
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing I−/formic acid as a green reagent for the reduction of sulfinates and sulfoxides†
J.
Armando Luján-Montelongo
 *,
Luis Javier
García de la Cuesta
,
Alicia E.
Cruz-Jiménez
,
Perla
Hernández
and
Alberto
Vela
*,
Luis Javier
García de la Cuesta
,
Alicia E.
Cruz-Jiménez
,
Perla
Hernández
and
Alberto
Vela
Departamento de Química, Centro de Investigación y de Estudios Avanzados, Avenida Instituto Politécnico Nacional 2508, San Pedro Zacatenco 07360, Ciudad de México, México. E-mail: jalujanm@cinvestav.mx; Tel: +52 55-57473729
First published on 22nd September 2023
Abstract
We present a convenient and efficient deoxygenation method for synthesizing disulfides and thioethers. The reaction involves an iodide-catalyzed reduction of methyl sulfinates or sulfoxides, respectively, employing formic acid as a stoichiometric reductant. The methodology demonstrates excellent yields and remarkable orthogonality towards other reducible functional groups. In silico explorations revealed the synergistic action of formic acid and in situ generated hydroiodic acid as Brønsted donors within a formic acid framework. This molecular arrangement activates the sulfinyl group and promotes subsequent deoxygenation. Notably, this approach enables the deoxygenation of sulfur-based functional groups using formic acid as the stoichiometric reductant without needing transition metals or strong acidic media such as hydroiodic acid. Overall, this methodology contributes to the advancement of the reduction of diverse oxygenated sulfur-containing compounds in a sustainable approach.
Organosulfur compounds are prevalent in both natural and synthetic settings. Among them, disulfides and thioethers play essential roles in diverse areas of chemistry, such as biological and synthetic organic chemistry.1,2 They are mainly synthesized by thiol oxidation, both industrially and in the laboratory.3,4 However, the formation of disulfides by the reduction of higher oxidation species is more limited to their role as intermediates in more complex synthetic processes,5 although direct syntheses have been developed.6 In contrast, sulfoxide deoxygenation is one of the most fundamental reactions in synthetic organic chemistry.1d,7 The sulfinyl and sulfanyl functional groups are frequently combined in synthetic efforts. One attractive feature of the sulfoxide/sulfide combination is the potential for an asymmetric structural elaboration based on the stereochemistry and activating properties of a sulfinyl group, which can be followed by reduction. This can be accomplished through the asymmetric induction capabilities of a sulfoxide, followed by deoxygenation, resulting in an asymmetrically functionalized thioether.8
During experiments on the Mannich-type synthesis of structurally diverse sulfonylmethyl formamides (3) from methyl sulfinates (1),9 we sporadically observed impurities in the form of disulfides (2, Scheme 1a). We speculated that high water content in certain formic acid (FA) batches or operational errors in the laboratory may have led to the formation of disulfides (2), as the concentration of N-formyl iminium species would not have been sufficient for successful Mannich reactions. After failing to reproduce the direct reduction of sulfinates to disulfides using sole formic acid (Scheme 1b) and exploring various possibilities, such as contamination from glassware (phantom reactivity),10 we observed that the incorporation of bromide salts resulted in the reproducible production of disulfides in modest yields (Scheme 1c, see the ESI†).11 However, a significant improvement was observed when substituting bromide with iodide (Scheme 1d). This substitution increased the yield and enhanced the overall reaction conditions and time. The remarkable efficacy of iodide as a reductant of sulfur-based functional groups is consistent with previous studies of its use under acidic conditions.12 Additionally, this method employs a sustainable sacrificial reductant, as formic acid can be derived from biomass processing and holds increasing significance in chemical synthesis.13,14 Furthermore, the successful application of this method to sulfoxides, an important sulfur-based organic family (vide supra), is worth noting.
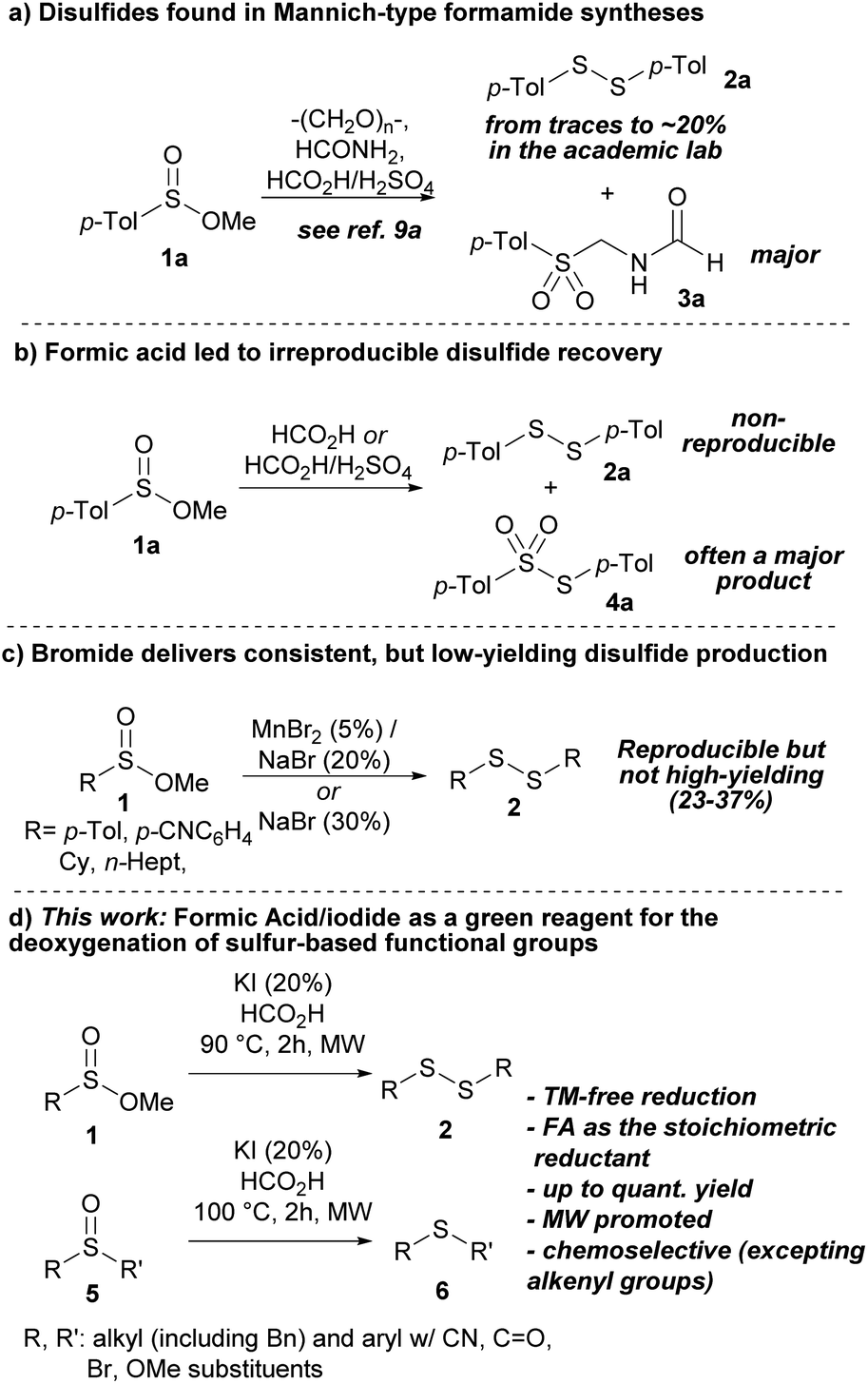 | ||
| Scheme 1 (a) Early findings of disulfides in the Mannich-type synthesis of p-toluenesulfonylmethylformamide 3a; (b) irreproducible synthesis of disulfide 2a by sole FA or FA/H2SO4; (c) utilization of bromide as a promoter for the reduction of methyl sulfinates 1 to disulfides 2; (d) reduction of alkyl sulfinates 1 for the synthesis of disulfides 2 using FA/I−, and its application to the deoxygenation of sulfoxides 5 to deliver thioethers 6. | ||
Our initial investigations into the reduction of methyl sulfinates (1) using I−/FA involved screening various iodide sources and examining sulfinates with alkyl groups of varying steric demand (see the ESI†). We first evaluated sodium iodide (NaI) and compared it to other iodide sources such as TBAI, ZnI2, and I2. However, none of these alternatives performed as well as sodium iodide (NaI, 88% conversion) and potassium iodide (KI, 95% conversion). Interestingly, we found that the combination of FA and either NaI or KI yielded better results than HI. The reaction with HI resulted in a less desirable outcome, with the yield of disulfide not exceeding 50%. As anticipated, the kinetics was slightly influenced by the steric demand of the alkoxy component of the sulfinate during the early stages of the reaction (t > 1 h). Nevertheless, complete conversion could be achieved in methyl (1a), ethyl, and isopropyl p-tolylsulfinates after 2 h (see the ESI†).
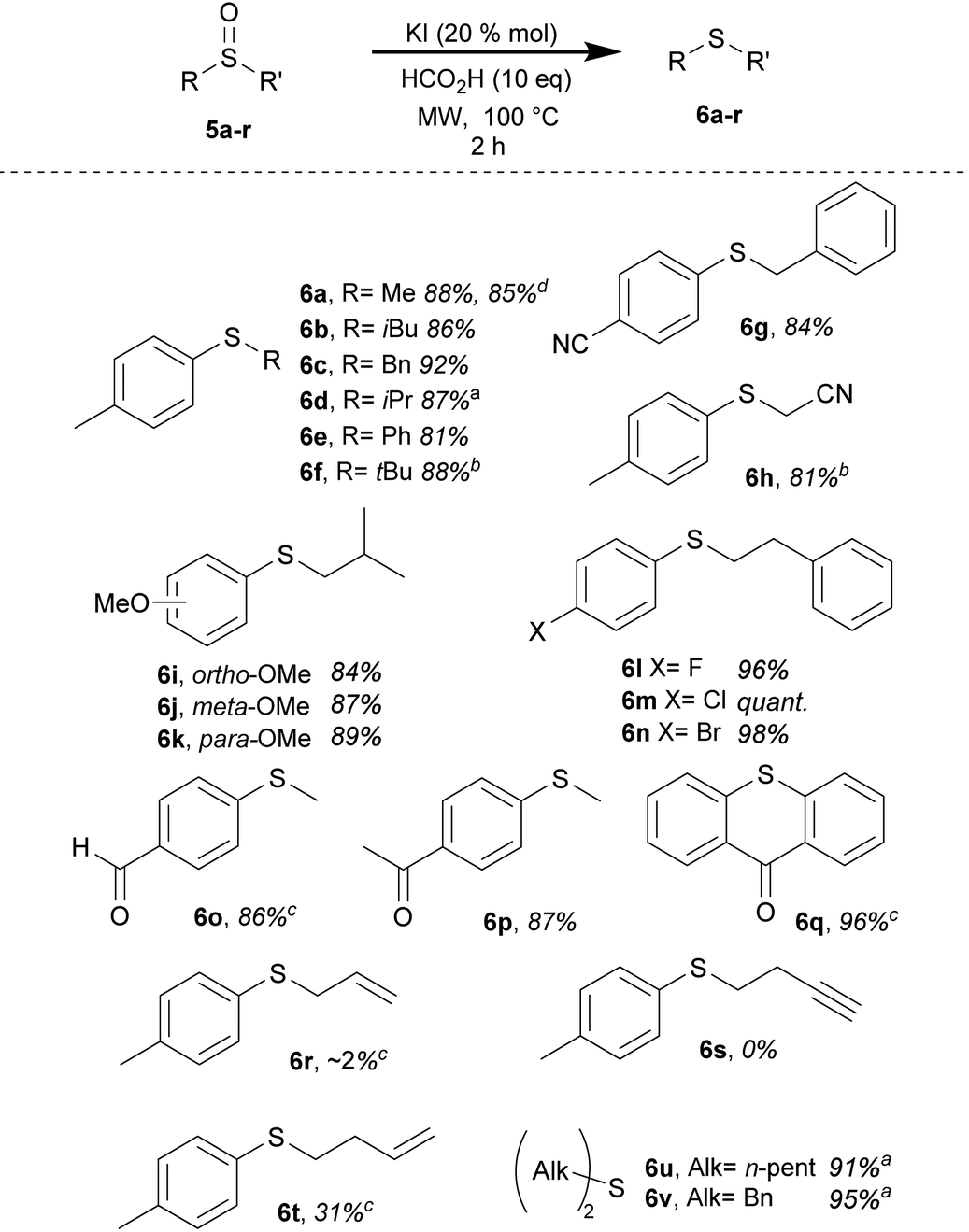
To maintain optimal atom economy, methyl sulfinates (1) were chosen to explore the scope of our disulfide synthesis method (Table 1). We found that both electron-withdrawing group (EWG) and electron-donating group (EDG)-bearing substrates were tolerant to the reduction. Notably, an aryl bromide (1h) and a cyano-based substrate (1i) were chemoselectively reduced exclusively at the sulfur center, with no reductive debromination15 or reduction or hydrolysis of the cyano group observed.16 Methoxy-substituted substrates (1b–d) were well tolerated, and satisfactory reductions were achieved for benzyl-based substrates (1j and k).17 Alkyl disulfides (2l and m) were obtained in fair yields, although higher reaction temperatures were required to achieve complete conversions in the 2 h timeframe. The need for a higher temperature (120 °C) for the cyclohexyl substrate (1l) can be attributed to the steric environment dictated by its bulky secondary alkyl scaffold.

The application of the I−/FA reductive method to another class of sulfinyl-based compounds proved to be successful. Various sulfoxides (5) containing both EWGs and EDGs exhibited complete chemoselectivity towards the sulfinyl group reduction, resulting in excellent yields of thioethers (6) (Table 2). The reduction of sulfoxides was mildly influenced by steric hindrance, as demonstrated by isopropyl and tert-butyl-substituted substrates (5d and f), requiring longer reaction times than the standard 2 h timeframe for complete conversion. Sulfoxides with cyano substituents (e.g., 5g), including one with an acetonitrile flanking fragment (5h), displayed complete chemoselective deoxygenation. Similarly, anisyl sulfoxides with para-, meta- or ortho-methoxy substitutions (5i–k) delivered good yields of thioethers (6i–k). Remarkably, no debromination occurred with a p-bromo substrate (5n), and additional halogenated sulfoxides (5l and m) provided excellent yields. The reaction exhibited complete orthogonality towards carbonyl compounds, including aldehydes and ketones (5o–q). This is particularly remarkable considering the abundant literature on carbonyl reductive transformations based on iodide.18 Although benzylic substrates such as 5c and 5g showed robustness under reductive conditions, an allyl-based sulfoxide (5r) yielded a minimal product (<2%), while a propargylic sulfoxide (5s) underwent complete decomposition. The alkenylic substrate that performed the best was an isolated and terminal olefin (5t) which exhibited a modest yield of 31% and needed appropriate reaction tuning. The deoxygenation of dialkyl sulfoxides was also effectively applicable in this method, as demonstrated by the successful transformation of dipentylsulfoxide (5u) and dibenzyl sulfoxide (5v), yielding outstanding results. Furthermore, a scaled-up experiment involving 2 g of the initial sulfoxide (5a) was conducted to explore the practicality of deoxygenation. The results obtained closely paralleled those from the microscale experiment, showing a yield of 85% compared to 88%. This concurrence underscores the practicality and viability of the method presented.
The mechanistic pathway for the reductive deoxygenation of the sulfinyl group was originally based on the known insight described by Hogeveen.19 In the case of methyl sulfinates (1), we first propose their conversion to the corresponding sulfinic acids (7) (Scheme 2a).9b,20 The sulfinic acid (7) could evolve reversibly towards an ephemeral sulfinylsulfone (8),21 which in turn could (a) disproportionate22 to generate a thiosulfonate (4) and a sulfonic species (9)23 or (b) follow an iodide-mediated deoxygenation assisted by Brønsted acid-activation of the sulfinyl group. Previous studies have proposed the formation of an iodosulfonium intermediate (IS+),6a,24 while others,25 including Hogeveen, have hinted that the reduction may proceed directly through a 10-electron sulfur intermediate. Regardless of the specific deoxygenation mechanism, the reduction involves an iodide attack on the sulfur atom, followed by reductive deiodination by another iodide unit. The generated iodine is then converted to iodide by FA under MW heating.14b,26 It is worth noting that when formic acid was substituted with acetic acid or acids of comparable pKa, such as glycolic or lactic acid, poor yields were observed due to the lack of regeneration of iodide (Scheme 2b). Interestingly, the reaction mixtures often displayed a reddish/brown hue, implying the presence of residual iodine, while also showing reactor pressurization, suggesting the generation of gaseous products such as CO2.27 However, it is noteworthy that under our specific reaction conditions, iodide ions are effectively regenerated at a synthetically adequate rate, as the complete conversion ensues without the requirement of elevated temperatures for iodide regeneration, as has been previously reported.14c
 | ||
| Scheme 2 (a) Plausible reaction pathway following Hogeveen's mechanism for the reduction of disulfides and sulfoxides using iodide (I−) and formic acid (FA). (b) Control experiments featuring other organic acids lacking reducing properties. Isolated yields. | ||
The orthogonality of the present method towards HI-sensitive functional groups, such as carbonyl, motivated us to investigate the mechanistic details using DFT calculations (ωB97XD/def2-TZVP;28 see section 11 of the ESI† for computational details, performed with Gaussian 09).29 We considered three scenarios for the activation of sulfinyl followed by reductive deiodination: (a) in situ HI formation involving two units for S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O activation and iodide donation, (b) multiple FA molecules acting within a formic acid/KI template for SO activation and iodide donation, and (c) a unit of HI and FA jointly promoting sulfinyl activation and iodide delivery (vide infra). The first scenario was deemed unlikely based on the thermodynamically unfavorable equilibrium between FA and iodide (cf. pKaHI = −9.530vs. pKaFA = 3.77),31 which would require the convergence of two HI units. This observation is consistent with our experimental findings on chemoselectivity. The second scenario appeared to lack sufficient acidity for sulfinyl activation. Intuitively, the plausibility of the third scenario became apparent, as it involved a single HI unit that was more likely to participate in the transition state compared to the first scenario. This scenario leveraged a formic acid network to bring relevant species (H+ and I−) into proximity, facilitating the desired transformation. Furthermore, a 20% mol iodide load was required to establish the necessary equilibria to generate sufficient HI. Nevertheless, explorations on the potential energy surface (PES) revealed a favorable configuration for the first scenario. By using dimethyl sulfoxide (DMSO)32 as a computational probe, we discovered that double Brønsted sulfinyl activation could occur effectively in a single step, with a negligible protonation barrier due to the high acidity of HI in Brønsted media (Scheme 3a). This results in a net activation barrier of ΔΔ‡GA-TS = 14.3 kcal mol−1 and a relative transition-state free energy of 50.8 kcal mol−1,33 which is significantly high and reflects the energy cost of generating two HI units from KI and FA. The second scenario involved the participation of two units of FA and one unit of KI (Scheme 3b). Also, in this case, there is only one activation/iodination stage, which is synergistically facilitated by the formic acid units anchored by a potassium ion. This arrangement positions the iodide quasi-apically relative to the ejecting oxygen, with double protonation occurring asynchronously. Regrettably, for this case, the relatively low acidity of the two FA units results in a considerably high energetic barrier of ΔΔ‡GB-TS = 36.5 kcal mol−1, despite the relatively lower transition state free energy of 36.9 kcal mol−1.34 In the third scenario, a “hybrid” FA/HI framework exhibited a favorable energetic profile (Scheme 3c). This case involves a progressive protonation process, cwith the first step effected by HI, followed by a subsequent protonation by FA, deoxygenation, and iodination. A detailed PES exploration revealed a first protonation barrier of ΔΔ‡GC-TS1 = 4.1 kcal mol−1, followed by a second one by FA with concurrent deoxygenation–iodination steps with an activation energy of ΔΔ‡GC-TS2 = 23.7 kcal mol−1.35 The associated relative transition-state free energies are 17.8 and 33.3 kcal mol−1, respectively. The third scenario appears to be the most plausible based on the thermochemical data. It is consistent with the formic acid framework/media without overlooking the possibility of discrete HI formation derived from KI/FA equilibria. It is worth noting that the three scenarios converge on forming a quasi-stable iodosulfonium intermediate (IS+), as suggested in previous works involving the iodide-mediated deoxygenation of sulfinyl groups (vide supra).
O activation and iodide donation, (b) multiple FA molecules acting within a formic acid/KI template for SO activation and iodide donation, and (c) a unit of HI and FA jointly promoting sulfinyl activation and iodide delivery (vide infra). The first scenario was deemed unlikely based on the thermodynamically unfavorable equilibrium between FA and iodide (cf. pKaHI = −9.530vs. pKaFA = 3.77),31 which would require the convergence of two HI units. This observation is consistent with our experimental findings on chemoselectivity. The second scenario appeared to lack sufficient acidity for sulfinyl activation. Intuitively, the plausibility of the third scenario became apparent, as it involved a single HI unit that was more likely to participate in the transition state compared to the first scenario. This scenario leveraged a formic acid network to bring relevant species (H+ and I−) into proximity, facilitating the desired transformation. Furthermore, a 20% mol iodide load was required to establish the necessary equilibria to generate sufficient HI. Nevertheless, explorations on the potential energy surface (PES) revealed a favorable configuration for the first scenario. By using dimethyl sulfoxide (DMSO)32 as a computational probe, we discovered that double Brønsted sulfinyl activation could occur effectively in a single step, with a negligible protonation barrier due to the high acidity of HI in Brønsted media (Scheme 3a). This results in a net activation barrier of ΔΔ‡GA-TS = 14.3 kcal mol−1 and a relative transition-state free energy of 50.8 kcal mol−1,33 which is significantly high and reflects the energy cost of generating two HI units from KI and FA. The second scenario involved the participation of two units of FA and one unit of KI (Scheme 3b). Also, in this case, there is only one activation/iodination stage, which is synergistically facilitated by the formic acid units anchored by a potassium ion. This arrangement positions the iodide quasi-apically relative to the ejecting oxygen, with double protonation occurring asynchronously. Regrettably, for this case, the relatively low acidity of the two FA units results in a considerably high energetic barrier of ΔΔ‡GB-TS = 36.5 kcal mol−1, despite the relatively lower transition state free energy of 36.9 kcal mol−1.34 In the third scenario, a “hybrid” FA/HI framework exhibited a favorable energetic profile (Scheme 3c). This case involves a progressive protonation process, cwith the first step effected by HI, followed by a subsequent protonation by FA, deoxygenation, and iodination. A detailed PES exploration revealed a first protonation barrier of ΔΔ‡GC-TS1 = 4.1 kcal mol−1, followed by a second one by FA with concurrent deoxygenation–iodination steps with an activation energy of ΔΔ‡GC-TS2 = 23.7 kcal mol−1.35 The associated relative transition-state free energies are 17.8 and 33.3 kcal mol−1, respectively. The third scenario appears to be the most plausible based on the thermochemical data. It is consistent with the formic acid framework/media without overlooking the possibility of discrete HI formation derived from KI/FA equilibria. It is worth noting that the three scenarios converge on forming a quasi-stable iodosulfonium intermediate (IS+), as suggested in previous works involving the iodide-mediated deoxygenation of sulfinyl groups (vide supra).
 | ||
| Scheme 3 Plausible molecular arrangements for the sulfinyl Brønsted activation and deoxygenation/iodination mechanisms. Energies in kcal mol−1 at 363.15 K. | ||
Further investigations into the involvement of a more complex network and the synergy between FA molecules were prompted by a recent study that recognized the enhancement of Brønsted acid catalysis through hydrogen bonding networks.35 To explore this for our reaction system, the transition state B-TS (B-TS-2FA) was gradually modified by adding an increased number of formic acid units connected through hydrogen bonding, up to four units (B-TS-3FA-HT, B-TS-3FA-HH, B-TS-4FA) (Scheme 4).
 | ||
| Scheme 4 Structures and related energy barriers for the concerted Brønsted activation of sulfinyl only by FA units, followed by deoxyiodination on the DMSO probe, showcasing their influence in an increasingly complex hydrogen bond network and its impact on the reduction of TS energies. Energies in kcal mol−1 at 363.15 K. | ||
Remarkably, the stabilization of the transition state in the sulfinyl activation/deoxygenation/iodination process through the increasing association of Brønsted acids via hydrogen bonds was evident. This led to a gradual decrease in the relative TS free energy from ΔGrel-B-TS-2FA = 36.9 kcal mol−1 to ΔGrel-B-TS-4FA = 32.3 kcal mol−1. More importantly, this increase in FA units’ participation also led to a gradual decrease in the energy barrier from ΔΔ‡GB-TS-2FA = 36.5 kcal mol−1 to ΔΔ‡GB-TS-4FA = 28.1 kcal mol−1. Interestingly, a “head-to-head” hydrogen bonding arrangement from the additional activating FA unit exhibited a more favorable kinetic profile, decreasing the activation barrier by ca. 1.5 kcal mol−1. This reduction is directly attributed to a slight increase in the energy of the precursory stationary arrangement. The stabilization of the TS remains unaffected by the specific mode of hydrogen bonding within the network. To validate that the stabilization energy provided by hydrogen bonding is independent of the acid–base Lewis association of FA units with the potassium cation, B-TS-4FA was modified by a dihedral rotation and subsequent optimization to obtain a new TS lacking the additional hydrogen bond (B-TS-4FAbis). Our findings reveal that the relative TS free energy increases by approximately 6.1 kcal mol−1, accompanied by an enthalpic increase of 6.6 kcal mol−1, which directly correlates to a significant contribution of the stabilizing enthalpic factor associated with the hydrogen bond within the formic acid network. A similar investigation was conducted to explore the preferential scenario (Scheme 3c) where both HI and FA synergistically participate in the activation of sulfinyl groups.
The results revealed relatively modest stabilization exerted by an additional FA unit, showing its preferential association with iodide (cf.C-TS2-3FA-FA and C-TS2-3FA-I). However, the introduction of a second additional hydrogen-bonded FA unit, H-bonded to either iodide (C-TS2-4FA-I-I) or FA (C-TS2-4FA-I-FA), did not lead to an improvement in the thermochemical profile. In fact, it resulted in a destabilizing effect, apparently of steric nature. Therefore, based on these findings, we establish that the third scenario either with 2 (C2-2FA) or 3 formic acid units, the one H-bonded to iodide (C2-3FA-I, Scheme 5), remains the most plausible scenario among all the cases considered. This scenario favorably combines the kinetic factors, such as a stronger acid (HI) and hydrogen bond interactions, to a lesser extent, contributing to the formation of the key iodosulfonium intermediate (IS+). Nonetheless, the role of other more complex hydrogen bond networks cannot be disregarded, as evidenced by the improved energetics when only FA units participated in the Brønsted activation of the sulfinyl group (see Scheme 4).
 | ||
| Scheme 5 Structures and related energy barriers for the concerted Brønsted activation by HI and FA, followed by deoxygenation and iodination on the DMSO probe featuring the influence of additional FA units. Energies in kcal mol−1 at 363.15 K. | ||
Conclusions
Formic acid, a sustainable reagent, plays multiple roles in the iodide-catalysed deoxygenation of sulfinates and sulfoxides, serving as a Brønsted catalyst, solvent, and reductant. This methodology yields excellent results and demonstrates remarkable tolerance towards reducible functional groups, including carbonyl, cyano, benzyl, and bromo. Using iodide as a reducing catalyst significantly enhances the reproducibility and performance of the reaction by preventing the decomposition of in situ generated sulfinic species through a known dismutation process. Although the mechanistic pathway can be rationalized based on the established knowledge of sulfinyl deoxygenation by iodide in Brønsted media, the tolerance towards HI-reducible groups prompted a DFT study to explore energetically-favorable molecular arrangements leading to a key iodosulfonium intermediate. Through in silico explorations, we discovered a molecular scenario where both formic acid and an HI unit (formed via a Brønsted equilibrium between KI and FA) synergistically participate. Additionally, more sophisticated models incorporating an elaborate formic acid network through hydrogen bonding were found to stabilize crucial transition states, where sulfinyl is Brønsted acid-activated and undergoes deoxygenation through iodide displacement. These findings support the notion of Brønsted catalyst activity enhancement through the development of hydrogen-bonding networks.36 Overall, our aim with this investigation was not only to contribute to sustainable and transition metal-free synthetic methodologies for relevant chemical transformations but also to stimulate mechanistic studies focusing on the relatively overlooked aspect of catalyst activation from the perspective of hydrogen bonding networks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from CONACYT/CONAHCYT (Mexico) through a Ciencia de Frontera grant (CF-2019-51493). A. C.-J. would like to express gratitude to CONACYT/CONAHCYT for the Ph. D. fellowship 708711 and L. J. G. C. thanks COMECYT for the undergraduate fellowship 19BTIIL0096. The authors would like to acknowledge the computing time provided by LANCAD and CONACYT at the supercomputer hybrid cluster Xiuhcoatl at the General Coordination of Information and Communication Technologies (CGSTIC) of CINVESTAV (https://clusterhibrido.cinvestav.mx). Special thanks are extended to Teresa Cortéz, Géiser Cuellar, Ma. Luisa Rodríguez and Víctor González for assistance in spectral acquisition. Additionally, the valuable technical assistance provided by Carolina Silva to J. G. C., as well as her support in conducting certain preliminary in vitro experiments, is greatly appreciated. Adabelia Tapia is also acknowledged for her early contributions to the project.References
- Selected literature: (a) D. Fass, Annu. Rev. Biophys., 2012, 41, 63–79 CrossRef CAS; (b) C. Wiedemann, A. Kumar, A. Lang and O. Ohlenschläger, Front. Chem., 2020, 8, 1–8 CrossRef; (c) T. M. Postma and F. Albericio, Eur. J. Org. Chem., 2014, 3519–3530 CrossRef CAS; (d) J. A. Lujan-Montelongo, J. B. Mateus-Ruiz and R. M. Valdez-García, Eur. J. Org. Chem., 2023, e202201156 CrossRef CAS , and references included within.
- Selected literature: (a) H. Chen, W. Jiang and Q. Zeng, Chem. Rec., 2020, 20, 1269–1296 CrossRef CAS PubMed; (b) M. I. Han and Ş. G. Küçükgüzel, Curr. Drug Targets, 2022, 23, 170–219 CrossRef CAS PubMed; (c) R. J. Huxtable, in Biochemistry of Sulfur, Wiley-VCH, Weinheim, 1996, vol. 2, pp. 10–20 Search PubMed.
- K.-M. Roy, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, vol. 36, pp. 629–655 Search PubMed.
- Selected literature: (a) C. S. Sauer, J. Köckenberger and M. R. Heinrich, J. Org. Chem., 2020, 85, 9331–9338 CrossRef CAS, and references included within. (b) C. Silva-Cuevas, E. Paleo, D. F. León-Rayo and J. A. Lujan-Montelongo, RSC Adv., 2018, 8, 24654–24659 RSC.
- Selected literature: (a) F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929–4932 CrossRef CAS; (b) T. T. Wang, F. L. Yang and S. K. Tian, Adv. Synth. Catal., 2015, 357, 928–932 CrossRef CAS; (c) C.-R. Liu and L.-H. Ding, Org. Biomol. Chem., 2015, 13, 2251–2254 RSC; (d) J. Chen, J. Mao, Y. He, D. Shi, B. Zou and G. Zhang, Tetrahedron, 2015, 71, 9496–9500 CrossRef CAS; (e) D. Wan, Y. Yang, X. Liu, M. Li, S. Zhao and J. You, Eur. J. Org. Chem., 2016, 55–59 CrossRef CAS; (f) Y. Guo, S. Zhong, L. Wei and J. P. Wan, Beilstein J. Org. Chem., 2017, 13, 2017–2022 CrossRef CAS; (g) X.-Q. Chu, T. Xie, Y.-W. Wang, X.-R. Li, W. Rao, H. Xu and Z.-L. Shen, Chem. Commun., 2020, 56, 8699–8702 RSC; (h) J. Wei, S. Liang, L. Jiang and W. Yi, J. Org. Chem., 2020, 85, 12374–12381 CrossRef CAS PubMed; (i) Y. Mu, M. Yang, F. Li, Z. Iqbal, R. Jiang, J. Hou, X. Guo, Z. Yang and D. Tang, New J. Chem., 2021, 45, 4934–4937 RSC.
- Selected literature: (a) H. Firouzabadi, J. Sulphur Chem., 2005, 26, 313–324 CrossRef CAS; (b) Q. Zhao, C. Qian and X.-z. Chen, Monatsh. Chem., 2013, 144, 1547–1550 CrossRef CAS; (c) Y. Zheng, F.-L. Qing, Y. Huang and X.-H. Xu, Adv. Synth. Catal., 2016, 358, 3477–3481 CrossRef CAS; (d) X.-Z. Yu, W.-L. Wei, Y.-L. Niu, X. Li, M. Wang and W.-C. Gao, Molecules, 2022, 27, 6232 CrossRef CAS PubMed.
- Selected literature: (a) J. Drabowicz, T. Numata and S. Oae, Org. Prep. Proced. Int., 1977, 9, 63–83 CrossRef CAS; (b) H. Firouzabadi and A. Jamalian, J. Sulfur Chem., 2008, 29, 53–97 CrossRef CAS; (c) L. Shiri and M. Kazemi, Res. Chem. Intermed., 2017, 43, 6007–6041 CrossRef CAS; (d) A.-C. Gaumont, M. Gulea, S. Perrio and V. Reboul, Reduction of S O and SO2 to S, S-X to S-H, and P O to P, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier Ltd, Amsterdam, 2014, vol. 8, pp. 535–563 Search PubMed; (e) H. Lin, L. Wu and M. Kazemi, Synth. Commun., 2021, 51, 1–27 CrossRef.
- For a concise example see: H. Kosugi, H. Konta and H. Uda, J. Chem. Soc., Chem. Commun., 1985, 211–213 RSC.
- (a) A. Tapia-Pineda, C. Perez-Arrieta, C. Silva-Cuevas, E. Paleo and J. A. Lujan-Montelongo, J. Chem. Educ., 2016, 93, 1470–1474 CrossRef CAS; (b) J. A. Lujan-Montelongo, A. O. Estevez and F. F. Fleming, Eur. J. Org. Chem., 2015, 1602–1605 CrossRef CAS.
- E. O. Pentsak, D. B. Eremin, E. G. Gordeev and V. P. Ananikov, ACS Catal., 2019, 9, 3070–3081 CrossRef CAS.
- (a) A. Tapia-Pineda, C. Pérez-Arrieta, C. Silva-Cuevas and J. A. Luján-Montelongo, presented in part at the 51th Chemistry Mexican Conference, Pachuca, September, 2016; (b) A. Tapia-Pineda, C. Pérez-Arrieta, C. Silva-Cuevas and J. A. Luján-Montelongo, Formic Acid as a Green Hydrogen Donor: Disulfides and Thiosulfonates from Alkyl Sulfinates, in Memorias de los trabajos Estudiantiles Presentados en el 51° Congreso Mexicano de Química, Sociedad Química de México, Mexico City, 2016, pp. 296–298. https://sqm.org.mx/wp-content/uploads/2021/05/Carteles-Estudiantiles.pdf (accessed June 2023) Search PubMed.
- Selected literature: (a) J. E. Milne, T. Storz, J. T. Colyer, O. R. Thiel, M. D. Seran, R. D. Larsen and J. A. Murry, J. Org. Chem., 2011, 76, 9519–9524 CrossRef CAS PubMed; (b) Q. Zhao, C. Qian and X.-z. Chen, Monatsh. Chem., 2013, 144, 1547–1550 CrossRef CAS; (c) C.-R. Liu and L.-H. Ding, Org. Biomol. Chem., 2015, 13, 2251–2254 RSC; (d) R. Rahaman, N. Devi, J. R. Bhagawati and P. Barman, RSC Adv., 2016, 6, 18929–18935 RSC; (e) L. Chen, J. Zhang, Y. Wei, Z. Yang, P. Liu, J. Zhang and B. Dai, Tetrahedron, 2019, 75, 130664 CrossRef; (f) M. Zarei, M. A. Ameri and A. Jamaleddini, J. Sulphur Chem., 2013, 34, 259–263 CrossRef CAS.
- X. Liu, S. Li, Y. Liu and Y. Cao, Chin. J. Catal., 2015, 36, 1461–1475 CrossRef CAS.
- (a) F. Xiao, S. Chen, J. Tian, H. Huang, Y. Liu and G.-J. Deng, Green Chem., 2016, 18, 1538–1546 RSC; (b) T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Green Chem., 2016, 18, 2966–2970 RSC; (c) J. Xu, X. Miao, L. Liu, Y. Wang and W. Yang, ChemSusChem, 2021, 14, 5311–5319 CrossRef CAS PubMed; (d) M. Gunawan, T. Hudaya and T. H. Soerawidjaja, J. Eng. Technol. Sci., 2021, 53, 210106 CrossRef CAS.
- A. V. Butin, A. S. Dmitriev, M. G. Uchuskin, V. T. Abaev and I. V. Trushkov, Synth. Commun., 2008, 38, 1569–1578 CrossRef CAS.
- A. Aramini, M. R. Sablone, G. Bianchini, A. Amore, M. Fanì, P. Perrone, A. Dolce and M. Allegretti, Tetrahedron, 2009, 65, 2015–2021 CrossRef CAS.
- K. Bao, A. Fan, Y. Dai, L. Zhang, W. Zhang, M. Cheng and X. Yao, Org. Biomol. Chem., 2009, 7, 5084–5090 RSC.
- Selected literature: (a) L. D. Hicks, J. K. Han and A. J. Fry, Tetrahedron Lett., 2000, 41, 7817–7820 CrossRef CAS; (b) M. Allukian, G. Han, L. Hicks and A. J. Fry, ARKIVOC, 2002, 76–79 Search PubMed; (c) J. E. Milne, T. Storz, J. T. Colyer, O. R. Thiel, M. D. Seran, R. D. Larsen and J. A. Murry, J. Org. Chem., 2011, 76, 9519–9524 CrossRef CAS PubMed; (d) L. Z. Yuan, D. Renko, I. Khelifi, O. Provot, J.-D. Brion, A. Hamze and M. Alami, Org. Lett., 2016, 18, 3238–3241 CrossRef CAS PubMed; (e) L.-Z. Yuan, G. Zhao, A. Hamze, M. Alami and O. Provot, Adv. Synth. Catal., 2017, 359, 2682–2691 CrossRef CAS.
- D. Landini, F. Montanari, H. Hogeveen and G. Maccagnani, Tetrahedron Lett., 1964, 5, 2691–2696 CrossRef.
- Selected literature: (a) C. Liu, J. Xu and G. Wu, RSC Adv., 2021, 11, 35156 RSC; (b) D. Wang, R. Zhang, W. Ning, Z. Yan and S. Lin, Org. Biomol. Chem., 2016, 14, 5136–5140 RSC; (c) S. Sun, J. Li, L. Pan, H. Liu, Y. Guo, Z. Gao and X. Bi, Org. Biomol. Chem., 2022, 20, 8885–8892 RSC.
- J. L. Kice and N. E. Pawlowski, J. Am. Chem. Soc., 1964, 86, 4898–4904 CrossRef CAS.
- H. Bredereck, A. Wagner, E. H. Beck, H. Herlinger and K.-G. Kottenhahn, Angew. Chem., 1958, 70, 268–269 CrossRef CAS.
- We assert that this method represents a significant secondary pathway that contributes to the limited efficiency of sulfinate reduction when bromide is employed. Despite achieving complete conversions, the yields typically fall below 30–40%. In contrast, the use of iodide yields significantly better results, ranging from good to excellent. This can be attributed to the superior nucleophilicity of iodide compared to bromide, which favors the reductive deoxygenation pathway instead of dismutation. While the impact of this secondary pathway is not a concern for sulfoxides, the yields obtained with iodide are considerably higher compared to those obtained with bromide.
- Selected literature: (a) K. Bahrami, M. M. Khodaei and A. Karimi, Synthesis, 2008, 2543–2546 CrossRef CAS; (b) B. W. Yoo, J. Park, H. J. Shin and C. M. Yoon, J. Sulfur Chem., 2017, 38, 597–603 CrossRef CAS; (c) C. P. R. Hackenberger, Org. Biomol. Chem., 2006, 4, 2291–2295 RSC. A bromosulfonium intermediate: (d) S. A. Pourmousavi and P. Salehi, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 803–807 CrossRef CAS.
- (a) M. Abbasi, M. R. Mohammadizadeh and Z. Moradi, Tetrahedron Lett., 2015, 56, 6610–6613 CrossRef CAS; (b) M. Abbasi, M. R. Mohammadizadeh and Z. Moradi, Bull. Chem. Soc. Jpn., 2016, 89, 405–407 CrossRef CAS.
- D. L. Hammick and M. Zvegintzov, J. Chem. Soc., 1926, 129, 1105–1108 RSC.
- Typically, a pressure of approximately 20 bar is recorded for 0.5–1 mmol reactions in a Biotage® 9 mL glass reactor vial, which is rated for 0.5–2 mL reactions (cat. #352016).
- The range-separated ωb97XD density functional approximation effectively captures dispersion forces, crucial for systems with S and I due to their polarizability and weak intermolecular interactions. Moreover, the def2-TZVP basis set provides a balanced treatment of electron correlation and quality, enabling accurate characterization of molecular geometry and electronic properties. See: C. de Souza Silva and F. d. C. Alves Lima, Comput. Theor. Chem., 2023, 1225, 114140 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. Trummal, L. Lipping, I. Kaljurand, I. A. Koppel and I. Leito, J. Phys. Chem. A, 2016, 120, 3663–3669 CrossRef CAS PubMed.
- K. F. Eidman and J. D. Belani, in Encyclopedia of Reagents for Organic Synthesis, ed. A. Charette, J. Bode, T. Rovis and R. Shenvi, John Wiley & Sons, Ltd, Chichester, UK, 2015, DOI:10.1002/047084289X.rf025.pub2.
- DMSO was reduced with complete conversion under the standard conditions; however, no attempts were made to isolate dimethylsulfide (DMS) or obtain a yield due to its high volatility.
- In the first scenario, the zero-energy reference was established by considering the reactants prior to the Brønsted equilibria. The reference system consisted of DMSO, two FA dimers, and three units of KI. Each released formate (from the Brønsted equilibria) was modeled in association with a single unit of FA to enhance the accuracy of the energetic profile.
- For the second scenario, DMSO, a FA dimer, and a unit of KI were employed as zero-energy benchmarks.
- For the third scenario, DMSO, a FA dimer, and two units of KI were employed as the zero-energy reference.
- T. Anh To, C. Pei, R. M. Koenigs and T. Vinh Nguyen, Angew. Chem., Int. Ed., 2022, 61, e202117366 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, spectroscopic data, copies of NMR spectra, and optimized structures in Cartesian coordinates and thermal corrections. See DOI: https://doi.org/10.1039/d3gc03213b |
| This journal is © The Royal Society of Chemistry 2023 |