
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically grown Fe2O3/Fe3O4 heterostructure nanotubes with In2O3 induced tandem internal electric fields for enhanced photoelectrochemical water oxidation†
Xiaohui
Yan
a,
Gang
Li
 *ac,
Kai
Shen
a,
Congwei
Wang
*b and
Kaiying
Wang
*ad
*ac,
Kai
Shen
a,
Congwei
Wang
*b and
Kaiying
Wang
*ad
aInstitute of Energy Innovation, Taiyuan University of Technology, Taiyuan 030024, China. E-mail: ligang02@tyut.edu.cn; kaiying.wang@usn.no
bCAS Key Laboratory of Carbon Materials, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China. E-mail: wangcongwei@sxicc.ac.cn
cCollege of Physics and Information Engineering, Minnan Normal University, Zhangzhou 361000, China
dDepartment of Microsystems, University of South-Eastern Norway, Horten, 3184, Norway
First published on 13th October 2023
Abstract
Nanostructured hematite (α-Fe2O3) shows promise as a semiconductor for photoelectrochemical (PEC) water oxidation. However, it suffers from inadequate charge separation, limited hole-collection efficiency and sluggish kinetics. Herein, a nanotubular Fe2O3/Fe3O4 p–n heterojunction is prepared via electrochemical anodization to in situ construct an internal electric field (IEF) that facilitates charge separation from photoactive hematite. Additionally, In2O3 clusters are introduced to form a second IEF with dual-phase iron oxides, exploiting their Fermi level difference. The unique configuration of the dual IEFs in a novel tandem way synergistically promotes charge carrier separation/migration, enhancing PEC performance. Specifically, the 1st IEF between Fe2O3 and Fe3O4 accelerates electron migration from Fe3O4 to Fe2O3 (with holes transporting in the opposite direction), while the 2nd IEF at the In2O3 and Fe2O3/Fe3O4 interface drives holes towards the In2O3 surface, enhancing the hole-collection efficiency. The composite photoanode achieves a state-of-the-art current density of 11.5 mA cm−2 at 1.55 VRHE and a superior applied bias photon-to-current efficiency of 0.44% at 0.95 V. DFT calculations reveal that In2O3 induces an electron-deficient surface, creating favorable adsorption sites for oppositely charged key intermediates (*OOH). This work presents a novel approach for modulating reaction kinetics via the construction of tandem IEFs and holds great significance for the rational design of efficient PEC catalysts.
Introduction
With the increasing demand for renewable hydrogen energy, significant attention has been directed towards harnessing solar energy. Apart from solar photovoltaics, which convert sunlight into electricity, photoactive semiconductors have emerged as highly promising alternatives for converting solar energy into storable and transportable chemicals. Comparatively, photoelectrochemical (PEC) water reduction into hydrogen and oxygen in aqueous electrolytes is regarded as a promising and feasible method for producing clean energy and storing renewable solar energy, providing a compelling solution.As the PEC reduction reaction occurs on a photocathode (hydrogen evolution reaction, HER), the electrons and protons consumed must be balanced by the oxidation of water on a photoanode (oxygen evolution reaction, OER). Nonetheless, significant challenges hinder the widespread adoption of this technique. Firstly, it is crucial to effectively separate the photogenerated charge carriers to ensure an adequate supply of targeted carriers towards the respective photoelectrodes (i.e., electrons towards the photocathode and holes towards the photoanode). Secondly, there is a need to overcome the sluggish kinetics associated with the four-electron OER process.1 Recent advancements have highlighted the potential of metal-oxide-based semiconductors, such as BiVO4,2,3 TiO2,4,5 WO3,6,7 and Fe2O3,8–10 in PEC water oxidation systems. These materials exhibit favourable characteristics such as tunable band gaps, visible-light activity, and excellent photochemical stability, enabling rapid charge transfer.11 Among them, α-Fe2O3 is the most stable state of iron oxide under ambient conditions and is considered as an environmentally friendly material. It is a naturally occurring compound derived from Earth's abundant iron resources and could be used in a number of applications, including catalysis, steel manufacturing, gas sensing, and optoelectronics. In particular, α-Fe2O3 stands out as a promising choice for a photoanode in PEC systems due to its abundance in nature, non-toxicity and suitable bandgap of approximately 2.1 eV.10,12 Without the employment of costly noble metals or environmentally sensitive elements, e.g., cadmium, α-Fe2O3 is a potential ideal photoanode candidate, which possesses the theoretical upper limit for solar-to-hydrogen efficiency up to 15%.13 Benefiting from the intrinsic advantages of the α-Fe2O3 semiconductor, solar energy could be effectively converted into hydrogen energy with the assistance of additional electricity, which could potentially provide a new green pathway for alleviating CO2 emission while storing intermittent renewable energy. Therefore, the application of α-Fe2O3 photoanodes could play a key role in achieving green and sustainable sunlight-generated hydrogen energy goals. However, the picosecond lifetime and short diffusion distance (2–4 nm) of photogenerated charge carriers inevitably lead to poor charge carrier (h+–e−) separation efficiency and limited carrier density. Additionally, a large overpotential arises from the unfavorable adsorption energy of key intermediates. Therefore, improving the separation efficiency of photogenerated charge carriers while simultaneously decreasing the energy barrier of rate-determining steps (RDS) is a compelling strategy for enhancing PEC performance. Recently, the construction of an internal electric field (IEF) has gained recognition as an effective strategy for enhancing reactivity in the realms of renewable energy.14 The generation of an IEF typically arises from polarization effects caused by imbalance of charge distributions across the interface of different components.14 More precisely, non-centrosymmetric materials, differences in Fermi levels (Ef) and external stress can induce shifts in the center of positive/negative charges, leading to the formation of space charge layers. These layers, known as the depletion and accumulation regions of electrons, result in the bending of the conduction band (CB) and valence band (VB), ultimately giving rise to a polarization field.15 For solar-driven PEC systems, the recombination of photogenerated charge carriers can be prevented by introducing a driving force from an IEF, allowing the directional migration of photoinduced electrons/holes. The IEF can also offer a favorable redox potential, sustaining electron migration paths, and optimal adsorption conditions for key intermediates.16 For instance, an electron accumulation region is beneficial for the HER, whereas an electron depletion region is more conducive to the OER. Therefore, a PEC system with a rationally designed IEF can not only facilitate the separation/migration of photogenerated charge carriers but also adjust intermediates’ adsorption energy for different elementary steps to accelerate the overall reaction kinetics.17 Magnetite (Fe3O4) coupled with Fe2O3 has been widely applied in capacitors and batteries due to its excellent electrical conductivity and alterable valence states. Significantly, the valence state transition between bivalent and trivalent oxidation states enables the in situ formation of a polarized interface characterized by uneven charge distributions. This phenomenon allows for the construction of an IEF and makes it well suited for implementation as a photoelectrode, thus achieving superior performance in PEC applications.18–21 Specifically, photoactive Fe2O3 generated charge carriers could be effectively separated and transported under the assistance of an Fe2O3/Fe3O4-induced IEF, while Fe3O4 can improve electrical conductivity and enhance electron transport.22 Though the Fe2O3/Fe3O4 heterostructure has shown significant progress in a PEC water splitting system, the intrinsic unfavorable water oxidation kinetics for key intermediates of either Fe2O3 or Fe3O4 still hinder its application.23 Therefore, secondary metal oxide cocatalysts with preferred adsorption properties for key intermediates could be implemented to improve PEC performance of iron-oxide-based photoelectrodes. Indium oxide (In2O3), renowned for its exceptional photo-permeability, high conductivity, and thermal transfer rate, has been widely used in solar cells and optoelectronics. The combination of dual-phase iron oxides (Fe2O3/Fe3O4) and indium oxide thus has significant potential for modulating the adsorption energy to facilitate the water oxidation kinetics. Furthermore, the disparity in Fermi levels and excellent compatibility between iron oxide and indium oxide can induce a second IEF to further improve the charge carrier separation efficiency for enhanced PEC performance.
