
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-glycerol hydrodeoxygenation to propylene: advancing knowledge on Mo-based catalyst characteristics and reaction pathways under flow conditions
Georgia
Ioannidou
a and
Angeliki A.
Lemonidou
 *ab
*ab
aDepartment of Chemical Engineering, Aristotle University of Thessaloniki, University Campus, Thessaloniki, 54124, Greece. E-mail: alemonidou@cheng.auth.gr
bChemical Process & Energy Resources Institute, Centre for Research and Technology-Hellas, Thessaloniki, 57001, Greece
First published on 9th November 2023
Abstract
In this work, the reaction pathways of one-step glycerol hydrodeoxygenation in the gas phase are exploited under flow conditions over molybdena-based catalysts (8.7 wt% Mo/black carbon). Hydrodeoxygenation (HDO) experiments with possible reaction intermediates along with temperature-programmed surface reaction spectroscopy (TPSR) experiments demonstrated two possible reaction pathways. The major one involves propylene formation via hydrodeoxygenation of 2-propen-1-ol which is considered to be the main intermediate of the reaction. In the secondary route, propanal formed via 2-propen-1-ol isomerization is further converted to 1-propanol. XPS measurements in fresh-reduced and reduced-used catalysts revealed that molybdena exists as multivalent species mostly as Mo5+, Mo4+ and Mo3+, the presence of which seems to be crucial for the desired pathway of successive HDOs towards propylene. TPSR tests using methanol as the probe molecule showed that redox and acid sites co-exist on the surface of reduced molybdena catalysts, and this bifunctionality is mostly served by the undercoordinated molybdena species. The long-term catalyst test at 280 °C and 60 bar pressure under glycerol hydrodeoxygenation conditions demonstrated that the catalyst remains stable in terms of activity and selectivity for the first 10 h with a gradual drop afterwards. Regeneration of the catalyst for a short time under hydrogen flow proved to be sufficient to fully recover the initial activity and product selectivity.
1. Introduction
Growing concerns about climate change along with the depletion of fossil resources have triggered the development of alternative and greener strategies to produce fuels and chemicals.1 Global emissions of carbon dioxide, a major greenhouse gas, have dramatically increased resulting in its concentration in the air of 414 ppm in 2021, with the power and industry sectors mostly contributing to them.2–5 More specifically, the chemical industry accounts for 20% of the total CO2 emissions.6 Propylene is considered an important chemical intermediate, the demand for which has undergone a booming growth as it is used to produce important chemicals (e.g., polypropylene, propylene oxide, acrylic acid, and acrylonitrile).7 Historically, the main routes for propylene production are energy consuming processes, such as steam cracking (SC) and fluid catalytic cracking (FCC), resulting in high CO2 emissions. Both the FCC and SC processes emit approximately 1.51 kgCO2 per kg of propylene produced.6 Attempting to fill the propylene gap between the market demand and propylene production, on purpose propylene technologies, such as propane dehydrogenation (PDH), olefin metathesis, and methanol to propylene (MTP), have been developed over the years. Among them, PDH appears very profitable and has been successfully industrialized worldwide.8–10 However, the main disadvantage of the PDH process is the origin of the feedstock as it is still derived from fossil fuels.The growing environmental issues along with the increased demand to fill the propylene gap can be tackled using a renewable feedstock. Biomass (e.g., lignocellulosic and algae) and its derivatives (glycerol, glucose, sorbitol) can pave environmentally friendly ways for producing alternative fuels and platform chemicals owing to their null carbon footprint.11,12 Biomass derived oxygenates, such as polyols, are considered promising and sustainable feedstocks to produce unsaturated hydrocarbons. Removal of their oxygen content to form olefinic compounds is important and can be realized via promising catalytic routes (hydrogenolysis, dehydration, etc.). Among them, hydrodeoxygenation (HDO) is considered the most appealing one to remove oxygen functional groups by using hydrogen as a reductant.13,14
Different types of HDO catalytic materials have been investigated in the last few years, such as noble metal catalysts (Pd, Pt, Ru, Rh),15 zeolites,16 non-noble metals (Ni, Cu)17,18 and catalysts based on transition metal oxides, such as Mo and W11. Among these, Mo-based catalysts have attracted much attention.19–21 Likozar's group has studied HDO of lignocellulosic biomass by using Mo-based catalysts, as well as Pd and Ni unsupported and/or supported on different supports. Among the different catalysts, Mo-based ones exhibited the best performance in terms of activity and selectivity.22,23 The promising performance of Mo-based catalysts has also been mentioned for the case of m-cresol conversion to toluene.24 It was found that the reducibility of Mo species as well as the type of support mostly affects the HDO activity. All the above results clearly point to the high potential of Mo-based catalysts for HDO reactions.
Glycerol is a non-toxic polyol derived from several biomass processes (biodiesel transesterification, hydrolysis, etc.) having three –OH groups and constitutes an ideal feedstock for valorization.25–29 The rapid expansion of biodiesel production, which generates substantial amounts of glycerol as a co-product, boosts the potential of bio-glycerol upgrading to value-added products, such as 1-propanol, 2-propen-1-ol, propanal, acrolein acrylic acid, C3-diols and others.30–37 Complete glycerol deoxygenation along with the formation of a double carbon bond is a challenging path that could lead to propylene. The choice of the catalytic material is of outmost importance as it should favor C–O rupture instead of C–C.
To the best of our knowledge, reports in the open literature on catalytic glycerol conversion to propylene either in one step or in a tandem process are limited (Table 1). Fadigas et al.38 explored the reaction of glycerol HDO to propylene over an Fe–Mo catalyst supported on activated carbon in the gas phase. Later, the same group39 studied the same reaction in the gas phase, nonetheless at ambient pressure and high temperature (300 °C), reaching high propylene selectivity (90%) for almost complete glycerol conversion. Very recently, M. El. Doukkali and co-workers systematically investigated the performance of Mo-based catalysts on hydrophobic-inert silica supports and in the form of carbide β-Mo2C.40 Other researchers have reported the use of bifunctional catalysts in a double bed (Ir/ZrO2 and ZSM-5-30, WO3-Cu/Al2O3 and SiO2/Al2O3, MoO3-Ni2/Al2O3 and ZSM-5-30, Pt/ZSM-5 and ZSM-5) for enhanced propylene formation.28,41–43 Even though this seems favorable for massive propylene production, the potentiality of these processes apart from not commercially acceptable propylene productivity (Table 1) still lies ahead as some issues should be first resolved. For example, the stability and reusability of the catalytic materials have not been reported, which are important and challenging aspects for a potential commercial application. The most complete study for propylene production via glycerol hydrodeoxygenation is the one authored by M. El Doukkali and coworkers40 as it provides insightful information covering most of the aforementioned issues. As there is a need to develop new processes that can significantly contribute to fill the propylene gap, more studies should focus not only on optimizing the current HDO processes, but also on improving catalytic performance that can pave the way for future scale-up studies for glycerol HDO to propylene and perhaps for similar complex molecules.
| Catalyst – reactor type | Operating conditions | Conversion (%) | Propylene selectivity (%) | Propylene productivity (kgC3H6 kgcat−1 h−1) | Ref. |
|---|---|---|---|---|---|
| Fe–Mo/activated carbon – fixed bed flow unit | 300 °C, 1 bar, H2/Glyc = 120, TOS∼h | 100 | 90 | 0.04 | 39 |
| Ir/ZrO2 & ZSM-5 – vertical fixed bed | 250 °C, 5 bar, H2/Glyc = 100, TOS = 2h | 73 | 88 | 0.17 | 28 |
| WO3-Cu/Al2O3 & SiO2/Al2O3 – fixed bed downflow glass reactor | 242 °C, 1 bar, H2/Glyc = 161, TOS = 2h | 100 | 84.8 | 0.0017 | 41 |
| MoO3-Ni2P/Al2O3 & ZSM-5 – fixed bed quartz reactor | 250 °C, 1 bar, H2/Glyc = 100, TOS∼h | 100 | 88 | 0.0016 | 42 |
| Pt/ZSM-5 & ZSM-5 – double bed reactor | 500 °C, 1 bar, H2/Glyc = 100, TOS = 500h | 100 | 63.7 | 0.0028 | 43 |
| MoOx@SBA-15, βMo2C@SBA-15 – single fixed bed | 318 °C, 50 bar, H2/Glyc = 98, TOS = 2.4h | 100 | 84.1 | 0.10 | 40 |
| 318 °C, 50 bar, H2/Glyc = 98, TOS = 14.5h | 100 | 64.8 | 0.07 | ||
| Fe–Mo/black carbon – batch reactor | 300 °C, 80 bar, H2/Glyc = 53, TOS∼h | 88.8 | 76.1 | 0.48 | 44 |
| Mo/black carbon – high pressure continuous flow reactor | 280 °C, 60 bar, H2/Glyc = 80, TOS = 4h | 100 | 71 | 0.10 | 45 |
| Mo/black carbon – high pressure continuous flow reactor | 280 °C, 60 bar, H2/Glyc = 80, TOS = 10h | 100 | 60 | 0.12 | Current study |
In a previous research study of our group, it was demonstrated that glycerol can be effectively converted to propylene, in one step, in the liquid phase over Mo-based catalysts.44 As mentioned earlier, molybdenum catalysts are widely applied in HDO reactions, and more specifically, they are also found to be attractive candidates for glycerol HDO to propylene, as apart from their deoxygenation ability, their mild hydrogenation activity does not favor the further conversion of propylene to propane. Considering that continuous flow operation is preferred for industrial applications, molybdenum catalysts were further evaluated in glycerol HDO in the gas phase employing a high-pressure continuous flow unit.45 The effect of reactor parameters was investigated over molybdenum catalysts supported on hydrophobic black carbon (Mo/BC). It was experimentally proved that increased temperature, hydrogen availability (expressed either as 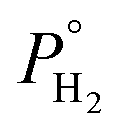 or
or  ) and W/F are key parameters for propylene production. The reaction conditions were further optimized with predictive modelling and under the optimum conditions (T = 280 °C,
) and W/F are key parameters for propylene production. The reaction conditions were further optimized with predictive modelling and under the optimum conditions (T = 280 °C, 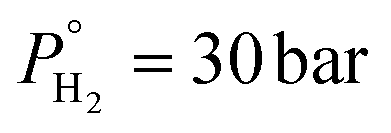 ,
,  , LHSV = 1 h−1 and W/F = 422 gcat molglyc−1 h−1) dictated by modelling, a propylene yield of 71% with almost complete glycerol conversion was experimentally validated over 8.7 wt% Mo/BC catalyst. The main byproduct was 1-propanol with 20% yield.
