
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cation–anion confined hydrogen-bonding catalysis strategy for ring-closing C–O/O–H metathesis of alkoxy alcohols under metal-free conditions†
Huan
Wang‡
ab,
Zhi-Hao
Zhao‡
ad,
Yanfei
Zhao
ac,
Fengtao
Zhang
ac,
Junfeng
Xiang
a,
Buxing
Han
 ac and
Zhimin
Liu
*ac
ac and
Zhimin
Liu
*ac
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuzm@iccas.ac.cn
bCollege of Chemistry & Chemical Engineering, Shaanxi Key Laboratory of Chemical Additives for Industry, Shaanxi University of Science & Technology, Xi'an, Shaanxi 710021, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dState Key Laboratory of Solidification Processing and School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an, Shaanxi 710072, P. R. China
First published on 29th September 2023
Abstract
Ring-closing metathesis (RCM) reactions of multiple bonds have seen considerable progress; however, RCM reactions involving single bonds, especially two different single bonds are scarce and extremely challenging. Herein, we present a cation–anion confined hydrogen bonding catalysis strategy for catalyzing the ring-closing C–O/O–H metathesis of alkoxy alcohols to O-heterocycles under metal-free conditions. Assisted with theoretical computation, the effective ionic liquid catalysts were first predicted. [HO-EtMIm][OTf] was found to display the highest activity, consistent with the predicted results. This catalyst could afford a series of O-heterocycles, including tetrahydrofurans, tetrahydropyrans, dioxanes, and some complex ethers that are difficult to access via conventional routes. Moreover, it was recyclable and reusable without activity loss after 5 recycles. Comprehensive investigations endorse that [HO-EtMIm]+ cation and [OTf]− anion selectively form hydrogen bonds with the ether O atom and hydroxyl H atom of alkoxy alcohol in opposite directions, respectively, which cooperatively catalyze the reaction in the cation–anion confined ionic microenvironment. The strategy presented here provides a novel and green route to access cyclic ethers.
Introduction
Metathesis reactions are widely applied to synthesize complex molecules, pharmaceuticals, and natural products.1–7 Particularly, ring-closing metathesis (RCM) reactions of multiple bonds, such as olefin-olefin metathesis2,8–11 and carbonyl-olefin metathesis5,12–16 have become powerful techniques for the preparation of various unsaturated rings in organic synthesis (Scheme 1a and b). In contrast, the RCM reactions of the C–X bond especially involving the C–O bond are rarely reported and still challenging owing to the low reactivity of single bonds.17–22 Recently, it has been reported that Lewis acid (e.g., Fe(OTf)3) and SO3H-functionalized ILs are capable of catalyzing ring-closing C–O/C–O bond metathesis reactions of aliphatic ethers to bis- and tris-ethers,13,23 and silicon could catalyze C–O bond RCM of polyethers.19 Notably, these RCM systems only involve the transformation of the substrates with two same single bonds (e.g., C–O bond). Compared to symmetrical compounds, asymmetric compounds are more difficult to be activated and transformed, and the catalytic system with two different active sites is usually required. The ring-closing C–O/O–H metathesis of alkoxy alcohols remains challenging and has not been reported so far, and therefore an efficient and green strategy for this reaction is highly desirable (Scheme 1c).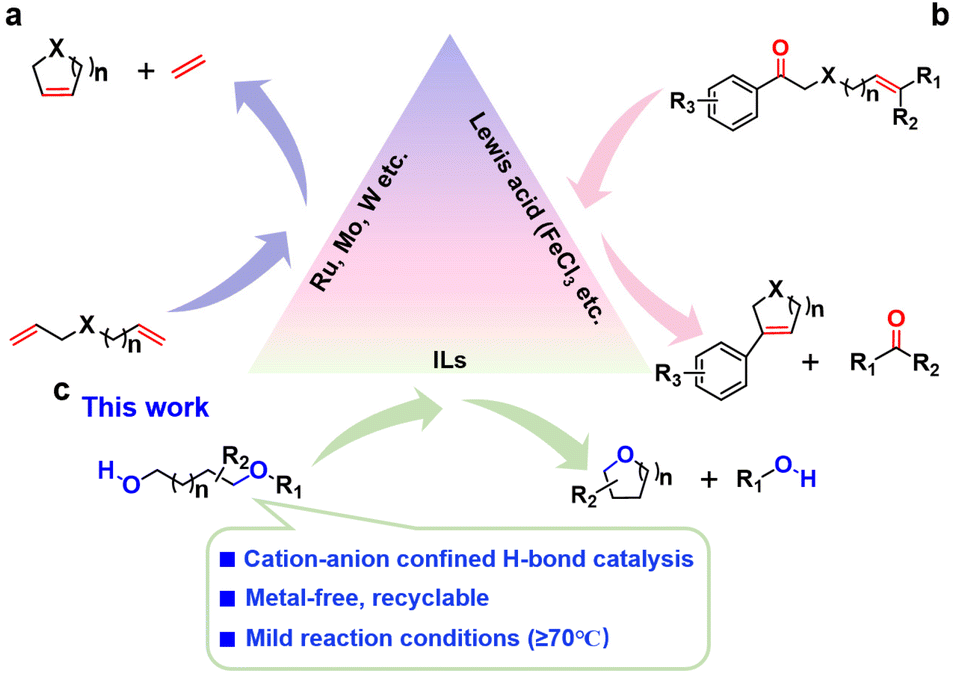 | ||
| Scheme 1 Ring-closing metathesis reactions. | ||
Since the past decades, ionic liquids (ILs) composed of organic cations and organic/inorganic anions have been emerging as promising alternatives to metal catalysts due to their unique properties such as structural designability, negligible volatility, strong H-bonding interactions, and easy recycling.24–29 Particularly, the strong hydrogen bonding interactions in IL systems make them show promising applications in catalyzing reactions under metal-free and mild conditions.30–35 For example, acetate-36 or trifluoroethanol-37 based ILs could activate 2-aminobenzonitriles via hydrogen bonding, thus catalyzing the reaction of CO2 with 2-aminobenzonitriles to quinazoline-2,4(1H,3H)-diones. Basic IL 1-ethyl-3-methylimidazolium acetate could catalyze the one-pot oxidative esterification of alcohols using O2 as an oxidant under catalysis of hydrogen bonding interaction.38 The cooperation of the IL cation as hydrogen bond (HB) donor and anion as acceptor could efficiently achieve dehydrative cyclization of diols to O-heterocycles.39
In this work, we for the first time report the ring-closing C–O/O–H bonds metathesis of alkoxy alcohols to O-heterocycles achieved over OH-functionalized ILs (Scheme 1 and Fig. S1†). Assisted with theoretical computations, the proper range of IL hydrogen bonding catalysts was first predicted. [HO-EtMIm][OTf] (1-hydroxyethyl-3-methyl imidazolium trifluorom-ethanesulfonate) was found to be very effective for the reaction, agreeing well with the prediction. A series of O-heterocycles including tetrahydrofurans, tetrahydropyrans, dioxanes, and some other complex ethers can be produced in high yields using this strategy. Comprehensive investigations indicate that a pair of cation and anion of [HO-EtMIm][OTf] serve as HB donor and acceptor (D–A), respectively, to form strong HBs with a molecule of 1,4-butanediol monomethyl ether (1a), which selectively activate the C–O and O–H bonds, thus synergistically catalyzing the transformation of alkoxy alcohol to cyclic ethers in the cation–anion confined electroneutral microenvironment.