In this work, highly ordered nanotubes composed of a dual-phase iron oxide heterojunction are directly grown via electrochemical anodization of stainless-steel foil. The indium oxide nanoparticles are thermally deposited onto the iron oxide surface as a cocatalyst to prepare a composite photoanode. The in situ constructed IEF between Fe2O3 and Fe3O4 effectively enhances the charge carrier separation from the photoactive hematite, while the second IEF between iron oxides with In2O3 further modulates the adsorption properties, lowering the energy barrier for the RDS. Benefiting from this distinctive tandem IEF and narrow depletion zones (∼10.1 nm), an excellent carrier density (1.33 × 1021 cm−3) is achieved with a superior photocurrent density of 11.5 mA cm−2 at 1.55 VRHE for PEC water oxidation. The DFT calculations further support the experimental findings by showing the electron redistribution at the In2O3 interface and revealing a reduced energy barrier for the formation of *OOH and thereby enhancing the water oxidation kinetics. Moreover, its unique nanotubular structures as three-dimensional self-supporting current collectors with low resistance can significantly enhance the electron migration efficiency. Therefore, ternary In2O3/Fe2O3/Fe3O4 heterostructures with rationally constructed tandem IEFs offer a promising integrated photoanode for efficient PEC water oxidation.
Results and discussion
Synthesis and characterization of photoanodes
The preparation routine for the iron-oxide-based photoanode is illustrated in Fig. 1a, and Fig. S1 and S2† provide additional details. Specifically, highly ordered iron-oxide nanotubes (NTs) were synthesized on stainless-steel (SSt) foil using an electrochemical anodization approach, defined as the ASSt precursor.24 Subsequently, the indium oxide cocatalyst was deposited onto the ASSt via a hydrothermal process in InCl3 alcoholic solution, followed by high-temperature stabilization (T-In@ASSt). The ASSt film displayed a highly ordered double-side nanotubular structure with relatively smooth inner nanowalls, as shown in the transversal and vertical cross-section SEM images in Fig. 1b and c respectively. The average diameter of the nanotubes was about 41.93 nm (inset of Fig. 1b). The blurred surface features and poor image quality at high magnification were attributed to the unfavored surface electron accumulation, implying that the surface electrical conductivity of the as-anodized ASSt was poor. The inset of Fig. 1c presents the selected area electron diffraction (SAED) pattern of ASSt, where the blurry spots combined with the broad diffraction peak in XRD (Fig. 1i) indicated the amorphous surface structure of ASSt. After annealing, the sample (referred to as T-ASSt) exhibited a needle-like morphology (Fig. 1d), and the corresponding energy-dispersive spectroscopy (EDS) mapping confirmed the presence of Fe and O signals (inset of Fig. 1d). The thicknesses of the T-ASSt NT layer and the nanowalls were about 6.19 μm and 22.61 nm, respectively (Fig. 1e). The flat SSt foil, indicated by the yellow arrow, served as the current collector, maintaining unimpaired electrical conductivity. The relatively smooth nanowalls of ASSt turned into rough surfaces with sub-nanoscale humps due to the crystallization of the initially amorphous iron species after annealing. Transmission electron microscopy (TEM) analysis of T-ASSt revealed an average NT diameter of ∼41.5 nm, as shown in Fig. 1f, which was consistent with the SEM observation. In order to gain more insights into the nanotubular structure of prepared photoanodes, high-resolution transmission electron microscopy (HRTEM) images were taken and the interlayer spacing was calculated based on the lattice fringe patterns (Fig. 1g, highlighted in red rectangles). Two sets of lattice patterns could be identified in the HRTEM image of T-ASSt. The Fast-Fourier Transform (FFT) values of interlayer fringe spacing for red rectangles 1 and 2 were found to be 0.252 and 0.246 nm, which correspond to the Fe3O4 (113) plane and α-Fe2O3 (110) plane, as shown in Fig. S3.† The SAED pattern of T-ASSt demonstrated a polycrystalline ring (Fig. 1h), verifying that the nanowalls of T-ASSt consisted of highly crystalline iron oxides. The XRD spectrum of T-ASSt further confirmed the coexistence of dual-phase α-Fe2O3 and Fe3O4 (Fig. 1i). | ||
| Fig. 1 (a) The schematics of the preparation process of the T-In@ASSt photoanode. Cross-section SEM images of as-anodized ASSt: (b) top view, the inset: summary of nanotube diameters and (c) vertically along the NT, the inset: SAED patterns. Cross-section SEM images of annealed T-ASSt: (d) top view, the inset: EDS mapping of Fe and O elements and (e) vertically along the NT, the inset: the whole T-ASSt layer with unanodized SSt. (f) TEM and (g) HRTEM images of T-ASSt. (h) SAED patterns of T-ASSt. (i) XRD spectra of ASSt and T-ASSt. SEM images of T-In@ASSt: (j) top view and (k) vertically along the NT, the inset: EDS elemental mapping. (l) TEM image of T-In@ASSt. (m) HRTEM image of T-In@ASSt, the inset: corresponding SAED patterns. | ||
After incorporation of In2O3 nanoparticles (NPs) as a cocatalyst, the obtained T-In@ASSt exhibited a similar nanotubular microstructure to T-ASSt (Fig. 1j). The diameters of T-In@ASSt remained around 40 nm with decorations of small NPs, and EDS elemental mapping illustrated the uniform dispersion of Fe, O and In elements (inset of Fig. 1j). The average size of the NPs was about 12–18 nm, uniformly distributed inside the nanowalls (Fig. 1k). TEM was employed to further demonstrate the crystalline structure of T-In@ASSt, which retained the original nanotubular structure with small NPs attached inside the nanowall (Fig. 1l). Well-resolved crystal lattice fringes of 0.246, 0.252 and 0.285 nm, corresponding to the (110), (113) and (222) planes of Fe2O3, Fe3O4 and In2O3, could be identified in the HRTEM image of T-In@ASSt, as shown in Fig. 1m and Fig. S4†. The potential interfacial nano-sintering between the attached In2O3 NPs and iron oxide NTs may lead to surface reconstruction due to nanoscale fusion during sintering. This could effectively reduce recombination sites and facilitate the migration of charge carriers.12 The SAED pattern obtained over the entire region of T-In@ASSt revealed a distinct polycrystalline ring, as shown in the inset of Fig. 1m, suggesting the high crystallinity of Fe2O3, Fe3O4 and In2O3. It is noted that due to the similarity of XRD patterns of Fe3O4 and γ-Fe2O3, it is difficult to differentiate Fe3O4 and γ-Fe2O3via XRD. For better analysis of various iron oxides, Raman spectroscopy was employed as shown in Fig. S6.† The spectrum of T-ASSt showed broad bands at 520–594 cm−1, corresponding to the presence of Fe2O3.25 The broad band from 1320 cm−1 to 1393 cm−1 is attributed to the second-order scattering of Fe2O3. Specifically, a pronounced peak at 671 cm−1 and two small broad peaks at 320–340 and 510–520 cm−1 in T-ASSt can be assigned to the A1g, T2g and Eg modes of Fe3O4, respectively.26,27 However, no γ-Fe2O3 characteristic peaks were observed near 200 and 800 cm−1 in both T-ASSt and T-In@ASSt, indicating the absence of γ-Fe2O3. In T-In@ASSt, characteristic bands appeared at 209 and 368 cm−1, representing the In–O vibrations and the In–O–In stretching vibration, respectively, indicating the presence of body-centered cubic In2O3.28,29 It is worth noting that Fe3O4 was partially transformed into α-Fe2O3, with the atomic ratio of α-Fe2O3/Fe3O4 adjusted from 0.33 to 0.70 after annealing to stabilize In2O3. This transition between Fe3+ and Fe2+ contributed to the different conductivity behaviours and alterable valence states between α-Fe2O3 and Fe3O4, thus endowing this heterostructure with unevenly distributed interface electrons.18,19,21 This uneven charge distribution leads to the formation of an IEF. The in situ formed IEF has the potential to enhance the separation and migration of photogenerated charge carriers from the photoactive hematite, thereby benefiting the PEC performance.
X-ray photoelectron spectroscopy (XPS) was employed to evaluate the surface chemical composition and valence states of the samples, namely ASSt, T-ASSt and T-In@ASSt. The survey scan illustrated the presence of Fe, O and C elements in all three samples, while the absence of the F signal in the latter two samples indicated the complete removal of residual fluorides after annealing (Fig. 2a). The relatively weak signal of the In element in T-In@ASSt demonstrated a limited loading amount of 0.1 at%. Analysis of the Fe 2p3/2 and Fe 2p1/2 peaks in ASSt (714.0 eV and 727.7 eV, respectively), T-ASSt (710.1 eV and 724.7 eV, respectively) and T-In@ASSt (710.6 eV and 724.5 eV, respectively) is shown in Fig. 2b. The spectrum of T-ASSt shifted to a lower binding energy compared to ASSt due to the improved crystallinity and the presence of more surface photo-responsive iron oxides. The broad peaks observed for Fe 2p1/2 and Fe 2p3/2 indicated the coexistence of dual-phase iron oxides (Fe2+/Fe3+). Specifically, the subpeaks centered at 712.5 and 722.8 eV in T-ASSt corresponded to spin-orbitals in Fe2O3, while the subpeaks at 708.6 eV and 718.9 eV were attributed to Fe2+ of Fe3O4. The Fe 2p1/2 subpeak in T-In@ASSt shifted to a lower binding energy compared to T-ASSt due to an increased Fe3+/Fe2+ atomic ratio. In the O 1s spectra (Fig. 2c), the broad peak of ASSt could be divided into three subpeaks, representing lattice O2− species (OL, 530.0 eV), metal–oxygen species (OV, 531.7 eV) and chemisorbed or dissociated oxygen species (OC, 533.3 eV), respectively.30,31 Both OL and OV in T-In@ASSt exhibited a nearly equal proportion, indicating an increased bonding of oxygen atoms with metal atoms. However, the movement and increase of the OV peak could be attributed to the formation of local defect sites. The In 3d peaks in T-In@ASSt were centered at 444.5 and 452.0 eV, respectively, indicating the presence of trivalent In3+ (Fig. 2d).
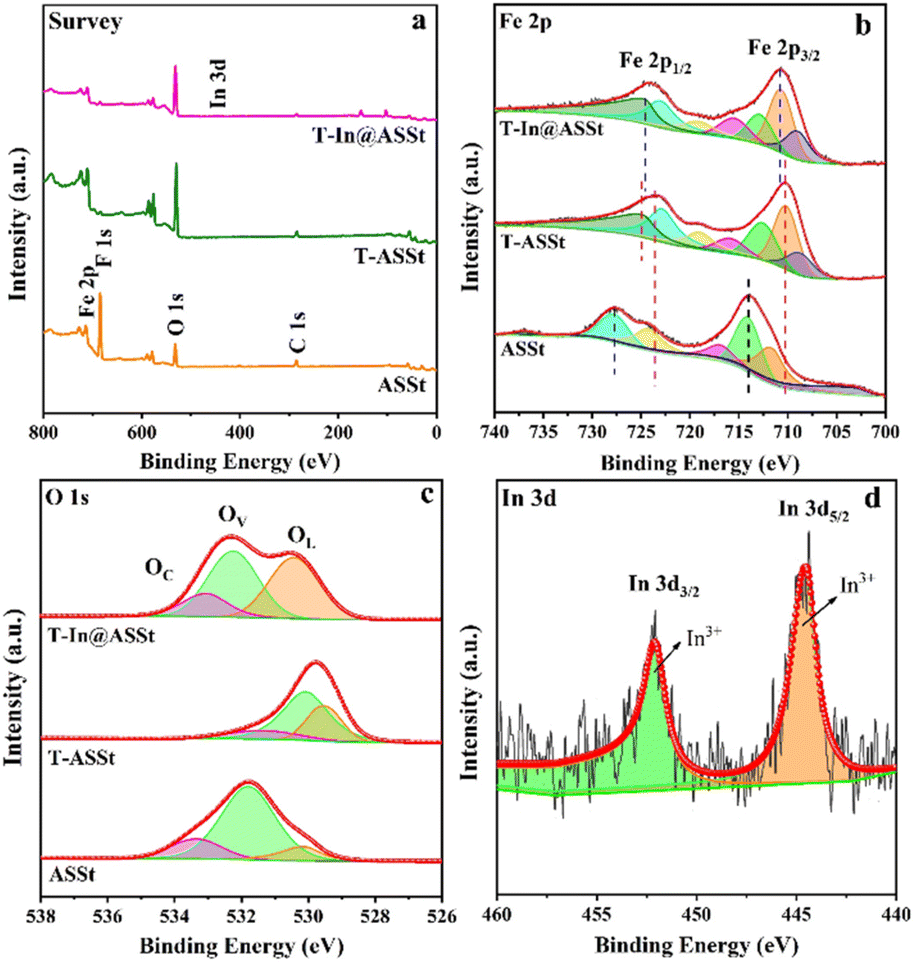 | ||
| Fig. 2 XPS spectra for (a) the survey scan and (b and c) the narrow scan of Fe 2p and O 2p of ASSt, T-ASSt and T-In@ASSt photoanodes. (d) The narrow scan of In 3d of the T-In@ASSt photoanode. | ||
Photoelectrochemical measurements for water oxidation
Linear sweep voltammetry (LSV) was employed to comprehensively study the PEC water oxidation performances of the photoanodes. The onset potential (Eonset) of commercial Fe2O3 under single illumination was 1.55 VRHE, and the dark current could be neglected due to its poor activity/conductivity (Fig. S7a†). It is noted that though the current density of Fe2O3 was jointly attributed to the photoinduced carrier migration and bias-induced electrochemical process, the increased current density above 1.55 V bias was mainly triggered via the illumination. Therefore, the current density at this bias was used to evaluate the PEC performances of prepared photoanodes.32 In comparison, the prepared photoanodes exhibited onset potentials of ∼1.38–1.46 V under dark conditions, as shown in Fig. S7b.† It has been proved that the illumination mode has a significant effect on the current density of Fe2O3 photoanodes.12 Therefore, the effects of illumination methods, specifically single- and double-(side) illumination, were evaluated for all prepared photoanodes (Fig. 3 and Fig. S7c†). Current density–voltage curves (J–V) measured under single- and double-illumination with simulated sunlight presented a two-stage behavior, as shown in Fig. 3a. Relatively gentle photocurrent slopes were observed at the initial stage below 1.23 V, indicating that the photoinduced charge carrier transportation dominantly contributed to the overall current densities.32 Comparatively, the current density increased rapidly after ∼1.23 V, demonstrating that the current densities were facilitated by the contribution of the applied bias voltage when the potential was above the redox potential of O2/H2O. The improved current density under double illumination could be attributed to the increased amounts of photoactive hematite and the improved ion diffusion rate. The Eonset values of single- and double-illuminated T-ASSt were 0.73 and 0.72 V vs. RHE, respectively, indicating that the dual-phase iron oxides’ heterostructure with an interfacial IEF could trigger water oxidation at a lower bias voltage. Comparatively, with the deposition of In2O3, T-In@ASSt exhibited improved onset potentials of 0.68 and 0.64 V under single and double illumination, respectively, demonstrating that the introduction of In2O3 could further facilitate the thermodynamic process of the OER. Moreover, an excellent current density of 6.4 mA cm−2 was achieved at 1.55 VRHE (corresponding to the Eonset of commercial Fe2O3) for T-ASSt under double-illumination, implying that the Fe2O3/Fe3O4-induced IEF could enhance charge carrier separation and suppress h+–e− recombination, as shown in Fig. S8.† More significantly, a nearly 80% improvement in the current density of 11.5 mA cm−2 at 1.55 VRHE was achieved for the T-In@ASSt photoanode, indicating the superior hole collection efficiency achieved by In2O3 modification, as shown in the inset of Fig. 3a. Tafel plots were calculated to assess the water oxidation kinetics of the photoanodes, as plotted in Fig. 3b. The Tafel slopes of T-ASSt were 110.2 and 104.3 mV dec−1 under single- and double-illumination, respectively. The slightly smaller Tafel slope under double-illumination infers the advantage of the double-side nanotubular morphology prepared via the electrochemical anodization, indicating faster OER kinetics. Similarly, T-In@ASSt presented Tafel slopes of 84.7 and 76.9 mV dec−1 under single- and double-illumination, respectively. This lowered Tafel slope suggested that T-In@ASSt possessed the most rapid reaction kinetics, expediting the formation of oxygenated species during the OER.33 | ||
| Fig. 3 PEC performances of T-ASSt and T-In@ASSt photoanodes under single/double illumination conditions in 1 M KOH: (a) LSV scans measured under single/double illumination with AM 1.5G simulated sunlight; the inset shows the current density of T-In@ASSt and commercial α-Fe2O3 at 1.55 VRHE. (b) Tafel plots. (c) Electrochemical impedance spectra; the inset shows the equivalent circuit. Photocurrent density without applying a bias potential: (d) single illumination and (e) double illumination. (f) Mott–Schottky plots collected at a frequency of 1 kHz in the dark. (g) ABPE spectra of T-ASSt and T-In@ASSt photoanodes. (h) IPCE spectra of T-ASSt and T-In@ASSt photoanodes under single/double illumination. (i) Gas evolved from the T-In@ASSt photoanode with an applied potential of 1.55 VRHE under double illumination. (Note: S and D in the graphs represent single- and double-illumination, respectively.) | ||
Charge transport efficiencies were evaluated through electrochemical impedance spectroscopy (EIS) without bias voltage (Fig. 3c). The EIS curves of all prepared photoanodes were composed of a small semicircle in the high-frequency region and a large semicircle in the low-frequency range. Nyquist plots were fitted using an equivalent circuit model, as shown in the inset of Fig. 3c, and the corresponding simulated parameters are listed in Table S2.† The series resistances (R1) at the interface between the photoactive materials (anodized film) and the current collector (SSt) were similar for T-ASSt under single- (1.2 Ω cm−2) and double-illumination (1.0 Ω cm−2). Similarly, the R1 values of T-In@ASSt were the same under single-/double-illumination as 0.8 Ω cm−2, indicating that the illumination method could hardly affect the resistance between the current collector and the photoactive materials. Interestingly, the resistance (R2) inside the photoanode of double-illuminated T-ASSt was larger (16.0 Ω cm−2) than that under single-illumination (12.2 Ω cm−2), while the R2 values of T-In@ASSt were 16.2 and 28.1 Ω cm−2 under single-/double-illumination, respectively. These R2 differences could be due to the attachment and shadowing of a large number of bubbles under double illumination, which affected the internal charge transfer.12 Moreover, the built-in IEFs could effectively facilitate the instant accumulation of charges under double illumination, which could hinder the charge transfer and thus increase internal transfer resistance. The R3 in the low-frequency range manifested the charge injection resistance near the electrolyte/photoanode interface, which was a significant factor affecting catalytic activity. Evidently, double-illuminated T-ASSt exhibited a smaller R3 of 233 Ω cm−2 compared to the single illuminated T-ASSt (309 Ω cm−2), indicating that double-illuminated T-ASSt possessed a higher carrier transfer efficiency at the photoanode/electrolyte interface. A much smaller R3 of 204 Ω cm−2 was achieved for double-illuminated T-In@ASSt, implying that the introduction of the In2O3 cocatalyst significantly improved the photogenerated charge injection efficiency, enhancing the catalytic activity. As shown in the Bode plots in Fig. S9,† the excellent fit curves were confirmed from the almost completely overlapped experimental data and calculated curves. The mod Z values for all samples were similar in the low-frequency region (<1 Hz), which indicated the analogous intrinsic conductivity and was in agreement with the Nyquist plots. Concretely, the low-frequency peak in the Bode phase plot is related to the charge transfer at the photoanode/electrolyte interface, while the high-frequency peak is associated with electron transport inside the photoanode.34 For single-illuminated T-ASSt, there was only one high-frequency peak, implying that the PEC performance was mainly affected by electron transport inside the iron oxide-based photoanode. For other samples, the presence of both low- and high-frequency peaks suggests that the water oxidation was synergistically controlled via the charge transport kinetics within the photoanode and photoanode/electrolyte interface. T-In@ASSt presented the smallest low-frequency peak, indicating the highest charge transfer rate across the photoanode/electrolyte interface.34,35 The high-frequency peaks of T-In@ASSt exhibited a positive shift compared with T-ASSt, demonstrating that the resistance inside the photoanode increases. These phenomena at the high-frequency and low-frequency peaks are consistent with the Nyquist plot results as mentioned above.
Steady-state photocurrent responses were observed without applying a bias potential. Upon illumination, all photoanodes presented an immediate light response, indicating the rapid emergence of photogenerated charge carriers. Subsequently, the rapid recombination of photogenerated charge carriers resulted in a reduction in photocurrent. Finally, a steady photocurrent was achieved as a result of the dynamic equilibrium between charge separation and recombination.36 A photocurrent density of 1 × 10−5 A cm−2 was achieved for single-illuminated T-ASSt (Fig. 3d), which was approximately 4 times that of ASSt (2.5 × 10−6 A cm−2). This significant increase indicated that the well-crystallized Fe2O3/Fe3O4 heterostructure could enhance the charge carrier separation via an induced directional IEF. Additionally, T-In@ASSt presented a six-fold enhancement in photocurrent density, reaching 6 × 10−5 A cm−2, compared to T-ASSt. This finding suggests that the modification of In2O3 could prevent charge carrier recombination via the second IEF introduced between In2O3 and iron oxides. Moreover, under double illumination, both T-ASSt (1.9 × 10−5 A cm−2) and T-In@ASSt (1.5 × 10−4 A cm−2) demonstrated higher photocurrent densities compared to single illumination, demonstrating that the materials being photoactivated under double illumination exhibited a significant increase, as shown in Fig. 3e.
Mott–Schottky (M–S) analysis, which represents changes in capacitance against applied potentials, is widely adopted to investigate semiconductor behaviors and the mechanism of charge separation and migration.37 The appearance of both positive and negative slopes (inverted “V-shape”) in all photoanodes indicated the coexistence of n- and p-type semiconductors within the dual-phase iron oxide heterostructures, as shown in Fig. 3f. Given that α-Fe2O3 is a well-known intrinsic n-type semiconductor, the coexisting Fe3O4 serves as the p-type component.38 When n- and p-type semiconductors come into contact, electrons would be spontaneously transferred to achieve equilibrium of Fermi levels. At the interface, band bending takes place due to the establishment of a spatial charge region resulting from the electron migration, resulting in a built-in IEF directing from the positive-charged to negative-charged semiconductor.39 Therefore, the in situ formed Fe2O3/Fe3O4 interface (n–p junction) could induce (the 1st) IEF, thus facilitating the separation of charge carriers generated in Fe2O3 in opposite directions as evidenced by the large photocurrent density shown in Fig. 3a. Moreover, the carrier density (Nd) could be calculated using the M–S slopes through the following equation:36
| Nd = (2/eεε0) [d(1/C2)/ dV]−1 | (1) |
| W = [(2εε0(V − Vfb))/(qNd)]1/2 | (2) |
The applied bias photon-to-current efficiency (ABPE) was calculated to evaluate the photoconversion efficiencies of photoanodes, as shown in Fig. 3g. The ABPE peaks for single- and double-illuminated T-ASSt were merely 0.02% at 0.96 V and 0.11% at 0.93 V, respectively. However, the single-illuminated T-In@ASSt achieved an ABPE of 0.36% at 0.97 V. Significantly, the ABPE of double-illuminated T-In@ASSt increased to as high as 0.44% at the lowest potential of 0.95 V, which was almost four-fold higher than that of double-illuminated T-ASSt. The incident photon-to-current efficiency (IPCE) spectra were shown as a wavelength function from 350 to 700 nm to confirm the proportion of the incident photons and photocurrent (Fig. 3h). Single-illuminated T-In@ASSt achieved a maximum IPCE of 16.7% at 350 nm, nearly 1.5 times higher than that of single-illuminated T-ASSt of 10.6%. The IPCE spectra of T-ASSt and T-In@ASSt under double illumination could be speculated based on previous photoresponsive optical properties, namely 42.5% at 350 nm for T-In@ASSt. The significant improvement in IPCE for T-In@ASSt indicates that the extraction efficiencies of charge carriers were significantly enhanced through the introduction of the In2O3 cocatalyst. This could be attributed to the second IEF that is expected to facilitate the migration of charge carriers from Fe2O3 to In2O3.43 To explore the PEC performance of T-In@ASSt, the production rates of evolved gases were measured at a constant bias of 1.55 VRHE with an effective illumination area of 1.0 cm2 using gas chromatography under AM 1.5G double illumination, as shown in Fig. 3i. Both hydrogen and oxygen were almost linearly generated with a stoichiometric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The gas evolution rates were 132.8 and 71.6 μmol cm−2 h−1 for hydrogen and oxygen, respectively. This water oxidation performance was among the state-of-the-art performances for iron-oxide-based photoanodes as summarized in Table S4.† Long-term durability is the prerequisite for photoelectrodes’ practical application. Therefore, the stability of the prepared T-In@ASSt photoanode was examined at 1.55 VRHE under double-illumination. T-In@ASSt exhibited a relatively stable current density of around 11.1 mA cm−2 after a long-term durability measurement, indicating its excellent stability with a remarkable retention of ∼97% as shown in Fig. S11.† It is noted that the current perturbation could be attributed to the aggregation and release of generated air bubbles.
:1. The gas evolution rates were 132.8 and 71.6 μmol cm−2 h−1 for hydrogen and oxygen, respectively. This water oxidation performance was among the state-of-the-art performances for iron-oxide-based photoanodes as summarized in Table S4.† Long-term durability is the prerequisite for photoelectrodes’ practical application. Therefore, the stability of the prepared T-In@ASSt photoanode was examined at 1.55 VRHE under double-illumination. T-In@ASSt exhibited a relatively stable current density of around 11.1 mA cm−2 after a long-term durability measurement, indicating its excellent stability with a remarkable retention of ∼97% as shown in Fig. S11.† It is noted that the current perturbation could be attributed to the aggregation and release of generated air bubbles.
Photoelectrochemical mechanism
Density functional theory (DFT) calculations were employed to investigate the effect of the In2O3 cocatalyst on the catalytic mechanism of iron oxide-based PEC water oxidation. Considering the positive potential environment, Fe2O3 would be more prone to distribute on the surface of the photoanode as evidenced from the TEM observation in Fig. 1m. The slab model of Fe2O3 (110) loaded with In2O3 clusters was analogically constructed. The differential charge density of Fe2O3 (110) loaded with In2O3 was calculated as shown in Fig. 4a, where the yellow region indicates electron accumulation and the cyan region indicates electron depletion. The results showed that there was distinctive interfacial charge redistribution as more electrons located at the Fe2O3/In2O3 interface, leading to an electron-deficient In2O3 surface. The deficit of electrons in In2O3 could be beneficial for the adsorption of oppositely charged hydroxyl in the alkaline electrolyte to enhance the hole-donating ability. The density of states (DOS) diagrams of both Fe2O3 (110) and the Fe2O3/In2O3 heterostructure are shown in Fig. 4b and c. The electronic states close to the Fermi level (Ef) were primarily contributed by the Fe d and In p orbitals. The calculated d-band center was −3.63 eV for Fe2O3 (110) and −3.16 eV for Fe2O3/In2O3. Generally, the bonding strength between adsorbates and transition metal oxide surfaces is determined through the occupancy rate of the bonding and anti-bonding states. Upshifting the d-band center towards Ef leads to the emptying of antibonding states. Consequently, in the case of introduction of In2O3 into Fe2O3, the upshift of the d-band center and the localized higher DOS intensity near the Ef of Fe2O3/In2O3 were beneficial for the adsorption of reactive species. The upshift of the d-band center for T-In@ASSt suggested a stronger chemisorption reactivity, facilitating the supply of holes for PEC water oxidation. | ||
| Fig. 4 (a) Differential charge density of Fe2O3(110) loaded with In2O3 clusters; density of states of (b) pristine Fe2O3(110) and (c) Fe2O3(110)/In2O3. (d) Gibbs free energy diagram of Fe2O3(110)/In2O3 with a 0.64 V bias voltage. (e) Adsorption configurations of key intermediates respectively. (f) Schematic diagram of IEF formation and band bending between pure Fe2O3 and Fe3O4, T-ASSt and In2O3. | ||
The Gibbs free energy diagram (Fig. 4d) was calculated and the elementary steps (Fig. 4e) of adsorption configurations of key intermediates (*OH/*O/*OOH) for the OER process were determined. The typical OER processes on both Fe2O3 (110) and Fe2O3 (110)/In2O3 were uphill when the electrode bias potential U was 0 V. Specifically, the rate-determining step (RDS) on Fe2O3 (110) was the last elementary step (*OOH → O2) with an energy barrier of 3.81 eV, whereas the RDS on Fe2O3 (110)/In2O3 was the formation of the *OOH intermediate (*O → *OOH) with an energy barrier of 3.27 eV. For Fe2O3 (110)/In2O3, the energy barrier for the *OOH → O2 step was 0.5 eV, which was 3.31 eV lower than that of the Fe2O3 (110) surface due to the intensified hole-donating ability. Though electron redistribution at the interfaces could decrease the energy barrier and lead to the transition of the RDS, the relatively large energy barrier still limited the OER performance. As the electrode potential increased to the experimentally measured onset potential of 0.64 V, the energy barrier of *OOH intermediate formation was significantly decreased to 2.63 eV, leading to the much enhanced PEC water oxidation performance. Interestingly, the energy barrier for the *OOH → O2 step in Fe2O3 (110)/In2O3 presented a downhill exothermal process after the bias potential was applied. A more efficient O2 generation was thereby expected for the Fe2O3 (110)/In2O3 photoelectrode.
To thoroughly investigate the mechanism behind the formation of tandem dual IEFs at the ternary In2O3/Fe2O3/Fe3O4 interfaces, ultraviolet photoelectron spectroscopy (UPS) spectra were employed to validate the DFT calculations. Pure Fe2O3, Fe3O4 and In2O3 were prepared as control samples, as verified by XRD analyses (Fig. S12†). By combining the UV-vis (Fig. S10†) and UPS (Fig. S13†) spectra, the detailed band positions and work functions of the composite photoanode components could be determined. Specifically, the work function (Φ) could be calculated using the equation Φ = hv − |Ecutoff − Ef|, where hv represents the photon source energy (He: 21.22 eV) and Ef and Ecutoff denote the Fermi level and cut-off energy, respectively.44 The work functions of pure Fe3O4 and Fe2O3 were thus calculated as 6.59 and 6.24 eV, respectively, implying a comparatively higher Ef for Fe2O3 than for Fe3O4. Therefore, when the dual-phase iron oxides emerged (in contact) after anodization as T-ASSt, the Fermi level was aligned to be 6.48 eV (Fig. S13c†), driving electron transfer from the high work function Fe2O3 to the low work function Fe3O4. The electron depletion layer and electron accumulation layer were formed at the Fe3O4/Fe2O3 interface accompanied by the upwards and downwards band bending with the in situ formed 1st IEF directing from Fe2O3 to Fe3O4, as schematically shown in Fig. 4f. This IEF could effectively separate the photogenerated charge carriers as electrons would be transported to the Fe2O3 side while holes would be transferred to the Fe3O4 side, as solidly evidenced by the enhanced photocurrent of T-ASSt as shown in Fig. 3d. Moreover, with the deposition of In2O3 NPs on T-ASSt, the differences in their Φ (6.63 eV for In2O3 and 6.48 eV for T-ASSt) and Ef facilitated electron migration from T-ASSt to In2O3. Similarly, a 2nd IEF from the positively-charged T-ASSt to the negatively-charged In2O3 was formed, accelerating the collection efficiency of the photogenerated holes on the surface of In2O3. Moreover, the presence of a potential barrier at the band edge effectively channeled the photogenerated holes of T-ASSt towards the high-energy band, significantly augmenting its ability to facilitate the OER. Benefiting from this unique tandem IEF mechanism, the charge carrier migration rate and hole collecting ability were maximized, thereby greatly enhancing the PEC performance.
As a crucial kinetic factor, the IEF could not only impact the spatial separation of charge carriers but also determine the charge carrier migration directions. In order to provide solid evidence for the formation of IEFs, an atomic force microscope with a Kelvin probe (KPFM) was employed to directly measure the surface potential variations on the photoanodes, as illustrated in Fig. 5. It has been demonstrated that the IEF can lead to the enhancement of surface potential.45,46 Concretely, the surface potential (ΔE) of commercial Fe2O3 was 30.1 mV, and with the introduction of the Fe3O4 phase, the ΔE of T-ASSt increased to 56.7 mV, indicating that the formed α-Fe2O3/Fe3O4 interface induced the 1st IEF. After the deposition of In2O3, the ΔE of T-In@ASSt increased to 83.6 mV, verifying the significantly pronounced IEF.47
 | ||
| Fig. 5 Surface potential of (a) commercial Fe2O3, (b) T-ASSt, and (c) T-In@ASSt detected with KPFM. | ||
Conclusions
In summary, densely ordered nanotubular ternary In2O3/Fe2O3/Fe3O4 heterostructures have been synthesized through electrochemical anodization followed by hydrothermal deposition and annealing. The in situ growth of a dual-phase Fe2O3/Fe3O4 p–n junction formed an IEF that effectively facilitated the separation of photogenerated charge carriers from the photoactive hematite. After the introduction of In2O3, a second IEF was constructed between iron oxides and In2O3, promoting efficient hole collection. These distinctive dual IEFs organized in a novel tandem way could synergistically facilitate the PEC water oxidation thermodynamically and kinetically. The optimal T-In@ASSt photoanode presented a remarkable current density of 11.5 mA cm−2 at 1.55 VRHE and an outstanding oxygen evolution rate of 71.6 μmol cm−2 h−1. Theoretical calculations indicated that the modification of In2O3 onto the nanowalls of the Fe2O3/Fe3O4 heterostructure reduced the energy barrier for the formation of the *OOH intermediate while simultaneously decreasing the binding strength of *OOH → O2, allowing for sufficient O2 evolution. This rational construction of a tandem IEF offers a promising strategy for preparing superior PEC water oxidation catalysts.Author contributions
C. W. Wang, G. Li and K. Y. Wang supervised the project. X. H. Yan and C. W. Wang conceptualized the idea. X. H. Yan performed the materials’ synthesis and photoelectrochemical measurements with the assistance of G. Li, K. Shen, C. W. Wang and K. Y. Wang. C. W. Wang performed the DFT calculations. X. H. Yan and C. W. Wang wrote the manuscript. G. Li, C. W. Wang and K. Y. Wang revised the manuscript. All authors contributed to the discussion and manuscript review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (U1810204, 61901293, 22002083, and 21905099), Natural Science Foundation of Shanxi Province (202103021224440), Innovative Research Team of Shanxi Hundred Talents Program, Shanxi “1331 Project” and Youth Innovation Promotion Association CAS (2020180). We thank Chunmei Feng from HZWTECH for help and discussions regarding the computations in this study and acknowledge HZWTECH for providing computation facilities.References
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- Q. G. Pan, C. Zhang, Y. J. Xiong, Q. X. Mi, D. D. Li, L. L. Zou, Q. H. Huang, Z. Q. Zou and H. Yang, ACS Sustainable Chem. Eng., 2018, 6, 6378 CrossRef CAS.
- W. J. Jo, J. W. Jang, K. J. Kong, H. J. Kang, J. Y. Kim, H. Jun, K. P. S. Parmar and J. S. Lee, Angew. Chem., Int. Ed., 2012, 51, 3147–3151 CrossRef CAS PubMed.
- H. i. Kim, D. Monllor-Satoca, W. Kim and W. Choi, Energy Environ. Sci., 2015, 8, 247–257 RSC.
- Y. J. Chen, S. F. Ji, W. M. Sun, Y. P. Lei, Q. C. Wang, A. Li, W. X. Chen, G. Zhou, Z. D. Zhang, Y. Wang, L. R. Zheng, Q. H. Zhang, L. Gu, X. D. Han, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2020, 59, 1295–1301 CrossRef CAS PubMed.
- M. Jadwiszczak, K. Jakubow-Piotrowska, P. Kedzierzawski, K. Bienkowski and J. Augustynski, Adv. Energy Mater., 2019, 10, 1903213 CrossRef.
- W. J. Li, P. M. Da, Y. Y. Zhang, Y. C. Wang, X. Lin, X. G. Gong and G. F. Zheng, ACS Nano, 2014, 8, 11770–11777 CrossRef CAS PubMed.
- Q. J. Bu, S. Li, Q. N. Wu, L. L. Bi, Y. H. Lin, D. J. Wang, X. X. Zou and T. F. Xie, ChemSusChem, 2018, 11, 3486–3494 CrossRef CAS PubMed.
- A. G. Tamirat, J. Rick, A. A. Dubale, W. N. Su and B. J. Hwang, Nanoscale Horiz., 2016, 1, 243–267 RSC.
- S. H. Shen, S. A. Lindley, X. Y. Chen and J. Z. Zhang, Energy Environ. Sci., 2016, 9, 2744–2775 RSC.
- S. Niu, W. J. Jiang, Z. Wei, T. Tang, J. Ma, J. S. Hu and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 7005–7013 CrossRef CAS PubMed.
- Z. J. Zhang, I. Karimata, H. Nagashima, S. Muto, K. Ohara, K. Sugimoto and T. Tachikawa, Nat. Commun., 2019, 10, 4832 CrossRef PubMed.
- Y. Q. Cong, M. M. Chen, T. Xu, Y. Zhang and Q. Wang, Appl. Catal., B, 2014, 147, 733–740 CrossRef CAS.
- Z. Jiang, X. Tan and Y. Huang, Sci. Total Environ., 2022, 806, 150924 CrossRef CAS PubMed.
- J. Ding, H. Zheng, H. Gao, Q. Liu, Z. Hu, L. Han, S. Wang, S. Wu, S. Fang and S. Chou, Adv. Energy Mater., 2021, 11, 2100973 CrossRef CAS.
- Y. Liu, Y. Chen, Y. Tian, T. Sakthivel, H. Liu, S. Guo, H. Zeng and Z. Dai, Adv. Mater., 2022, 34, 2203615 CrossRef CAS PubMed.
- Q. Xu, J. Zhang, H. Zhang, L. Zhang, L. Chen, Y. Hu, H. Jiang and C. Li, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- C. Ratnasamy and J. P. Wagner, Catal. Rev.: Sci. Eng., 2009, 51, 325–440 CrossRef CAS.
- G. K. Reddy, P. Boolchand and P. G. Smirniotis, J. Phys. Chem. C, 2012, 116, 11019–11031 CrossRef CAS.
- H. Topsoe and M. Boudart, J. Catal., 1973, 31, 346–359 CrossRef CAS.
- S. Nie, E. Starodub, M. Monti, D. A. Siegel, L. Vergara, F. El Gabaly, N. C. Bartelt, J. de la Figuera and K. F. McCarty, J. Am. Chem. Soc., 2013, 135, 10091–10098 CrossRef CAS PubMed.
- C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, John Wiley & Sons, Hoboken, Canada, 2nd edn, 2006 Search PubMed.
- J. Leduc, Y. Goenuellue, P. Ghamgosar, S. J. You, J. Mouzon, H. Choi, A. Vomiero, M. Grosch and S. Mathur, ACS Appl. Nano Mater., 2019, 2, 334–342 CrossRef CAS.
- Y. G. Wang, G. Li, K. Y. Wang and X. Y. Chen, Appl. Surf. Sci., 2020, 505, 144497 CrossRef CAS.
- H. X. Li, J. Q. Wan, Y. W. Ma, M. Z. Huang, Y. Wang and Y. M. Chen, Chem. Eng. J., 2014, 250, 137–147 CrossRef CAS.
- H. J. Wu, G. L. Wu and L. D. Wang, Powder Technol., 2015, 269, 443–451 CrossRef CAS.
- P. Kumar, H. N. Lee and R. Kumar, J. Mater. Sci.: Mater. Electron., 2014, 25, 4553–4561 CrossRef CAS.
- F. Zhang, X. Y. Li, Q. D. Zhao and A. C. Chen, J. Phys. Chem. C, 2016, 120, 19113–19123 CrossRef CAS.
- H. C. Xu, Y. Wang, X. L. Dong, N. Zheng, H. C. Ma and X. F. Zhang, Appl. Catal., B, 2019, 257, 117932 CrossRef CAS.
- L. Li, G. M. Zhou, Z. Weng, X. Y. Shan, F. Li and H. M. Cheng, Carbon, 2014, 67, 500–507 CrossRef CAS.
- F. Yan, C. L. Zhu, S. Wang, Y. Zhao, X. T. Zhang, C. Y. Li and Y. J. Chen, J. Mater. Chem. A, 2016, 4, 6048–6055 Search PubMed.
- J. S. Kang, Y. Noh, J. Kim, H. Choi, T. H. Jeon, D. Ahn, J. Y. Kim, S. H. Yu, H. Park, J. H. Yum, W. Choi, D. C. Dunand, H. Choe and Y. E. Sung, Angew. Chem., Int. Ed., 2017, 56, 6583–6588 CrossRef CAS PubMed.
- J. Y. Zhang, Y. Yan, B. B. Mei, R. J. Qi, T. He, Z. T. Wang, W. S. Fang, S. Zaman, Y. Q. Su, S. J. Ding and B. Y. Xia, Energy Environ. Sci., 2021, 14, 365–373 RSC.
- R. Yalavarthi, A. Naldoni, R. Zbořil and Š. Kment, Catal. Today, 2021, 361, 117–123 CrossRef CAS.
- F. Malara, F. Fabbri, M. Marelli and A. Naldoni, ACS Catal., 2016, 6, 3619–3628 CrossRef CAS.
- S. C. Zhang, Z. F. Liu, M. G. Ruan, Z. G. Guo, E. Lei, W. Zhao, D. Zhao, X. F. Wu and D. M. Chen, Appl. Catal., B, 2020, 262, 118279 CrossRef CAS.
- S. He, C. Yan, X. Z. Chen, Z. Wang, T. Ouyang, M. L. Guo and Z. Q. Liu, Appl. Catal., B, 2020, 276, 119138 CrossRef CAS.
- B. Lei, D. Xu, B. Wei, T. F. Xie, C. Y. Xiao, W. L. Jin and L. L. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 4785–4795 CrossRef CAS PubMed.
- L. Chen, J. T. Ren and Z. Y. Yuan, Adv. Energy Mater., 2023, 2203720 CrossRef CAS.
- Z. H. Zhou, J. Liu, R. Long, L. G. Li, L. J. Guo and O. V. Prezhdo, J. Am. Chem. Soc., 2017, 139, 6707–6717 CrossRef CAS PubMed.
- Z. L. Wang, X. Mao, P. Chen, M. Xiao, S. A. Monny, S. C. Wang, M. Konarova, A. J. Du and L. Z. Wang, Angew. Chem., Int. Ed., 2019, 58, 1030–1034 CrossRef CAS PubMed.
- R. Ovcharenko, E. Voloshina and J. Sauer, Phys. Chem. Chem. Phys., 2016, 18, 25560–25568 RSC.
- E. Kecsenovity, B. Z. Endródi, P. T. S. Tóth, Y. Zou, R. A. Dryfe, K. Rajeshwar and C. Janáky, J. Am. Chem. Soc., 2017, 139, 6682–6692 CrossRef CAS PubMed.
- X. J. Zou, Y. Y. Dong, J. Ke, H. Ge, D. Chen, H. J. Sun and Y. B. Gui, Chem. Eng. J., 2020, 400, 125919 CrossRef CAS.
- J. Li, L. J. Cai, J. Shang, Y. Yu and L. Z. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed.
- J. Li, G. G. Zhan, Y. Yu and L. Z. Zhang, Nat. Commun., 2016, 7, 11480 CrossRef CAS PubMed.
- J. Yang, J. F. Jing, W. L. Li and Y. F. Zhu, Adv. Sci., 2022, 9, 2201134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03466f |
| This journal is © The Royal Society of Chemistry 2023 |