, LHSV = 1 h−1 and W/F = 422 gcat molglyc−1 h−1) dictated by modelling, a propylene yield of 71% with almost complete glycerol conversion was experimentally validated over 8.7 wt% Mo/BC catalyst. The main byproduct was 1-propanol with 20% yield.
The scope of the current work and its novelty span from the efforts to shed more light on the mechanistic pathways of glycerol HDO to propylene under flow conditions to the determination of the nature of the active sites of the promising 8.7 wt% Mo/BC catalyst and to its stability and reusability as well as the effect of the prereduction step. The identification of the primary and secondary routes of glycerol HDO is attempted by performing experiments in the gas phase under flow conditions with the intermediate products formed, as well as by applying temperature programmed surface reaction spectrometry. Additional studies to determine the nature of the catalyst active sites by using methanol oxidation as the chemical probe reaction along with XPS measurements are performed. The stability and reusability of Mo/BC, as well as the effect of the pre-reduction step are also assessed. The results obtained lead to productivity values (Table 1) equal to 0.10–0.12 kgC3H6 kgcat−1 h−1 which are among the highest compared to others reported in the literature and can pave the way for further techno-economic assessment that might contribute to finding a sustainable way towards an industrial accelerated glycerol to propylene route in the future.
2. Materials and methods
2.1. Catalyst preparation
A molybdenum catalyst with 8.7 wt% Mo loading supported on a commercially available black carbon (BC) Vulcan XC72 was prepared following a procedure previously described45 using MoO3 as the molybdenum precursor. The obtained catalyst was calcined at 200 °C for 2 h under synthetic air flow (100 cm3 min−1) and then prior to the reaction was reduced at 500 °C for 30 min under pure hydrogen flow (40 cm3 min−1). The calcined and reduced samples were named fresh and reduced, respectively, and labeled as ‘fresh Mo/BC’ and ‘reduced Mo/BC’, respectively.2.2. Catalyst characterization
The basic characterization of fresh and reduced Mo/BC samples (BET surface area, XRD, H2-TPR, TGA) can be found in our earlier work.45 The Brunauer–Emmett–Teller (BET) surface area of the used samples was determined by N2 physisorption at 77 K with an Autosorb-1 (Quantachrome, Florida, UK) flow apparatus. Thermogravimetric analysis of the reduced and reduced-used catalysts was conducted on a TG 209 F3 Tarsus (NETZSGH, Germany) apparatus. A small amount of the sample was placed in an aluminum sample cup and heated up to 800 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere (50 cm3 min−1).TPSR experiments using pure methanol (CH3OH-TPSR) as the probe molecule were performed to extract information on the nature of the catalyst active sites. Fresh and reduced samples (0.2 g) were placed in the reactor and were further impregnated with a drop of liquid methanol to cover the catalyst surface. Then, the reactor was heated up to 100 °C and held for 2 h under helium flow (60 cm3 min−1). After cooling to room temperature, the catalytic samples were heated up to 800 °C at a constant rate of 10 °C min−1 under helium flow (30 cm3 min−1). The following masses (m/z) were recorded: 4 (He), 15 (CH4), 18 (H2O), 29 (HCHO), 31 (CH3OH), 44 (CO2), 46 (dimethyl ether, DME), and 60 (methyl formate, HCOOCH3) and the overlapping fragmentations of the compounds were appropriately considered.
Temperature programmed surface reaction tests under H2 flow (H2-TPSR) were conducted over the reduced catalyst to elucidate the reaction routes of glycerol to propylene. Prior to measurements, the catalytic samples were impregnated with 10 wt% aqueous solution of the desired feed. Apart from glycerol, 2-propen-1-ol, 2-propanol, 1-propanol, propanal and acetone were also used as feeds. The catalytic samples (0.15 g) were then flushed under flowing He, for 0.5 h at 60 °C. Afterwards, the temperature was increased up to 600 °C at a constant rate of 5 °C min−1 under 50% H2/He flow (total flow 20 cm3 min−1). Overlapping fragmentation contributions of all the abovementioned compounds were taken into consideration. Furthermore, TPSR experiments were also performed following the same procedure under pure He flow instead of H2 using glycerol as the feed. Table 2 displays the main fragment ions of all components that were considered in the CH3OH-TPSR and TPSR tests of glycerol, products and intermediates.
| Product | Molecular Weight | Main fragment ions |
|---|---|---|
| a The m/z signals recorded appear in bold. | ||
| H2 | 2 | 2(100)a |
| He | 4 | 4(100) |
| Propane | 44 | 29(100), 28(59), 27(42), 44(27), 43(23), 39(18) |
| 1-Propanol | 60 | 31(100), 29(17), 27(16), 42(13) |
| Propylene | 42 | 41(100), 39(73), 42(71), 27(38), 40(29), 38(20), 37(12) |
| Acetone | 58 | 43(100), 58(25), 42(9), 41(3), 44(2.5) |
| CO2 | 44 | 44(100), 28(10) |
| 2-Propanol | 60 | 45(100), 43(19), 27(16), 29(12.5), 41(8), 39(6), 42(4), 44(4) |
| Propanal | 58 | 58(100), 29(88), 28(58), 27(57), 57(30), 26(16) |
| 2-Propen-1-ol | 58 | 57(100), 31(41), 39(28), 29(25), 27(23.2), 58(23) |
| Glycerol | 92 | 61(100), 43(77), 44(43), 31(39), 29(30), 15(22) |
| Methane | 16 | 16(100), 15(89), 14(20) |
| Methanol | 32 | 31(100), 32(74), 29(45) |
| Formaldehyde | 30 | 29(100), 30(58), 28(24) |
| Dimethyl ether | 46 | 45(100), 46(60), 29(38), 15(24) |
| Methyl formate | 60 | 31(100), 32(46), 29(45), 60(38), 15(19) |
| H2O | 18 | 18(100), 17(21) |
2.3 Catalytic activity tests
Activity measurements were conducted in the gas-phase in a continuous flow high-pressure reactor unit (FlowCat, HEL) equipped with a fixed bed reactor (din = 6 mm), electronic pressure and temperature controllers, a H2 mass flow controller and a HP liquid pump. Typically, a certain amount of the reduced sample with the particle size between 45 and 75 μm was loaded in the middle of the reactor packed with quartz particles at both ends. Then the system temperature and pressure were set at the desired values equal to 280 °C and 60 bar, respectively. Following this step, an aqueous feed solution containing 10 wt% (unless otherwise stated) of the oxygenate and the rest water was fed to the top of the reactor together with hydrogen flow. Before entering the reactor, the feed was preheated at 200 °C by passing it through an electrically heated line.The reactor exit stream was separated in the liquid and gas phases by passing it through a condenser at 0 °C. The gaseous products were online analyzed in a GC (Agilent 7890A) equipped with a TCD detector and two columns in series-bypass configuration (Molecular Sieve and Plot Q). Apart from propylene, which was the main product detected in the gas phase, propane at low concentrations was found under specific conditions, while other lower alkanes and/or COx were not detected. In addition to gaseous products, liquid products were collected hourly and analyzed offline using a GC (Agilent 8890A) with an FID detector and a capillary column (DB-Wax, UI 30 m × 530 μm × 1.0 μm). The carbon balance of all experiments was 85 ± 5%. Most of the experiments have been performed at least twice, others even three times. A simple statistical analysis was performed using the Minitab statistical program, while the analysis of variance (ANOVA) was used to evaluate the results via the calculation of the p-value and coefficient R2. A small p-value (<0.05) and a high value of R2 (>90%) were observed confirming the reliability of the current results.
The reaction pathways of glycerol HDO to propylene were investigated by performing experiments with the possible intermediate products formed, such as 2-propen-1-ol, propanal, 2-propanol, 1-propanol and acetone, at a constant time on stream equal to 4 h and by maintaining the same values of the reaction parameters (T = 280 °C, Psystem = 60 bar, LHSV = 1.2 h−1, W/F = 211 gcat molfeed−1 h−1), unless otherwise noted.
The following expressions were used to determine the catalytic activity. The conversion of the reactant (eqn (1)), selectivity towards a product (based on the reactant consumed) (eqn (2)), propylene yield (eqn (3)), propylene rate (eqn (4)) and carbon balance (eqn (5)) were calculated as:
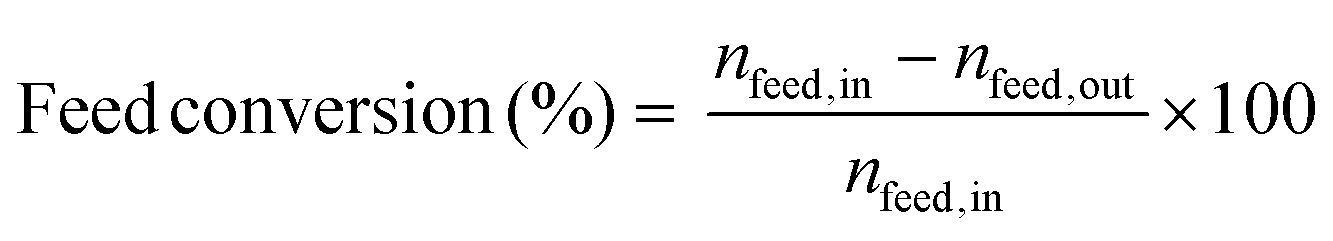 | (1) |
 | (2) |
 | (3) |
 | (4) |
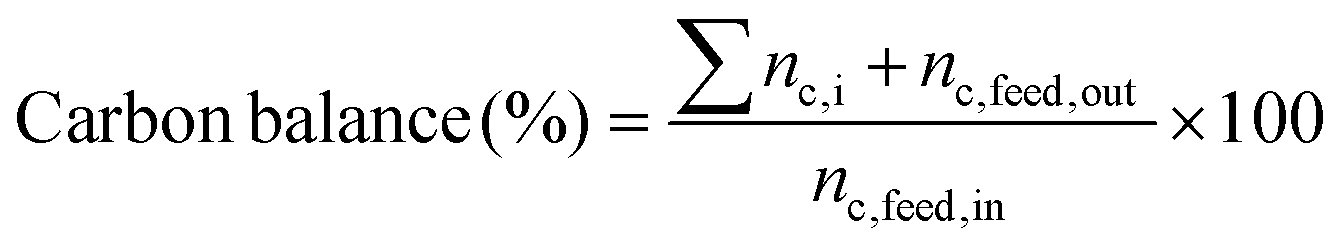 | (5) |
3. Results and discussion
3.1 Catalyst characterization
Temperature programmed surface reaction spectroscopy (TPSR) is considered a powerful technique providing valuable information about the nature of catalyst active sites, the reaction mechanism and kinetics using chemical probe molecules. Among them, methanol has attracted much attention, especially for identifying the nature of active sites, due to its ability to easily adsorb on catalytic surfaces, as well as its reactivity to metal oxides compared to the other molecules used.46,47 Methanol conversion can lead to various products reflecting the nature of the catalyst active sites. Redox centers oxidatively convert methanol towards formaldehyde, acid sites yield dimethyl ether (DME) and methyl formate, while basic sites produce CO2.48The CH3OH-TPSR spectra for both fresh and reduced catalysts are presented in Fig. 1 and 2, respectively. It is interesting to see that the same reaction products, nonetheless at different reaction temperatures, were detected indicating the presence of the same active sites with different chemical activity. Another common feature of both spectra is the appearance of methanol at relatively elevated temperatures with a broad desorption peak maximizing at 400–450 °C. Considering that molecularly physiosorbed methanol releases at low temperature (∼100 °C),49 the elevated temperature methanol peak in both samples derives more likely from self-hydrogenation of surface methoxy species (*OCH3) which undergo protonation and desorb as CH3OH.50–52 This is mostly associated with DME and HCHO formation due to the high concentration of the surface methoxy species.51 A closer look at both graphs confirms this assumption, as the CH3OH peak centers at approximately the same temperature where the production of both HCHO and DME occur. Similarly, it is noteworthy that CH4 production appears at the spectrum of both materials, which can be attributed either to decomposition reactions of both CH3OH and DME and/or their hydrogenolysis53 with hydrogen which is a decomposition product of CH3OH along with HCHO.54,55 In fact, two TPSR CH4 peaks appear in both cases, one at 100 °C and the other one at 650 °C. The desorption peak at low temperature can be ascribed to hydrogenolysis reactions, while the other at high temperature to decomposition ones.53
 | ||
| Fig. 1 CH3OH-TPSR spectrum of fresh catalyst. Desorption products corresponding to (a) methanol, redox sites (HCHO) and CH4 and (b) acid sites (DME, HCOOCH3) and water. The following masses were used: m/z = 31 for CH3OH, m/z = 30 for HCHO, m/z = 45 for DME, m/z = 60 for HCOOCH3, m/z = 15 for CH4 and m/z = 18 for H2O. | ||
 | ||
| Fig. 2 CH3OH-TPSR spectrum of reduced catalyst. Desorption peaks corresponding to (a) methanol and redox sites and (b) acid sites and water. The following masses were used: m/z = 31 for CH3OH, m/z = 30 for HCHO, m/z = 45 for DME, m/z = 60 for HCOOCH3, m/z = 15 for CH4 and m/z = 18 for H2O. | ||
The CH3OH-TPSR spectrum of fresh catalytic material is shown in Fig. 1. It can be easily seen that the chemisorbed surface methoxy species yield HCHO, DME and methyl formate reflecting the presence of both redox and acid active sites. Even though the present results were not fully quantified, based on almost similar sensitivity factors of the above products,52 it can be inferred from the relevant intensities of the peaks that the fresh catalytic material possesses high selectivity towards dehydration products (DME and methyl formation). The production of DME and methyl formate occurs at three different temperature zones indicating the presence of three different intermediate methoxy species which are adsorbed on catalyst active sites with different acid strengths. The surface methoxy species at the lowest temperature correspond to the Mo-OCH3 species formed on the catalyst surface, while the other peaks reflect the high stability of the Mo-OCH3 intermediates implying that they have had their redox and/or acidic properties retarded.51 This is mostly the case for the redox sites which appear to be less active, as HCHO, the product of methanol reduction, desorbs at very high temperature >600 °C.
The CH3OH-TPSR experiment for the reduced Mo/BC catalyst (Fig. 2) presents the same product spectrum as that for the fresh one except for different desorption temperature, indicating the existence of both redox and acid sites. Specifically, the main desorption peak of HCHO formed on the redox sites, have shifted towards lower temperatures reflecting the higher activity of the reduced molybdenum sites than the oxidized ones.50 The same is also valid for the desorption temperature of DME and methyl formate produced on acid sites. In both Fig. 1 and 2, water, which is associated with the formation of both DME and methyl formate, is also presented. As shown in Fig. 1b, three intense water peaks appear in the same temperature range with those of DME and methyl formate. Even though the resolution of the water signal in the MS analyser used (Omnistar) was not high, the recorded signals at m/z = 18 are more pronounced for the fresh Mo/BC catalyst where DME and methyl formate are more favored compared to the reduced Mo/BC (Fig. 2b).
A rough calculation was performed on the number of active (both redox and acid) sites that can be made by integrating the desorption peaks of the methanol products. The integration revealed that the total number of actives sites increases by 37% upon reduction. More specifically, reduction favors product formation proving that the reduced Mo3+, Mo4+, and Mo5+ species enhance methanol chemisorption more efficiently than the fully oxidized Mo6+ ones.50 However, the most significant difference between the fresh and reduced catalysts is in the acid/redox ratio which in the fresh catalyst is roughly 1 while it drops to 0.15 in the reduced catalyst. The high decrease of acid in favor of redox sites on the surface of the reduced catalyst might affect the production of the target product by altering the reaction pathway as will be discussed further on. As the catalytic activity of the surface molybdenum oxide was found to increase upon reduction, it can be suggested that the reduced molybdena species which behave as both redox and acid sites are the most active ones capable of catalyzing the hydrodeoxygenation steps during glycerol HDO.
3.2. Catalytic performance
The catalytic results of our previous studies for glycerol HDO to propylene in the liquid phase in a batch reactor44 and in the gas phase in a fixed bed reactor under flow conditions45 revealed the outstanding catalytic activity of the Mo-based catalyst supported on black carbon. Employing standard reaction conditions (280 °C, 60 bar system pressure), under flow conditions, propylene yield exceeded 60% for almost complete glycerol conversion for the steady state TOS = 4 h. In most cases, 1-propanol was the main byproduct formed reaching a maximum yield value of 20%. Apart from 1-propanol, propanal and 2-propen-1-ol were also identified, with varying yields depending on the applied reaction conditions. After having extensively tested the effect of the reaction parameters,45 the performance of the catalyst is further explored by testing its stability for extended reaction time along with its reusability at the selected optimum reaction conditions (T = 280 °C, Psystem = 60 bar,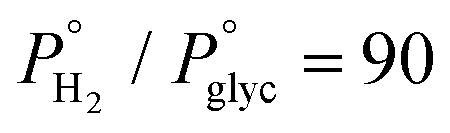 , LHSV = 1.2 h−1, W/F = 211 gcat molglyc−1 h−1), under which both the maximum propylene yield and formation rate were achieved.
, LHSV = 1.2 h−1, W/F = 211 gcat molglyc−1 h−1), under which both the maximum propylene yield and formation rate were achieved.
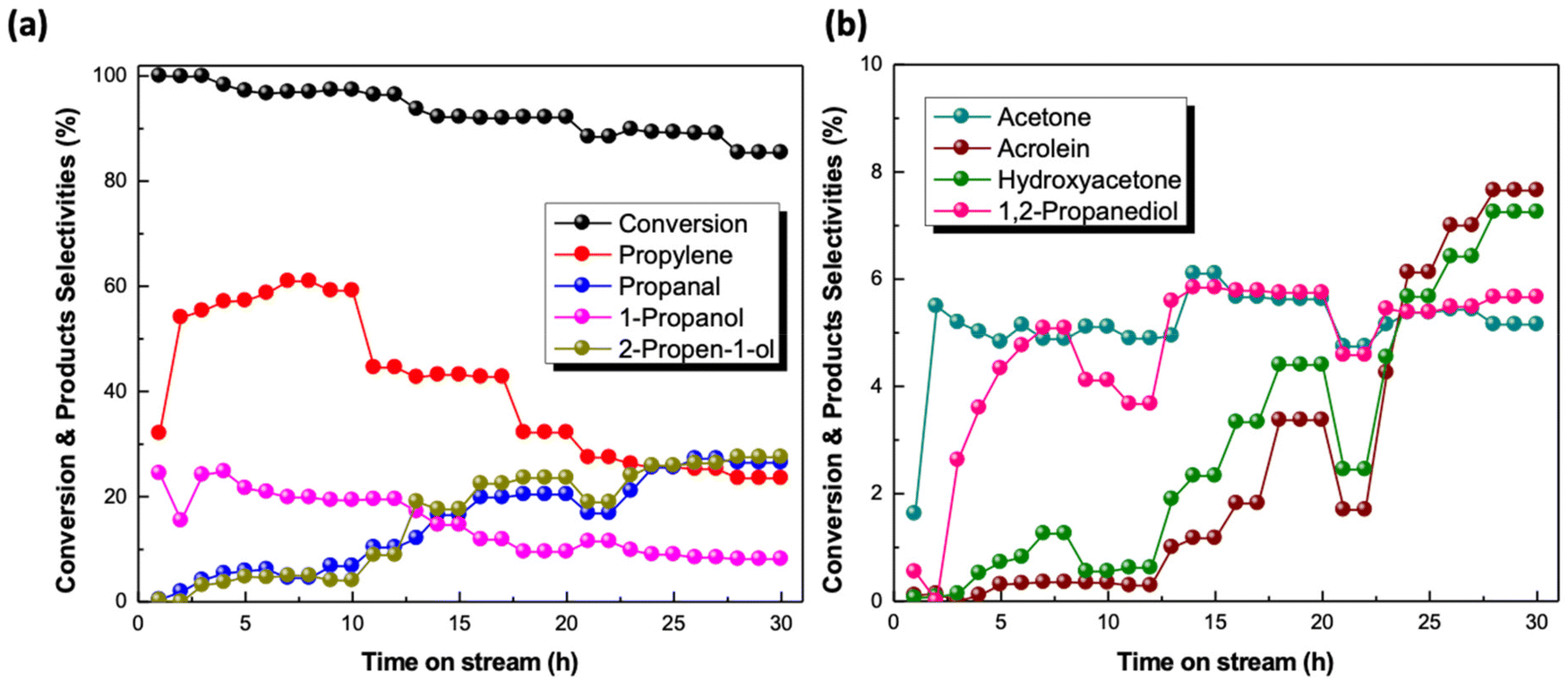 | ||
Fig. 3 Stability test in the reaction of glycerol hydrodeoxygenation over reduced Mo/BC catalyst at T = 280 °C, Ps = 60 bar,  , LHSV = 1.2 h−1 and W/F = 211 gcat molglyc−1 h−1. , LHSV = 1.2 h−1 and W/F = 211 gcat molglyc−1 h−1. | ||
These results suggest that not only the number of active sites, but their functionality also changes during extended catalyst testing under reaction conditions. BET measurements revealed almost 50% loss of the surface area after 30 h (BET surface area of the reduced and reduced-used catalyst: 131 and 67 m2 g−1, respectively). Indication about the loss of active sites is also provided by TGA measurements of the reduced-used catalyst after 30 h TOS under inert nitrogen flow. Fig. 4 illustrates the variation in the catalyst weight as a function of temperature. For comparison, the profiles of the reduced and reduced-used after 4 h TOS for Mo-based catalysts are also included. As expected, under inert conditions the profile of the freshly reduced catalyst is almost flat. The catalyst which was used for a typical test of 4 h TOS shows a gradual loss of 4% in total, which is extended from 100–600 °C. However, the profile of the catalyst weight change after 30 h TOS is different. The loss in weight up to 200 °C, which is considered as the temperature range where water desorbs, amounts to 2.5 wt%. Further 6% weight loss up to 450 °C is seen and this can be ascribed to adsorbed intermediate species and/or products limiting thus the number of available sites for the reactions to proceed.
 | ||
| Fig. 4 Thermogravimetric analysis of Mo/BC catalysts under nitrogen flow. | ||
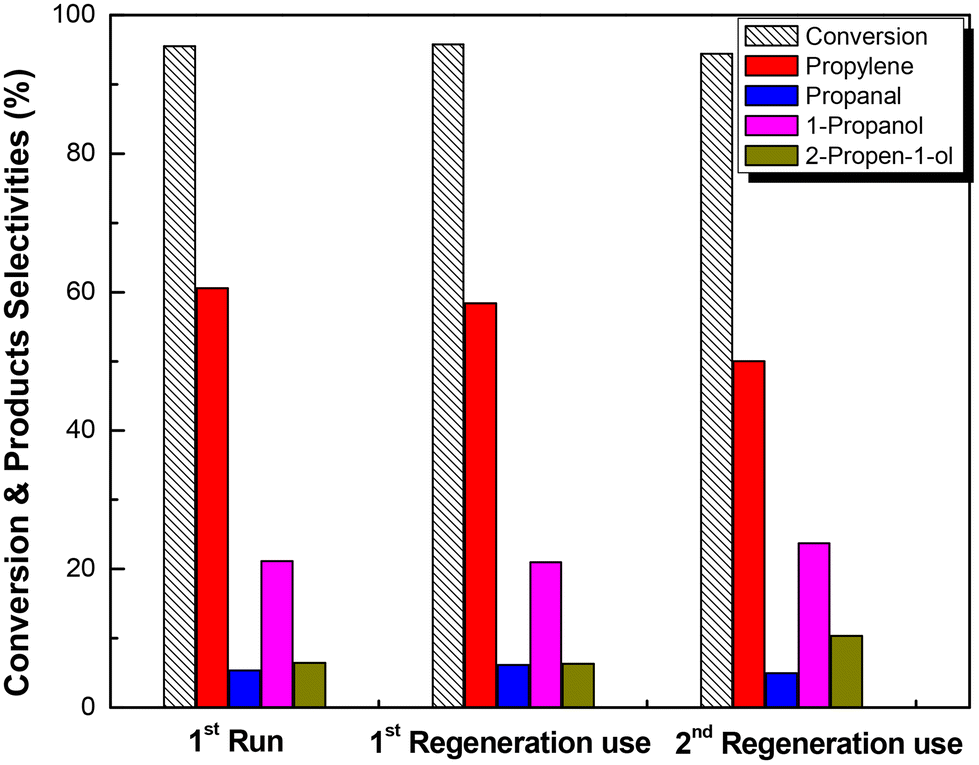 | ||
Fig. 5 Reusability test of Mo/BC catalyst in glycerol hydrodeoxygenation reaction at T = 280 °C, Ps = 60 bar,  , LHSV = 1.2 h−1, W/F = 211 gcat molglyc−1 h−1 and TOS = 4 h. Regeneration procedure: the same with prereduction step (T = 500 °C, 30 min in H2 flow). , LHSV = 1.2 h−1, W/F = 211 gcat molglyc−1 h−1 and TOS = 4 h. Regeneration procedure: the same with prereduction step (T = 500 °C, 30 min in H2 flow). | ||
 | ||
| Fig. 6 Thermogravimetric analysis of Mo/BC catalysts under nitrogen flow. | ||
 ) the performance of the catalyst does not change during 5 h TOS. The catalyst shows high activity mostly towards dehydration products with hydroxyacetone being the product with the highest selectivity ∼54%, while glycerol conversion does not surpass 50%. Production of acrolein, a product of two consecutive dehydration steps from glycerol, is also highly favored with 28% selectivity. As acidity is considered a key factor promoting HDO reactions of glycerol, it can be suggested that the Mo6+ oxidized species provide the required acidity towards dehydration products.56,57 However, their hydrogenation ability is negligible. It has also been reported that glycerol generates hydroxyacetone through Lewis acid sites, while acrolein via Brønsted acid sites. Thus, as the formation of both is highly favored, it can be assumed that both Lewis and Brønsted active sites co-exist on the catalyst surface which favor almost exclusively the formation of dehydration products.58,59 Even though the production of both hydroxyacetone and acrolein require the presence of acid sites, their production from redox and/or basic sites is also feasible. In the open literature, catalytic materials having redox sites, such as copper oxide, have been applied for the dehydration reaction of glycerol.59,60 These results further verify the results obtained in CH3OH-TPSR experiments according to which the ratio of the acid/redox sites is significantly lower in the reduced catalyst compared to the fresh one. Performance data suggest that the reduced catalyst with the low acid/redox ratio promotes the complete deoxygenation to propylene while samples with higher ratios lead to partially deoxygenated products in agreement with the work of M. El Doukkali et al.40 Summarizing, catalyst hydrogen pre-treatment step is of utmost importance to obtaining the active sites for the reaction to proceed towards propylene production.
) the performance of the catalyst does not change during 5 h TOS. The catalyst shows high activity mostly towards dehydration products with hydroxyacetone being the product with the highest selectivity ∼54%, while glycerol conversion does not surpass 50%. Production of acrolein, a product of two consecutive dehydration steps from glycerol, is also highly favored with 28% selectivity. As acidity is considered a key factor promoting HDO reactions of glycerol, it can be suggested that the Mo6+ oxidized species provide the required acidity towards dehydration products.56,57 However, their hydrogenation ability is negligible. It has also been reported that glycerol generates hydroxyacetone through Lewis acid sites, while acrolein via Brønsted acid sites. Thus, as the formation of both is highly favored, it can be assumed that both Lewis and Brønsted active sites co-exist on the catalyst surface which favor almost exclusively the formation of dehydration products.58,59 Even though the production of both hydroxyacetone and acrolein require the presence of acid sites, their production from redox and/or basic sites is also feasible. In the open literature, catalytic materials having redox sites, such as copper oxide, have been applied for the dehydration reaction of glycerol.59,60 These results further verify the results obtained in CH3OH-TPSR experiments according to which the ratio of the acid/redox sites is significantly lower in the reduced catalyst compared to the fresh one. Performance data suggest that the reduced catalyst with the low acid/redox ratio promotes the complete deoxygenation to propylene while samples with higher ratios lead to partially deoxygenated products in agreement with the work of M. El Doukkali et al.40 Summarizing, catalyst hydrogen pre-treatment step is of utmost importance to obtaining the active sites for the reaction to proceed towards propylene production.
 | ||
Fig. 7 Effect of hydrogen reduction as a catalyst pre-treatment step at T = 280 °C, Ps = 60 bar, 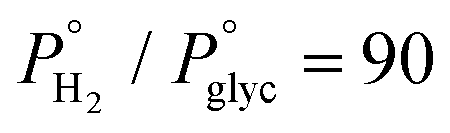 , LHSV = 1.2 h−1 and W/F = 211 gcat molglyc−1 h−1 and TOS = 4h for reduced and TOS = 5h for non-reduced catalyst. , LHSV = 1.2 h−1 and W/F = 211 gcat molglyc−1 h−1 and TOS = 4h for reduced and TOS = 5h for non-reduced catalyst. | ||
Considering the above results, along with that of the reusability test, reduced Mo species are most likely the active species for the selective HDO of glycerol to propylene. To further confirm this, measurements of catalysts by XPS, which provides valuable information about the composition and the oxidation state of metal species in the upper surface layers, were performed.
Fig. 8 visualizes the Mo 3d XPS spectra of fresh, reduced Mo/BC catalyst, while Fig. 9 that of reduced-used and non-reduced-used. Mo 3d levels are split into 3d5/2 and 3d3/2 due to the spin–orbital coupling.61 In the case of fresh sample, the observed binding energy of Mo 3d5/2 and 3d3/2 levels correspond to 232.5 and 236 eV, respectively. Based on the literature data61–65 and as presented in Table 3, these values are close to the ones typically referred to Mo6+ ions indicating that in the fresh sample molybdenum species are only present at the highest oxidation state 6+. The XPS data fitting for both reduced (Fig. 8b) and reduced-used (Fig. 9a) sample indicates that the formed species are related with numerous Mo oxidation states at different percentages, as shown in Table 3, whereas the peak deconvolution indicates that four oxidation states of Mo (Mo6+, Mo5+, Mo4+ and Mo3+) coexist on the surface. The appearance of new molybdenum species, namely Mo4+ and Mo5+, which is associated with a notable diminution of Mo6+ species suggests that during reduction almost half part of MoO3 in the upper surface layers is reduced to MoO2 (Table 3). A small amount of Mo3+ is also formed as shown by the peaks located at 228.6 and 231.7 eV, suggesting that a part of Mo4+ species undergoes an over hydrogen-reduction to Mo3+. XRD measurements of the catalytic materials45 showed that all the detected peaks of both reduced and reduced-used sample correspond to MoO2, while those of fresh to MoO3. Taking into account both XRD and XPS results it can be assumed that the bulk oxide is different than the composition of the upper surface layer of the catalyst. On the other hand, comparing reduced (Fig. 8b) and reduced-used samples (Fig. 9a), the population of Mo6+ and Mo5+ species further decreased from 50.10 to 40.20% and from 29.90 to 24.10%, respectively, while that of Mo4+ and Mo3+ increases indicating that a reductive atmosphere during experiments favors the reduction of Mo6+ to Mo5+ and its further reduction to Mo4+ over that of Mo4+ to Mo3+. XPS data for non-reduced-used catalyst (Fig. 9b) indicated the formation of Mo4+ and Mo5+ species which can be attributed to the reductive environment during experiments. Nonetheless, their formation is very limited not surpassing 8% which was not enough to promote propylene production. Comparing the catalytic performance data with CH3OH-TPSR and XPS results, it can be suggested that the hydrodeoxygenating activity is attributed to the reduced Mo3+, Mo4+, and Mo5+ species.
 | ||
| Fig. 8 XPS Mo 3d spectra (black line) and the fitted (green line) for (a) fresh and (b) reduced Mo/BC catalyst. | ||
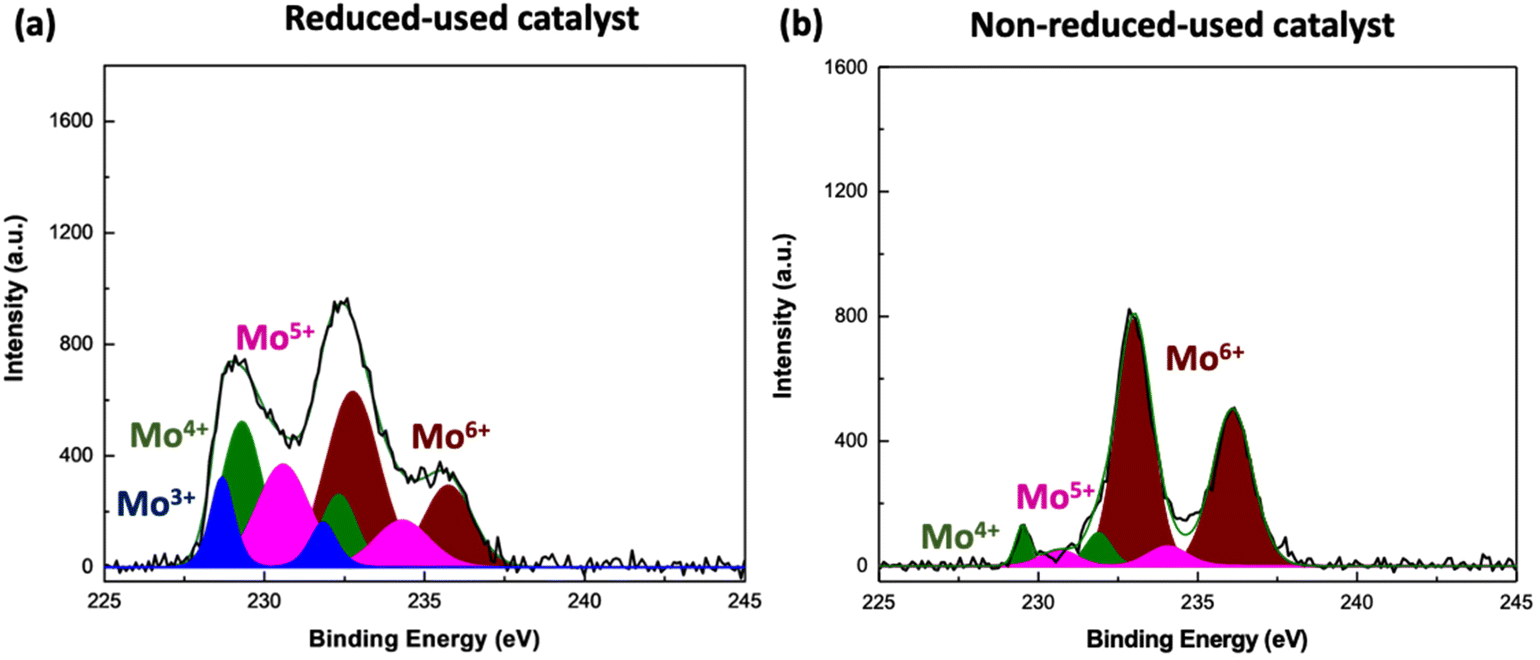 | ||
| Fig. 9 XPS Mo 3d spectra (black line) and the fitted (green line) for (a) reduced-used and (b) non-reduced-used Mo/BC catalyst. | ||
| Population of oxidation state of catalytic materials (%) | Standard reported values (eV) | |||||
|---|---|---|---|---|---|---|
| Oxidation state | Fresh catalyst | Reduced catalyst | Reduced-used catalyst | Non-reduced-used catalyst | Mo 3d5/2 | Mo 3d3/2 |
| Mo6+ | 100 | 50.10 | 40.20 | 84.40 | 232.5 | 235.7 |
| Mo5+ | 29.90 | 24.10 | 7.60 | 231.5 | 234.7 | |
| Mo4+ | 12.40 | 25.30 | 7.90 | 230.1 | 232.2 | |
| Mo3+ | 7.60 | 10.40 | — | 229.3 | 232.6 | |
3.3. Glycerol to propylene reaction routes
 | ||
| Fig. 10 Propylene formation rate and propylene productivity using glycerol and possible intermediate products as feedstocks at T = 280 °C, Ps = 60 bar, LHSV = 1.2 h−1and TOS = 4 h. *2 wt% aqueous solution was used as the feed. | ||
Fig. 11 illustrates the variation of conversion levels and product distribution using each intermediate product as feedstock. The results show that 2-propen-1-ol is the most selective over Mo/BC achieving the highest selectivity, equal to 66% for 73% conversion. It should be mentioned that this test was conducted using four times lower W/F than the other tests with the intermediates confirming the high activity of the catalyst towards conversion of this compound. The product mixture consists also of propanal and 1-propanol with selectivity of 22 and 12% respectively. Apart from hydrodeoxygenation, 2-propen-1-ol undergoes isomerization to propanal, a part of which undergoes sequential hydrogenation forming 1-propanol. 2-Propanol also enhances propylene production (57% selectivity), nonetheless at relatively lower rates as was mentioned previously. Notwithstanding the above, 2-propanol formation from glycerol was not detected at the tests performed at constant reaction temperature 280 °C and variable pressure and flow conditions applied in the previous45 and in the present studies, suggesting that the route of propylene formation via 2-propanol dehydration can be neglected. In addition, propylene was not detected using propanal and 1-propanol as feedstocks under the standard reaction conditions. The test with propanal was performed using a more dilute aqueous solution (2 wt%), instead of 10 wt% applied in other tests, due to difficulties in handling this volatile oxygenate. Propanal over Mo/BC catalyst shows high activity towards 1-propanol (∼50% selectivity) at high conversion levels (72%). A few unidentified peaks were detected at short retention times in the analysis of gas products, which might be attributed to ethers and/or products with less than 3 carbon atoms. In fact, it was mentioned that over transition metal oxides, propanal can follow ketonization, condensation and in few cases C–C bond cracking.63 The high stability of 1-propanol under the present reaction conditions was confirmed with the test in which it was used as feedstock which revealed a conversion marginally surpassing 10%. The products with the highest selectivity were its isomer, 2-propanol, and the dehydrogenation product, propanal, with 25 and 20% selectivity, respectively. Acetone, the most thermodynamically favored isomer out of 2-propen-1-ol and propanal, has also been used as feedstock. The activity of the Mo-based catalyst to convert acetone was low (15%), producing 2-propanol (58% selectivity) and propylene (26%). Acetone has been proposed as the main intermediate for propylene production from glycerol.39 However, the present results do not support this reaction pathway for the formation of propylene because of the absence of acetone in the product mixture and the extremely low acetone conversion activity of the catalyst under the conditions studied.
 | ||
| Fig. 11 Catalytic performance of glycerol and possible reaction intermediates products at T = 280 °C, Ps = 60 bar, LHSV = 1.2 h−1and TOS = 4 h. *W/F applied in 2-propen-1-ol test was 4 times lower than the other tests for the same bed volume. **2 wt% aqueous solution of propanal was used as the feed. | ||
The distribution of the products is also presented in tabular form (Table 4) as formation rates together with the consumption rate of each reactant. Under current reaction conditions, 2-propen-1-ol consumption rate is the highest among all (21.04 mmol2-propen-1-ol gcat−1 h−1) with propylene being the main product formed followed by propanal and 1-propanol. Comparing that with entry 1, both propanal's and 1-propanol's formation rates are noticeably higher (5.00 and 3.00 mmolproduct gcat−1 h−1) implying that a part of 2-propen-1-ol undergoes isomerization to propanal, which sequentially hydrogenates partially to 1-propanol due to the reductive environment. The lower rate of formation of 1-propanol from propanal (entry 5) can be ascribed to the much lower concentration of propanal as pointed also in the previous paragraph.
| Feed | Consumption rate (mmolfeed gcat−1 h−1) | Formation rate (mmolproduct gcat−1 h−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Propylene | Propane | Propanal | 1-Propanol | 2-Propen-1-ol | 2-Propanol | Acetaldehyde | |||
| a 2 wt% aqueous solution was used as the feed. Some unidentified peaks were detected which are more likely ethers without being quantified. | |||||||||
| 1 | Glycerol | 4.68 | 3.10 | — | 0.17 | 0.94 | — | — | — |
| 2 | 2-Propen-1-ol | 21.04 | 13.00 | — | 5.00 | 3.00 | — | — | — |
| 3 | 2-Propanol | 4.42 | 3.25 | 0.30 | — | — | — | — | — |
| 4 | 1-Propanol | 0.53 | — | — | 0.17 | — | — | 0.17 | 0.10 |
| 5 | Propanala | 0.98 | — | — | — | 0.65 | — | — | — |
| 6 | Acetone | 1.10 | 0.28 | — | — | — | — | 0.71 | — |
In this study, reaction pathways of glycerol conversion are also examined by TPSR under hydrogen flow. Attempting to get a general and adaptive approach to simulate the reaction, TPSR tests in H2 flow were performed over reduced Mo/BC catalyst following the procedure mentioned in section 2.2.2.
The TPSR profiles of products formed following the adsorption of glycerol are depicted in Fig. 12, that of the intermediates 2-propen-1-ol, propanal and 1-propanol are presented in Fig. 13, while that of 2-propanol and acetone are shown in Fig. 14. It is noteworthy that the desorbed species agree qualitatively with those observed during steady state HDO experiments. The reduced Mo/BC catalyst shows an ability to convert all feeds to propylene, apart from propanal and 1-propanol in agreement with the steady state tests as mentioned in the previous section. In few cases, more than one desorption peak appears for the same component (2-propen-1-ol double desorption peak during glycerol TPSR experiment) implying types of active sites with varying activity66 as also concluded from the CH3OH-TPSR experiments.
 | ||
| Fig. 12 Glycerol TPSR spectrum using hydrogen over reduced Mo/BC catalyst. | ||
 | ||
| Fig. 13 TPSR spectrum of (a) 2-propen-1-ol, (b) propanal and (c) 1-propanol using hydrogen over reduced Mo/BC catalyst. | ||
 | ||
| Fig. 14 TPSR spectrum of (a) acetone and (b) 2-propanol using hydrogen over reduced Mo/BC catalyst. | ||
The TPSR-spectrum of glycerol (Fig. 12a and b) shows that the reaction network highly depends on temperature and can be roughly divided into three different zones. Glycerol molecular desorption occurs at 200 °C with an extremely low intensity peak implying the high activity of the reduced Mo surface species to convert this oxygenate with the aid of H2 to various products. Temperatures below 250 °C mostly favor the production of partially deoxygenated products, with 2-propen-1-ol and 2-propanol being the major products desorbed with extremely high intensity. Other intermediates like propanal, 1-propanol and acetone appear in the same temperature range but with significantly lower intensity. The appearance of 2-propanol in comparable intensity with 2-propen-1-ol, highlights the difference in the reaction sequence at lower temperatures. Further temperature increase up to 350 °C, primarily promotes propylene formation and desorption. Nevertheless, at temperatures higher than 400 °C propylene production is restricted in favor of propane, while the reaction pathway changes promoting the formation and the desorption of partially deoxygenated intermediates (acetone, propanal and 1-propanol).
The evolution of products following the adsorption of 2-propen-1-ol on the catalyst surface is remarkably interesting with the appearance of propylene, which dominates in the whole temperature range, indicating the high activity of reduced Mo/BC in converting 2-propen-1-ol to propylene. This result agrees with the high rate of propylene formation obtained in the steady state runs using 2-propen-1-ol as the feed. Despite the higher activity to propylene, 2-propen-1-ol isomerization towards propanal also occurs above 300 °C with the latter being converted to 1-propanol on account of the reductive environment. Comparing 2-propen-1-ol and propanal TPSR-spectra (Fig. 13a and b), it can be seen that the activity of the catalyst surface sites towards propanal conversion is much lower as it desorbs intact in the temperature range 200–400 °C with a high intensity. The main products formed from the partial conversion of propanal are its isomer 2-propen-1-ol which desorbs up to 450 °C and propanols, 1- and 2-. No propylene formation was detected using either propanal (Fig. 13b) or its hydrogenation product 1-propanol (Fig. 13c) as feedstock.
TPSR of acetone and 2-propanol were also conducted. As depicted in Fig. 14a, adsorbed acetone starts to hydrogenate at temperatures as low as 100 °C as evidenced by the desorption of 2-propanol, which dominates in the product spectrum. In the test with 2-propanol as the feedstock, propylene appears as the main product desorbing at low temperature (onset 100 °C). In the temperature range 300–500 °C, the adsorbed species are further hydrogenated to propane. Even though propylene formation might also be feasible using both acetone and 2-propanol as the feedstock (even at low temperatures), when glycerol is used as the feedstock, their formation is possible only at lower temperatures than the one applied during HDO experiments (280 °C). In fact, steady state experiments that took place at lower temperatures (240 °C) favored both acetone and 2-propanol production, nonetheless without being further converted to the desired product, propylene, which was not detected.45 The above results confirm that the reaction pathways through which glycerol is converted to propylene are highly dependent on temperature.
TPSR of glycerol was also conducted under inert conditions using helium instead of hydrogen, to study the reaction pathway under an inert atmosphere. The profiles of the desorbed compounds as a function of temperature are presented in Fig. 15. Glycerol is converted to various products, but the extent of its conversion is much lower as depicted by the intensity of its desorption peak especially when compared with the corresponding glycerol peak under hydrogen flow (Fig. 12). Lower conversion of glycerol (69%) under inert conditions compared to 99% under hydrogen was also observed in the liquid phase under batch conditions over molybdena catalysts.62 In addition, under an inert atmosphere glycerol desorbs at 50 degrees lower than that in the presence of H2 with a maximum intensity at 150 °C. Further comparison of Fig. 15 with Fig. 12 shows that the products formed, and their distribution vary significantly. As expected, in the absence of hydrogen, propylene and 1-propanol appear with lower intensities (selectivity) compared to that in the presence of hydrogen. However, the striking difference is that under inert conditions, acetone dominates in the product spectrum desorbing at 200 °C. Its isomer, 2-propen-1-ol, also appears as a product at a slightly higher temperature (Tmax = 250 °C) followed by propylene, which is the product of 2-propen-1-ol hydrogenolysis. The formation of propylene under inert conditions might also be possible, implying that intermediate products formed function as hydrogen donors enhancing hydrogenation reactions.67 However, under inert conditions or low hydrogen availability, propylene's formation is restricted in favor of dehydration and/or partially deoxygenated products (acetone, propanal, 2-propen-1-ol).
 | ||
| Fig. 15 Glycerol TPSR spectrum under inert gas flow (He) over reduced Mo/BC catalyst. | ||
 | ||
| Scheme 1 Proposed reaction pathways of glycerol HDO towards propylene over reduced Mo/BC catalyst. | ||
In parallel to the desired pathway of 2-propen-1-ol re-adsorption on oxygen vacancies leading to the formation of propylene with a high rate, another pathway also proceeds. A part of 2-propen-1-ol undergoes isomerization to propanal which is sequentially hydrogenated to 1-propanol. The experimental results using glycerol as well as the intermediates 2-propen-1-ol, propanal and 1-propanol under steady state and dynamic (TPSR) conditions support this pathway as well. It was also proved that the rate of propylene produced via this route is negligible. The probe reaction of CH3OH-TPSR showed that both acidic and redox sites exist on the surface of the Mo catalyst; however over the reduced catalyst the ratio of acid/redox sites is much lower than that of the fresh fully oxidized catalyst. As in all the tests under typical conditions the catalyst was pre-reduced, we can propose that isomerization proceeds on Mo acid sites that exist on the surface and due to their low density, the extent of this pathway is limited.
4. Conclusions
In this study, an in-depth effort to elucidate the reaction pathways of bio-glycerol hydrodeoxygenation to propylene over molybdena catalysts supported on black carbon under flow conditions was made. For this, the catalytic results were obtained by performing experiments with the main intermediate products formed as well as by applying a powerful technique, temperature programmed surface reaction spectroscopy (TPSR). The results showed that the presence of hydrogen is essential for the formation of 2-propen-1-ol and the final product propylene. Nonetheless, 2-propen-1-ol isomerization towards propanal is considered a secondary route with the latter being converted to 1-propanol due to the reductive environment. The presence of acid and redox sites on the surface of the catalyst was examined using CH3OH-TPSR as the probe reaction. It was found that both sites exist on the surface and that over the reduced catalyst surface, redox sites prevail confirming the higher extent of the desired pathway of the intermediate 2-propen-1-ol hydrodeoxygenation to propylene over its isomerization which takes place over acid sites. The catalyst H2-reduction step at a high temperature (500 °C) for 0.5 h prior to the reaction is vital for the formation of the active sites, which according to XPS measurements are Mo5+, Mo4+ and Mo3+. Despite the reducing environment during the reaction, the catalyst without pre-reduction showed a 50% decrease in activity and more importantly poor hydrodeoxygenation selectivity with almost negligible propylene formation.The performance of the reduced Mo/BC catalyst was further investigated during the stability test for TOS = 30 h. The selectivity to propylene was not affected for the first 10 h, while a total loss of approximately 17% of its initial activity was observed. The loss of the activity was mostly ascribed to the coverage of the active sites by adsorbed intermediate species. In addition to the stability test, the reusability of the catalyst was also evaluated. Both activity and selectivity to propylene showed no significant differences after two regeneration tests using H2 treatment proving that the catalytic active sites are fully recovered.
