
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly transparent polyurethane thermosets with tunable properties and enzymatic degradability derived from polyols originating from hemicellulosic sugars†
Nejib
Kasmi
 *a,
Yosra
Chebbi
a,
Alessandra
Lorenzetti
b and
Minna
Hakkarainen
*a
*a,
Yosra
Chebbi
a,
Alessandra
Lorenzetti
b and
Minna
Hakkarainen
*a
aDepartment of Fibre and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden. E-mail: nejib@kth.se; minna@kth.se
bDepartment of Industrial Engineering, Padova University, Via F. Marzolo 9, IT 35131 Padova, Italy
First published on 27th October 2023
Abstract
Polyols made entirely from biomass could play a substantial role in producing greener polyurethanes (PUs) with lower environmental impact. Here, six fully biobased branched polyester polyols derived from hemicellulosic sugars and dicarboxylic acids were prepared by melt polycondensation and further utilized for the synthesis of twelve highly transparent and malleable PU thermosets. The latter were prepared by solvent-free step growth polymerization under extremely mild reaction conditions, easily applicable industrially. This included a very short reaction time (125–335 s), low operating temperatures (40–65 °C) and excellent yields (100%). In addition, no purification step was required. All the obtained PUs possessed very good thermal stability exceeding 235 °C, Tg (3.6–70.4 °C), and a broad hot-pressing window up to 192 °C above the respective Tg. These amorphous materials demonstrated a wide range of stress–strain behaviors, from hard to ductile, with elongation at break and tensile strength appearing in the 15–188% and 3.3–31.1 MPa range, respectively, comparable or superior to those of common commercially available fossil-based PU thermosets on the market. The enzymatic hydrolysis behaviour of the synthesized PUs was assessed using lipases from Candida rugosa and Aspergillus niger. All PUs showed some susceptibility to enzymatic attack, with a maximum mass loss up to 35% after 30 days. Most importantly, it was found that compositional control by tailoring of the length of the diacid unit in the branched polyol backbone and/or the chemical crosslinking degree of the resulting PU networks can be used as a practical method for effective ‘on demand’ tuning of the thermal properties, mechanical performance and enzymatic degradation rate. Taking advantage of these promising features in combination with the optical transparency, the developed polyurethane thermosets with easily adjustable properties show great potential as innovative materials for a wide application range.
Introduction
The production of polyurethanes (PUs) reached 24 million metric tons in 2018 and continues to grow annually by 4%, making PUs the sixth most produced plastic-type worldwide.1 PUs are renowned for their versatility with applications ranging from appliances to adhesives, furniture, insulation, coatings, elastomers, flexible and rigid foams, and automotive applications.1 The global market size of PU materials accounted for 69 billion USD in 2020, and is forecast to reach a market cap of 89 billion USD by 2030.2 However, most of the PUs are still produced from petrochemical resources.3 Additionally, conventional PU thermosets that account for 31% of the thermoset market4 are not easily biodegraded or recycled due to their permanent crosslinked structures, which can escalate their adverse environmental impact.3 With increasing reliance on PUs and in response to the growing awareness of global challenges related to dwindling fossil resources, greenhouse gas emissions, environmental pollution and surging energy demand,5 much work has been devoted to the development of more environmentally friendly PUs with reduced carbon footprint.3,6–17The prodigious scientific and technical upsurge in the conversion of biomass into new monomers and building blocks offers the possibility of obtaining a plethora of sustainable and greener polyurethanes. In this vein, and with the aim of improving the long-term sustainability of the PU industry, extensive efforts have been recently made in the academia and chemical industry to exploit biomass-derived feedstocks as precursors for different polyols.6–8,18–22 Various renewable resources such as vegetable oils, polysaccharides (starch, agro-wastes, etc.), microalgae and lignocellulose have been explored for the production of biobased polyols, the main constituent of PUs.18 Indeed, polysaccharides and lignocellulose were used to produce polyethers and aromatic polyols, respectively, while lipids have been the main source for the preparation of polyester polyols.18 Among them, the most abundant biobased polyols used in the PU industry are obtained from vegetable oils, produced by the esterification of fatty acids with glycerol.23–25
The global market for polyols, employed as the main component in PU synthesis, was 26 billion USD in 2019 and is forecast to reach 34.4 billion USD by 2024 due to the continuously growing demand for polyurethanes.18 In contrast, the global biobased polyol market was valued at 4.7 billion USD in 2017.23 In more detail, castor oil-based polyols are produced for various PU applications by Vandeputte in Belgium,23 Nivapol Technology Aps in Denmark23 and the American Company Holdings LLC with several sites in the UK.23 Other biobased polyols from soybean oil, sunflower oil and rapeseed oil were produced by several German companies.23,26,27 Renewable polyester polyols derived from castor and soybean oils can be obtained by converting their corresponding oil through ozonation, transesterification and epoxidation reactions.28 The Cargill company in the USA produces a series of soy-based polyols under the trade name BiOH® for the production of flexible polyurethane foams for carpet cushions, mattress toppers, automotive seats and upholstered furniture.23 The same company – Cargill – purchased in 2017 another company developing soy-based polyols marketed under the trade name Agrol®.23 Several renewable polyols extracted from rapeseed oil were also commercialized by the spin-off company PolyLabs Ltd, founded by Latvian State Institute of Wood Chemistry (LSIWC).23 This demonstrates the huge potential of biobased polyols as building blocks for PUs.
Hemicelluloses are an important renewable feedstock for the preparation of a wide range of sustainable building blocks including diacids, polyols, diamines, diols, etc. through biotechnological and catalytic pathways.29,30 1,2,4-Butanetriol (BTO) and 1,2,6-hexanetriol (HTO) are biobased triols, which can be obtained from xylose with the respective 5-hydroxymethylfurfural (HMF) obtained from derived-hemicellulose sugar.31,32 Succinic (SucA) and adipic (AdiA) acids are further examples of promising biobased C2 and C4 monomers derivable from lignocellulosic biomass,33,34 while azelaic acid (AzeA) can be industrially obtained from oleic acid through ozonolysis or extracted from plant sources.35,36 A systematic combination of these sugar-derived triols and biobased diacids through melt polycondensation enabled recently the synthesis of six new fully biobased branched polyester polyols with different structural features.37
We envisioned that the appealing characteristics of the above-mentioned hydroxyl-ended polyester polyols, such as their renewability, very low Tg (from −57.9 to −18.6 °C) and good thermal stability (Td,5% in the 168–236 °C range), would endow them with great potential to be employed as starting materials in polyurethane synthesis. Furthermore, compared to polyurethanes based on polyether polyols, PUs prepared from polyester polyols have typically higher mechanical properties and are much more susceptible to hydrolytic and enzymatic degradation due to the presence of hydrolysable ester moieties.7,38 Thus, as a concrete contribution towards greener PU materials, we utilized these 100% biomass-derived branched polyester polyols for step growth polymerization under mild solvent-free conditions, aiming at a novel portfolio of highly transparent aliphatic PU thermosets with different crosslinking degrees. The effect of compositional and structural control on the properties of the resulting PU networks, including their thermal and mechanical properties as well as their sensitivity to enzymatic degradation, was assessed in detail.
Experimental
Materials
1,2,4-Butanetriol (BTO, purum >90%), 1,2,6-hexanetriol (HTO, purum 96%), azelaïc acid (AzeA, purum 98%), succinic acid (SucA, purum 99%), adipic acid (AdiA, purum 99.5%), tertrabutyl titanate (TBT, purum 97%), dibutyltin dilaurate catalyst (DBTL, purum 95%), lipase from Candida rugosa, lipase from Aspergillus niger, and hexamethylene diisocyanate (HDI, purum 99%) reagents were purchased from Sigma-Aldrich. All other solvents and materials used were of analytical grade.Synthesis of branched polyols
Six renewable branched polyesters bearing free hydroxyl end groups were synthesized from sugar-derived triols (BTO and HTO) and biobased diacids (SucA, AdiA and AzeA). The synthesis proceeded via a two-stage melt polycondensation method (esterification and polycondensation) as previously described in detail by us.37 The structural features of these polyester polyols were scrutinized by 1D and 2D NMR including COSY, HSQC and HMBC spectroscopy.37 Briefly, predetermined amounts of BTO or HTO triol and SucA, AdiA or AzeA diacid with a molar ratio of diacid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :triol of 1:3 were fed in the presence of 400 ppm of TBT catalyst into an autoclave reactor equipped with a mechanical stirrer. In the first step, esterification reaction was carried out under a N2 flow at the temperature range of 160–190 °C for 4 h. In the second step of solvent-free synthesis of polyols (polycondensation), the temperature of the mixture was raised to 210–230 °C stepwise under a high vacuum of 100 mbar. The reaction lasted for 3 h (1.5 h at 210 °C, 1 h at 220 °C, and for an additional 0.5 h at 230 °C). The target polyols were purified by dissolution in DMSO followed by precipitation in water (using 10× volume of DMSO). These low Tg polyols derived from fully renewable resources are tagged as: SucA/BTO, AdiA/BTO, AzeA/BTO, SucA/HTO, AdiA/HTO, and AzeA/HTO.
:triol of 1:3 were fed in the presence of 400 ppm of TBT catalyst into an autoclave reactor equipped with a mechanical stirrer. In the first step, esterification reaction was carried out under a N2 flow at the temperature range of 160–190 °C for 4 h. In the second step of solvent-free synthesis of polyols (polycondensation), the temperature of the mixture was raised to 210–230 °C stepwise under a high vacuum of 100 mbar. The reaction lasted for 3 h (1.5 h at 210 °C, 1 h at 220 °C, and for an additional 0.5 h at 230 °C). The target polyols were purified by dissolution in DMSO followed by precipitation in water (using 10× volume of DMSO). These low Tg polyols derived from fully renewable resources are tagged as: SucA/BTO, AdiA/BTO, AzeA/BTO, SucA/HTO, AdiA/HTO, and AzeA/HTO.
The method used in this work for determining the hydroxyl number of the produced polyester polyols is based on ISO 4629-2 (2016).39 Hydroxyl number values of 552, 810, 497, 522, 720 and 612 mg KOH per g were obtained for SucA/HTO, AdiA/HTO, AzeA/HTO, SucA/BTO, AdiA/BTO and AzeA/BTO polyols, respectively.
Synthesis of crosslinked polyurethane networks
As presented in Scheme 1 (in the Results and discussion section), a series consisting of twelve new aliphatic polyurethane thermosets with different crosslinking degrees were successfully synthesized on gram-scale (see Table S1†) via a “solvent-free” step growth polymerization approach as follows: a predetermined amount of biobased branched polyester bearing hydroxyl end groups (used as polyol) and an HDI crosslinker with molar ratios of polyol/HDI of 1/0.75 and 1/1 were charged in a round bottom flask in the presence of DBTL catalyst (0.01 wt%). The reaction was carried out under solvent-free conditions and a mild N2 flow and heated for only 125–335 s at very low operating temperature in the 40–65 °C range, depending on the polyol involved and the crosslinking degree of the targeted PU. After cooling to room temperature, the prepared partially and fully crosslinked polymer networks were characterized as they are not subjected to any purification step. All the resulting PU materials were labeled as PU X Y, where X is the polyol used to form the crosslinked polyurethane (e.g., AzeA/HTO or AdiA/BTO polyol) and Y is the polyol/HDI molar ratio (1/0.75 or 1/1). | ||
| Scheme 1 Synthesis route of (A) sugar-derived branched polyols and (B) their corresponding polyurethane thermosets with different crosslinking degrees. | ||
Fourier transform infrared (FTIR) spectroscopy
The infrared spectra of the branched polyols and their corresponding PUs were obtained using a PerkinElmer Spectrum 2000 Fourier transform infrared (FTIR) spectrometer (Norwalk, CT), in the wavenumber range of 750–4000 cm−1 and in transmittance mode. The FTIR spectra were recorded at a resolution of 4 cm−1.Differential scanning calorimetry (DSC)
The thermal transition behaviour of all the developed biobased polyols and polyurethanes was studied by means of a Mettler Toledo DSC820 (Mettler Toledo, USA). The polyol monomers and PU samples, sealed in an aluminum pan, were heated under nitrogen flow (50 mL min−1) from 30 to 150 °C at a heating rate of 10 °C min−1. The polymer sample was then cooled down to −30 °C (or to −70 °C for polyols) at 10 °C min−1 and maintained at this temperature for 3 min to erase any thermal history before heating again for the second heating cycle from −30 °C (PU) or −70 °C (polyol) to 150 °C with a heating rate of 10 °C min−1. The glass transition temperature (Tg) was determined from the second heating scan.Thermogravimetric analysis (TGA)
To investigate the thermal stability of the renewable branched polyols and the PU thermosets with different crosslinking degrees synthesized from these monomers, thermogravimetric (TGA) analysis was performed under a N2 atmosphere with a TGA/SDTA851e (Mettler Toledo, USA). The sample with a weight around 8–10 mg was inserted into 70 μL ceramic crucibles and heated from 30 °C to 600 °C at a constant heating rate of 10 °C min−1.Mechanical properties
The mechanical properties, i.e. Young's Modulus, strain at break and stress at break, of the synthesized polyurethane networks were investigated by means of an Instron 5944 universal testing machine equipped with a 500 N load cell, with an effective gauge length of 15 mm and at a crosshead speed of 5 mm min−1. The PUs were compression molded to films with an average thickness ∼0.3 mm, length = 20 mm and width = 5 mm as follows: for fully cross-linked PUs made from AdiA and AzeA diacids and all partially crosslinked PU materials: 180 °C for 15 min, force 100 kN followed by cooling to room temperature and for SucA-based PUs in a completely crosslinked state: 190 °C for 15 min and a force of 130 kN followed by cooling to room temperature. The thickness of each analyzed polyurethane film was considered as the mean value of 4 different measurements performed along the specimen's length. The polymer samples were kept in an environmental chamber at 22 °C and 50% relative humidity for at least 48 h before testing. The reported properties presented average values of 4 determinations.Enzymatic degradation test
Polyurethane thermosets were compression molded to films 2.5 cm2 in size and approximately 0.3 mm thickness, following the same film preparation method used for the tensile tests. These films were placed in flasks containing 10 mL of phosphate buffer solution (pH = 7.4, 0.1 M) with 0.1 mg mL−1Candida rugosa lipase and 0.05 mg mL−1 of lipase from Aspergillus niger.40–42 The enzymatic hydrolysis was carried out at 37 ± 1 °C in an oven up to 30 days, while the enzymes and the buffered solution were replaced every 120 h in order to maintain the enzyme activity. After a scheduled period of incubation (every 5 days), the films were removed from the medium, washed carefully with distilled water, and then dried in a vacuum oven at 36 °C until constant weight. The degree of enzymatic degradation was estimated from the mass loss of the polymer samples. The surface morphologies of the PU films after different enzymatic hydrolysis times were examined through a Hitachi S-4800 ultrahigh resolution field emission scanning electron microscope (FE-SEM). Before analysis, the films were coated with a platinum/palladium (Pt/Pd) coater target at a 2 nm thickness using a Cressington 208HR sputter coater.Results and discussion
Synthesis of branched polyols and their corresponding polyurethane thermosets
Six fully biobased branched polyester-type polyols were successfully synthesized in the bulk according to the Bimolecular Non-Linear Polymerization (BMNLP) approach as depicted in Scheme 1A.37 Briefly, the two-step synthesis consisted of an esterification step, where predetermined amounts of triol (BTO or HTO) and diacid (SucA, AdiA or AzeA) with a molar ratio of diacid:triol of 1:3 were charged into a reactor equipped with a mechanical stirrer in the presence of the TBT catalyst. In the second step of the solvent-free synthesis of branched polyols, the polycondensation step was conducted under high vacuum. The developed low-Tg oligomers were recovered in high yields (80–85%) and exhibited low molecular weights in the 2730–3680 g mol−1 range.37 As shown in Scheme 1B, the renewable polyols were then reacted with the bifunctional diisocyanate (HDI) at polyol:HDI molar ratios of 1:0.75 and 1:1 to yield a series of partially and fully crosslinked polyurethane networks, respectively (composed of six polymers in each series). The synthesis of these polyurethane-based thermosetting materials with very high yield (≈100%) was successfully performed via a solvent-free step growth polymerization method under mild conditions; the curing temperatures ranged from 40 to 65 °C and curing times from 125 to 335 s (Table 1).
| PU sample | Curing temperature (°C) | Reaction time (s) |
|---|---|---|
| PU SucA/HTO 1/1 | 65 | 300 s |
| PU SucA/HTO 1/0.75 | 64 | 226 s |
| PU AdiA/HTO 1/1 | 49 | 200 s |
| PU AdiA/HTO 1/0.75 | 46 | 160 s |
| PU AzeA/HTO 1/1 | 48 | 270 s |
| PU AzeA/HTO 1/0.75 | 41 | 180 s |
| PU SucA/BTO 1/1 | 58 | 125 s |
| PU SucA/BTO 1/0.75 | 54 | 261 s |
| PU AdiA/BTO 1/1 | 46 | 195 s |
| PU AdiA/BTO 1/0.75 | 58 | 335 s |
| PU AzeA/BTO 1/1 | 44 | 220 s |
| PU AzeA/BTO 1/0.75 | 40 | 165 s |
As an example, for illustrative purposes, the solvent-free and fast polymerization reaction to synthesize PU AdiA/BTO 1/1 was recorded and is provided in Video S1,† and the resulting rigid PU foam material produced is shown in Video S2† and Fig. 1. Photographs of all as-received PUs are presented in Fig. S1.† These PU materials vary widely in physical properties, from soft, malleable and rigid foams (Fig. 1) to tough and hard networks, depending on the branched polyol involved and/or the crosslinking degree of the target polyurethane.
 | ||
| Fig. 1 Photographs of some of the prepared biobased polyurethane thermosets. Photographs of the other synthesized PU networks are shown in Fig. S1 of the ESI.† | ||
Most importantly, the new flexible PU AzeA/HTO 1/1 foam having the longest aliphatic diacid (AzeA) and triol (HTO) in its backbone exhibited impressive elastic compressive behaviour. Indeed, as clearly shown in Fig. 2 and Video S3† and regardless of the number of consecutive compression cycles applied, this polyurethane thermoset completely recovers its original shape when the compression force is released, thus showing excellent resilient performance.
 | ||
| Fig. 2 Photo-demonstration of the resilience of fully cross-linked PU AzeA/HTO 1/1 foam before, under and after compression. | ||
Such a feature is highly attractive for many demanding applications. All the prepared PU networks were completely insoluble in common solvents, showing excellent chemical resistance. From an economical perspective, the mild synthesis conditions applied for obtaining these PU thermosets, in combination with no purification or separation steps required, are very beneficial due to low energy consumption. Therefore, this solvent-free synthesis approach has good potential to provide a greener, viable and cost-effective route for the production of renewable PU materials also at the industrial scale.
Structural characterization of the prepared PU thermosets
The prepared PU networks were highly resistant to organic solvents; therefore FTIR spectroscopy was used to confirm the successful synthesis of the targeted PU materials. The FTIR spectra of SucA/HTO polyol and its corresponding PUs are shown as relevant examples in Fig. 3, while the FTIR spectra of the other biobased polyols and polyurethane thermosets are provided in the ESI (Fig. S2†). None of the PU FTIR spectra show the stretching vibration band characteristic of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO at 2200–2300 cm−1, indicating that all isocyanate groups have completely reacted with the polyols during the polymerization process. Compared with the FTIR spectra of polyols, two new bands arising from the formed urethane linkages, i.e. ν(C–N) and in-plane bending vibrations of δ(N–H), were respectively observed in the spectra of all produced polyurethanes at 1240 cm−1 and 1535 cm−1.43 Another piece of evidence for the successful synthesis of PU networks is the shift of the characteristic peak of the CO bond in the branched polyester polyols (1725 cm−1) to lower wavenumbers (1696 cm−1) in the FTIR spectra of PU materials. This can be explained by the formation of strong hydrogen-bonding interactions between the carbonyl of the ester moiety and –N–H of the urethane linkage. This hence results in a pronounced downshift of ∼25 cm−1 in (vCO) stretching frequency. The broad band at 1696 cm−1 is attributed to the urethane carbonyl stretching (vCO) overlapping with the H-bonded ester carbonyl band in the polyol. Comparison with polyols producing a broad band centered at around 3340 cm−1 shows that all crosslinked PUs revealed a band at a slightly lower wavenumber of 3290 cm−1 assigned to the –N–H stretching vibrations of the urethane group (Fig. 3 and S2†).44
CO at 2200–2300 cm−1, indicating that all isocyanate groups have completely reacted with the polyols during the polymerization process. Compared with the FTIR spectra of polyols, two new bands arising from the formed urethane linkages, i.e. ν(C–N) and in-plane bending vibrations of δ(N–H), were respectively observed in the spectra of all produced polyurethanes at 1240 cm−1 and 1535 cm−1.43 Another piece of evidence for the successful synthesis of PU networks is the shift of the characteristic peak of the CO bond in the branched polyester polyols (1725 cm−1) to lower wavenumbers (1696 cm−1) in the FTIR spectra of PU materials. This can be explained by the formation of strong hydrogen-bonding interactions between the carbonyl of the ester moiety and –N–H of the urethane linkage. This hence results in a pronounced downshift of ∼25 cm−1 in (vCO) stretching frequency. The broad band at 1696 cm−1 is attributed to the urethane carbonyl stretching (vCO) overlapping with the H-bonded ester carbonyl band in the polyol. Comparison with polyols producing a broad band centered at around 3340 cm−1 shows that all crosslinked PUs revealed a band at a slightly lower wavenumber of 3290 cm−1 assigned to the –N–H stretching vibrations of the urethane group (Fig. 3 and S2†).44
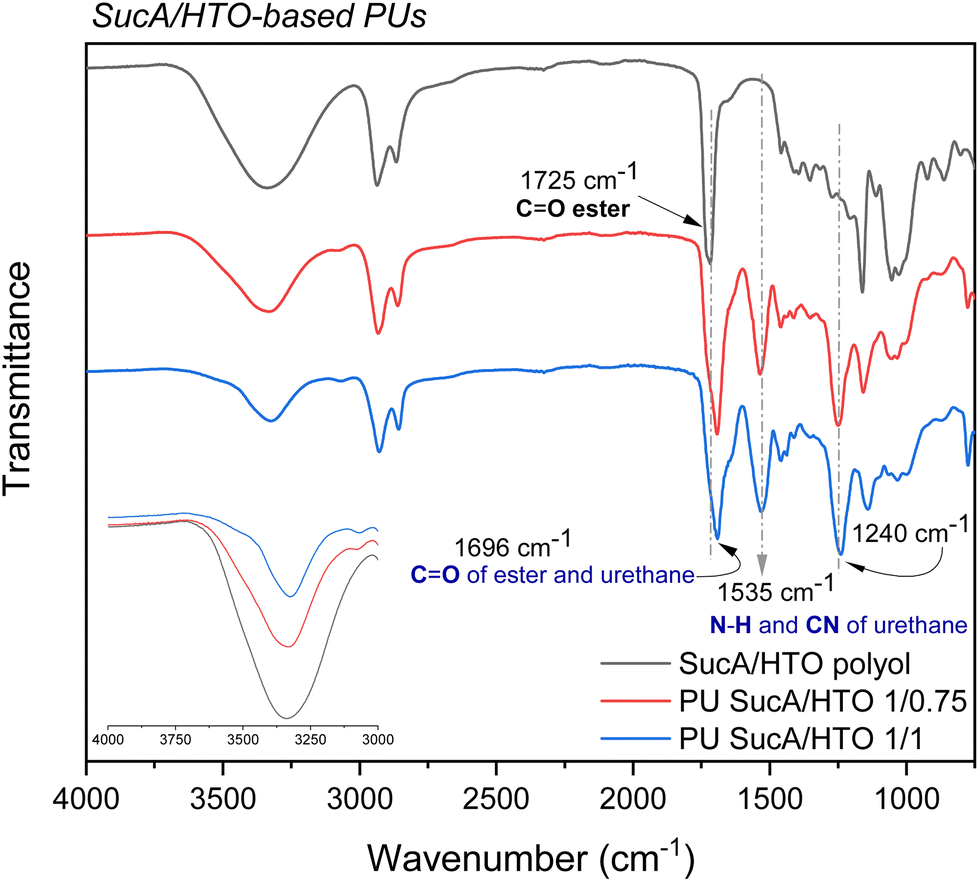 | ||
| Fig. 3 FTIR spectra of hemicellulose-based SucA/HTO polyol and its corresponding biobased PU thermosets. | ||
Importantly, the aforementioned N–H peak gradually decreases in relative intensity as the HDI crosslinker content increases. Similar findings were recently reported by Kasprzyk et al. for biobased polyurethanes, where the intensity of the –N–H peak of urethane groups in the FTIR spectra decreased continuously upon increasing the isocyanate/polyol molar ratio.45 Additionally, typical bands at 2860 and 2930 cm−1 are due to the aliphatic C–H symmetric and asymmetric stretching modes of the CH2 groups of the polyol and long-chain HDI. To recapitulate, FTIR data prominently corroborate that a series of partially and fully cross-linked polyurethane thermosets was successfully synthesized from biobased aliphatic polyols with branched molecular architectures.
Thermal properties of polyurethane networks
The assessment of the thermal properties of polymers is of high importance in order to determine their potential application domains. Therefore, the thermal transitions of the polyester-type polyols and their corresponding polyurethane networks with different cross-linking degrees were thoroughly investigated by DSC analyses. The DSC thermograms of the branched polyols and their derivative PU thermosets are presented in Fig. 4 and Fig. S3.† The DSC traces of BTO-based PUs are shown as examples for illustrative purposes (Fig. 4). For all the PU networks, regardless of their crosslinking density (whether partially or fully cross-linked) or the used polyol type, the DSC traces revealed the presence of only one Tg and no peaks associated with cold crystallization. Neither melting endotherms nor crystallization phenomena could be detected during cooling/heating scans, hence confirming the fully amorphous nature of the materials. The recorded DSC data related to the glass transition temperatures are presented in Table S2.† The partially and fully crosslinked polyurethanes showed Tg values in the range of 3.6–18.5 °C and 18.0–70.4 °C, respectively. These values are significantly higher compared to those of the corresponding amorphous polyols (Tg = −57.9 °C to −18.6 °C). The observed trends mainly depended on the level of crosslinking with HDI. As shown in Fig. 5A and Fig. S4A, B,† it is worth noting that regardless of their compositions, the most densely crosslinked PU materials (1/1 polyol to HDI) possess higher glass transition temperatures than the partially crosslinked ones (1/0.75 polyol to HDI), wherein Tg values of 61.4 vs. 5.8, 18.0 vs. 12.7, 20.4 vs. 3.6, 70.4 vs. 10.9, 45.1 vs. 6.3, and 23.5 vs. 18.5 °C were found for the polyurethane samples PU SucA/HTO, PU AdiA/HTO, PU AzeA/HTO, PU SucA/BTO, PU AdiA/BTO, and PU AzeA/BTO, respectively. These could be ascribed to the increasingly crosslinked architectures of the PU thermosets, leading to increasing Tg values.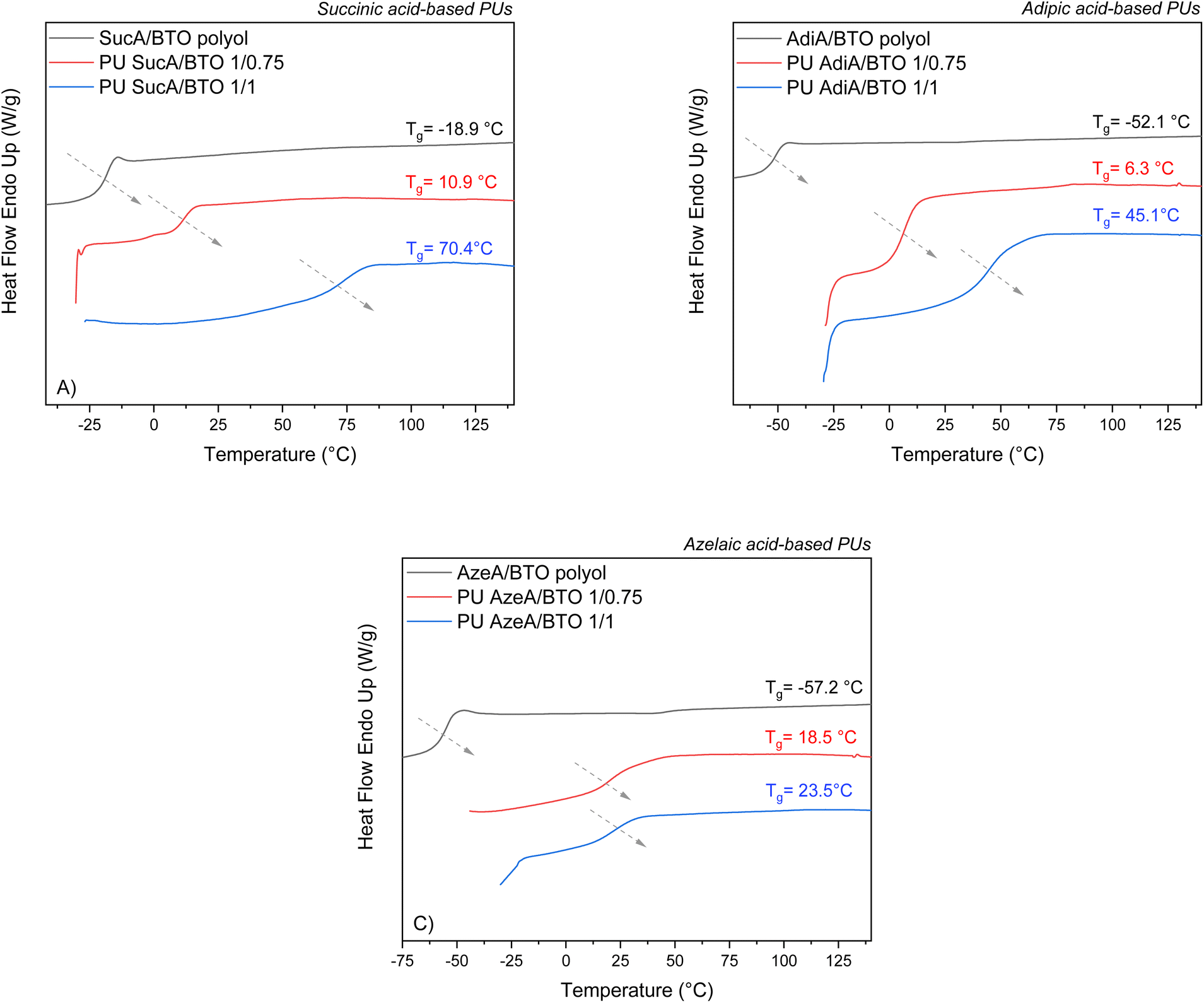 | ||
| Fig. 4 DSC thermograms of the PU thermosets with different degrees of crosslinking based on: (A) SucA/BTO polyol, (B) AdiA/BTO polyol, and (C) AzeA/BTO polyol. | ||
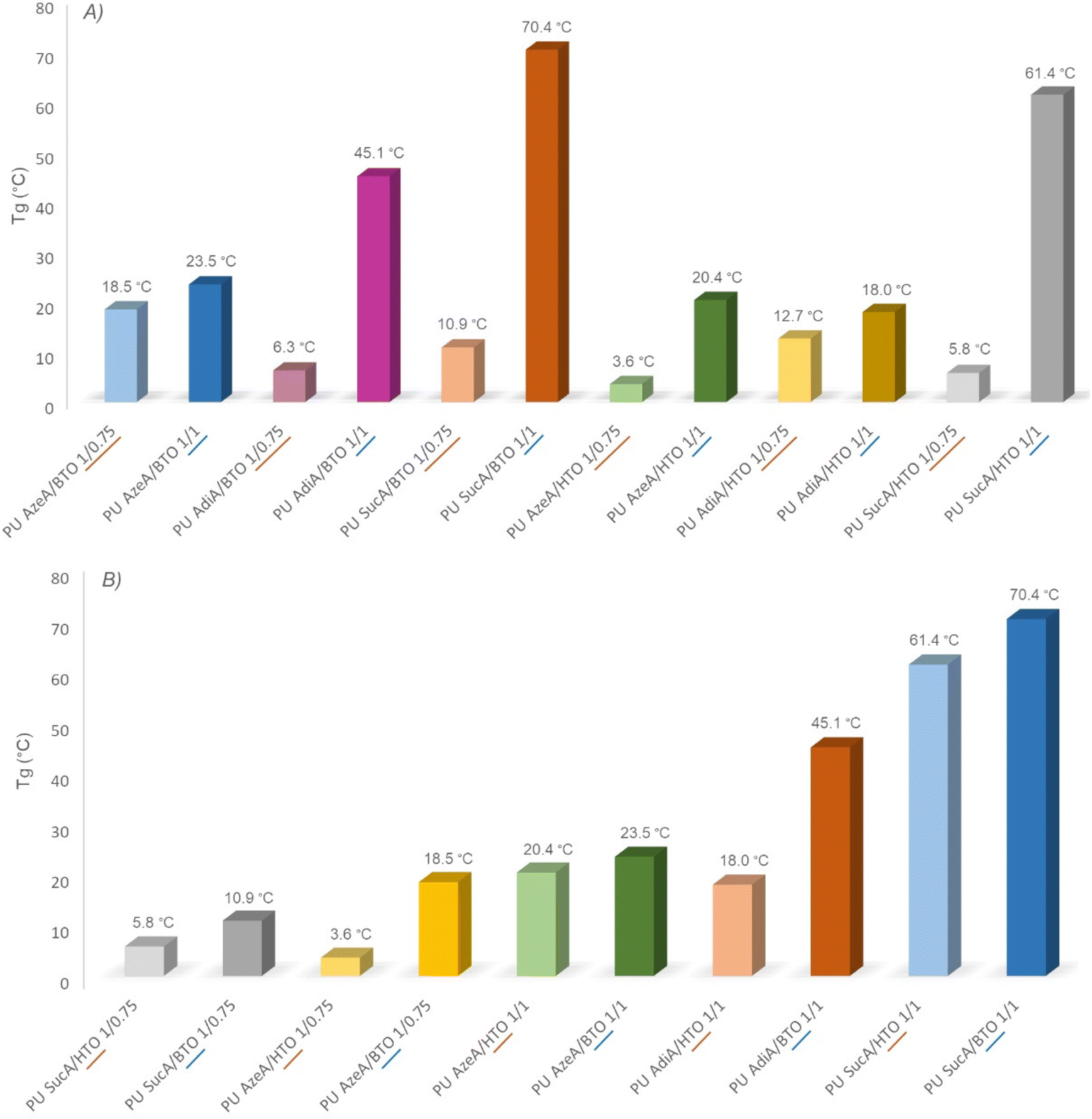 | ||
| Fig. 5 Effects of (A) the crosslinking degree of PUs and (B) the chain length of the used triols (BTO and HTO) on the Tg of PU networks. | ||
DSC results also demonstrated an obvious correspondence between the Tg value and the chain length of the triols incorporated into the PU thermosets produced with different crosslinking densities. PU networks derived from HTO triol, whether partially or fully crosslinked, mostly showed higher Tg values than those containing BTO triol with shorter aliphatic carbon chains as elucidated in Fig. 5B.
For fully cross-linked PU networks derived from BTO-based polyols, a discernible trend in Tg values was observed depending on the aliphatic diacid type incorporated into the backbone of the polyols involved (see Fig. S4C†). Indeed, upon decreasing the diacid chain length from C7 (azelaic acid) to C2 (succinic acid), Tg steadily increased from 23.5 °C to 70.4 °C. The compositional control of the branched polyester polyols used in PU synthesis and/or the adjustment of the HDI molar ratio to these polyols can, thus, be utilized for the synthesis of cross-linked polyurethane networks with tailored thermal performances.
Thermal degradation of polyurethane networks
The thermal stability of polymers is crucial for determining their potential applications and processability. For that, TGA analysis was carried out under a nitrogen atmosphere to assess the resistance to heat of the synthesized PU thermosets. The TGA thermograms in the temperature range from 30 to 600 °C of HTO-based PU materials are depicted in Fig. 6 as representative examples and the remaining TGA thermograms are shown in Fig. S5.† The most representative degradation parameters, such as 5% mass loss-temperatures (Td,5%) and the char yields at 500 °C, are listed in Table S2.† It was found that all the prepared biobased polyurethanes exhibited high thermal stabilities over 235.0 °C with onset decomposition temperatures (Td,5%) appearing in the 235–275 °C range, leaving just 0.5–6.1% of residual weight upon heating to 500 °C. The decomposition profiles of the PU materials illustrate that they degrade by a multi-stage process. As shown in Fig. 7 and Fig. S6,† AzeA-based PU thermosets prepared from BTO-based polyols, irrespective of their degree of crosslinking, exhibited higher heat resistance than their counterparts obtained with shorter chain SucA or AdiA diacid. Indeed, the Td,5% values of the partially crosslinked polymers PU AzeA/BTO 1/0.75, PU AdiA/BTO 1/0.75 and PU SucA/BTO 1/0.75 were 267.8 °C, 247.0 °C, and 235.0 °C, respectively, against 275.5 °C, 265.0 °C, and 263.1 °C for their fully crosslinked homologues (1/1 polyol to HDI), respectively.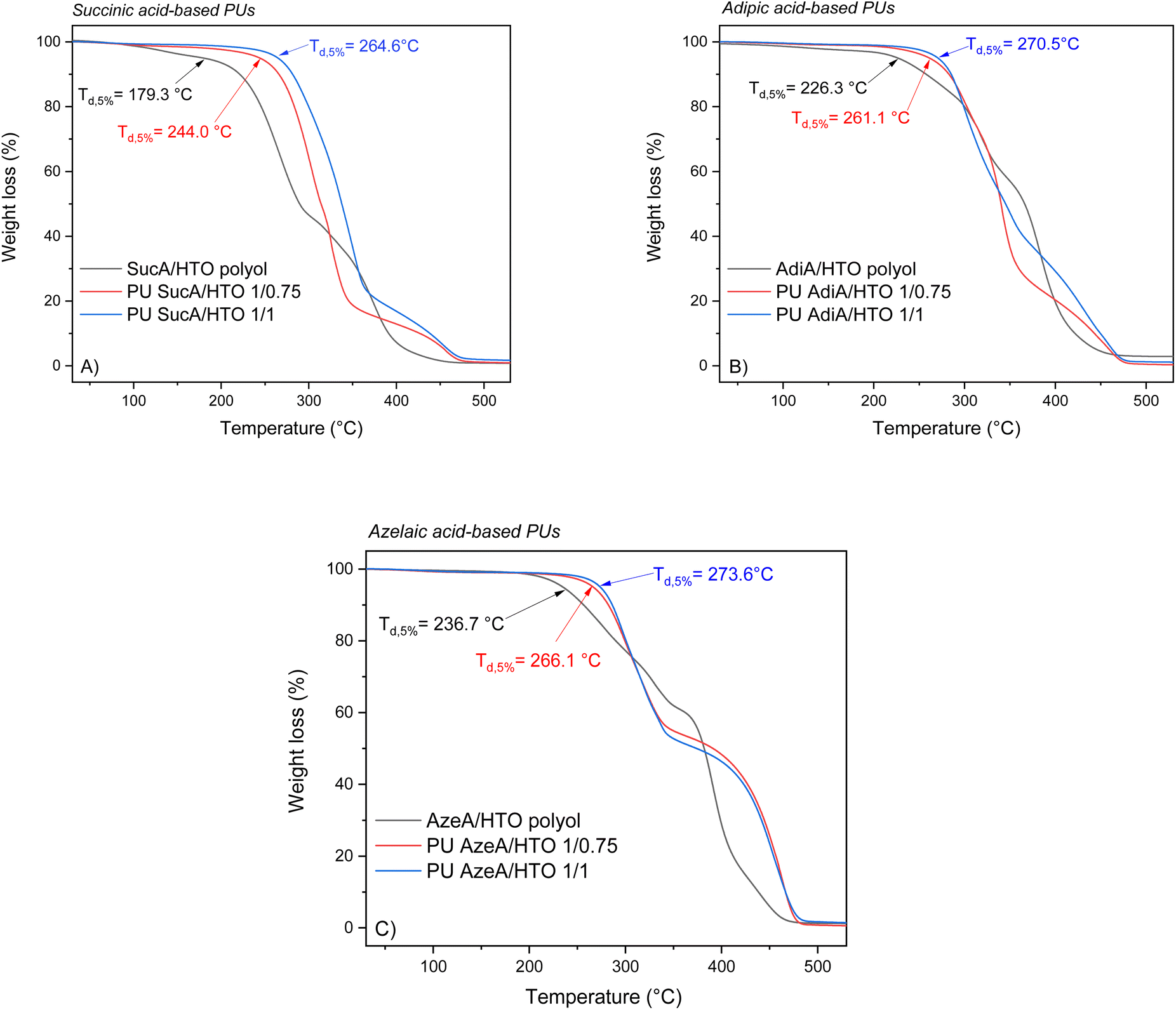 | ||
| Fig. 6 TGA thermograms of the PU networks with different degrees of crosslinking based on: (A) SucA/HTO polyol, (B) AdiA/HTO polyol, and (C) AzeA/HTO polyol. | ||
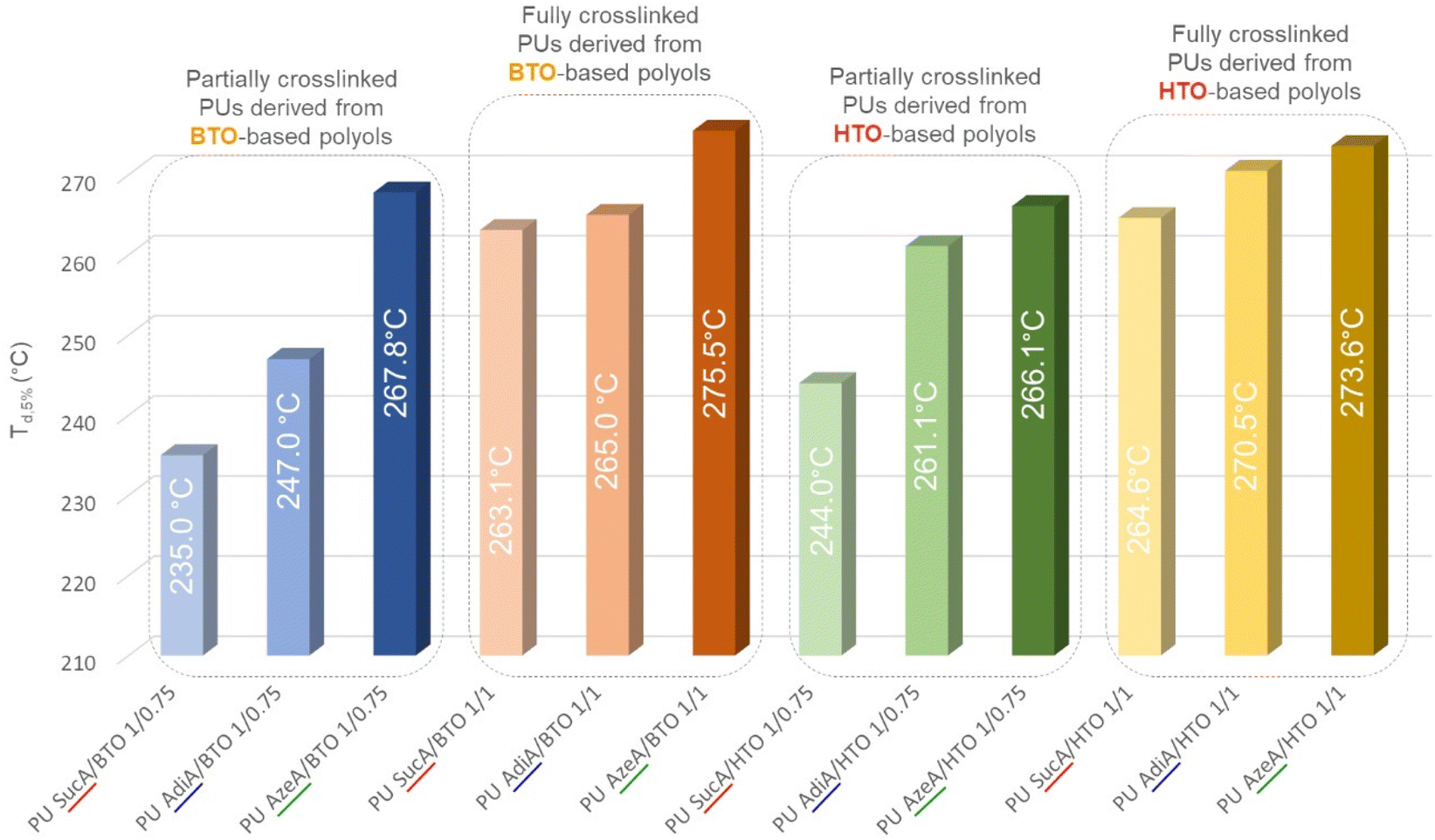 | ||
| Fig. 7 Effect of the diacid type in the polyols on the heat resistance of PU thermosets. | ||
It is worth noting that HTO-based PU networks (either partially or fully branched) present a similar trend to BTO-based samples, where polyurethane having the longest aliphatic diacid (AzeA) showed higher thermal stability compared with polyurethanes synthesized from shorter diacids, AdiA or SucA. In more detail, the partially crosslinked thermoset PU AzeA/HTO 1/0.75 turned out to be more thermally stable than PU AdiA/HTO 1/0.75 and PU SucA/HTO 1/0.75 (showing a Td,5% of 266.1 °C vs. 261.1 °C and 244.0 °C, respectively). The fully crosslinked HTO-based thermosets followed the same trend, that is, upon increasing the diacid chain length from C2 (SucA) to C7 (AzeA), the Td,5% increased from 264.6 °C to 273.6 °C. These discernible trends are most likely attributed to the presence of a long-chain aliphatic diacid incorporated into the PU thermosets’ macromolecular chain, which improved their heat resistance by delaying the onset of degradation (Td,5%). Such findings are in agreement with a previous study reporting that chain segments originating from longer aliphatic diacids are less vulnerable to thermal degradation.46
As shown in Fig. S7,† the HTO-based PU networks prepared from SucA and AdiA diacids (either partially or fully crosslinked samples) exhibited higher heat resistance than their BTO-based homologues. These results confirmed that the incorporation of aliphatic diacids, flexible comonomers used in the synthesis of branched polyols, with different chain lengths (SucA, AdiA, and AzeA) in the corresponding polyurethane networks can be utilized to tailor the thermal stability of PUs. The demonstrated excellent resistance to heat ensures good thermal stability at temperatures much higher than the glass transition of the materials. This provides a broad thermal processing window up to 192–262 °C above the respective Tg, thereby making the materials suitable candidates for hot-pressing.
Mechanical properties of the polyurethane thermosets
The synthesized PUs demonstrated a wide range of stress–strain behaviors, ranging from hard to ductile. The mechanical properties of compression molded PU films investigated by tensile testing are depicted in Fig. 8, 9 and S8.† The typical stress–strain curves of these novel biobased polyurethane networks are illustrated in Fig. 8A and B, while their main tensile properties including Young's modulus (E), tensile strength (σ) and elongation at break (ε) are summarized in Table S3.†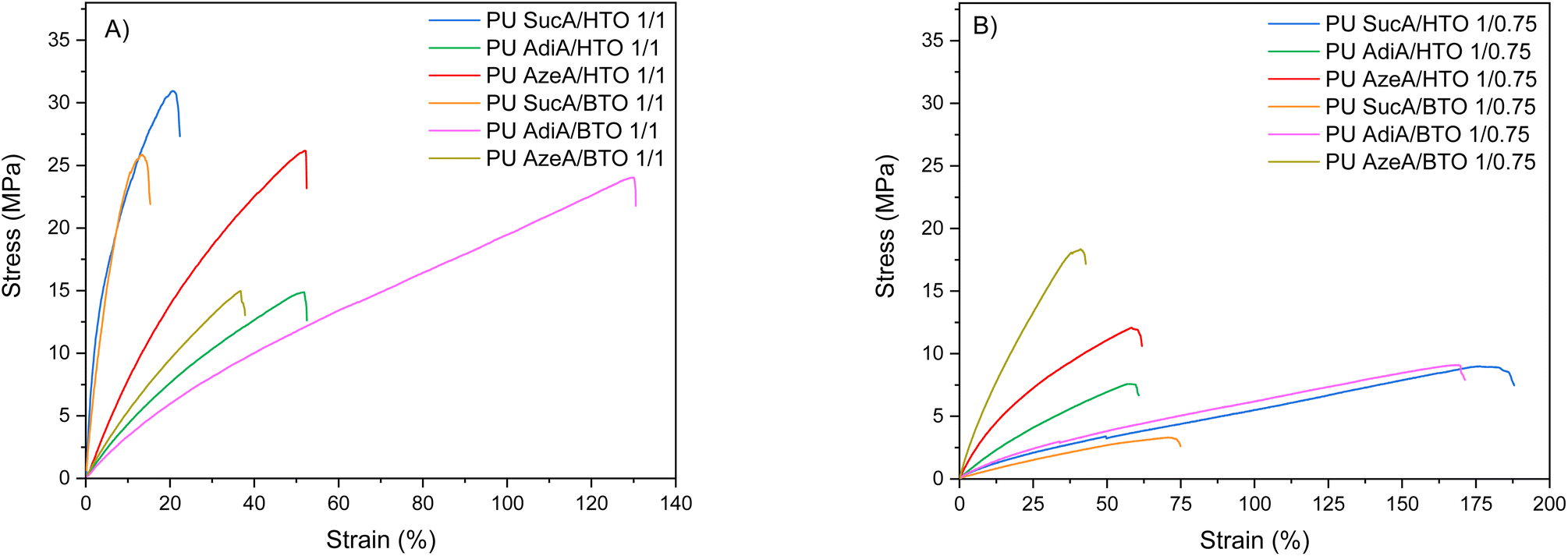 | ||
| Fig. 8 Stress–strain curves of the prepared polyurethane thermosets: (A) fully crosslinked PUs, and (B) partially crosslinked PUs. | ||
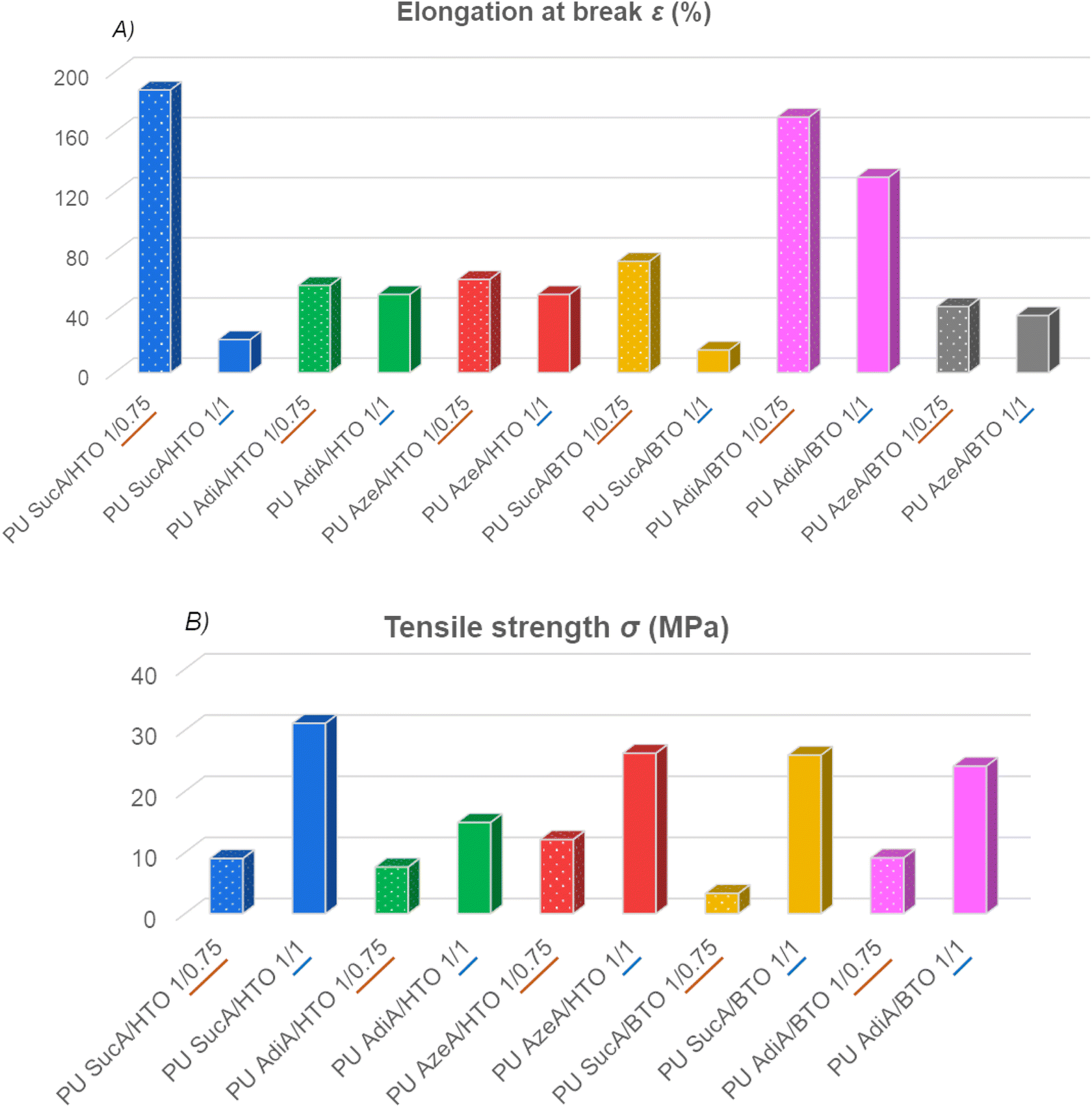 | ||
| Fig. 9 Comparison of (A) elongation at break and (B) tensile strength of prepared polyurethane thermosets as a function of their crosslinking degree. | ||
All the PU networks with partially crosslinked structures (bearing free –OH functional end-groups in combination with lower crosslinking density) were found to be significantly more flexible compared to the same polyurethane materials with fully crosslinked structures (absence of terminal –OH functions and higher crosslinking density). The presence of such free reactive hydroxyl groups has also been reported to greatly facilitate the malleability and reprocessability of polyurethane thermosets.47 For partially crosslinked PU networks, the ε, σ and E appeared in the 44–188%, 3.3–18.3 MPa and 6.1–84.9 MPa range, respectively. Importantly, the partially crosslinked polymers PU AdiA/BTO 1/0.75 and PU SucA/HTO 1/0.75 exhibited the highest elongations at break, 170% and 188%, respectively (Fig. S9†). The latter with low Tg and high elasticity possessed the ability to recover after being stretched, thus behaving like elastomeric materials at room temperature. Chemical crosslinking has been widely used to alter the physical properties of polymers including the improvement of their mechanical performance. This is consistent with the obtained E and σ data in the present study, where the fully crosslinked PU thermosets with strain varying in the 15–130% range possessed higher σ (14.9–31.1 MPa) and E values (34.9–648.8 MPa) compared to those of the partially crosslinked polyurethanes (see Table S3†).
Regardless of the chemical compositions of the prepared PU networks, discernible trends in their ε, σ and E values were observed as a function of their crosslinking degrees. In more detail, as clearly shown in Fig. 9A, the elongation at break increased when the branched polyol/HDI molar ratio in the macromolecular chains of PU thermosets decreased from 1/1 (completely crosslinked state) to 1/0.75 (partially crosslinked polymers). In contrast, the tensile strength and modulus follow the opposite trend, that is, upon increasing the branching degree from partially to fully crosslinked materials (except PU AzeA/BTO), a remarkable increase in the tensile strength and modulus was observed (Fig. 9B and S8†). This could be mainly ascribed to the greater content of crosslinking agent (HDI), which reduces the mobility of polymer chain segments due to shorter distance between the crosslinks and therefore enhances the stiffness of the resulting PU networks, leading to an increase in E and σ. These findings demonstrated that controlling the chemical crosslinking density of the prepared networks by tailoring the polyol/HDI molar ratio can be used as a practical method to effectively tune the mechanical properties of the final polyurethane materials.
It is also worth mentioning that the macroscopic observation of all renewable polyurethane materials revealed optically transparent networks. PU SucA/BTO 1/1 (having the lowest elongation at break of 15%) was slightly yellow, while the rest of the fully crosslinked PUs and all the partially crosslinked polymers were colorless (see the photographs in Fig. 10 and S10†). In Videos S4 and S5,† PU AdiA/BTO 1/1 and PU AzeA/BTO 1/1 are shown as representative examples illustrating the high transparency of the developed fully crosslinked PUs.
 | ||
| Fig. 10 Photographs of partially crosslinked polyurethane films. | ||
As can be observed from Fig. 11, the PU networks produced in this work exhibited comparable or higher tensile strength and elongation at break than most biobased PU thermosets reported in the literature or non-renewable commercially available crosslinked polyurethanes.3,6,48–59 The attractive features of the developed PU thermosets such as good and easily tunable mechanical performance, including a very broad range of elongation at break (15–188%), Young's moduli (6.1–648.8 MPa) and tensile strength (3.3–31.1 MPa), together with their high heat resistance and good optical transparency could cement these PUs as a promising class of high-performance amorphous polyurethane materials.
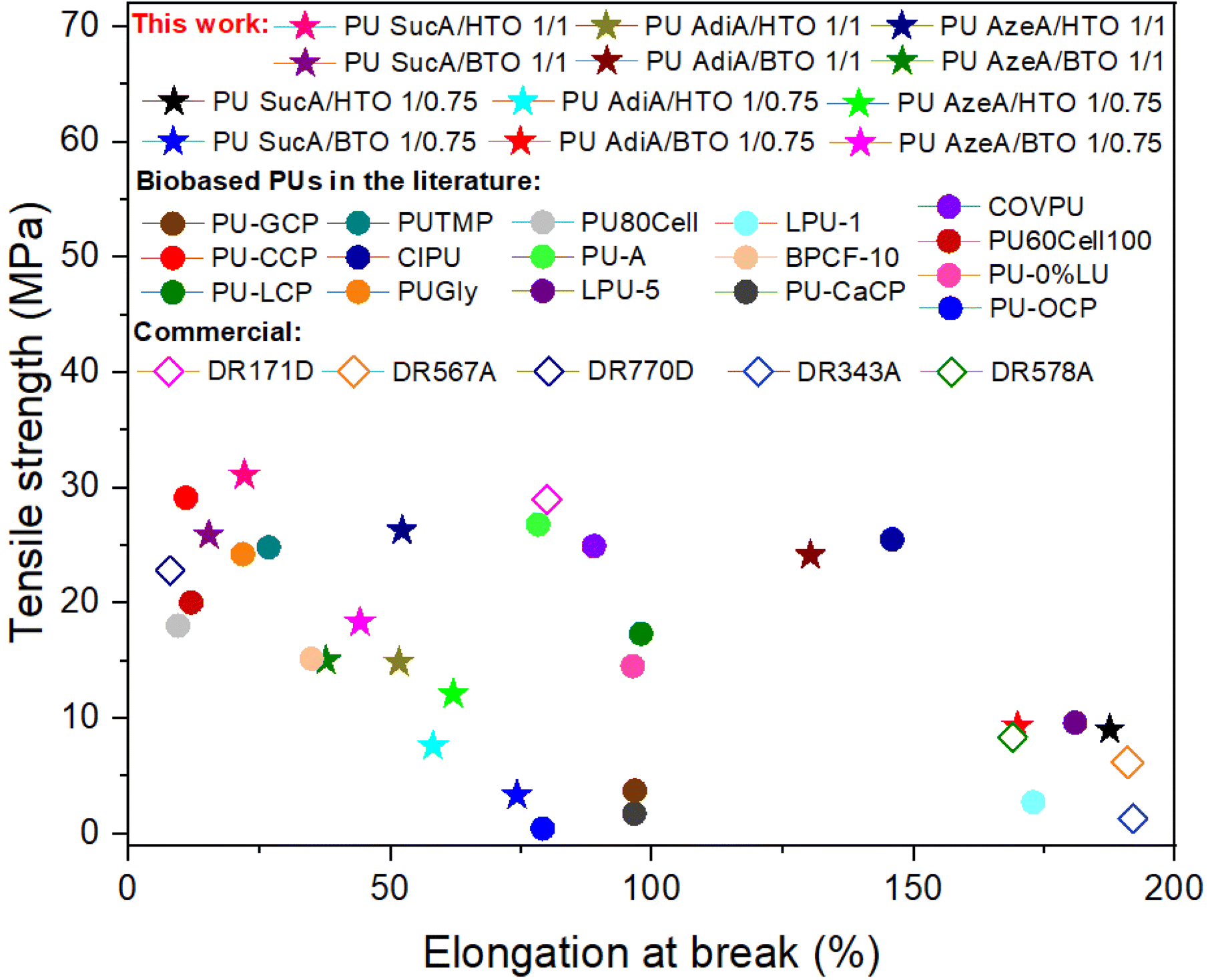 | ||
| Fig. 11 Comparison of the mechanical performance of synthesized PU networks in this work with other biobased crosslinked polyurethanes in the literature and commercial non-renewable crosslinked polyurethanes.3,6,48–59 | ||
Enzymatic degradation of PU thermosets
In specific applications, the biodegradability of the materials can help reduce environmental pollution. Here, aliphatic condensation polymers are expected to have higher susceptibility to biodegradation compared to aromatic ones.60,61 For preliminary evaluation of the susceptibility of our aliphatic PU networks to biodegradation, all synthesized aliphatic PU networks with different crosslinking degrees were subjected to enzymatic degradation assays in aqueous solution using Candida rugosa lipase and lipase from Aspergillus niger as enzymes. These enzymes have been commonly used to evaluate the enzymatic degradability of polyesters, polyurethanes and polyester-urethanes.42,62,63 The choice to use a mixture of both enzymes is due to the fact that all the developed PU networks contain ester and urethane linkages in their backbones. It should also be pointed out that lipases are activated only with the help of a substrate binding domain, as they are too large to penetrate deeply into the polymer.64,65 The enzymatic degradation rate of polymers is sensitive to many parameters in addition to the chemical structure, such as the molecular weight, chain mobility, degree of crystallinity and hydrophobicity of the main chain.64 The extent of biodegradation of the PUs was assessed by measuring their mass loss during incubation in the presence of enzymes and by monitoring microscopic changes on their surfaces. The mass loss percentage of the biobased thermosetting polyurethanes developed herein as a function of incubation time is depicted in Fig. 12.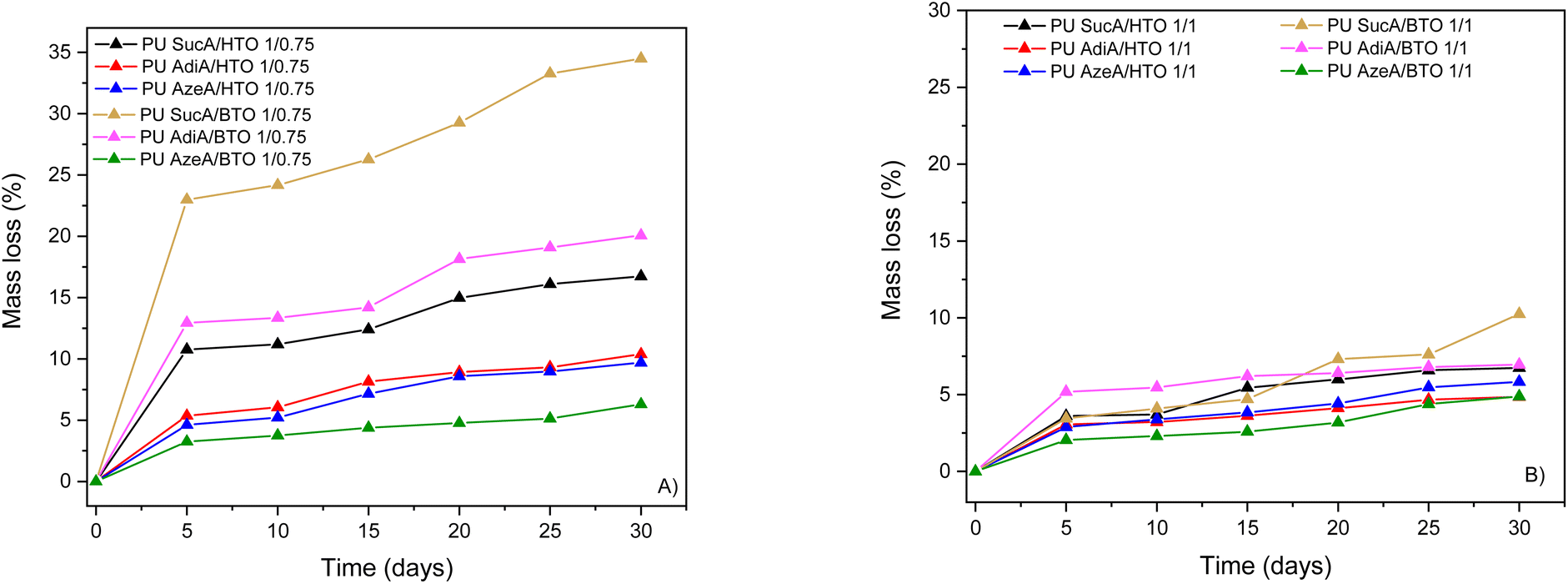 | ||
| Fig. 12 Mass loss of the films against time of the enzymatic hydrolysis of (A) partially crosslinked and (B) fully crosslinked polyurethane networks. | ||
All the fully cross-linked PU materials degraded quite slowly and showed similar degradation rates with the total mass loss in the range of ≈5–10% after 30 days (Fig. 12B). In contrast, it can be clearly seen in Fig. 12A that the partially crosslinked polyurethanes bearing free hydroxyl groups degrade faster and the enzymatic hydrolysis rate is dramatically accelerated and highly dependent on the incorporated diacid (Fig. 13A), revealing higher mass loss oscillating between 6% and 35%.
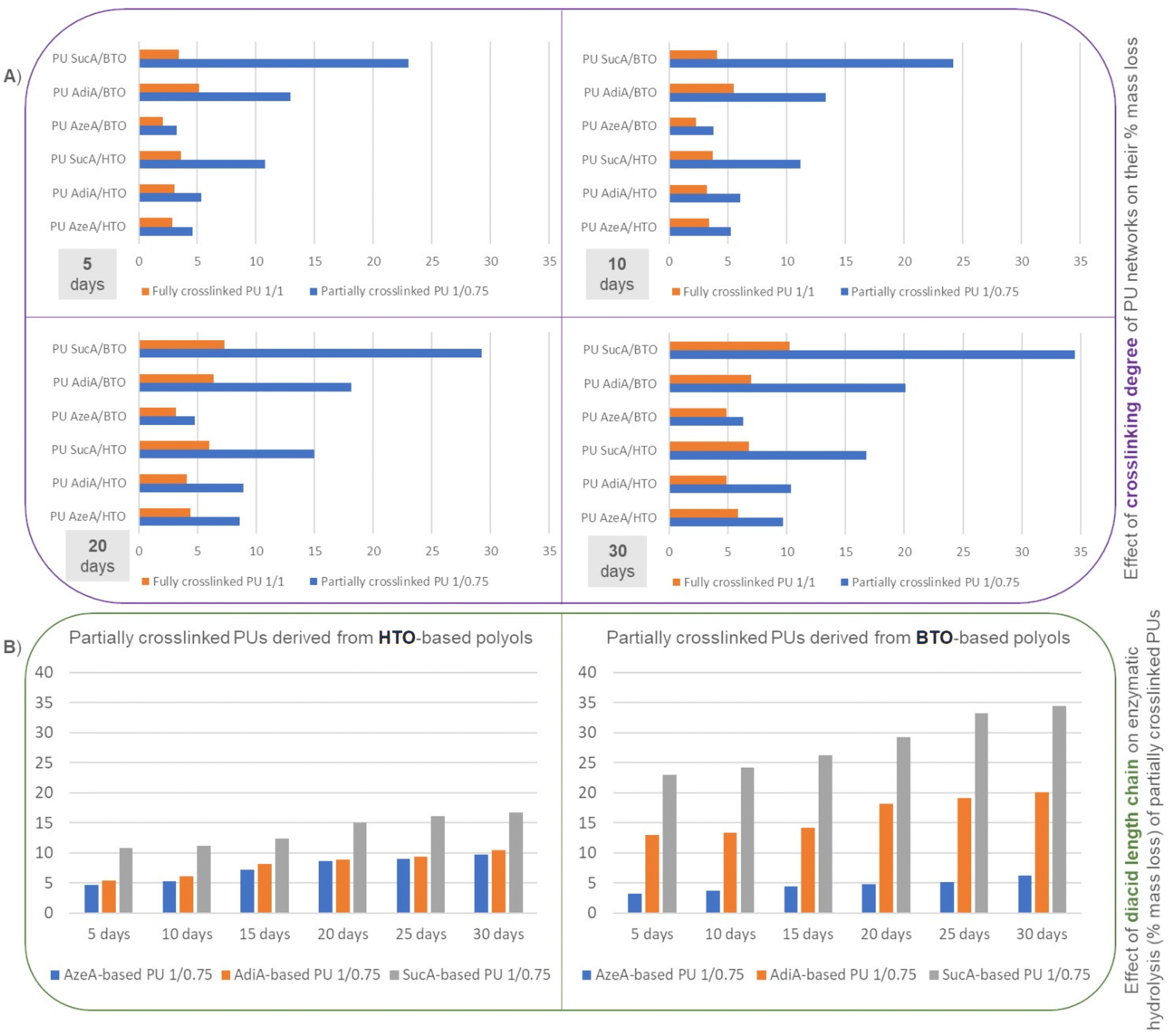 | ||
| Fig. 13 (A) Effect of the crosslinking degree of the synthesized PU thermosets and (B) the effect of the diacid type (SucA, AdiA, and AzeA) incorporated into the backbone of partially crosslinked PUs on their enzymatic hydrolysis (% mass loss). | ||
As illustrative examples for comparative purposes, mass loss values close to 20% and 35% were observed for PU AdiA/BTO 1/0.75 and PU SucA/BTO 1/0.75 after 30 days, respectively, i.e. approximately 3 times higher values compared to their fully crosslinked counterparts (vs. ≈7 and 10% for PU AdiA/BTO 1/1 and PU SucA/BTO 1/1, respectively). Therefore, the results presented in Fig. 13A clearly corroborate that the existence of free –OH functional groups on the surface of the polyurethane network in combination with the lower degree of crosslinking enhances the enzymatic hydrolysis rate, regardless of the incubation time. This is perfectly consistent with previous studies reporting that the amorphous character of polyesters and free –OH groups on their surface play a prominent role in inducing biodegradation by enhancing hydrophilicity and uptake of water into amorphous regions, thus accelerating the scission of ester bonds which leads in turn to increased mass loss.66,67 This is fully in line with the results obtained for the biobased PU thermosets developed here. These findings support that the enzymatic degradation rate of the synthesized PU networks can be tuned “on demand” by varying the feed molar ratio of branched polyol to HDI. In other words, changing the crosslinking degree of the polyurethane thermosets can be utilized as a key parameter to adjust their enzymatic degradation rate and thereby potentially also their susceptibility to biodegradation. Such a feature is highly desirable to design sustainable PU materials with controlled biodegradation behaviour depending on their target practical applications.
For all the partially crosslinked PUs, discernible trends in mass loss values were observed as a function of the diacid chain length incorporated into their backbone. As clearly shown in Fig. 13B, the enzymatic degradation rate of these PU thermosets mainly depends on their chemical compositions, that is, the longer the methylene units in the diacid within the PU's molecular chain, the lower the mass loss. This could be mainly ascribed to increasing hydroxyl and ester-group density going from diacid with 7 (AzeA) or 4 (AdiA) CH2 groups to the shorter diacid consisting of 2 CH2 units (SucA). This is in very good agreement with similar outcomes recently reported by Yu et al. for biobased polyesters derived from succinic, adipic, suberic and sebacic acids, where the mass loss of polyesters gradually increased with decreasing aliphatic unit length in diacids.66 This behavior is understandable owing to the presence of higher ester and hydroxyl group density, which in turn leads to higher amounts of hydrolysable groups and higher hydrophilicity.66
Additionally, the effect of the triol type on the enzymatic degradation is more pronounced in the case of partially crosslinked PU materials. As can be seen from Fig. 13B, for all partially crosslinked SucA and Adid-based PU thermosets, obvious trends in % mass loss were found depending on the triol type inserted in the polyurethane chain. Indeed, PUs containing shorter-chain BTO triol showed a significantly higher enzymatic degradation rate (higher % mass loss) than their HTO-derived counterparts. In more detail, for partially crosslinked SucA-based PUs, % mass loss values of 23 vs. 11, 24 vs. 11, 26 vs. 12, 29 vs. 15, 33 vs. 16 and 35 vs. 17% were found after 5, 10, 15, 20, 25 and 30 days of incubation for PU SucA/BTO 1/0.75 and PU SucA/HTO 1/0.75, respectively. The partially crosslinked polyurethane networks based on AdiA follow the same trend as was observed for SucA-based PUs, where mass loss values of 13 vs. 5, 13 vs. 6, 14 vs. 8, 18 vs. 9, 19 vs. 9 and 20 vs. 10% were obtained after 5, 10, 15, 20, 25 and 30 days of incubation for PU AdiA/BTO 1/0.75 and PU AdiA/HTO 1/0.75, respectively. Such findings are fully consistent with what is reported in the literature for polyesters, where the enzymatic hydrolysis rate is generally shown to be reduced upon increasing the chain length of the alkyl diols within their molecular chain.68
SEM observations provided complementary evidence for the morphological modifications taking place on the PU films after 30 days of incubation, hence verifying the gravimetric analysis results. The micrographs of the exposed surfaces of partially and fully crosslinked PUs are presented at different magnifications in Fig. 14 and Fig. S11,† respectively.
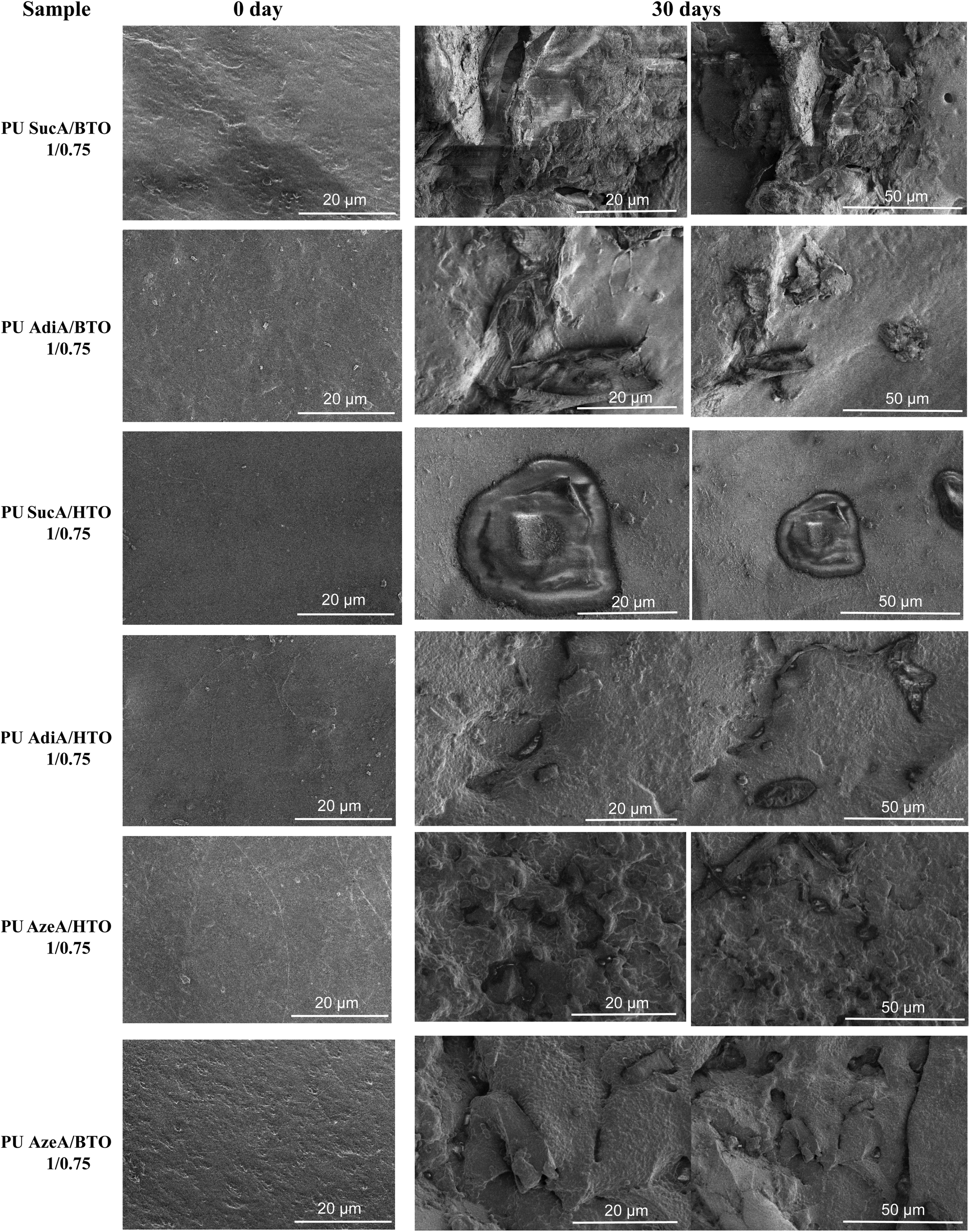 | ||
| Fig. 14 SEM micrographs of partially crosslinked polyurethane networks before (0 days) and after 30 days of enzymatic degradation. | ||
All PU films subjected to SEM observation revealed a smooth surface before enzymatic hydrolysis without the appearance of holes. A clear surface erosion of varying degrees is visible for all PU thermoset samples incubated for 30 days, witnessing their sensitivity to enzymatic biodegradation. The extension and the number of eroded areas depend on the polyurethane composition and its crosslinking degree. The surface erosion is obviously more recurrent in the case of partially crosslinked polyurethane samples as shown in Fig. 14. Conversely, fewer changes are observed for the fully crosslinked PUs (Fig. S11†).
In summary, the structural and compositional control by tailoring either the length of the aliphatic carbon chain in the diacid (SucA, AdiA, and AzeA) or the crosslinking degree of the polyurethane materials can be used as a strategy to effectively tune “on demand” the biodegradability of the developed PUs. This opens new horizons for the development of biodegradable polyurethane thermosets with customized degradation rates at specified environments, prepared from hemicellulosic sugar-derived branched polyols.
Conclusions
We demonstrate a facile, industrially viable, and cost-effective synthesis route for a novel portfolio of highly transparent aliphatic polyurethane thermosets made from 100% renewable branched polyesters bearing free hydroxyl end groups. These low-Tg polyols were prepared by melt polycondensation using a combination of biobased diacids (succinic, adipic, and azelaic acids) and sugar-derived triols (1,2,4-butanetriol and 1,2,6-hexanetriol), while step growth polymerization was utilized to synthesize the PUs under mild solvent-free conditions with very short reaction times of 2–5 min and moderate temperatures of 40–65 °C. Furthermore, no purification or separation steps were needed. The developed PU materials with different crosslinking degrees are fully amorphous, revealing excellent and tunable thermal properties (Tg oscillating from 3.6 to 70.4 °C, high resistance to heat up to 235 °C, and a broad hot-pressing window up to 192 °C above their Tg). They also exhibited mechanical performance comparable to commercial petrochemical-based thermosetting polyurethanes, with elongation at break and tensile strength in the range of 15–188% and 3.3–31.1 MPa, respectively. Enzymatic hydrolysis experiments indicated that all the synthesized PU thermosets are susceptible to enzymatic degradation, with the highest recorded mass loss up to 35% within 30 days. Interestingly, all the thermal and mechanical properties as well as the enzymatic degradability of the prepared PU networks could be widely tuned “on demand”, either by altering the crosslink density or/and by simply varying the length of the aliphatic chain in the branched polyols. In conclusion, next-generation polyurethane thermosets with readily tailored performance and high transparency were demonstrated, using a very simple synthetic approach that is highly sought for industrial scalability. The impressive characteristics of the produced PUs endow them with the potential to serve as greener alternatives for a wide application range.Author contributions
Nejib Kasmi: conceptualization, writing – original draft, investigation, formal analysis, validation, methodology, and data curation. Yosra Chebbi: writing – original draft. Alessandra Lorenzetti: formal analysis. Minna Hakkarainen: supervision, resources, funding acquisition, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.References
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, V. Vlcek, P. Christopher and M. M. Abu-Omar, Opportunities in Closed-Loop Molecular Recycling of End-of-Life Polyurethane, ACS Sustainable Chem. Eng., 2023, 11, 6114–6128 CrossRef CAS.
- Global Polyurethane Market. MENAFN Prophecy Market Insights. https://menafn.com/1103037291/Global-Polyurethane-Market-worth-US-8876-billion-by-2030-with-a-CAGR-of-250 (accessed 23 August 2023).
- D. Zhao, X. Liang, J. Wang, J. Du and Y. Zhu, Vanillin-Based Degradable Polyurethane Thermosets Demonstrating High Bio-Content and Mechanical Properties, ACS Appl. Polym. Mater., 2023, 5, 4536–4545 CrossRef CAS.
- X.-Z. Wang, M.-S. Lu, J.-B. Zeng, Y. Weng and Y.-D. Li, Malleable and thermally recyclable polyurethane foam, Green Chem., 2021, 23, 307–313 RSC.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Plant oil renewable resources as green alternatives in polymer science, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- S. Laurichesse, C. Huillet and L. Averous, Original Polyols Based on Organosolv Lignin and Fatty Acids: New Bio-Based Building Blocks for Segmented Polyurethane Synthesis, Green Chem., 2014, 16, 3958–3970 RSC.
- A. Mouren and L. Avérous, Sustainable cycloaliphatic polyurethanes: from synthesis to applications, Chem. Soc. Rev., 2023, 52, 277 RSC.
- C. Becquet, M. Ferreira, H. Bricout, B. Quienne, S. Caillol, E. Monflier and S. Tilloy, Synthesis of diols from jojoba oil via rhodium-catalyzed reductive hydroformylation: a smart way to access biobased polyurethanes, Green Chem., 2022, 24, 7906–7912 RSC.
- S. Lemouzy, A. Delavarde, F. Lamaty, X. Bantreil, J. Pinaud and S. Caillol, Lignin-based bisguaiacol diisocyanate: a green route for the synthesis of biobased polyurethanes, Green Chem., 2023, 25, 4833–4839 RSC.
- C. Wang, J. Xie, X. Xiao, S. Chen and Y. Wang, Development of Nontoxic Biodegradable Polyurethanes Based on Polyhydroxyalkanoate and L-lysine Diisocyanate with Improved Mechanical Properties as New Elastomers Scaffolds, Polymers, 2019, 11, 1927 CrossRef CAS PubMed.
- R. Kaur, P. Singh, S. Tanwar, G. Varshney and S. Yadav, Assessment of Bio-Based Polyurethanes: Perspective on Applications and Bio-Degradation, Macromolecules, 2022, 2, 284–314 CAS.
- B. Wang, S. Ma, X. Xu, Q. Li, T. Yu, S. Wang, S. Yan, Y. Liu and J. Zhu, High-Performance, Biobased, Degradable Polyurethane Thermoset and Its Application in Readily Recyclable Carbon Fiber Composites, ACS Sustainable Chem. Eng., 2020, 8, 11162–11170 CrossRef CAS.
- Y. Zhang, J. Liao, X. Fang, F. Bai, K. Qiao and L. Wang, Renewable High-Performance Polyurethane Bioplastics Derived from Lignin–Poly(ε-caprolactone), ACS Sustainable Chem. Eng., 2017, 5, 4276–4284 CrossRef CAS.
- H. E. Wray, S. Luzzi, P. D'Arrigo and G. Griffini, Life Cycle Environmental Impact Considerations in the Design of Novel Biobased Polyurethane Coatings, ACS Sustainable Chem. Eng., 2023, 11, 8065–8074 CrossRef CAS.
- T. Dong, E. Dheressa, M. Wiatrowski, A. P. Pereira, A. Zeller, L. M. L. Laurens and P. T. Pienkos, Assessment of Plant and Microalgal Oil-Derived Nonisocyanate Polyurethane Products for Potential Commercialization, ACS Sustainable Chem. Eng., 2021, 9, 12858–12869 CrossRef CAS.
- J. Peyrton, C. Chambaretaud, A. Sarbu and L. Averous, Biobased Polyurethane Foams Based on New Polyol Architectures from Microalgae Oil, ACS Sustainable Chem. Eng., 2020, 8, 12187–12196 CrossRef CAS.
- S. Gogoi and N. Karak, Biobased Biodegradable Waterborne Hyperbranched Polyurethaneas an Ecofriendly Sustainable Material, ACS Sustainable Chem. Eng., 2014, 2, 2730–2738 CrossRef CAS.
- H. Sardon, D. Mecerreyes, A. Basterretxea, L. Avérous and C. Jehanno, From Lab to Market: Current Strategies for the Production of Biobased Polyols, ACS Sustainable Chem. Eng., 2021, 9, 10664–10677 CrossRef CAS.
- T. A. R. Silva, A. C. Marques, R. G. dos Santos, R. A. Shakoor, M. Taryba and M. F. Montemor, Development of BioPolyurethane Coatings from Biomass-Derived Alkylphenol Polyols—A Green Alternative, Polymers, 2023, 15, 2561 CrossRef CAS PubMed.
- O. Gómez-Jiménez-Aberasturi and J. R. Ochoa-Gómez, New approaches to producing polyols from biomass, J. Chem. Technol. Biotechnol., 2017, 92, 705–711 CrossRef.
- P. Furtwengler and L. Averous, Renewable polyols for advanced polyurethane foams from diverse biomass resources, Polym. Chem., 2018, 9, 4258–4287 RSC.
- M. Ghasemlou, F. Daver, E. P. Ivanova and B. Adhikari, Polyurethanes from seed oil-based polyols: A review of synthesis, mechanical and thermal properties, Ind. Crops Prod., 2019, 142, 111841 CrossRef CAS.
- A. Fridrihsone, F. Romagnoli, V. Kirsanovs and U. Cabulis, Life Cycle Assessment of vegetable oil based polyols for polyurethane Production, J. Cleaner Prod., 2020, 266, 121403 CrossRef CAS.
- M. Desroches, M. Escouvois, R. Auvergne, S. Caillol and B. Boutevin, From Vegetable Oils to Polyurethanes: Synthetic Routes to Polyols and Main Industrial Products, Polym. Rev., 2012, 52, 38–79 CrossRef CAS.
- L. Maisonneuve, G. Chollet, E. Grau and H. Cramail, Vegetable Oils: A Source of Polyols for Polyurethane Materials, OCL: Oilseeds Fats, Crops Lipids, 2016, 23, D508 CrossRef.
- Neukapol polyols, Altropol Kunststoff GmbH, 2023. Available online: https://www.altropol.de/en/produkte/neukapol-polyole/, (accessed 23 August 2023).
- T. A. McKeon, D. G. Hayes, D. F. Hildebrand and R. J. Weselake, Industrial Oil Crops, Elsevier Inc., 2016. DOI:10.1016/C2015-0-00068-5.
- Y. Ma, Y. Xiao, Y. Zhao, Y. Bei, L. Hu, Y. Zhou and P. Jia, Biomass based polyols and biomass based polyurethane materials as a route towards sustainability, React. Funct. Polym., 2022, 175, 105285 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers, Polym. Chem., 2015, 6, 4497–4559 RSC.
- X.-J. Ji, H. Huang, Z.-K. Nie, L. Qu, Q. Xu and G. T. Tsao, Fuels and Chemicals from Hemicellulose Sugars, in Biotechnology in China III: Biofuels and Bioenergy, ed. F.-W. Bai, C.-G. Liu, H. Huang and G. T. Tsao, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 199–224 Search PubMed.
- J. Ohyama and A. Satsuma, Reductive Conversion of 5-Hydroxymethylfurfural in Aqueous Solutions by Furan Ring Opening and Rearrangement, in Production of Biofuels and Chemicals with Bifunctional Catalysts, ed. Z. Fang, R. L. Smith Jr and H. Li, Springer Singapore, Singapore, 2017, pp. 159–185 Search PubMed.
- A. Lachke, Biofuel fromD-xylose—The second most abundant sugar, Resonance, 2002, 7, 50–58 CrossRef CAS.
- J. Han, A bio-based ‘green’ process for catalytic adipic acid production from lignocellulosic biomass using cellulose and hemicellulose derived γ-valerolactone, Energy Convers. Manage., 2016, 129, 75–80 CrossRef CAS.
- H. Choudhary, S. Nishimura and K. Ebitani, Metal-free oxidative synthesis of succinic acid from biomass-derived furan compounds using a solid acid catalyst with hydrogen peroxide, Appl. Catal., A, 2013, 458, 55–62 CrossRef CAS.
- A. Köckritz and A. Martin, Synthesis of azelaic acid from vegetable oil-based feedstocks, Eur. J. Lipid Sci. Technol., 2011, 113, 83–91 CrossRef.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Oxidative Cleavage of Fatty Acid Derivatives for Monomer Synthesis, Catalysts, 2018, 8, 464 CrossRef.
- N. Kasmi, C. Pinel, D. Da Silva Perez, R. Dieden and Y. Habibi, Synthesis and characterization of fully biobased polyesters with tunable branched architectures, Polym. Chem., 2021, 12, 991 RSC.
- P. Lozano and E. Carcia-Verdugo, From green to circular chemistry paved by Biocatalysis, Green Chem., 2023, 25, 7041–7057 RSC.
- ISO 4629-2:2016. Binders for paints and varnishes—Determination of hydroxyl value—Part 2: Titrimetric method using a catalyst. https://www.iso.org/standard/63237.html.
- S.-L. Li, F. Wu, Y. Z. Wang and J.-B. Zeng, Biobased thermoplastic poly(ester urethane) elastomers consisting of poly(butylene succinate) and poly(propylene succinate), Ind. Eng. Chem. Res., 2015, 54, 6258–6268 CrossRef CAS.
- R. Gautam, A. S. Bassi and E. K. Yanful, Candida rugosa lipase-catalyzed polyurethane degradation in aqueous medium, Biotechnol. Lett., 2007, 29, 1081–1086 CrossRef CAS PubMed.
- A. Magnin, E. Pollet, V. Phalip and L. Avérous, Evaluation of biological degradation of polyurethanes, Biotechnol. Adv., 2020, 39, 107457 CrossRef CAS PubMed.
- Y. Kurimoto, M. Takeda, A. Koizumi, S. Yamauchi, S. Doi and Y. Tamura, Mechanical properties of polyurethane films prepared from liquefied wood with polymeric MDI, Bioresour. Technol., 2000, 74, 151–157 CrossRef CAS.
- J. Datta and E. Głowińska, Effect of hydroxylated soybean oil and bio-based propanediol on the structure and thermal properties of synthesized bio-polyurethanes, Ind. Crops Prod., 2014, 61, 84–91 CrossRef CAS.
- P. Kasprzyk, E. Sadowska and J. Datta, Investigation of Thermoplastic Polyurethanes Synthesized via Two Different Prepolymers, J. Polym. Environ., 2019, 27, 2588–2599 CrossRef CAS.
- N. Kasmi, L. Papadopoulos, Y. Chebbi, G. Z. Papageorgiou and D. N. Bikiaris, Effective and facile solvent-free synthesis route to novel biobased monomers from vanillic acid: Structure–thermal property relationships of sustainable polyesters, Polym. Degrad. Stab., 2020, 181, 109315 CrossRef CAS.
- Y. Jin, Z. Lei, P. Taynton, S. Huang and W. Zhang, Malleable and Recyclable Thermosets: The Next Generation of Plastics, Matter, 2019, 1, 1456–1493 CrossRef.
- C. Zhang, S. A. Madbouly and M. R. Kessler, Biobased Polyurethanes Prepared from Different Vegetable Oils, ACS Appl. Mater. Interfaces, 2015, 7, 1226–1233 CrossRef CAS PubMed.
- B. Xu, Q. Yin, F. Han, J. Cheng, J. Zhao and J. Zhang, A Bio-Based Healable/Renewable Polyurethane Elastomer Derived from L-Tyrosine/Vanillin/Dimer Acid, Chem. Eng. Sci., 2022, 258, 117736 CrossRef CAS.
- Y. Hu, Y. Tian, J. Cheng and J. Zhang, Synthesis of Eugenol-Based Polyols via Thiol-Ene Click Reaction and High-Performance Thermosetting Polyurethane Therefrom, ACS Sustainable Chem. Eng., 2020, 8, 4158–4166 CrossRef CAS.
- C. Meiorin, T. Calvo-Correas, M. A. Mosiewicki, M. I. Aranguren, M. A. Corcuera and A. Eceiza, Comparative Effects of Two Different Crosslinkers on the Properties of Vegetable Oil-Based Polyurethanes, J. Appl. Polym. Sci., 2020, 137, 48741 CrossRef CAS.
- Y. Sun, D. Sheng, H. Wu, X. Tian, H. Xie, B. Shi, X. Liu and Y. Yang, Bio-Based Vitrimer-Like Polyurethane Based on Dynamic Imine Bond with High-Strength, Reprocessability, Rapid-Degradability and Antibacterial Ability, Polymer, 2021, 233, 124208 CrossRef CAS.
- J. Chen, D. Hu, Y. Li, J. Zhu, A. Du and J. Zeng, Castor Oil- Based High Performance and Reprocessable Poly(Urethane Urea) Network, Polym. Test., 2018, 70, 174–179 CrossRef CAS.
- D. S. Porto, A. Cassales, H. Ciol, N. M. Inada and E. Frollini, Cellulose as a Polyol in the Synthesis of Bio-Based Polyurethanes with Simultaneous Film Formation, Cellulose, 2022, 29, 6301–6322 CrossRef CAS.
- C. Zhang, H. Wu and M. R. Kessler, High Bio-Content Polyurethane Composites with Urethane Modified Lignin as Filler, Polymer, 2015, 69, 52–57 CrossRef CAS.
- Z. Yu, D. Cheng, B. Gao, Y. Yao, C. Liu, J. Li, C. Wang, J. Xie, S. Zhang, Z. Li and Y. Yang, Bio-Based Polyurethane Based on a Dynamic Covalent Network with Damage Tolerance for Controlled Release of Fertilizers, ACS Appl. Mater. Interfaces, 2022, 14, 56046–56055 CrossRef CAS PubMed.
- D. S. Porto, C. M. G. de Faria, N. M. Inada and E. Frollini, Polyurethane Films Formation from Microcrystalline Cellulose as a Polyol and Cellulose Nanocrystals as Additive: Reactions Favored by the Low Viscosity of the Source of Isocyanate Groups Used, Int. J. Biol. Macromol., 2023, 236, 124035 CrossRef CAS PubMed.
- H. Wang and Q. Zhou, Synthesis of Cardanol-Based Polyols Via Thiol-Ene/Thiol-Epoxy Dual Click-Reactions and Thermosetting Polyurethanes Therefrom, ACS Sustainable Chem. Eng., 2018, 6, 12088–12095 CrossRef CAS.
- Durethane® R. Available online: https://www.mearthane.com/products/durethane-r/ (accessed on 27 June 2023).
- K. A. Fransen, S. H. M. Av-Ron, T. R. Buchanan, D. J. Walsh, D. T. Rota and L. Van Note, High-throughput experimentation for discovery of biodegradable polyesters, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220021120 CrossRef CAS PubMed.
- Y. Chen, L. Tan, L. Chen, Y. Yang and X. Wang, Study on biodegradable aromatic/aliphatic copolyesters, Braz. J. Chem. Eng., 2008, 25, 321–335 CrossRef CAS.
- R. Gautam, A. S. Bassi and E. K. Yanful, Candida rugosa lipase-catalyzed polyurethane degradation in aqueous medium, Biotechnol. Lett., 2007, 29, 1081–1086 CrossRef CAS PubMed.
- S. Khruengsa, T. Sripahco and P. Pripdeevech, Biodegradation of Polyester Polyurethane by Embarria clematidis, Front. Microbiol., 2022, 13, 874842 CrossRef PubMed.
- N. Kasmi, M. Wahbi, L. Papadopoulos, Z. Terzopoulou, N. Guigo, N. Sbirrazzuoli, G. Z. Papageorgiou and D. N. Bikiaris, Synthesis and characterization of two new biobased poly(pentylene 2,5-furandicarboxylate-co-caprolactone) and poly(hexamethylene 2,5-furandicarboxylate-co-caprolactone) copolyesters with enhanced enzymatic hydrolysis properties, Polym. Degrad. Stab., 2019, 160, 242–263 CrossRef CAS.
- R. Gautam, A. S. Bassi and E. K. Yanful, Candida rugosa lipase-catalyzed polyurethane degradation in aqueous medium, Biotechnol. Lett., 2007, 29, 1081–1086 CrossRef CAS PubMed.
- Y. Yu, H. Liu and Z. Wei, Synthesis, physical properties, and functionalization of biobased unsaturated polyesters derived from Cis-2-butene-1,4-diol, Polym. Degrad. Stab., 2022, 206, 110203 CrossRef CAS.
- D. N. Bikiaris, G. Z. Papageorgiou and D. S. Achilias, Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s, Polym. Degrad. Stab., 2006, 91, 31–43 CrossRef CAS.
- K. Haernvall, S. Zitzenbacher, M. Yamamoto, M. B. Schick, D. Ribitsch and G. M. Guebitz, Polyol structure and ionic moieties influence the hydrolytic stability and enzymatic hydrolysis of bio-based 2, 5-furandicarboxylic acid (FDCA) copolyesters, Polymers, 2017, 9, 403 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03302c |
| This journal is © The Royal Society of Chemistry 2023 |