Results and discussion
Taking 1a as a model of alkoxy alcohols, we set out to select the ILs with cation and anion as HB donor and acceptor, respectively, through density functional theory (DFT) calculations. Considering that the strengths of HBs between the cations or anions of ILs and the reactant reflect the activity of the IL catalyst in our previous work,23,39 we performed DFT calculations to estimate the lengths of HBs between 1a and various cations or anions, respectively. As shown in Fig. 1a and S2,† the cations [HO-EtN111]+, [HO-EtMIm]+, and [EMIm]+ serve as HB donors to form HBs with the ether O atom of 1a with the HB lengths following the order: [HO-EtN111]+ < [HO-EtMIm]+ < [EMIm]+; especially, the HB lengths of 1a with [HO-EtN111]+ and with [HO-EtMIm]+ are less than 2.0 Å, suggesting very strong hydrogen bonding interaction.40,41 The anions [OTf]−, [PF6]−, Cl− serve as HB acceptors to form HBs with the hydroxyl H of 1a, and the HB lengths are as follows: [OTf]− < [PF6]− < Cl−, indicating the strongest ability of [OTf]− to form HB with 1a. Additionally, the NBO charges of the methylene C atom in –CH2OCH3 and the hydroxyl O atom in 1a were calculated to get information on electron transfer caused by hydrogen bonding (Fig. 1b and S2†). Compared to those of 1a, both NBO charges of methylene C atom in –CH2OCH3 and hydroxyl O atom in 1a change remarkably as 1a interacts with these cations or anions, following the order: [EMIm]+ < [HO-EtN111]+ < [HO-EtMIm]+ and [PF6]− < [OTf]− < Cl−, respectively. From the above results, it is obvious that all the selected cations and anions can interact with 1avia hydrogen bonding interaction, which results in changes in the NBO charges of methylene C atom in –CH2OCH3 and hydroxyl O atom in 1a accordingly. Therefore, it may be deduced that a pair of cation and anion could jointly interact with 1avia hydrogen bonding due to the presence of electrostatic force. It is generally accepted that the shorter the HB length, the stronger the hydrogen bonding interaction, and that the larger change in the NBO charge suggests more electron transfer. Hence, the ILs composed of OH-functionalized cation and anion containing halogen/O atoms may be capable of catalyzing the RCM of 1a.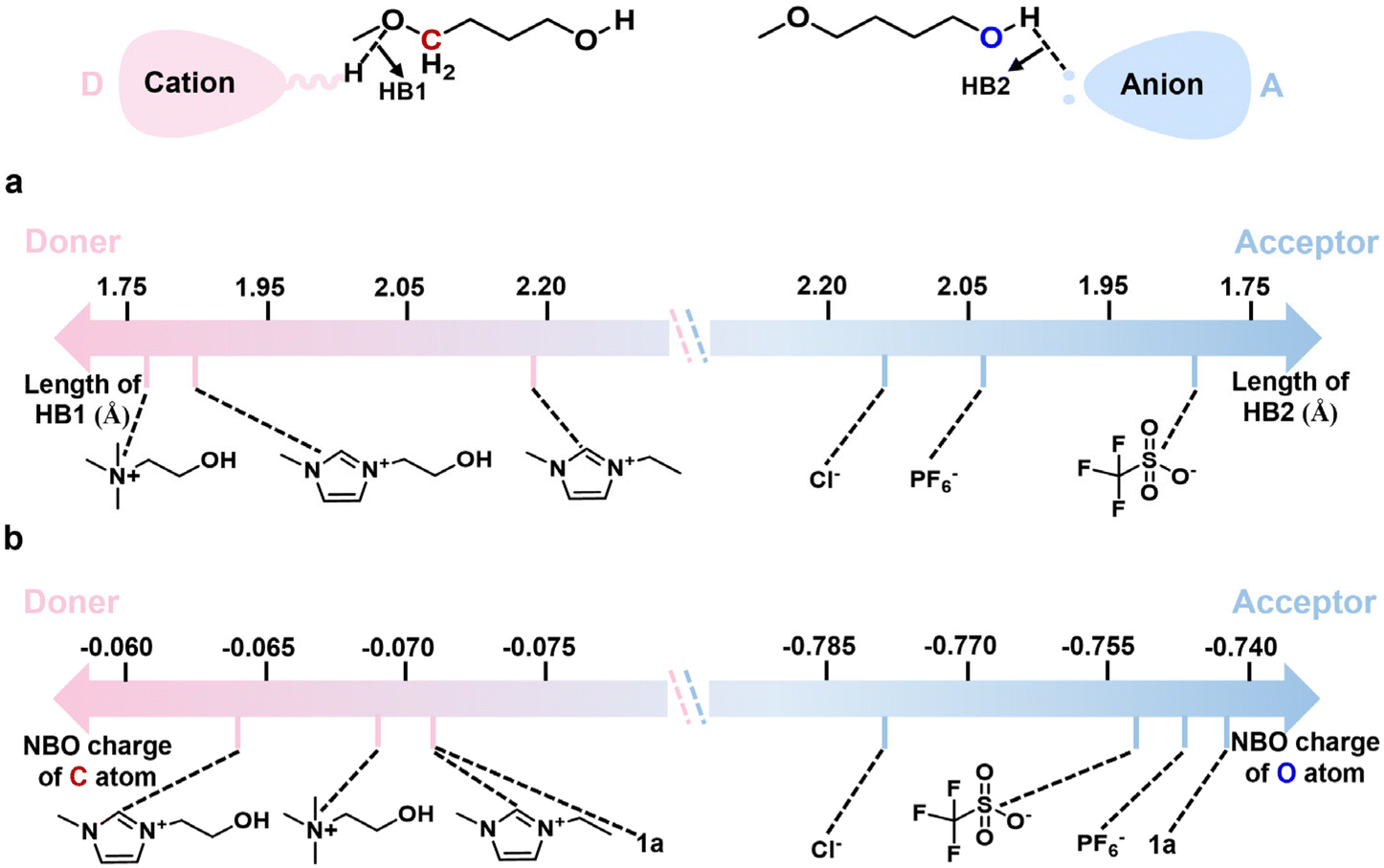 | ||
| Fig. 1 Hydrogen bonding interaction of 1a with various cations or anions. (a) The lengths of HBs between 1a and various cations or anions, respectively; (b) the natural bond orbital (NBO) charges of methylene C atom of –CH2OCH3 and hydroxyl O atom in 1a when 1a interacts with and without various cations or anions, respectively. | ||