The current results can be considered as very promising ones offering interesting feedback that advances the scientific knowledge not only of the whole process, but of catalysts as well, strengthening the potential use of molybdenum-based catalysts in large-scale application of bioglycerol HDO to propylene, contributing at the same time for future green processes in general. In the current process, 1-propanol is the main by-product with the highest yield (20%). Nonetheless, this should not be considered as a limitation for a future scale-up scenario, as the separation of 1-propanol and propylene can easily take place via a distillation step due to their different boiling points. 1-Propanol, which is still considered a green chemical, can be further used as a raw material for the production of a wide variety of chemicals, and/or by applying a different catalytic material to be dehydrated to propylene, further increasing the propylene yield and improving the feasibility of the HDO process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been funded by the European Regional Development Fund of the European Union and Greek National Funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – _CREATE – _INNOVATE (project code: T1EDK-02864). Ms Georgia Ioannidou acknowledges the Helmholtz Association of German Research Centers for the doctoral scholarship through the European program Helmholtz European Partnership for Technological Advancement (HEPTA).The authors would like to acknowledge Mr. Dimitrios Karfaridis from the Physics Department of the Aristotle University of Thessaloniki for the XPS measurements. In addition, Cabot company is acknowledged for supplying the catalyst support Carbon Black (VULCAN XC72).
References
- A. M. Ruppert, K. Weinberg and R. Palkovits, Hydrogenolysis Goes Bio: From Carbohydrates and Sugar Alcohols to Platform Chemicals, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- R. Rehan and M. Nehdi, Carbon Dioxide Emissions and Climate Change: Policy Implications for the Cement Industry, Environ. Sci. Policy, 2005, 8(2), 105–114 CrossRef CAS.
- M. T. Ravanchi, S. Sahebdelfar and F. T. Zangeneh, Carbon Dioxide Sequestration in Petrochemical Industries with the Aim of Reduction in Greenhouse Gas Emissions, Front. Chem. Sci. Eng., 2011, 5(2), 173–178 CrossRef CAS.
- A. Mikhaylov, N. Moiseev, K. Aleshin and T. Burkhardt, Global Climate Change and Greenhouse Effect, Entrep. Sustain. Issues, 2020, 7(4), 2897–2913 Search PubMed.
- P. Friedlingstein, M. O'Sullivan, M. W. Jones, R. M. Andrew, L. Gregor, J. Hauck, C. le Quéré, I. T. Luijkx, A. Olsen, G. P. Peters, W. Peters, J. Pongratz, C. Schwingshackl, S. Sitch, J. G. Canadell, P. Ciais, R. B. Jackson, S. R. Alin, R. Alkama, A. Arneth, V. K. Arora, N. R. Bates, M. Becker, N. Bellouin, H. C. Bittig, L. Bopp, F. Chevallier, L. P. Chini, M. Cronin, W. Evans, S. Falk, R. A. Feely, T. Gasser, M. Gehlen, T. Gkritzalis, L. Gloege, G. Grassi, N. Gruber, Ö. Gürses, I. Harris, M. Hefner, R. A. Houghton, G. C. Hurtt, Y. Iida, T. Ilyina, A. K. Jain, A. Jersild, K. Kadono, E. Kato, D. Kennedy, K. Klein Goldewijk, J. Knauer, J. I. Korsbakken, P. Landschützer, N. Lefèvre, K. Lindsay, J. Liu, Z. Liu, G. Marland, N. Mayot, M. J. McGrath, N. Metzl, N. M. Monacci, D. R. Munro, S.-I. Nakaoka, Y. Niwa, K. O'Brien, T. Ono, P. I. Palmer, N. Pan, D. Pierrot, K. Pocock, B. Poulter, L. Resplandy, E. Robertson, C. Rödenbeck, C. Rodriguez, T. M. Rosan, J. Schwinger, R. Séférian, J. D. Shutler, I. Skjelvan, T. Steinhoff, Q. Sun, A. J. Sutton, C. Sweeney, S. Takao, T. Tanhua, P. P. Tans, X. Tian, H. Tian, B. Tilbrook, H. Tsujino, F. Tubiello, G. R. van der Werf, A. P. Walker, R. Wanninkhof, C. Whitehead, A. Willstrand Wranne, R. Wright, W. Yuan, C. Yue, X. Yue, S. Zaehle, J. Zeng and B. Zheng, Global Carbon Budget, Earth Syst. Sci. Data, 2022, 14(11), 4811–4900 CrossRef.
- R. C. Müller, A. Schiessl, R. Volk and F. Schultmann, Assessment of Site-Specific Greenhouse Gas Emissions of Chemical Producers: Case Studies of Propylene and Toluene Diisocyanate, J. Cleaner Prod., 2021, 317, 128086–128116 CrossRef.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Propane to Olefins Tandem Catalysis: A Selective Route towards Light Olefins Production, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- T. K. Phung, T. L. M. Pham, K. B. Vu and G. Busca, (Bio)Propylene Production Processes: A Critical Review, J. Environ. Chem. Eng., 2021, 9, 105673–105686 CrossRef CAS.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Propane Dehydrogenation: Catalyst Development, New Chemistry, and Emerging Technologies, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- Z. Xu, C. Fang and T. Ma, Analysis of China's Olefin Industry Using a System Optimization Model Considering Technological Learning and Energy Consumption Reduction, Energy, 2020, 191, 116462–116474 CrossRef.
- P. Sudarsanam, N. K. Gupta, B. Mallesham, N. Singh, P. N. Kalbande, B. M. Reddy and B. F. Sels, Supported MoOx and WOx Solid Acids for Biomass Valorization: Interplay of Coordination Chemistry, Acidity, and Catalysis, ACS Catal., 2021, 11(21), 13603–13648 CrossRef CAS.
- A. Hommes, H. J. Heeres and J. Yue, Catalytic Transformation of Biomass Derivatives to Value-Added Chemicals and Fuels in Continuous Flow Microreactors, ChemCatChem, 2019, 11(19), 4671–4708 CrossRef CAS.
- Y. S. Yun, C. E. Berdugo-Díaz and D. W. Flaherty, Advances in Understanding the Selective Hydrogenolysis of Biomass Derivatives, ACS Catal., 2021, 11(17), 11193–11232 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Recent Advances in Hydrodeoxygenation of Biomass-Derived Oxygenates over Heterogeneous Catalysts, Green Chem., 2019, 21(14), 3715–3743 RSC.
- A. Gutierrez, R. K. Kaila, M. L. Honkela, R. Slioor and A. O. I. Krause, Hydrodeoxygenation of Guaiacol on Noble Metal Catalysts, Catal. Today, 2009, 147(3–4), 239–246 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado, M. Olazar and J. Bilbao, Transformation of Oxygenate Components of Biomass Pyrolysis Oil on a HZSM-5 Zeolite. II. Aldehydes, Ketones, and Acids, Ind. Eng. Chem. Res., 2004, 43(11), 2619–2626 CrossRef CAS.
- S. Sitthisa, T. Pham, T. Prasomsri, T. Sooknoi, R. G. Mallinson and D. E. Resasco, Conversion of Furfural and 2-Methylpentanal on Pd/SiO2 and Pd-Cu/SiO2 Catalysts, J. Catal., 2011, 280(1), 17–27 CrossRef CAS.
- S. Sitthisa and D. E. Resasco, Hydrodeoxygenation of Furfural over Supported Metal Catalysts: A Comparative Study of Cu, Pd and Ni, Catal. Lett., 2011, 141(6), 784–791 CrossRef CAS.
- D. R. Moberg, T. J. Thibodeau, F. G. Amar and B. G. Frederick, Mechanism of Hydrodeoxygenation of Acrolein on a Cluster Model of MoO3, J. Phys. Chem. C, 2010, 114(32), 13782–13795 CrossRef CAS.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Effective Hydrodeoxygenation of Biomass-Derived Oxygenates into Unsaturated Hydrocarbons by MoO3 Using Low H2 Pressures, Energy Environ. Sci., 2013, 6(6), 1732–1738 RSC.
- 2014_Hydrodeoxygenation of Liquefied Biomass on Urchin-like MoS2.
- M. Grilc, B. Likozar and J. Levec, Hydrodeoxygenation and Hydrocracking of Solvolysed Lignocellulosic Biomass by Oxide, Reduced and Sulphide Form of NiMo, Ni, Mo and Pd Catalysts, Appl. Catal., B, 2014, 150(151), 275–287 CrossRef.
- 2014_Hydrodeoxygenation of Solvolysed Lignocellulosic Biomass by Unsupported MoS2, MoO2, Mo2C and WS2 Catalysts.
- 2015_Reactivity and Stability Investigation of Supported Molybdenum Oxide Catalysts for the Hydrodeoxygenation (HDO) of m-Cresol.
- A. Rodrigues, J. C. Bordado and R. G. Dos Santos, Upgrading the Glycerol from Biodiesel Production as a Source of Energy Carriers and Chemicals - A Technological Review for Three Chemical Pathways, Energies, 2017, 10(11), 1817–1853 CrossRef.
- Z. Y. Zakaria, N. A. S. Amin and J. Linnekoski, A Perspective on Catalytic Conversion of Glycerol to Olefins, Biomass Bioenergy, 2013, 55, 370–385 CrossRef CAS.
- G. M. Lari, G. Pastore, M. Haus, Y. Ding, S. Papadokonstantakis, C. Mondelli and J. Pérez-Ramírez, Environmental and Economical Perspectives of a Glycerol Biorefinery, Energy Environ. Sci., 2018, 11(5), 1012–1029 RSC.
- L. Yu, J. Yuan, Q. Zhang, Y. M. Liu, H. Y. He, K. N. Fan and Y. Cao, Propylene from Renewable Resources: Catalytic Conversion of Glycerol into Propylene, ChemSusChem, 2014, 7(3), 743–747 CrossRef CAS PubMed.
- Y. Nakagawa and K. Tomishige, Heterogeneous Catalysis of the Glycerol Hydrogenolysis, Catal. Sci. Technol., 2011, 1(2), 179–190 RSC.
- M. Wang, H. Yang, Y. Xie, X. Wu, C. Chen, W. Ma, Q. Dong and Z. Hou, Catalytic Transformation of Glycerol to 1-Propanol by Combining Zirconium Phosphate and Supported Ru Catalysts, RSC Adv., 2016, 6(35), 29769–29778 RSC.
- M. El Doukkali, A. Iriondo and I. Gandarias, Enhanced Catalytic Upgrading of Glycerol into High Value-Added H2 and Propanediols: Recent Developments and Future Perspectives, Mol. Catal., 2020, 490, 110928–110957 CrossRef CAS.
- A. Kostyniuk, D. Bajec, P. Djinović and B. Likozar, Allyl Alcohol Production by Gas Phase Conversion Reactions of Glycerol over Bifunctional Hierarchical Zeolite-Supported Bi- and Tri-Metallic Catalysts, Chem. Eng. J., 2020, 397, 125430–125450 CrossRef CAS.
- B. Katryniok, S. Paul, M. Capron and F. Dumeignil, Towards the Sustainable Production of Acrolein by Glycerol Dehydration, ChemSusChem, 2009, 2(8), 719–730 CrossRef CAS PubMed.
- E. S. Vasiliadou and A. A. Lemonidou, Glycerol Transformation to Value Added C3 Diols: Reaction Mechanism, Kinetic, and Engineering Aspects, Wiley Interdiscip. Rev.: Energy Environ., 2015, 4(6), 486–520 CAS.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Glycerol as a Potential Renewable Raw Material for Acrylic Acid Production, Green Chem., 2017, 19(14), 3186–3213 RSC.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, Selective Hydrogenolysis of Glycerol to 1,3-Propanediol over Rhenium-Oxide-Modified Iridium Nanoparticles Coating Rutile Titania Support, ACS Catal., 2019, 10913–10930 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, Catalytic Materials for the Hydrogenolysis of Glycerol to 1,3-Propanediol, J. Mater. Chem. A, 2014, 2(19), 6688–6702 RSC.
- J. C. Fadigas, R. Gambetta, C. J. A. Mota and V. L. C. Gonçalves, PatentUS2011/0184216, 2011 Search PubMed.
- C. J. A. Mota, V. L. C. Gonçalves, J. E. Mellizo, A. M. Rocco, J. C. Fadigas and R. Gambetta, Green Propene through the Selective Hydrogenolysis of Glycerol over Supported Iron-Molybdenum Catalyst: The Original History, J. Mol. Catal. A: Chem., 2016, 422, 158–164 CrossRef CAS.
- M. El Doukkali, F. Dumeignil and S. Paul, New Insights in Single-Step Hydrodeoxygenation of Glycerol to Propylene by Coupling Rational Catalyst Design with Systematic Analysis, Appl. Catal., B, 2023, 324, 122280–122302 CrossRef CAS.
- D. Sun, Y. Yamada and S. Sato, Efficient Production of Propylene in the Catalytic Conversion of Glycerol, Appl. Catal., B, 2015, 13(20), 174–175 Search PubMed.
- Z. Wu, H. Yan, S. Ge, J. Gao, T. Dou, Y. Li, A. C. K. Yip and M. Zhang, MoO3 Modified Ni2P/Al2O3 as an Efficient Catalyst for Crude Glycerol to Propylene, Catal. Commun., 2017, 92, 80–85 CrossRef CAS.
- Z. Wu, K. Zhao, S. Ge, Z. Qiao, J. Gao, T. Dou, A. C. K. Yip and M. Zhang, Selective Conversion of Glycerol into Propylene: Single-Step versus Tandem Process, ACS Sustainable Chem. Eng., 2016, 4(8), 4192–4207 CrossRef CAS.
- V. Zacharopoulou, E. S. Vasiliadou and A. A. Lemonidou, One-Step Propylene Formation from Bio-Glycerol over Molybdena-Based Catalysts, Green Chem., 2015, 17(2), 903–912 RSC.
- G. Ioannidou, V.-L. Yfanti and A. A. Lemonidou, Optimization of Reaction Conditions for Hydrodeoxygenation of Bio-Glycerol towards Green Propylene over Molybdenum-Based Catalyst, Catal. Today, 2023, 423, 113902 CrossRef CAS.
- M. Badlani and I. E. Wachs, Methanol: A “Smart” Chemical Probe Molecule, Catal. Lett., 2001, 75(3–4), 137–149 CrossRef CAS.
- J. M. Jehng, I. E. Wachs, G. S. Patience and Y. M. Dai, Experimental Methods in Chemical Engineering: Temperature Programmed Surface Reaction Spectroscopy—TPSR, Can. J. Chem. Eng., 2021, 99(2), 423–434 CrossRef CAS.
- Z. Skoufa, E. Heracleous and A. A. Lemonidou, On Ethane ODH Mechanism and Nature of Active Sites over NiO-Based Catalysts via Isotopic Labeling and Methanol Sorption Studies, J. Catal., 2015, 322, 118–129 CrossRef CAS.
- L. E. Briand, J. M. Jehng, L. Cornaglia, A. M. Hirt and I. E. Wachs, Quantitative Determination of the Number of active Surface Sites and the Turnover Frequencies for methanol Oxidation over Metal Oxide Catalysts. Fundamentals of the Methanol Chemisorption and Application to Monolayer Molybdenum Oxide Catalysts, Catal. Today, 2003, 78(1–4), 257–268 CrossRef CAS.
- I. E. Wachs, J. M. Jehng and W. Ueda, Determination of the Chemical Nature of Active Surface Sites Present on Bulk Mixed Metal Oxide Catalysts, J. Phys. Chem. B, 2005, 109(6), 2275–2284 CrossRef CAS PubMed.
- X. Wang and I. E. Wachs, Designing the Activity/Selectivity of Surface Acidic, Basic and Redox Active Sites in the Supported K2O-V2O5/Al 2O3 Catalytic System, Catal. Today, 2004, 96, 211–222 CrossRef CAS.
- E. Cheng and J. Notestein, Molybdenum Oxide and Sulfide Active Sites for Isobutane Dehydrogenation with Methanol as a Probe Molecule, J. Catal., 2022, 413, 498–508 CrossRef CAS.
- W. Zhao, B. Zhang, G. Wang and H. Guo, Methane Formation Route in the Conversion of Methanol to Hydrocarbons, J. Energy Chem., 2014, 23(2), 201–206 CrossRef CAS.
- Y. Shen, Z. Xu, L. Wang and Y. Zhan, Hydrogen Production from Bioinspired Methanol Reforming at Room Temperature, Green Chem., 2021, 23(15), 5618–5624 RSC.
- Y. Liu, F. M. Kirchberger, S. Müller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Critical Role of Formaldehyde during Methanol Conversion to Hydrocarbons, Nat. Commun., 2019, 10, 1462 CrossRef PubMed.
- B. Katryniok, S. Paul, M. Capron and F. Dumeignil, Towards the Sustainable Production of Acrolein by Glycerol Dehydration, ChemSusChem, 2009, 2(8), 719–730 CrossRef CAS PubMed.
- C. H. Zhou, J. N. Beltramini and G. Q. Lu, Chemoselective Catalytic Conversion of Glycerol as a Biorenewable Source to Valuable Commodity Chemicals, Chem. Soc. Rev., 2008, 37(3), 527–549 RSC.
- K. Bhaduri, A. Ghosh, A. Auroux, S. Chatterjee, A. Bhaumik and B. Chowdhury, Soft-Templating Routes for the Synthesis of Mesoporous Tantalum Phosphates and Their Catalytic Activity in Glycerol Dehydration and Carbonylation Reactions, Mol. Catal., 2022, 518, 112074–112090 CrossRef CAS.
- F. Fernandes Barbosa and T. Pinheiro Braga, Catalytic Conversion of Glycerol to Acetol and Acrolein Using Metal Oxides: Surface Reactions, Prospects and Challenges, ChemCatChem., 2023, 15, e202200950 CrossRef CAS.
- S. Erfle, U. Armbruster, U. Bentrup, A. Martin and A. Brückner, Impact of Redox Properties on Dehydration of Glycerol to Acrolein over Heteropolyacids Assessed by Operando-EPR Spectroscopy, Appl. Catal., A, 2011, 391(1–2), 102–109 CrossRef CAS.
- S. S. Sunu, E. Prabhu, V. Jayaraman, K. I. Gnanasekar, T. K. Seshagiri and T. Gnanasekaran, Electrical Conductivity and Gas Sensing Properties of MoO3, Sens. Actuators, B, 2004, 101(1–2), 161–174 CrossRef CAS.
- V. Zacharopoulou, E. S. Vasiliadou and A. A. Lemonidou, Exploring the Reaction Pathways of Bioglycerol Hydrodeoxygenation to Propene over Molybdena-Based Catalysts, ChemSusChem, 2018, 11(1), 264–275 CrossRef CAS PubMed.
- H. Ren, Y. Chen, Y. Huang, W. Deng, D. G. Vlachos and J. G. Chen, Tungsten Carbides as Selective Deoxygenation Catalysts: Experimental and Computational Studies of Converting C3 Oxygenates to Propene, Green Chem., 2014, 16(2), 761–769 RSC.
- X. Wang, F. Zhao and L. Huang, Low Temperature Dehydration of Glycerol to Acrolein in Vapor Phase with Hydrogen as Dilution: From Catalyst Screening via TPSR to Real-Time Reaction in a Fixed-Bed, Catalysts, 2020, 10(1), 43–55 CrossRef CAS.
- W. Suprun, R. Gläser and H. Papp, Reaction Pathways for Catalytic Gas-Phase Oxidation of Glycerol over Mixed Metal Oxides, in Catalysis- Innovative Applications in Petrochemistry and Refining, 2011 Search PubMed.
- X. Wang, F. Zhao and L. Huang, Low Temperature Dehydration of Glycerol to Acrolein in Vapor Phase with Hydrogen as Dilution: From Catalyst Screening via TPSR to Real-Time Reaction in a Fixed-Bed, Catalysts, 2020, 10(1), 43–55 CrossRef CAS.
- Y. Liu, H. Tüysüz, C. J. Jia, M. Schwickardi, R. Rinaldi, A. H. Lu, W. Schmidt and F. Schüth, From Glycerol to Allyl Alcohol: Iron Oxide Catalyzed Dehydration and Consecutive Hydrogen Transfer, Chem. Commun., 2010, 46(8), 1238–1240 RSC.
- A. V. Mironenko and D. G. Vlachos, Conjugation-Driven “Reverse Mars-van Krevelen”-Type Radical Mechanism for Low-Temperature C-O Bond Activation, J. Am. Chem. Soc., 2016, 138(26), 8104–8113 CrossRef CAS PubMed.
- P. Mars and D. W. Van Krevelen, Oxidations Carried out by Means of Vanadium Oxide Catalysts, Chem. Eng. Sci., 1954, 8, 41–59 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |