMF. Additionally, the DOSY results strongly indicate the information of intermolecular hydrogen bonds between carbazole and DMF. The proton signals on the carbazole benzene ring moves downfield in varying degrees as affected by hydrogen bonding (CO⋯H–N). The peak shape changes of carbazole between 7.15 and 7.45 ppm are suggested to be caused by DMF constructing a hydration shell around the carbazole molecule. It is envisaged that the interaction mechanism between carbazole/anthracene and DMF will provide guidance for efficient screening of carbazole/anthracene separation solvents, and play an important role in promoting the development of coal tar separation industry in the near future.
4 Experimental
4.1 Materials
Carbazole (96%), anthracene (98%) and tetramethylsilane (99%) were purchased from Aladdin Reagent Company (Shanghai). N,N-Dimethylformamide was obtained from Tianjin Tianli Chemical Reagent Co., Ltd. Benzene-D6 (C6D6, 99.5% atom D) was supplied by Cambridge Isotope Laboratories. All samples were used without further purification.4.2 NMR experiments
NMR spectra were acquired on a Bruker AV-III 400 MHz NMR spectrometer (9.39 T), using a 5 mm PABBO BB/19F–1H/D probe with a z gradient coil producing a maximum gradient strength of 0.5 T m−1. Carbazole (2.0 mg) and anthracene (2.0 mg) were dissolved in C6D6 (0.4 mL), and 0, 2.5 and 4.0 mg of DMF were added into three NMR tubes for preliminary experiments. In the chemical shift titration experiments, where the changes in the chemical shifts of carbazole with increasing DMF concentrations are observed, samples were prepared by adding different amounts of DMF into solutions of 2.0 mg carbazole in 0.4 mL C6D6. The 1D pure shift NMR data can be reconstructed using the “pshift” function from the Topspin software pulse program. The “psyche.mf” file was set as an acquisition pulse sequence. Other acquisition parameters were set as follows: sweep width (SWH) 10000 Hz or 24 ppm, number of dummy scans (DS) 2 and number of scans (NS) 8, respectively. The spectra of carbazole, anthracene and DMF in C6D6 were acquired at variable temperatures, i.e. 5, 8, 15, and 25 °C. The NOE spectra were determined using the noesygpphpp pulse sequence. 1H DOSY experiments were performed with the Bruker standard bipolar pulse longitudinal eddy current delay (BPPLED) pulse sequence. Each DOSY NMR experiment collected 16 BPPLED spectra with 32k data points. The diffusion time (Δ) was 50 ms. The duration of the pulse field gradient (δ/2) was adjusted to be between 730 and 1100 μs and to obtain 2–95% residual signals with the maximum gradient strength. The delay for gradient recovery was 0.2 ms and the eddy current delay was 5 ms. The gradient strength was incremented in 16 steps from 2% to 95% of its maximum value in a linear ramp. The data were processed with Bruker Topspin 3.1 software and the DOSY plots were obtained using Dynamics Center 2.2.4 software.
Author contributions
Hui Cao was responsible for article writing and data analysis and completed some experiments. Mengyu Dou finished writing the original draft. Zexiang Lyu was responsible for designing and completing part of the experiments. Yingxiong Wang was responsible for the logical analysis and revision of the manuscript. Christian Marcus Pedersen was in charge of critical review and revision. Yan Qiao received funding support for this project and has oversight and leadership responsibility for the planning and execution of research activities. All authors who contributed to this article are listed.Conflicts of interest
We state that the manuscript is original and credible and not under consideration for publication and has not been published elsewhere in any medium including electronic journals and computer databases of public nature. All authors are aware of the submission and agree to its publication.Acknowledgements
The authors thank the Natural Science Foundation of Shanxi Province (202103021224439) and the CAS “Light of West China” Program, Local Science and Technology Development Fund Projects Guided by the Central Government (2020ZY0010).References
- X. S. Cong, Separting techniques and application of coal tar, J. Zaozhuang Univ., 2009, 26, 69–72 Search PubMed.
- H. C. Yan, W. Y. Fang, P. Cui, Z. H. Liao, L. X. Wang and Q. S. Li, Current status of processing and utilization of medium and low temperature coaltar, Chem. & Ind., 2019, 48, 4 Search PubMed.
- E. Coutiño-González, B. Hernández-Carlos, R. Gutiérrez-Ortiz and L. Dendooven, The earthworm Eisenia fetida accelerates the removal of anthracene and 9, 10-anthraquinone, the most abundant degradation product, in soil, Int. Biodeterior. Biodegrad., 2010, 64, 525–529 CrossRef.
- N. C. Scherrer, S. Zumbuehl, F. Delavy, A. Fritsch and R. Kuehnen, Synthetic organic pigments of the 20th and 21st century relevant to artist's paints: Raman spectra reference collection, Spectrochim. Acta, Part A, 2009, 73, 505–524 CrossRef PubMed.
- A. Mohammed, M. A. Ibrahim, N. Tajuddeen, A. B. Aliyu and M. B. Isah, Antidiabetic potential of anthraquinones: A review, Phytother. Res., 2020, 34, 486–504 CrossRef CAS PubMed.
- E. M. Malik and C. E. Muller, Anthraquinones As Pharmacological Tools and Drugs, Med. Res. Rev., 2016, 36, 705–748 CrossRef CAS PubMed.
- J. Cervantes-Gonzalez, D. A. Vosburg, S. E. Mora-Rodriguez, M. A. Vazquez, L. G. Zepeda, C. Villegas Gomez and S. Lagunas-Rivera, Anthraquinones: Versatile Organic Photocatalysts, ChemCatChem, 2020, 12, 3811–3827 CrossRef CAS.
- Y. Liu, J. He, L. Han and J. Gao, Influence of the auxiliary acceptor and π-bridge in carbazole dyes on photovoltaic properties, J. Photochem. Photobiol., A, 2017, 332, 283–292 CrossRef CAS.
- L. J. Gut, K. Lee, J. A. Juvik, C. C. Rebeiz, C. E. Bouton and C. A. Rebeiz, Porphyric insecticides. IV: Structure-activity study of substituted phenanthrolines Pest Manag. Sci, Pest Manage. Sci., 1993, 39, 19–30 CrossRef CAS.
- C. P. Vlaar, L. Castillo-Pichardo, J. I. Medina, C. M. Marrero-Serra, E. Velez, Z. Ramos and E. Hernandez, Design, synthesis and biological evaluation of new carbazole derivatives as anti-cancer and anti-migratory agents, Bioorg. Med. Chem., 2018, 26, 884–890 CrossRef CAS PubMed.
- Y. D. Zhang, T. Wada and H. Sasabe, Carbazole photorefractive materials, J. Mater. Chem., 1998, 8, 809–828 RSC.
- D. W. Later, R. B. Lucke, E. K. Chess and J. A. Franz, Separation and identification of carbazole, benz[e]indole and benz[g]indole in coal-derived materials, Fuel, 1987, 66, 1347–1352 CrossRef CAS.
- O. N. Pavlovich, Separation of high-purity phenanthrene and anthracene from the anthracene fraction of coal tar, Coke Chem., 2012, 55, 247–248 CrossRef.
- A. Burel, S. J. T. Brugman, M. Mignot, Y. Cartigny, S. Tisse, N. Couvrat, V. Peulon-Agasse, P. Cardinael and G. Coquerel, Phenanthrene Purification: Comparison of Zone Melting and Co-Crystallization, Chem. Eng. Technol., 2016, 39, 1317–1325 CrossRef CAS.
- N. Couvrat, A. Burel, S. Tisse, Y. Cartigny and G. Coquerel, Combining zone melting and preparative chromatography to purify Phenanthrene, J. Therm. Anal. Calorim., 2012, 112, 293–300 CrossRef.
- F. Esmaeilzadeh and I. Goodarznia, Supercritical extraction of phenanthrene in the crossover region, J. Chem. Eng. Data, 2005, 50, 49–51 CrossRef CAS.
- I. Goodarznia and F. Esmaeilzadeh, Solubility of an anthracene, phenanthrene, and carbazole mixture in supercritical carbon dioxide, J. Chem. Eng. Data, 2002, 47, 333–338 CrossRef CAS.
- C. Ye, X. Ding, W. Li, T. Wu, M. Fan and J. Feng, Highly Efficient Solvent Screening for Separating Carbazole from Crude Anthracene, Energy Fuels, 2016, 30, 3529–3534 CrossRef CAS.
- A. A. Umar, I. B. M. Saaid, A. A. Sulaimon and R. B. M. Pilus, A review of petroleum emulsions and recent progress on water-in-crude oil emulsions stabilized by natural surfactants and solids, J. Pet. Sci. Eng., 2018, 165, 673–690 CrossRef CAS.
- D. Fischer, Study on efficiency of zone-melting of aromatic-hydrocarbons, Mater. Res. Bull., 1973, 8, 385–392 CrossRef CAS.
- M. Tachibana and M. Furusawa, Selective separations of small amounts of naphthacene and 5H-benzo[b]carbazole from analogous polycyclic aromatic-compounds using bibenzyl as a zone-melting medium, J. Mol. Liq., 1991, 251, 241–246 CAS.
- L. Yi, J. Feng, M. Gauthier and W. Li, Effect of the addition of deep eutectic solvent to the anthracene separation, J. Mol. Liq., 2021, 339, 116762 CrossRef CAS.
- K. M. Harmon, S. H. Gill, P. G. Rasmussen and G. L. Hardgrove, Hydrogen bonding. Part 69. Inter- and intramolecular hydrogen bonding effects on the structure, solubility, and reactivity of 4,5-dicarboxyimidazoles, J. Mol. Struct., 1999, 478, 145–154 CrossRef CAS.
- A. Swartjes, P. B. White, M. Lammertink, J. Elemans and R. J. M. Nolte, Host-Guest Exchange of Viologen Guests in Porphyrin Cage Compounds as Studied by Selective Exchange Spectroscopy (1D EXSY) NMR, Angew. Chem., Int. Ed., 2021, 60, 1254–1262 CrossRef CAS PubMed.
- X. Gao, M. Ma, C. M. Pedersen, R. Liu, Z. Zhang, H. Chang, Y. Qiao and Y. Wang, Interactions between PAMAM-NH2 and 6-Mercaptopurine: Qualitative and Quantitative NMR studies, Chem. – Asian J., 2021, 16, 3658–3663 CrossRef CAS PubMed.
- P. Liu, C. M. Pedersen, J. Zhang, R. Liu, Z. Zhang, X. Hou and Y. Wang, Ternary deep eutectic solvents catalyzed d-glucosamine self-condensation to deoxyfructosazine: NMR study, Green Energy Environ., 2021, 6, 261–270 CrossRef.
- Q. Zhao, C. M. Pedersen, J. Wang, R. Liu, Y. Zhang, X. Yan, Z. Zhang, X. Hou and Y. Wang, NMR diffusion analysis of catalytic conversion mixtures from lignocellulose biomass using PSYCHE-iDOSY, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.02.003.
- S. Nagaoka, T. Terao, F. Imashiro, A. Saika, N. Hirota and S. Hayashi, An NMR relaxation study on the proton transfer in the hydrogen bonded carboxylic acid dimers, J. Chem. Phys., 1983, 79, 4694–4703 CrossRef CAS.
- H. B. Seba and B. Ancian, 2-dimensional intermolecular heteronuclear C-13, H-1 overhauser effect as a probe for hydration of the amide group, J. Chem. Soc., Chem. Commun., 1990, 996–997 RSC.
- G. S. Kapur, E. J. Cabrita and S. Berger, The qualitative probing of hydrogen bond strength by diffusion-ordered NMR spectroscopy, Tetrahedron Lett., 2000, 41, 7181–7185 CrossRef CAS.
- C. Sandström, H. Baumann and L. Kenne, The use of chemical shifts of hydroxy protons of oligosaccharides as conformational probes for NMR studies in aqueous solution. Evidence for persistent hydrogen bond interaction in branched trisaccharides, Chem. Commun., 1998, 2385–2393 Search PubMed.
- Y. L. Yang, Y. C. Ho, Y. A. Dyakov, W. H. Hsu, C. K. Ni, Y. L. Sun, W. C. Tsai and W. P. Hu, Effects of intramolecular hydrogen bonding on the excited state dynamics of phenol chromophores, Phys. Chem. Chem. Phys., 2013, 15, 7182–7190 RSC.
- Y. H. Pei, L. Y. Kong, X. Li and T. R. Zhu, Studies on the effect of intramolecular hydrogen bonding on ~ 1H chemical shift of aromatic proton, Bopuxue Zazhi, 1992, 9, 303–306 CAS.
- J. C. Heng, L. S. Liao, P. Zheng and X. T. Xie, Pπ-Conjugation of the aromatic compounds with the elements of group VA or VIA, Chin. J. Org. Chem., 1996, 16, 474–480 Search PubMed.
- S. Xiang, G. Yu, Y. Liang and L. Wu, 1H NMR and DFT studies of steric effects on intermolecular C–H⋯O hydrogen bonding in solution, J. Mol. Struct., 2006, 789, 43–51 CrossRef CAS.
- D. Laage, T. Elsaesser and J. T. Hynes, Water Dynamics in the Hydration Shells of Biomolecules, Chem. Rev., 2017, 117, 10694–10725 CrossRef CAS PubMed.
- W. Wei, H. Gou, X. Sun, X. Fei, X. Li and X. Liu, Imidazole derivative with an intramolecular hydrogen bond as thermal latent curing accelerator for epoxy/phenolic resins, J. Appl. Polym. Sci., 2021, 139, e5911 Search PubMed.
- T. Brand, E. J. Cabrita and S. Berger, Intermolecular interaction as investigated by NOE and diffusion studies, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 159–196 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2im00020b |
| ‡ These authors contributed equally to this work. |
| This journal is © Institute of Process Engineering of CAS 2023 |