Based on the calculated results, we selected the ILs composed of the above cations and anions to examine their catalytic activities for 1a RCM (Fig. 2a). To our delight, the ILs with [OTf]− anion and –OH functionalized cations, e.g., [HO-EtMIm][OTf], [HO-EtMMIm][OTf] and [HO-EtN111][OTf], were very effective for the reaction, even better than the commonly applied H2SO4 (22 wt%) catalyst. Especially, [HO-EtMIm][OTf] showed the best performance, generating tetrahydrofuran (1b) as the sole product with an excellent yield of 91% and an E factor of 0.4442 (Fig. 2a and S3†). To explore the stability of 1a under the experimental conditions, the reaction of 1b with MeOH was tried, and no 1a was detected, suggesting that the formation of 1b was irreversible (Fig. S4†).
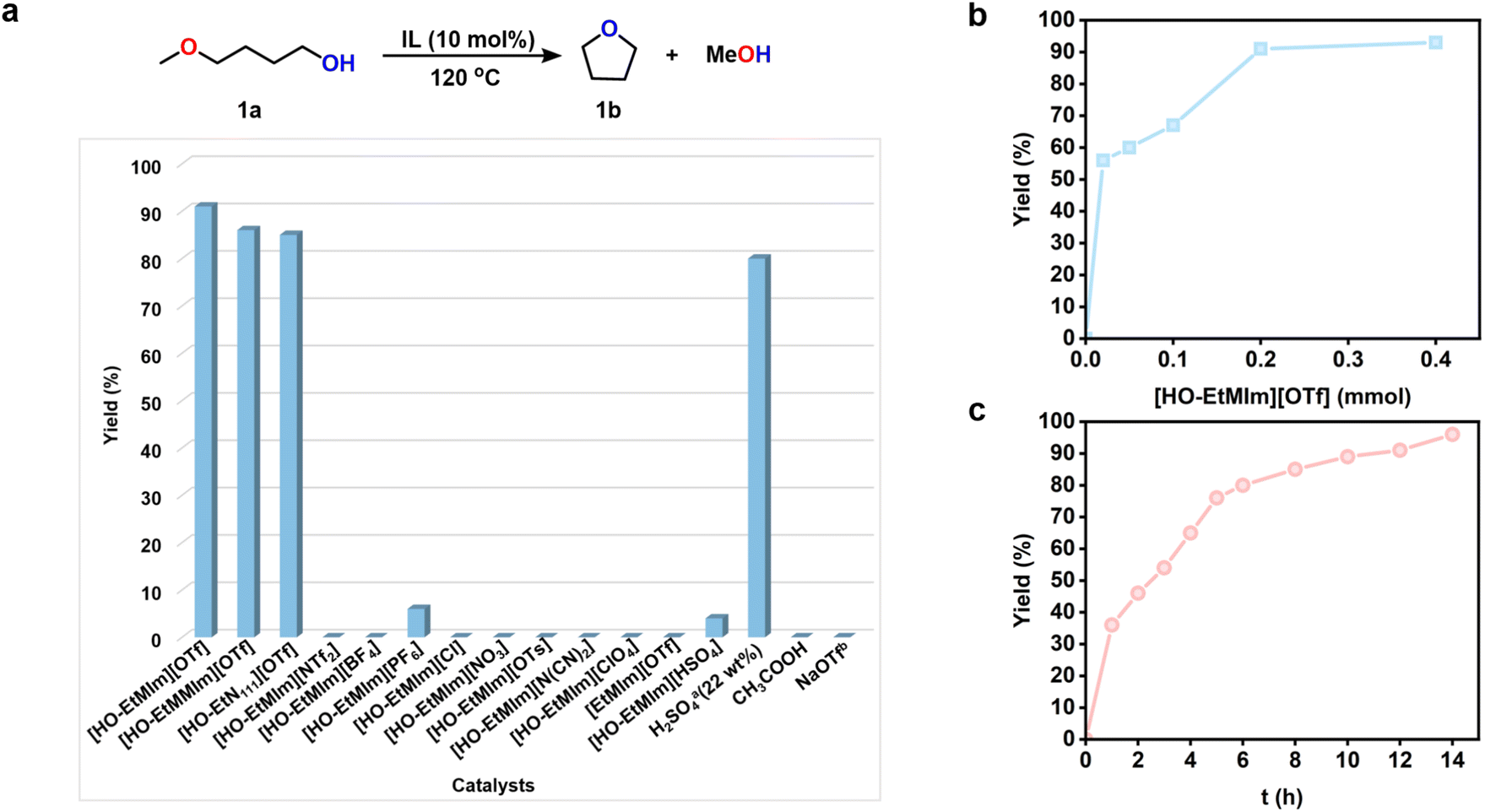 | ||
Fig. 2 Screening conditions for ring-closing metathesis of 1a. (a) Screening for ILs; effect of (b) the catalyst loadings and (c) reaction time on the ring-closing metathesis of 1a. Reaction conditions: 1a (2 mmol), 120 °C of (a) 10 mol% of IL, 12 h; (b) 12 h; (c) 10 mol% of [HO-EtMIm][OTf]. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The amount of catalyst was based on H2SO4. b10 mol% of NaOTf, n-hexane (0.45 M). Yields were determined by 1H NMR, and the data were the average of three replicates with reproducibility of ±3%. The amount of catalyst was based on H2SO4. b10 mol% of NaOTf, n-hexane (0.45 M). Yields were determined by 1H NMR, and the data were the average of three replicates with reproducibility of ±3%. | ||
However, the OH-functional ILs with other anions, including [HO-EtMIm][NTf2], [HO-EtMIm][BF4], [HO-EtMIm][PF6], [HO-EtMIm][Cl], [HO-EtMIm][NO3], [HO-EtMIm][OTs], [HO-EtMIm][N(CN)2] and [HOEtMIm][ClO4] were ineffective (Fig. 2a), suggesting that the [OTf]− anion of the effective ILs played an important role in this reaction. On the other hand, the presence of –OH in the cation was also crucial deduced from the fact that the reaction did not occur using [EtMIm][OTf] as the catalyst (Fig. 2a). In comparison, the experimental results worked out to be consistent with the calculated prediction. The cooperation of the cation and anion of the IL is responsible for the catalytic performance. Additionally, Brønsted acid IL [HO-EtMIm][HSO4] was also examined for this reaction, showing much lower activity than [HO-EtMIm][OTf]. CH3COOH that has a similar pKa value to [HO-EtMIm][OTf]39 was found to be ineffective. These findings suggest that the RCM reaction of 1a may not follow an acid-catalysis mechanism. Notably, no 1-methoxy-4-(4-methoxybutoxy)butane was detected in the reaction solution, indicating that no intermolecular dehydration reaction of 1a occurred, which may be attributed to the cation–anion confinement effect. In addition, NaOTf was examined as a catalyst, and no 1b was obtained, implying that the effect of the residual metal (Na+, 0.012 wt%) in the ILs on this reaction could be negligible (Fig. 2a).
Subsequently, the effects of temperature, the amount of IL, reaction time on the RCM of 1a over [HO-EtMIm][OTf], and recycling test were investigated (Table S1† and Fig. 2, 3). As shown in Table S1,† the reaction could take place even at 70 °C, and the yield of 1b increased with temperature, achieving 94% at 140 °C within 12 h. As indicated, even with the amount of IL at 1 mol%, a high 1b yield of 56% was still reached with a turnover frequency (TOF) of 112 h−1 (Fig. 2b); a 1b yield of 36% was obtained within 1 h at 120 °C, and it reached up to 96% for 14 h (Fig. 2c). All these findings indicate that this catalyst was very efficient for 1a RCM. Moreover, since [HO-EtMIm][OTf] is immiscible with product 1b, it could be easily restored through phase separation and vacuum drying to remove the generated MeOH and unreacted 1a. The recycling tests indicate that the product yield did not decrease obviously after the IL was reused for five runs (Fig. 3). The 1H and 19F NMR spectra of IL before and after the reaction remained unchanged (Fig. S5 and S6†). These results indicate that this IL has good stability and reusability under experimental conditions. Notably, a small amount of dimethyl ether was obtained, which was originated from the dehydrative etherification of the generated MeOH during the reaction process (Fig. S3†).39
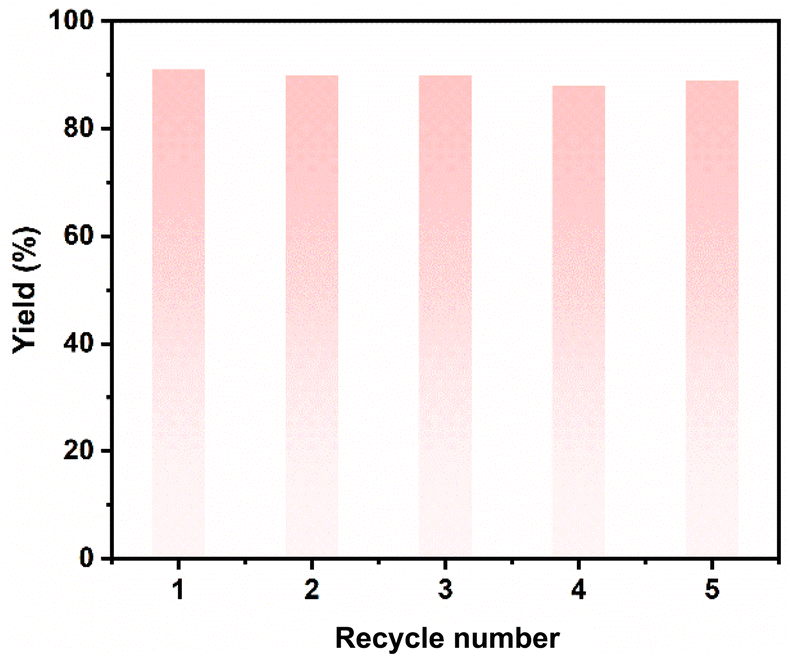 | ||
| Fig. 3 The IL recycling tests for ring-closing metathesis of 1a. Reaction conditions: 1a (2 mmol), [HO-EtMIm][OTf] (10 mol%), 120 °C, 12 h. | ||
Under the optimized experimental conditions, the RCM reactions of a series of alkoxy alcohols over [HO-EtMIm][OTf] were conducted. As illustrated in Table 1 and Fig. S7–10,† five- and six-membered aliphatic and aromatic O-heterocycles were produced in high yields from 81% to ∼99% in most cases under similar experimental conditions (1b–4b). However, only a 50% yield of 5b was obtained, probably due to the steric hindrance effect of 5a. 1,4-Dioxane (6b), dimethyl-substituted 1,4-dioxane (7b) as well as 3-hydroxytetrahydrofuran (8b) that are usually applied in drug design were also obtained in good yields. It should be pointed out that no byproducts from intermolecular dehydration of alkoxyl alcohols were detectable during the above reactions, although [HO-EtMIm][OTf] performed well in catalyzing the intermolecular dehydration of alcohols in our previous study.39 This supports the cation–anion confinement effect on the RCM reactions of alkoxyl alcohols.












Clearly, OH-functional ILs with [OTf]− anion are suitable catalysts for the RCM reactions of alkoxyl alcohols according to either the DFT prediction or the experimental results. However, why do both anion and cation of the IL have such an important effect on this reaction, and how do the anion and cation of the IL synergistically catalyze reactions?
To answer these questions, the hydrogen bonding interaction between these ILs and 1a was investigated by DFT calculations and NMR analysis. For comparison, five ILs including [HO-EtMIm][OTf], [HO-EtMMIm][OTf], [HOEtN111][OTf], [HO-EtMIm][Cl] and [EMIm][OTf] were selected to perform the DFT calculations. From the optimized figurations of these ILs interacting with 1a (Fig. 4 and S11†), it is obvious that 1a could form dual HBs with both the cation and anion of the ILs. The estimated lengths of HBs between the ether O atom of 1a and the hydroxyl H or C2–H in the cations are 1.77 Å for [HO-EtMIm][OTf], 1.78 Å for [HO-EtN111][OTf], 1.80 Å for [HO-EtMMIm][OTf], 1.81 Å for [HO-EtMIm][Cl] and 2.13 Å for [EMI-m][OTf], meanwhile, those of HBs between the hydroxyl H atom of 1a and the O atom in [OTf]− or Cl− atom are estimated to be 1.86 Å, 1.91 Å, 1.93 Å, 1.97 Å and 2.13 Å for [HO-EtN111][OTf], [EMIm][OTf], [HO-EtMIm][OTf], [HO-EtMMIm][OTf] and [HO-EtMIm][Cl], respectively. These results demonstrate that as the HB donor the OH-functionalized cations including [HO-EtMIm]+, [HO-EtMMIm]+, and [HO-EtN111]+ have similar ability to form strong HBs with the ether O atom of 1a in the presence of the [OTf]− anion, while as HB acceptor the [OTf]− anion shows a much stronger ability to form HB with hydroxyl H atom of 1a than Cl−. The NBO charges of the methylene C atom of –CH2OCH3 and the hydroxyl O atom in 1a were also calculated, which changed remarkably as 1a interacted with the ILs. The differences of NBO charges between the methylene C atom of –CH2OCH3 and the hydroxyl O atom in 1a were estimated to be 0.718, 0.693, 0.691, 0.689 and 0.686 as 1a interacted with [HO-EtMIm][Cl], [HO-EtMIm][OTf], [HO-EtN111][OTf], [HO-EtMIm] [OTf] and [EMIm][OTf], respectively (Fig. 4 and S11†). Obviously, the stronger dual hydrogen bonding interaction of the IL with 1a and the larger difference of NBO charges may reflect the higher reactivity of 1a under the catalysis of the IL. Therefore, it could be deduced that strong dual HBs, appropriate synergy and electrostatic confinement effect of the IL cation and anion are responsible for their catalytic activity.
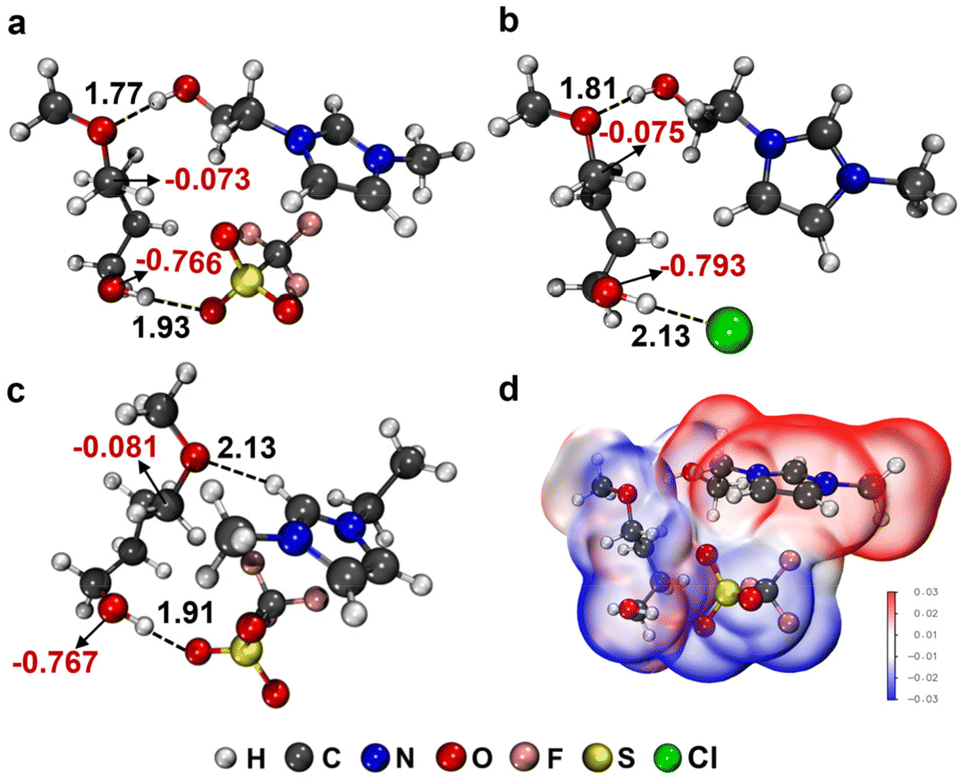 | ||
| Fig. 4 DFT calculations. The interaction figurations of 1a with ILs: (a) [HO-EtMIm][OTf], (b) [HO-EtMIm][Cl] and (c) [EMIm][OTf] optimized at M062X-D3/def2-TZVP level [black word: atom distance (Å), red word: NBO charges]; (d) ESP distribution of [HO-EtMIm][OTf] with 1a. | ||
Furthermore, the electrostatic potential (ESP) distribution of [HO-EtMIm][OTf] with 1a was performed for an in-depth study on the hydrogen bonding between them (Fig. 4d). It is obvious that at the areas where the HBs are formed, the positive surface potential (red area) of –OH in [HO-EtMIm]+ overlaps with the negative surface potential (blue area) of ether O atom in 1a, meanwhile, the positive surface potential of –OH in 1a overlaps with the negative surface potential of O atoms in [OTf]−.43–45
Moreover, the hydrogen bonding interaction between [HO-EMIm][OTf] and 1a was further evidenced by NMR spectroscopic experiments. As shown in Fig. 5, the chemical shift assigned to the hydroxyl H atom of [HO-EtMIm]+ shifted upfield, meanwhile the signal to the ether O atom in 1a became wider and shifted to 1.61 ppm from the initial 0.76 ppm. These results provide evidence for the formation of HB between 1a and [HO-EtMIm]+ in the form of CH3–(CH2)O⋯[HO-EtMIm]+, with the electron transfer from the ether O atom of 1a to the hydroxyl H atom of [HO-EtMIm]+, which may activate the ether C–O bond of CH3OCH2– in 1a. Meanwhile, the chemical shifts assigned to the O atoms of hydroxyl in 1a and in the [OTf]− anion shifted from −17.26 to −18.31 ppm and from 163.31 to 163.78 ppm, respectively. The signal belonged to the hydroxyl H atom of 1a shifted downfield, and the chemical shift of F atoms in [OTf]− shifted upfield (Fig. 5 and S12†). These results indicate that the hydroxyl H atom of 1a may form a HB with the O atom of [OTf]− promoted by the F atoms (see ESI† for details).46
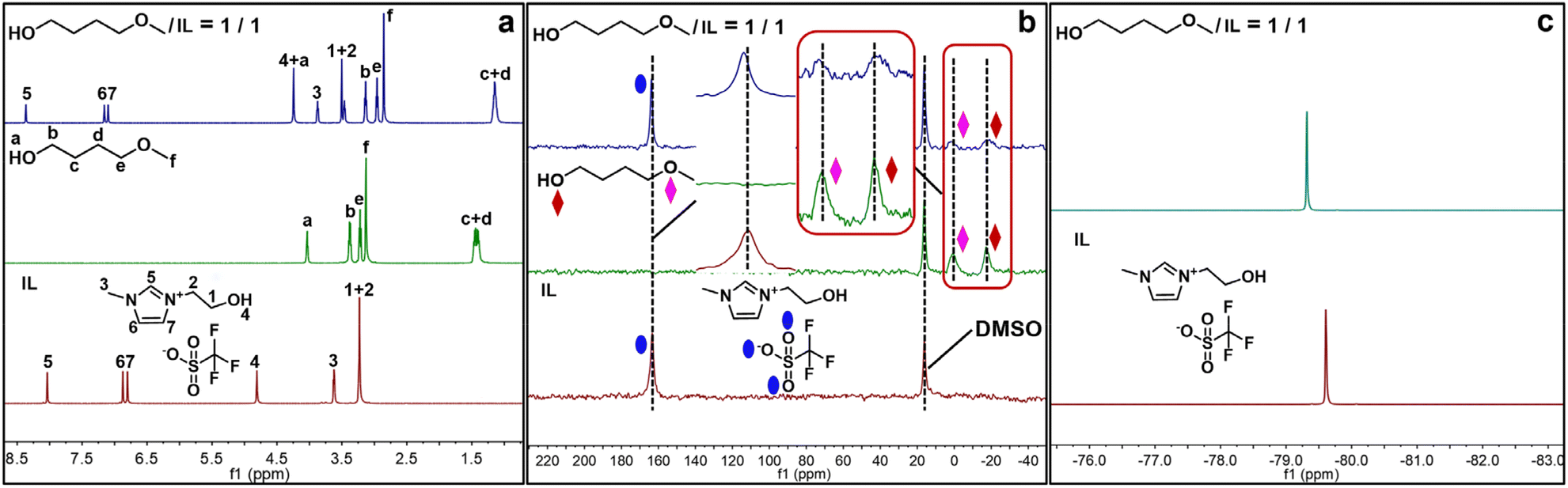 | ||
| Fig. 5 NMR analysis. 1H (a), 17O (b) and 19F (c) NMR spectra of [HO-EMIm][OTf], 1a and their mixture recorded at 333.15 K. | ||
To get an insight into the reaction mechanism, in situ1H NMR experiments were conducted (Fig. 6 and S13†). Interestingly, two new peaks appeared at δ = 4.21 and 4.77 ppm as the reaction occurred, which are assigned to the H atom of –OMe and –OH in [HO-EtMIm]-OMe intermediate, respectively. At the beginning of the reaction, a signal appeared at 4.98 ppm, belonging to the hydroxyl H atoms in the state of TS1. Additionally, as the reaction proceeded, the amount of 1a (the peaks appeared at δ = 2.00 and 3.99 ppm) declined with increases in the amounts of 1b (the peaks appeared at δ = 2.21 and 4.06 ppm) and MeOH (the peak appeared at δ = 3.75 ppm) (Fig. S13†).
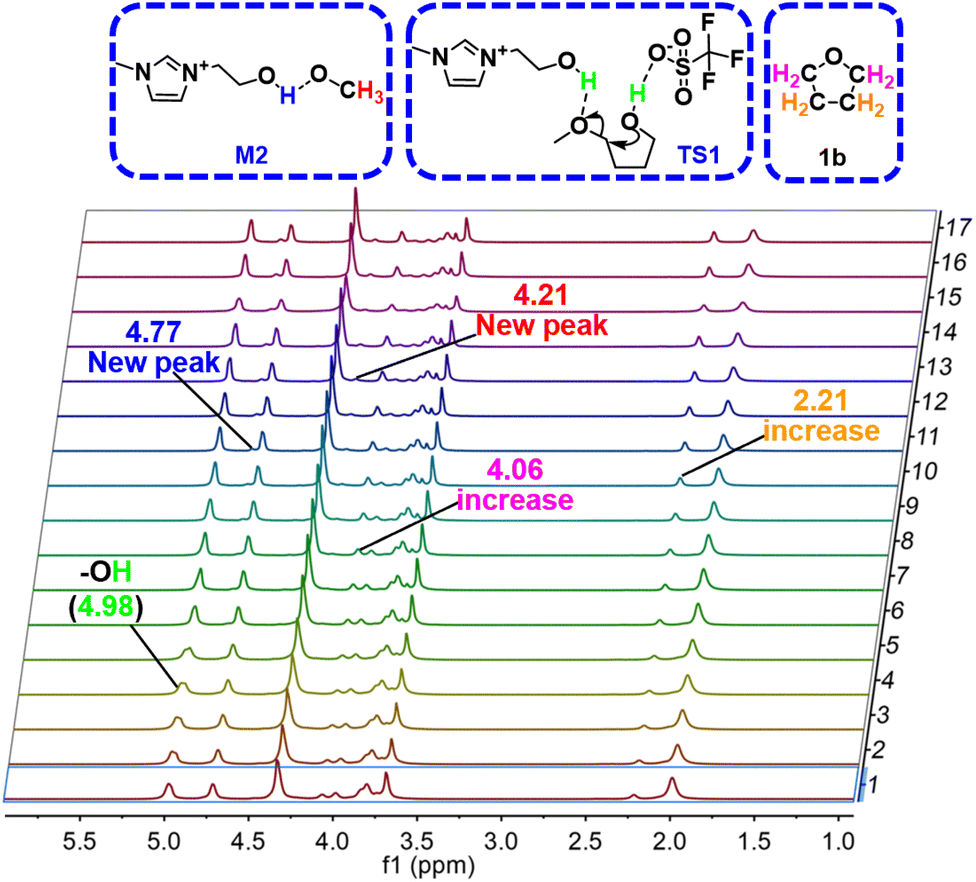 | ||
| Fig. 6 The in situ1H NMR spectra were recorded in 1a transformation process over [HO-EMIm][OTf] at 393.2 K. | ||
Based on the experimental results and previous reports,23,39 a possible reaction pathway is proposed as illustrated in Fig. S14.† First, both C–O and O–H bonds of 1a are simultaneously activated by a pair of IL cation and anion via strong H-bonding interaction. Then, the activated C atom linked to –OCH3 is attacked by the activated hydroxyl O atom of 1a, furnishing cyclic oxonium intermediate (M1) stabilized by the [OTf]− anion via the transition state of TS1, together with [HO-EtMIm]-OMe complex (M2). Finally, 2b and MeOH are generated via dehydrogenation of intermediate M1 by [HO-EtMIm]-OMe, with the regeneration of the IL.
Conclusions
In summary, assisted with DFT calculations a cation–anion confined hydrogen-bonding-catalysis strategy for ring-closing C–O/O–H bond metathesis of alkoxy alcohols to O-heterocycles is presented. [HO-EtMIm][OTf] can achieve this reaction under mild conditions, and affords various five-, six- and seven-membered aliphatic and aromatic O-heterocycles with/without alkyl, hydroxyl substituents in high yields. Mechanism studies indicate that the IL cation and anion serve as HB donor and acceptor, respectively, to form dual strong HBs with an alkoxy alcohol molecule in opposite directions, which cooperatively catalyze the reaction with high efficiency. This novel and green way to access O-heterocycles would be employed in the late-stage diversification of complex molecules and synthesis of various drugs and natural product derivatives from diverse substrates with target function groups. We believe that the cation–anion confined H-bonding catalysis strategy has promising applications in more reactions.Author contributions
H. W. and Z. L. designed the project and prepared the manuscript for publication. H. W. and Z. Z. carried out the experiments and collected the data. Z. Z. performed the DFT calculations. All authors analyzed the data and contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the Natural Science Basic Research Plan in Shaanxi Province of China (2023-JC-QN-0113), Scientific Research Plan Projects of Shaanxi Education Department (22JK0294), National Key Research and Development Program of China (2020YFA0710201), National Natural Science Foundation of China (22121002, 21890761, 22233006) and China Postdoctoral Science Foundation (2022M722597).References
- H. Albright, A. J. Davis, J. L. Gomez-Lopez, H. L. Vonesh, P. K. Quach, T. H. Lambert and C. S. Schindler, Chem. Rev., 2021, 121, 9359–9406 CrossRef CAS PubMed.
- X. Shen, T. T. Nguyen, M. J. Koh, D. Xu, A. W. Speed, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 541, 380–385 CrossRef CAS PubMed.
- A. J. Davis, R. B. Watson, D. J. Nasrallah, J. L. Gomez-Lopez and C. S. Schindler, Nat. Catal., 2020, 3, 787–796 CrossRef CAS.
- P. K. Quach, J. H. Hsu, I. Keresztes, B. P. Fors and T. H. Lambert, Angew. Chem., Int. Ed., 2022, e202203344 CAS.
- H. Albright, P. S. Riehl, C. C. McAtee, J. P. Reid, J. R. Ludwig, L. A. Karp, P. M. Zimmerman, M. S. Sigman and C. S. Schindler, J. Am. Chem. Soc., 2019, 141, 1690–1700 CrossRef CAS PubMed.
- A. Koner, B. Morgenstern and D. M. Andrada, Angew. Chem., Int. Ed., 2022, 61, e202203345 CrossRef CAS PubMed.
- M. R. Becker, R. B. Watson and C. S. Schindler, Chem. Soc. Rev., 2018, 47, 7867–7881 RSC.
- D. L. Nascimento, A. Gawin, R. Gawin, P. A. Gunka, J. Zachara, K. Skowerski and D. E. Fogg, J. Am. Chem. Soc., 2019, 141, 10626–10631 CrossRef CAS PubMed.
- E. F. van der Eide and W. E. Piers, Nat. Chem., 2010, 2, 571–576 CrossRef CAS PubMed.
- H. F. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2009, 48, 2085–2089 CrossRef CAS PubMed.
- N. Lemcoff, N. B. Nechmad, O. Eivgi, E. Yehezkel, O. Shelonchik, R. S. Phatake, D. Yesodi, A. Vaisman, A. Biswas and N. G. Lemcoff, Nat. Chem., 2023, 15, 475–482 CrossRef CAS PubMed.
- C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef CAS PubMed.
- L. Ma, W. Li, H. Xi, X. Bai, E. Ma, X. Yan and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 10410–10413 CrossRef CAS PubMed.
- Y. Zhang, J. Jermaks, S. N. MacMillan and T. H. Lambert, ACS Catal., 2019, 9, 9259–9264 CrossRef CAS PubMed.
- E. K. Cho, P. K. Quach, Y. Zhang, J. H. Sim and T. H. Lambert, Chem. Sci., 2022, 13, 2418–2422 RSC.
- S. R. Todtz, C. W. Schneider, T. Malakar, C. Anderson, H. Koska, P. M. Zimmerman and J. J. Devery III, J. Am. Chem. Soc., 2023, 145, 13069–13080 CrossRef CAS PubMed.
- T. Biberger, S. Makai, Z. Lian and B. Morandi, Angew. Chem., Int. Ed., 2018, 57, 6940–6944 CrossRef CAS PubMed.
- J. Zhu, R. Zhang and G. Dong, Nat. Chem., 2021, 13, 836–842 CrossRef CAS PubMed.
- N. Ansmann, T. Thorwart and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202210132 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, J. Liu, C. Zhang, Z. Zhang, J. Xu, S. Xu, F. Wang and F. Wang, ChemSusChem, 2015, 8, 2066–2072 CrossRef CAS PubMed.
- Y. Ma, L. Zhang, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 12434–12437 CrossRef CAS PubMed.
- Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, F. Zhang, Y. Wu, R. Li, J. Xiang, Z. Wang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 11850–11855 CrossRef CAS PubMed.
- M. Zhang, R. Ettelaie, T. Yan, S. Zhang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2017, 139, 17387–17396 CrossRef CAS PubMed.
- J. Hu, J. Ma, Q. Zhu, Z. Zhang, C. Wu and B. Han, Angew. Chem., Int. Ed., 2015, 54, 5399–5403 CrossRef CAS PubMed.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113–7131 CrossRef CAS PubMed.
- Y. Zhao, B. Han and Z. Liu, Acc. Chem. Res., 2021, 54, 3172–3190 CrossRef CAS PubMed.
- T. Yang, J. Yang, X. Deng, E. Franz, L. Fromm, N. Taccardi, Z. Liu, A. Görling, P. Wasserscheid and O. Brummel, Angew. Chem., Int. Ed., 2022, 61, e202202957 CrossRef CAS PubMed.
- S. Dongare, O. K. Coskun, E. Cagli, K. Y. Lee, G. Rao, R. D. Britt, L. A. Berben and B. Gurkan, ACS Catal., 2023, 13, 7812–7821 CrossRef CAS PubMed.
- K. Chen, G. Shi, W. Zhang, H. Li and C. Wang, J. Am. Chem. Soc., 2016, 138, 14198–14201 CrossRef CAS PubMed.
- M. Hulla, S. M. A. Chamam, G. Laurenczy, S. Das and P. J. Dyson, Angew. Chem., Int. Ed., 2017, 56, 10559–10563 CrossRef CAS PubMed.
- Y. Wu, Y. Zhao, R. Li, B. Yu, Y. Chen, X. Liu, C. Wu, X. Luo and Z. Liu, ACS Catal., 2017, 7, 6251–6255 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- F. Wu, Y. Wang, Y. Zhao, M. Tang, W. Zeng, Y. Wang, X. Chang, J. Xiang, B. Han and Z. Liu, Sci. Adv., 2023, 9, eade7971 CrossRef CAS PubMed.
- H. Wang, Y. Wu, Y. Zhao and Z. Liu, Acta Phys.-Chim. Sin., 2021, 37, 2010022 Search PubMed.
- W. Lu, J. Ma, J. Hu, J. Song, Z. Zhang, G. Yang and B. Han, Green Chem., 2014, 16, 221–225 RSC.
- Y. Zhao, B. Yu, Z. Yang, H. Zhang, L. Hao, X. Gao and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 5922–5925 CrossRef CAS PubMed.
- M. Liu, Z. Zhang, H. Liu, Z. Xie, Q. Mei and B. Han, Sci. Adv., 2018, 4, eaas9319 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, F. Zhang, Z. Ke, B. Han, J. Xiang, Z. Wang and Z. Liu, Sci. Adv., 2021, 7, eabg0396 CrossRef CAS PubMed.
- D. A. Decato, A. M. S. Riel, J. H. May, V. S. Bryantsev and O. B. Berryman, Angew. Chem., Int. Ed., 2021, 60, 3685–3692 CrossRef CAS PubMed.
- R. R. Annapureddy, F. Burg, J. Gramüller, T. P. Golub, C. Merten, S. M. Huber and T. Bach, Angew. Chem., Int. Ed., 2021, 133, 7999–8005 CrossRef.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Wu, W. Zhang, L. Qi, Y. Ren and H. Ma, J. Mol. Struct., 2019, 1197, 171–182 CrossRef CAS.
- J. Zhang, J. Chem. Theory Comput., 2018, 14, 572–587 CrossRef CAS PubMed.
- S. M. Banik, A. Levina, A. M. Hyde and E. N. Jacobsen, Science, 2017, 358, 761–764 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03041e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |